

Abstract
One goal of genomic medicine is to uncover an individual’s genetic risk for disease, which generally requires data connecting genotype to phenotype, as done in genome-wide association studies (GWAS). While there may be clinical promise to employing prediction tools such as polygenic risk scores (PRS), it currently stands that individuals of non-European ancestry may not reap the benefits of genomic medicine because of underrepresentation in large-scale genetics studies. Here, we discuss why this inequity poses a problem for genomic medicine and the reasons for the low transferability of PRS across populations. We also survey the ancestry representation of published GWAS and investigate how estimates of ancestry diversity in GWAS participants might be biased. We highlight the importance of expanding genetic research in Africa, one of the most underrepresented regions in human genomics research, and discuss issues of ethics, resources, and technology for equitable advancement of genomic medicine.
Keywords: precision medicine, human genomics, diversity, polygenic risk score, GWAS, Africa
THE PROMISE AND CHALLENGES OF GENOMIC MEDICINE
From its initial objective of identifying all the genes in the human genome, the Human Genome Project (HGP) has led to a multitude of discoveries and outcomes related to human health and disease prevention. Genomic analyses have led to the discovery of new trait-associated loci (1) and, in some cases, the identification of causal variants impacting common disease risk (2). Clinically, genomics has had some success in improved diagnoses of rare, often congenital, diseases (3); treatments for chronic conditions (4); and even cures for certain monogenic diseases (5). Precision medicine (also known as personalized medicine) has benefited from developments in genomics, which has the potential to improve healthcare.
Precision medicine considers an individual’s genetics and environment for a tailored treatment and prevention strategy to promote health. Genomic medicine, a subset of precision medicine, has benefited from the massive growth in datasets with genetic and phenotypic data. The growth of participants included in genome-wide association studies (GWAS) has been tremendous (6). Many complex traits and diseases have a polygenic basis and a considerable genetic variance component based on heritability studies (7, 8). Thus, applying knowledge gained from large genetic datasets to quantify risk can potentially benefit patients through both preventative and treatment approaches.
One tool used in genomic medicine is the polygenic risk score (PRS). A PRS aggregates the effects of independent genetic variants, which typically have small effects on the phenotype, to quantify genetic risk or burden. While in recent years there has been a flurry of new methods for PRS calculation (9), most of them rely on GWAS summary statistics to estimate weights for alleles to estimate an individual’s genetic risk. The field of human genetics has entered an era of GWAS sample sizes on the order of hundreds of thousands, and even millions, of individuals, as in the case of height and type 2 diabetes (10, 11). With growing sample sizes, GWAS are estimating the effect sizes of common variants more accurately, which can be useful for risk stratification for clinically relevant phenotypes (12, 13).
Despite these numbers, the populations that form some of the largest GWAS cohorts do not adequately represent all of the genetic variation in the world. This inadequate representation in biomedical research participation has resulted in an incomplete catalog of human genetic variation and an incomplete understanding of the relationship between genetic and phenotypic variation. The extent of the disparity presents a major roadblock toward equitably advancing genomic medicine. For example, a potential issue lies in the equitable use of PRS for disease risk prediction, which has been shown to be less useful in non-European groups (14, 15). However, there have been efforts to broaden our knowledge of human genetic variation, including the National Heart, Lung, and Blood Institute’s TOPMed initiative, the PAGE (Population Architecture Using Genomics and Epidemiology) Study, the H3Africa Consortium, and the National Institutes of Health’s All of Us initiative (16–19).
In this review, we discuss why the lack of ancestry diversity in large human genetic studies poses a problem for genomic medicine. We survey the ancestries represented by participants in GWAS and how these estimates are influenced by resampling individuals across GWAS. Finally, we discuss the barriers to equitable representation in human genetic data, with a focus on African genomics given its underrepresentation in human genetics, and we outline future directions to help ameliorate inequities in precision medicine.
WHAT IS THE CURRENT LANDSCAPE OF ANCESTRY REPRESENTATION IN GENOME-WIDE ASSOCIATION STUDIES?
The ancestry of individuals in GWAS is heavily biased toward European populations (6, 20–22). Previous surveys of ancestry diversity in GWAS have generally relied on metadata provided by the GWAS Catalog, a publicly accessible database of GWAS (23). The GWAS Catalog metadata contain entries for individual studies, where one study is one GWAS for a phenotype, and each entry contains information about the study, such as the number of individuals and their ancestry, if known. A statistic often reported is the proportion of individuals belonging to a particular ancestry group across all GWAS up to some timepoint. For example, Morales et al. (22) reported 78% of all individuals in GWAS were of European ancestry up to 2017, and Sirugo et al. (20) reported 78% up to 2019. The GWAS Diversity Monitor, an online web resource, updates these ancestry diversity statistics in real time, and reported that 83.8% of all participants in GWAS were of European ancestry as of November 2021 (24).
We downloaded metadata of published GWAS from the GWAS Catalog and conducted several analyses related to the ancestry composition of studies and individuals used in studies to examine changes over time.1 The numbers of studies and participants in GWAS have grown tremendously since 2005 (Figure 1a). Examining yearly trends, we observed that the European bias in GWAS has changed little over the decade of 2011–2020, whether considering the proportion of studies using European compared to non-European participants or considering the ancestry of individual participants (Figure 1b). After 2012, which had the lowest proportion of European ancestry participants, there was a rise to a stable proportion of ~80% over the period of 2015–2020 (Figure 1b).
Figure 1.
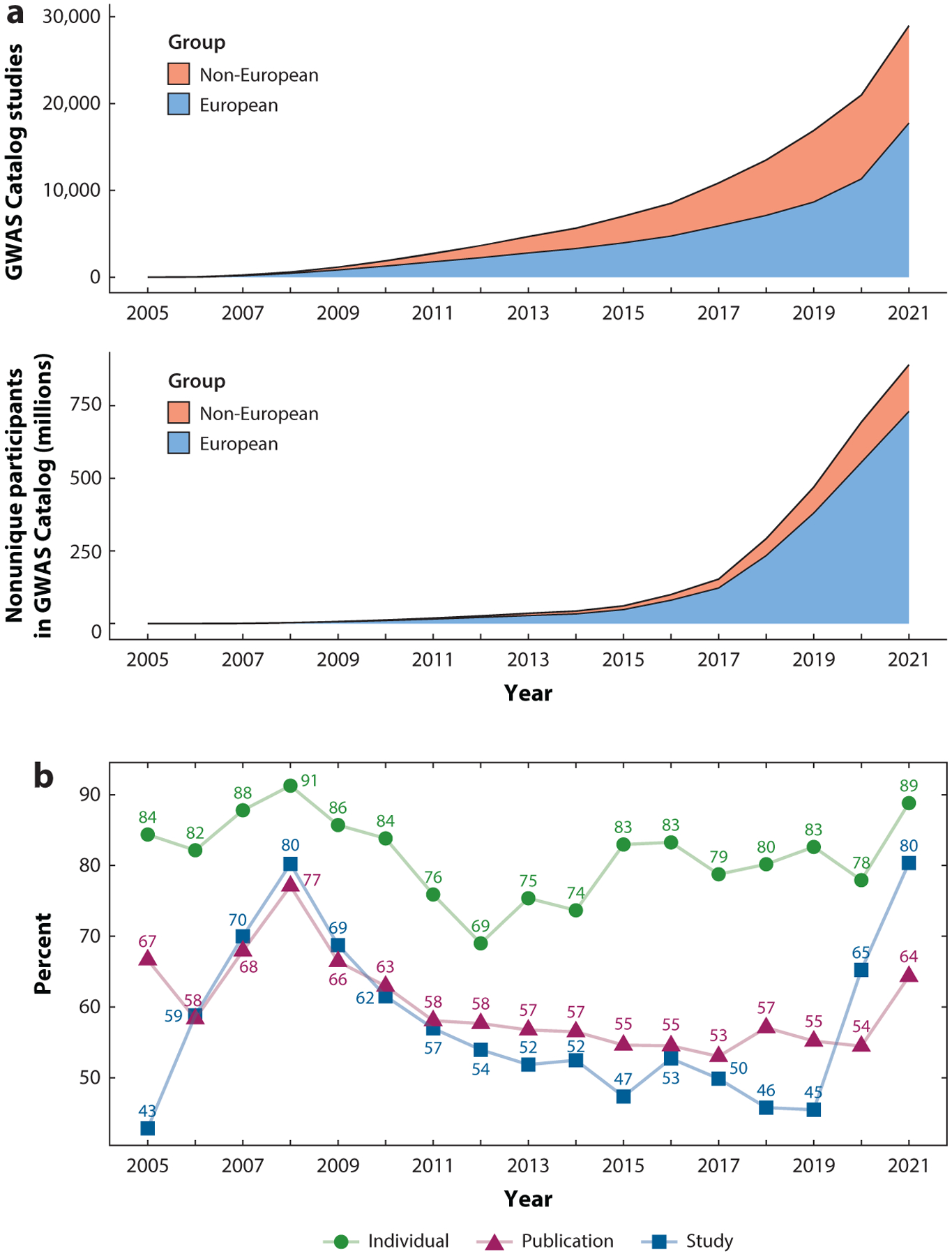
(a) The cumulative number of European and non-European ancestry studies and participants each year from the GWAS Catalog. (b) The percentage per year of participants of European ancestry and of studies and publications that used exclusively European ancestry individuals. The estimates of numbers of individuals here were calculated without accounting for repeated sampling.
Previous reports of the cumulative proportion of European individuals involved in GWAS are affected by the issue of repeated sampling of individuals, which may lead to biased estimates if one cares about counting only independent GWAS participants. To investigate how repeated sampling of the same individuals might affect estimates of European bias in GWAS, we examined all the height GWAS publications and manually kept track of repeated sampling of the same individual under the assumption that the same cohort across publications contains overlapping individuals. Several large resources for height GWAS such as the GIANT (Genetic Investigation of Anthropometric Traits) Consortium and the UK Biobank have been repeatedly used across studies (Figure 2a). We found that after correcting for repeated sampling, Europeans still constituted the majority of GWAS participants for height (68%); this percentage is lower than the percentage of Europeans in such studies when not considering repeated sampling of individuals (87%) (Figure 2b). The next most common ancestries of height GWAS were East Asian (17%), African American/Afro-Caribbean (7.5%), and Hispanic or Latin American (3.8%) (Figure 2c). Therefore, when one considers the number of independent individuals used in GWAS, the proportion of participants of European ancestry may be somewhat smaller than estimates that do not consider resampling of individuals across multiple GWAS. Nonetheless, the overall message remains the same: that European ancestry individuals still constitute a large majority of participants in GWAS.
Figure 2.
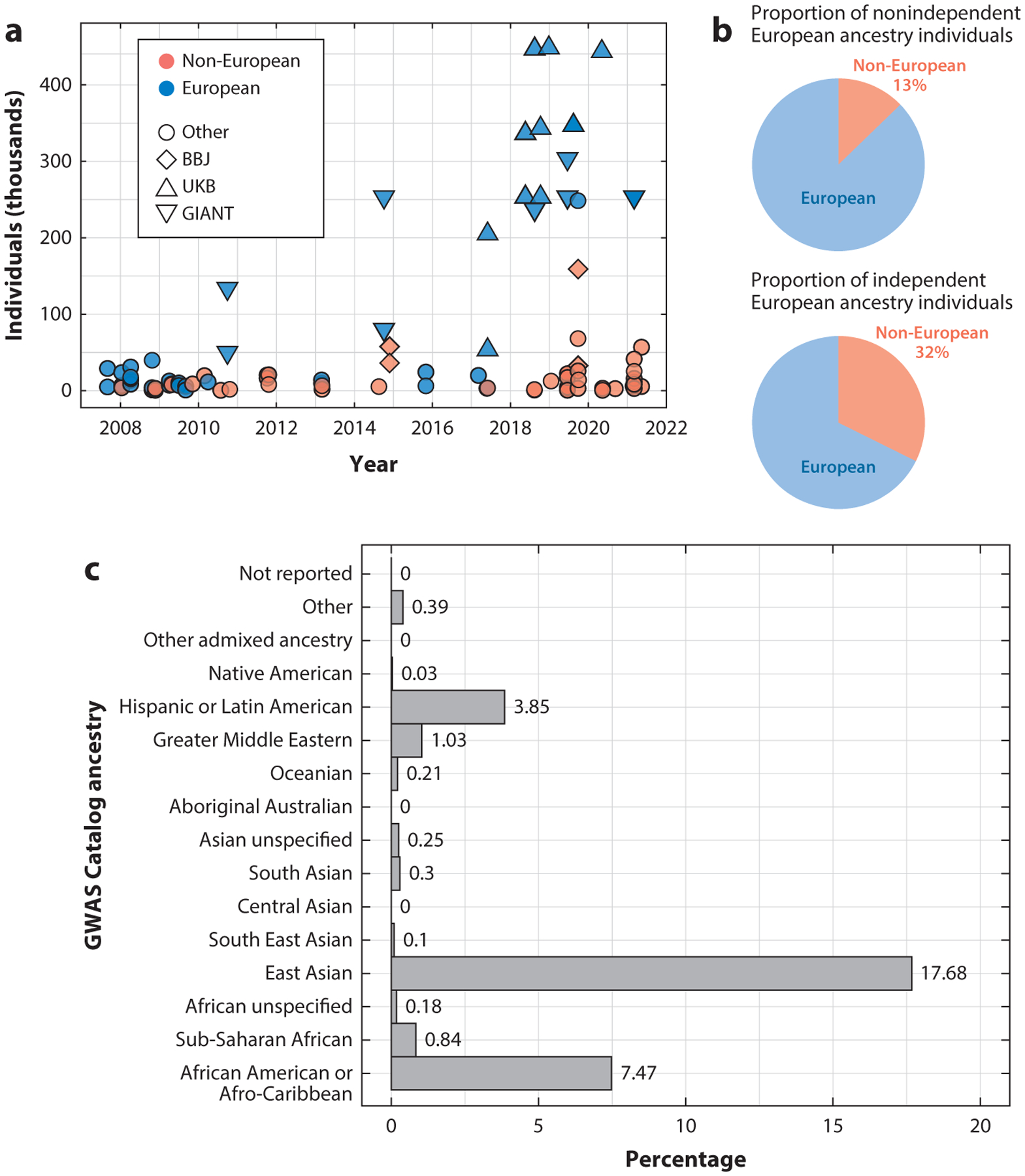
(a) The number of participants in individual height GWAS over time. Points are colored by whether the participants in the study were of European ancestry and assigned a shape based on whether the data are from a major cohort or consortium. (b) The proportion of all participants of height GWAS that are of European and non-European ancestry based on (top) a naïve (i.e., not accounting for repeated sampling of individuals) estimate from the GWAS Catalog and (bottom) an estimate after removing repeatedly sampled individuals across height studies. (c) Proportion of all unique participants from height GWAS for each GWAS Catalog ancestry category other than European. Abbreviations: BBJ, BioBank Japan; GIANT, Genetic Investigation of Anthropometric Traits; GWAS, genome-wide association study; UKB, UK Biobank.
In order to generate more accurate PRS for individuals of diverse ancestries, researchers need to increase the sample sizes of independent participants, since the number of independent participants helps drive the performance of predictive models. Because most large biobanks are located in North America and Europe (Figure 3), they are generally enriched for individuals with European ancestry. For several traits of biomedical interest with the largest number of GWAS Catalog studies [e.g., Alzheimer’s disease, asthma, body mass index (BMI), coronary artery disease, systolic blood pressure, schizophrenia, type 2 diabetes], we recorded the GWAS with the highest sample size available across several ancestry categories (Figure 4a–g) (22). We observed that European ancestry individuals are vastly more represented across the surveyed phenotypes. Among non-European groups, East Asian participants were the largest group included in GWAS for the surveyed phenotypes. However, even among the GWAS we examined, the largest East Asian sample size (n = 433,540) is not even half of the European sample size (n = 1,114,458) (Figure 4g). In addition, there were consistently lower numbers of sub-Saharan African participants compared to African American or Afro-Caribbean participants across the phenotypes we examined. For type 2 diabetes, for example, we see extreme disparities, with the European GWAS sample size at n = 1,114,458, the African American or Afro-Caribbean sample size at n = 56,092, and the sub-Saharan African sample size at n = 7,809. Other groups, such as Native American, Oceanian, and Southeast Asian, were also severely underrepresented for type 2 diabetes GWAS.
Figure 3.
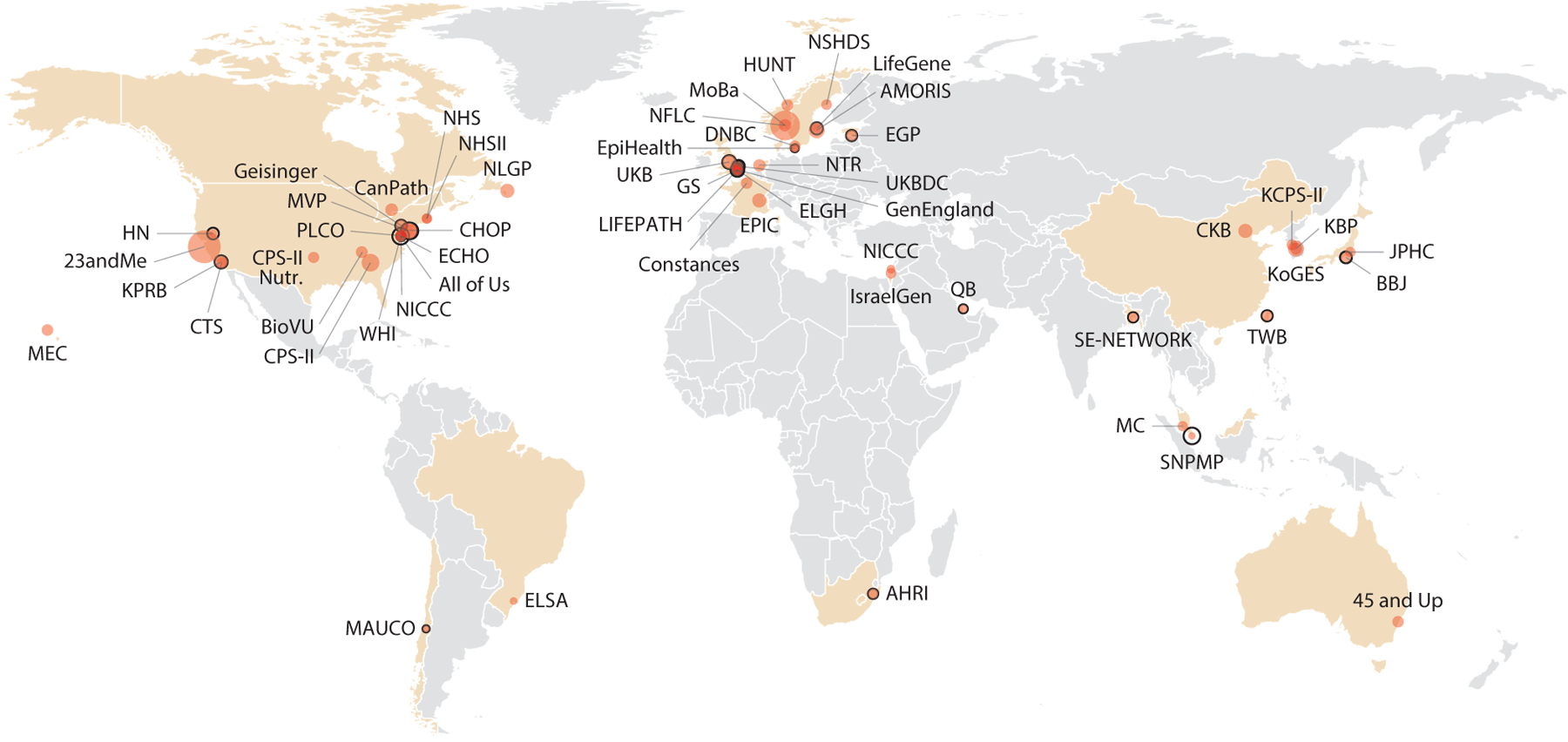
World map of cohorts with genetic and phenotypic data that are part of the International HundredK+ Cohorts Consortium (IHCC). Countries with cohorts are highlighted and individual cohorts are sized according to the number of individuals enrolled. For cohorts with ongoing enrollment, empty circles are drawn according to the target number of individuals. Abbreviations: AHRI, Africa Health Research Institute Population Cohort; AMORIS, Apolipoprotein Mortality Risk Study; BBJ, BioBank Japan; BioVU, Biobank Vanderbilt University; CanPath, Canadian Partnership for Tomorrow’s Health; CHOP, Children’s Hospital of Philadelphia Biorepository; CKB, China Kadoorie Biobank; CPS-II, Cancer Prevention Study II; CPS-II Nutr., CPS-II Nutrition Cohort; CTS, California Teachers Study; DNBC, Danish National Birth Cohort; ECHO, Environmental Influences on Child Health Outcomes Cohort; EGP, Estonian Genome Project; ELGH, East London Genes and Health; ELSA, Estudo Longitudinal de Saúde do Adulto; EPIC, European Prospective Investigation into Cancer, Chronic Diseases, Nutrition and Lifestyle; Geisinger, Geisinger MyCode Community Health Initiative; GenEngland, Genomics England/100,000 Genomes Project; GS, Generations Study; HN, Healthy Nevada; HUNT, Trøndelag Health Study; IsraelGen, Israel Genome Project; JPHC, Japan Public Health Center–Based Prospective Study; KBP, Korea Biobank Project; KCPS-II, Korean Cancer Prevention Study; KoGES, Korean Genome and Epidemiology Study; KPRB, Kaiser Permanente Research Bank; LIFEPATH, Lifecourse Biological Pathways Underlying Social Differences in Healthy Aging Study; MAUCO, Maule Cohort; MC, Malaysian Cohort; MEC, Multiethnic Cohort Study; MoBa, Norwegian Mother and Child Cohort Study; MVP, Million Veteran Program; NFLC, Norwegian Family-Based Life Course Study; NHS, Nurses’ Health Study; NHSII, Nurses’ Health Study II; NICCC, National Israeli Cancer Control Center; NLGP, Newfoundland 100K Genome Project; NSHDS, Northern Sweden Health and Disease Study; NTR, Netherlands Twin Registry; PLCO, Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial; QB, Qatar Biobank; SE-NETWORK, South(east) Asian Cohorts NETWORK; SNPMP, Singapore National Precision Medicine Program; TWB, Taiwan Biobank; UKB, UK Biobank; UKBDC, UK Blood Donor Cohorts; WHI, Women’s Health Initiative.
Figure 4.
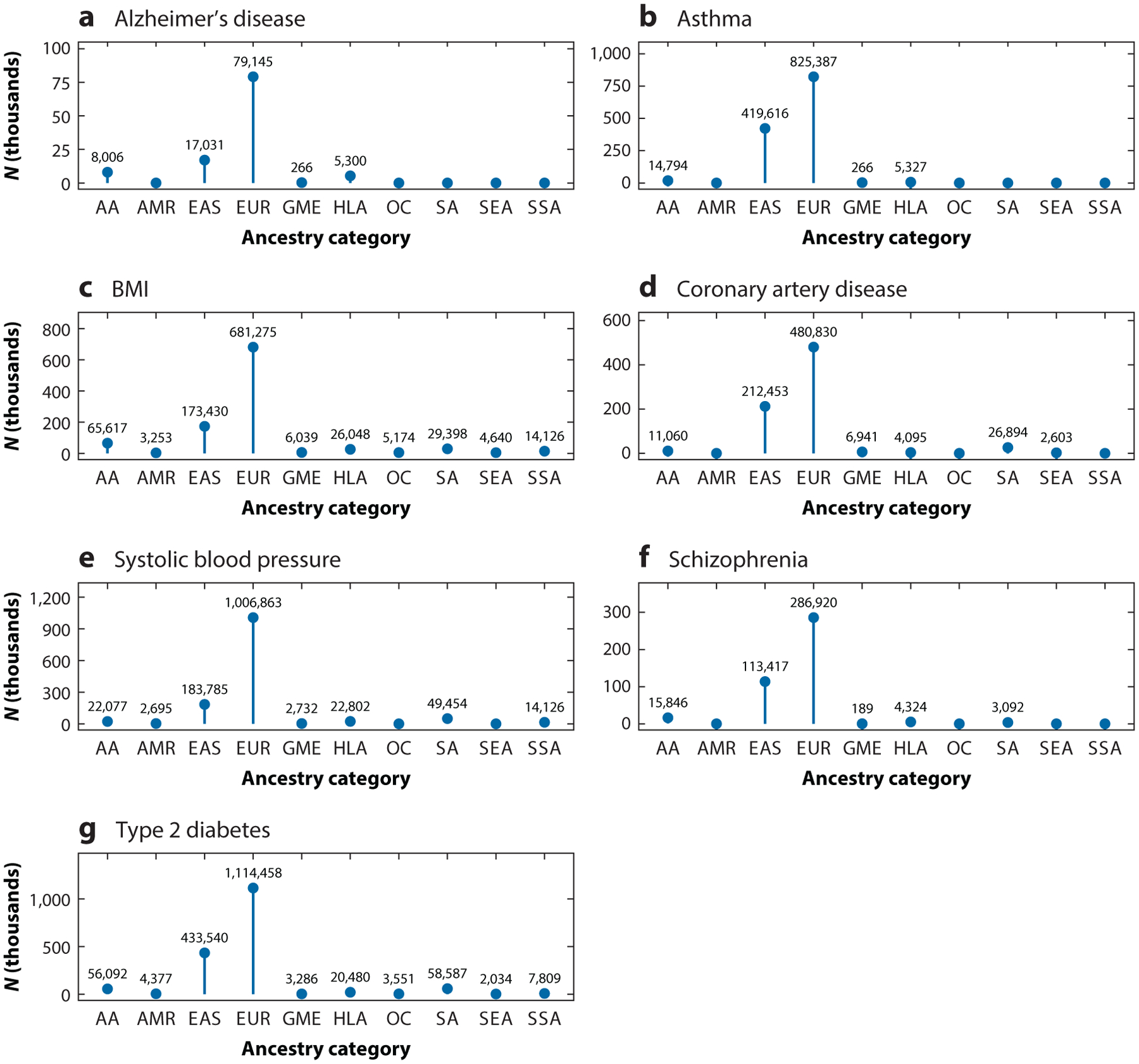
The sample sizes of the largest GWAS, at the time of writing, for (a) Alzheimer’s disease, (b) asthma, (c) BMI, (d) coronary artery disease, (e) systolic blood pressure, (f) schizophrenia, and (g) type 2 diabetes, across ancestry categories based on the GWAS Catalog ancestry ontology. Abbreviations: AA, African American or Afro-Caribbean; AMR, Native American; BMI, body mass index; EAS, East Asian; EUR, European; GME, Greater Middle Eastern; GWAS, genome-wide association studies; HLA, Hispanic or Latin American; OC, Oceanian; SA, South Asian; SEA, Southeast Asian; SSA, sub-Saharan African.
BUILDING BLOCKS OF POLYGENIC RISK SCORES: TRANSFERABILITY OF GENOME-WIDE ASSOCIATION STUDIES RESULTS ACROSS POPULATIONS
A current issue in genomic medicine is understanding the differences in the accuracy of PRS across populations of different ancestries and reducing these disparities. While the quantitative difference in predictive power for PRS, often based on GWAS of primarily European ancestry individuals, varies according to phenotype, generally the same trend appears of reduced prediction accuracy for non-European groups compared to that for European groups (14, 15). For example, PRS for people of African descent that predict the likelihood of cardiomyopathies can be unreliable, or even misleading, if developed using data that are largely from European ancestry populations (25).
Various causes of the nontransferability of PRS across populations of different ancestries have been explored, but it should be noted that the heritability (i.e., how much genetic variance explains the phenotypic variance) places an upper bound on the predictive power of PRS. Heritability can vary widely across phenotypes, within traits across populations, and even within ancestry groups for the same phenotype (7). Reasons for heritability differences include differences in the environmental variance component or in genetic variation at trait-influencing loci. However, differences in heritability cannot fully account for losses of explanatory power of PRS across ancestry groups.
One of the causes of PRS’ lack of portability across ancestry groups is that there are differences in the patterns of linkage disequilibrium (LD) across ancestry groups due to differences in their demographic histories (20). Single-nucleotide polymorphisms (SNPs) associated with a phenotype in GWAS typically are tag SNPs that are in LD with the causal variant(s). A SNP that tags a causal variant in one population may not tag the same causal variant in another population. Under one model, differences in LD and allele frequencies can account for up to ~86% of the loss in prediction accuracy between Europeans and Africans on average over several traits (26). Several methods have tried to address limited PRS transferability by using cohorts of different ancestry, with some success (27–29).
Differences in effect sizes between populations (effect size heterogeneity) at causal variants can also contribute to the portability problem of PRS across populations. For some phenotypes, effect size correlations across populations can fall significantly under 1, suggesting the magnitude and perhaps even direction of effects of some variants might differ across populations (30, 31). However, in practice, detecting actual trans-ancestry effect size heterogeneity for causal variants presents a challenge. For example, a tag SNP in high LD with the causal variant in one population would have a larger effect on the phenotype than it would in another population in which the LD between it and the causal variant is lower. Additionally, given the generally higher LD in non-African populations (32), a tag SNP ascertained in a non-African group, which could capture the effects of multiple causal variants, may have a different effect size compared to the same variant in an African population that captures the effect of a single causal variant. One study examined correlations of causal effect sizes for shared common variants across populations from different continents while accounting for LD; across nine traits the average correlation of effect sizes was 0.55 between East Asians and Europeans (33). In functionally important regions of the genome (e.g., conserved sites), the trans-ancestry correlation appears to be further diminished (34).
Effect size heterogeneity could also stem from interaction effects, such as gene-by-environment (GxE), gene-by-gene (GxG; i.e., epistasis), and dominance effects. Interaction effects could impact PRS transferability due to cross-population differences in environment and allele frequencies (or genotype frequencies in the case of dominance effects) at the causal variants. Evidence of the importance of GxE in the genetic architecture of complex traits has been growing, particularly for some traits such as BMI (35–38), blood pressure (39, 40), and psychiatric traits (41). It has also been shown that sample characteristics, such as sex and age, of the GWAS and test cohorts can reduce PRS portability across groups even of the same genetic ancestry (42). The most consistent GxE signal for BMI, and arguably for any complex trait, comes from the FTO locus, but like most genotype–phenotype associations we know little about the mechanism underlying this signal. A difference across populations in the contribution of direct genetic effects (variants carried by the individual that influence the individual’s phenotype) and indirect genetic effects (variants carried by relatives, such as parents, that act indirectly on an individual’s phenotype) can also be a consequence of GxE interactions. Marginal GWAS effect sizes include both indirect and direct effects, and it is difficult to partition these effects without family data (43).
In contrast, robust evidence for epistasis playing a significant role in the genetic architecture of complex traits in human populations is less apparent. It is challenging to evaluate the replicability of myriad GxG signals claimed in the literature, especially given the rich variety of approaches for detecting epistasis (44).There are, however, some convincing examples of GxG effects in humans, such as the APOE ε4 variant, which confers different risks for Alzheimer’s disease across ancestry groups (45). To control for environmental confounding, several studies have modelled the interaction between risk variants and local ancestry background and observed significant interaction effects, which could be due to frequency differences in nearby interacting variants present on different ancestry backgrounds (46, 47). Whether similar kinds of epistatic effects, but of a smaller magnitude, occur broadly remains to be shown. Leveraging local ancestry in GWAS might be helpful for discovering GxG effects, and some efforts have been made in this space (48).
Aside from interaction effects (GxG, GxE, and dominance), observed effect size heterogeneity can also arise from uncontrolled population stratification in GWAS, which biases effect size estimates from one population and, thus, makes them less applicable in other populations. This issue has resurfaced in recent years with the advent of very large GWAS that rely on meta-analysis of many different cohorts or biobank-scale cohorts. In the case of height, it has been demonstrated that typical methods for accounting for population structure, such as incorporating principal components from genome-wide data, do not fully correct stratification (49–51). For GWAS designs in which cohorts of small size are meta-analyzed, the bias introduced by population stratification is possibly exacerbated (52). This situation was the case for the height GWAS by the GIANT Consortium, which affected downstream analyses that used these summary statistics by, for example, inflating signals of selection exhibited by height-associated alleles (53, 54).
Population-specific variants related to a trait of interest could also limit PRS transferability, manifested in either heterogeneous effects at the causal variant or highly disparate allele frequencies. Allele frequency differences between the PRS test population and the GWAS population can either increase or decrease the phenotypic variance explained by GWAS SNPs in the test population depending on the minor allele frequencies (MAFs). There are some examples of population-specific variants implicated in GWAS—that is, some alleles segregate at appreciable frequencies in one population but are rare in others (55–57). In general, however, only a modest fraction of genetic variants segregate at common frequencies in a single population while remaining rare in other populations (58). This observation has led to the assumption that causal variants are shared among populations with diverse ancestries, motivating the use of multiancestry datasets for GWAS. Multiancestry GWAS have grown more common in order to increase genetic diversity and boost overall sample sizes, resulting in the detection of associated loci that would not necessarily have been discovered just by studying Europeans (59, 60). Furthermore, the inclusion of African populations, which have lower levels of LD relative to non-African populations, can help facilitate fine-mapping, a process of narrowing down a set of variants likely to contain the causal variant(s) (61, 62).
Very large European cohorts could possibly overcome challenges of statistical power in GWAS with low MAF variants, but there are still fundamental issues beyond detecting an association. For example, the frequency of the derived light skin pigmentation allele (rs1426654) of SLC24A5 stands at ~99.9% in the European subset of the UK Biobank due to positive selection for the light allele (63). Despite the rarity of the minor allele, the large sample size (n = 356,530) and large effect size allow for detection of this SNP in a European cohort for a GWAS of skin pigmentation (63). This variant also reaches genome-wide significance in a cohort of only 1,570 sub-Saharan Africans, as the small sample size is compensated by high cohort MAF and the large effect size (64). Even if sheer sample size can overcome the challenges of detecting an association for a low MAF variant, there is still the fundamental problem of the transferability of the effect size across populations. Even if one could detect all causal variants for a phenotype in a population, the generalizability of a model based on these data to different populations is ultimately limited by the extent of effect size differences across populations.
The genetic architecture for a trait may differ across ancestry groups due to differences in demographic history and selection regimes. In this case, it will be critical to include populations of diverse ancestry in GWAS. Furthermore, as demonstrated by the GWAS of skin pigmentation in sub-Saharan Africans (64), even GWAS with small sample sizes can produce novel insights. In this study, MFSD12 was identified to play a role in skin pigmentation, which may have implications for melanoma risk (65). We argue that efforts should still be made to include underrepresented populations in human genomics research, even if sample sizes are not as large as European samples, as they can still aid in elucidating the etiology of disease and biological processes.
ISSUES FOR TRANS-ANCESTRY TRANSFERABILITY OF RARE VARIANTS
There is robust evidence that rare variants (e.g., MAF < 0.1%) contribute substantially to complex trait heritability (66–69). However, most analyses and methods to date involving polygenic prediction do not include rare variants. The inclusion of rare variants poses additional, or a different set of, issues in terms of trans-ancestry transferability compared to those posed by common variants.
Discovery and accurate measurement of rare variant associations and their effects are generally more prone to issues of population stratification compared to those of common variants (50, 70–72), as rare variants are typically newer than common variants and so should have a finer-scale population structure than common variants have. As rare, large-effect variants are often population specific or even singletons (69, 73), currently it is largely unclear how robust these associations are, how well they will replicate across ancestries, and how much practical utility these variants have for trait prediction, as they may often be entirely nonexistent in a cohort in which the prediction is performed.
Due to difficulties in accurately measuring effects of individual rare variants, association tests that aggregate a set of variants below a frequency threshold at a genic region or genome-wide (often according to biological significance, e.g., putative loss-of-function or missense variants with high predicted deleteriousness) are commonplace (74, 75). One popular method for aggregating variants, the burden test, relies on the premise that all variants in the set are associated with the phenotype and have the same direction of effect (75). Rare variant sets ascertained in one population may be less useful for prediction in other populations if some of the variants are absent in other populations or if some of the variants in the set are not actually associated with the phenotype.
In the case of true, well-calibrated rare variant associations, however, there are factors that facilitate transferability across populations, especially when compared to common variants. Rare variants typically have little LD with other variants (at least when measured using r2), implying that true associations may be more likely to be causal, and consequently that effect sizes may be more similar across ancestries (76–79). It may be more likely that rare variant associations in coding regions that have clear functional annotations (e.g., loss-of-function) are indeed causal across ancestries, assuming the same functional impact in different populations (80, 81).
Nonadditive effects are mentioned as potentially limiting the transferability of genetic effects across populations. But for rare variants, differences in effects due to dominance or a variety of different epistatic models are likely extremely small even if the true underlying effect is nonadditive (82). As the amount of sequencing datasets and individuals used in imputation panels increases in size and diversity (83, 84), and as methods that include rare variants for use in polygenic prediction improve, practical usage of rare variants may increase and performance of polygenic predictions may approach estimates of heritability even for ancestrally diverse populations.
BARRIERS TOWARD EQUITY IN GENOMIC MEDICINE IN AFRICA
Despite technological advances and a drop in the cost of sequencing technology, inequalities and deficits in resources for conducting genomics research have limited human genomics knowledge from becoming a global public good, particularly in Africa (85). A 2002 World Health Organization report recognized this concern as far back as two decades ago (86), and there are a variety of issues that drive this disparity. First, while genomic data from African populations are increasingly becoming available, in most African countries there is a shortage of trained genetics professionals who can assist in unraveling the significance of genetic results and translating them in a meaningful way to research participants (87, 88). Second, there is limited public understanding of concepts in genomics, as evidenced during consent processes for genomics studies and in clinical practice (89, 90). Third, there is a near absence of empirical data and frameworks for ethical, legal, and social implications to support regulation of data governance, such as intellectual property rights and commercialization of products associated with African genomic research (91).
Genomics research initiatives have called for the development of ethics and legal frameworks on data sharing, data governance, intellectual property, patent regulations, and product commercialization associated with African genomic research. This call has become even more urgent in recent years following international controversies on the use of genomic and health data from Africa (92) and the creation of private initiatives that aim to use African genomic data for both research and commercialization purposes (93). While there are emerging data-use legislation and guidelines, such as the San code of research ethics (94) and the Protection of Personal Information Act of 2013 in South Africa, their impacts on data-driven health and genomics research have still not been fully explored. These research and ethics guidelines bear relevance for genomics projects in Africa, such as the H3Africa Consortium (19), which is generating vast quantities of genetic data and is involved in sharing personal health findings with study participants. Despite some progress toward diagnosis and management of monogenic conditions in high-income nations, the lack of local expertise in genetics as well as funding issues in sub-Saharan Africa have stymied progress in genomic medicine. Furthermore, research topic biases do not always reflect the urgent public health needs of the African continent, particularly when driven by investigators from Europe and North America (95, 96).
With advances in precision medicine, global health actors are increasingly seeking ways to aggregate diverse health data (e.g., clinical, genomic, and behavioral) within populations, with the goal of enabling new models for using genomic data to support health research, clinical care, and public health programs (97, 98). While the use of genomic data in healthcare and innovation offers new opportunities of clinical benefit, it also raises important ethical, legal, and social challenges (99).
TECHNICAL CHALLENGES AND FUTURE DIRECTIONS
As we embark on the third decade since the completion of the HGP, there is increasing evidence that research studies focused on genetic diversity, especially in African individuals, are a scientific imperative (100, 101). Approximately 300 million base pairs, or 10% of a pan-genome, generated from 910 individuals of African descent were not found in the current human reference genome (101). Recent analyses on whole-genome sequencing data of 426 African individuals, comprising 50 ethnolinguistic groups across Africa, uncovered more than 3 million previously undescribed variants (102). Moreover, the African continent follows a north–south axis, spanning approximately 70° of latitude. This range has a role in the enormous diversity of climates and natural environments in Africa, and so the selection pressures can vary considerably across the African continent (103–105). African genomes can reveal genes and variants that contribute to health and disease not found in previous Euro-centric studies, as African ancestry individuals collectively have more genetic variation. For example, certain nonsense variants in the gene PCSK9 are extremely rare in Europeans (<0.1% frequency) but relatively common in African Americans (~2% frequency) (106). These PCSK9 variants are correlated with much lower levels of low-density lipoprotein cholesterol (106), a finding that has led to a new medication (evolocumab) for dyslipidemia, which is associated with heart attack and stroke. A study of 909 Africans of Xhosa ancestry with schizophrenia and 917 matched African controls found many rare mutations that contribute to schizophrenia, along with mechanistic insights (107). A 2016 study in a Swedish population identified many of these same mutations but required a sample more than four times the size (108).
High levels of both genetic and phenotypic diversity make African populations particularly informative for GWAS. For example, although only 2.4% of participants in large GWAS were of African ancestry (as of 2017), 7% of known associations at the time came from GWAS conducted in African populations (22). In addition, factors such as high fertility rates, high levels of consanguinity, and local genetic bottlenecks for some populations in sub-Saharan Africa could lead to the discovery of novel single-gene disease-causing variants. Indeed, with only about 25% of human genes found to have disease-associated variants to date (109), more disease genes remain to be identified, especially in understudied populations. For example, a targeted diagnosis gene panel testing a range of variants associated with inherited hearing impairment had much lower yield in African populations than it did in European and Asian populations, and the data suggest that novel variants in known genes, as well as genes not yet implicated in hearing impairment, are more likely to be found in monogenic conditions in African populations (110–113).
The underrepresentation of African genomic data in the development of health and genomic medical tools, such as sequencing chips and the Affymetrix DMET™ (Drug Metabolism Enzymes and Transporters) platform, yields biases against their potential applications to African populations and constitutes a potential source of error. In recent years, efforts to develop genotype arrays more suited to diverse populations have led to the development of the H3Africa and MEGAEX (Expanded Multi-Ethnic Genotyping Array) arrays (19, 114), which have a focus on sites that may be common in African ancestry individuals but not necessarily in European ancestry individuals. Genotype imputation reference panels have continued to increase in size and diversity, with the recent TOPMed release expanding public imputation reference panels to 97,256 deeply sequenced individuals of diverse ancestries (17, 115), increasing imputation accuracy for increasingly rare variants across worldwide populations. As the sizes of exome and whole-genome sequence datasets increase, progress will be made on uncovering the impact of rare and private mutations on disease. Advances in the past several years on reference graph genome representations (116, 117) may serve to increase read mappability, especially for diverged populations and regions of the genome that are highly variable but clinically important, such as the region coding for the human leukocyte antigen genes.
While there is a scientific imperative for increasing resources for African genomic data, there are concerns that this effort might not translate into enhancing sustainable genomic medicine in Africa due to overburdened and under-resourced African genomics workforce and healthcare systems (88). Initiatives such as H3Africa have laid a foundation for capacity development, but this foundation needs to be nurtured with additional efforts from both the international community and African governments. We offer three general recommendations to facilitate further development in African genomics. First, the construction of an African genomics workforce can be achieved most effectively through the development of academic and industrial partnerships between high- and low-income countries (which may be between African countries themselves) and by providing incentives for the private sector to invest in genomics research directed toward neglected diseases of the world’s poorest people. Second, African countries must develop a critical mass of expertise in computer science and bioinformatics to utilize the vast quantities of genomic data that are being generated through initiatives such as H3Africa. Coupled with clinical data, population-scale databases of genomic data will fuel the application of genomic findings for lab-based diagnosis, as well as the development of bioinformatics tool sets for better genetic risk prediction. Third, African countries need to adopt appropriate national frameworks to consider the ethical implications of genomics research and its applications in their own unique social, cultural, economic, and religious contexts, and to develop policies and guidelines based on recent experience with large-scale genomic research, including datasets from Africa (118, 119).
Finally, for traits with significant GxE effects, focusing only on ancestry diversity in GWAS study cohorts may not be enough to promote equity in genomic medicine across worldwide populations. For instance, socioeconomic status has been shown to have significant GxE interactions for BMI (36, 120) and myriad psychiatric disorders (121). As socioeconomic status is often associated with ancestry (at least in the United States) (122), accounting for interactions between socioeconomic status and genetics could help address issues in PRS transferability, as differences in environment influence marginal effect size estimates. Deep phenotyping in large-scale biobanks, as done in the UK Biobank, is instrumental in assessing GxE interactions, and efforts in hospital-based biobanks will offer more accurate phenotyping compared to data sources that rely mainly on self-reports (123). With evidence for genome-wide GxE interactions in health-relevant traits becoming more substantial in recent years, it is apparent that diversity in terms of ancestry and environment is important for increasing equity in genomic medicine.
CONCLUDING REMARKS
In the past several years, there has rightfully been an increased focus on generating diverse ancestry genetic data, as well as on developing methods suited to analyze and utilize these data. This movement toward greater inclusion in genomics research should be sustained, with resources allocated for data collection and for training individuals to conduct research on members of underrepresented ancestry groups. Tracking progress in this endeavor, say, in terms of the number of GWAS participants, must be done thoughtfully so that the metric for quantifying ancestry diversity accords with the specific question or goal at hand. In addition to sampling more widely from populations of diverse ancestries, attention must also be paid to differences in environments, as this may generate differences in genetic effects due to GxE effects. Proper infrastructure is needed in low- and middle-income countries to facilitate research and the utilization of genomic medicine, which could benefit a wide segment of society. For monogenic (and often rare) diseases, genomics has led to improved diagnoses and even treatments. One hope for genomic medicine is extending such successes toward common, polygenic diseases. It is imperative that future tools, such as genetic prediction, are effective across ancestry groups to ensure equity in precision medicine.
ACKNOWLEDGMENTS
We thank Alexander Platt for comments on and suggestions for our manuscript. This work was supported by funding from the Penn Center for Global Genomics and Health Equity, as well as from NIH (National Institutes of Health) grants 1R35GM134957, ADA 1-19-VSN-02, R01AR076241 (to S.A.T.), and 1F31HG011813-01A1 (to D.J.).
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Footnotes
We performed analysis of the GWAS Catalog metadata using R version 4.0.3. Code and associated data files for the analysis can be found in our GitHub repository (https://github.com/TishkoffLab/GWAScatalogAnalysis). Specific details about steps in the analyses and data curation can be found in the R markdown file.
LITERATURE CITED
- 1.Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, et al. 2017. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet 101(1):5–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lawlor DA, Harbord RM, Sterne JAC, Timpson N, Davey Smith G. 2008. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat. Med 27(8):1133–63 [DOI] [PubMed] [Google Scholar]
- 3.100,000 Genomes Proj. Pilot Investig. 2021. 100,000 Genomes pilot on rare-disease diagnosis in health care—preliminary report. N. Engl. J. Med 385(20):1868–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sabatine MS. 2019. PCSK9 inhibitors: clinical evidence and implementation. Nat. Rev. Cardiol 16(3):155–65 [DOI] [PubMed] [Google Scholar]
- 5.Frangoul H, Altshuler D, Cappellini MD, Chen Y-S, Domm J, et al. 2021. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med 384(3):252–60 [DOI] [PubMed] [Google Scholar]
- 6.Mills MC, Rahal C. 2019. A scientometric review of genome-wide association studies. Commun. Biol 2:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ge T, Chen C-Y, Neale BM, Sabuncu MR, Smoller JW. 2017. Phenome-wide heritability analysis of the UK Biobank. PLOS Genet. 13(4):e1006711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Golan D, Lander ES, Rosset S. 2014. Measuring missing heritability: inferring the contribution of common variants. PNAS 111(49):E5272–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma Y, Zhou X. 2021. Genetic prediction of complex traits with polygenic scores: a statistical review. Trends Genet. 37(11):995–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vujkovic M, Keaton JM, Lynch JA, Miller DR, Zhou J, et al. 2020. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet 52(7):680–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, et al. 2018. Meta-analysis of genome-wide association studies for height and body mass index in ~700,000 individuals of European ancestry. Hum. Mol. Genet 27(20):3641–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, et al. 2018. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet 50(9):1219–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khera AV, Chaffin M, Wade KH, Zahid S, Brancale J, et al. 2019. Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell 177(3):587–96.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. 2019. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet 51(4):584–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duncan L, Shen H, Gelaye B, Meijsen J, Ressler K, et al. 2019. Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun 10:3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bien SA, Wojcik GL, Hodonsky CJ, Gignoux CR, Cheng I, et al. 2019. The future of genomic studies must be globally representative: perspectives from PAGE. Annu. Rev. Genom. Hum. Genet 20:181–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, et al. 2021. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590(7845):290–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.All Us Res. Prog. Investig. 2019. The “All of Us” Research Program. N. Engl. J. Med 381(7):668–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.H3Africa Consort., Rotimi C, Abayomi A, Abimiku A, Adabayeri VM, et al. 2014. Enabling the genomic revolution in Africa. Science 344(6190):1346–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sirugo G, Williams SM, Tishkoff SA. 2019. The missing diversity in human genetic studies. Cell 177(1):26–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Popejoy AB, Fullerton SM. 2016. Genomics is failing on diversity. Nature 538(7624):161–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morales J, Welter D, Bowler EH, Cerezo M, Harris LW, et al. 2018. A standardized framework for representation of ancestry data in genomics studies, with application to the NHGRI-EBI GWAS Catalog. Genome Biol. 19:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, et al. 2019. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47(D1):D1005–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mills MC, Rahal C. 2020. The GWAS Diversity Monitor tracks diversity by disease in real time. Nat. Genet 52:242–43 [DOI] [PubMed] [Google Scholar]
- 25.Curtis D. 2018. Polygenic risk score for schizophrenia is more strongly associated with ancestry than with schizophrenia. Psychiatr. Genet 28(5):85–89 [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Guo J, Ni G, Yang J, Visscher PM, Yengo L. 2020. Theoretical and empirical quantification of the accuracy of polygenic scores in ancestry divergent populations. Nat. Commun 11:3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Márquez-Luna C, Loh P-R, South Asian Type 2 Diabetes (SAT2D) Consort., SIGMA Type 2 Diabetes Consort., Price AL. 2017. Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet. Epidemiol 41(8):811–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weissbrod O, Kanai M, Shi H, Gazal S, Peyrot WJ, et al. 2021. Leveraging fine-mapping and non-European training data to improve cross-population polygenic risk scores. medRxiv 10.1101/2021.01.19.21249483. 10.1101/2021.01.19.21249483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruan Y, Lin Y-F, Feng Y-CA, Chen C-Y, Lam M, et al. 2021. Improving polygenic prediction in ancestrally diverse populations. medRxiv 10.1101/2020.12.27.20248738. 10.1101/2020.12.27.20248738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown BC, Ye CJ, Price AL, Zaitlen N. 2016. Transethnic genetic-correlation estimates from summary statistics. Am. J. Hum. Genet 99(1):76–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veturi Y, de los Campos G, Yi N, Huang W, Vazquez AI, Kühnel B. 2019. Modeling heterogeneity in the genetic architecture of ethnically diverse groups using random effect interaction models. Genetics 211(4):1395–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, et al. 2002. The structure of haplotype blocks in the human genome. Science 296(5576):2225–29 [DOI] [PubMed] [Google Scholar]
- 33.Galinsky KJ, Reshef YA, Finucane HK, Loh P-R, Zaitlen N, et al. 2019. Estimating cross-population genetic correlations of causal effect sizes. Genet. Epidemiol 43(2):180–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi H, Gazal S, Kanai M, Koch EM, Schoech AP, et al. 2021. Population-specific causal disease effect sizes in functionally important regions impacted by selection. Nat. Commun 12:1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abadi A, Alyass A, Robiou du Pont S, Bolker B, Singh P, et al. 2017. Penetrance of polygenic obesity susceptibility loci across the body mass index distribution. Am. J. Hum. Genet 101(6):925–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rask-Andersen M, Karlsson T, Ek WE, Johansson Å. 2017. Gene-environment interaction study for BMI reveals interactions between genetic factors and physical activity, alcohol consumption and socioeconomic status. PLOS Genet. 13(9):e1006977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robinson MR, English G, Moser G, Lloyd-Jones LR, Triplett MA, et al. 2017. Genotype-covariate interaction effects and the heritability of adult body mass index. Nat. Genet 49(8):1174–81 [DOI] [PubMed] [Google Scholar]
- 38.Wang H, Zhang F, Zeng J, Wu Y, Kemper KE, et al. Genotype-by-environment interactions inferred from genetic effects on phenotypic variability in the UK Biobank. Sci. Adv 5(8):eaaw3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sung YJ, Winkler TW, de las Fuentes L, Bentley AR, Brown MR, et al. 2018. A large-scale multi-ancestry genome-wide study accounting for smoking behavior identifies multiple significant loci for blood pressure. Am. J. Hum. Genet 102(3):375–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waken RJ, de las Fuentes L, Rao DC. 2017. A review of the genetics of hypertension with a focus on gene-environment interactions. Curr. Hypertens. Rep 19(3):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arnau-Soler A, Macdonald-Dunlop E, Adams MJ, Clarke T-K, MacIntyre DJ, et al. 2019. Genome-wide by environment interaction studies of depressive symptoms and psychosocial stress in UK Biobank and Generation Scotland. Transl. Psychiatry 9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mostafavi H, Harpak A, Agarwal I, Conley D, Pritchard JK, Przeworski M. 2020. Variable prediction accuracy of polygenic scores within an ancestry group. eLife 9:e48376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Young AI, Benonisdottir S, Przeworski M, Kong A. 2019. Deconstructing the sources of genotype-phenotype associations in humans. Science 365(6460):1396–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei W-H, Hemani G, Haley CS. 2014. Detecting epistasis in human complex traits. Nat. Rev. Genet 15(11):722–33 [DOI] [PubMed] [Google Scholar]
- 45.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, et al. 1997. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. JAMA 278(16):1349–56 [PubMed] [Google Scholar]
- 46.Griswold AJ, Celis K, Bussies PL, Rajabli F, Whitehead PL, et al. 2021. Increased APOE ε4 expression is associated with the difference in Alzheimer’s disease risk from diverse ancestral backgrounds. Alzheimers Dement. 17(7):1179–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marca-Ysabel MV, Rajabli F, Cornejo-Olivas M, Whitehead PG, Hofmann NK, et al. 2021. Dissecting the role of Amerindian genetic ancestry and the ApoE ε4 allele on Alzheimer disease in an admixed Peruvian population. Neurobiol. Aging 101:298.e11–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Atkinson EG, Maihofer AX, Kanai M, Martin AR, Karczewski KJ, et al. 2021. Tractor uses local ancestry to enable the inclusion of admixed individuals in GWAS and to boost power. Nat. Genet 53(2):195–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kerminen S, Martin AR, Koskela J, Ruotsalainen SE, Havulinna AS, et al. 2019. Geographic variation and bias in the polygenic scores of complex diseases and traits in Finland. Am. J. Hum. Genet 104(6):1169–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zaidi AA, Mathieson I. 2020. Demographic history mediates the effect of stratification on polygenic scores. eLife 9:e61548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rajabli F, Feliciano BE, Celis K, Hamilton-Nelson KL, Whitehead PL, et al. 2018. Ancestral origin of ApoE ε4 Alzheimer disease risk in Puerto Rican and African American populations. PLOS Genet. 14(12):e1007791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patterson N, Price AL, Reich D. 2006. Population structure and eigenanalysis. PLOS Genet. 2(12):e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berg JJ, Harpak A, Sinnott-Armstrong N, Joergensen AM, Mostafavi H, et al. 2019. Reduced signal for polygenic adaptation of height in UK Biobank. eLife 8:e39725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sohail M, Maier RM, Ganna A, Bloemendal A, Martin AR, et al. 2019. Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies. eLife 8:e39702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koyama S, Ito K, Terao C, Akiyama M, Horikoshi M, et al. 2020. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat. Genet 52(11):1169–77 [DOI] [PubMed] [Google Scholar]
- 56.Asgari S, Luo Y, Akbari A, Belbin GM, Li X, et al. 2020. A positively selected FBN1 missense variant reduces height in Peruvian individuals. Nature 582(7811):234–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takeuchi F, Akiyama M, Matoba N, Katsuya T, Nakatochi M, et al. 2018. Interethnic analyses of blood pressure loci in populations of East Asian and European descent. Nat. Commun 9:5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.1000 Genomes Proj. Consort. 2015. A global reference for human genetic variation. Nature 526(7571):68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wojcik GL, Graff M, Nishimura KK, Tao R, Haessler J, et al. 2019. Genetic analyses of diverse populations improves discovery for complex traits. Nature 570(7762):514–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Klarin D, Damrauer SM, Cho K, Sun YV, Teslovich TM, et al. 2018. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat. Genet 50(11):1514–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Asimit JL, Hatzikotoulas K, McCarthy M, Morris AP, Zeggini E. 2016. Trans-ethnic study design approaches for fine-mapping. Eur. J. Hum. Genet 24(9):1330–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Graham SE, Clarke SL, Wu K-HH, Kanoni S, Zajac GJM, et al. 2021. The power of genetic diversity in genome-wide association studies of lipids. Nature 600(7890):675–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Canela-Xandri O, Rawlik K, Tenesa A. 2018. An atlas of genetic associations in UK Biobank. Nat. Genet 50(11):1593–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Crawford NG, Kelly DE, Hansen MEB, Beltrame MH, Fan S, et al. 2017. Loci associated with skin pigmentation identified in African populations. Science 358(6365):eaan8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wei C-Y, Zhu M-X, Lu N-H, Peng R, Yang X, et al. 2019. Bioinformatics-based analysis reveals elevated MFSD12 as a key promoter of cell proliferation and a potential therapeutic target in melanoma. Oncogene 38(11):1876–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wainschtein P, Jain D, Zheng Z, TOPMed Anthropom. Work. Group, TOPMed Consort., et al. 2021. Recovery of trait heritability from whole genome sequence data. bioRxiv 10.1101/588020. 10.1101/588020 [DOI] [Google Scholar]
- 67.Marouli E, Graff M, Medina-Gomez C, Lo KS, Wood AR, et al. 2017. Rare and low-frequency coding variants alter human adult height. Nature 542(7640):186–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li X, Kim Y, Tsang EK, Davis JR, Damani FN, et al. 2017. The impact of rare variation on gene expression across tissues. Nature 550(7675):239–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hernandez RD, Uricchio LH, Hartman K, Ye C, Dahl A, Zaitlen N. 2019. Ultrarare variants drive substantial cis heritability of human gene expression. Nat. Genet 51(9):1349–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mathieson I, McVean G. 2012. Differential confounding of rare and common variants in spatially structured populations. Nat. Genet 44(3):243–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Persyn E, Redon R, Bellanger L, Dina C. 2018. The impact of a fine-scale population stratification on rare variant association test results. PLOS ONE 13(12):e0207677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ma S, Shi G. 2020. On rare variants in principal component analysis of population stratification. BMC Genet. 21:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Momozawa Y, Mizukami K. 2021. Unique roles of rare variants in the genetics of complex diseases in humans. J. Hum. Genet 66(1):11–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nicolae DL. 2016. Association tests for rare variants. Annu. Rev. Genom. Hum. Genet 17:117–30 [DOI] [PubMed] [Google Scholar]
- 75.Lee S, Abecasis GR, Boehnke M, Lin X. 2014. Rare-variant association analysis: study designs and statistical tests. Am. J. Hum. Genet 95(1):5–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Asimit JL, Hatzikotoulas K, McCarthy M, Morris AP, Zeggini E. 2016. Trans-ethnic study design approaches for fine-mapping. Eur. J. Hum. Genet 24(9):1330–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Spain SL, Barrett JC. 2015. Strategies for fine-mapping complex traits. Hum. Mol. Genet 24(R1):R111–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marigorta UM, Navarro A. 2013. High trans-ethnic replicability of GWAS results implies common causal variants. PLOS Genet. 9(6):e1003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Evans DM, Cardon LR. 2005. A comparison of linkage disequilibrium patterns and estimated population recombination rates across multiple populations. Am. J. Hum. Genet 76(4):681–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Amariuta T, Ishigaki K, Sugishita H, Ohta T, Koido M, et al. 2020. Improving the trans-ancestry portability of polygenic risk scores by prioritizing variants in predicted cell-type-specific regulatory elements. Nat. Genet 52(12):1346–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kichaev G, Pasaniuc B. 2015. Leveraging functional-annotation data in trans-ethnic fine-mapping studies. Am. J. Hum. Genet 97(2):260–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hill WG, Goddard ME, Visscher PM. 2008. Data and theory point to mainly additive genetic variance for complex traits. PLOS Genet. 4(2):e1000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barton AR, Sherman MA, Mukamel RE, Loh P-R. 2021. Whole-exome imputation within UK Biobank powers rare coding variant association and fine-mapping analyses. Nat. Genet 53(8):1260–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, et al. 2016. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet 48(10):1279–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Røttingen J-A, Chamas C, Goyal LC, Harb H, Lagrada L, Mayosi BM. 2012. Securing the public good of health research and development for developing countries. Bull. World Health Organ 90(5):398–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.WHO (World Health Organ.). 2002. Genomics and world health: report of the Advisory Committee on Health Research. Tech. Rep., WHO, Geneva [Google Scholar]
- 87.Wonkam A, Tekendo CN, Sama DJ, Zambo H, Dahoun S, et al. 2011. Initiation of a medical genetics service in sub-Saharan Africa: experience of prenatal diagnosis in Cameroon. Eur. J. Med. Genet 54(4):e399–404 [DOI] [PubMed] [Google Scholar]
- 88.Kromberg JGR, Sizer EB, Christianson AL. 2013. Genetic services and testing in South Africa. J. Commun. Genet 4(3):413–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kengne Kamga K, De Vries J, Nguefack S, Munung NS, Wonkam A. 2021. Explanatory models for the cause of Fragile X Syndrome in rural Cameroon. J. Genet. Couns 30(6):1727–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kengne-Ouafo JA, Millard JD, Nji TM, Tantoh WF, Nyoh DN, et al. 2016. Understanding of research, genetics and genetic research in a rapid ethical assessment in north west Cameroon. Int.Health 8(3):197–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Baynam GS, Groft S, van der Westhuizen FH, Gassman SD, du Plessis K, et al. 2020. A call for global action for rare diseases in Africa. Nat. Genet 52:21–26 [DOI] [PubMed] [Google Scholar]
- 92.Moodley K, Kleinsmidt A. 2021. Allegations of misuse of African DNA in the UK: Will data protection legislation in South Africa be sufficient to prevent a recurrence? Dev. World Bioeth 21(3):125–30 [DOI] [PubMed] [Google Scholar]
- 93.Adepoju P. 2019. Africa’s first biobank start-up receives seed funding. Lancet 394(10193):108. [DOI] [PubMed] [Google Scholar]
- 94.Callaway E. 2017. South Africa’s San people issue ethics code to scientists. Nature 543(7646):475–76 [DOI] [PubMed] [Google Scholar]
- 95.Adedokun BO, Olopade CO, Olopade OI. 2016. Building local capacity for genomics research in Africa: recommendations from analysis of publications in Sub-Saharan Africa from 2004 to 2013. Glob. Health Action 9:31026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wonkam A, Kenfack MA, Muna WFT, Ouwe-Missi-Oukem-Boyer O. 2011. Ethics of human genetic studies in sub-Saharan Africa: the case of Cameroon through a bibliometric analysis. Dev. World Bioeth 11(3):120–27 [DOI] [PubMed] [Google Scholar]
- 97.Heitmueller A, Henderson S, Warburton W, Elmagarmid A, Pentland AS, Darzi A. 2014. Developing public policy to advance the use of big data in health care. Health Aff. 33(9):1523–30 [DOI] [PubMed] [Google Scholar]
- 98.Horton RH, Lucassen AM. 2019. Recent developments in genetic/genomic medicine. Clin. Sci 133(5):697–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ienca M, Ferretti A, Hurst S, Puhan M, Lovis C, Vayena E. 2018. Considerations for ethics review of big data health research: a scoping review. PLOS ONE 13(10):e0204937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Green ED, Gunter C, Biesecker LG, Di Francesco V, Easter CL, et al. 2020. Strategic vision for improving human health at The Forefront of Genomics. Nature 586(7831):683–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sherman RM, Forman J, Antonescu V, Puiu D, Daya M, et al. 2019. Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat. Genet 51:30–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Choudhury A, Aron S, Botigué LR, Sengupta D, Botha G, et al. 2020. High-depth African genomes inform human migration and health. Nature 586(7831):741–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shriner D, Rotimi CN. 2018. Whole-genome-sequence-based haplotypes reveal single origin of the sickle allele during the Holocene Wet Phase. Am. J. Hum. Genet 102(4):547–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, et al. 2010. Association of trypanolyticApoL1 variants with kidney disease in African Americans. Science 329(5993):841–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sierra B, Triska P, Soares P, Garcia G, Perez AB, et al. 2017. OSBPL10, RXRA and lipid metabolism confer African-ancestry protection against dengue haemorrhagic fever in admixed Cubans. PLOS Pathog. 13(2):e1006220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. 2005. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet 37(2):161–65 [DOI] [PubMed] [Google Scholar]
- 107.Gulsuner S, Stein DJ, Susser ES, Sibeko G, Pretorius A, et al. 2020. Genetics of schizophrenia in the South African Xhosa. Science 367(6477):569–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Genovese G, Fromer M, Stahl EA, Ruderfer DM, Chambert K, et al. 2016. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat. Neurosci 19(11):1433–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.OMIM (Online Mendel. Inherit. Man). 2022. OMIM gene map statistics. Web Resour., OMIM, Baltimore, MD, accessed Dec. 15, 2021. https://www.omim.org/statistics/geneMap [Google Scholar]
- 110.Lebeko K, Sloan-Heggen CM, Noubiap JJN, Dandara C, Kolbe DL, et al. 2016. Targeted genomic enrichment and massively parallel sequencing identifies novel nonsyndromic hearing impairment pathogenic variants in Cameroonian families. Clin. Genet 90(3):288–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yan D, Tekin D, Bademci G, Foster J, Cengiz FB, et al. 2016. Spectrum of DNA variants for non-syndromic deafness in a large cohort from multiple continents. Hum. Genet 135(8):953–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wonkam A, Manyisa N, Bope CD, Dandara C, Chimusa ER. 2021. Whole exome sequencing reveals pathogenic variants in MYO3A, MYO15A and COL9A3 and differential frequencies in ancestral alleles in hearing impairment genes among individuals from Cameroon. Hum. Mol. Genet 29(23):3729–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, et al. 2016. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet 135(4):441–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bien SA, Wojcik GL, Zubair N, Gignoux CR, Martin AR, et al. 2016. Strategies for enriching variant coverage in candidate disease loci on a multiethnic genotyping array. PLOS ONE 11(12):e0167758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Das S, Forer L, Schönherr S, Sidore C, Locke AE, et al. 2016. Next-generation genotype imputation service and methods. Nat. Genet 48(10):1284–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. 2019. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol 37(8):907–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rakocevic G, Semenyuk V, Lee W-P, Spencer J, Browning J, et al. 2019. Fast and accurate genomic analyses using genome graphs. Nat. Genet 51(2):354–62 [DOI] [PubMed] [Google Scholar]
- 118.Wonkam A. 2021. Sequence three million genomes across Africa. Nature 590(7845):209–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wonkam A, de Vries J. 2020. Returning incidental findings in African genomics research. Nat. Genet 52:17–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sulc J, Mounier N, Günther F, Winkler T, Wood AR, et al. 2020. Quantification of the overall contribution of gene-environment interaction for obesity-related traits. Nat. Commun 11:1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ye J, Wen Y, Sun X, Chu X, Li P, et al. 2021. Socioeconomic deprivation index is associated with psychiatric disorders: an observational and genome-wide gene-by-environment interaction analysis in the UK Biobank cohort. Biol. Psychiatry 89(9):888–95 [DOI] [PubMed] [Google Scholar]
- 122.Shrider EA, Kollar M, Chen F, Semega J. 2021. Income and poverty in the United States: 2020. Gov. Rep., U.S. Census Bur., Washington, DC [Google Scholar]
- 123.Fort D, Wilcox AB, Weng C. 2014. Could patient self-reported health data complement EHR for phenotyping? AMIA Annu. Symp. Proc 2014:1738–47 [PMC free article] [PubMed] [Google Scholar]