

Summary
Hundreds of loci in human genomes have alleles that are methylated differentially according to their parent of origin. These imprinted loci generally show little variation across tissues, individuals, and populations. We show that such loci can be used to distinguish the maternal and paternal homologs for all human autosomes without the need for the parental DNA. We integrate methylation-detecting nanopore sequencing with the long-range phase information in Strand-seq data to determine the parent of origin of chromosome-length haplotypes for both DNA sequence and DNA methylation in five trios with diverse genetic backgrounds. The parent of origin was correctly inferred for all autosomes with an average mismatch error rate of 0.31% for SNVs and 1.89% for insertions or deletions (indels). Because our method can determine whether an inherited disease allele originated from the mother or the father, we predict that it will improve the diagnosis and management of many genetic diseases.
Keywords: parent of origin, phasing, haplotype, DNA methylation, epigenetics, imprinting, DMR, Strand-seq, nanopore
Graphical abstract
Highlights
-
•
Genomic imprinting can help infer the parent of origin of homologs
-
•
Imprinted DNA methylation must be combined with chromosome-length haplotypes
-
•
The method was validated for five trios with diverse genetic backgrounds
-
•
In future, parent-of-origin phasing will improve cascade genetic testing
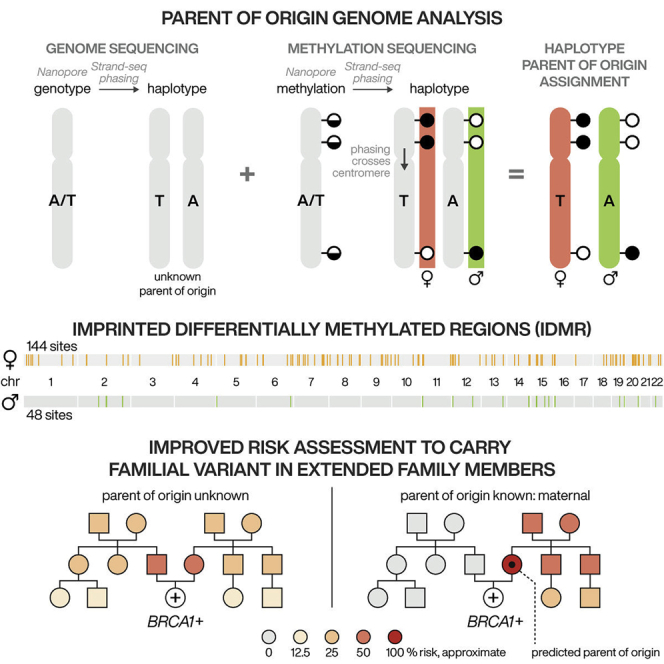
Akbari et al. present a method for identifying homologous chromosomes inherited from the mother or the father without using data from the parents. The method relies on phased DNA methylation at maternally and paternally imprinted loci, as well as chromosome-length phasing of DNA sequence. Testing on five trios showed that the method can correctly infer the parent of origin of all autosomes with a mismatch error rate of 0.31% for SNVs.
Introduction
Although phasing is conventionally defined as the task of distinguishing alleles from maternal and paternal homologs, in practice most current phasing methods neglect parental information entirely. Instead, they group alleles from each chromosome or subchromosomal phase block into two haplotypes (for diploids), which correspond to different homologous chromosomes but are not assigned a parent of origin (PofO). These haplotypes can only be assigned a PofO if trio information of some kind is available, for example by comparing previously discovered alleles for the mother or the father with the child’s alleles at heterozygous loci. In this sense, true phase information is largely out of reach for current genomic methods that do not incorporate sequence data from at least one parent next to the child.1,2,3,4
A striking exception to this paradigm is the parental information provided by consistent differences in DNA methylation between maternally and paternally inherited alleles at imprinted differentially methylated regions (iDMRs). This differential methylation is either established in gametes and escapes the epigenetic reprogramming that follows fertilization or it is established after fertilization,5,6 and it persists through adulthood. iDMRs reliably suppress the expression of either maternal or paternal alleles at nearby genes or gene clusters and, crucially, can be detected in cell lines or fresh samples using the unique ion current signature of 5-methyl-cytosine by nanopore sequencing (Oxford Nanopore Technologies).7,8,9,10 Long nanopore reads can be used to call both sequence variation and DNA methylation to detect genome-wide allele-specific methylation.9,10 Despite the fact that phasing using nanopore reads can achieve megabase-scale phase blocks, full chromosome haplotypes cannot be obtained, and each chromosome is represented in several phase blocks with likely switches between the paternal and maternal origin of the blocks along the chromosome.9
Conversely, some phasing techniques lack parental information but produce phase blocks that span centromeres, repetitive regions, and runs of homozygosity.11,12 Single-cell template strand sequencing (Strand-seq) is a library preparation method that captures parental DNA template strands in daughter cells cultured for one DNA replication round in the presence of BrdU.13 Reads from Watson template strands map to the reference genome in the minus orientation and reads from Crick template strands map in the plus orientation, meaning that alleles covered by reads with different orientations belong to different homologs. This approach enables the construction of global, chromosome-length haplotypes.11 Because Strand-seq phase blocks are generally sparse (i.e., they do not phase all single-nucleotide variants [SNVs]), Strand-seq often serves as a scaffold upon which reads or subchromosomal phase blocks from other sequencing techniques are combined, effectively phasing them relative to each other.14,15,16,17
Determining a PofO for germline variants can aid in clinical genetics through variant curation, the efficient screening of relatives for genetic disease, and is essential to evaluate disease risk when a pathogenic variant has PofO effects, that is, when a patient’s risk of disease depends on from which parent it is inherited (e.g., hereditary paraganglioma-pheochromocytoma syndrome due to pathogenic variants in SDHD or SDHAF2).18,19,20,21,22 Cascade genetic testing is used for pathogenic variants associated with diseases such as hereditary cancers with the goal of preventing or catching cancers early in family members.23 In the absence of PofO information due to parents being unavailable, deceased, or declining genetic testing, cascade genetic testing must be offered to both sides of the family until segregation is confirmed. This may be costly and burdensome to patients and families, exacerbating already low rates of uptake of cascade genetic testing.24,25 Eliminating the need to test one side of the family is a clear benefit and a major clinical utility of defining a PofO for pathogenic variants, and more broadly, establishing chromosome-length haplotypes with accurate parental segregation of genomic variation has widespread applications.
We report that alleles along the full length of each human autosome can be assigned to the maternal or paternal homolog when nanopore methylation and iDMRs are integrated with Strand-seq chromosome-length haplotypes (Figure 1). This method does not require parental sequence data (trio information) or SNV linkage analysis but instead relies on the fact that all human autosomes have at least one iDMR of known parental origin. The only input required is a sample of fresh whole blood or other viable cells that can be cultured. We validated PofO assignment for heterozygous SNVs and insertions or deletions (indels) against five gold-standard trios from the Genome in a Bottle Consortium (GIAB), the Human Genome Structural Variation Consortium (HGSVC), and the 1000 Genomes Project (1KGP).16,26,27 By tracing pathogenic variants through families with sequencing efforts directed toward select family members, our method has the potential to transform cascade genetic testing and improve screening for genetic disease.
Figure 1.

Overview of the parent-of-origin (PofO) phasing method
(A) The inputs for the workflow are nanopore long reads and data from single-cell Strand-seq libraries. Nanopore data are used to call variants, some of which are phased with Strand-seq in an inversion-aware manner. These phased variants are then used to phase the nanopore reads, which are used to phase more variants and DNA methylation. Finally, the DNA methylation status of iDMRs is used to identify the PofO for each homologous chromosome.
(B) Without examining DNA methylation, Strand-seq and nanopore reads can be combined to construct chromosome-length haplotypes,14,16 but the assignment of each homolog (i.e., chromosome-length haplotype) to haplotype 1 or haplotype 2 (HP1 or HP2) is random with respect to its PofO, as shown by this cartoon. However, iDMRs can be used to distinguish maternal and paternal homologs. Lollipops mark the locations of all 144 maternal iDMRs used in this study (methylated on the maternal homolog) and all 48 paternal iDMRs.
For iDMR names and locations shown relative to cytobands, see Figure S7.
Results
Nanopore and Strand-seq enable chromosome-scale haplotyping
We used five human genomes to demonstrate our approach, including NA12878; HG002 and HG005 from GIAB; HG00733 from HGSVC; and NA19240 from 1KGP.16,26,27 For all the samples, we used nanopore sequencing data at 24–38× depth of coverage (Figure S1) and 42–220 Strand-seq libraries with 2.78–9.46× combined depth of coverage per sample (Figure S2). Nanopore raw signals were base called and mapped to the human reference genome GRCh38. SNVs and indels (“variants”) were called from nanopore reads using Clair328 (Figure S3). The SNV callsets for each individual included nearly all SNVs in the five corresponding ground truth callsets (mean [M]: 97.98%, standard deviation [SD]: 1.67%, range: 95.51%–99.64%; Table 1), while fewer indels were recovered (M: 64.01%, SD: 8.43%, range: 52.69%–78.18%). For both SNVs and indels, we recovered the greatest proportion of ground-truth variants in the individual with the greatest nanopore coverage, while we recovered the smallest proportion in the individual with the least coverage. This suggests that including more nanopore data would be one way to address the high false-negative rate for indels.
Table 1.
Phasing of heterozygous variants and comparison with the ground-truth callset
| Heterozygous SNVs | HG002 | HG005 | HG00733 | NA12878 | NA19240 |
|---|---|---|---|---|---|
| Total in ground-truth callset | 2,117,525 | 1,922,666 | 2,168,512 | 2,027,097 | 2,787,148 |
| Common between nanopore and ground truth | 2,100,326 | 1,915,821 | 2,071,156 | 2,009,263 | 2,688,200 |
| Strand-seq switch rate | 0.0202 | 0.0078 | 0.0087 | 0.002 | 0.0055 |
| Strand-seq switch/flip rate | 0.0112 | 0.0044 | 0.0049 | 0.0012 | 0.003 |
| Strand-seq mismatch rate | 0.0136 | 0.006 | 0.0067 | 0.0014 | 0.0048 |
| Strand-seq number of correctly phased | 1,496,173 | 1,516,727 | 1,255,486 | 1,906,619 | 1,903,540 |
| Strand-seq number of incorrectly phased | 20,560 | 9,155 | 8,457 | 2,730 | 9,239 |
| Combined Strand-seq and nanopore switch rate | 0.0016 | 0.0011 | 0.0027 | 0.0008 | 0.0029 |
| Combined Strand-seq and nanopore switch/flip rate | 0.001 | 0.0007 | 0.0016 | 0.0005 | 0.0016 |
| Combined Strand-seq and nanopore mismatch rate | 0.0054 | 0.0024 | 0.0034 | 0.0007 | 0.0035 |
| Combined Strand-seq and nanopore number of correctly phased | 2,076,204 | 1,903,642 | 2,061,700 | 2,001,412 | 2,676,235 |
| Combined Strand-seq and nanopore number of incorrectly phased | 11,256 | 4,634 | 7,017 | 1,489 | 9,414 |
| Heterozygous indels | HG002 | HG005 | HG00733 | NA12878 | NA19240 |
|---|---|---|---|---|---|
| Total in ground-truth callset | 326,220 | 250,169 | 286,492 | 292,829 | 335,801 |
| Common between nanopore and ground truth | 215,614 | 195,590 | 150,941 | 186,625 | 199,359 |
| Combined Strand-seq and nanopore switch rate | 0.0409 | 0.0397 | 0.0237 | 0.0334 | 0.0323 |
| Combined Strand-seq and nanopore switch/flip rate | 0.0213 | 0.0206 | 0.0124 | 0.0172 | 0.0167 |
| Combined Strand-seq and nanopore mismatch rate | 0.0243 | 0.0218 | 0.0133 | 0.0174 | 0.0177 |
| Combined Strand-seq and nanopore number of correctly phased | 202,815 | 184,615 | 147,100 | 178,390 | 193,356 |
| Combined Strand-seq and nanopore number of incorrectly phased | 5,061 | 4,106 | 1,986 | 3,161 | 3,477 |
We include only biallelic heterozygous variants in the ground-truth callset (0/1, 1/0, 0|1, or 1|0) for consistency with the phasing error rate calculations. See also Table S1.
Before iDMRs can be used to assign a PofO to homologs, chromosome-length haplotypes must be constructed. This is because iDMRs cover only a small fraction of the autosomal human genome (estimated in this study to be 0.014%) and are not necessarily phased relative to variants. While nanopore reads alone can be used to phase nearly all called variants for each sample, the resulting phase blocks are relatively short (N50 M ± SD = 4.85 ± 3.66 Mb), do not span full chromosomes, and do not all contain iDMRs (Figures S4 and S5). We therefore applied inversion-aware Strand-seq phasing to the nanopore SNVs first and constructed sparse, chromosome-length haplotypes. Strand-seq phased 60.62–94.89% of the common heterozygous SNVs between the ground-truth and nanopore callsets with 0.14%–1.36% mismatch error rates (number of incorrectly phased variants/number of all phased variants), with each chromosome spanned by a single phase block (Tables 1, S1, and S2; Figures S4 and S6). Strand-seq-phased SNVs were then used to phase nanopore reads (fraction of reads with at least MAPQ 20 that were successfully phased, M ± SD: 71% ± 9.6%), which were, in turn, used to re-phase all variants and achieve dense, chromosome-scale haplotypes containing nearly all heterozygous SNVs and most indels (Tables 1 and S1). Combining Strand-seq and nanopore in this way allowed us to phase 99.39%–99.91% of the heterozygous SNVs and 96.41%–98.77% of the heterozygous indels that were present in both the ground-truth and nanopore callsets, with mismatch error rates 0.07%–0.54% for SNVs and 1.33%–2.43% for indels (Tables 1 and S1). Our phasing approach used the GRCh38 reference genome, and we did not perform de novo genome assembly of any kind.
iDMRs assign PofO to haplotypes
PofO-specific DNA methylation at iDMRs provides a unique source of information to determine the PofO of homologs, represented by chromosome-length haplotypes, without using parental sequence data. We assembled a list of 192 iDMRs from previous genome-wide studies6,29,30,31,32 (see STAR Methods; Figure S7; Table S3). Chromosome X was ignored as it is not generally thought to have iDMRs. We combined DNA methylation information from phased nanopore reads with the known PofO information at the imprinted intervals to assign the PofO to each homolog (see STAR Methods). On average, 5.7 iDMRs (median: 5, SD: 5.2, range: 1–29) were used for PofO assignment of each chromosome, and each chromosome was assigned to its parental origin with an average of 95.7% confidence score (median: 99%, SD: 7.2%, range: 57.5%–100%; see STAR Methods; Figures 2 and S8–S11; Table S4). On average, 7% of iDMRs conflicted with the majority PofO assignment. However, because iDMRs are weighted by the degree of differential methylation in each sample, conflicting iDMRs represented only 3% of the PofO contribution values (; see STAR Methods; Table S4).
Figure 2.

CpG methylation at paternal and maternal iDMRs used for PofO assignment in HG002
Maternally methylated iDMRs are red and upward and paternally methylated iDMRs are blue and downward. Bars represent the fraction of CpGs with methylation differences ≥0.35 (differential methylation) between haplotypes (HP1 – HP2 for haplotype 1, which is on the left side, and HP2 – HP1 for haplotype 2, which is on the right side) at each iDMR for each haplotype. The chromosomes themselves are colored according to Giemsa banding (white, gray, and black), with the centromeres in red and the stalks of acrocentric chromosomes in blue-gray.
See also Figures S8–S11.
We examined 220 autosomal homologs across five individuals in this study (5 individuals × 22 autosomes × 2 ploidy) and compared the inferred PofO with the trio-assigned PofO in the ground-truth-phased variant callsets. All 220 homologs were correctly assigned a PofO, that is, the chromosome-length haplotype was correctly identified as either maternal or paternal and had few phasing errors (chromosome-level mismatch error rates for SNVs, M ± SD: 0.34% ± 0.53%, range: 0.03%–4.86%; for indels, M ± SD: 1.93% ± 0.58%, range: 0.98%–5.35%; Figures 3 and S12; Table S1).
Figure 3.

Per-chromosome results for PofO assignment of heterozygous (het)-SNVs
PofO could be assigned to all homologs. The small fraction of variants with incorrect PofO are sporadic phasing errors in the Strand-seq or nanopore data or local clusters of phasing errors on chromosomes 8 and 9, in the case of HG002.
See also Figure S12.
Roughly half of the iDMRs used were reported in at least two previous studies, while the rest were reported in just one study and confirmed by partial methylation observed among 179 WGBS datasets from 119 blood and 60 tissue samples (see STAR Methods). One potential weakness of the single-study iDMRs is that 38 of them (19.8%) come from a previous study of 12 trios that included the same five trios examined here,32 and it is possible that some of these iDMRs might provide misleading or insufficient PofO information when examined in new individuals (i.e., if they are not truly imprinted). We tested the dependence of PofO phasing on the single-study iDMRs by re-running PofO assignment using only the 93 iDMRs found in at least two studies: 208 of 220 autosomal homologs were correctly assigned a PofO (94.5%), while chromosome 5 was not assigned a PofO for NA19240 and chromosome 12 was not assigned a PofO for any individual because it did not have an iDMR. This suggests that PofO phasing is not reliant on poorly characterized iDMRs, likely because all autosomes have at least three iDMRs (in the full set of 192), with the exception of chromosome 17, which has one, and chromosome 3, which has two. This redundancy helps maintain robust PofO assignment even when some putative iDMRs provide weak or conflicting parental information. At a few iDMRs in some samples, for example at TRPC3 on chromosome 4 in NA12878, we detected slight hypermethylation on the parental allele that is reported to be unmethylated: this might be due to inaccuracies in methylation calling or phasing of nanopore reads, or it could reflect random allelic methylation rather than imprinting. Such methylation discrepancies could conceivably assign the wrong PofO to homologs. In this instance, additional iDMRs on the same chromosome enabled correct PofO assignment, albeit with the lowest confidence score (57.5%).
For additional confirmation that PofO phasing extracts reliable parental information, we calculated Mendelian error rates between each child’s inferred parental haplotypes and ground-truth variant genotypes for their parents (Figures 4 and S13–S16; see STAR Methods). Local phasing errors are indicated by elevated Mendelian error rates at a large common inversion on chromosome 8 for HG002 (mismatch rate 99.86% for SNVs and 97.05% for indels inside the inversion at chr8: 8120810–12362538), which is the individual with the most phasing errors overall (SNV mismatch rate 0.54%; Table 1), as well as at the centromere for chromosome 9. The latter is in fact a single bin of 1,000 SNVs stretched across the centromere, which exaggerates its importance in Figure S13. Globally, Mendelian error rates for maternal-mother and paternal-father comparisons were low (M ± SD: 0.27% ± 2.69%; calculated for non-overlapping bins of 1,000 variants), while they were high for maternal-father and paternal-mother comparisons (representing misassigned PofO; M ± SD: 25.75% ± 14.14%). For maternal-mother and paternal-father comparisons, the highest mean error rate for any chromosome was 2.29% for chromosome 8 in HG002. This is less than one-eighth of the lowest mean error rate for any chromosome in maternal-father and paternal-mother comparisons (19.69% for chromosome 21 in NA12878), suggesting that the PofO assignment is correct for all chromosomes.
Figure 4.

Mendelian error rates show that PofO phasing correctly infers parental haplotypes
(A) The inferred maternal haplotype for HG005 (child) is compared with the ground-truth variant genotypes for HG007 (mother). The Mendelian error rate is low across all chromosomes.
(B) The inferred paternal haplotype for HG005 (child) is compared with the ground-truth variant genotypes for HG006 (father).
(C) The inferred maternal haplotype for HG005 (child) is compared with the ground-truth variant genotypes for HG006 (father). This is the expected pattern if a PofO is misassigned for all chromosomes.
(D) We artificially produced chromosomes and regions with incorrect PofO assignments in the comparison of HG005’s maternal haplotype with HG007. The lack of such regions is evidence that the PofO phasing method correctly distinguishes maternal and paternal homologs. We switched the maternal and paternal haplotypes for chromosomes 1, 2, and 7 to simulate erroneous iDMR inferences, and we created a large switch error on chromosome 15 by reducing the bin size in the BreakpointR step.33
See also Figures S13–S16.
Discussion
We show that homologous chromosomes, represented by chromosome-length haplotypes of SNVs and indels, can be assigned a PofO without using parental sequence data. Long nanopore reads provide DNA sequence information along with PofO information in the form of DNA methylation differences between maternal and paternal alleles at known iDMRs. Strand-seq libraries provide sparse global haplotype information that phases variants and nanopore reads to reconstruct individual homologs. The PofO of each homolog can then be determined based on the consensus of one or more embedded iDMRs (Figure 1).
PofO phasing has the potential to address immediate clinical needs in the diagnosis and management of genetic disease. These include improving variant curation and estimates of disease penetrance through co-segregation of variants to each side of the family with and without relevant disease phenotypes, determining which parent may have a risk for mosaicism in the context of a de novo variant, and establishing appropriate screening recommendations for pathogenic variants in genes with known PofO effects—as seen with SDHD and SDHAF2.18,19,20,21,22 Furthermore, PofO phasing provides a considerable advantage over current clinical testing in facilitating cascade genetic testing that allows opportunities for intervention in actionable genetic diseases.34 Contacting, counseling, and testing relatives is a significant logistical and financial burden to patients and healthcare systems, especially when considering adult-onset conditions, where testing of parents is frequently not possible. Cascade genetic testing may be hindered by limited intrafamily communication and fractured family structures and has low uptake in ethnic minority populations.25 PofO phasing stands to enable focused approaches to cascade genetic testing throughout families, bringing goals of optimal cascade genetic testing rates within reach.35 Of importance, the ability of PofO phasing to infer the pathogenic variant status of a patient’s parent with a high degree of certainty is likely to place an even greater emphasis on the duty to warn at-risk individuals of actionable genomic findings that may have been primarily or secondarily sought throughout the course of genetic testing. Similar issues are already familiar to clinical genetics in the setting of obligate carriers, but because this approach need only test a single person to reconstruct the complete genomic contribution from each parent, there will be ethical considerations if PofO phasing is integrated into mainstream clinical genetic testing due to the unprecedented scale.
Other sequencing technologies could perhaps provide the DNA methylation, DNA sequence, and long-range phase information required for PofO phasing, or different methods could be used to combine them. For instance, PofO could be assigned to de novo trio-free diploid assemblies15,36 rather than chromosome-length haplotypes of small variants. SMRT sequencing (Pacific Biosciences), which now provides accurate DNA methylation as well as DNA sequence,37 might be a substitute for nanopore data that provides better indel detection, and long-range phasing with Hi-C36 could perhaps be used instead of Strand-seq, although if phase switches occur at centromeres,16 then chromosome arms that lack iDMRs (16q, 17q, 18p, and 20p) may not be assigned to a PofO.
Sequencing costs for PofO phasing are relatively low, with as little as 24× nanopore and 3× Strand-seq coverage used in this study. The DNA methylation information that underlies PofO assignment is robust and can easily be extracted from nanopore sequence data, while formerly rare Strand-seq libraries can now be produced in large numbers (>1,000) at a reduced cost.38 In principle, genomic regions that are identical by descent in distant relatives could also be leveraged to partially assign PofOs with large SNV datasets, using either the sex chromosomes or the ethnicity of the parents, but such bioinformatic approaches would require that parents differ substantially in genetic background and would be subject to well-known ethnic biases in genomic datasets.39 Given the simplicity and accuracy of PofO phasing, the lack of trio-free alternatives at present for extracting PofO information from genomic data, and the method’s remarkable clinical applications, PofO phasing has the potential to become a routine component of genomic analysis.
Limitations of the study
In addition to the possibility that some putative iDMRs may not in fact be imprinted (see results), true iDMRs may display biological variability that could prevent PofO assignment for some chromosomes in new individuals or in other tissues or cell types that have fewer or different somatic iDMRs than the cell lines used in this study. Even though the paternal or maternal origin of methylation at iDMRs is consistent whenever just one allele is methylated, imprinted methylation can be variable in the sense that the two parental alleles may have similar amounts of DNA methylation in some tissues and individuals.6,40 In rare cases, epimutation or genomic imprinting disorders may also alter DNA methylation at iDMRs.41 Although redundant iDMRs (see results) should generally allow PofO assignment even in the presence of such limited inter-individual and inter-tissue variability, ultimately, our method must be tested on additional trios from diverse genetic populations to determine which chromosomes are troublesome for PofO phasing. Advances in characterizing human DNA methylation may further improve PofO phasing by identifying additional iDMRs on iDMR-poor chromosomes (e.g., chromosome 17), by removing spurious iDMRs, and perhaps even by enabling PofO assignment for the X chromosome in females.42
Even when a homolog is assigned the correct PofO overall, local phasing errors can cause incorrect PofO assignment for some variants. The chromosome-length haplotypes constructed in this study are highly accurate, however, with mean mismatch error rates of 0.31% for SNVs and 1.89% for indels. Although we identified only 64.01% of the indels in the ground-truth dataset, this reflects a limitation of current nanopore technology and would be straightforward to improve with the addition of short Illumina reads.28,43 We observed rare switch errors for SNVs and indels primarily at centromeres and at inversions (e.g., an inversion on chromosomes 8 in HG002 caused the largest mismatch error rates; Figures 3 and S13; Table S1), but these generally contain few variants. Phasing errors at centromeres are likely due to misaligned reads in repetitive sequences, while errors at inversions are due to changes in sequence orientation that disrupt the directional information Strand-seq exploits for phasing.11 Inversion-related phasing errors can be partially addressed with a new StrandPhaseR function that re-phases variants inside known inversions.44 This is essential when iDMRs fall inside inversions, where they may support the wrong PofO if phasing is not corrected (e.g., iDMRs RIMBP3 and CDRT15P6) or when genes of interest fall inside inversions (e.g., PMS2 in inversion chr7: 5850673–6795880).
DNA-methylation-based (canonical) imprinting has been described in all placental mammals, and genomic maps of iDMRs have been established for a number of species, notably mice and primates.10,45,46,47 Although our approach can potentially be expanded to other organisms, it would be limited to chromosomes with known iDMRs (e.g., not chromosomes 4, 5, 13, 14, 16, and 19 in mice).10 This would primarily require adjusting cell culture and flow cytometry conditions for Strand-seq library preparation to suit non-human cells.48
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| Ligation sequencing kit | Oxford Nanopore Technologies | Cat. #: SQK-LSK109 |
| PromethION R9.4.1 pore flow cell | Oxford Nanopore Technologies | Cat. #: FLO-PRO002 |
| Deposited data | ||
| HG002 nanopore FAST5 files | This study | SRA: SRP395905 |
| HG005 and HG00733 nanopore FAST5 files |
The Human Pangenome Reference Consortium |
s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=NHGRI_UCSC_panel/ |
| NA12878 nanopore FAST5 files | Ref. Jain et al.,33 | nanopore-wgs-consortium NA12878 rel6 https://github.com/nanopore-wgs-consortium/NA12878 |
| NA19240 nanopore FAST5 files | Ref. De Coster et al.,49 | ENA: PRJEB26791 |
| HG002 and HG005 Strand-seq FASTQ files |
The Genome in a Bottle Consortium |
ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/ |
| HG00733 and NA19240 Strand-seq FASTQ files |
The Human Genome Structural Variation Consortium |
www.internationalgenome.org/ |
| NA12878 Strand-seq FASTQ files | Ref. Hanlon et al.,38 | SRA: SRP326369; BioProject: PRJNA742746 |
| PofO phased VCF files | This study | https://doi.org/10.5281/zenodo.7052113 |
| Experimental models: Cell lines | ||
| Human: EBV-transformed B cells for GM24385 Coriell RRID: CVCL_7526 |
Coriell | CVCL_1C78 |
| Software and algorithms | ||
| Clair3 v0.1-r10 | Ref. Zheng et al.,28 | N/A |
| f5c v0.6 | Ref. Gamaarachchi et al.,50 | N/A |
| Nanopolish v0.13.3 | Ref. Simpson et al.,8 | N/A |
| NanoMethPhase v1.0 | Ref. Akbari et al.,9 | https://doi.org/10.5281/zenodo.4474430 |
| ASHLEYS QC | Ref. Eimer et al.,51 | N/A |
| BreakpointR | Ref. Porubsky et al.,52 | GitHub: daewoooo/breakpointR commit 58cce0b09d0104089 2b3f6abf0b11caeb403d3f5 |
| StrandPhaseR | Ref. Porubsky et al.,14 | GitHub: daewoooo/StrandPhaseR commit bb19557235de3d82092 abdc11b3334f615525b5b |
| InvertypeR | Ref. Hanlon et al.,53 | N/A |
| correctInvertedRegionPhasing | Ref. Porubsky et al.,44 | N/A |
| PatMat v1.1.1 | This study | https://doi.org/10.5281/zenodo.7308808; GitHub: https://github.com/vahidAK/PatMat |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Steven J.M. Jones (sjones@bcgsc.ca).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
NA12878: female, age unknown. HG002: male, 45 years. HG005: male, 33 years. HG00733: female, age unknown. NA19240: female, age unknown. This study involved five trios, and PofO was assigned for the five children. HG002 cells were purchased from Coriell for nanopore sequencing. They were cultured in RPMI 1640 medium supplemented with 15% FBS (not heat inactivated) at 37°C with 5% CO2 at a density of 106 cells/mL with a split ratio of 1:4. The cell line was not authenticated, except insofar as HG002’s small variant haplotypes match those of his parents (Figure S13).
Method details
Nanopore sequencing and data
We sequenced native DNA from an Ashkenazi son (GM24385, or HG002) at 33-fold coverage. We first extracted high molecular weight genomic DNA from an EBV-transformed B-lymphocyte cell line and sheared it to an average size of 6–20 kb using g-TUBE shearing (Covaris). We then repaired potential nicks using the NEBNext FFPE DNA Repair kit (PreCR treatment) and performed end-repair and A-tailing using the NEBNext Ultra II kit. We ligated to the Oxford Nanopore Technologies adapters using NEBNext Quick Ligase, and we removed small library fragments using a 0.4:1 bead:library ratio. We sequenced the library on an Oxford Nanopore Technologies PromethION 24 instrument using an R9.4.1 pore flow cell and software version 19.06.9 (MinKNOW GUI v4.0.23). A nuclease flush was performed after 48 h of sequencing and prior to library re-loading for a further 24 h.
In addition to HG002, we used public nanopore data for HG005, HG00733, NA12878 and NA19240 (Figure S1). Raw nanopore fast5 files for HG005 and HG00733 were downloaded from the Human Pangenome Reference Consortium54 (https://github.com/human-pangenomics); NA12878 was obtained from Jain et al. 201833; and NA19240 from De Coster et al. 2019.49 For HG002, HG005 and NA12878, ground truth phased variants were obtained through GIAB v4.2.1 (hifiasm_v11_phasetransfer), and for NA19240 and HG00733 phased variants were obtained from 1KGP shapeit2 v2a (shapeit2_integrated_snvindels_v2a_related_samples).26,27
Nanopore data analysis
Base calling and mapping
Nanopore signal-level data were basecalled using Oxford Nanopore Technologies guppy basecaller v6.0.1 and the super accuracy model (dna_r9.4.1_450bps_sup) with default settings. Basecalled nanopore reads were mapped to the human reference genome (GRCh38; with the EBV genome but no ALT contigs) using minimap2 v2.24 with the -ax map-ont --MD -L options selected.55
Variant calling
Upon alignment, Clair3 v0.1-r10 with trained model r941_prom_sup_g5014 and default settings was used to call variants from nanopore alignment data.28 High quality variant calls (marked as “PASS” by the software) from Clair3 were then used for Strand-seq phasing (see the next section).
Methylation calling
To call DNA methylation and obtain per-read CpG methylation information from nanopore data, we first indexed fastq file using f5c v0.650 and then called methylation using nanopolish v0.13.3 with default settings8. Per-read methylation call data were then preprocessed using NanoMethPhase v1.0 methyl_call_processor module with --callThreshold 1.5 for downstream analysis and PofO phasing.9,56
Strand-seq phasing and inversion correction
We obtained 45 public Strand-seq libraries for HG005 and 66 for HG002 from GIAB26,57 (ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/), and we obtained 230 libraries for HG00733 and 234 libraries for NA19240 from HGSVC.16 We used the 96 high-depth OP-Strand-seq libraries for NA12878 described previously (clusters 5 and 6).38
We trimmed adapters from paired-end FASTQ files and removed short reads (<30 bp) and low-quality bases (<15) with Cutadapt.58 We used Bowtie2 to align reads to the GRCh38 human reference genome and discarded reads that had MAPQ less than 10 or that did not map to chromosomes 10–22, X, and Y.59 We used Picard to mark duplicate reads (github.com/broadinstitute/picard/) and then ran ASHLEYS QC with default settings and window sizes 5000000, 2000000, 1000000, 800000, 600000, 400000, and 200000 to discard libraries with a Strand-seq quality score below 0.5.51
We ran BreakpointR (commit 58cce0b09d01040892b3f6abf0b11caeb403d3f5 of github.com/daewoooo/breakpointR) with background set to 0.1, chr set to the autosomes, and maskRegions set to a previously described blacklist.52,53 We used 8 Mb bins because we found they linked phasing across difficult regions such as inversions more readily and prevented large switch errors (Figure 4D). We used the function exportRegions with default settings to identify regions of the genome with both Watson and Crick reads that are suitable for phasing. We phased biallelic heterozygous SNVs called from the nanopore data for each sample using StrandPhaseR with num.iterations set to 3, with splitPhasedReads and assume.biallelic set to TRUE, with R v4.0.5, and with v1.0.1 or higher of the dependency rlang (commit bb19557235de3d82092abdc11b3334f615525b5b of the devel branch of github.com/daewoooo/StrandPhaseR).14
Inversions disrupt Strand-seq’s directional phase information. We called inversions for each sample using the R package InvertypeR (commit a5fac3b6b8264db28de1a997ad0bc062badea883 of github.com/vincent-hanlon/InvertypeR/commits/main).53 In brief, we used the nanopore SNVs to create a pair of composite files for each sample, with the addition of the genomic coordinates chr8: 8231088–12039415 in the blacklist to ensure that the common large inversion at those coordinates was correctly represented. We genotyped a catalog of published inversion coordinates with adjust_method set to ‘all’ and with priors as previously described, as well as a list of de novo sample-specific strand switches identified by running BreakpointR three times on the composite files with different bin sizes.52,53 For the latter, we used prior probabilities of 0.9, 0.05, and 0.05 for reference, heterozygous, and homozygous genotypes, respectively. We combined inversions with posterior probabilities above 0.95 from the two callsets by discarding any inversions from the catalog callset that intersected the de novo callset (bedtools intersect -v -r -f 0.1). We did not remove misoriented reference contigs, which appear as homozygous inversions in all samples, because they disrupt phasing in the same way that inversions do.
The function correctInvertedRegionPhasing in the StrandPhaseR package switches the phase of heterozygous SNVs within homozygous inversions and re-phases SNVs within heterozygous inversions.44 We used sample-specific inversion calls larger than 10 kb along with the nanopore sample-specific SNV positions, recall.phased and assume.biallelic set to TRUE, het.genotype set to ‘lenient’, lookup.bp set to 1000000, background set to 0.1, and lookup.blacklist set to the blacklist above. The resulting chromosome-length inversion-corrected SNV haplotypes were used to phase nanopore reads relative to each other.
iDMR selection
We gathered the list of previously reported iDMRs from five prior genome-wide studies.6,29,30,31,32 iDMRs with overlap between two or more studies were merged into the largest total interval. Moreover, iDMRs within 1 kb and with the same PofO were merged. This resulted in 93 iDMRs reported in at least two studies and 306 iDMRs reported in only a single study and supported by at least two probes if they came from array studies. We previously surveyed imprinted methylation genome-wide using 12 nanopore-sequenced cell lines with their trio sequencing information from 1KGP.32 We used the same cell lines to examine the 306 iDMRs from a single study, above. At each allele for each CpG site with coverage >4× within the iDMRs, methylation frequency (the fraction of reads methylated at a CpG) was calculated. We then calculated the difference between average methylation frequencies for the paternal and maternal alleles for each iDMR in each cell line. Ninety-four iDMRs with between alleles and with conflicting PofO between any of the 12 cell lines and the corresponding prior study were excluded. To further validate the 212 remaining iDMRs reported in a single study, we used WGBS datasets for 119 blood samples from 87 individuals in the Blueprint consortium and 60 tissue samples for 29 tissue types in ENCODE and the RoadMap consortium60,61,62 (Tables S5 and S6). At iDMRs only one allele is methylated, therefore, the aggregated methylation frequency from both alleles at these regions is expected to be ∼50% (partial methylation). Thus, we examined partial methylation at the 212 iDMRs in the WGBS datasets. For each WGBS sample, we used CpGs with at least five mapped WGBS reads and at each iDMR we counted the number of CpGs with partial methylation (methylation frequency between 0.35 and 0.65 among mapped reads). An iDMR with 0.35–0.65 methylation is then considered partially methylated if it had at least five CpGs in the WGBS sample and more than 60% of the CpGs showed partial methylation (see similar requirements used elsewhere32). We only included iDMRs that were partially methylated in at least two and 5% of individuals or tissues in which the iDMR could be examined (i.e., the iDMR had at least five CpGs with a coverage of ≥5). Out of the 212 iDMRs, 113 iDMRs were excluded because they did not meet the inclusion criteria, suggesting that they rarely provide parental information. Unlike true iDMRs, randomly selected loci are unlikely to meet these criteria. To test this, we randomly selected 100 genomic intervals with at least 15 CpGs and lengths of 1, 2, or 3 kb and repeated the experiment 100 times. On average, 2.4% of intervals met the inclusion criteria. Overall, we gathered a list of 192 known iDMRs of which 93 were reported in multiple studies and 99 were reported in a single study (Table S3).
Chromosome-scale haplotypes and PofO detection
We then integrated several steps to detect chromosome-scale haplotypes with their PofO.
-
1.
Strand-seq phasing provides sparse chromosome-scale haplotypes. Phased SNVs from Strand-seq were used to phase nanopore reads to either HP1 or HP2 haplotypes. We used a minimum mapping quality of 20 and base quality of 7 to tag each read with the phased SNVs. We used somewhat similar criteria as NanoMethPhase9 to assign reads to haplotypes: we tag a read as HP1 if # HP1 SNVs > # HP2 SNVs in the read, the read has at least one phased SNV from HP1, and , and vice versa.
-
2.
Phased nanopore reads from step 1 were then used to re-phase all the variants (SNVs and indels) to each haplotype. Variants assigned to the haplotype supported by more reads and at least two phased reads needed to support a variant to assign it as HP1 or HP2.
-
3.
Nanopore reads were then phased a second time using all the phased variants from step 2 with the conditions mentioned in step 1.
-
4.
Per-read methylation information for each nanopore read at known iDMRs were extracted and integrated to its phase information from step 3. This enabled us to phase each CpG methylation in each read to either HP1 or HP2 and calculate the methylation frequency () at each CpG site for each haplotype. Methylation frequencies were then used to assign haplotypes to their PofO for each sample as follows:
At each of the 192 known iDMRs we considered CpGs detected in both haplotypes and counted CpGs with ≥0.35 difference in methylation frequency between haplotypes (differential methylation). Tuning this parameter by trial and error allowed us to identify informative CpGs while avoiding erroneous differential methylation. We then calculated the contribution/detection value of the iDMR to the PofO detection of each haplotype as follows:
where is the average methylation frequency for the haplotype, is the number of differentially methylated CpGs that support PofO for the haplotype, and is the number of all CpGs at the iDMR. For maternally methylated iDMRs, indicates the contribution value for HP1 as the maternal and HP2 as the paternal allele and indicates the contribution value for HP2 as the maternal and HP1 as the paternal allele, vice versa for paternally methylated iDMRs. The length of an iDMR can vary between individuals, and different studies report different start and end positions for the same iDMR. We wished to capture PofO information even when just a small part of an iDMR is imprinted in an individual, while avoiding inferences based on very few CpGs. Therefore, we only used iDMRs with comprising at least 10% of all detected CpGs and with more than 11 detected CpGs in total (i.e., and ). As an example, for a maternally methylated iDMR with 20 CpGs detected in both haplotypes and 0.8 average methylation frequency at HP1 and 0.3 at HP2, if 12 CpGs show ≥0.35 methylation in HP1 compared to HP2 and two CpGs show ≥0.35 methylation in HP2 compared to HP1, then:for HP1 as maternal and HP2 as paternal is , and for HP2 as maternal and HP1 as paternal is .
On each chromosome for each haplotype as being maternal or paternal, the value of will be (similarly for HP2):
Where is the number of iDMRs considered for the chromosome. If, for example, (i.e., ) is greater than (i.e., ) then HP1 is the maternal homolog and HP2 is the paternal homolog and we calculated the confidence score for PofO assignment of the chromosome as follows: -
5.
Finally, phased variants from step 2 were assigned to their PofO with the results from step 4.
All the steps are integrated into our workflow and tool, PatMat, and a tutorial is provided on GitHub (https://github.com/vahidAK/PatMat; Zenodo: https://doi.org/10.5281/zenodo.7308808).
Mendelian errors
To verify the PofO assignments, we calculated the frequency of one kind of Mendelian error between the PofO-assigned haplotypes and the genotypes of the parents. We obtained genotypes from GIAB for the parents of HG002 and HG005 (v4.2.1), from 1KGP for the parents of HG00733 and NA19240 (v2a), and from Byrska-Bishop et al. 2021 for the parents of NA12878.26,27,57,63 For each parent-child pair, we examined loci at which we found a phased heterozygous genotype for the child and either a heterozygous or homozygous alternate genotype for the parent. Where the child had a maternal reference allele and the mother was homozygous alternate, we called a Mendelian error (similarly for the child’s paternal allele and the father’s genotype). We did this for non-overlapping bins of 1000 variants and calculated the error rate as the number of such Mendelian errors divided by the number of variants examined. We plotted the resulting error rates on chromosomes using RIdeogram.64
Quantification and statistical analysis
PofO phasing was validated for 110 chromosomes from 5 individuals. No statistical tests were performed. However, we report non-probabilistic confidence scores that reflect the strength of differential methylation for each chromosome.
Acknowledgments
We thank Yanni Wang and Daniel Chan for help with making Strand-seq libraries. We are grateful for discussions related to inversion-aware phasing with David Porubsky. We thank Martin Krzywinski and Katherine Dixon for their work on the graphical abstract. V.A. acknowledges funding from the University of British Columbia Four Year Doctoral Fellowship. Work in the Lansdorp laboratory is funded by a Program Project Grant (#1074) from the Terry Fox Research Institute; a Project Grant (#PJT-159787) from the Canadian Institutes of Health Research; and a grant (#40044) from the Canadian Foundation for Innovation and the government of British Columbia. L.L. acknowledges funding from a Project Grant (#PJT-165992) from the Canadian Institutes of Health Research. K.A.S. acknowledges funding from the Canada Research Chairs program and Canadian Institutes of Health Research. S.J.M.J. acknowledges funding from the Canada Research Chairs program and the Canadian Foundation for Innovation.
Author contributions
Conceptualization and methodology, all authors; software, validation, visualization, and writing – original draft, V.A. and V.C.T.H.; writing – review & editing, all authors.
Declaration of interests
The authors declare no competing interests.
Published: December 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100233.
Contributor Information
Peter M. Lansdorp, Email: plansdor@bccrc.ca.
Steven J.M. Jones, Email: sjones@bcgsc.ca.
Supplemental information
related to “Nanopore and Strand-seq enable chromosome-scale haplotyping” and “Strand-seq phasing and inversion correction”
Inversions with low read density warnings are likely in repetitive regions at centromeres or telomeres. Note that only inversions on the autosomes and larger than 10 kb were used for phasing correction. Genotypes 0|1 and 1|0 are not PofO phased.
Transparent peer review records for Akbari et al.
Data and code availability
-
•
Nanopore data for HG002 have been deposited at the NCBI Sequence Read Archive and are publicly available as of the date of publication (SRA: SRP395905). Phased variant call files are also deposited at Zenodo (https://doi.org/10.5281/zenodo.7052113). The accession numbers are also listed in the key resources table.
-
•
All original code has been deposited at Zenodo (https://doi.org/10.5281/zenodo.7308808) and on GitHub (https://github.com/vahidAK/PatMat) and is publicly available. Tools and DOIs are also listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Zheng G.X.Y., Lau B.T., Schnall-Levin M., Jarosz M., Bell J.M., Hindson C.M., Kyriazopoulou-Panagiotopoulou S., Masquelier D.A., Merrill L., Terry J.M., et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 2016;34:303–311. doi: 10.1038/nbt.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wenger A.M., Peluso P., Rowell W.J., Chang P.-C., Hall R.J., Concepcion G.T., Ebler J., Fungtammasan A., Kolesnikov A., Olson N.D., et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019;37:1155–1162. doi: 10.1038/s41587-019-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng H., Jarvis E.D., Fedrigo O., Koepfli K.-P., Urban L., Gemmell N.J., Li H. Haplotype-resolved assembly of diploid genomes without parental data. Nat. Biotechnol. 2022;40:1332–1335. doi: 10.1038/s41587-022-01261-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jarvis E.D., Formenti G., Rhie A., Guarracino A., Yang C., Wood J., Tracey A., Thibaud-Nissen F., Vollger M.R., Porubsky D., et al. Automated assembly of high-quality diploid human reference genomes. bioRxiv. 2022 doi: 10.1101/2022.03.06.483034. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henckel A., Arnaud P. Genome-wide identification of new imprinted genes. Brief. Funct. Genomics. 2010;9:304–314. doi: 10.1093/bfgp/elq016. [DOI] [PubMed] [Google Scholar]
- 6.Zink F., Magnusdottir D.N., Magnusson O.T., Walker N.J., Morris T.J., Sigurdsson A., Halldorsson G.H., Gudjonsson S.A., Melsted P., Ingimundardottir H., et al. Insights into imprinting from parent-of-origin phased methylomes and transcriptomes. Nat. Genet. 2018;50:1542–1552. doi: 10.1038/s41588-018-0232-7. [DOI] [PubMed] [Google Scholar]
- 7.Laszlo A.H., Derrington I.M., Brinkerhoff H., Langford K.W., Nova I.C., Samson J.M., Bartlett J.J., Pavlenok M., Gundlach J.H. Detection and mapping of 5-methylcytosine and 5-hydroxymethylcytosine with nanopore MspA. Proc. Natl. Acad. Sci. USA. 2013;110:18904–18909. doi: 10.1073/pnas.1310240110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simpson J.T., Workman R.E., Zuzarte P.C., David M., Dursi L.J., Timp W. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods. 2017;14:407–410. doi: 10.1038/nmeth.4184. [DOI] [PubMed] [Google Scholar]
- 9.Akbari V., Garant J.-M., O’Neill K., Pandoh P., Moore R., Marra M.A., Hirst M., Jones S.J.M. Megabase-scale methylation phasing using nanopore long reads and NanoMethPhase. Genome Biol. 2021;22:68. doi: 10.1186/s13059-021-02283-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gigante S., Gouil Q., Lucattini A., Keniry A., Beck T., Tinning M., Gordon L., Woodruff C., Speed T.P., Blewitt M.E., et al. Using long-read sequencing to detect imprinted DNA methylation. Nucleic Acids Res. 2019;47:e46. doi: 10.1093/nar/gkz107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porubský D., Sanders A.D., van Wietmarschen N., Falconer E., Hills M., Spierings D.C.J., Bevova M.R., Guryev V., Lansdorp P.M. Direct chromosome-length haplotyping by single-cell sequencing. Genome Res. 2016;26:1565–1574. doi: 10.1101/gr.209841.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Selvaraj S., R Dixon J., Bansal V., Ren B. Whole-genome haplotype reconstruction using proximity-ligation and shotgun sequencing. Nat. Biotechnol. 2013;31:1111–1118. doi: 10.1038/nbt.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falconer E., Hills M., Naumann U., Poon S.S.S., Chavez E.A., Sanders A.D., Zhao Y., Hirst M., Lansdorp P.M. DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nat. Methods. 2012;9:1107–1112. doi: 10.1038/nmeth.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porubsky D., Garg S., Sanders A.D., Korbel J.O., Guryev V., Lansdorp P.M., Marschall T. Dense and accurate whole-chromosome haplotyping of individual genomes. Nat. Commun. 2017;8:1293. doi: 10.1038/s41467-017-01389-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porubsky D., Ebert P., Audano P.A., Vollger M.R., Harvey W.T., Marijon P., Ebler J., Munson K.M., Sorensen M., Sulovari A., et al. Fully phased human genome assembly without parental data using single-cell strand sequencing and long reads. Nat. Biotechnol. 2021;39:302–308. doi: 10.1038/s41587-020-0719-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaisson M.J.P., Sanders A.D., Zhao X., Malhotra A., Porubsky D., Rausch T., Gardner E.J., Rodriguez O.L., Guo L., Collins R.L., et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019;10:1784. doi: 10.1038/s41467-018-08148-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebert P., Audano P.A., Zhu Q., Rodriguez-Martin B., Porubsky D., Bonder M.J., Sulovari A., Ebler J., Zhou W., Serra Mari R., et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science. 2021;372 doi: 10.1126/science.abf7117. eabf7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Mey A.G., Maaswinkel-Mooy P.D., Cornelisse C.J., Schmidt P.H., van de Kamp J.J. Genomic imprinting in hereditary glomus tumours: evidence for new genetic theory. Lancet. 1989;2:1291–1294. doi: 10.1016/s0140-6736(89)91908-9. [DOI] [PubMed] [Google Scholar]
- 19.Knowles J.W., Rader D.J., Khoury M.J. Cascade screening for familial hypercholesterolemia and the use of genetic testing. JAMA. 2017;318:381–382. doi: 10.1001/jama.2017.8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hensen E.F., Jordanova E.S., van Minderhout I.J.H.M., Hogendoorn P.C.W., Taschner P.E.M., van der Mey A.G.L., Devilee P., Cornelisse C.J. Somatic loss of maternal chromosome 11 causes parent-of-origin-dependent inheritance in SDHD-linked paraganglioma and phaeochromocytoma families. Oncogene. 2004;23:4076–4083. doi: 10.1038/sj.onc.1207591. [DOI] [PubMed] [Google Scholar]
- 22.Hao H.X., Khalimonchuk O., Schraders M., Dephoure N., Bayley J.P., Kunst H., Devilee P., Cremers C.W.R.J., Schiffman J.D., Bentz B.G., et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139–1142. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hampel H. Genetic counseling and cascade genetic testing in Lynch syndrome. Fam. Cancer. 2016;15:423–427. doi: 10.1007/s10689-016-9893-5. [DOI] [PubMed] [Google Scholar]
- 24.Lee P.W.C., Bedard A.C., Samimi S., Beard V.K., Hong Q., Bedard J.E.J., Gilks B., Schaeffer D.F., Wolber R., Kwon J.S., et al. Evaluating the impact of universal Lynch syndrome screening in a publicly funded healthcare system. Cancer Med. 2020;9:6507–6514. doi: 10.1002/cam4.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Braley E.F., Bedard A.C., Nuk J., Hong Q., Bedard J.E.J., Sun S., Schrader K.A. Patient ethnicity and cascade genetic testing: a descriptive study of a publicly funded hereditary cancer program. Fam. Cancer. 2022;21:369–374. doi: 10.1007/s10689-021-00270-0. [DOI] [PubMed] [Google Scholar]
- 26.Zook J.M., McDaniel J., Olson N.D., Wagner J., Parikh H., Heaton H., Irvine S.A., Trigg L., Truty R., McLean C.Y., et al. An open resource for accurately benchmarking small variant and reference calls. Nat. Biotechnol. 2019;37:561–566. doi: 10.1038/s41587-019-0074-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.1000 Genomes Project Consortium. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng Z., Li S., Su J., Leung A.W.-S., Lam T.-W., Luo R. Symphonizing pileup and full-alignment for deep learning-based long-read variant calling. bioRxiv. 2021 doi: 10.1101/2021.12.29.474431. Preprint at. [DOI] [PubMed] [Google Scholar]
- 29.Court F., Tayama C., Romanelli V., Martin-Trujillo A., Iglesias-Platas I., Okamura K., Sugahara N., Simón C., Moore H., Harness J.V., et al. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014;24:554–569. doi: 10.1101/gr.164913.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joshi R.S., Garg P., Zaitlen N., Lappalainen T., Watson C.T., Azam N., Ho D., Li X., Antonarakis S.E., Brunner H.G., et al. DNA methylation profiling of uniparental disomy subjects provides a map of parental epigenetic bias in the human genome. Am. J. Hum. Genet. 2016;99:555–566. doi: 10.1016/j.ajhg.2016.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hernandez Mora J.R., Tayama C., Sánchez-Delgado M., Monteagudo-Sánchez A., Hata K., Ogata T., Medrano J., Poo-Llanillo M.E., Simón C., Moran S., et al. Characterization of parent-of-origin methylation using the Illumina Infinium MethylationEPIC array platform. Epigenomics. 2018;10:941–954. doi: 10.2217/epi-2017-0172. [DOI] [PubMed] [Google Scholar]
- 32.Akbari V., Garant J.-M., O’Neill K., Pandoh P., Moore R., Marra M.A., Hirst M., Jones S.J.M. Genome-wide detection of imprinted differentially methylated regions using nanopore sequencing. Elife. 2022;11 doi: 10.7554/eLife.77898. e77898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jain M., Koren S., Miga K.H., Quick J., Rand A.C., Sasani T.A., Tyson J.R., Beggs A.D., Dilthey A.T., Fiddes I.T., et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018;36:338–345. doi: 10.1038/nbt.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller D.T., Lee K., Chung W.K., Gordon A.S., Herman G.E., Klein T.E., Stewart D.R., Amendola L.M., Adelman K., Bale S.J., et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG) Genet. Med. 2021;23:1381–1390. doi: 10.1038/s41436-021-01172-3. [DOI] [PubMed] [Google Scholar]
- 35.Offit K., Tkachuk K.A., Stadler Z.K., Walsh M.F., Diaz-Zabala H., Levin J.D., Steinsnyder Z., Ravichandran V., Sharaf R.N., Frey M.K., et al. Cascading after peridiagnostic cancer genetic testing: an alternative to population-based screening. J. Clin. Oncol. 2020;38:1398–1408. doi: 10.1200/JCO.19.02010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garg S., Fungtammasan A., Carroll A., Chou M., Schmitt A., Zhou X., Mac S., Peluso P., Hatas E., Ghurye J., et al. Chromosome-scale, haplotype-resolved assembly of human genomes. Nat. Biotechnol. 2021;39:309–312. doi: 10.1038/s41587-020-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tse O.Y.O., Jiang P., Cheng S.H., Peng W., Shang H., Wong J., Chan S.L., Poon L.C.Y., Leung T.Y., Chan K.C.A., et al. Genome-wide detection of cytosine methylation by single molecule real-time sequencing. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2019768118. e2019768118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanlon V.C.T., Chan D.D., Hamadeh Z., Wang Y., Mattsson C.-A., Spierings D.C.J., Coope R.J.N., Lansdorp P.M. Construction of Strand-seq libraries in open nanoliter arrays. Cell Rep. Methods. 2022;2:100150. doi: 10.1016/j.crmeth.2021.100150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ledford H. Cancer geneticists tackle troubling ethnic bias in studies. Nature. 2019;568:154–155. doi: 10.1038/d41586-019-01080-2. [DOI] [PubMed] [Google Scholar]
- 40.Prickett A.R., Oakey R.J. A survey of tissue-specific genomic imprinting in mammals. Mol. Genet. Genomics. 2012;287:621–630. doi: 10.1007/s00438-012-0708-6. [DOI] [PubMed] [Google Scholar]
- 41.Monk D., Mackay D.J.G., Eggermann T., Maher E.R., Riccio A. Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 2019;20:235–248. doi: 10.1038/s41576-018-0092-0. [DOI] [PubMed] [Google Scholar]
- 42.Jima D.D., Skaar D.A., Planchart A., Motsinger-Reif A., Cevik S.E., Park S.S., Cowley M., Wright F., House J., Liu A., et al. Genomic map of candidate human imprint control regions: the imprintome. Epigenetics. 2022;17:1920–1943. doi: 10.1080/15592294.2022.2091815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shafin K., Pesout T., Chang P.-C., Nattestad M., Kolesnikov A., Goel S., Baid G., Kolmogorov M., Eizenga J.M., Miga K.H., et al. Haplotype-aware variant calling with PEPPER-Margin-DeepVariant enables high accuracy in nanopore long-reads. Nat. Methods. 2021;18:1322–1332. doi: 10.1038/s41592-021-01299-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Porubsky D., Höps W., Ashraf H., Hsieh P., Rodriguez-Martin B., Yilmaz F., Ebler J., Hallast P., Maria Maggiolini F.A., Harvey W.T., et al. Recurrent inversion polymorphisms in humans associate with genetic instability and genomic disorders. Cell. 2022;185:1986–2005.e26. doi: 10.1016/j.cell.2022.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Renfree M.B., Hore T.A., Shaw G., Graves J.A.M., Pask A.J. Evolution of genomic imprinting: insights from marsupials and monotremes. Annu. Rev. Genomics Hum. Genet. 2009;10:241–262. doi: 10.1146/annurev-genom-082908-150026. [DOI] [PubMed] [Google Scholar]
- 46.Cheong C.Y., Chng K., Ng S., Chew S.B., Chan L., Ferguson-Smith A.C. Germline and somatic imprinting in the nonhuman primate highlights species differences in oocyte methylation. Genome Res. 2015;25:611–623. doi: 10.1101/gr.183301.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie W., Barr C.L., Kim A., Yue F., Lee A.Y., Eubanks J., Dempster E.L., Ren B. Base-Resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012;148:816–831. doi: 10.1016/j.cell.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hills M., Falconer E., O’Neill K., Sanders A.D., Howe K., Guryev V., Lansdorp P.M. Construction of whole genomes from scaffolds using single cell strand-seq data. Int. J. Mol. Sci. 2021;22:3617. doi: 10.3390/ijms22073617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Coster W., De Rijk P., De Roeck A., De Pooter T., D’Hert S., Strazisar M., Sleegers K., Van Broeckhoven C. Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome. Genome Res. 2019;29:1178–1187. doi: 10.1101/gr.244939.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gamaarachchi H., Lam C.W., Jayatilaka G., Samarakoon H., Simpson J.T., Smith M.A., Parameswaran S. GPU accelerated adaptive banded event alignment for rapid comparative nanopore signal analysis. BMC Bioinf. 2020;21:343. doi: 10.1186/s12859-020-03697-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eimer C., Sanders A.D., Korbel J.O., Marschall T., Ebert P. ASHLEYS: automated quality control for single-cell Strand-seq data. Bioinformatics. 2021;37:3356–3357. doi: 10.1093/bioinformatics/btab221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Porubsky D., Sanders A.D., Taudt A., Colomé-Tatché M., Lansdorp P.M., Guryev V. breakpointR: an R/Bioconductor package to localize strand state changes in Strand-seq data. Bioinformatics. 2020;36:1260–1261. doi: 10.1093/bioinformatics/btz681. [DOI] [PubMed] [Google Scholar]
- 53.Hanlon V.C.T., Mattsson C.-A., Spierings D.C.J., Guryev V., Lansdorp P.M. InvertypeR: bayesian inversion genotyping with Strand-seq data. BMC Genom. 2021;22:582. doi: 10.1186/s12864-021-07892-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang T., Antonacci-Fulton L., Howe K., Lawson H.A., Lucas J.K., Phillippy A.M., Popejoy A.B., Asri M., Carson C., Chaisson M.J.P., et al. The Human Pangenome Project: a global resource to map genomic diversity. Nature. 2022;604:437–446. doi: 10.1038/s41586-022-04601-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Akbari V., Garant J.-M., O’Neill K., Pandoh P., Moore R., Marra M., Hirst M., Jones S. Zenodo; 2021. NanoMethPhase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wagner J., Olson N.D., Harris L., Khan Z., Farek J., Mahmoud M., Stankovic A., Kovacevic V., Yoo B., Miller N., et al. Benchmarking challenging small variants with linked and long reads. Cell Genom. 2022;2:100128. doi: 10.1016/j.xgen.2022.100128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10. [Google Scholar]
- 59.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stunnenberg H.G., International Human Epigenome Consortium. Hirst M., de Almeida M., Altucci L., Amin V., Amit I., Antonarakis S.E., Hirst M. The international human epigenome consortium: a Blueprint for scientific collaboration and discovery. Cell. 2016;167:1145–1149. doi: 10.1016/j.cell.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 61.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bernstein B.E., Stamatoyannopoulos J.A., Costello J.F., Ren B., Milosavljevic A., Meissner A., Kellis M., Marra M.A., Beaudet A.L., Ecker J.R., et al. The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Byrska-Bishop M., Evani U.S., Zhao X., Basile A.O., Abel H.J., Regier A.A., Corvelo A., Clarke W.E., Musunuri R., Nagulapalli K., et al. High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. bioRxiv. 2021 doi: 10.1101/2021.02.06.430068. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hao Z., Lv D., Ge Y., Shi J., Weijers D., Yu G., Chen J. RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ. Comput. Sci. 2020;6:e251. doi: 10.7717/peerj-cs.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
related to “Nanopore and Strand-seq enable chromosome-scale haplotyping” and “Strand-seq phasing and inversion correction”
Inversions with low read density warnings are likely in repetitive regions at centromeres or telomeres. Note that only inversions on the autosomes and larger than 10 kb were used for phasing correction. Genotypes 0|1 and 1|0 are not PofO phased.
Transparent peer review records for Akbari et al.
Data Availability Statement
-
•
Nanopore data for HG002 have been deposited at the NCBI Sequence Read Archive and are publicly available as of the date of publication (SRA: SRP395905). Phased variant call files are also deposited at Zenodo (https://doi.org/10.5281/zenodo.7052113). The accession numbers are also listed in the key resources table.
-
•
All original code has been deposited at Zenodo (https://doi.org/10.5281/zenodo.7308808) and on GitHub (https://github.com/vahidAK/PatMat) and is publicly available. Tools and DOIs are also listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.