

Summary
We combined functional genomics and human genetics to investigate processes that affect type 1 diabetes (T1D) risk by mediating beta cell survival in response to proinflammatory cytokines. We mapped 38,931 cytokine-responsive candidate cis-regulatory elements (cCREs) in beta cells using ATAC-seq and snATAC-seq and linked them to target genes using co-accessibility and HiChIP. Using a genome-wide CRISPR screen in EndoC-βH1 cells, we identified 867 genes affecting cytokine-induced survival, and genes promoting survival and up-regulated in cytokines were enriched at T1D risk loci. Using SNP-SELEX, we identified 2,229 variants in cytokine-responsive cCREs altering transcription factor (TF) binding, and variants altering binding of TFs regulating stress, inflammation, and apoptosis were enriched for T1D risk. At the 16p13 locus, a fine-mapped T1D variant altering TF binding in a cytokine-induced cCRE interacted with SOCS1, which promoted survival in cytokine exposure. Our findings reveal processes and genes acting in beta cells during inflammation that modulate T1D risk.
Keywords: type 1 diabetes, beta cell, proinflammatory cytokines, functional genomics, accessible chromatin, 3D chromatin interactions, gene expression, human genetics, CRISPR screen, high-throughput reporter assay
Graphical abstract
Highlights
-
•
Genomic maps of beta cells exposed to inflammatory cytokines IL-1β, IFN-γ, and TNF-α
-
•
CRISPR-KO screen identified 867 genes affecting cytokine-induced beta cell loss
-
•
High-throughput TF binding assay mapped regulatory variants in beta cell sites
-
•
Identified SOCS1 as a T1D risk gene mediating cytokine-induced beta cell loss
Benaglio et al. investigated molecular processes and genes that affect type 1 diabetes by mediating pancreatic beta cell loss in response to inflammatory stimuli using functional genomics and human genetics. These results provide supportive evidence that the beta cell plays an intrinsic role in the development of type 1 diabetes. Furthermore, the data and results represent a useful resource to broaden understanding of type 1 diabetes pathogenesis and identify novel therapies targeting beta cell function.
Introduction
Type 1 diabetes (T1D) is a complex disease caused by autoimmune destruction of the insulin-producing beta cells in the pancreas. The pathophysiology of T1D is characterized by aberrant immune response to antigens leading to the development of islet autoimmunity and T cell-mediated destruction of beta cells.1 As part of T1D progression, immune cell infiltration and inflammation occurs in the local environment around islets, through which beta cells are exposed to external stimuli such as proinflammatory cytokines produced by immune cells.2 Beta cells themselves have been argued to intrinsically contribute to T1D in response to these stimuli, for example by promoting cell death. Studying beta cell function during T1D directly is challenging, however, due to the limited availability of donor samples and the difficulty in capturing the window in which beta cells are under immune attack. An alternate strategy is to use in vitro models, for example by culturing islets or beta cells with interleukin 1β (IL-1β), interferon γ (IFN-γ), and tumor necrosis factor α (TNF-α),3,4,5,6,7 which are proinflammatory cytokines produced by antigen-producing cells and T cells during T1D that signal to beta cells. Previous studies using this in vitro model have revealed effects on beta cell regulation, function, and survival3,5,6,7,8; however, the genes and processes in beta cells that may influence T1D in response to these cytokines are poorly defined.
Human genetics represents an avenue through which to identify genes and processes in beta cells that play a causal role in T1D. Genome-wide association studies have identified over 90 loci associated with T1D, the majority of which are non-coding and likely affect gene regulation.9,10 Variants at T1D risk loci are enriched in islet cis-regulatory elements (cCREs) induced by proinflammatory cytokines,7 but not islet regulatory elements in the basal state, which supports that risk of T1D in beta cells acts downstream of external stimuli during T1D progression. Genes at several T1D loci affect beta cell function in cytokine signaling, such as PTPN2 and DEXI.7,11,12 At most T1D loci, however, whether risk genes affect beta cell function in cytokines is unknown. More broadly, determining the pathways through which these risk genes operate can help to converge on mechanisms through which beta cells intrinsically affect disease.
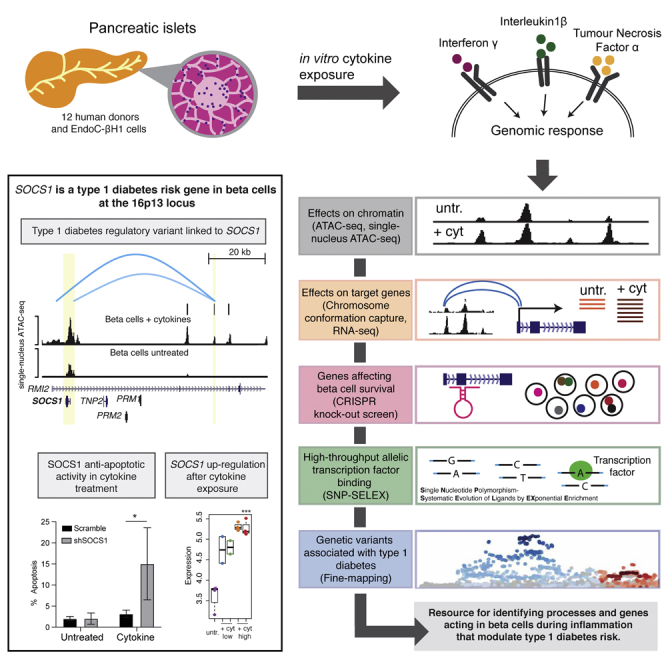
In this study, we used functional genomics to map cis-regulatory programs in pancreatic beta cells as well as identify genes that affect beta cell survival upon exposure to the cytokines IL-1β, IFN-γ and TNF-α. We then integrated these data with T1D fine mapping to identify risk variants regulating beta cell survival during cytokine exposure.
Results
Overview of study design
We combined human genetics and functional genomics to identify genes that affect T1D risk by modulating beta cell survival in response to cytokines (Figure 1). First, we created a map of cytokine-responsive cCREs in beta cells using bulk and single nuclear assay for transposase-accessible chromatin with sequencing (snATAC-seq). Second, we linked cytokine-responsive beta cell cCREs to target genes using co-accessibility and HiChIP. Third, we identified genes affecting beta cell survival in cytokines using a genome-wide CRISPR knockout screen in EndoC-βH1 cells. Fourth, we identified variants in cytokine-responsive beta cell cCREs affecting in vitro TF binding using high-throughput SNP-SELEX. Finally, we used fine mapping to annotate T1D risk variants regulating genes involved in cytokine-induced beta cell survival.
Figure 1.

Overview of study design
Schematic representation of the experimental design of the study.
Map of pancreatic beta cell chromatin in response to cytokines
We first performed bulk ATAC-seq in seven primary islets cultured in vitro with the cytokines IL-1β, IFN-γ, and TNF-α as well as in untreated conditions (Table S1). We performed these assays across multiple dimensions (35 assays in total), including treatment dose (high-dose, low-dose), duration (6, 24, 48, 72 h), and cytokines used (all three, or just IL-1β and IFN-γ). We determined the effects of cytokine signaling on islet chromatin by performing principal-component analysis (PCA) of normalized read counts (Figure S1A), which revealed reproducible changes in cytokine treatment as well as patterns across treatment dimensions such as an intermediate effect of low-dose compared with high-dose treatment.
We next identified specific islet cCREs responsive to cytokine stimulation. We defined a set of 165,884 genome-wide cCREs in islets, and then identified cCREs with differential accessibility in cytokines using DESeq2.13 There were 22,877 cCREs with increased activity in any treatment and 22,092 cCREs with decreased activity in any treatment (false discovery rate [FDR] <0.1, Figures 2A and S1B, Table S2). Consistent with previous reports,7 cCREs with increased activity in cytokines were enriched for interferon regulatory factor (IRF) (IRF1 p < 10−300), STAT (STAT1 p = 2.8 × 10−130), and nuclear factor-κB (NFKB-P65-REL p = 2.1 × 10−279) motifs, whereas cCREs with decreased activity were enriched for FOXA (p = 5.8 × 10−63), NKX6.1 (p = 1.1 × 10−28), and other motifs (Figures 2B and 2C, Table S3). We observed marked differences in cCREs that respond to cytokines across dose and duration (Figures S1A−S1E), as well as stronger effects on cCRE activity when including TNF-α (Figure S1C). Sequence motifs also showed variable enrichment across dimensions of treatment, for example SMAD transcription factors (TFs) were more enriched at longer durations (SMAD2 6 h p = 0.24, 24 h p = 0.03, 48 h p = 8.6 × 10−4, 72 h p = 2.2 × 10−6) (Figure S1F).
Figure 2.

Map of islet accessible chromatin in inflammatory cytokine exposure
(A) Genome browser of the CXCL10/CXCL11 locus showing ATAC-seq across cytokine treatments at 24 h s. 2cyt: IL-1b and IFN-γ, 3cyt: IL-1b, IFN-γ, and TNF-α, lo: low-dose, hi: high-dose, untr: untreated.
(B and C) Sequence motifs enriched in up-regulated (B) and down-regulated (C) cCREs across all treatments, compared with all tested cCREs.
(D) UMAP of snATAC-seq profiles of islet samples from four individuals.
(E) Barplot showing the proportion of cytokine-treated and untreated cells in each cell type.
(F) Genome browser showing cytokine-responsive cCREs shared across cell types (left) or beta cell-specific (right).
(G) Scatterplot showing effect of cytokine-responsive cCREs in bulk ATAC (x-axis) and in beta cell snATAC (y-axis). Spearman correlation and p-values are indicated. Bottom: density plot showing effect size in beta cells for cytokine-responsive cCREs significant in both beta cells and bulk islets. P value from two-sided Wilcoxon signed rank test is shown.
The effects of cytokine exposure on individual islet cell types are obscured from assays of bulk tissue. Therefore, we next performed snATAC-seq in cytokine-treated and untreated islets from four donors at 24 h post-treatment. We used a high dose of all cytokines for these assays, as this produced the strongest effects in bulk. After removal of low-quality and doublet cells (see STAR Methods), we performed UMAP dimensionality reduction and clustering on a total of 7,829 nuclei (Figure 2D). Each of the resulting clusters contained cells from all four donors and was represented by untreated (total nuclei = 3,947) and cytokine-treated (total nuclei = 3,882) cells (Figures 2E and S2A). We assigned clusters cell type identity based on promoter accessibility of known marker genes (Figures S2B and S2C), which revealed endocrine, exocrine, endothelial, and stellate cells. In addition, we identified two clusters of beta cells that were enriched for genes related to hormone production and stress response, respectively, suggesting the clusters represent distinct states in line with previous findings14 (Figure S2D).
We next defined cCREs in beta cells and other cell types and used the resulting cCREs to annotate cytokine-responsive cCREs identified in bulk (Figure S3A). We identified 38,931 cytokine-responsive islet cCREs active in beta cells, a small percentage (8.2%) of which were specific to beta cells relative to other endocrine cell types (Figure 2F). We further used snATAC data from cytokine-treated and untreated cells to identify differential sites in beta cells directly. There were 2,412 cytokine-responsive beta cell cCREs (FDR <0.1 Figure S3B, Table S2), almost all of which (99%, 2,388) had significant, concordant effects in bulk islets. The effects of cytokine treatment on cCRE activity were generally stronger on beta cells relative to bulk islets, although there were fewer cCREs overall with significant changes in beta cells (Figure 2G). Compared with alpha cells, there were many more cCREs with cytokine-responsive activity in beta cells (2,412 versus 226) (Figure S3C). Furthermore, the effects of cytokine treatment on cCRE activity were consistently stronger in beta cells compared with alpha cells (p = 1.2 × 10−255, two-sided Wilcox signed rank test) (Figure S3D). These results suggest that beta cell chromatin may be more responsive to the cytokines IL-1β, IFN-γ, and TNF-α compared with alpha cells.
Finally, we identified TF motifs differentially enriched in cytokine-responsive beta cell accessible chromatin. We identified motifs differentially enriched in single cytokine-treated and untreated beta cells using ChromVAR.15 The most enriched motifs in beta cells were broadly consistent with those identified in bulk data, with IRF-family TFs showing highest enrichment in cytokine-treated beta cells and FOXA TFs the strongest depletion (Figure S3E, Table S3). However, when comparing motif enrichments in alpha and beta cells, there was more significant enrichment of IRF- and STAT-family motifs in cytokine-treated beta cells (Figure S3E).
In summary, we generated a comprehensive catalog of cCREs that respond to proinflammatory cytokines in pancreatic islets and beta cells.
Linking cytokine-responsive beta cell cCREs to gene targets
As most cytokine-responsive beta cell cCREs are distal to gene promoters, we next sought to link cytokine-responsive cCREs to the gene targets they regulate.
We first identified cytokine-responsive cCREs correlated with the activity of gene promoters using co-accessibility in cytokine-treated and untreated beta cells with Cicero.16 In total, we identified 400,403 and 277,447 pairs of co-accessible cCREs (score >0.05) in cytokine-treated and untreated beta cells, respectively, 30% of which involved a promoter. We then annotated cytokine-responsive beta cell cCREs co-accessible with at least one promoter. There were 11,124 and 8,434 cytokine-responsive cCREs co-accessible with a putative target gene in cytokine-treated and untreated beta cells, respectively, while ∼10% of cytokine-responsive cCREs were at promoters directly (Figure 3A). As co-accessibility represents a correlation between cCREs that may not always reflect direct cis regulation, we next mapped 3D interactions between cCREs using HiChIP in cytokine-treated and untreated EndoC-βH1 cells. We used an H3K27ac antibody for HiChIP assays to identify 3D interactions involving active regulatory elements such as enhancers and promoters, and thus likely did not effectively capture interactions between other classes of elements. Co-accessible sites were significantly enriched for 3D interactions compared with non-co-accessible sites (cytokine odds ratio [OR] = 3.6, 95% confidence interval [CI] = 3.49-3.67; untreated OR = 3.2, 95% CI = 3.13-3.30, both p < 2.2 × 10−16, Fisher’s exact test). In total, 2,520 and 2,063 distal cCREs co-accessible with a promoter in cytokine-treated and untreated cells, respectively, had a significant (FDR <0.10) interaction.
Figure 3.

Target genes of beta cells cCREs in inflammatory cytokine exposure
(A) Fraction of cytokine-responsive cCREs (CR-cREs) co-accessible with at least one promoter in beta cells in untreated, cytokine-stimulated, or pooled cells; or proximal (<10 kb) to a promoter.
(B and C) Enrichment of (B) distal cytokine-responsive cCREs or (C) promoter-proximal cytokine-responsive cCREs (<10 kb) for co-accessibility to genes with concordant effects. Odds ratios and uncorrected p-values from Fisher’s exact test are shown.
(D) Example of an up-regulated cytokine-responsive cCRE linked to cytokine-up-regulated gene BCL6. Top to bottom: co-accessibility in beta cells in cytokine or untreated conditions, virtual 4C from HiChIP in EndoC-βH1 in cytokine or untreated conditions, snATAC in beta cells in cytokine or untreated conditions, gene annotations. Virtual 4C counts are scaled between 0 and 1. Only co-accessibility arcs that link the highlighted distal peak and promoter are shown.
(E) Normalized expression of BCL6 in human islets in cytokines. Log2 fold change and uncorrected p values shown are from DESeq2 analysis comparing high-dose three-cytokine-treated islets (red) versus untreated (purple).
(F−G) Same as (D) and (E) but showing an example of a down-regulated cCRE linked to the promoter of a down-regulated gene MNX1. Treatment abbreviations as in Figure 1.
We next assessed the relationship between cytokine-responsive beta cell cCREs and the expression of target genes linked to the cCREs in cytokines. We performed RNA sequencing (RNA-seq) in islets treated with cytokines and identified differentially expressed genes (DEGs) in cytokine-treated compared with untreated cells using DESeq2.13 High-dose exposure to all three cytokines produced the largest changes in expression, where 3,367 genes had increased and 3,414 had decreased expression in cytokines (Figures S4A−S2F, Table S4). High-dose treatment using just IL-1β and IFN-γ resulted in 5,051 DEGs. As with bulk ATAC-seq, low-dose treatment resulted in fewer DEGs overall, and these genes were largely a subset of the genes identified in high-dose treatment (Figures S4B and S4C, Table S4).
We determined whether genes co-accessible with cytokine-responsive distal cCREs had directionally concordant changes in expression. In these analyses, we used just genes differentially expressed in high-dose cytokines. Distal cCREs (>10 kb from TSS) with up- or down-regulated activity in cytokine treatment were significantly enriched for co-accessibility to genes with increased or decreased expression, respectively (Figure 3B). We observed similar patterns when considering distal cCREs with 3D interactions to genes (Figure S4G). Cytokine-responsive cCREs proximal to gene promoters shown even stronger enrichment for concordant effects on expression (Figure 3C). At the 3q27 locus, a cytokine-induced beta cell cCRE was co-accessible with BCL6 in cytokine-treated beta cells, and BCL6 attenuates the proinflammatory response but induces apoptosis in beta cells17 (Figure 3D). The cCRE interacted with the BCL6 promoter in cytokine-treated cells only (FDR = 6.2 × 10−6), and BCL6 had increased expression in cytokines (Figures 3D and 3E). Similarly, at the 7q36 locus, a beta cell cCRE with decreased activity in cytokines was co-accessible with the promoter of MNX1, which maintains beta cell fate (Figure 3F). We observed an interaction between the cCRE and the MNX1 promoter in untreated beta cells only (FDR = 5.4 × 10−6) and MNX1 had decreased expression in cytokines (Figures 3F and 3G).
Together these results implicate target genes of cytokine-responsive distal cCREs in beta cells.
Genes affecting beta cell survival in response to cytokine exposure
Given target genes of cytokine-responsive cCREs in beta cells, we next determined which genes had cellular functions directly relevant to T1D. As beta cell loss is the primary pathogenic endpoint of T1D, we sought to identify genes affecting beta cell survival in response to cytokines. We performed a genome-wide pooled CRISPR loss-of-function screen in the EndoC-βH1 beta cell line using cell survival under high-dose cytokine exposure for 72 h as an endpoint (Figure 4A). We selected a longer duration of treatment than for chromatin and gene expression assays to effectively capture cell loss in response to cytokines. In brief, after transfecting cells with the single guide RNA (sgRNA) library, we split and cultured cells in either high-dose cytokine or control. The representation of sgRNAs between cytokine and untreated conditions was compared to identify genes promoting or preventing beta cell loss in response to cytokines.
Figure 4.

Genes affecting beta cell survival in cytokine exposure
(A) Design of the genome-wide CRISPR loss-of-function screen in cytokine-treated EndoC-βH1 cells.
(B) Volcano plot of gene effects on beta cell survival from the screen. Effect sizes and uncorrected −log10 p values are shown from MAGeCK, and genes with significant (FDR < 0.1) enrichment and depletion are in bold. The most significant genes with TPM > 1 in islets are labeled.
(C) Enrichment of known T1D risk loci for genes enriched and depleted in screen, partitioned by expression (+/− exp = FDR < 0.1, ++/−− exp = FDR < 1 × 10−5) in islets after high-dose three-cytokine stimulation. Values are odds ratios, and error bars are 95% CI from Fisher’s exact test.
(D) Scatterplot showing the effect size of genes promoting beta cell survival in the screen and differential expression of the gene in islets after cytokine treatment. Genes mapping within 1 MB of a known T1D locus or within 1 MB of a variant with nominal (p < 1 × 10−4) T1D association are colored.
(E) Pathways from gene ontology (GO) and KEGG enriched in genes with increased expression in cytokine-treated islets and promoting beta cell survival. P values are from GSEA analysis. A subset of genes mapping to known T1D loci or with nominal T1D association are shown with corresponding pathways. Only pathways that contain at least one T1D gene are shown, and the full list is in Table S6.
Among 18,703 genes targeted by sgRNAs after transduction, 867 genes had significant (FDR <0.10) differences in recovered sgRNAs between cytokine-treated and untreated cells. Among these, sgRNAs for 427 genes were enriched in cytokine-treated compared with untreated cells and thus these genes promoted beta cell loss (“pro-death”) in response to cytokines (Figure 4B, Table S5). Conversely, sgRNAs for 440 genes were depleted in cytokine-treated compared with untreated cells and thus these genes prevented beta cell loss (“pro-survival”) in response to cytokines (Figure 4B, Table S5). More than half of genes affecting beta cell loss (57% pro-death, 60% pro-survival) were linked to a cytokine-responsive cCRE, and a quarter of genes affecting beta cell loss (20% pro-death, 27% pro-survival) also had cytokine-induced changes in expression (Figure S4H). Our screen identified genes known to affect beta cell survival, for example XIAP,18 JUND,19 PTPN2,11 and SOCS1.20,21 To annotate the function of pro-death and pro-survival genes, we performed gene ontology enrichment analyses (Table S6). As expected, pro-death genes were enriched for DNA damage response, apoptosis, and protein folding, and pro-survival genes were enriched for autophagy, which protects against beta cell stress, and phosphorylation and kinase activity, which suppress inflammatory responses. Pro-survival genes were also enriched for RNA metabolism and splicing, and pro-death genes were enriched for lipid metabolism, which have been implicated in beta cell function and survival.22,23
Interestingly, genes regulating processes related to mitochondrial function were enriched among both pro-death and pro-survival genes. We found that pro-survival mitochondria-related genes were primarily involved in mitochondria organization and mitophagy, such as USP36, VDAC1, MFF, TIMM9, YME1L1, SIRT5, and SPATA18. Conversely, mitochondria-related genes in the pro-death category were mostly electron transport chain components, such as NDUFA6, NDUFB2, ACAD9, CYCS and SDHD. A key mitophagy regulator, CLEC16A, has been previously shown to protect beta cells against inflammatory damage, mediated in part by reactive oxygen species (ROS) generated in mitochondria.24 Our data suggest that mitophagy and mitochondria quality control are important pro-survival processes in beta cells in response to cytokines and provide novel regulators of beta cell mitophagy.
Given genes and molecular processes affecting cytokine-induced beta cell loss, we next determined which genes and processes might be relevant to T1D. We tested for enrichment of genes affecting cytokine-induced beta cell loss at loci involved in genetic risk of T1D and observed no evidence for enrichment among the full set of either pro-survival or pro-death genes. Next, we segregated pro-survival and pro-death genes based on whether their expression was significantly up-regulated or down-regulated, or had no change, in cytokines. Pro-survival genes that had up-regulated expression in cytokines (n = 84 genes) were significantly enriched at known T1D loci (+exp OR = 1.82, 95% CI = 0.97,3.23, p = 0.048, Fisher’s exact test), and no other subset showed enrichment (Figure 4C). This enrichment was stronger when considering only genes with the most significant increases (FDR <1 × 10−5) in cytokine-induced expression (++exp OR = 3.28, 95% CI = 1.33,7.37, p = 5.1 × 10−3, Fisher’s exact test). Numerous genes with highly induced expression mapped to known T1D risk loci (Figure 4D), and this subset of genes also included several with roles in mitophagy.
We next characterized the molecular functions of pro-survival genes with up-regulated expression in cytokines. These genes were broadly enriched for molecular processes related to modulation of the inflammatory response, ubiquitination and proteasomal degradation, translation, and autophagy (Table S6, Figure 4E). Among genes at T1D loci were negative regulators of cytokine signaling PTPN2 and SOCS1, both of which inhibit JAK/STAT signaling to suppress inflammatory responses and promote beta cell survival.11 Other beta cell survival genes function in protein ubiquitination, which targets proteins for degradation by the proteasome. Proinflammatory cytokines induce endoplasmic reticulum (ER) stress in beta cells,25 and proteasome-mediated ER-associated protein degradation (ERAD) resolves ER stress in beta cells.26 Ubiquitin-mediated proteolysis may therefore protect beta cells from cytokine-induced stress, although the function of most of these genes in beta cells is unknown. We also observed enrichment of class I MHC antigen-related terms, although these genes were largely overlapping with other terms.
Together these results identify genes and molecular processes that affect beta cell loss in response to cytokines and reveal that T1D risk is specifically enriched for pro-survival genes highly induced in cytokines.
Identifying functional regulatory variants in beta cell cCREs with SNP-SELEX
Given that beta cell pro-survival genes up-regulated in cytokines were enriched at T1D risk loci, we next sought to determine the transcriptional regulators of gene activity in beta cells during cytokine exposure through which T1D risk is mediated.
Because variants often affect gene regulation via TF binding, we systematically determined the effects of genetic variants in cytokine-responsive beta cell cCREs on TF binding. A total of 184,086 variants were selected and tested for in vitro differential TF binding using a highly multiplexed assay SNP-SELEX.27 We designed a library of 44 base pair oligos surrounding each variant containing each of the four possible alleles for SNPs, or the two observed alleles for indels. We then tested oligos for binding to 530 E. coli-expressed TF proteins by sequencing recovered oligos across four binding cycles, where the entire experiment was performed in duplicate (Figure 5A; Table S7).
Figure 5.

Identifying transcriptional regulators affecting T1D risk in beta cell cCREs with SNP-SELEX
(A) Design of HT-SELEX-seq experiment.
(B) Top: Example of enrichment profiles of bound oligos within an experiment and of an SNP with preferential binding. Bottom: Distribution of the number of variants with allelic binding per TF across 489 TFs and table summarizing the number of bound variants and allelic binding variants across TFs.
(C) Enrichment of variants with allelic binding for T1D association among all tested variants, variants in beta cell cCREs, and variants in cytokine-responsive beta cell cCREs. Values are odds ratios and error bars are 95% CI from Fisher’s exact test.
(D) Enrichment of variants with allelic binding of specific TF sub-families for T1D association among variants in cytokine-responsive beta cell cCREs. Values represent odds ratios by Fisher’s exact test, and points are colored by p value.
(E) Regional plot of T1D association, with variants with p < 10−4 in black; bulk ATAC-seq from human islets. Treatment abbreviations as in Figure 1.
(F) EMSA using nuclear extract (NE) from cytokine-treated MIN6 with probes for each allele of rs10483809.
(G) Luciferase assays for rs10483809 alleles in MIN6 in untreated or high-dose cytokines compared with empty vector. Values are mean and error bars SD from n = 9 transfections, with uncorrected p values shown from two-sided t tests.
After quality filtering (Figures S5A−S5D), 130,225 variants were bound by at least one TF and were further analyzed for allelic binding. We identified variants with allelic differences in TF binding by calculating a preferential binding score (PBS) score between alleles (Figure 5B, Table S8). There were 28,972 variants affecting binding of at least one TF (p < 0.05 by Monte Carlo randomization27), with a mean of 2 TFs per variant and of 123 variants per TF (Figure 5B). TFs from the same family often clustered together based on correlation in variant effects on binding (Figure S5E).27,28,29 Variant effects on TF binding from SNP-SELEX were correlated with predicted effects from DeepSEA30 (mean r = 0.81, Figure S5F) and position weight matrix (PWM) models (r = 0.91, Figure S5G), although this was highly variable across TFs (Figure S5H). Consistent with previous findings,27 a minority of variants (29% on average per TF) with allelic effects from SNP-SELEX had a corresponding PWM prediction (Figure S5I), which highlights the benefit of this experimental resource.
There were 8,424 variants in beta cell cCREs affecting TF binding, including 2,229 in cytokine-responsive beta cell cCREs. T1D-associated variants in beta cell cCREs were enriched for allelic effects on TF binding, and this enrichment was stronger for variants in cytokine-responsive cCREs (Figure 5C). By comparison, there was limited enrichment among all tested variants for allelic effects on TF binding (Figure 5C). We next grouped TFs into 220 sub-families using TFClass,31 and tested for enrichment of T1D association among variants in cytokine-responsive beta cell cCREs disrupting each TF sub-family. Sub-families with strongest enrichment (OR >2, Fisher’s exact test) included BCL6, POU3, PBX, MYC, ARX, and PDX1 (Figure 5D). We also observed enrichment for sub-families regulating stress, mitophagy, and immune responses, such as ATF3-like, IRF, NR4, and GLI-like TFs (Figure 5D). To identify specific TFs likely regulating cytokine-induced beta cell cCREs, we annotated TF genes in each sub-family with differential expression in cytokine exposure. TF genes within enriched sub-families with cytokine-induced expression included BCL6, GLIS3, IRF1/2/7/9, PBX1, PDX1, and ATF3 (Table S4).
We then identified specific variants at T1D risk loci affecting TF binding in cytokine-responsive beta cell cCREs. In total 380 variants in cytokine-responsive beta cell cCREs mapped within 1 MB of a T1D locus and affected TF binding. At the RAD51B locus, variant rs10483809 (T1D p = 8.1 × 10−6) mapped in a cytokine-induced beta cell cCRE (Figure 5E) and the T1D risk allele had preferential binding to IRF- (p = 0.0066) and CUX-family (p = 0.048) TFs (Table S8). As SNP-SELEX is based on in vitro interactions, we validated allelic effects on regulatory activity in beta cells. Electrophoretic mobility shift assay (EMSA) using nuclear extract from the beta cell line MIN6, a rodent line that has been used extensively for reporter assays of variant activity,32,33,34,35 demonstrated stronger binding to the T1D risk allele (Figure 5F). We also identified increased enhancer activity for the risk allele in luciferase reporter assays in MIN6 cells, which was more pronounced in cytokines (Figure 5G). This variant maps in RAD51B, which is a pro-apoptotic protein involved in DNA recombination36 and up-regulated in cytokines (Table S4).
Together these results identify functional variants altering TF binding in beta cell cCREs and reveal TFs through which variants in cytokine-responsive beta cell cCREs broadly affect T1D risk.
T1D risk variants linked to genes affecting beta cell survival in cytokines
Finally, given molecular processes and regulatory networks enriched for T1D risk in cytokine-induced beta cells, we layered functional genomics and human genetics to annotate T1D loci that regulate genes affecting beta cell loss in cytokines.
We intersected cytokine-responsive beta cell cCREs with fine mapping 99% credible sets of 136 T1D signals.9 At 77 T1D signals, at least one credible set variant overlapped a beta cell cCRE, and at 52 signals a credible set variant overlapped a cytokine-responsive beta cell cCRE (Table S9). Among these were variants at loci previously implicated in beta cells, such as PTPN2, DEXI, GLIS3, and DLK1.11,12,37,38,39 We next linked credible set variants in cytokine-induced beta cell cCREs at 38 signals to putative target genes using co-accessibility (Table S9). Genes linked to credible set variants in cytokine-responsive beta cell cCREs included 22 genes affecting beta cell loss from the CRISPR screen in addition to key stress response genes (Table S9). We did not find evidence that credible set variants were islet expression QTLs (eQTLs) for these genes,40 although current eQTL maps have not been generated from islets under cytokine stimulation.
At the 16p13 locus, which has two independent T1D risk signals, seven credible set variants overlapped cytokine-induced beta cell cCREs (Figures 6A–6C, Table S9). Among these, only one variant rs35342456 affected TF binding from SNP-SELEX (p = 2.4 × 10−5), and therefore is a functional candidate for underlying T1D association (Figure 6D). A previous study identified a functional variant rs193778 in cytokine-stimulated islets at this locus,7 but this variant was not present in our 99% credible sets. To validate that rs35342456 has regulatory effects in beta cells, we performed an EMSA to measure TF binding to each allele using nuclear extract from cytokine and untreated MIN6 cells (Figures 6E and S6). Consistent with SNP-SELEX, we observed allele-specific TF binding in beta cells (Figure 6E).
Figure 6.

T1D locus 16p13 regulates beta cell survival gene SOCS1 in cytokine exposure
(A) Regional plot showing T1D association for two independent signals at the DEXI/SOCS1 locus.
(B) Fine mapping probabilities of the secondary signal. Variants with SNP-SELEX effect on differential TF binding and within a cytokine-responsive cCRE are highlighted in red.
(C) Genome browser of the locus showing bulk ATAC-seq. Treatment abbreviations as in Figure 1.
(D) SNP-SELEX results for variant rs35342456.
(E) EMSA with nuclear extract (NE) from MIN6 cells showing preferential binding to the reference allele.
(F) Zoom in of the locus showing location of variant rs35342456 (yellow line) in a cytokine-responsive beta cell cCRE with co-accessibility and 3D interaction to the SOCS1 promoter in cytokine-treated and untreated EndoC-βH1 cells. For 3D interactions, virtual 4C counts are scaled between 0 and 1.
(G) Counts of each sgRNA in the CRISPR-KO screen targeting SOCS1 in untreated and high-dose cytokine EndoC-βH1, normalized to the sequencing depth of each sample. Effect size and uncorrected p value from MAGeCK.
(H) Normalized expression of SOCS1 in human islet samples in cytokines. Log2 fold change and uncorrected p values from DESeq2 comparing high-dose three-cytokine (red) and untreated (purple).
(I) Quantification of apoptotic EndoC-βH1 cells with shRNA targeting SOCS1 or scramble in either high-dose cytokine or vehicle (0.1% BSA). Values are mean and error bars are SD from n = 4 transductions, and p values are shown from two-way ANOVA followed by pairwise comparisons using Tukey’s HSD.
The cytokine-induced cCRE harboring rs35342456 was co-accessible with SOCS1, which had up-regulated expression and promoted beta cell survival in cytokines, implicating SOCS1 as a candidate causal gene for T1D (Figures 6F–6H). We confirmed SOCS1 as a target of cytokine-dependent cCRE activity using HiChIP in cytokine-treated and untreated EndoC-βH1 cells. We observed a significant interaction (FDR = 0.068) between the cCRE and SOCS1 promoter in cytokine-treated cells only (Figure 6F). Furthermore, there was no evidence of interaction between the cCRE and other genes, including previously implicated genes DEXI and CLEC16A, the expression of which was also not affected by cytokines (Table S4). These results reveal that SOCS1 is a likely target of T1D variant activity in cytokine-induced beta cells at the 16p13 locus.
In the CRISPR screen SOCS1 promoted beta cell survival after cytokines, and SOCS1 had significant increase in cytokine-induced expression (Figures 6G and 6H). We determined the effects of SOCS1 on cytokine-induced beta cell survival using an independent assay that measures apoptosis using a fluorogenic probe. We performed knockdown of SOCS1 via short hairpin RNA (shRNA) in EndoC-βH1 cells cultured in untreated or cytokines and measured staining for apoptosis using flow cytometry (Figure S7). We observed a significant increase in apoptosis in SOCS1 shRNA compared with scramble control shRNA, and this effect was stronger in cytokine-treated cells (shRNA F = 7.45, p = 0.01827, shRNA:treatment interaction F = 7.43, p = 0.01835, two-way ANOVA; shRNA in control p = 1, shRNA in cytokine, p = 0.0106, Tukey’s honestly significant difference [HSD] test; Figure 6I).
These results reveal that the induction of SOCS1 activity in response to cytokine exposure promotes human beta cell survival and may play a causal role in T1D.
Discussion
Genes with highly induced expression that promoted beta cell survival in response to cytokines were specifically enriched at T1D loci. These genes broadly reflect two classes of intrinsic mechanisms that protect beta cells against cytokines: direct inhibition of the inflammatory response, and resolution of stress-induced damage due to the inflammatory response. The activity of these pro-survival genes is induced by distal beta cell cCREs that respond to cytokine signaling, and these cCREs in turn often harbor T1D risk variants. As a result, T1D risk may be explained in part by reduced induction of pro-survival genes in beta cells in response to proinflammatory cytokines during T1D progression.
Pro-survival genes involved in modulating the immune response included PTPN2 and SOCS1, which map to known T1D risk loci. Both genes suppress the inflammatory response by inhibiting the JAK/STAT pathway. Previous studies in model systems demonstrated that knockdown of PTPN2 in beta cells led to increased phosphorylation of STAT1/3 upon activation by IFN-γ as well as phosphorylation of the pro-apoptotic BIM,11,41 which in turn increased beta cell death. Studies in model systems have shown that SOCS1 promotes beta cell survival by blocking the phosphorylation of JAK to suppress the inflammatory response.20,21,42 In line with these findings, our study reveals a role for PTPN2 and SOCS1 in promoting human beta cell survival in cytokines. Furthermore, based on links to a T1D risk variant, the regulation of SOCS1 activity in beta cells after cytokine exposure potentially plays a causal role in T1D.
Pro-survival genes with highly induced expression in cytokines were also involved in ubiquitin-mediated proteolysis. Among these were LM O 7, PPP1R11, and PSMD2, which mapped to T1D loci.42,43,44 Cytokine signaling in beta cells induces proteasomal activity,45 and the proteasome is involved in cell survival.45,46,47,48 As proinflammatory cytokines induce ER stress in beta cells in the context of T1D,3 which can lead to cell death,49 and ER stress is resolved in part through protein degradation,50 these genes may function in resolving ER stress. Cytokines also cause beta cell death through the production of ROS in mitochondria.51,52,53 Mitophagy is induced by ROS production downstream of inflammation to prevent beta cell damage,54 and our analyses revealed pro-survival genes affecting mitophagy. Moreover, many pro-survival genes were involved in class I MHC antigen processing and presentation. Class I MHC activity in beta cells is necessary for T1D progression,55 likely by promoting an immune response via exposure of antigens. While it is possible that some genes affect T1D via antigen presentation, other models will be needed to test this hypothesis.
While our study identifies SOCS1 as a novel candidate gene for T1D, several other genes at 16p13 have been implicated, including DEXI and CLEC16A. Inhibition of DEXI in beta cells reduces the activation of STAT and chemokine production and promotes survival in response to viral double-stranded RNA.12 T1D variants at 16p13 were also previously shown to interact with DEXI in cytokine-treated beta cells, although we did not find corresponding evidence in our HiChIP data, nor did DEXI have differential expression in cytokines. For CLEC16A, pancreas-specific deletion in mice led to decreased mitophagy and abnormal mitochondria,38 although we did not identify CLEC16A in our screen and CLEC16A expression was not affected in cytokines. Another candidate at this locus, CIITA, is an MHC class II trans-activator that has induced expression in cytokines and is expressed in beta cells from T1D donors.56 Ultimately, it is likely that several genes mediate T1D risk in beta cells at this locus.
Our study also provided novel insight into regulators of gene activity in beta cells through which T1D variants act. Variants altering TF binding within cytokine-responsive beta cell cCREs were broadly enriched for T1D association, supporting that a subset of T1D risk disrupts beta cell regulatory programs that respond to cytokines. The function of TF sub-families enriched for T1D association largely mirrored processes enriched in pro-survival genes, providing orthogonal support for the role of these processes in T1D. Furthermore, we pinpointed specific TFs within these sub-families likely driving altered beta cell regulatory activity in T1D. For example, in beta cells, IRF TFs and BCL6 regulate inflammation,17,57,58 ATF3, GLIS3, and MYC regulate stress response and apoptosis,59,60,61,62 and NR4A1/3, PDX1, and MYC regulate mitochondrial function and mitophagy.61,63,64 Although we did not identify evidence that T1D variants affected expression of target genes of cytokine-responsive cCREs, mapping eQTLs in islet donors exposed to cytokines will help uncover these effects.
In summary, we identified regulators, genes, and pathways linked to T1D risk that modulate beta cell survival after cytokine exposure, providing new avenues to preserve beta cell mass in T1D.
Limitations of the study
Multiple genes had opposite effects on beta cell survival compared with previous reports. DEXI is a pro-survival gene in our screen but was previously shown to induce beta cell death in response to viral double-stranded RNA (dsRNA).12 In another example, NDRG2 is a pro-death gene but was previously shown to protect beta cells from lipotoxicity.65 In such cases, opposing effects on survival could arise from differences in cellular responses to different stressors such as viral dsRNA, cytokines, or lipids, or from differences between species.
Our CRISPR screen also identified genes affecting beta cell proliferation.66 The relevance to primary beta cells in cytokine exposure is unclear, however, as these genes might reflect the transformed nature of EndoC-βH1 cells. At present, EndoC-βH1 is the only human beta cell option for a genome-wide CRISPR screen, which requires large numbers of cells for sufficient coverage. Study designs that compare sgRNAs recovered from fluorescence-activated cell sorting (FACS)-sorted cells based on a cellular marker may also complement our cell loss design. Human pluripotent stem cell-derived islet organoids could be a future platform for screens but will require a differentiation-compatible lentivirus transduction method and scalable beta cell purification strategy.
The cytokines IL-1β, IFN-γ, and TNF-α have been extensively used as an in vitro model of T1D, but beta cells are exposed to additional stimuli during T1D. For example, a study revealed changes in beta cell regulation upon exposure to IFN-α.67 Generation of genomic maps in beta cells exposed to other cytokines will therefore be informative, and these maps will benefit from profiling modalities such as DNA methylation and histone modifications. Beta cells are also exposed to other stimuli beyond cytokines in T1D. ER stress,68 oxidative stress,69 hypoxia,70 and hyperglycemia71 have all been used as in vitro models of beta cell function, but the genomic response to these stressors and their role in T1D risk is largely unknown. As in vitro models only partially re-capitulate disease biology, genomic mapping in beta cells from individuals in pre-T1D or early-stage T1D will also help in interpreting disease risk. In addition, cells within the islet may have heterogeneity in their exposure to stimuli that is not fully captured by in vitro models.
Finally, as we profiled a relatively small number of donors, larger sample sizes will enable detecting more subtle changes in gene regulation in response to cytokine exposure, as well as identifying cytokine responses that interact with phenotype, such as age and sex, or genotype.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human pancreatic islets (See Table S1 for sample list and identifiers) | University of Alberta | http://www.bcell.org/ |
| Human pancreatic islets (See Table S1 for sample list and identifiers) | IIDP | https://iidp.coh.org/ |
| Human pancreatic islets (See Table S1 for sample list and identifiers) | City of Hope | https://www.cityofhope.org/ |
| Critical commercial assays | ||
| Tagment DNA TDE1 Enzyme and Buffer Kits | Illumina | Cat#20034197 |
| Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.1 | 10× Genomics | Cat#1000175 |
| TruSeq Stranded Total RNA Library Prep Gold | Illumina | Cat#20020599 |
| LightShift™ Chemiluminescent EMSA Kit | ThermoFisher | Cat#20148 |
| Dual-Luciferase Reporter System assay | Promega | Cat#E1910 |
| Deposited data | ||
| Bulk ATAC-seq | This paper | GEO:GSE205853 |
| Bulk RNA-seq | This paper | GEO:GSE205853 |
| snATAC-seq | This paper | GEO:GSE205853 |
| HiChIP | This paper | GEO:GSE205853 |
| CRISPR screen | This paper | GEO:GSE205853 |
| SELEX-Seq data | This paper and Yan J. et al., 202127 | GEO:GSE118725 |
| Processed summary files | This paper | https://doi.org/10.5281/zenodo.7084136 |
| Custom code | This paper | https://doi.org/10.5281/zenodo.7183895 |
| Experimental models: Cell lines | ||
| EndoC-βH1 cell line | UniverCell Biosolutions | N/A |
| MIN6 cell line | Miyazaki et al., 199072 | N/A |
| Oligonucleotides | ||
| sgRNA library amplification: see STAR Methods details | This paper | N/A |
| Luciferase gene reporter primers: see STAR Methods details | This paper | N/A |
| rs10483809:5′Biotin–ATCTTTCACTTTCCCT[A/G] TCGATACTTCATATGT |
This paper | N/A |
| rs35342456:5′Biotin-GCTGGGCGTG GTGGCTCACGCCTGT[A/C]ATCTTGTTG |
This paper | N/A |
| SOCS1_1_FWD_qPCR CACGCA CTTCCGCACATTC |
This paper | N/A |
| SOCS1_1_REV_qPCR TAAGGGC GAAAAAGCAGTTCC |
This paper | N/A |
| Recombinant DNA | ||
| GeCKO-V2 CRISPR gRNA library | Sanjana et al., 201473 | Addgene #1000000048 |
| Lentiviral packing vector pMD2.G | Dr. Didier Trono | Addgene #12259 |
| Lentiviral packing vector psPAX2 | Dr. Didier Trono | Addgene #12260 |
| pGL4.23 Luciferase reporter vector | Promega | Cat#E8411 |
| pRL-SV40 Renilla reporter vector | Promega | Cat#E2231 |
| Lentiviral shSOCS1 (SHCLNG MISSION shRNA) | Sigma-Aldrich | Cat#TRCN0000356244 |
| Software and algorithms | ||
| MACS2 | Zhang et al., 200874 | https://github.com/macs3-project/MACS |
| Samtools | Li and Durbin, 200975 | http://samtools.sourceforge.net |
| HOMER | Heinz et al., 201076 | http://homer.ucsd.edu/homer |
| BWA | Li & Durbin, 200975 | http://bio-bwa.sourceforge.net |
| 10× Genomics Cell Ranger ATAC v1.1 | 10× Genomics | https://support.10xgenomics.com/single-cell-atac/software/downloads/latest |
| SCANPY | Wolf et al., 201877 | https://scanpy.readthedocs.io/en/stable |
| Cicero | Pliner et al., 201816 | https://cole-trapnell-lab.github.io/cicero-release |
| ChromVAR | Schep et al., 201715 | https://github.com/GreenleafLab/chromVAR |
| STAR | Dobin et al., 201378 | https://github.com/alexdobin/STAR |
| RSEM | Li & Dewey, 201179 | http://deweylab.github.io/RSEM |
| DESeq2 | Love et al., 201413 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| MAPS | Juric et al., 201980 | https://github.com/HuMingLab/MAPS |
| MAGeCK | Wang et al., 201981 | https://github.com/WubingZhang/MAGeCKFlute |
| GSEA | Subramanian et al., 200582 | http://www.gsea-msigdb.org/gsea/msigdb/annotate.jsp |
Resource availability
Lead contact
Further information and requests for resources, reagents and analytical results should be directed to the lead contact, Kyle J Gaulton (kgaulton@health.ucsd.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Human islet samples
Human islet samples were obtained through the Integrated Islet Distribution Program (IIDP), the University of Alberta and the City of Hope National Medical Center. Information on the donors such as sex, age, BMI, and ethnicity can be found in Table S1. All islet donors were non-diabetic and no other clinical information was provided for these samples. Islets were enriched using a dithizone stain and cultured in CMRL 1066 supplemented with 10% FBS, 1X pen-strep, 8 mM glucose, 2 mM L-glutamine, 1 mM sodium pyruvate, 10 mM HEPES, and 250 ng/mL Amphotericin B. For cytokine-treated samples, human cytokines were added to the culture media for 24 h as follows: for high doses, 10 ng/mL and IFN-g, 0.5 ng/mL IL-1B (two cytokines), with 1 ng/mL TNF-α where indicated (three cytokines); for low doses, 0.2 ng/mL IFN-g and 0.01 ng/mL IL-1B (two cytokines), with 0.02 ng/mL TNF-α where indicated (three cytokines). Islet studies were approved by the Institutional Review Board of the University of California San Diego. The sample size for human islet studies was determined based on previous literature of genomic studies in primary islets and otherwise no statistical methods were used to determine sample size. This study used a paired design where islets from the same donor were split and included in both treated and untreated experimental groups.
EndoC-βH1 cell line
EndoC-βH1 cells were obtained from UniverCell Biosolutions. EndoC-βH1 cells were cultured at 9×104 cells/cm2 of cell culture surface area pre-coated with ECM (Sigma, E1270) and Fibronectin (Sigma, F1141). Cell culture media containing DMEM (Gibco,11885084), 2% BSA (Sigma, A1470), 3.5 × 10−4% 2-mercaptoethanol (Gibco, 21985023), 0.12% Nicotinamide (Calbiochem, 481907), 5.5 ng/mL transferrin (Sigma, T8158), 6.7 pg/mL Sodium Selenite (Sigma, 214485) and 1% Penicillin-Streptomycin (Gibco, 15140122) were refreshed every 2 days. Cells were passaged weekly using 0.25% Trypsin-EDTA for dissociation, which was quenched with an equal volume of FBS and two volumes of IMDM media (Gibco, 12440053). Dissociated cells were spun down at 1200 rpm for 5 min and counted before seeding with the above-mentioned density. The EndoC-βH1 cells are routinely tested to confirm no contamination with mycoplasma.
MIN6 cell line
MIN6 cells72 were obtained from the Jhala lab at the University of California San Diego. MIN6 mouse insulinoma cells were cultured in DMEM containing 1.5 g/L sodium bicarbonate, 4% heat inactivated FBS, gentamicin, and 50uM beta-mercaptoethanol. Detail on cell passaging and treatment for luciferase reporter and electrophoretic mobility shift assays are reported in the section below. The MIN6 cells are routinely tested to confirm no contamination with mycoplasma and authenticated using morphology and Western blotting of key marker proteins.
Method details
Islet nuclei isolation
Human islets were collected from culture, centrifuged at 500xg for 3 min and washed twice in HBSS. Islets were resuspended in nuclei permeabilization buffer consisting of 5% BSA, 0.2% IGEPAL-CA630, 1 mM DTT, and 1X cOmplete EDTA-free protease inhibitor (Sigma) in 1X PBS. Islets were then homogenized using a chilled dounce homogenizer, incubated on a tube rotator for 10 min and filtered using a 30μM filter (sysmex), and centrifuged in a 4C microcentrifuge at 500xg. Isolated nuclei were resuspended in 1X TDE1 buffer (Illumina) and quantified using a Countess II Automated Cell Counter (Thermo).
Bulk ATAC-seq data generation
We performed ATAC-seq assays using nuclei isolated from islets in the following conditions: n = 5 24 h high-dose three cytokine, n = 5 24 h high-dose two cytokine, n = 3 24 h low-dose three cytokine, n = 7 24 h low-dose two cytokine, n = 3 24 h untreated, n = 2 6 h high-dose three cytokine, n = 2 6 h untreated, n = 2 48 h high-dose three cytokine, n = 2 48 h untreated, n = 2 72 h high-dose three cytokine, n = 2 72 h untreated, for a total of 35 assays (Table S1). For each assay, approximately 50,000 islet nuclei were tagmented in a 25uL reaction volume containing Tagmentation buffer and 2.5uL TDE1 (Illumina), which was mixed using gentle pipetting. Transposition reactions were then carried out for 30 min at 37C in a thermal cycler. Tagmentation reactions were cleaned using a 2X reaction volume of Ampure XP beads (Beckman Coulter) and then eluted in 20uL Buffer EB (Qiagen). We prepared libraries with 10uL of tagmented DNA in a PCR reaction of 25uL total volume using the Nextera XT Dual-Indexed primer system (Nextera) and NEBNext High-Fidelity PCR Master Mix (New England Biolabs). The PCR protocol used for tagmentation was as follows: 72°C for 5 min; 98°C for 30 s; 12 rounds of 98°C for 10 s followed by 63°C for 30 s; 72°C for 1 min. Libraries were double size selected using Ampure XP beads (Beckman Coulter) by adding 0.55X library volume of AMPure beads, incubating for 15 min, and transferring the supernatant to a new tube. Next, 0.65X library volume of AMPure beads was added to the supernatant and incubated at RT for 15 min. Finally, the samples were washed twice with 80% ethanol and eluted in Buffer EB to a final volume of 20uL. Libraries were analyzed using Quibit HS DNA kit (Thermo) and a 2200 Bioanalyzer (Agilent Biosciences), and sequenced by the UCSD Institute for Genomic Medicine on an Illumina HiSeq 4000 using paired end reads of 100 bp to an average of 72.5 M read pairs sequenced per sample.
Bulk RNA-seq data generation
We isolated RNA using the RNeasy Mini system (Qiagen) from a total of 16 samples of human islets including n = 4 distinct islet donors exposed for 24 h to either high-dose three cytokine, high-dose two cytokine or untreated conditions, and n = 2 distinct islet donors exposed for 24 h to low-dose three cytokine or low-dose two cytokine conditions. Approximately 500–1000 islets were used for RNA isolation per sample. The RNA quality was assessed using a 2200 TapeStation to confirm RNA integrity, and all samples had a RIN score of >7. Ribodepleted total RNA libraries were prepared using TruSeq Stranded Total RNA Library Prep Gold (Cat#20020599) and sequenced at the UCSD Institute for Genomic Medicine on an Illumina HiSeq 4000 using paired-end reads of 100 bp to an average of 34.5 M read pairs sequenced per sample.
Single nucleus ATAC-seq data generation
We performed single nucleus ATAC-seq (snATAC-seq) assays from n = 3 distinct islet donors cultured in untreated and high-dose cytokine treatment and n = 1 additional islet donor in untreated conditions only. Isolated nuclei (described in the above section) were adjusted to a concentration of approximately 3,000 nuclei/μL in 1X nuclei buffer (10X Genomics). We targeted 5,000 nuclei per assay for use in the 10X Genomics Chromium Single Cell ATAC assay using v1 chemistry. Nuclei from two donors and same treatment conditions (SAMN12833535, SAMN12889245) were pooled in equal amount prior to snATAC library preparation to a final concentration of 3,000 nuclei/μL in 1X nuclei buffer (10X Genomics) and were de-multiplexed after sequencing (described in the analysis section below). Tagmentation reactions were carried out in a total volume of 15 μL containing nuclei resuspended in 5 μL of 1X Nuclei Buffer (10× Genomics), 10 μL ATAC buffer (10× Genomics) and ATAC enzyme (10× Genomics), and incubated for 60 min at 37 °C. Single-cell ATAC–seq libraries were generated using Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.1 (Cat#1000175, 10× Genomics), Chromium Next GEM Chip H Single Cell Kit (Cat# 1000161, 10× Genomics) and Single Index Kit N Set A (Cat# 1000212, 10× Genomics), following the manufacturer’s instructions. The resulting libraries were quantified using a Qubit fluorimeter (Life Technologies) and run on a TapeStation (High Sensitivity D1000, Agilent) to verify the laddering patterns of nucleosomes. Sequencing was performed at the UCSD Institute for Genomic Medicine on an Illumina NovaSeq 6000 using a specific 50 bp paired-end 10X ATAC run configuration (50 bp + 8 bp + 16 bp + 50 bp for Read1 + Index1 + Index2 + Read2) to an average of 60 M read pairs per sample.
We performed snATAC assays from n = 3 distinct islet donors in untreated conditions using a similar procedure as above with modifications. Approximately 1,000 islet equivalents were resuspended in 1 mL nuclei permeabilization buffer (10 mM Tris-HCL (pH 7.5), 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20 (Sigma), 0.1% IGEPAL-CA630 (Sigma), 0.01% Digitonin (Promega) and 1% fatty acid-free BSA (Proliant 68700), homogenized using 1 mL dounce homogenizer, filtered with 30 μm filter (CellTrics) and incubated for 10 min at 4°C. Nuclei were pelleted with a swinging bucket centrifuge for 5 min (500 × g, 4°C), washed with Wash buffer (10 mM Tris-HCL (pH 7.5), 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20, and 1% BSA (Proliant 7500804), resuspended in 30 μL of 1X Nuclei Buffer (10× Genomics) and counted using a hemocytometer. 15,360 nuclei were used for tagmentation. Libraries were generated using the procedure described above, using Chromium Chip E Single Cell ATAC kit (10× Genomics, 1000086) and indexes (Chromium i7 Multiplex Kit N, Set A, 10× Genomics, 1000084). Library quantification was performed as above and libraries were sequenced on NextSeq 500 and NovaSeq 6000 sequencers (Illumina) with the same read lengths as above, to an average of 117 M reads per sample.
HiChIP data generation
To collect samples for HiChIP assays, 10 million EndoC-βH1 cells were treated with either control (0.1% BSA) or high-dose three cytokines (0.5 ng/mL IL1β, 1 ng/mL TNFα and 10 ng/mL IFNγ) for 72 h. Treated cells were cross-linked with 1% formaldehyde for 15 min with shaking at room temperature, followed by a 5-min quenching step with 1.25 M glycine/PBS. Cross-linked EndoC-βH1 cells in both control and cytokine-treated conditions were washed three times with ice-cold PBS and collected from the dish with a cell scraper. Cells were then pelleted, flash frozen with liquid nitrogen and shipped to Arima Genomics (Carlsbad, CA) for assay. HiChIP assays were performed by Arima Genomics using the Arima-HiC + kit (P/N A101020) according to manufacturer’s protocols with a HiChIP-validated antibody for H3K27ac (Active Motif Cat # 91193). Libraries were prepared by Arima Genomics using the Accel-NGS 2 S Plus DNA library kit (Swift Biosciences). The resulting libraries were sequenced at the UCSD Institute for Genomic Medicine on an Illumina NovoSeq with 150 bp paired end reads.
Lentiviral human GeCKO-V2 library preparation, transduction, and titration
To package lentivirus encoding the human GeCKO-V2 CRISPR screen library,73 plasmids containing the gRNA library (Addgene, 1000000048) were transfected into the HEK 293 T cells together with the lentiviral packing vectors, pMD2.G (Addgene, 12259) and psPAX2 (Addgene, 12260), using a PolyJetTM DNA transfection reagent (Signagen Laboratories, 504788). Transfected cells were kept in the culture to allow virus to be released. Lentivirus-containing media was collected at 36, 48, 72 h post transfection and filtered through a 0.45 μm cell strainer to remove cell debris. Lentiviral particles were pelleted at 20,000 rcf for 2 h, using an Optima L-80 XP Ultracentrifuge machine (Beckman Coulter). The same media for EndoC-βH1 cell culture was used to resuspend the virus. A spin-inoculation method was adopted to transduce the viral library into the EndoC-βH1 cell line. Cells were pre-treated with 8 μg/mL polybrene (Sigma, TR-1003) in the culture media for 30 min; virus was added to the cells and the entire plate was spun in a swing-bucket centrifuge machine at 930 g for 45 min. After 48 h, the sgRNA and Cas9 protein were expressed in the EndoC-βH1 cells.
CRISPR loss-of-function screen for regulators of β-cell survival under cytokine stress
The EndoC-βH1 cells were expanded to a total of 300 million cells before spin-inoculation with the lentiviral human GeCKO-V2 library at an MOI = 0.3. To enrich for successfully transduced cells, a 3-day puromycin (5 μg/mL, Sigma, P8833) selection was performed 48 h after the spin-inoculation. Approximately 60 M (500X genome coverage) cells were harvested as a representation control for the GeCKO-V2 sgRNA library. The rest of the cells were kept in culture for an additional 14 days with puromycin (1 μg/mL) to achieve sufficient gene deletion and were subsequently treated with either 0.1% BSA or a combination 0.5 ng/mL IL1β (PerroTech, 200–01B), 1 ng/mL TNFα (PerroTech, 300–01A) and 10 ng/mL IFNγ (PerroTech, 300-02) for 72 h. A time-point experiment was performed to evaluate which treatment duration was necessary to induce cell death. EndoC-βH1 cells were seeded 24 h before the cytokine treatment and residual cell number was counted at 24, 48 and 72 h of treatment (n = 3). Cell numbers are shown in Table S5. We harvested another 60 M (500X genome coverage) cells from the control (0.1% BSA) treated cells and 30 M (250X genome coverage) cells from the cytokine treated cells, although they started with the same number.
Genomic DNA from all three conditions were purified with a Quick-gDNA™ MidiPrep kit (Zymo Research, D3100). gRNA libraries were amplified from the genomic DNA using a two-step nested PCR method modified from a previous published protocol.83 In brief, guide RNA inserts were amplified from the genomic DNA with the following primers:
F1-1:TCCCTACACGACGCTCTTCCGATCTNNNNNGGAAAGGACGAAACACCG
F1-2:TCCCTACACGACGCTCTTCCGATCTNNNNNHGGAAAGGACGAAACACCG
F1-3:TCCCTACACGACGCTCTTCCGATCTNNNNNHHGGAAAGGACGAAACACCG
F1-4:TCCCTACACGACGCTCTTCCGATCTNNNNNHHYGGAAAGGACGAAACACCG
R1-1:GGAGTTCAGACGTGTGCTCTTCCGATCNNNNNTGCTATTTCTAGCTCTAAAAC
R1-2:GGAGTTCAGACGTGTGCTCTTCCGATCNNNNNVTGCTATTTCTAGCTCTAAAAC
R1-3:GGAGTTCAGACGTGTGCTCTTCCGATCNNNNNVMTGCTATTTCTAGCTCTAAAAC
R1-4:GGAGTTCAGACGTGTGCTCTTCCGATCNNNNNVMAATGCTATTTCTAGCTCTAAAAC. Pooled F1 primers (F1-1 to F1-4) and R1 primers (R1-1 to R1-4) were used in each PCR reaction to avoid cluster registration failure on Illumina machines. Amplicons from the first step of PCR were gel purified and subjected to a second round of PCR to add Illumina sequencing adaptors and TruSeq indexes. Primers used in the second PCR step are the following: F2:AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGA; R2:CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCG. Sequencing library amplified from the second round of PCR were size-selected and purified with a magnetic bead-based SPRIselect reagent (Beckman Coulter, B23318), and subjected to HiSeq4000 Illumina NGS platform using a single read (SR75) method at the UCSD Institute for Genomic Medicine.
Electrophoretic mobility shift assay
Electrophoretic Mobility Shift Assay (EMSA) was carried out using LightShift™ Chemiluminescent EMSA Kit (20148, ThermoFisher Scientific). Untreated and cytokine treated MIN6 nuclear extracts (NEs) were prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents as per manufacturer’s recommendation (78833, ThermoFisher Scientific), supplemented with 1× protease inhibitors (40694200, Roche Diagnostics GmbH). For cytokine-treated condition, MIN6 cells were cultured in T75 flasks to 70% confluency and treated with 10 ng/mL IFN-γ, 0.5 ng/mL IL-1β, and 1 ng/mL TNF-α cytokine mixture prepared fresh 24 h prior to NE preparation. NE protein concentration was determined using a NanoDrop (ThermoFisher Scientific) and samples were stored at −80°C until analyses. Sense single-stranded EMSA oligonucleotides for reference and alternate alleles were purchased from Integrated DNA Technologies, with the following sequences:
rs10483809 (RAD51B): 5′Biotin–ATCTTTCACTTTCCCT[A/G]TCGATACTTCATATGT
rs35342456 (SOCS1): 5′Biotin–GCTGGGCGTGGTGGCTCACGCCTGT[A/C]ATCTTGTTG.
Binding reaction mixtures were prepared for each allele and contained 10× Binding Buffer, 50% glycerol, 0.1 M MgCl2, 1 μg/μL in 10 mM Tris Poly(dI∗dC), 1% NP-40 (20148, ThermoFisher Scientific), 100 and 25fmol of labeled probe for rs10483809 and rs35342456 respectively, and 8–17 μg NE. For corresponding competition reaction(s), 200-fold excess of unlabeled probe at (20 or 5 pmol) was used. Competition reactions were incubated at RT for 10 min with NE and unlabeled probe prior to adding biotin-labeled probe. Reaction mixtures were further incubated for 20 min at RT, and 5× Loading Buffer was added to each mixture to stop the reaction. Empty 6% TBE gel (EC62655BOX, Invitrogen) was run at 100 V in 0.5× UltraPure TBE Buffer (15581-044, Invitrogen, Life Technologies) at 4 ˚C prior to loading samples. Samples were subsequently run on the same gel at 100 V for 90 min at 4˚C. DNA-protein complexes on the gel were transferred to 0.45 mm Biodyne™ Pre-Cut Modified Nylon Membrane (77016, Thermo Scientific) at 380 mÅ for 45 min, and were crosslinked for 15 min using UV Transilluminator (VWR, VWR International). Complexes were detected using Chemiluminescent Nucleic Acid Detection Module (20148, ThermoFisher Scientific) after blocking for 1 h. Images were captured using a C-DiGit Blot Scanner (Model 3600, Li-Cor Biosciences).
Gene reporter assays
We cloned a 400 bp insert containing the rs10483809 reference allele using human DNA (Coriell) as a template into the pGL4.23 luciferase reporter vector (Promega) upstream of the minimal promoter in the forward direction using the restriction enzymes KpnI and SacI. A pGL4.23 reporter containing the alternate allele was then generated through site-directed mutagenesis (SDM) using the Q5 Site-Directed Mutagenesis kit (New England Biolabs). The primer sequences used for SDM were as follows: rs10483809_cloning_FWD CCATGGTTTCTTCCTGGGTA; rs10483809_cloning_REV GCACAAAATAGAAGAAAGATCAAGAA; rs10483809_SDM_P1 TTTCTCTTTCgCAAACTCCTC; rs10483809_SDM_P2 TGTCACTGACTGAGTTGC. For gene reporter assays, MIN6 cells between passages 17–21 were plated at a density of 250,000 viable cells/cm2 in a 48-well plate. The day after plating, MIN6 cells were co-transfected with 500 ng of experimental pGL4.23 vector containing the reference insert, alternate insert, or no insert (empty vector) and 1 ng pRL-SV40 Renilla luciferase reporter vector (Promega) using Lipofectamine 3000 (Thermo Fisher). We also included MIN6 cells that were not transfected as a control. At 24 h post-transfection, cells were fed culture media and, for the cytokine-stimulated cells, high-dose two cytokines or three cytokines were added to the media. At 48 h post-transfection, cells were lysed and used in the Dual-Luciferase Reporter System assay (Promega).
Flow cytometry analysis of EndoC-βH1 apoptosis
Lentivirus construct expressing SOCS1 shRNA (shSOCS1) was obtained from Sigma-Aldrich (TRC# TRCN0000356244, TTTCGCCCTTAGCGTGAAGAT). A none-targeting scramble shRNA (Scramble, CCTAAGGTTAAGTCGCCCTCG) construct in the same vector was used as control. Lentivirus expressing shSOCS1 and Scramble were packaged in 293 T cells and introduced into EndoC-βH1 cells using the spin-inoculation protocol described above. Transduced EndoC-βH1 cells were cultured for two days before a 72-h treatment of vehicle (0.1%BSA) and cytokine (10 ng/mL IFN-γ, 0.5 ng/mL IL-1β, 1 ng/mL TNF-α). Treated EndoC-βH1 cells were then dissociated into single cell suspension using 0.25% Trypsin-EDTA. Cells were washed with 1 mL ice-cold flow buffer comprised of 0.2% BSA in PBS and centrifuged at 200 × gravity for 5 min. Cells were then resuspended in flow buffer containing ApotrackerTM-Green (Biolegend) and Propidium iodide and stained following manufacture’s instruction. Cells were washed twice with 1 mL ice-cold flow buffer and centrifuged at 4 °C and 200 × gravity for 5 min. Cell pellets were resuspended in 300 μL ice-cold flow buffer and analyzed in a FACS LSRFortessa™ system (BD Biosciences). To confirm SOCS1 knock-down, approximately 1.2 million EndoC-βH1 cells were collected and washed before RNA isolation using the RNeasy Micro kit (QIAGEN) according to the manufacturer’s instructions and RT-qPCR was performed. 500 ng for total RNA was converted to cDNA using iScript™ cDNA Synthesis Kit (Bio-Rad). Gene expression was quantified with iQ™ SYBR® Green Supermix (Bio-Rad), using the following primers: SOCS1_1_FWD_qPCR CACGCACTTCCGCACATTC; SOCS1_1_REV_qPCR TAAGGGCGAAAAAGCAGTTCC.
SNP-SELEX variant selection
Variants were selected and classified based on 4 criteria. (1) T1D loci: We selected 86,067 variants from 57 known T1D loci, including the MHC region. Variants at these 57 loci were selected based on: credible set variants from fine mapping data for 36 loci,84 all variants in 1,000 Genomes Project (1KGP) phase 3 EUR in LD (r2>0.2) with index variants at the remaining 21 loci, and all variants in 1KGP with EUR MAF >0.5% in regulatory elements within 250 kb of index variants at all 57 loci. (2) T2D loci: We selected 33,354 variants at known T2D loci, which include lead variants and variants in LD with r2≥0.6 in EUR and non-EUR, and credible variants from fine mapping studies. (3) Islet enhancers: We included 56,796 variants in 1KGP phase 3, filtered for Hardy-Weinberg Equilibrium p-value ≥1e-5 and MAF ≥0.5% that intersected with islet enhancers, defined using published ATAC-Seq and H3K27ac ChIP-Seq data from human islets.85,86 (4) Random: 7,869 negative control variants from filtered 1KGP SNPs, but randomly chosen from the genome were included. Variants from categories 2, 3 and 4 have been included as a validation set in a previous publication.27 The total number of selected variants is 184,086, including 183,373 SNPs and 713 indels. A small subset of variants overlaps between the 4 different selection methods, and therefore in total there were 182,226 distinct variants selected.
SNP-SELEX data generation
The generation of the SNP-SELEX data used in this study was described in a previous study27 although details of how these data were generated are also included here. The oligonucleotide design for each variant consisted of a target sequence of 44 nt containing the variant, flanked by illumina TruSeq dual-index system adapters and barcodes. Three hundred and eighty-four pools of oligonucleotides were synthesized by CustomArray (Seattle, WA), each pool carrying a unique sequence barcode. To control for PCR duplicates, the 3 nucleotides at each end of the 44 nt sequence were synthesized Ns, which generated random combination of nucleotides tagging each molecule. For SNPs, the central position was substituted by an N, resulting in synthesis of all 4 nucleotidic variations (97,758 oligos), while for indels (maximum 3bp-long) both a long (44 nt) and a short (41–43 nt) form were synthesized (259 × 2 oligos). The oligos were double stranded using 20 cycles of PCR and sequenced for 2 × 50 paired-end cycles with Illumina Hiseq 2500 as input references.
The cDNA of 530 distinct TF proteins were cloned into pET20a plasmids87 and expressed using Rosetta (DE3) pLysS E. coli strains using auto-inducing ZYP5052 medium as described in Jolma et al.29 The protein sequences and source for each TF are reported in Yan et al.27 The SNP-SELEX experiments were carried out according to the bead-based SELEX using glutathione Sepharose method described in detail by Jolma et al. 2013,29 which was adapted to a high-throughput liquid handler system (Beckman FXP integrated with a Biotek Plate Washer). For each TF, 6xHis-tagged TF protein was immobilized to Ni Sepharose 6 Fast Flow beads (GE, 17-5318-01) in Promega binding buffer (10 mM Tris pH7.5, 50 mM NaCl, 1 mM MgCl2, 4% glycerol, 0.5 mM EDTA, 5 μg/ml poly-dIdC) across 8 × 96-well plates. Oligos from input were added into the protein beads mixture and incubated at RT for 30 min. Beads were washed for 12 times with the Promega binding buffer and re-suspended in TE (10 mM Tris pH 8.0, 1 mM EDTA). The eluted DNA was amplified by PCR and purified (Qiagen, 28004), and one aliquot was taken for library preparation and sequencing and another aliquot of the same product was added to the protein beads mixtures for a new binding cycle. We performed n = 2 independent replicates of the entire experiment consisting of four binding cycles each. The results of the experiment were sequenced using two flow cells of 2 × 50 paired end illumina Hiseq 2500. To reduce confounding due to systematic synthesis bias, in the second replicate experiment the order of the input pools was inverted (i.e. the same TF protein was hybridized to an oligo pool synthesized with a different barcode).
Quantification and statistical analysis
Details on tests, effect sizes, error and significance estimates are reported in the main text and figure legends for statistical analyses, and additional details including number and unit of replicate samples and correction for multiple tests for each analysis are provided in this section.
ATAC-seq data analysis
Processing
FASTQ reads were trimmed using Trim Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) with flags ‘--paired’ and ‘--quality 10’ and aligned to the hg19 reference genome with BWA mem75 using the ‘-M’ flag. We used Picard to mark duplicate reads and filtered, sorted, indexed, and aligned reads using samtools88 with flags ‘-q 30’, ‘-f 3’, ‘-F 3332’. Mitochondrial reads were also removed. Peaks were called on the filtered reads using MACS274 with parameters ‘--extsize 200 --keep-dup all --shift −100 --nomodel’. We generated bigWig tracks normalized by RPKM for each experiment using bamCoverage.89 TSS enrichment scores for each ATAC-seq experiment were calculated using ‘tssenrich’ (https://pypi.org/project/tssenrich/), as the aggregate read distribution in a 4 kb window centered on the TSS and normalized to an extended region of 1.9 kb on each side, according to the Encyclopedia of DNA Elements (ENCODE) guidelines.
PCA
We identified all peaks identified in at least two individual samples and constructed a read count matrix using edgeR.90 We then calculated normalization factors using the ‘calcNormFactors’ function and used limma to apply the voom transformation and regress out batch effects and sample quality as measured by TSS enrichment scores. We then calculated principal components (PCs) using the top 10,000 most variable peaks using the ‘prcomp’ function with rank 2. The software used to generate PCs is located at https://rdrr.io/github/anthony-aylward/exploreatacseq.91
Differential chromatin accessibility
We generated a ‘master’ set of consensus ATAC-seq peaks by merging reads from all experiments and calling peaks on these merged reads using MACS2 as described above. The peaks were filtered to remove sites found in less than three individual samples and the ENCODE hg19 blacklist v2.92 A count matrix of reads from each sample mapping to this list of peaks was created using featureCounts93 and used for differential accessibility analysis using DESeq2.13 We used the experimental design ‘∼treatment + donor’ where donor is included as a covariate to enable comparing the effects of treatment within donor. p-values were corrected using FDR as computed by the Benjamini-Hochberg method, and we considered sites significant at FDR<0.10. The numbers of distinct islet donors per treated and untreated group were 24 h high-dose three cytokine n = 5, 24 h high-dose two cytokine n = 5, 24 h low-dose three cytokine n = 3, 24 h low-dose two cytokine n = 3, 6 h high-dose three cytokine n = 2, 48 h high-dose three cytokine n = 2, 72 h high-dose three cytokine n = 2; Table S1). To compare the effects of treatment with and without TNF-α (n = 5 distinct islet donors per group), we compared the absolute log2 fold changes from DESeq using a two-sided Wilcoxon signed rank test in R. To identify differentially accessible sites across treatment durations we performed linear regression using the lm() function in R using log2 fold changes as a function of duration and obtained p-values from the regression model. The p-values reported are un-corrected, and a nominal p-value of 0.01 was considered differential across duration.
Motif enrichment analysis
We used the ‘findMotifsGenome’ tool from HOMER76 to test differentially accessible chromatin sites for motif enrichment compared to a background of consensus ATAC-seq peaks, and using the masked hg19 genome as reference.
RNA-seq data analysis
We used STAR (2.5.3a)78 to align paired-end RNA-Seq reads to hg19 genome with a splice junction database built from the Gencode v19 gene annotation94 and the following parameters: --outFilterMultimapNmax 20 --outFilterMismatchNmax 999 --alignIntronMin 20 --alignIntronMax 1000000 --alignMatesGapMax 1000000 --outSAMtype BAM Unsorted --quantMode TranscriptomeSAM. Gene expression values were quantified using the RSEM package (1.3.1)79 with default parameters and loaded into R for further processing. Genes were filtered for >0.1 TPM on average per sample with 22,175 genes remaining after filtering. Raw expression counts were normalized using voom transformation from limma package and corrected for sample batch effects using limma removeBatchEffect. The R prcomp function was used to perform principal component analysis for the top 500 most variable genes. We identified differentially expressed genes between each cytokine treatment (high-dose three cytokine n = 4 distinct islet donors, high-dose two cytokine n = 4 distinct islet donors, low-dose three cytokine n = 2 distinct islet donors, low-dose two cytokine n = 2 islet donors) and untreated conditions using DESeq213 with default settings using design = ∼ donor + condition where donor is included as a covariate to enable comparing treatment effects within donors. p-values were corrected using FDR, and an FDR threshold of 0.10 was used for significance. Metascape (metascape.org) was used to perform gene ontology enrichment analysis with default settings.
Single nuclei ATAC-Seq data analysis
Processing
10× Genomics Cell Ranger ATAC v1.1 (cellranger-atac count) was used to process 10× fastq files for each sample and perform alignment to the hg19 reference genome. For each assay, we then removed barcode multiplets using Cell Ranger’s multiplet removal script (version 1.1). BAM files were filtered for PCR duplicates, converted into tagAlign files, and intersected with a reference set of islet ATAC-seq peaks14 to construct a sparse matrix containing read counts in peaks for each cell. Cells with a minimum of 500 (sample SAMN15337453, untreated), 1,000 (samples SAMN12833535 and SAMN12889245), or 4,000 (sample SAMN15337453, treated and sample SAMN15314807) total mapped reads were retained for further analysis.
Clustering
Prior to combining all samples, each assay was clustered separately using scanpy v.1.6.0.77 First, we extracted highly variable peaks using mean read depth and dispersion. Read depth was normalized and log-transformed counts were regressed out within highly variable peaks. We then performed PCA analysis and obtained the top 50 principal components. We calculated the nearest 30 neighbors using cosine metric to perform UMAP dimensionality reduction (min_dist = 0.3) and clustering using the Leiden algorithm. For each assay, cells with low usable counts and fraction of reads in peaks were iteratively removed. In order to obtain more accurate clustering and cell type assignment, the filtered assays were then merged and combined with 3 independent islet snATAC datasets from non-diabetic individuals (A0019, C0025, C0026) filtered using the same criteria as above, and the top 50 PCs were obtained from the merged experiments. Harmony95 was then used to batch correct PCs for experiments. Using the corrected PCs, we applied the UMAP dimensionality reduction method, and clustered cells using the Leiden algorithm (Resolution = 0.5), and sub-clustered using the Louvain algorithm (Resolution = 1.5). Low-quality cells from the merged clusters were iteratively removed and manual doublet removal was performed on sub-clusters with above average high usable read depth or those that expressed multiple marker genes. After the entire filtering process, 28,853 cells were removed in total, and the final merged cluster contained 25,200 cells (untreated cells: 21,318; cytokine-treated cells: 3882) mapping to 10 clusters. Cell type of each cluster was assigned based on chromatin accessibility at promoter regions of known marker genes14 and verified through UCSC genome browser tracks. The 3 independent islet samples were removed from the final clustering and the remaining cells (untreated cells: 3,947, cytokine-treated cells: 3,882) were used for downstream analysis. To identify sub-populations of beta and alpha cells, the remaining cells mapping to either cell type were re-clustered separately, after Harmony batch correction for experiment, and using Leiden algorithm with resolution 0.1. The scanpy rank_genes_groups() function was used to identify the most different accessible sites between the two sub-clusters, which were then assigned on the closest gene TSS (max distance 50,000 bp). GO analysis was performed using the R library enrichR against the GO_Biological_Process_2021 database, on genes corresponding to peaks with logFC >0.5 and p.adjust<0.05.
snATAC pooled sample demultiplexing
To assign the pooled assays to the two sample donors, we genotyped non-islet tissue from the two samples. During islet picking, non-islet cells were collected separately from the islets, washed with 1X HBSS, pelleted at 500rcf for 5 min, and snap frozen with liquid nitrogen until genomic DNA extraction. We extracted genomic DNA from the non-islet cells using the PureLink Genomic DNA mini kit. Samples were genotyped by the UCSD Institute for Genomic Medicine using the Illumina Infinium Omni 2.5–8 assay. Genotypes were called using GenomeStudio (v2.0.4) with default settings. Using PLINK,96 we filtered out rare variants with MAF <0.01 in the Haplotype Reference Consortium panel r1.1 and ambiguous alleles with MAF >0.4. Filtered variants were used to impute genotypes into the HRC r1.1 panel using the Michigan Imputation Server with minimac4. Genotypes with high imputation quality (R2>0.3) were used to demultiplex pooled snATAC samples using Demuxlet97 with default settings.
Peak calling
To identify chromatin accessibility peaks in each islet cell type, we extracted the reads from all cells within a given cluster and generated separate tagAlign files for each cell type. To correct for the 9-nt duplication created by Tn5 transposase, we shifted the reads aligned to the positive strand by +4 bp and reads aligned to the negative strand by -5 bp. We then called peaks using MACS274 with the parameters ‘q 0.05’, ‘--nomodel’, ‘--keep-dup all’, and ‘g hs’. Blacklisted regions (v.2) from ENCODE were removed. The bedgraph output by MACS2 was sorted, normalized to counts per million (CPM), and converted to bigwig for visualization on UCSC genome browser. The peak calls from the individual cell types were then used to annotate the consensus set of peaks identified in bulk islet ATAC using bedtools intersect (v2.26.0).
Differential chromatin accessibility in islet cell types
We generated distinct BAM files for each cell type, donor and condition, using the barcodes to extract reads from the filtered and duplicate-removed BAM files from each assay using ‘samtools’ and ‘grep’. There were n = 3 and n = 4 islet donors in treated and untreated groups, respectively. For each cell type, we generated a matrix of read counts mapping to bulk ATAC consensus peaks using featureCounts.93 Each matrix was filtered for an average read depth of 1 per sample/condition. We used DESeq2 to identify differentially accessible sites between cytokine treated and untreated samples with donor as a covariate (design = ∼ treatment + donor). p-values were corrected using FDR, and an FDR threshold of 0.10 was used for significance. To visualize results as heatmap and hierarchical clustering, we concatenated the matrices for each cell types, normalized the raw counts using DESeq variance stabilizing transformation (vst) function, filtered for peaks with differential accessibility in at least one cell type and plotted the resulting matrix using ‘pheatmap’. To compare the effects of cytokine treatment between beta cells and bulk islets and between beta cells and alpha cells, we compared the absolute log2 fold changes from DESeq at the same peaks using a two-sided Wilcoxon signed rank test in R.
Co-accessibility
Using Cicero (version 1.4.4),16 we calculated co-accessibility between pairs of snATAC peaks. To indicate which cells were accessible in which peak, we created a sparse m x n binary matrix by encoding cells from a given cell type (n) and merged peaks across all cell types (m), obtained using bedtools merge. We calculated Cicero co-accessibility scores following the recommended analysis protocol (https://cole-trapnell-lab.github.io/cicero-release/docs/#recommended-analysis-protocol), using the 30 nearest neighbors of UMAP coordinates to aggregate cells, and a window size of 1Mb to calculate cicero models. We then set a threshold of 0.05 and a minimum distance of 10 kb to define pairs co-accessible for a given cell type. Co-accessibility was calculated for either untreated beta cells, cytokine-treated beta cells or merged treated-untreated beta cells. To annotate co-accessibility links between distal and promoter peaks, we categorized peaks within a 5 kb window of a transcription start site (+/− 2.5 kb from TSS (GENCODE version 19) as ‘promoter’, and otherwise as ‘distal’. To calculate enrichment in cytokine-responsive cCRE for concordant effects with distal genes, we annotated each bulk ATAC consensus peak with results of differential accessibility in islets (24 h high-dose three cytokines) and co-accessibility in beta cells (merged treated-untreated conditions) with at least one gene with differential expression (24 h high-dose three cytokines). We then performed Fisher exact test on each combination of direction of effects (upregulated cCRE vs upregulated gene, upregulated cCRE vs downregulated gene, downregulated cCRE vs upregulated gene and downregulated cCRE vs downregulated gene). The same test was performed for cCREs proximal to gene promoters (<10 kb from TSS).
Motif enrichment analysis
Using ChromVAR15 (version 1.8.0) we calculated the deviation in accessibility from expected accessibility within islet cell types. We used a binary sparse matrix of accessible cells in each ATAC peak (see above) as input, and add GC bias using the ‘BSgenome.Hsapiens.UCSC.hg19’ library for genome sequence input. We then filtered cells with a minimum depth of 1500 and a minimum proportion of reads in peaks of 0.15, and filtered peaks for non-overlapping coordinates. The remaining peaks were annotated for motif occurrence from the JASPAR database, using the matchMotifs function from the motifmatchr package. We then computed deviations and variability for each cell type separately (alpha and beta) with the provided ChromVAR functions. For each transcription factor (n = 386) we then plotted the absolute difference of the average deviation scores of cytokine treated and untreated cells in alpha and beta cells using a scatterplot.
HiChIP data analysis
Data was processed with the MAPS v2.0 pipeline using default settings.80 We used hg19 as the reference genome and H3K27ac ChIP-seq peaks in EndoC-βH1 cells from a published study.98 The p-values obtained from MAPS were corrected using FDR, and we retained all interactions between 5 kb windows both containing a H3K27ac peak at FDR<0.10. Significant interactions were intersected with promoter regions of genes from GENCODE to identify enhancer-promoter interactions. Contact matrices were generated using the pre command from Juicer tools.99 For virtual 4C analyses, we extracted all contacts which included the 5 kb window around the site of interest.
Analysis of CRISPR screen results
Adaptor sequences ggaaaggacgaaacaccg and gttttagagctagaaatagca flanking the 19–20 base pair of sgRNA sequences were trimmed using cutadapt. Trimmed sequencing reads were then aligned to the reference sgRNA library with bowtie2 with default settings, resulting in a BAM file that can be used for sgRNA counting with the MAGeCK model-based tool for CRISPR-Cas9 knockout screens.81 Statistical significance of guide RNA representation in control and cytokine treated datasets for n = 18,703 genes was estimated with the mle subcommand in the MAGeCK package and p-values were corrected using FDR. An effect (beta) for each gene from this analysis was extracted as an indicator of enrichment (positive beta) or depletion (negative beta) of sgRNAs targeting this gene in the cytokine-treated cells. miRNA genes and genes with less than 3 sgRNA guides were excluded from further analysis. Significantly enriched (427) or depleted (440) genes at FDR<0.10 were further filtered for expression in islet (average sample TPM>=1). Gene ontology analysis was performed using GSEA82 (http://www.gsea-msigdb.org/gsea/msigdb/annotate.jsp) against the REACTOME and GO biological process gene sets, including only gene sets with more than 20 and less than 1000 genes. A two-sided Fisher exact test was used to calculate enrichment of pro-cell survival and pro-cell death genes segregated by up-regulated, down-regulated or no change in expression in high-dose cytokine treatment within 1Mb of known T1D risk loci including MHC9 compared to other genes expressed in islets and tested in the screen, The p-values are reported as un-corrected.
SNP-SELEX sequencing data analysis
Sequence processing
FASTQ files from each cycle and input were first filtered for identical sequences using FastUniq (v1.1),100 which removed on average 10% of reads in each experiment, to a final median depth of 3 and 0.64 million paired-end reads for the input and the selected oligos respectively. Sequencing reads were then aligned using BWA-MEM (version 0.7.12)75 to the oligo library fasta files. For each oligo, the number of read pairs carrying each nucleotide was counted, only counting reads that were uniquely mapped, correctly paired, with quality = 60 and with the same sequence at the SNP position. Oligos with less than 8 read pairs for SNPs and 4 reads pairs for indels were excluded for further analysis. To estimate the consistency between the two experimental replicates we calculated the correlation between the proportion of reads aligning to a given SNP over the total number of reads in each experiment. The proportion of reads aligning to each oligo in each experiment was well correlated between the two replicates (median Pearson coefficient r = 0.86), and the correlation increased over the cycles, indicating that the selection for the same oligo by a given protein was reproducible between the two replicates. We excluded that this result was due to the starting oligonucleotide stoichiometry in the pools, as the different replicates had different input material.
Motif analysis was performed on the oligo sequences (40 nt) that were selected at cycle 4 of each experiment to determine the enrichment for the expected motif or family of motifs. For motif enrichment we used HOMER (library 4.7)87 and MEME 4.12.0 (libraries: JASPAR_CORE_2014_vertebrates, jolma2013, encode_known, Mariani_2017 and Barrera_2016).101 A positive motif match was determined if the expected motif (matching with the first three letters of the name) or a motif from the same structural family (defined by homer classification) were found among the top 20 enriched motifs. For 564 experiments, we found a positive motif match in both replicates, for 90 in either of the two replicates and for 114 in none of the replicates. Because for some analyzed TFs the motif is not known, for example for Zinc Finger proteins, we did not consider failed experiment only based on the motif enrichment, but also on the correlation between replicates. If the correlation between replicates was <0.5 and one of the two replicates was enriched for the expected motif, then remove only the replicate that did not contain the motif (16 experiments removed). If the correlation was <0.5 and both replicates did not have motif in the corresponding family, we removed both replicates (53 experiments removed).
Identification of variants with differential TF binding
For each experimental replicate, allelic counts were tabulated for each oligo at each cycle, including only those variants covered by at least 8 read pairs for SNPs, or 4 reads pairs for indels, in all five cycles (0–4). Furthermore, variants with less than 2 read pairs in the input for both the reference and alternate alleles and composing <5% of the total reads in the pool were removed, as potentially biased inputs. To quantifying the magnitude of the difference between reference and alternate allele binding across all cycles, we used the “Preferential Binding Score” or PBS, which has been previously described.27 The PBS corresponds to the AUC between the differences of log odds ratios of the two alleles compared to cycle 0 (the input), and is calculated as follows:
-
1)
For a given oligo, the odds of allele a at cycle c is defined by the frequency P of allele a at cycle c, divided by 1-Pa,c, which is equal to the read counts of allele a divided by the sum of read counts of all other nucleotides r: Odds a,c = P a,c/(1-P a,c) = counts (a,c)/counts(r,c)
-
2)
The odds ratio is calculated as the ratio between the odds of allele a at cycle c and the odds of allele a at cycle 0: OR a,c = Odds a,c/Odds a,0
-
3)
LogOR are calculated for reference and alternate allele for each cycle: LogORa,c = log10(counts (a,c)) + log10(counts (rest, 0)) - log10(counts(a,0)) -log10(counts (rest,c))
-
4)
The PBS is the AUC of the difference between LogORref and LogORalt (ΔLogOR), calculated with the formula:
For each experiment replicate, to determine the statistical significance of the observed values, a Monte Carlo randomization was conducted, which consisted of 250,000 randomly generated PBS measurements. The randomizations consisted of shuffling the SNP labels 250,000 times within each cycle and one PBS measurement was extracted each time. We observed that experiments with fewer than 25 oligos generated non-normal PBS random distributions, therefore experiments with less than 25 variants remaining after the above filtering steps were excluded.
After calculating preferential binding statistics in each individual experiment (same “well”, two technical replicates), results of the two replicates for each experiment were combined using meta-analysis of p-values, weighted on the total number or reads for reference and alternate allele in cycles 1 to 4, and the average of effect sizes (PBS). Further, experimental replicates of the same TF protein (different “wells”, variable number of replicates) were meta-analyzed to obtain a unique value for each TF. The p-values reported are un-corrected, and variants at a nominal p-value of 0.05 were considered to affect TF binding.
Correlation of SNP-SELEX results between transcription factors
To compare variant effects on binding of different TFs, we first computed a matrix of PBS scores where each row corresponded to an SNP and each column to a TF. After filtering the matrix to retain only TFs with at least 50 bound variants, TF families with at least 3 components and variants that were pbSNPs in at least one TF (27,655 variants and 457 TFs), we calculated a pairwise correlation matrix using the cor() function in R, using the “pairwise” option. To perform hierarchical clustering on TFs, we filtered the pairwise correlation matrix, retaining only rows and columns with non-missing values (264 TFs). The dendrogram of the hierarchical clustering of distances was obtained using the R command as.dendrogram(hclust(as.dist(1-correlation_matrix))) and plotted using functions form the “circlize” R package.
Correlation between SNP-SELEX and SNP effect predictions
Predictions of TF motif alteration by SNPs were calculated using the package motifbreakR.102 All variants analyzed by SNP-SELEX were first formatted and filtered using the function snps.from.file (search.genome = BSgenome.Hsapiens.UCSC.hg19) and the resulting 181,540 variants were tested against the H.sapiens 'HOCOMOCOv10' library of motifs PWM (640 TFs, including most of those tested by SNP-SELEX), and using the parameters: filterp = TRUE, method = "ic", threshold = 5e-4, BPPARAM = BiocParallel:bpparam("SerialParam"). 177,270 variants were predicted to alter at least one of the motifs of the library. For each SNP, the difference of PWM scores from the two alleles was compared with the PBS scores from SELEX from the corresponding TFs (500 unique TFs, 129,842 unique variants, 1,896,977 combinations). Pearson correlation coefficient was calculated for 234 s TF that had a minimum of 10 testable, bound SNPs with PWM predicted effects, or 146 when only considering pbSNPs. Similarly, predictions of SNP effect on TF binding were obtained from DeepSEA calculations (http://deepsea.princeton.edu/job/analysis/create/) and filtered for E-value <0.01. For each TF in the database, the predicted allelic log2 fold change of each SNP was averaged across the different cell types and then compared with SELEX PBS scores, for TFs having a minimum of 10 bound SNP (37 TFs: ATF2, ATF3, BATF, CEBPB, CTCF, E2F4, ELF1, ELK1, ELK4, ETS1, FOSL1, FOXA1, FOXA2, FOXM1, FOXP2, GATA2, GATA3, IRF3, IRF4, MEF2C, MYBL2, NANOG, NFATC1, NFIC, POU2F2, POU5F1, PRDM1, RFX5, RUNX3, RXRA, SRF, TCF12, TCF7L2, USF1, USF2, YY1, ZBTB7A) or pbSNPs (24 TFs).
Genetic association enrichment analysis
We tested variants with allelic effects on TF binding for enrichment of T1D association using genome-wide summary statistic data.9 We defined three categories of variants: (i) all variants, (ii) mapping in beta cell cCREs, (iii) mapping in cytokine-responsive beta cell cCREs. For each variant category, we identified several different p-value thresholds and segregated SNP-SELEX variants based on (i) allelic effects of TF binding, or no allelic effect on TF binding, (ii) reaching p-value threshold or not, and then for each comparison performed a two-sided Fisher exact test to obtain odds ratios and 95% CI.
Gene reporter assays
Luciferase activity from the experimental plasmids, normalized by dividing by the corresponding Renilla activity, was compared to the normalized activity of the empty pGL4.23 vector. For each allele there were n = 9 distinct transfections per group. A two-sided t-test was used to compare activity between the alternate and reference allele. The p-values reported are un-corrected. We determined whether the data met the assumptions of the test prior to statistical analysis using Shapiro-Wilk and quantile-quantile (QQ) plots.
Electrophoretic mobility shift assay
For rs35342456, n = 3 distinct EMSA experiments were performed and for one of these experiments a technical replicate of the binding reaction was also performed (Figure S6). For rs10483809 n = 1 EMSA experiment was performed. Images were visually inspected for concordance across replicates and no statistical analyses were performed.
qPCR and flow cytometry analysis of EndoC-βH1 apoptosis
The qPCR analysis of SOCS1 knock-down in EndoC-βH1 cells was carried out using the ΔΔCt method using n = 3 transductions with lentivirus expressing scramble or SOCS1 shRNA. p-values were calculated by two-sided t-test (Figure S7A). To measure apoptosis, for each group we performed n = 4 distinct transductions and performed flow cytometry analysis using FlowJo v10. In brief, intact, singlet cells were gated based on forward and side scatter intensity of epifluorescence. Propidium iodide and ApotrackerTM-Green signals from the gated cells were plotted on a scatterplot. Apoptotic cells were quantified by Apotracker+ and Propidium iodide-staining (Figures S7B and S7C). A two-way ANOVA was used to evaluate the effect of shRNA as well as an interaction between shRNA and treatment status, and Tukey’s HSD test was used for pairwise comparisons. We determined whether the data met the assumptions of the test prior to statistical analysis using Shapiro-Wilk and quantile-quantile (QQ) plots.
Acknowledgments
The work performed in this study was supported by NIH grants DK122607 and DK120429 to K.J.G. and M.S.; DK112155 to M.S., K.F., K.J.G., and B.R.; and DK105541 to M.S., K.F., and B.R.
Author contributions
K.J.G. and M.S. conceived of and supervised the study. K.J.G., P.B., H.Z., M.O., and M.S. wrote the manuscript. J.T. and B.R. supervised the design and generation of SNP-SELEX data. K.F. supervised the design of SNP-SELEX and analyses of genomic data. S.P. supervised generation of single-cell data. P.B. performed statistical data analysis of genomic, CRISPR-KO screen, and genetic data; H.Z. performed the CRISPR-KO screen and interpreted the results; M.O. and M.M. performed genomic assays. J.Y. performed the SNP-SELEX assay. P.B., M.O., R.E., E.B., K.K., A.A., J.C., G.W., and J.N. performed bulk and single-cell data analysis. M.O., R.E., J.K., and S.C. performed functional validation experiments. P.B., N.N., Y.Q., A.A., and M.K.D. performed design and analysis of SNP-SELEX data. All authors contributed to and approved of the final version of the manuscript.
Declaration of interests
K.J.G. is a consultant of Genentech and holds stock in Neurocrine Biosciences. B.R. is a consultant of Arima Genomics and a co-founder of Epigenome Technologies. P.B. is an employee of Shoreline Bioscience. N.N. is an employee of Guardant Health. E.B. is an employee and shareholder of Aetion. K.K. is an employee of Cartography Bio. Y.Q. is an employee of Sana Biotechnology. M.D. is an employee and shareholder of Seer. J.C. is an employee and shareholder of Pfizer.
Published: November 11, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100214.
Supplemental information
Data and code availability
-
•
The ATAC-seq, RNA-seq, snATAC-seq, HiChIP and CRISPR screen data are available at the NCBI Gene Expression Omnibus (GEO) repository under accession number GSE205853 at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE205853. The SELEX-seq data are in GEO under accession number GSE118725 at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE118725. Supplementary summary-level files from genomics experiments are available in Zenodo under accession number https://doi.org/10.5281/zenodo.7084136 at https://zenodo.org/record/7106728.
-
•
Custom code for data processing and statistical analyses are publicly available on Github at https://github.com/Gaulton-Lab/BetaCells_cytokines_T1D and deposited in Zenodo under https://doi.org/10.5281/zenodo.7183895.
-
•
Any additional information related to analysis of the data reported in this paper is available from the lead contact upon request.
References
- 1.Atkinson M.A. The pathogenesis and natural history of type 1 diabetes. Cold Spring Harb. Perspect. Med. 2012;2:a007641. doi: 10.1101/cshperspect.a007641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eizirik D.L., Colli M.L., Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat. Rev. Endocrinol. 2009;5:219–226. doi: 10.1038/nrendo.2009.21. [DOI] [PubMed] [Google Scholar]
- 3.Brozzi F., Nardelli T.R., Lopes M., Millard I., Barthson J., Igoillo-Esteve M., Grieco F.A., Villate O., Oliveira J.M., Casimir M., et al. Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia. 2015;58:2307–2316. doi: 10.1007/s00125-015-3669-6. [DOI] [PubMed] [Google Scholar]
- 4.Nunemaker C.S. Considerations for defining cytokine dose, duration, and milieu that are appropriate for modeling chronic low-grade inflammation in type 2 diabetes. J. Diabetes Res. 2016;2016:1–9. doi: 10.1155/2016/2846570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ortis F., Naamane N., Flamez D., Ladrière L., Moore F., Cunha D.A., Colli M.L., Thykjaer T., Thorsen K., Orntoft T.F., Eizirik D.L. Cytokines interleukin-1 and tumor necrosis factor- regulate different transcriptional and alternative splicing networks in primary -cells. Diabetes. 2010;59:358–374. doi: 10.2337/db09-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eizirik D.L., Sammeth M., Bouckenooghe T., Bottu G., Sisino G., Igoillo-Esteve M., Ortis F., Santin I., Colli M.L., Barthson J., et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 2012;8:e1002552. doi: 10.1371/journal.pgen.1002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramos-Rodríguez M., Raurell-Vila H., Colli M.L., Alvelos M.I., Subirana-Granés M., Juan-Mateu J., Norris R., Turatsinze J.-V., Nakayasu E.S., Webb-Robertson B.-J.M., et al. The impact of proinflammatory cytokines on the β-cell regulatory landscape provides insights into the genetics of type 1 diabetes. Nat. Genet. 2019;51:1588–1595. doi: 10.1038/s41588-019-0524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rabinovitch A., Suarez-Pinzon W.L. Cytokines and their roles in pancreatic islet beta-cell destruction and insulin-dependent diabetes mellitus. Biochem. Pharmacol. 1998;55:1139–1149. doi: 10.1016/s0006-2952(97)00492-9. [DOI] [PubMed] [Google Scholar]
- 9.Chiou J., Geusz R.J., Okino M.-L., Han J.Y., Miller M., Melton R., Beebe E., Benaglio P., Huang S., Korgaonkar K., et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature. 2021;594:398–402. doi: 10.1038/s41586-021-03552-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robertson C.C., Inshaw J.R.J., Onengut-Gumuscu S., Chen W.-M., Santa Cruz D.F., Yang H., Cutler A.J., Crouch D.J.M., Farber E., Bridges S.L., et al. Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes. Nat. Genet. 2021;53:962–971. doi: 10.1038/s41588-021-00880-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore F., Colli M.L., Cnop M., Esteve M.I., Cardozo A.K., Cunha D.A., Bugliani M., Marchetti P., Eizirik D.L. PTPN2, a candidate gene for type 1 diabetes, modulates interferon-gamma-induced pancreatic beta-cell apoptosis. Diabetes. 2009;58:1283–1291. doi: 10.2337/db08-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dos Santos R.S., Marroqui L., Velayos T., Olazagoitia-Garmendia A., Jauregi-Miguel A., Castellanos-Rubio A., Eizirik D.L., Castaño L., Santin I. DEXI, a candidate gene for type 1 diabetes, modulates rat and human pancreatic beta cell inflammation via regulation of the type I IFN/STAT signalling pathway. Diabetologia. 2019;62:459–472. doi: 10.1007/s00125-018-4782-0. [DOI] [PubMed] [Google Scholar]
- 13.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiou J., Zeng C., Cheng Z., Han J.Y., Schlichting M., Miller M., Mendez R., Huang S., Wang J., Sui Y., et al. Single-cell chromatin accessibility identifies pancreatic islet cell type– and state-specific regulatory programs of diabetes risk. Nat. Genet. 2021;53:455–466. doi: 10.1038/s41588-021-00823-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schep A.N., Wu B., Buenrostro J.D., Greenleaf W.J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods. 2017;14:975–978. doi: 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pliner H.A., Packer J.S., McFaline-Figueroa J.L., Cusanovich D.A., Daza R.M., Aghamirzaie D., Srivatsan S., Qiu X., Jackson D., Minkina A., et al. Cicero predicts cis-regulatory DNA interactions from single-cell chromatin accessibility data. Mol. Cell. 2018;71:858–871.e8. doi: 10.1016/j.molcel.2018.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Igoillo-Esteve M., Gurzov E.N., Eizirik D.L., Cnop M. The transcription factor B-cell lymphoma (BCL)-6 modulates pancreatic {beta}-cell inflammatory responses. Endocrinology. 2011;152:447–456. doi: 10.1210/en.2010-0790. [DOI] [PubMed] [Google Scholar]
- 18.Wu H., Panakanti R., Li F., Mahato R.I. XIAP gene expression protects β-cells and human islets from apoptotic cell death. Mol. Pharm. 2010;7:1655–1666. doi: 10.1021/mp100070j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Good A.L., Cannon C.E., Haemmerle M.W., Yang J., Stanescu D.E., Doliba N.M., Birnbaum M.J., Stoffers D.A. JUND regulates pancreatic β cell survival during metabolic stress. Mol. Metabol. 2019;25:95–106. doi: 10.1016/j.molmet.2019.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chong M.M.W., Chen Y., Darwiche R., Dudek N.L., Irawaty W., Santamaria P., Allison J., Kay T.W.H., Thomas H.E. Suppressor of cytokine signaling-1 overexpression protects pancreatic β cells from CD8 + T cell-mediated autoimmune destruction. J. Immunol. 2004;172:5714–5721. doi: 10.4049/jimmunol.172.9.5714. [DOI] [PubMed] [Google Scholar]
- 21.Suo G.J., Qin J., Zhong C.P., Zhao Z.X. Suppressor of cytokine signaling 1 inhibits apoptosis of islet grafts through caspase 3 and apoptosis-inducing factor pathways in rats. Transplant. Proc. 2010;42:2658–2661. doi: 10.1016/j.transproceed.2010.04.039. [DOI] [PubMed] [Google Scholar]
- 22.Sharma R.B., Alonso L.C. Lipotoxicity in the pancreatic beta cell: not just survival and function, but proliferation as well? Curr. Diabetes Rep. 2014;14:492. doi: 10.1007/s11892-014-0492-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moss N.D., Sussel L. mRNA processing: an emerging frontier in the regulation of pancreatic β cell function. Front. Genet. 2020;11:983. doi: 10.3389/fgene.2020.00983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kowluru A. Oxidative stress in cytokine-induced dysfunction of the pancreatic beta cell: known knowns and known unknowns. Metabolites. 2020;10:E480. doi: 10.3390/metabo10120480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hara T., Mahadevan J., Kanekura K., Hara M., Lu S., Urano F. Calcium efflux from the endoplasmic reticulum leads to β-cell death. Endocrinology. 2014;155:758–768. doi: 10.1210/en.2013-1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu T., Zhang S., Xu J., Zhang Y., Sun T., Shao Y., Wang J., Tang W., Chen F., Han X. HRD1, an important player in pancreatic β-cell failure and therapeutic target for type 2 diabetic mice. Diabetes. 2020;69:940–953. doi: 10.2337/db19-1060. [DOI] [PubMed] [Google Scholar]
- 27.Yan J., Qiu Y., Ribeiro Dos Santos A.M., Yin Y., Li Y.E., Vinckier N., Nariai N., Benaglio P., Raman A., Li X., et al. Systematic analysis of binding of transcription factors to noncoding variants. Nature. 2021;591:147–151. doi: 10.1038/s41586-021-03211-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jolma A., Kivioja T., Toivonen J., Cheng L., Wei G., Enge M., Taipale M., Vaquerizas J.M., Yan J., Sillanpää M.J., et al. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 2010;20:861–873. doi: 10.1101/gr.100552.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jolma A., Yan J., Whitington T., Toivonen J., Nitta K.R., Rastas P., Morgunova E., Enge M., Taipale M., Wei G., et al. DNA-binding specificities of human transcription factors. Cell. 2013;152:327–339. doi: 10.1016/j.cell.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 30.Zhou J., Troyanskaya O.G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods. 2015;12:931–934. doi: 10.1038/nmeth.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wingender E., Schoeps T., Haubrock M., Krull M., Dönitz J. TFClass: expanding the classification of human transcription factors to their mammalian orthologs. Nucleic Acids Res. 2018;46:D343–D347. doi: 10.1093/nar/gkx987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khetan S., Kales S., Kursawe R., Jillette A., Ulirsch J.C., Reilly S.K., Ucar D., Tewhey R., Stitzel M.L. Functional characterization of T2D-associated SNP effects on baseline and ER stress-responsive β cell transcriptional activation. Nat. Commun. 2021;12:5242. doi: 10.1038/s41467-021-25514-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spracklen C.N., Shi J., Vadlamudi S., Wu Y., Zou M., Raulerson C.K., Davis J.P., Zeynalzadeh M., Jackson K., Yuan W., et al. Identification and functional analysis of glycemic trait loci in the China Health and Nutrition Survey. PLoS Genet. 2018;14:e1007275. doi: 10.1371/journal.pgen.1007275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kulzer J.R., Stitzel M.L., Morken M.A., Huyghe J.R., Fuchsberger C., Kuusisto J., Laakso M., Boehnke M., Collins F.S., Mohlke K.L. A common functional regulatory variant at a type 2 diabetes locus upregulates ARAP1 expression in the pancreatic beta cell. Am. J. Hum. Genet. 2014;94:186–197. doi: 10.1016/j.ajhg.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fogarty M.P., Cannon M.E., Vadlamudi S., Gaulton K.J., Mohlke K.L. Identification of a regulatory variant that binds FOXA1 and FOXA2 at the CDC123/CAMK1D type 2 diabetes GWAS locus. PLoS Genet. 2014;10:e1004633. doi: 10.1371/journal.pgen.1004633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suwaki N., Klare K., Tarsounas M. RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin. Cell Dev. Biol. 2011;22:898–905. doi: 10.1016/j.semcdb.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 37.Aylward A., Chiou J., Okino M.-L., Kadakia N., Gaulton K.J. Shared genetic risk contributes to type 1 and type 2 diabetes etiology. Hum. Mol. Genet. 2018 doi: 10.1093/hmg/ddy314. [DOI] [PubMed] [Google Scholar]
- 38.Soleimanpour S.A., Gupta A., Bakay M., Ferrari A.M., Groff D.N., Fadista J., Spruce L.A., Kushner J.A., Groop L., Seeholzer S.H., et al. The diabetes susceptibility gene Clec16a regulates mitophagy. Cell. 2014;157:1577–1590. doi: 10.1016/j.cell.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kameswaran V., Golson M.L., Ramos-Rodríguez M., Ou K., Wang Y.J., Zhang J., Pasquali L., Kaestner K.H. The dysregulation of the DLK1-MEG3 locus in islets from patients with type 2 diabetes is mimicked by targeted epimutation of its promoter with TALE-DNMT constructs. Diabetes. 2018;67:1807–1815. doi: 10.2337/db17-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Viñuela A., Varshney A., van de Bunt M., Prasad R.B., Asplund O., Bennett A., Boehnke M., Brown A.A., Erdos M.R., Fadista J., et al. Genetic variant effects on gene expression in human pancreatic islets and their implications for T2D. Nat. Commun. 2020;11:4912. doi: 10.1038/s41467-020-18581-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santin I., Moore F., Colli M.L., Gurzov E.N., Marselli L., Marchetti P., Eizirik D.L. PTPN2, a candidate gene for type 1 diabetes, modulates pancreatic -cell apoptosis via regulation of the BH3-only protein bim. Diabetes. 2011;60:3279–3288. doi: 10.2337/db11-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liau N.P.D., Laktyushin A., Lucet I.S., Murphy J.M., Yao S., Whitlock E., Callaghan K., Nicola N.A., Kershaw N.J., Babon J.J. The molecular basis of JAK/STAT inhibition by SOCS1. Nat. Nat. Commun. 2018;9:1558. doi: 10.1038/s41467-018-04013-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Powell J.A., Pitman M.R., Zebol J.R., Moretti P.A.B., Neubauer H.A., Davies L.T., Lewis A.C., Dagley L.F., Webb A.I., Costabile M., Pitson S.M. Kelch-like protein 5-mediated ubiquitination of lysine 183 promotes proteasomal degradation of sphingosine kinase 1. Biochem. J. 2019;476:3211–3226. doi: 10.1042/BCJ20190245. [DOI] [PubMed] [Google Scholar]
- 44.Xie Y., Ostriker A.C., Jin Y., Hu H., Sizer A.J., Peng G., Morris A.H., Ryu C., Herzog E.L., Kyriakides T., et al. LMO7 is a negative feedback regulator of transforming growth factor β signaling and fibrosis. Circulation. 2019;139:679–693. doi: 10.1161/CIRCULATIONAHA.118.034615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lundh M., Bugliani M., Dahlby T., Chou D.H.-C., Wagner B., Ghiasi S.M., De Tata V., Chen Z., Lund M.N., Davies M.J., et al. The immunoproteasome is induced by cytokines and regulates apoptosis in human islets. J. Endocrinol. 2017;233:369–379. doi: 10.1530/JOE-17-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Concannon C.G., Koehler B.F., Reimertz C., Murphy B.M., Bonner C., Thurow N., Ward M.W., Villunger A., Strasser A., Kögel D., Prehn J.H.M. Apoptosis induced by proteasome inhibition in cancer cells: predominant role of the p53/PUMA pathway. Oncogene. 2007;26:1681–1692. doi: 10.1038/sj.onc.1209974. [DOI] [PubMed] [Google Scholar]
- 47.Grimm L.M., Osborne B.A. Apoptosis and the proteasome. Results probl. Results Probl. Cell Differ. 1999;23:209–228. doi: 10.1007/978-3-540-69184-6_10. [DOI] [PubMed] [Google Scholar]
- 48.Drexler H.C. Programmed cell death and the proteasome. Apoptosis. 1998;3:1–7. doi: 10.1023/a:1009604900979. [DOI] [PubMed] [Google Scholar]
- 49.Ghosh R., Colon-Negron K., Papa F.R. Endoplasmic reticulum stress, degeneration of pancreatic islet β-cells, and therapeutic modulation of the unfolded protein response in diabetes. Mol. Metabol. 2019;27S:S60–S68. doi: 10.1016/j.molmet.2019.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu Y., Gao Y., Zhang M., Deng K.-Y., Singh R., Tian Q., Gong Y., Pan Z., Liu Q., Boisclair Y.R., Long Q. Endoplasmic reticulum-associated degradation (ERAD) has a critical role in supporting glucose-stimulated insulin secretion in pancreatic β-cells. Diabetes. 2019;68:733–746. doi: 10.2337/db18-0624. [DOI] [PubMed] [Google Scholar]
- 51.Padgett L.E., Broniowska K.A., Hansen P.A., Corbett J.A., Tse H.M. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann. N. Y. Acad. Sci. 2013;1281:16–35. doi: 10.1111/j.1749-6632.2012.06826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Imoto K., Kukidome D., Nishikawa T., Matsuhisa T., Sonoda K., Fujisawa K., Yano M., Motoshima H., Taguchi T., Tsuruzoe K., et al. Impact of mitochondrial reactive oxygen species and apoptosis signal-regulating kinase 1 on insulin signaling. Diabetes. 2006;55:1197–1204. doi: 10.2337/db05-1187. [DOI] [PubMed] [Google Scholar]
- 53.Hou N., Torii S., Saito N., Hosaka M., Takeuchi T. Reactive oxygen species-mediated pancreatic beta-cell death is regulated by interactions between stress-activated protein kinases, p38 and c-Jun N-terminal kinase, and mitogen-activated protein kinase phosphatases. Endocrinology. 2008;149:1654–1665. doi: 10.1210/en.2007-0988. [DOI] [PubMed] [Google Scholar]
- 54.Sidarala V., Pearson G.L., Parekh V.S., Thompson B., Christen L., Gingerich M.A., Zhu J., Stromer T., Ren J., Reck E.C., et al. Mitophagy protects β cells from inflammatory damage in diabetes. JCI Insight. 2020;5:141138. doi: 10.101172/jci.insight.141138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hamilton-Williams E.E., Palmer S.E., Charlton B., Slattery R.M. Beta cell MHC class I is a late requirement for diabetes. Proc. Natl. Acad. Sci. USA. 2003;100:6688–6693. doi: 10.1073/pnas.1131954100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Russell M.A., Redick S.D., Blodgett D.M., Richardson S.J., Leete P., Krogvold L., Dahl-Jørgensen K., Bottino R., Brissova M., Spaeth J.M., et al. HLA class II antigen processing and presentation pathway components demonstrated by transcriptome and protein analyses of islet β-cells from donors with type 1 diabetes. Diabetes. 2019;68:988–1001. doi: 10.2337/db18-0686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gysemans C., Callewaert H., Moore F., Nelson-Holte M., Overbergh L., Eizirik D.L., Mathieu C. Interferon regulatory factor-1 is a key transcription factor in murine beta cells under immune attack. Diabetologia. 2009;52:2374–2384. doi: 10.1007/s00125-009-1514-5. [DOI] [PubMed] [Google Scholar]
- 58.Thomas H.E. Interferon signalling in pancreatic beta cells. Front. Biosci. 2009;644 doi: 10.102741/3270. [DOI] [PubMed] [Google Scholar]
- 59.Gurzov E.N., Barthson J., Marhfour I., Ortis F., Naamane N., Igoillo-Esteve M., Gysemans C., Mathieu C., Kitajima S., Marchetti P., et al. Pancreatic β-cells activate a JunB/ATF3-dependent survival pathway during inflammation. Oncogene. 2012;31:1723–1732. doi: 10.1038/onc.2011.353. [DOI] [PubMed] [Google Scholar]
- 60.Hartman M.G., Lu D., Kim M.-L., Kociba G.J., Shukri T., Buteau J., Wang X., Frankel W.L., Guttridge D., Prentki M., et al. Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol. Cell Biol. 2004;24:5721–5732. doi: 10.1128/MCB.24.13.5721-5732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosselot C., Baumel-Alterzon S., Li Y., Brill G., Lambertini L., Katz L.S., Lu G., Garcia-Ocaña A., Scott D.K. The many lives of Myc in the pancreatic β-cell. J. Biol. Chem. 2021;296:100122. doi: 10.101074/jbc.REV120.011149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dooley J., Tian L., Schonefeldt S., Delghingaro-Augusto V., Garcia-Perez J.E., Pasciuto E., Di Marino D., Carr E.J., Oskolkov N., Lyssenko V., et al. Genetic predisposition for beta cell fragility underlies type 1 and type 2 diabetes. Nat. Genet. 2016;48:519–527. doi: 10.1038/ng.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reynolds M.S., Hancock C.R., Ray J.D., Kener K.B., Draney C., Garland K., Hardman J., Bikman B.T., Tessem J.S. β-Cell deletion of Nr4a1 and Nr4a3 nuclear receptors impedes mitochondrial respiration and insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2016;311:E186–E201. doi: 10.1152/ajpendo.00022.2016. [DOI] [PubMed] [Google Scholar]
- 64.Soleimanpour S.A., Ferrari A.M., Raum J.C., Groff D.N., Yang J., Kaufman B.A., Stoffers D.A. Diabetes susceptibility genes Pdx1 and Clec16a function in a pathway regulating mitophagy in β-cells. Diabetes. 2015;64:3475–3484. doi: 10.2337/db15-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shen L., Liu X., Hou W., Yang G., Wu Y., Zhang R., Li X., Che H., Lu Z., Zhang Y., et al. NDRG2 is highly expressed in pancreatic beta cells and involved in protection against lipotoxicity. Cell. Mol. Life Sci. 2010;67:1371–1381. doi: 10.1007/s00018-010-0258-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Keller M.P., Paul P.K., Rabaglia M.E., Stapleton D.S., Schueler K.L., Broman A.T., Ye S.I., Leng N., Brandon C.J., Neto E.C., et al. The transcription factor Nfatc2 regulates β-cell proliferation and genes associated with type 2 diabetes in mouse and human islets. PLoS Genet. 2016;12:e1006466. doi: 10.1371/journal.pgen.1006466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Colli M.L., Ramos-Rodríguez M., Nakayasu E.S., Alvelos M.I., Lopes M., Hill J.L.E., Turatsinze J.-V., Coomans de Brachène A., Russell M.A., Raurell-Vila H., et al. An integrated multi-omics approach identifies the landscape of interferon-α-mediated responses of human pancreatic beta cells. Nat. Commun. 2020;11:2584. doi: 10.101038/s41467-020-16327-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oslowski C.M., Urano F. In: Methods in Enzymology. Conn P. Michael., editor. Elsevier; 2011. Measuring ER stress and the unfolded protein response using mammalian tissue culture system; pp. 71–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gille J.J., Joenje H. Cell culture models for oxidative stress: superoxide and hydrogen peroxide versus normobaric hyperoxia. Mutat. Res. 1992;275:405–414. doi: 10.1016/0921-8734(92)90043-O. [DOI] [PubMed] [Google Scholar]
- 70.Muñoz-Sánchez J., Chánez-Cárdenas M.E. The use of cobalt chloride as a chemical hypoxia model. J. Appl. Toxicol. 2019;39:556–570. doi: 10.1002/jat.3749. [DOI] [PubMed] [Google Scholar]
- 71.Brereton M.F., Rohm M., Shimomura K., Holland C., Tornovsky-Babeay S., Dadon D., Iberl M., Chibalina M.V., Lee S., Glaser B., et al. Hyperglycaemia induces metabolic dysfunction and glycogen accumulation in pancreatic β-cells. Nat. Commun. 2016;7:13496. doi: 10.101038/ncomms13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miyazaki J., Araki K., Yamato E., Ikegami H., Asano T., Shibasaki Y., Oka Y., Yamamura K. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- 73.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.101186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Juric I., Yu M., Abnousi A., Raviram R., Fang R., Zhao Y., Zhang Y., Qiu Y., Yang Y., Li Y., et al. MAPS: model-based analysis of long-range chromatin interactions from PLAC-seq and HiChIP experiments. PLoS Comput. Biol. 2019;15:e1006982. doi: 10.1371/journal.pcbi.1006982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang B., Wang M., Zhang W., Xiao T., Chen C.-H., Wu A., Wu F., Traugh N., Wang X., Li Z., et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat. Nat. Protoc. 2019;14:756–780. doi: 10.1038/s41596-018-0113-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Diao Y., Fang R., Li B., Meng Z., Yu J., Qiu Y., Lin K.C., Huang H., Liu T., Marina R.J., et al. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat. Nat. Methods. 2017;14:629–635. doi: 10.1038/nmeth.4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Onengut-Gumuscu S., Chen W.-M., Burren O., Cooper N.J., Quinlan A.R., Mychaleckyj J.C., Farber E., Bonnie J.K., Szpak M., Schofield E., et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat. Genet. 2015;47:381–386. doi: 10.1038/ng.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jin W., Mulas F., Gaertner B., Sui Y., Wang J., Matta I., Zeng C., Vinckier N., Wang A., Nguyen-Ngoc K.-V., et al. A network of microRNAs acts to promote cell cycle exit and differentiation of human pancreatic endocrine cells. iScience. 2019;21:681–694. doi: 10.1016/j.isci.2019.10.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang A., Yue F., Li Y., Xie R., Harper T., Patel N.A., Muth K., Palmer J., Qiu Y., Wang J., et al. Epigenetic priming of enhancers predicts developmental competence of hESC-derived endodermal lineage intermediates. Cell Stem Cell. 2015;16:386–399. doi: 10.1016/j.stem.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yin Y., Morgunova E., Jolma A., Kaasinen E., Sahu B., Khund-Sayeed S., Das P.K., Kivioja T., Dave K., Zhong F., et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017;356 doi: 10.101126/science.aaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aylward A., Okino M.-L., Benaglio P., Chiou J., Beebe E., Padilla J.A., Diep S., Gaulton K.J. Glucocorticoid signaling in pancreatic islets modulates gene regulatory programs and genetic risk of type 2 diabetes. PLoS Genet. 2021;17:e1009531. doi: 10.1371/journal.pgen.1009531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 94.Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S., et al. GENCODE: the reference human genome annotation for the ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.-R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kang H.M., Subramaniam M., Targ S., Nguyen M., Maliskova L., McCarthy E., Wan E., Wong S., Byrnes L., Lanata C.M., et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 2018;36:89–94. doi: 10.1038/nbt.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lawlor N., Márquez E.J., Orchard P., Narisu N., Shamim M.S., Thibodeau A., Varshney A., Kursawe R., Erdos M.R., Kanke M., et al. Multiomic profiling identifies cis-regulatory networks underlying human pancreatic β cell identity and function. Cell Rep. 2019;26:788–801.e6. doi: 10.1016/j.celrep.2018.12.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Durand N.C., Shamim M.S., Machol I., Rao S.S.P., Huntley M.H., Lander E.S., Aiden E.L. Juicer provides a one-click system for analyzing loop-resolution hi-C experiments. Cell Syst. 2016;3:95–98. doi: 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xu H., Luo X., Qian J., Pang X., Song J., Qian G., Chen J., Chen S. FastUniq: a Fast de novo duplicates removal tool for paired short reads. PLoS One. 2012;7:e52249. doi: 10.1371/journal.pone.0052249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Machanick P., Bailey T.L. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011;27:1696–1697. doi: 10.1093/bioinformatics/btr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Coetzee S.G., Coetzee G.A., Hazelett D.J. motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics. 2015;31:3847–3849. doi: 10.1093/bioinformatics/btv470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The ATAC-seq, RNA-seq, snATAC-seq, HiChIP and CRISPR screen data are available at the NCBI Gene Expression Omnibus (GEO) repository under accession number GSE205853 at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE205853. The SELEX-seq data are in GEO under accession number GSE118725 at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE118725. Supplementary summary-level files from genomics experiments are available in Zenodo under accession number https://doi.org/10.5281/zenodo.7084136 at https://zenodo.org/record/7106728.
-
•
Custom code for data processing and statistical analyses are publicly available on Github at https://github.com/Gaulton-Lab/BetaCells_cytokines_T1D and deposited in Zenodo under https://doi.org/10.5281/zenodo.7183895.
-
•
Any additional information related to analysis of the data reported in this paper is available from the lead contact upon request.