

Abstract
A diverse group of intracellular microorganisms, including Listeria monocytogenes, Shigella spp., Rickettsia spp., and vaccinia virus, utilize actin-based motility to move within and spread between mammalian host cells. These organisms have in common a pathogenic life cycle that involves a stage within the cytoplasm of mammalian host cells. Within the cytoplasm of host cells, these organisms activate components of the cellular actin assembly machinery to induce the formation of actin tails on the microbial surface. The assembly of these actin tails provides force that propels the organisms through the cell cytoplasm to the cell periphery or into adjacent cells. Each of these organisms utilizes preexisting mammalian pathways of actin rearrangement to induce its own actin-based motility. Particularly remarkable is that while all of these microbes use the same or overlapping pathways, each intercepts the pathway at a different step. In addition, the microbial molecules involved are each distinctly different from the others. Taken together, these observations suggest that each of these microbes separately and convergently evolved a mechanism to utilize the cellular actin assembly machinery. The current understanding of the molecular mechanisms of microbial actin-based motility is the subject of this review.
A diverse group of intracellular microorganisms, including Listeria monocytogenes, Shigella spp., spotted fever group Rickettsia spp., and vaccinia virus, utilize actin-based motility to move within and spread between mammalian host cells. L. monocytogenes is a gram-positive bacillus that enters the human host via the intestine and can cause meningitis, fetal death, and diarrhea. Shigella spp. infect cells of the intestine and cause diarrhea and dysentery. Spotted fever group Rickettsia spp. are fastidious obligate intracellular coccobacillary organisms that cause Rocky Mountain spotted fever and related diseases. Vaccinia virus is a poxvirus that is the vaccine against smallpox.
These organisms have in common a pathogenic life cycle that involves a stage within the cytoplasm of mammalian host cells (Fig. 1). The bacterial organisms induce uptake into an endocytic vacuole, while vaccinia virus enters by fusion. Bacteria gain access to the cell cytoplasm by lysing the vacuole, whereas vaccinia virus enters directly into the cytoplasm. Once in the cytoplasm, each of these microbes recruits to its surface host actin and other cytoskeletal proteins and activates the assembly of an actin tail.
FIG. 1.

Pathogenesis of Shigella (representative of the pathogenesis of Listeria and Rickettsia as well). 1, Shigella organisms (solid ellipses) enter mammalian host cells by inducing phagocytosis. 2 to 4, After entry, the bacterium is within a phagocytic vacuole (step 2), which it lyses (step 3), thereby releasing it into the cytoplasm of the host cell (step 4). 5, the bacterium assembles an actin tail on one pole. Assembly of the actin tail propels it through the cell cytoplasm. 6, Actin tail assembly also enables it to form a protrusion from the cell surface. The protrusion contacts the membrane of the adjacent cell and is taken up, along with the bacterium within it. 7 to 9, The bacterium is then within a double-membrane vacuole, which it lyses, thereby releasing it into the cytoplasm of the adjacent cell. 10, The bacterium again assembles an actin tail that propels it through the cell.
The continuous assembly of an actin tail provides sufficient force to propel the organisms through the cytoplasm of the infected cell and into adjacent cells. Passage of Listeria, Shigella, and Rickettsia into adjacent cells occurs via membrane protrusions that form when the bacterium pushes out against the cell membrane (Fig. 1). These protrusions are engulfed by the adjacent cell, placing the bacterium into a double-membrane-bound vacuole (Fig. 1). The bacterium lyses the double membranes and is thereby released into the cytoplasm of the adjacent cell. Vaccinia virus also forms protrusions from the cell; however, in contrast to Listeria, Shigella, and Rickettsia, the viral particle fuses with the membrane at the tip of the protrusion and is thereby released into the extracellular space.
Actin-based motility is essential to the virulence of Listeria, Shigella, and vaccinia virus, and although its role in virulence has not been directly tested for Rickettsia, it is presumably important there as well. For Listeria, Shigella, and vaccinia virus, genetic deletion or modification of the genes that encode proteins known to be required for actin-based motility markedly attenuates the organism. For Rickettsia, genetic systems for manipulating genes do not yet exist.
As will become evident in this review, each of these organisms utilizes preexisting mammalian pathways of actin rearrangement to induce its own actin-based motility. In particular, each organism has evolved a protein(s) that binds and activates one or more of the components of a mammalian actin assembly pathway, thereby inducing the cascade of activation of all downstream effector molecules, with the end result being de novo actin nucleation, polymerization, and cross-linking. Particularly remarkable is that while all of these microbes use the same or overlapping pathways, each intercepts the pathway at a different step. In addition, the microbial molecules involved are all distinctly different. Taken together, these observations suggest that each of these microbes separately and convergently evolved a mechanism to utilize the cellular actin assembly machinery. The current understanding of the molecular mechanisms of microbial actin-based motility is the subject of this review.
ACTIN CYTOSKELETON: DYNAMICS AND FUNCTION
The network of elements known as the actin cytoskeleton provides the supportive framework of the three-dimensional structure of eukaryotic cells. The actin cytoskeleton is dynamic. It provides the forces that enable the cell to adopt a variety of shapes and to undertake directed movements. All cells are able to form various types of extensions or retractions of the cell membrane in response to external stimuli. Certain cell types, such as polymorphonuclear leukocytes, monocyte/macrophages, and metastatic cells, are able to move rapidly through tissues. These movements are mediated by the actin cytoskeleton. The microbial pathogens Listeria, Shigella, vaccinia virus, and Rickettsia have evolved mechanisms to utilize preexisting pathways of actin cytoskeleton rearrangements to generate their own motility within cells. Only in the last several years have we begun to understand the molecular mechanism of actin assembly. Study of actin-based motility of Listeria and Shigella has been a central component of the work that has led to our current understanding of the process.
To better understand microbial actin-based motility, the current understanding of the principles of actin cytoskeletal dynamics will be reviewed here. Several excellent reviews on this subject have recently been published (8, 19, 29, 31, 62, 125, 129, 184, 199).
Actin forms the scaffold of the cell's supportive structures. It is assisted in this by a large number of proteins known collectively as actin-binding proteins or actin-associated proteins. In many cell types, actin is the most abundant protein, constituting more than 5% of total cellular protein. Actin exists in two forms, monomeric (G-actin), which is soluble in the cytosol, and filamentous (F-actin). “Filamentous actin” refers to thin, flexible, helically arranged homopolymers of actin (Fig. 2A) that may be up to hundreds of micrometers in length.
FIG. 2.

Actin filament structure and assembly dynamics. (A) Asymmetric homopolymer of helically arranged actin. The pointed or minus end (left) and barbed or plus end (right) are indicated. ATP bound to the actin monomer is hydrolyzed to ADP shortly after addition to the filament. (B) Actin polymerization curve. An idealized curve from a pyrene actin assay is shown. Actin is polymerized in vitro under experimental test conditions. At time zero, potassium and magnesium are added. The lag phase represents the time required for actin nucleation. The rapid polymerization phase represents the time during which short filaments elongate. Steady state represents an equilibrium between growth of the filaments due to monomer addition and shortening of the filaments due to loss of monomer.
Assembly is such that actin filaments are asymmetric, with two structurally distinct ends. ATP-bound monomers polymerize onto the barbed or plus end of the filament at up to 10 times the rate at which they polymerize onto the pointed or minus end. A large pool of filamentous actin is in dynamic equilibrium with the pool of monomeric actin, while certain actin filaments (such as those in microvilli) are quite stable. Trimers of actin (actin nuclei) are the nucleating structures for the spontaneous polymerization of monomeric actin into filaments. Actin filaments are generally found associated with other actin filaments, cross-linked into bundles or aggregates by one of several actin cross-linking proteins.
Each actin molecule is a single polypeptide of 395 amino acid residues. Actin filaments are approximately 8 nm in diameter. Each actin monomer is tightly bound by a molecule of ATP. Following addition of the monomer to the barbed end of the filament, ATP is hydrolyzed to ADP (Fig. 2A). Monomers that dissociate from the pointed end of the filament are therefore bound by ADP, which can then be exchanged for ATP in a process that is slow. The regenerated ATP-bound monomer is then competent for addition to a filament barbed end. The process of continually adding monomers to the barbed end while continually dissociating monomers from the pointed end is known as treadmilling.
The free actin monomer concentration at which the proportion of cellular actin that is polymerized is constant (i.e., the critical concentration) is approximately 0.2 μM (approximately 8 μg/ml). The concentration of monomeric actin in the cell is actually much greater than this, typically about 50 to 200 μM (2 to 8 mg/ml). To maintain this high concentration of unpolymerized actin, the pool of monomeric actin is largely sequestered by actin monomer-binding proteins, principally thymosin and profilin, such that it is not free. In addition, an actin monomer that dissociates from a filament is bound by ADP, which must be exchanged for ATP before it is competent to bind a filament barbed end, and, as noted above, ADP-ATP exchange on actin monomers is slow.
Addition or dissociation of monomers from filaments can be blocked by proteins that bind specifically to a filament end (capping proteins). Filaments can be broken into short filaments by severing proteins. Thus, modulation of actin monomer-binding proteins, capping proteins, and severing proteins enables the cell to regulate the rate of actin assembly or dissociation in localized regions of the cell. Because of its ability to rapidly polymerize into filaments, to rapidly dissociate into monomers, or to be severed into short filaments, actin provides the cell with a dynamic structural scaffold that can be rearranged quickly in response to appropriate stimuli.
Assay of Actin Polymerization
Actin polymerization is commonly measured in vitro by a spectrofluorometric assay in which fluorescently tagged actin (pyrene-actin) gives a wavelength-specific signal on polymerization (30, 80). Polymerization of pure actin requires monovalent and divalent cations, typically potassium and magnesium. An example of the readout from the spetrophotometric assay is shown in Fig. 2B. In a typical experiment, potassium and magnesium are added to an experimental sample that contains monomeric actin and ATP and polymerization is monitored spectrophotometrically over several minutes. Initially there is a lag phase, which represents the time required for actin nucleation, the process in which actin nuclei (trimers), which serve as the starters for actin filaments, are formed. The lag represents a kinetic barrier to nucleation that is due to the metastable character of nuclei; only some will go on to form filaments. The lag phase is followed by a rapid polymerization phase, during which short filaments elongate, which is seen as a steep rise in the curve. The slope of the curve represents the rate of actin polymerization. Following the rapid polymerization phase, a steady-state equilibrium is reached as the growth of the filaments due to monomer addition is balanced by the loss of monomer from the filaments; the equilibrium is seen as a leveling off of the curve. Thus, the spectrophotometric assay permits the analysis of the extent of the kinetic barrier to polymerization, the rate of polymerization, and the relative steady-state amount of polymerized actin under a given set of experimental conditions. The dissociation of actin filaments can be monitored in a similar assay system.
Cytoplasmic extracts
A tool that is commonly used in the study of actin assembly in vitro is cytoplasmic extracts isolated from living cells. Extracts allow the investigator to study the motility of objects that are not able to enter intact mammalian cells, such as Sepharose beads coated with a protein of interest (18) or Escherichia coli cells that express a protein of interest (56, 78). Extracts also allow the investigator to deplete specific factors or add purified proteins, antibodies, or peptides. Most commonly used are extracts from Xenopus laevis oocytes, human platelets, and bovine brain. Variations in the methods of preparation exist; depending on the details of preparation, extracts may contain somewhat different concentrations of various cytoskeletal proteins.
Lamellipodia and Filopodia (Microspikes)
Mammalian cells have the capacity to form several types of cell extensions that are dependent on de novo actin polymerization. These extensions are mediated by local rearrangements of the actin cytoskeleton in the cell cortex, the region just beneath the plasma membrane. Actin filaments assemble near the leading edge of the membrane and thereby push it forward (109); they disassemble at some distance away from the membrane (174). Lamellipodia are sheet-like extensions and filopodia (microspikes) are long thin extensions of the cortex and plasma membrane. Formation of these extensions is regulated by a complex set of signaling pathways involving the small GTPases Cdc42 and Rac (discussed below and summarized in Fig. 3). As will become apparent in this review, microbes that utilize actin-based motility do so by means of these preexisting cellular actin rearrangement signaling pathways.
FIG. 3.

Signaling pathways involving the small GTPases Cdc42 and Rac. Shown is an overview of the Cdc42 and Rac signal transduction pathways that lead to actin rearrangements in the form of lamellipodia (membrane ruffles) and filopodia (microspikes). (Adapted from reference 11 with permission of the publisher.)
Rho Family GTPases and Regulation of the Actin Cytoskeleton
Rho GTPases are a family of small molecules that function as molecular switches in the regulation of a variety of cellular processes, including actin cytoskeletal dynamics, transcription, cell adhesion, and cell cycle progression (reviewed in references 11 and 71). Ten members of the family, many with multiple isoforms, have been identified in mammalian systems. Rho, Rac, and Cdc42 are the most extensively characterized and appear to be most important in actin cytoskeletal signaling pathways. Rho GTPases cycle between an active GTP-bound state and an inactive GDP-bound state. The GTP-bound form is able to interact with downstream effector molecules, while an intrinsic GTPase activity returns the molecule to its inactive state.
The principal function of Rho GTPases is regulation of the assembly and organization of the actin cytoskeleton (Fig. 3). Extracellular factors induce cytoskeletal changes through these switches. Filopodium formation, which can be induced by bradykinin, is thought to be regulated by Cdc42, since expression of a dominant negative Cdc42 inhibits this response (81). Lamellipodium formation, which can be induced by several growth factors (platelet-derived growth factor, insulin, or epidermal growth factor) is thought to be regulated by Rac, since expression of a dominant negative Rac specifically inhibits this response (141). Stress fiber and focal-adhesion formation are thought to be regulated by Rho, since exoenzyme C3 transferase, which inactivates Rho, blocks these responses (140). Thus, three relatively distinct signal transduction pathways are thought to be involved in the regulation of the actin cytoskeleton.
Since the microbial pathogens discussed in this review utilize the Cdc42 pathway for actin-based motility, components of this pathway are described in detail in the following sections.
Arp2/3 Complex
Each of the microbial pathogens that utilizes actin-based motility activates actin assembly through the Arp2/3 complex. The Arp2/3 complex is localized to areas of dynamic actin assembly in the cell (97, 116, 118, 173, 185). Data from the last several years suggest that the Arp2/3 complex is probably essential to the actin rearrangements that mediate the formation of lamellipodia and filopodia, as well as the formation of microbial actin tails.
Arp2/3 is a stable complex of seven polypeptides, present in 1:1 stoichiometry with each other (93, 97, 118, 185, 189). The complex is present in all eukaryotes examined, and each of the subunits is evolutionarily highly conserved. That the complex is functionally important has been demonstrated in yeast, where deletion of subunits is lethal or severely deleterious to cell growth (6, 88, 103, 112, 113, 155, 189, 190).
Two of the seven subunits can be modeled to fold similarly to conventional actin (73) and have therefore been designated Arp2 and Arp3 (for “actin-related protein”). The surface of the Arp2 and Arp3 subunits that corresponds to the barbed end of conventional actin is relatively similar to conventional actin, while the surface that corresponds to the pointed end of conventional actin is relatively divergent (73). The similarities at the barbed ends suggest that the barbed ends might function similarly to the barbed ends of conventional actin. The other five polypeptides in the complex are novel, and their designations vary among species (94).
The Arp2/3 complex is the only known cellular factor that stimulates the nucleation of actin filaments that grow from their barbed ends. It increases the rate of nucleation of new actin filaments, both in vitro (115, 187) and on the surface of Listeria monocytogenes (186). It is thought that the barbed ends of the Arp2 and Arp3 subunits serve as the surface on which nucleation is initiated.
Arp2 and Arp3 are not known to interact within an unactivated Arp2/3 complex (reviewed in reference 117). A model for the mechanism of Arp2/3 complex stimulation of actin nucleation suggests that on activation, the conformation of the complex is altered such that Arp2 and Arp3 are brought together to form a stable dimer within the complex. The barbed ends of Arp2 and Arp3 are thought to be exposed, so that polymerization of a new actin filament can be initiated on them. The filament would then extend by polymerization off the barbed end of this nucleus. This model predicts that the Arp2/3 complex caps the pointed end of the growing filament, to which it has been shown to bind with high (nanomolar) affinity in vitro (14, 115).
In addition to nucleating new actin filaments, the Arp2/3 complex links filaments at 70° angles (12, 115, 124). Branched structures similar to those observed in vitro are seen at the leading edge of motile cells (Fig. 4A to C) (173). Arp3 and one of the smaller subunits, p35 (or its human homolog p34), appear to bind actin directly (118). The 70° angle and the actin filaments themselves are quite stiff (12); as a result, the polymerizing, cross-linked network could generate force against the cell membrane, thereby pushing it forward. Extension of the filaments is halted by the binding of capping protein to the free barbed ends, which probably occurs at a distance from the leading edge or the microbial surface.
FIG. 4.

Dendritic network at the leading edge of a lamellopodium of a motile cell. (A) Branching of actin filaments at approximately 70° angles, consistent with Arp2/3-mediated cross-linking. (B and C) Higher magnification of boxes b and c, respectively, in panel A. (D) Dendritic nucleation model. The proposed model of Arp2/3 complex-mediated nucleation and branching of actin filaments is shown. The Arp2/3 complex binds to the side of an actin filament and is bound by a WASP family member (in this diagram, Scar). Both binding to the side of the filament and binding by a WASP family member are thought to play a role in activation of Arp2/3 complex nucleation of the new filament on the side of the mother filament. (E) Proposed model for the formation of branched networks of actin filaments in lamellipodia at the leading edge of motile cells. The Arp2/3 complex, in conjunction with a WASP family member (not shown), nucleates actin filaments on the sides of existing filaments. Cofilin severs filaments to generate additional uncapped barbed ends that can polymerize quickly. These processes lead to the rapid extension of an actin filament network that generates the force to push the cell membrane forward. (Panels A, B, C, and E reprinted from reference 173 with permission of the publisher. Panel D reprinted from reference 96 with permission of the publisher.
Branching of filaments by the Arp2/3 complex appears to occur by binding of the Arp2/3 complex to the sides of filaments. The bulk of evidence supports a model in which the Arp2/3 complex binds the side of an actin filament with direct interactions between the Arp3 and p34 subunits and the exposed regions of actin subunits within the mother filament (2, 12, 115), although an alternative model has been proposed (124). Binding of the Arp2/3 complex to the side of actin filaments stimulates actin nucleation (2, 5). Side binding is thought to activate the Arp2/3 complex so that a new filament is initiated from the barbed ends of the Arp2 and Arp3 subunits. This mechanism has been designated the dendritic nucleation model of Arp2/3-mediated actin assembly (Fig. 4D) (115).
Wiskott-Aldrich Syndrome Protein Family of Proteins
In isolation, the Arp2/3 complex activates actin nucleation only weakly (187). Arp2/3 complex activation of actin nucleation is stimulated (i) by other cellular proteins, in particular members of the Wiskott-Aldrich syndrome protein (WASP) family (194), and (ii) by filamentous actin (2, 5). As described in detail below (see “Listeria”), L. monocytogenes has evolved to directly stimulate Arp2/3 complex-activated actin nucleation by mimicking WASP, while Shigella and vaccinia virus have evolved to indirectly stimulate Arp2/3 complex-activated actin nucleation by activating WASP family members on their surfaces. Mimicking of WASP or activation of the WASP family member leads to stimulation of the Arp2/3 complex and rapid actin assembly.
The WASP family includes WASP, which is expressed only in hematopoeitic cells and is mutated in Wiskott-Aldrich syndrome (35); N-WASP, which is expressed ubiquitously and particularly enriched in the brain (104); Scar (WAVE), which is expressed widely (95); and the yeast isoform Las17p/Bee1p (90). Wiskott-Aldrich syndrome is a rare inherited immunodeficiency (reviewed in reference 163). Patients suffer from impaired immunity, thrombocytopenia, eczema, and hematopoietic malignancies. Cytoskeletal abnormalities have been demonstrated in lymphocytes. However, the wide range of clinical abnormalities have not yet been explained at the molecular level.
Members of the WASP family are structurally similar, sharing particular functional domains and domain organization (Fig. 5). Near the carboxy terminus of all of these proteins is one or two verprolin homology (V) domains (also known as WASP homology 2 [WH2] domains) and an acidic (A) domain. Between these two domains is a region that has been referred to as a cofilin homology (C) domain. In WASP and N-WASP, the verprolin homology, cofilin homology, and acidic domains are together referred to as the VCA domain. In Scar, they are referred to as the WA domain. The VCA (or WA) domain stimulates Arp2/3 complex activation of actin nucleation (96, 145, 188). The verprolin homology domain binds monomeric actin, and the acidic domain binds the Arp2/3 complex (95, 100, 107, 145). The cofilin homology domain is involved in intramolecular interactions with the amino-terminal regulatory region of the protein (see below). Despite its name, the structure of this domain appears to be dissimilar from that of the related sequences in cofilin, and no cofilin-like activity has been demonstrated for it or the intact proteins (39, 63). Experimental data suggest that this region participates in actin binding (100, 107).
FIG. 5.

Domain structure of WASP family members. Black, WH1 domain; blue, GBD; yellow, proline-rich region (number indicates the number of motifs containing a stretch of five or more prolines); green, WH2 or verprolin homology domain; pink, acidic domain (numbers indicate the numbers of acidic/basic residues). The cofilin homology domain lies between the WH2 and acidic domains and is not specifically indicated. Numbers to the right indicate length in amino acids. (Adapted from reference 63 with permission of the publisher.)
In all WASP family members, amino-terminal to the WH2 domain(s) is a proline-rich region that binds the actin monomer-binding protein profilin (106, 169). This proline-rich region enhances stimulation of the Arp2/3 complex by the WH2 and acidic domains (96). However, the role of profilin in Arp2/3 complex activity is uncertain. Deletion of the proline-rich region of Scar has minimal effects on actin assembly (106), and profilin inhibits rather than stimulates polymerization in the presence of a Scar fragment that contains the proline-rich, WH2, and acidic domains (96). However, in the presence of N-WASP and the Arp2/3 complex, Cdc42-stimulated nucleation of actin is enhanced by profilin (193). Of note, if the concentration of free monomeric actin is held constant, the stimulation of actin assembly by both Cdc42 and a carboxy-terminal fragment of N-WASP is enhanced by profilin, even though the carboxy-terminal fragment of N-WASP does not contain the proline-rich sequences that bind profilin (193), suggesting that a part of the enhancement that is mediated by profilin may be independent of binding to N-WASP.
In WASP and N-WASP, amino-terminal to the proline-rich region is a consensus binding site for the GTPase Cdc42 (105), known as the GTPase-binding domain (GBD). Within this domain is a 16-residue conserved motif, known as the Cdc42/Rac-interactive binding (CRIB) motif, which is also present in unrelated Cdc42 ligands. Near the amino terminus of WASP and N-WASP is a pleckstrin homology (PH) or WASP homology 1 (WH1) domain, which is involved in the binding of phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) (104). Adjacent to the PH domain in N-WASP but not in WASP is a calmodulin-binding (IQ) motif (Fig. 5). Recently, a lysine-rich basic motif that lies just amino-terminal of the GBD has also been shown to specifically bind PI(4,5)P2 (131).
In the inactive state of N-WASP or WASP, the GBD forms an intramolecular interaction with the so-called cofilin homology domain (74, 105). The carboxy-terminal end of the GBD (C-terminal to the CRIB motif) and the cofilin homology domain are each both necessary and sufficient for this autoinhibitory interaction (74). Binding of Cdc42 to WASP, and by extension to N-WASP, disrupts this intramolecular linkage and autoinhibition (74). One of the GBD residues involved in autoinhibition is known to be mutated (Leu270 to Pro) in the naturally occurring human disease X-linked severe congenital neutropenia, which is characterized by severe recurrent bacterial infections (36).
The GBD, to which Cdc42 binds, and the basic motif, to which PI(4,5)P2 binds, together constitute the essential regulatory region of N-WASP and, by extension, WASP (131, 144). Whereas N-WASP that is not bound by Cdc42 and PI(4,5)P2 or that is bound by only one of these molecules stimulates Arp2/3 complex-mediated actin nucleation only weakly, binding of both molecules synergistically stimulates N-WASP activation of Arp2/3-mediated actin nucleation (39, 131, 145). Consistent with these observations, in the absence of Cdc42 and PI(4,5)P2, an N-WASP fragment that consists only of the WH2, cofilin, and acidic domains, thereby lacking the regulatory region, stimulates actin polymerization markedly more than full-length N-WASP does (39, 145).
It has been suggested that in their folded autoinhibited conformations, WASP and N-WASP are unable to bind the Arp2/3 complex. Structural studies further suggest that residues that are involved in binding to the Arp2/3 complex may be inaccessible (74). However, data addressing whether the Arp2/3 complex binds autoinhibited N-WASP are conflicting (131, 144).
Cofilins
Cofilins are actin-associated proteins that have been described to have both actin-severing and -depolymerizing activities (21, 23, 67). The family consists of highly homologous members that have essentially identical activities. Depending on the species, cofilins have been designated cofilin, actin-depolymerizing factor (ADF), actophorin, or destrin. In motile cells, cofilin contributes to actin polymerization at the leading edge by severing actin filaments (23, 196), thereby generating a pool of uncapped barbed ends that can rapidly extend. In its phosphorylated state, cofilin is inactive. Inactivation of cofilin by overexpression of the kinase that phosphorylates it completely inhibits the generation of actin barbed ends and abolishes cell motility (196). Thus, cofilin is essential to the motility of eukaryotic cells.
Debranching and Depolymerization
Arp2/3 complex-mediated linkages between filaments are metastable. At a distance from the leading edge, the actin filament branches dissociate from the mother filaments and depolymerize. Dissociation of the Arp2/3 complex from the mother filaments is accelerated by the conversion of ATP-actin to ADP-actin in the filaments, a process that is enhanced by cofilin (14). The Arp2/3 complex is thought to dissociate from the pointed ends of the filaments, either before or after debranching, allowing rapid depolymerization of actin from the free pointed ends.
Summary
The WASP family of proteins and the Arp2/3 complex appear to be essential to the pathway of de novo actin filament nucleation that induces lamellipodium and filopodium formation, as well as microbial motility (described in detail below). At the cell cortex, activated Arp2/3 complex nucleates new actin filaments by stabilizing and capping the barbed end of actin oligomers. It does so after binding to the sides of existing actin filaments and/or by forming branches at the uncapped barbed ends of existing filaments. Actin filaments extend rapidly by polymerization on uncapped barbed ends. Also at the cell cortex, cofilin severs existing and/or newly polymerized filaments, thereby generating additional uncapped barbed ends off which polymerization occurs. As a result, branched networks, which provide the force to push the cell membrane forward, are rapidly formed (Fig. 4E). The Arp2/3 complex is activated by WASP family members. Autoinhibition of WASP family members, which is mediated by an intramolecular interaction, is relieved by the binding of Cdc42 and PI(4,5)P2.
LISTERIA
L. monocytogenes and the related L. ivanovii are facultative intracellular pathogens that move through cells and spread directly from one cell into an adjacent cell by means of actin-based motility. This section of the review will focus on actin assembly induced by L. monocytogenes; relatively little is known about that induced by L. ivanovii (22, 50, 57, 72, 82). L. monocytogenes is widespread in nature and causes disease in a variety of herd animals as well as in humans. In humans it can cause meningitis, fetal death, and diarrhea. The ability to spread directly between cells allows Listeria to avoid many components of the host immune response. In the murine model of infection, the organism colonizes and replicates in the liver and spleen. The 50% lethal dose for mice of an L. monocytogenes mutant defective in actin assembly is 4 log units higher than that of wild-type L. monocytogenes (76).
L. monocytogenes is able to enter and assemble actin in all types of adherent cells that have been tested. In the cytoplasm of infected cells, it has been documented to move at rates of 13.2 to 22 μm/min (34, 58), which are approximately the same rates of movement as those documented for Shigella (5.4 to 26 μm/min [58, 158, 197]), threefold higher than the rates of movement documented for Rickettsia (4.8 to 8 μm/min [58, 59]), and five- to eightfold higher than the rates of movement documented for vaccinia virus (2.8 μm/min [32]) (Table 1). Dabiri et al. reported individual L. monocytogenes cells moving at 88 μm/min in J774 macrophages (34), but such speeds have not been documented by other investigators. In cytoplasmic extracts, L. monocytogenes has been documented to move at 6.4 to 13 μm/min, which is about half the rate of movement of intracellular L. monocytogenes (58, 78, 99, 176).
TABLE 1.
Reported rates of microbial actin-based motilitya
| Environment | Motility (μm/min) of:
|
||||
|---|---|---|---|---|---|
| L. monocytogenes | S. flexneri | E. coli expressing Shigella IcsA | Vaccinia virus | Rickettsiab | |
| Cells | 13.2 ± 0.24 (34) | 5.4 ± 4.2 (197) | NAc | 2.8 ± 0.5 (32) | 8 ± 1 (58) |
| 22 ± 5 (58) | 12.3 ± 7.7 (158) | 4.8 ± 0.6 (59) | |||
| 26 ± 5 (58) | |||||
| Xenopus extracts | 6.4 (176) | NAd | 12.9 ± 7.8 (56) | NDe | 2.0 ± 0.2 (58) |
| 11 ± 2.4 (78) | 29.5 ± 5 (78) | ||||
| 11 ± 2 (99) | |||||
| 13 ± 5 (58) | |||||
References are given in parentheses.
R. rickettsii or R. conorii.
NA, not applicable. E. coli cannot be readily introduced into the cytoplasm of mammalian cells.
NA, not applicable. S. flexneri does not move in Xenopus extracts, apparently because IcsA expression is relatively low in vitro (Magdalena and Goldberg, submitted).
ND, not determined. Vaccinia virus motility has not been assayed in cytoplasmic extracts.
Morphology of the Listeria Actin Tail
When visualized by electron microscopy, Listeria actin tails in the cytoplasm of infected cells consist of multiple short actin filaments that are arranged in a disorganized-appearing nonparallel network (Fig. 6) (177, 180). Decoration of tails with the S1 subfragment of myosin, which binds to subunits within actin filaments in an asymmetric fashion, demonstrates that the barbed ends of the filaments within the tails are oriented toward the bacterial body (177). Actin tails formed on the surface of beads coated with the Listeria actin assembly protein ActA have the dendritic appearance of actin filaments near the leading edge of lamellipodia (Fig. 4A) (20).
FIG. 6.

Thin-section electron microscopy of an actin tail formed by an intracellular L. monocytogenes bacterium. Note that the tail consists of short actin filaments that are bundled in a nonparallel fashion. (Reprinted from reference 180 with permission of the publisher.)
In Vero cells, Listeria actin tails average 1 μm in diameter and 5 μm in length, compared to 0.7 μm in diameter and 7 μm in length for Shigella and 1.5 μm in diameter and 5 to 17 μm in length for Rickettsia (58, 75) (Table 2). Of note, the length and diameter of actin tails for any particular organism vary among different cell lines, such that comparisons can not be made reliably across cell lines.
TABLE 2.
Reported morphological characteristics of microbial intracellular actin tailsa
| Organism | Tail length (μm) | Tail width (μm) | Length of filaments within tail (μm) |
|---|---|---|---|
| L. monocytogenes | 4–12 (58) | 1 (58) | 0.1 (58)b |
| <0.3 (179)b | |||
| S. flexneri | 5–15 (58) | 0.7 (58) | 0.1 (58) |
| Vaccinia virus | 6.4–9.6 (32) | NDc | 0.74 (33) |
| Rickettsiac | 4–6 (58) | 1.5 (58) | 0.3–3 (58) |
| 16.7 (59) | >1 (75) |
References are given in parentheses.
Other investigators have reported that filaments within the tails of L. monocytogenes are severalfold longer than 0.1 μm (Coughlin and Mitchison, unpublished).
ND, not determined.
R. rickettsii or R. conorii.
Near to the bacterium, filaments in the Listeria tail are longer than those at a distance from the bacterium and appear to lie parallel to the sides of the bacterial body, with relatively few filaments immediately adjacent to the pole (58, 78). This differs from the tails of Shigella, where the filaments appear short throughout the tail and, near to the bacterium, are adjacent to the pole and not the sides of the bacterial body. These differences in patterns of actin filaments on Listeria and Shigella mirror the differences in localization of the actin-polymerizing proteins of the two organisms, ActA and IcsA, as described below. The filaments within the tails of Listeria have been reported to be short, generally about 0.1 μm in length (58), although other data suggest that they may be several fold longer (M. Coughlin and T. J. Mitchison, unpublished data). The filaments within the tails of Shigella have also been reported to be about 0.1 μm in length, whereas those within the tails of Rickettsia and vaccinia virus have been reported to be longer, 0.3 to 3 μm (or >1 μm) and 0.74 μm, respectively (33, 58, 75).
To spread from one cell into an adjacent cell, Listeria pushes against the cell membrane to form a finger-like protrusion that contains the bacterium at its tip. The protrusion, along with the bacterium within it, is then taken up into the adjacent cell by an endocytic process. The actin tails that trail bacteria within protrusions differ in morphology from the tails within the body of the cell. Those within the protrusion consist predominantly of extremely long filaments of actin that are bundled in a parallel array (Fig. 7) (156). They contain relatively few short actin filaments of the type seen in actin tails within the body of the cell; the few short filaments that are present are oriented at angles to the long filaments (156). Near to the bacterial body within protrusions, the filaments splay out and are less tightly bundled (156). The reason that tails within the protrusions differ in morphology from those within the cell body is uncertain, although a possible explanation is that factors involved in generating the branched array or in capping the growing filaments near to the bacterial body may be partially excluded from the protrusions.
FIG. 7.

Negative-staining electron microscopy of an actin tail formed by an L. monocytogenes bacterium within a cell surface protrusion. Note that the tail consists of long actin filaments that are bundled in a parallel fashion. (Reprinted from reference 156 with permission of the publisher)
L. monocytogenes Protein ActA
The L. monocytogenes surface protein ActA mediates actin assembly. L. monocytogenes strains that carry a gene disruption in ActA are unable to assemble actin tails or spread from cell to cell in a tissue culture monolayer (16, 37, 76) and are 3 log units less virulent in mice (16). Heterologous expression of ActA in L. innocua, a nonpathogenic Listeria species that does not normally polymerize actin, enables this organism to assemble actin tails (78). Purified ActA that is linked to the surface of streptococci (161) or to the surfaces of beads (18) enable the streptococci or beads to form actin tails, demonstrating that ActA is sufficient to mediate actin-based motility in the absence of other Listeria factors.
ActA is a 639-amino-acid protein that is anchored in the bacterial membrane by the carboxy-terminal 26 amino acids, such that following cleavage of the 29-amino-acid amino-terminal secretion signal, the 584-amino-acid residue mature amino terminus is exposed on the bacterial surface (76). ActA has a calculated molecular mass of 67 kDa, although it migrates on sodium dodecyl sulfate-polyacrylamide gel electrophoresis at 90 kDa (76). ActA expression is markedly increased inside mammalian cells (16, 43, 110). Several strains that constitutively overexpress ActA have facilitated its purification and been used in motility assays (160). ActA from Listeria-infected cells migrates as three distinct polypeptides, as a result of its phosphorylation during intracellular growth (16). Phosphorylation appears to occur on residues within the proline-rich repeat region (see below), since an ActA derivative with this region deleted is not phosphorylated (85). The role of ActA phosphorylation in Listeria motility is unknown.
The functions of sequences within the surface-exposed portion of ActA have been characterized by several groups, largely through extensive deletion analysis and site-directed mutagenesis. Native and recombinant ActA are readily purified, facilitating the analysis of mutant ActA proteins in vitro. Removal of the carboxy-terminal transmembrane domain of ActA leads to its secretion, allowing its easy purification from culture supernatants. Note that in the published literature, some authors designate as amino acid 1 the amino-terminal residue of mature ActA (after cleavage of the signal peptide); in this review, we designate as amino acid 1 the amino-terminal residue of full-length immature ActA, which includes the signal peptide.
The entire amino-terminal portion of the mature protein, amino acids 30 to 263, is essential for actin polymerization in Listeria-infected cells. Deletion of amino acids 50 to 260 leads to loss of the ability to assemble actin filaments in Listeria-infected mammalian cells (85), and deletion of amino acids 30 to 265 leads to loss of association of actin with a derivative of ActA that anchors on the mitochondria of transfected cells (126). A fragment that consists of ActA amino acids 30 to 263 is sufficient for motility in cytoplasmic extracts when artificially bound to the bacterial surface (86).
Requirement of Host Arp2/3 Complex for ActA-Mediated Actin Assembly
ActA-mediated stimulation of actin polymerization requires the mammalian host protein complex Arp2/3 (186, 187). As discussed in detail above, the known biochemical activities of the Arp2/3 complex are (i) cross-linking or branching of actin filaments at 70° angles (12, 115, 124) and (ii) weak nucleating activity (116, 118). By immunofluorescence, the Arp2/3 complex is located throughout the actin tail of motile Listeria (185, 186). Sequestration of Arp2/3 complex by addition to cytoplasmic extracts of the Arp2/3-binding domain of Scar1 (WAVE), a WASP family member, or depletion of the Arp2/3 complex from platelet or bovine brain extracts inhibits Listeria motility (39, 101, 194). Similar inhibition of Listeria motility is seen on overexpression of the same domain of Scar1 in infected cells (101). Motility is rescued by the addition of purified Arp2/3 complex (39, 101, 194), specifically indicating that sequestration of the Arp2/3 complex interferes with Listeria actin tail formation. Reconstitution of Listeria motility using purified cytoskeletal proteins requires the Arp2/3 complex (91). Using pure proteins in vitro, in the absence of the Arp2/3 complex the mature surface-exposed portion of ActA (amino acids 29 to 613) or ActA amino acids 29 to 263 has no effect on the nucleation of actin polymerization, but in the presence of Arp2/3 complex each has a marked stimulatory effect (187) (Fig. 8). Finally, the actin filaments in the tail of intracytoplasmic Listeria are branched at acute angles, similar to the acute angles at which the Arp2/3 complex branches actin in vitro (12), and, by electron microscopy, the Arp2/3 complex is localized to the Y junctions of the dendritic network of tails formed by ActA-coated beads in extracts (20). These data suggest that the Arp2/3 complex mediates some or all cross-linking or branching of actin filaments in the Listeria tail.
FIG. 8.
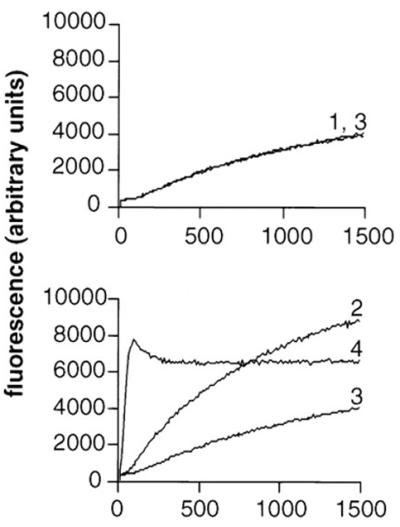
ActA stimulation of Arp2/3 complex-mediated actin polymerization. Fluorescence intensity is plotted as a function of time in pyrene-actin polymerization assays. 1, actin; 2, actin and Arp2/3 complex; 3, actin and ActA (amino acids 29 to 263); 4, actin, Arp2/3 complex, and ActA (amino acids 29 to 263). (Reprinted from reference 187 with permission of the publisher.)
After entry into the cell cytoplasm or placement in cytoplasmic extracts, the association of Listeria with actin that is first visualized is the assembly of a “cloud” of filamentous actin that surrounds the bacterium. This cloud evolves into an actin tail that extends exclusively from one pole of the bacterium. An area of uncertainty at present is whether the Arp2/3 complex or another factor(s) is responsible for the conversion of actin clouds into actin tails. Whereas actin tails consist of filaments that have been shown to be cross-linked (20), it is not known whether the filaments within the actin cloud are cross-linked. The data are confusing. In vitro, in the presence of the Arp2/3 complex and actin, wild-type Listeria forms clouds of polymerized actin on its surface but rarely forms actin tails (187). In addition, sequestration of the Arp2/3 complex by overexpression of the Arp2/3-binding domain of Scar1 eliminates actin tail assembly but not the formation of actin clouds on the bacterial surface (101). These data suggest that either (i) the affinity of ActA for the Arp2/3 complex is adequately high that, in the second set of experiments, sufficient Arp2/3 complex remains associated with ActA to nucleate the polymerization of filaments on the Listeria surface but inadequate amounts are present to branch or cross-link filaments into a tail, or (ii) as pertains to the first set of experiments, while the Arp2/3 complex is required for cross-linking or branching of the filaments into a tail, other host factors must also be present.
Alanine mutagenesis shows that an extensive region of ActA, stretching from amino acids 166 to 256, appears to be involved in the ability of ActA to convert actin clouds into actin tails. At 4 h of infection, wild-type Listeria is associated with an actin tail about twice as often as it is associated with an actin cloud. Mutation of clustered charged amino acids within amino acids 166 to 256 leads to an inversion of this ratio as well as up to a 50% decrease in rates of movement, by a mechanism that is unknown (86a). Whether these residues in ActA are involved in modifying the interaction with the Arp2/3 complex or in binding another host factor is not known.
Domains of ActA That Function in Actin Assembly
Since the WH2 and acidic domains of WASP family members are required for WASP-mediated Arp2/3 complex activation of actin nucleation, investigators have looked to identify and characterize homologous domains in ActA, as well as other domains that might function in actin assembly. A summary of the results of these works is shown in a schematic diagram of the functional domains of ActA in Fig. 9A.
FIG. 9.

Functional domains of L. monocytogenes ActA. (A) Schematic of ActA. SP, signal peptide; A, WASP-like acidic domain, possibly involved in Arp2/3 complex binding; WH2, WASP homology 2 domain; PI, phosphoinositide binding region; Pro-rich repeats, proline-rich repeats; TM, transmembrane anchor; AB, monomeric actin-binding region, amino acids 60 to 100; dimer, region required for ActA dimerization, amino acids 126 to 155; clouds/tails, region important for formation of actin tails, rather than actin clouds, amino acids 166 to 256. (B) Alignment of ActA amino acids 85 to 99 with the first of two WH2 domains of N-WASP and of ActA amino acids 121 to 170 with the WH2 and Arp2/3 complex-binding domains of WASP family members. (C) Alignment of ActA amino acids 32 to 45 with the acidic domains of WASP family members. Sequences used for alignment: mouse WASP, human N-WASP, and human Scar1. (Panel B adapted from reference 195 with permission of the publisher. Panel C adapted from reference 159 with permission of the publisher.)
Regions of ActA That Bind Monomeric Actin: Amino Acids 60 to 101.
The ability of ActA amino acids 29 to 263 to stimulate Arp2/3 activation of actin assembly in vitro suggested that this region of ActA could interact with both Arp2/3 and monomeric actin. ActA amino acids 30 to 263 fused to a linker and expressed on the bacterial surface mediate actin tail assembly in cytoplasmic extracts (86), which demonstrates that the region is sufficient for actin tail assembly. When ActA is purified and bound to beads, the same region of ActA polymerizes actin in cytoplasmic extracts (28).
ActA binds the Arp2/3 complex with moderately high affinity (Kd, 0.7 ± 0.2 μM) (195). ActA inhibition of the spontaneous polymerization of actin in a pyrene-actin assay best fits a model in which ActA binds two actin monomers (195). One of these actin-binding sites appears to lie between ActA amino acids 60 and 101. Deletion of amino acids 60 to 101 leads to a marked decrease in the binding of ActA to monomeric actin, as measured by the loss of ability of the construct to inhibit the spontaneous polymerization of actin in both a pyrene-actin assay and an actin-pelleting assay (in which monomeric actin remains soluble whereas polymerized actin pellets) (159). A similar effect is seen using ActA lacking the entire amino-terminal region (amino acids 31 to 262) (159). An ActA fragment that consists of amino acids 60 to 100 binds actin, as measured in the same pyrene-actin assay, and a glutathione S-transferase (GST) fusion to ActA amino acids 62 to 103 binds fluorescently labeled monomeric actin (28, 195). Mutagenesis of charged residues to alanine within amino acids 60 to 66, 70 to 77, or 80 to 88 decreases the binding of the mutant ActA to monomeric actin (86a), indicating that the entire region between amino acids 60 and 88 is involved in binding to monomeric actin (86a). Amino acids 85 to 99 align with the first of two WH2 domains of N-WASP (195). WH2 domains are known to bind monomeric actin.
A second actin-binding domain in ActA has been identified at amino acids 121 to 138, immediately amino-terminal to the Arp2/3-binding domain (see below) (195). An ActA fragment that consists of amino acids 121 to 152 binds actin, as measured in the pyrene-actin assay (195). However, alanine mutagenesis of clustered charged residues within this region does not affect binding to monomeric actin in the same assay (86a), raising the possibility that actin binding of this region is context specific. Within this region, amino acids 121 to 138 align with the actin-binding WH2 domain of WASP family members, most closely with the second of the two WH2 domains of N-WASP (Fig. 9B) (195).
Regions of ActA That Activate Arp2/3-Mediated Actin Nucleation: Amino Acids 121 to 170
Actin polymerization assays of ActA derivatives in the presence of the Arp2/3 complex suggest that ActA amino acid residues 121 to 170 contain an Arp2/3-binding and activation domain. Amino acids 121 to 170 of ActA activate Arp2/3-mediated actin polymerization (195), and deletion of amino acids 136 to 165 from an active fragment of ActA leads to loss of this activity (159). Furthermore, deletion of amino acids 136 to 165 leads to loss of ActA-mediated actin assembly by Listeria in the cytoplasm of mammalian cells (159). Within this region, ActA amino acids 143 to 169 align with the Arp2/3-binding acidic domains of WASP family members (Fig. 9B) (195), suggesting that these residues are responsible for the observed Arp2/3-activating activity. In fact, deletion of five basic amino acids within this region (amino acids 146 to 150, KKRRK) leads to loss of actin tail assembly on Listeria in cytoplasmic extracts (86) and to a marked reduction in activation of Arp2/3-mediated actin polymerization (159). When all of these five amino acids are changed to alanine, similar results are obtained (86a). Moreover, mutation of either Arg148 to Lys or Arg149 to Ser leads to loss of actin polymerization on the bacterial surface (128). Of note, the Arp2/3 complex still colocalizes with the bacterium, as determined by immunfluorescence of the 21-kDa subunit, suggesting that Arg148 and Arg149 are required for activation of the Arp2/3 complex but not for its binding (128). Alanine mutagenesis of the charged residues within the overlapping stretch of amino acids 140 to 147 leads to a more modest effect, whereas alanine mutagenesis of amino acids 156 to 160 leads to a moderate effect, as seen in actin polymerization assays (86a). Taken together, these data suggest that Arg148 and Arg149 are absolutely critical and that amino acids 156 to 160 are important for the activation of Arp2/3-mediated actin polymerization.
The entire amino terminus of ActA (amino acids 30 to 612) binds Arp2/3 with moderately high affinity (Kd, 0.5 ± 0.2 uM). Deletion of ActA amino acids 136 to 165 does not eliminate binding to the Arp2/3 complex (159), suggesting that the stretch between ActA amino acids 143 to 169 is not the sole region of ActA that binds Arp2/3. The amino terminus of mature ActA (amino acids 31 to 58) contains a stretch of acidic amino acids that aligns with a carboxy-terminal acidic domain of WASP family proteins (Fig. 9C). Deletion of this domain leads to a decrease in ActA activation of Arp2/3-mediated actin nucleation (159). In addition, alanine mutagenesis of charged residues within amino acids 32 to 42 or 44 to 54 results in significantly decreased rates of Listeria movement (86a). Mutations in amino acids 44 to 54 result in an approximately twofold decrease in activation of Arp2/3-mediated actin polymerization, whereas mutations in amino acids 32 to 42 do not alter the activation of Arp2/3-mediated actin polymerization (86a). In WASP family members, the carboxy-terminal acidic domain is known to be involved in binding to the Arp2/3 complex. Taken together, these data suggest that in ActA, amino acids 31 to 58 may be a second region, in addition to amino acids 143 to 169, that is involved in Arp2/3 binding, although the exact limits of the binding region are still poorly defined.
Binding Site for ActA on the Arp2/3 Complex
ActA amino acids 121 to 170 chemically cross-link with a zero-length cross-linker to the Arp2/3 complex subunits p40, Arp2, and Arp3 (195). The WASP family member Scar1 chemically cross-links to the same Arp2/3 complex subunits (195). Furthermore, Scar1 and N-WASP each compete with the binding of ActA to the Arp2/3 complex in vitro (195).
ActA binding to the Arp2/3 complex competes with the binding of profilin but not cofilin (actophorin) (195). The protein sequence of Arp2 is 50% identical to that of conventional actin, and the profilin- and cofilin-binding domains of conventional actin have been identified (13, 53, 154). These data, along with the chemical cross-linking studies described above, have permitted the proposal of a binding site of ActA on Arp2 within the Arp2/3 complex (Fig. 10) (195). This binding site would place ActA or other nucleation-promoting factors (i.e., WASP family members) at the site on Arp2 that is analogous to the barbed end of conventional actin (195). This may enable ActA or another nucleating factor to assist in the addition of actin monomers to the Arp2/3 complex.
FIG. 10.
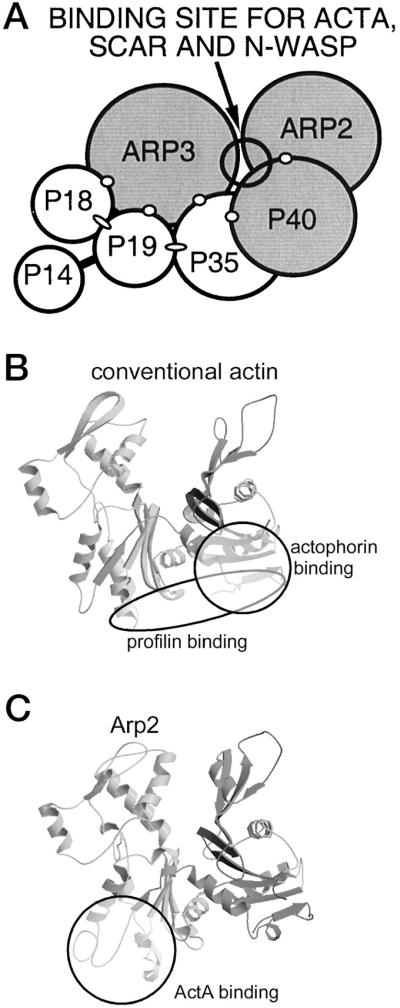
Proposed location of the ActA-binding site on the Arp2/3 complex and on the Arp2 subunit. (A) Proposed site for binding of ActA, Scar, and N-WASP to the nearest-neighbor model of the Arp2/3 complex. Shading indicates subunits to which ActA, Scar, and N-WASP cross-link. Open circles and dark lines indicate subunits that cross-link within the complex (118). Ovals indicate interactions demonstrated by two-hybrid analysis (95). (B) Profilin- and actophorin (cofilin)-binding sites on conventional actin (13, 53, 154). (C) Proposed structure of the Arp2 subunit, based on homology to conventional actin (73). The binding site of ActA that has been proposed on the basis of competition experiments (see the text) is indicated. (Reprinted from reference 195 with permission of the publisher.)
The data on ActA interactions with the Arp2/3 complex and monomeric actin provide compelling evidence that ActA is structurally and functionally similar to members of the WASP family of nucleation-promoting factors. This is the first example of several in this review of a microbial protein that has evolved to mimic a mammalian one.
Dissociation of the Arp2/3 Complex from ActA
The Arp2/3 complex is distributed throughout the length of the actin tail on intracellular Listeria (185, 186). The Arp2/3 complex, bound to ActA on the bacterial surface, forms the pointed end of newly nucleated actin filaments, which rapidly polymerize on the free barbed ends. Recent data indicate that the Arp2/3 complex dissociates from ActA by the time approximately 10 actin monomers have added to the barbed end of the new filament. As described above, the Arp2/3 complex also links filaments at 70° angles, most probably by initiating new filaments after binding to the side of an existing filament or by cross-linking the Arp2/3-capped pointed end to the side of an existing filament (2, 115). Thus, it appears that in conjunction with its dissociation from ActA, the Arp2/3 complex forms a cross-linked or branched network of rapidly growing actin filaments at the bacterial surface. The 70° angle and the actin filaments themselves are quite stiff (12); as a result, the polymerizing, cross-linked network can generate force against the bacterial surface, thereby pushing the bacterium forward. Extension of the filaments is halted by the binding of capping protein to the free barbed ends (see below). Free Arp2/3 complex probably rapidly binds the available Arp2/3 binding site on ActA, where it is activated and initiates further de novo actin nucleation.
ActA Proline-Rich Repeats and the Enabled and Vasodilator-Stimulated Phosphoprotein Family Proteins
The central region of ActA (amino acids 293 to 390) contains four 11-amino-acid proline-rich repeats, the first three of which contain the motif FPPPP and the fourth of which contains the motif FPPIP (37, 76). These proline-rich repeats are separated by three 24- to 33-amino-acid sequences, the first two of which are almost identical. Proline-rich sequences similar to those found in ActA are also present in the mammalian cytoskeletal proteins zyxin and vinculin (138). Zyxin is present in focal contacts, microfilaments, and dynamic regions of the membrane (138), and vinculin is present in focal contacts (17, 47) and intercellular adherens junctions (48, 49).
The vasodilator-stimulated phosphoprotein (VASP) and the related protein Mena are present on the surface of intracytoplasmic Listeria (22, 52). On motile bacteria, VASP and Mena localize to the bacterial pole at the junction of the bacterium with the actin tail (22, 52), in the same distribution as ActA. Consistent with this localization, purified VASP binds ActA (22).
VASP and Mena are members of the Ena/VASP family of proteins, which also includes Ena and Evl. VASP, Mena, and Evl are mammalian proteins, while Ena is the Drosophila protein Enabled (51). These proteins localize to focal contacts and regions of dynamic actin rearrangements, such as the leading edge of motile cells (52, 137). The rate of extension of the membrane at the leading edge correlates with the local concentration of VASP (147).
The polypeptides of members of this family contain three domains: highly homologous amino- and carboxy-terminal domains, called Ena/VASP homology domains 1 and 2 (EVH1 and EVH2), and a central more divergent proline-rich region. The amino-terminal EVH1 domain, which is similar to the WH1 domain of WASP family members (see “Actin cytoskeleton: dynamics and function” above), mediates interactions with focal contact proteins, including zyxin and vinculin (15, 52, 119, 138, 139). The proline-rich region mediates interactions with profilin, SH3 domains, and WW domains (1, 41, 52, 136). The carboxy-terminal EVH2 domain mediates tetramerization of the protein and interactions with actin filaments (4, 87). Of note, the Ena/VASP proteins and the WASP family of proteins have proline-rich motifs that contain the consensus (G/A/L/S)PPPPP (132), that are distinct in both structure and function from the proline-rich motifs of ActA, and that do not bind VASP (119). VASP is enriched in platelets, and Mena and Evl are enriched in brain extracts. In Listeria motility assays, VASP, Mena, and Evl functionally complement one another, although not to wild-type rates of movement (87).
The proline-rich repeats of ActA are required for VASP binding. Deletions or site-directed mutagenesis of these repeats leads to loss of detectable VASP by immunofluorescence microscopy (119, 127, 162). Peptide scanning of ActA identified only the proline-rich motifs as VASP ligands (119). The binding site of the ActA proline-rich motifs on VASP, Mena, and Evl, is the EVH1 domain. ActA binds the EVH1 polypeptide of Mena or Evl fused to GST in a solid-phase binding assay (119). Preincubation of Listeria with the EVH1 polypeptide of VASP, Mena, or Evl fused to GST inhibits bacterial actin assembly on addition to extracts (87). A single tryptophan residue within the EVH1 domain (Trp23 of Mena and Ena) is critical for binding to the ActA proline-rich region (42, 130).
ActA-derived peptides that contain residues flanking the FPPPPT core bind with higher affinity than do truncated subsequences (119). The ActA dodecapeptide 333FEFPPPPTEDEL344 binds with the highest affinity of all peptides tested (Kd, 19 μM) and with significantly higher affinity than zyxin or vinculin peptides, indicating that determinants of both specificity and affinity reside in these flanking residues (7). This peptide binds the VASP EVH1 domain with significantly higher affinity than does a zyxin-derived peptide (7), and a similar ActA-derived peptide binds VASP with higher affinity than does intact zyxin or vinculin (119). Moreover, a variety of other proline-rich peptides do not bind VASP (119). The phenylalanine at position 333, the glutamic acid at position 343, the leucine at position 344, and the prolines at positions 336 and 339 are essential for the observed high-affinity binding to EVH1 (7). The extremely high affinity of ActA for VASP suggests that intracytoplasmic Listeria is able both to readily recruit VASP from its mammalian ligands zyxin and vinculin and to remain tightly bound to it.
Role of VASP Binding in Listeria Actin-Based Motility
Several roles for the interaction of Ena/VASP proteins with ActA in Listeria actin-based motility have been proposed and are supported to different extents by the existing data. VASP is known to bind the actin monomer-binding protein profilin (136), leading to the suggestion that VASP serves to increase the local pool of actin monomers in the vicinity of ActA. Profilin is present on the surface of intracytoplasmic Listeria, and the amount of profilin associated with the bacteria decreases proportionally with the amount of associated VASP (162). Consistent with this, in depletion/add-back experiments, VASP is required for profilin to have an effect on Listeria movement (87). However, data on the requirement of profilin for wild-type rates of Listeria motility are inconsistent. Two groups report differing results in assays of Listeria motility in extracts depleted of profilin (87, 99, 176). When Listeria motility is reconstituted using purified protein components, profilin increases the speed of motile Listeria but is not absolutely required for movement (91). Fairly consistent among these studies is that profilin accelerates Listeria movement modestly, that it accelerates movement less significantly than VASP, and that it requires VASP for its effect.
A second role proposed for VASP in Listeria motility is the bundling of actin filaments into a tail. In VASP-depleted extracts, actin polymerizes on the surface of Listeria into loosely organized networks but does not bundle to form tails (87). Also, in vitro, VASP can bundle actin filaments (87). However, since ActA amino acids 166 to 256, which lie outside of the proline-rich repeats, appear to also be required for the conversion of actin clouds into actin tails (see “Requirement of Host Arp2/3 complex for ActA-mediated actin assembly” above), if VASP is important in this process, it is probably not the only factor.
A third role proposed for VASP in Listeria motility is the initiation of actin polymerization on the bacterial surface. The data on this are conflicting. Deletion of the ActA proline-rich repeats, to which VASP binds, leads to decreased speeds of Listeria motility, with a linear relationship between the number of repeats and the speed (162). The frequency of actin tail formation is also lower in these strains, although in a nonlinear relationship that is suggestive of a role for the long repeats located between the proline-rich motifs in initiation of movement (162). In the complete absence of any proline-rich motifs, Listeria forms actin tails, albeit at low frequencies (162). Deletion of the proline-rich repeats also leads to a slowing of bacterial spread between cells, as measured by the plaque assay, and an increase of the 50% lethal dose in a mouse model of listeriosis (162). In the absence of the proline-rich repeats, no VASP or profilin is detected on the bacterial surface by immunofluorescence (162). When Listeria motility is reconstituted using purified protein components, Listeria is able to assemble actin tails in the absence of VASP, but the presence of VASP increases the speed of moving bacteria about 10-fold (91). In parallel experiments, in the presence of VASP, profilin increases the rate of movement about twofold (91).
In contrast, microinjection of a synthetic peptide that matches the second ActA proline-rich repeat leads to a complete halt of bacterial actin tail assembly and motility, not simply a decrease in speed (165). In addition, immunodepletion of VASP from platelet extracts or Evl from brain extracts of Mena−/− mice leads to a complete loss of Listeria actin tail formation that is reconstituted by the addition of recombinant VASP (87).
In extracts, the affinity of intact VASP for ActA appears to be higher than that of the VASP EVH1 domain alone, suggesting that VASP domains outside of the EVH1 domain interact with other regions of ActA or with other molecules in the assembly complex (87). In addition, as mentioned above, a large region of ActA (amino acids 166 to 256), which is just amino-terminal to the proline-rich repeats (amino acids 263 and 390), appears to be important in the conversion of actin clouds to actin tails (86a).
Whether VASP is required for the initiation of actin polymerization on the bacterial surface remains unresolved. Possible explanations for the observed discrepancies in the data include the possibility that in addition to the proline-rich region, VASP binds another site on ActA and the possibility that another factor that binds VASP and is essential to VASP-mediated activity is titrated out of the extracts or cytosol on immunodepletion of VASP. Such a factor might interact within ActA amino acids 166 and 256. Evidence against VASP binding to another site on ActA is that no ActA peptides outside of the proline-rich region bind VASP (119) and no VASP is detectable by immunofluorescence on the surface of ActA derivatives lacking the proline-rich region (119), although this latter assay could miss small amounts of VASP. Evidence against an essential factor being immunodepleted along with VASP is that repletion with micromolar amounts of recombinant VASP restores motility to wild-type rates (87).
It has been thought that Ena/VASP proteins serve to accelerate actin assembly by recruiting monomeric actin to sites of actin reorganization and perhaps also by bundling actin filaments. Recent data on the effect of overexpression and deletion of Ena/VASP proteins on cell motility have raised questions about the exact mechanism of action of these proteins in Listeria actin-based motility (9). Overexpression of Mena inhibits the motility of fibroblasts in a dose-dependent manner (9). Cells that genetically lack all Ena/VASP proteins move more quickly than do the same cells complemented with Mena (9). Also, constitutive targeting of Ena/VASP proteins to the cell membrane decreases cell motility and the rates of membrane protrusion and retraction (9). Taken together, these data indicate that Ena/VASP proteins negatively regulate fibroblast movement (9). However, this study addresses the effect of Ena/VASP proteins on fibroblast motility and not on actin dynamics per se. Fibroblast motility is mediated by membrane protrusion and retraction, which requires a delicate balance of factors. The precise way in which this balance is disrupted by alteration in Ena/VASP protein concentration is unclear. Reconciliation of these data with the observed effect of Ena/VASP proteins in Listeria motility will require further work, including the analysis of Listeria motility in cells lacking all Ena/VASP proteins.
Cofilin in Listeria Motility
Cofilin (ADF) depolymerizes actin. While its mode of action has been controversial, the data suggest that it (i) severs actin, thereby increasing the number of filament ends and consequently the number of sites at which dissociation of monomers can occur (67), and (ii) increases the dissociation of monomers from the pointed end (21). Possibly important to the controversy is the observation that the ability of cofilin to sever actin filaments is dependent on the method of its preparation (67).
In highly diluted platelet extracts, addition of cofilin leads to an increase in the rate of movement of Listeria and a shortening of the Listeria actin tail (21). These effects probably result at least in part from the generation of an increased pool of monomeric actin under conditions in which it is limiting. Reconstitution of Listeria motility using purified cytoskeletal proteins also requires cofilin, perhaps for similar reasons, since the reconstitution experiments were conducted using filamentous actin and not monomeric actin (91). Thus, whether cofilin is important in Listeria motility other than to contribute to the maintenance of an intracytoplasmic pool of monomeric actin remains uncertain. Of note, the rates of movement achieved in the reconstitution experiments are three- to fivefold lower than those achieved in intact cytoplasmic extracts (78, 99, 176), suggesting that the reconstitutive mixture used in these experiments may be incomplete.
Capping Protein in Listeria Motility
Capping protein binds to the barbed ends of actin filaments and blocks actin assembly. Marchand et al. (99) suggested that some factor might maintain the filaments near the bacterial surface uncapped to allow their rapid elongation, although such a factor has not been identified. Recent evidence suggests that the regulated capping of these growing filaments is essential to Listeria motility. Capping protein is required for reconstitution of Listeria motility with purified components in the presence of filamentous actin (91). In the absence of capping protein, unregulated extension of the branched network of actin filaments generated at the bacterial surface may occur. Such unregulated polymerization can lead to the extension of filaments beyond the vicinity of the bacterial body to form a fishbone-like web (124), which would be inefficient at providing force behind the bacterial body. Unregulated polymerization would also lead to depletion of the local stores of monomeric actin, further impinging on bacterial movement.
Interaction of Phosphoinositides with ActA
Phosphoinositides bind to and play a role in the regulation of several actin-associated proteins, including capping protein, vinculin, profilin, and gelsolin. Phosphoinositides also bind ActA in vitro (28, 166). Binding of phosphoinositides to ActA induces a conformational change in the amino-terminal portion of the protein (amino acids 29 to 267) (28). The binding site appears to be located toward the carboxyl end of this region, between amino acids 217 and 266 (28). Within this binding site is a stretch of amino acids with significant homology to a domain found within cecropins. Cecropins are peptide antibiotics, and the cecropin domain that is homologous to ActA forms an amphipathic α-helix that is thought to associate with membranes (28). Several phosphoinositides bind ActA with different affinities (28). It has been proposed that phosphoinositide bound to ActA might inhibit the capping activity of capping protein near the bacterial surface (99). The data are not convincing, however, since ActA binds the D-3 phosphoinositides better than PI(4,5)P2 (28), and the D-3 phosphoinositides that have been directly tested do not inhibit capping protein activity (152). Moreover, ActA binding diminishes the PI(4,5)P2-mediated inhibition of capping protein activity (166).
Asymmetry of ActA
Several investigators have examined the distribution of ActA on the Listeria surface. Kocks et al. found that ActA is asymmetrically distributed (77). These investigators observed a gradient of ActA from one pole down the sides of the bacillus, such that ActA is present on one pole and the sides but essentially absent from the second pole, after labeling with a rabbit polyclonal antiserum and imaging by confocal and immunogold electron microscopy (77). The actin tail forms at the Listeria pole that has the higher density of ActA (77). Other investigators have found ActA to be uniformly distributed on the surface, after labeling with rabbit polyclonal antiserum and imaging by indirect immunofluorescence microscopy (22, 120). It seems likely that differences in antibody purification, Listeria strains, or microscopic technique explain the observed differences in distribution. In any case, the observed asymmetry is significantly less marked than that of IcsA on the surface of Shigella (see below) (55).
To characterize the role that ActA asymmetry might play in actin assembly, purified ActA has been attached asymmetrically to various scaffolds and actin assembly and motility has been analyzed in cytoplasmic extracts. ActA bound to the surface of the diplococcus Streptococcus pneumoniae becomes asymmetrically distributed as the S. pneumoniae organism undergoes cell division. All ActA-coated S. pneumoniae cells form actin clouds, indicating that nucleation of actin polymerization is occurring on the S. pneumoniae surface (161). However, only S. pneumoniae cells on which ActA is asymmetrically distributed exhibit unidirectional actin-based motility (161). Because of random fluctuations and thermal dynamics of the cytoplasmic extract, the requirement for asymmetry depends on the size of the ActA-coated object (18). ActA-coated microspheres of approximately the same diameter as a bacterium (1 to 2 μm) require ActA to be asymmetric to form actin tails, whereas smaller beads (diameter, ≤0.5 μm) form actin tails in the absence of ActA asymmetry (18). Thus, in experimental systems, ActA asymmetry is essential to unidirectional actin-based motility.
ActA Is a Dimer
ActA can dimerize, and at least some ActA on the surface of Listeria appears to be dimerized (114). Deletion of ActA amino acids 126 to 155 results in loss of dimerization. This region encompasses both the potential second actin monomer-binding domain (amino acids 121 to 138) and the Arp2/3 complex-binding domain (amino acids 143 to 169 [see above]). Whether actin assembly by Listeria is mediated by ActA that is dimerized and what function ActA dimerization might have in actin assembly are not known.
Model of Listeria Actin Tail Assembly
Based on current knowledge, the following model of Listeria actin tail assembly can be constructed (Fig. 11). ActA, present asymmetrically on the bacterial surface, binds and activates the Arp2/3 complex. The weak actin-nucleating activity of the Arp2/3 complex is greatly enhanced by binding to ActA. Thus, the interaction of ActA with the Arp2/3 complex initiates de novo actin polymerization at the bacterial surface. In addition, ActA binds VASP, which may also enhance the initiation of actin polymerization by unknown mechanisms. The barbed ends of the newly nucleated actin filaments remain uncapped for a short period, permitting rapid addition of actin monomers onto these ends. The filament ends are then capped, and polymerization is thus halted by capping protein or another protein with capping protein-like activity. The Arp2/3 complex forms a network of filaments that are cross-linked at 70° angles, in conjunction with its release from ActA. The binding of the Arp2/3 complex to the sides of filaments also stimulates its nucleating activity. Thus, a network of short actin filaments, linked at 70° angles, is rapidly generated at the bacterial surface. Whether cofilin also severs filaments near the bacterial surface, thereby generating uncapped barbed ends, is unknown. The branched network is metastable, since debranching occurs with conversion of ATP-actin in the filaments to ADP-actin, which is accelerated by cofilin. Debranched filaments depolymerize rapidly. A local pool of actin monomers is generated by cofilin severing and depolymerization from the barbed ends of locally released filaments. Monomeric ATP-actin is drawn into the polymerization machinery by its binding to ActA and/or the binding of profilin-ATP-actin monomers to VASP. The assembly of this actin filament network at the Listeria surface provides the force to propel the bacterium through the cytosol of infected cells.
FIG. 11.

Model of actin tail assembly by Listeria ActA. (A) ActA (light blue bar) binds the Arp2/3 complex (orange) at its Arp2/3 complex-binding domain (amino acids 143 to 169, designated Arp2/3) and possibly at its acidic domain (amino acids 31 to 58, designated A). ActA binds an actin monomer (black hexagon) at its actin-binding domain (amino acids 60 to 100, designated AB). ActA binds VASP (pink oval) at its proline-rich repeat region (amino acids 293 to 390, designated by four bands), and VASP binds profilin (P, green rectangle)-ATP-actin. (B) The actin nucleation activity of the Arp2/3 complex is stimulated by its binding to ActA. It mediates the addition of actin monomers to the barbed end and caps the pointed end of a new actin filament. (C) As the filament extends, the original Arp2/3 complex is released from ActA and another Arp2/3 complex binds both ActA and the side of an existing actin filament. Each interaction stimulates the actin-nucleating activity of the Arp2/3 complex. (D) Repeated rounds of filament branching, filament nucleation, filament extension, and Arp2/3 complex release generate a network of actin filaments linked at 70° angles. At a distance from the bacterial surface, filament barbed ends are capped, thereby halting the addition of more actin monomers. Also at a distance from the bacterial surface, filaments debranch and, with the assistance of the actin-severing protein cofilin, depolymerize, thereby maintaining a local pool of actin monomers. Whether cofilin also severs filaments near the bacterial surface, thereby generating uncapped barbed ends, is unknown.
SHIGELLA
Shigella moves through the cytoplasm of infected cells by actin-based motility. Shigella motility was first observed by Ogawa et al. in the 1960s (122). Disease caused by Shigella involves invasion and spread through the lining of the human colon. Mutants of Shigella that do not assemble actin are greatly attenuated in human volunteers and in monkey and mouse experimental models (10, 79, 89, 98, 149), which indicates that actin-based motility is essential to disease pathogenesis. Histologic analysis of experimentally infected monkeys demonstrates that Shigella mutants that do not assemble actin cause smaller ulcerations and abscesses in the intestinal mucosa (149), directly implicating actin-based motility and cell-to-cell spread in the disease process.
In infected cells, Shigella moves at rates of 5.4 to 26 μm/min (58, 158, 197), which is similar to the rate of movement of Listeria, approximately 2.5-fold faster than Rickettsia movement, and approximately 5- to 8-fold faster than vaccinia virus movement (Table 1). In Xenopus laevis oocyte cytoplasmic extracts, wild-type Shigella does not assemble actin tails (78). This lack of actin assembly appears to be due to relatively low levels of expression of IcsA (VirG), the Shigella protein that mediates actin assembly, under in vitro conditions and can be rescued by expression of IcsA at high levels in either Shigella or Escherichia coli (J. Magdalena and M. B. Goldberg, submitted for publication). Thus, most work with Xenopus cytoplasmic extracts, platelet extracts, and bovine brain extracts has involved E. coli expressing IcsA from a high-copy-number plasmid.
Morphology of Actin Tails Formed by Shigella
The actin tail formed by Shigella is morphologically similar to that formed by Listeria (58). Shigella actin tails average 7 μm in length and 0.7 μm in width, slightly longer and thinner than those of Listeria or Rickettsia (Table 2) (58). The filaments within the Shigella tail have been reported to be uniformly short (approximately 0.1 μm in length), bundled in a branching network, and oriented with the barbed ends toward the bacterial body, as are filaments within the Listeria actin tail (58). A significant difference from Listeria is that actin filaments are restricted to the bacterial old pole and are absent from the sides of the bacillus (58), consistent with the relatively restricted localization of IcsA to the bacterial pole (see below) (55).
Shigella spreads from cell to cell by pushing out against the cell membrane to form a protrusion, with the bacterium at its tip, that is taken up by the adjacent cell. By light microscopy, these protrusions appear similar to those of Listeria. To our knowledge, a detailed electron microscopic analysis of the morphology of Shigella actin tails within protrusions has not been performed.
Shigella IcsA (VirG) Protein
The Shigella protein IcsA (first identified as and currently also known as VirG [89]) mediates actin tail assembly. Deletion of icsA leads to loss of actin tail assembly and a marked reduction in spread between cells (10, 89, 98, 123). Purified IcsA bound to inert particles and E. coli strains that express IcsA are able to form actin tails in cytoplasmic extracts (Fig. 12 and 13) (56, 78, J. Magdalena and M. B. Goldberg, unpublished data). Taken together, these data indicate that IcsA is both required and sufficient for actin tail assembly by Shigella.
FIG. 12.

Actin tail formation by IcsA-expressing E. coli. Time-lapse microscopy of IcsA-expressing E. coli in X. laevis oocyte cytoplasmic extracts is shown. IcsA is sufficient to enable E. coli to assemble actin tails. Bar, 10 μm. (Reprinted from reference 56 with permission of the publisher.)
FIG. 13.

Actin tail formation by IcsA-coated particles. Fluorescence (top) and phase (bottom) microscopy of IcsA-coated irregularly shaped silica particles in X. laevis oocyte cytoplasmic extracts are shown. Rhodamine actin, which fluoresces on polymerization, has been added to extracts. A single particle breaks away from the clump of particles as it assembles an actin tail. The clump of particles polymerizes actin on its surface (Magdalena and Goldberg, unpublished).
A schematic diagram of IcsA is shown in Figure 14. IcsA is a 1,102-amino-acid, 110-kDa protein that is anchored in the bacterial outer membrane by a carboxy-terminal domain (the β domain). Approximately 750 amino acids of the mature amino terminus (the α domain) are exposed on the bacterial surface. IcsA is a member of the autotransporter family of gram-negative bacterial virulence proteins. Members of this family are thought to mediate their own translocation across the outer membrane. Translocation across the outer membrane is thought to occur by insertion of the β domain into the outer membrane to form an amphipathic β-barrel and passage of the mature amino-terminal α domain through the β-barrel to the bacterial surface (reviewed in reference 61).
FIG. 14.

Domains of IcsA. α domain (amino acids 53 to 758), portion of mature IcsA that is exposed on the bacterial surface; β domain (amino acids 759 to 1102), outer membrane anchor; SP (amino acids 1 to 52), signal peptide; actin assembly, region that is sufficient to polymerize actin in in vitro assays (see the text).
The α domain of IcsA, which is exposed on the bacterial surface, contains all regions that are required for actin assembly. Inert particles bound with the purified α domain polypeptide form actin tails in cytoplasmic extracts. (Magdalena and Goldberg, unpublished). Moreover, IcsA amino acids 53 to 508 are sufficient to enhance actin polymerization in in vitro assays, suggesting that the residues involved in actin tail formation are within this region. Functional regions within the α domain have otherwise been poorly characterized. The amino acid sequence of the α domain shows minimal similarity to known proteins and, in particular, no similarity to Listeria ActA or vaccinia virus A36R. Purification of IcsA has been more difficult than the purification of ActA, since investigators have been unable to generate significant quantities of stable protein, thereby hampering in vitro analyses.
IcsA is localized predominantly on the old pole of the bacterium (Fig. 15) (55). As the bacterial cell begins to divide, IcsA begins to be expressed on the second pole as well (Fig. 15) (55). As would be predicted, actin assembly occurs on the pole at which IcsA is most highly expressed (Fig. 16) (55). IcsA is targeted directly to the old pole and is inserted into the outer membrane at the old pole (24, 143, 167). Once inserted, it diffuses laterally in the outer membrane (143). The extracellular portion of IcsA is cleaved from the bacterial surface by the IcsA-specific protease IcsP (SopA) (39, 158). Expression of IcsP enhances the gradient of IcsA in the outer membrane (143), but the regulation of its expression has not been worked out.
FIG. 15.

Unipolar IcsA on the surface of Shigella. Indirect-immunofluorescence (left) and phase (right) microscopy of S. flexneri are shown. Arrows indicate unipolar IcsA on nondividing bacteria; arrowheads indicate bipolar IcsA on a dividing bacterium.
FIG. 16.

Fluorescence microscopy showing that actin assembly occurs at the Shigella pole on which IcsA is most highly expressed. IcsA (red) and polymerized actin (green).
Requirement of N-WASP for IcsA-Mediated Actin Assembly
N-WASP is absolutely required for Shigella actin assembly. Overexpression of either of two dominant-negative derivatives of N-WASP (which contain either a deletion in the cofilin homology domain or a deletion of the entire VCA domain) leads to about a 90% reduction in actin tail formation on intracellular Shigella (111, 170). Immunodepletion of N-WASP from Xenopus cytoplasmic extracts leads to a marked reduction in actin tail assembly by E. coli expressing IcsA, which is restored by repletion of N-WASP (170). Platelet extracts, which naturally lack N-WASP, support actin tail formation by E. coli expressing IcsA only after preincubation of the bacteria with N-WASP (39). Expression of only the N-WASP CRIB domain inhibits actin tail formation (111).
Finally, in N-WASP−/− cells, Shigella is unable to assemble actin tails (163a). In these cells, the bacteria are also unable to make protrusions from the cell surface and unable to spread between cells, while initial entry into the cell is unaffected (163a). The defects in actin tail formation, protrusion formation, and intercellular spread in N-WASP−/− cells is rescued by expression of N-WASP from an integrated retroviral vector, demonstrating that the disruption of actin assembly is due exclusively to the absence of N-WASP in these cells (163a). Notably, WASP, which has approximately 50% homology to and similar domain structure to N-WASP (104), does not rescue Shigella actin assembly in N-WASP−/− cells (163a). This is consistent with the observation that E. coli expressing IcsA does not assemble actin tails in platelet extracts, which are rich in WASP but lack N-WASP (39). The inability of WASP to functionally complement the N-WASP deficiency indicates that residue or domain differences between N-WASP and WASP are important to Shigella binding and/or activation of N-WASP.
Interaction of IcsA with N-WASP
IcsA binds directly to N-WASP. Initial experiments show that N-WASP is recruited to the pole at which actin tails form on intracellular Shigella (Fig. 17) (170). N-WASP is restricted to the bacterial pole and is absent from the actin tail itself (Fig. 17) (170), suggesting that during polymerization and bundling of filaments into the tail, it is not released from the bacterial surface.
FIG. 17.

Fluorescence microscopy of Shigella-infected cells showing localization of N-WASP to the Shigella pole. (A) Actin (phalloidin); (B) N-WASP antibody; (C) IcsA antibody; (D) overlay of actin and N-WASP antibody, where yellow indicates colocalization. (Reprinted from reference 170 with permission of the publisher.)
Analysis of the domain of N-WASP that binds IcsA has generated conflicting data. Egile et al. found that full-length N-WASP, but not the carboxy-terminal VCA fragment of N-WASP, binds IcsA-coated beads (39). In addition, E. coli that expresses IcsA formed actin tails in platelet extracts after preincubation with full-length N-WASP but not after preincubation with the VCA fragment (39). Taken together, these data strongly suggest that the domain of N-WASP that binds to IcsA is outside of the VCA fragment, despite prior reports that the verprolin homology domain (i.e., the V within the VCA fragment) is required for N-WASP binding to IcsA-coated beads (170).
The Arp2/3 Complex Is Required for Shigella Actin-Based Motility
N-WASP is known to bind to the Arp2/3 complex and activate Arp2/3-mediated actin nucleation and polymerization (see “Actin cytoskeleton: dynamics and function,” above). Consistent with this, recent data indicate that the Arp2/3 complex is required for IcsA-mediated actin polymerization. The Arp2/3 complex is present throughout the length of Shigella actin tails, as shown by immunolocalization of Arp3 and the p34 subunit in Shigella-infected cells (Fig. 18) (44, 58, M. B. Goldberg, unpublished data). Depletion of the Arp2/3 complex from platelet extracts leads to loss of motility of N-WASP-coated IcsA-expressing E. coli, and addition of 15 μM pure Arp2/3 complex restores motility to 30% of initial rates, indicating that the Arp2/3 complex is required for motility (39). The observed lack of restoration of motility to initial rates suggests that factors other than the Arp2/3 complex are important in motility and may have been depleted during Arp2/3 depletion. Incubation of IcsA-coated beads with N-WASP, F-actin, and pure Arp2/3 complex leads to the formation of actin clouds around the beads (39).
FIG. 18.

Fluorescence microscopy of Shigella-infected cells showing localization of the Arp2/3 complex to actin tails formed by Shigella. (A) Arp3 antibody; (B) actin (phalloidin); (C) p34 antibody; (D) actin (phalloidin).
In in vitro polymerization assays with purified components, IcsA has a minor stimulatory effect on Arp2/3 complex activation of actin polymerization (Fig. 19) (39). In the presence of N-WASP, the stimulatory effect of IcsA is markedly increased and is IcsA concentration dependent (Fig. 19) (39). These data are consistent with the model that IcsA binding to N-WASP stimulates N-WASP-mediated activation of the Arp2/3 complex (Fig. 20).
FIG. 19.

IcsA stimulates N-WASP activation of Arp2/3 complex-mediated actin polymerization. Fluorescence intensity is plotted as a function of time in pyrene-actin polymerization assays. 1, actin alone; 2, actin and Arp2/3 complex; 3, actin and 0.25 uM IcsA; 4, actin and N-WASP; 5, actin, Arp2/3 complex, and 0.25 μM IcsA; 6, actin, Arp2/3 complex, and N-WASP; 7, actin, Arp2/3 complex, N-WASP, and 30 nM IcsA; 8, actin, Arp2/3 complex, N-WASP, and 0.25 μM IcsA. (Reprinted from reference 39 with permission of the publisher.)
FIG. 20.

Model of actin tail assembly by Shigella IcsA. (A) IcsA (blue bar) on the surface of Shigella binds N-WASP (maroon). (B) IcsA binding disrupts the intramolecular bonds and activates N-WASP, thereby stimulating N-WASP activation of the Arp2/3 complex (orange), N-WASP binding to monomeric actin (black hexagon), and possibly N-WASP binding to profilin-actin (P, green rectangle). The Arp2/3 complex nucleates actin, mediates the addition of actin monomers to the barbed end, and caps the pointed end of a new actin filament. (C) As the filament extends, the original Arp2/3 complex is released from N-WASP and another Arp2/3 complex binds both N-WASP and the side of an existing actin filament. Each interaction stimulates the actin-nucleating activity of the Arp2/3 complex. (D) Repeated rounds of filament branching, filament nucleation, filament extension, and Arp2/3 complex release generate a network of actin filaments linked at 70° angles. At a distance from the bacterial surface, filament barbed ends are capped, thereby halting the addition of more actin monomers. Also at a distance from the bacterial surface, filaments debranch and, with the assistance of the actin-severing protein cofilin, depolymerize, thereby maintaining a local pool of actin monomers. Whether cofilin also severs filaments near the bacterial surface, thereby generating uncapped barbed ends, is unknown. V, verprolin homology domain; C, cofilin homology domain; A, acidic domain; Pro, proline-rich region; GBD, GTPase-binding domain; PH, pleckstrin homology domain.
Role of Profilin in Shigella Actin-Based Motility
Profilin, an actin monomer-binding protein, is capable of delivering monomeric actin to sites of actin assembly. Profilin binds proline-rich sequences. Whereas Shigella IcsA lacks proline-rich sequences, N-WASP has some near the carboxy-terminal VCA domains. Of note, there are conflicting data about the role of these proline-rich sequences and profilin in N-WASP activation of Arp2/3-mediated actin assembly (see “Actin cytoskeleton: dynamics and function” above). Thus, the role of profilin in Shigella motility may not be mediated via its binding to the N-WASP proline-rich sequences.
Although profilin appears to not be absolutely required for Shigella actin tail assembly, recent data suggest that it contributes to the attainment of maximal rates of movement by the bacterium. Two isoforms of profilin exist, profilin I and profilin II. Profilin I has an approximately sevenfold-higher affinity for N-WASP than does profilin II (169). Profilin I colocalizes with the Shigella actin tail (108). Overexpression of a profilin I mutant that is unable to bind monomeric actin (His133 to Ser) leads to a marked decrease in Shigella motility in infected cells, and overexpression of a profilin I mutant that is unable to bind proline-rich sequences (His119 to Glu) leads to a moderate decrease (108). Overexpression of either leads to a reduction in intercellular spreading of Shigella (108).
Depletion of profilin from Xenopus cytoplasmic extracts leads to an approximately 30% decrease in the motility of E. coli expressing IcsA, which is rescued by the addition of recombinant wild-type profilin I but not by the addition of the profilin I His133-to-Ser or His119-to-Glu mutant (108). Overexpression of an N-WASP mutant that lacks the proline-rich domain and hence binds profilin poorly leads to a 90% reduction in actin tail formation by Shigella but no reduction in tail formation by Listeria (108). In cytoplasmic extracts immunodepleted of N-WASP, addition of the N-WASP mutant that lacks the proline-rich domain did not restore motility of E. coli expressing IcsA whereas addition of wild-type N-WASP did (108). Finally, in motility assays of E. coli expressing IcsA with purified cytoskeletal proteins, profilin was not absolutely essential for actin tail formation but was required for maximal rates of movement (91).
Role of Vinculin and VASP in Shigella Actin-Based Motility
The proline-rich repeats of Listeria ActA bind VASP, which in turn binds profilin bound to monomeric actin. Since IcsA lacks proline-rich sequences, it has been postulated that vinculin, which contains proline-rich sequences to which VASP is known to bind, might dock on IcsA and serve to link it with VASP and profilin-actin. Vinculin is a component of focal contacts (17, 47) and intercellular adherens junctions (48, 49). Vinculin contains two domains, a 95-kDa amino-terminal “head” domain and a 30-kDa carboxy-terminal “tail” domain, between which lies the proline-rich region. In its inactive state, vinculin is folded such that an intramolecular interaction between the head and tail domains blocks binding of the tail domain to filamentous actin (68, 69).
The role of vinculin in Shigella actin-based motility is controversial. Vinculin localizes to the surface of intracellular Shigella and throughout Shigella actin tails within cell surface protrusions (44, 70, 84, 172). A monoclonal antibody to the head domain of vinculin recognizes vinculin on the surface of Shigella (84), indicating that the vinculin associated with the bacterium includes the head domain. In in vitro assays, the α domain of IcsA binds the vinculin head domain but not the tail domain (172). In Shigella-infected cells, a small amount of vinculin is cleaved to yield isolated head domain that contains a proline-rich sequence at its carboxy terminus (84). Microinjection of purified vinculin head domain accelerates Shigella motility, while microinjection of full-length vinculin, which is probably inactive due to an intramolecular interaction between the head and tail domains, does not (84). These observations have led to the suggestion that Shigella IcsA cleaves vinculin and the released vinculin head domain serves as a linker between IcsA and VASP on the bacterial surface (84).
In F9 5.51 cells, which are deficient in vinculin, Shigella actin-based motility is normal; the rate of intracellular movement and the rate of protrusion formation are not different from those of Shigella in congenic wild-type F9 cells (54). However, small amounts of truncated vinculin can be detected in F9 5.51 cells, leading investigators to suggest that the small amount of vinculin that is present is sufficient to support Shigella motility (164). Finally, vinculin is not required to reconstitute IcsA-mediated motility using purified cytoskeletal proteins (91).
VASP, a ligand of vinculin, is present on the surface of intracellular Shigella and in Shigella actin tails (22, 44, 58). However, localization of a protein to sites of actin assembly does not necessarily mean that it plays role in the process. To our knowledge, no studies have been performed to address whether VASP or other VASP family members play a functional role in Shigella actin-based motility. Further investigation, particularly using cells that totally lack vinculin or Ena/VASP proteins, will be useful in definitively determining the role of these proteins in Shigella actin-based motility.
Role of Cofilin in Shigella Actin-Based Motility
Cofilin, which depolymerizes actin, both severs actin (67) and accelerates the dissociation of monomers from the pointed end (21). To date, few studies have examined its role in Shigella motility. Cofilin is present in Shigella actin tails, as detected by immunofluorescence (58). Reconstitution of IcsA-mediated actin-based motility using purified cytoskeletal proteins requires cofilin (91); however, since the actin used in the reconstitution experiments was filamentous rather than monomeric, the requirement for cofilin may simply result from the need to generate actin monomers from the filamentous pool. Thus, it remains uncertain whether cofilin is important in Shigella motility, other than to contribute to the maintenance of an intracytoplasmic pool of monomeric actin.
Role of Cdc42 in Shigella Actin-Based Motility
Cdc42 is a Rho family GTPase that activates N-WASP by binding to its CRIB domain (see “Actin cytoskeleton: dynamics and function” above). Experimental evidence on the role of Cdc42 in Shigella actin-based motility is conflicting. Microinjection of a dominant-negative form of Cdc42 into Shigella-infected cells leads to a decrease in the rate of movement of motile bacteria, and microinjection of a dominant active form leads to an increase (171). Inhibition of Shigella motility in cytoplasmic extracts by the addition of the Rho family inhibitor RhoGDI is partially rescued by the addition of purified Cdc42 (171). One group has observed colocalization of green fluorescent protein (GFP)-Cdc42 to Shigella with actin tails (171), while another group found no detectable GFP-Cdc42 on Shigella with actin tails (111).
In extracts depleted of N-WASP, addition of wild-type N-WASP rescued Shigella motility whereas addition of an N-WASP mutant (His208 to Asp) known to not bind Cdc42 did not rescue motility (171). Overexpression of the same N-WASP mutant in Shigella-infected cells leads to fewer actin tails on intracellular bacteria and decreased intercellular spread of bacteria in a plaque assay (171). In in vitro actin polymerization assays, in the presence of N-WASP and the Arp2/3 complex, the addition of Cdc42 increased the level of actin polymerization induced by IcsA to an extent that appears more than simply additive (171).
Due to theoretical indirect effects of dominant negative or constituitively active Cdc42 on other elements involved in actin assembly, these data should be interpreted cautiously, particularly given that data from another investigator suggest that Cdc42 plays no role in Shigella motility (T. Shibata, F. Takeshima, F. Chen, F. W. Alt, and S. B. Snapper, submitted for publication).
Roles of Nck and WIP in Shigella Actin-Based Motility
As described in detail below (see “Vaccinia virus”), WIP and the adaptar protein Nck both bind WASP family members and appear to be required for vaccinia virus actin-based motility. Whether either might play a role in Shigella actin-based motility has therefore been examined. Neither overexpression of a dominant negative form of Nck (Nck lacking its SH2 domain) nor overexpression of a dominant negative form of WIP (the isolated WASP-binding domain of WIP) alters Shigella actin tail formation, although both were recruited to the bacterial surface (111). These data suggest that while Nck and WIP may bind N-WASP (163a) (or another factor) on Shigella, neither is required for Shigella actin-based motility.
Model of Shigella Actin-Based Motility
The Shigella protein IcsA is localized to the old pole of the bacterium, where it mediates assembly of the actin tail (Fig. 15 and 16). IcsA recruits N-WASP to the bacterial surface. Binding of IcsA to N-WASP is postulated to disrupt the intramolecular bond that is present in inactive N-WASP, thereby activating it (Fig. 20).
Activated N-WASP stimulates Arp2/3 complex activation of actin assembly (Fig. 20). As in the Listeria model, activated Arp2/3 complex probably mediates de novo nucleation of actin filaments, rapid elongation of filaments at the barbed ends, and cross-linking of filaments at 70° angles. At a distance from the bacterial surface, filaments are likely to become capped, thereby preventing further elongation and leading to the observed short length of filaments within the tail.
The roles of VASP, vinculin, profilin, and cofilin are uncertain. It has been hypothesized that Shigella cleaves vinculin to release the vinculin head domain, which then binds IcsA and serves as a linker for VASP. Profilin, which is known to bind VASP, would then deliver monomeric actin to the site of actin assembly at the Shigella pole. Cofilin has been postulated to be important in the disassembly of actin filaments within the tail, at a distance from the bacterial body, which would liberate actin monomers for incorporation into newly generated filaments. Cofilin might also sever newly generated filaments adjacent to the bacterial body, thereby increasing the number of uncapped barbed ends on which rapid polymerization can occur.
VACCINIA VIRUS
Vaccinia virus is a double-stranded DNA virus and a member of the poxvirus family, which also includes variola virus, the causative agent of smallpox. Vaccinia virus was successfully used as the vaccine for smallpox. It does not cause disease in immunocompentent humans. However, it uses actin-based motility and has been extensively studied in experimental systems.
Vaccinia virus is the only virus known to use actin-based motility; several others that have been examined do not. Actin-based motility has been proposed to facilitate vaccinia virus spread between cells, although no data specifically demonstrate a role for actin-based motility in vaccinia virus pathogenesis. It is not essential to viral spread, since virus particles are able to use microtubules for anterograde movement to reach the cell periphery in the absence of actin assembly (129, 147). Vaccinia virus mutants that are unable to assemble actin yet able to move along microtubules and fuse with the plasma membrane do not currently exist, thus making it difficult to directly test the role of actin-based motility in pathogenesis.
Vaccinia virus replicates in the cytoplasm of the host cell. It has an unusually complex development involving the formation of two distinct intracellular forms, the so-called intracellular mature virus (IMV) and the intracellular enveloped virus (IEV). IMV form about 4 h after cellular infection. A small proportion of the IMV later develop into IEV by becoming wrapped in the membrane cisternae derived from the trans-Golgi network. The IEV forms actin tails that propel it toward the cell periphery (Fig. 21A). Only the IEV form utilizes actin-based motility, since blockage of IEV formation with the drug N1-isonicotinoyl-N2-3-methyl-4-chlorobenzoylhydrazine (IMCBH) prevents actin tail formation (32). The IEV form is also thought to be the source of infectious extracellular enveloped virus, which are thought to be released after fusion of the outermost membrane of IEV with the plasma membrane. Treatment with cytochalasin D, which blocks de novo actin assembly, does not interfere with IMV assembly but prevents the release of IEV from the cell, which indicates that actin assembly is important for viral movement to the cell periphery and/or for viral release.
FIG. 21.

Fluorescence microscopy of vaccinia virus-infected HeLa cells, labeled for actin (red) and vaccinia virus IMV particles (green), showing actin tail formation by vaccinia virus. (A) Actin tails on intracellular vaccinia virus. Bar, 50 μm. (B) Protrusions from the cell surface, formed by vaccinia virus, that contain actin tails and have viral particles at the tips. Bar, 10 μm. (Adapted from reference 32 with permission of the publisher.)
Morphology of Actin Tails Formed by Vaccinia Virus
Like Listeria, Shigella, and Rickettsia, vaccinia virus assembles actin tails that propel it through the cell cytoplasm. Vaccinia virus particles move at 2.8 ± 0.5 μm/min (32), which is similar to the speeds of Listeria and Shigella and slower than that of Rickettsia (Table 1). Like the other three organisms, the barbed ends of the tail filaments are facing the microbial body, as demonstrated by decorating tails with the S1 subfragment of myosin, which binds to subunits within actin filaments in an asymmetric fashion (33) (Fig. 22). Actin monomers are nucleated at the surface of the viral particle (33). Filaments near the viral particle are splayed at approximately 45° angles (33) (Fig. 22). Filaments within the tail at a distance from the viral particle appear to be capped, since they do not incorporate actin monomers (33). Individual filaments within the tail are longer than those documented for any of the other three organisms, averaging 0.74 μm (33).
FIG. 22.

Electron microscopy of filaments within the actin tail that have been decorated with the S1 subfragment of myosin show actin tail formation by vaccinia virus. Stars, viral particles. Bar, 400 nm. (Reprinted from reference 33 with permission of the publisher.)
Vaccinia virus also forms actin tail-containing protrusions from the cell surface (32, 64–66, 83) (Fig. 21B), which differ from those of Listeria and Shigella in that during extension of the protrusion, the viral particle fuses with the plasma membrane, thereby placing the viral particle on the outside of the cell membrane rather than within it (33) (Fig. 23A).
FIG. 23.

(A) Proposed model of vaccinia virus fusion with the membrane of a cell surface protrusion. (Left) Actin tail assembly pushes the viral particle against the cell membrane, forming a protrusion from the cell surface; (middle), the membrane of the viral particle fuses with the cell membrane at the tip of the protrusion; (right) the viral particle is released from the tip of the protrusion. (B) Diagram of the topology and interaction of vaccinia virus actin assembly protein A36R with other vaccinia virus membrane proteins. The cytoplasmic and luminal faces of the vaccinia virus outer IEV membrane and the amino (N) and carboxy (C) termini of A33R, A36R, A34R, and B5R are shown. (C) Diagram of SH2 and SH3 domains of the adapter protein Nck. A partial list of proteins with which these domains have been shown to interact is indicated. (Panel A adapted from reference 33 with permission of the publisher. Panel B reprinted from reference 147 with permission of the American Society for Microbiology. Panel C adapted from reference 102 with permission of the publisher.)
Vaccinia Virus Protein A36R
Actin assembly on vaccinia virus is mediated by the viral protein A36R (45, 147). A36R is a type 1b membrane protein that has a 195-amino-acid carboxy-terminal domain located outside the viral particle and exposed to the host cell cytoplasm, with a short amino-terminal domain of the protein in the particle lumen (147, 182a) (Fig. 23B). During vaccinia virus infection, it is acquired by the viral particle after wrapping of the IMV with Golgi membrane to form the IEV. When expressed independently of viral infection, it localizes to the Golgi and to sites of actin assembly (92). Variola virus encodes a homolog of A36R, suggesting that it may have used actin-based motility, while other poxviruses—myxoma, Shope fibroma, and fowlpox viruses—do not encode homologs (129).
Deletion of A36R leads to loss of actin tail formation, without interfering with IEV formation (148, 191). A36R is tyrosine phosphorylated on the viral particle by cellular Src kinases on Tyr112 and to a much lesser extent on Tyr132 (45). Tyrosine phosphorylation of A36R is required for viral actin assembly (45). A36R activity, but not incorporation into the viral particle, is dependent on its phosphorylation (45).
It is not yet known whether A36R is the only vaccinia virus protein required for actin assembly. The additional vaccinia virus proteins A34R, A33R, or F12L may also play a role in actin assembly. A34R is indirectly required for actin tail assembly, since it is required for IEV assembly and organization (147), and a more direct role has not been ruled out. A33R may be required for actin tail assembly, since an A33R mutation leads to loss of actin assembly (146), and it is not required for IEV assembly (147). Data suggest that A33R may function in part as a chaperone for transfer of A36R from the Golgi membrane to the IEV (192). Deletion of F12L leads to a twofold reduction in IEV formation and a 99% reduction in actin tail formation (198), suggesting that it may play a role in actin assembly. Further studies will be required to definitively determine whether any of these viral proteins are directly required for actin assembly.
Requirement for the Cellular Adapter Protein Nck
The ubiquitous host cell protein Nck colocalizes with tyrosine-phosphorylated A36R (45). Nck appears to be required for vaccinia virus actin assembly, since overexpression of the Nck SH2 domain leads to a marked decrease in actin tail formation (45). Nck belongs to a family of adapter proteins that function to link components within a signal transduction cascade (102). These adapter proteins consist of SH2 and SH3 domains that bind to specific signal transduction components. SH2 domains bind specific phosphotyrosine-containing regions, generally on proteins in pathways that utilize phosphorelays. SH3 domains bind proline-rich regions, generally on proteins in pathways involved in cytoskeletal rearrangements. Nck contains one SH2 domain, which binds several factors, including one that leads to an association with c-Src tyrosine kinase (26, 27, 102, 153) (Fig. 23C). Nck also contains three SH3 domains, which bind a variety of factors (Fig. 23C) (102). Of particular relevance to vaccinia virus biology, the second Nck SH3 domain binds WIP and the third binds the sequence GRSGPXPPXP within the proline-rich region of WASP (3, 133, 142, 157). The Nck SH3 domains activate N-WASP-mediated actin nucleation in vitro in a manner that appears to be redundant with Cdc42 activation of N-WASP-mediated actin nucleation (121).
Nck recruitment to vaccinia virus is dependent on the presence of N-WASP (163a), as well as both the presence and phosphorylation of A36R (45). Fusions of the N-WASP proline-rich region to GFP were able to bind cellular Nck but were not present at sites of actin assembly on vaccinia virus (111). These data suggest that Nck and N-WASP may exist as a preformed complex that binds vaccinia and that Nck is not clearly upstream of N-WASP in this process. In vitro, a peptide consisting of amino acids 105 to 116 of A36R binds Nck, dependent on the phosphorylation of Tyr112 (45). Given the dependence of Nck on N-WASP for recruitment to vaccinia virus, the significance of these binding studies for in vivo actin assembly is unclear, although it is possible that Nck binds A36R in an N-WASP-dependent manner.
Requirement for WASP-Interacting Protein
The cellular protein WIP is also recruited to the surface of vaccinia virus and appears to be the linker between Nck and N-WASP (111). WIP is a proline-rich protein that was first identified in lymphocytes and is thought to be involved in T-cell receptor signaling via the actin cytoskeleton (135, 150). In fact, WIP is widely expressed and contains features suggesting that it may be involved in actin rearrangements in all cells. Near its amino terminus, WIP contains two WASP homology 2 (WH2) domains (also known as verprolin homology [VH] domains). Verprolin is a yeast protein that is involved in cytoskeletal control of cell polarity (38, 181), and WIP can functionally complement the cell polarity pathway in verprolin-deficient yeast (182). The first WH2 domain in WIP contains a KLKK motif; KLKK and similar motifs are critical for actin binding in known actin-binding proteins (181, 183). WIP also contains three XPPPPP motifs (where X is G, A, L, or S); XPPPPP motifs are known to bind profilin (132). Thus, although there are no experimental data, the primary sequence of WIP suggests that it may bind both actin and profilin.
WIP interacts with WASP family members via its WASP-binding domain (WBD). WIP WBD binds the WH1 domain of WASP and N-WASP (3, 111, 134, 168); the WH1 domain includes both the PH domain and the IQ motif. The single tryptophan residue within the WH1 domain of Mena and Ena is required for binding to the proline-rich region of ActA (42, 130). The analogous tryptophan residue within the WH1 domain of N-WASP (Trp54) appears to be essential to N-WASP binding to WIP, since its mutation to alanine leads to loss of recruitment of the N-WASP WH1 domain to vaccinia virus and loss of its binding to the N-WASP-binding domain of WIP (111). Additional point mutations in the N-WASP WH1 domain that interfere with its binding to the WIP N-WASP binding domain include Cys35 to Trp, Arg76 to Cys, and Glu123 to Lys, each of which corresponds to a missense mutation that naturally occurs in Wiskott-Aldrich syndrome, suggesting that this interaction might be important in the pathogenesis of the disease (111).
WIP is present on vaccinia virus particles that have actin tails (111). In addition, overexpression of the WIP WBD significantly inhibits vaccinia virus actin tail assembly, suggesting that WIP may be required for vaccinia virus actin-based motility (111).
WIP recruitment to vaccinia virus is dependent on N-WASP (163a), which suggests that, like Nck, WIP may exist in a preformed complex with N-WASP that binds vaccinia virus and that WIP is not clearly upstream of N-WASP in this process. Nck recruitment to vaccinia virus may be dependent on WIP, since overexpression of the WIP WBD, which is not itself recruited to vaccinia virus, leads to an absence of both endogenous WIP and endogenous Nck on vaccinia virus (111). An alternative explanation of this latter observation is that overexpression of the WIP WBD may sequester N-WASP, preventing its recruitment to vaccinia virus. Future work in this area, in particular the development of Nck- and WIP-deficient cell lines, will aid in the clarification of their roles in vaccinia virus actin assembly.
Requirement for WASP Family Members
WASP is a downstream effector of Nck and probably also of WIP (3, 102, 111, 134, 142, 163, 168). N-WASP, a WASP family member (see “Actin cytoskeleton: dynamics and function,” above), is recruited to the viral particle (45). In N-WASP−/− cells, vaccinia viruses are unable to assemble actin tails (163a). Complementation of the N-WASP defect with either N-WASP or WASP rescues vaccinia virus actin tail formation (163a), which suggests that the protein(s) that links the vaccinia virus particle to the actin assembly machinery binds WASP family members nonspecifically. Overexpression of dominant negative N-WASP–ΔWA inhibits actin tail formation (45). Taken together, these data indicate that a WASP family member is absolutely required for vaccinia virus actin assembly.
Recruitment of WIP and Nck to vaccinia virus particles appears to be N-WASP-dependent (163a). However, a dominant negative N-WASP construct that contains only the WH1 domain and a mutated CRIB domain (His208 to Asp) is recruited to the vaccinia virus surface and prevents vaccinia virus actin assembly and recruitment of endogenous N-WASP and VASP to the vaccinia virus surface but does not interfere with recruitment of Nck or WIP (111). Of note, vaccinia virus actin-based motility is independent of Cdc42 (111), an upstream activator of N-WASP.
Role of the Arp2/3 Complex
N-WASP is known to activate Arp2/3-mediated actin assembly (see “Actin cytoskeleton: dynamics and function” above). It is therefore reasonable to conclude that N-WASP (or WASP) recruited to vaccinia virus activates Arp2/3 complex-mediated actin assembly, as in Shigella, although this has not been directly demonstrated in either case. Curiously, the Arp2/3 complex does not localize to the vaccinia virus particle, although it is present throughout the vaccinia virus actin tail (44). The significance of this is unknown.
Asymmetry of Actin Assembly on Vaccinia Virus
Actin assembly is asymmetric on the viral particle. The particle is oblong, and assembly is generally along the long side of the particle (Fig. 22) and always on only one side (33). The nature of what generates this asymmetry is unknown, although the phosphotyrosine signal on the viral particle is also asymmetric (44), suggesting that tyrosine-phosphorylated A36R is asymmetric. By electron microscopic immunostaining, A36R appears to be present over the entire surface of IEV (40), which suggests that the phosphorylation of A36R may be asymmetric.
Model of Vaccinia Virus Actin-Based Motility
Vaccinia virus mimics the receptor tyrosine kinase (Src family kinase-dependent) signaling pathway responsible for actin assembly at the plasma membrane. This is in contrast to actin assembly by Listeria and Shigella, where there is no evidence that tyrosine phosphorylation plays a role (44). Downstream of the tyrosine phosphorylation of A36R on vaccinia virus the pathways in vaccinia virus and Shigella are similar in that both require N-WASP (or, in the case of vaccinia virus WASP). WASP family proteins appear to be required for recruitment of WIP and Nck to the viral surface.
A model of vaccinia virus actin assembly is shown in Fig. 24 (46). Nck binds WIP via its second SH3 domain, and WIP binds N-WASP via its WBD. Tyrosine phosphorylation of A36R by cellular Src family kinases leads to the binding of Nck to A36R. Data suggest that Nck, WIP, and N-WASP may exist as a preformed complex that binds A36R. On binding, N-WASP is activated, binds monomeric actin, and activates Arp2/3 complex-mediated actin assembly. Cross-linking of actin filaments within the vaccinia virus tail is probably mediated by the Arp2/3 complex, as is probably the case for Listeria and Shigella tails. The filaments within the vaccinia virus tail have been observed to be at 45° angles, while the Arp2/3 complex links actin filaments at 70° angles; whether these differences imply distinct mechanisms of cross-linking or are merely within the range of experimental measurement is unclear.
FIG. 24.

Model of actin tail assembly by vaccinia virus A36R. Nck, WIP, and N-WASP bind vaccinia A36R. Amino acid Tyr112 of vaccinia virus A36R is phosphorylated (Ph) by cellular Src family kinases. Phosphorylated A36R interacts with the adapter protein Nck. Nck, WIP, and N-WASP may exist as a preformed complex. Stimulated N-WASP activates the Arp2/3 complex. WBD, WASP-binding domain; NBD, Nck-binding domain; Pro, proline-rich region; N-WASP abbreviations as in the legend to Fig. 20.
RICKETTSIA
Rickettsia spp. are obligate intracellular coccobacillary gram-negative bacteria. All members of the spotted fever group of Rickettsia species utilize actin-based motility to move inside and between cells (Fig. 25). The spotted fever group includes R. rickettsii, which is the agent of Rocky Mountain spotted fever, R. conorii, R. akari, R. australis, R. montana, and R. parkeri. In addition, at least one member of the typhus group of Rickettsia (R. typhi, the agent of murine typhus) forms actin tails, albeit shorter than those of spotted fever group Rickettsia species (Fig. 25C) (75). Schaechter et al. first observed Rickettsia moving inside cells in 1957 (151). Genetic systems have not yet been developed for Rickettsia. Hence, the understanding of its mode of actin-based motility is based exclusively on morphologic and microscopic studies, of which there have been relatively few. Herein, we will review the current knowledge in the field.
FIG. 25.

Actin tails formed by Rickettsia spp. Fluorescence microscopy of Rickettsia-infected Vero cells, labeled for actin (red) and Rickettsia (green). (A) Actin tails formed by spotted fever group species R. rickettsii. (B) Higher magnification of the inset from panel A, showing distinct actin bundles of the tail wrapped in a helical fashion. (C) Short hook-shaped actin tails formed by typhus group species R. typhi. (D) Absence of actin tails on typhus group species R. prowazekii. (Adapted from reference 75 with permission of the American Society for Microbiology.)
Following uptake into mammalian cells, Rickettsia escapes from the endocytic vacuole within about 15 min. In the cytoplasm, an actin cloud polymerizes around the bacterial body (60). As early as 30 min after infection, the actin cloud is reorganized into an actin tail at the bacterial pole (60). Rickettsia forms protrusions of the cell membrane that are somewhat shorter than those of Listeria and Shigella; these protrusions are thought to permit intercellular spread, much as they do for these other two organisms.
In intact mammalian cells, Rickettsia moves at rates of 4.8 to 8 μm/min (58, 59), which is approximately 2.5-fold lower than the rate of Listeria and Shigella movement (Table 1). In Xenopus oocyte cytoplasmic extracts, Rickettsia moves at 2 ± 0.2 μm/min (58), which is 3- to 15-fold slower than the rate of movement of Listeria or E. coli expressing IcsA (Table 1). Rickettsia appears to move in a straighter trajectory than Listeria (59).
Morphology of the Rickettsia Actin Tail
The morphology of the actin tails formed by spotted fever group Rickettsia species is strikingly different from the morphology of those formed by Listeria, Shigella, or vaccinia virus. Whereas the tails formed by Listeria, Shigella, and vaccinia consist of short filaments of actin cross-linked at acute angles, the tails formed by Rickettsia consist of longer actin filaments arranged in a parallel array (Fig. 26) (58, 59, 75). Filaments within Rickettsia tails are 0.3 to 3 μm long, while those of Shigella and Listeria have been reported to be generally 0.1 μm long (Table 2) (58, 75). The morphology of the actin tail formed by Rickettsia in the body of the cell, with long parallel filaments, is similar to that of the actin tail formed by Listeria within cell surface protrusions. Whether similar mechanisms are operative in these two settings is unknown.
FIG. 26.

Transmission electron microscopy showing actin tails formed by R. rickettsii. (A) Long parallel filaments extending from the sides of the bacterium and absent from the bacterial pole. (B) Decoration of the filaments within the tail with the S1 subfragment of myosin, demonstrating that the barbed ends of the filaments face the bacterial body. (C) Higher magnification of filaments decorated with the S1 subfragment of myosin. The inset shows the decorated filament marked by an asterisk. Bars, 0.5 μm. (Reprinted from reference 75 with permission of the American Society for Microbiology.)
Filaments within the tail are oriented with their barbed ends toward the bacterium, as determined by decoration with the S1 subfragment of myosin (Fig. 26), similar to what has been observed for Listeria and Shigella (58, 75, 178, 179). The density of filaments in Rickettsia tails is lower than that of filaments in Listeria or Shigella tails (58). The filaments in Rickettsia tails are significantly more stable than those in Listeria tails, with a half-life of 100 ± 19.2 versus 33.0 ± 7.6 s for Listeria (59). The half-life of the filaments is approximately the same along the entire length of the tail (59).
Tails formed by spotted fever group Rickettsia species often appear to consist of two or more distinct actin bundles, which may appear to be wrapped around one another in a helical fashion (Fig. 25A and B) (59, 60, 75). This pattern suggests that the organism might be rotating around its long axis as it moves forward. As the bacterium moves forward, the gaps in the helix remain stationary, suggesting that the filaments within the tail are stationary, as they are for Listeria and Shigella (56, 59, 175).
Close to the bacterial body, the actin filaments within the tail are present along the sides of the bacteria and generally absent from the bacterial poles (Fig. 26) (58, 75). One group has reported that the overall length of tails formed by Rickettsia is 4 to 6 μm, which is similar to the length of the tails formed by Listeria and slightly shorter than the length of the tails formed by Shigella. Another group has reported that tails formed by Rickettsia in the same cell line are severalfold longer than tails formed by Listeria, averaging 16.7 μm long (59, 75). The tails average 1.5 μm in width, which is 2-fold wider than Shigella tails and 1.5-fold wider than Listeria tails (58).
In contrast to Listeria, Shigella, and vaccinia virus, Rickettsia has the capacity to enter and multiply within the cell nucleus. Intranuclear organisms also assemble actin tails, which can grow to great lengths and wind through the nucleus. Unlike actin tails associated with cytoplasmic Rickettsia organisms, tails associated with intranuclear Rickettsia organisms are not fixed in space but instead move and may generate force on the nuclear membrane (59).
Actin tails formed by R. typhi are generally hook shaped and are shorter than those formed by spotted fever group Rickettsia species averaging 3 μm long (Fig. 25C) (60, 75). R. prowazekii, another member of the typhus group, does not form actin tails (Fig. 25D) (75). To our knowledge, no other members of the typhus group, including R. canada and Orientia tsutsugamushi, nor the related organisms Ehrlichia chaffeensis, E. phagocytophila, and Coxiella burnetii have been analyzed for the ability to assemble actin tails.
Rickettsial Proteins Involved in Actin-Based Motility
Rickettsial protein synthesis is required for actin-based motility (60). The protein(s) involved in actin-based motility has not been identified. It has been postulated that the outer membrane protein OmpA may be important to actin-based motility, since it is present in spotted fever group Rickettsia species and absent in other Rickettsia species, but there is no direct evidence to support or refute this.
Absence of the Arp2/3 Complex in Rickettsia Actin Tails
Two lines of evidence suggest that the Arp2/3 complex may be uninvolved in Rickettsia actin assembly or may play a very different role from the role it plays in the other microbes discussed in this review. First, the Arp2/3 complex is not associated with actin tails formed by Rickettsia, as determined by immunofluorescence microscopy using antibody to the Arp3 subunit (58). Second, whereas the Arp2/3 complex cross-links actin filaments at 70° angles (see “Actin cytoskeleton: dynamics and function” above), the filaments within actin tails formed by Rickettsia are arranged in parallel.
Host Proteins Associated with Rickettsia Actin Tails
Both VASP and its ligand profilin are associated with actin tails formed by Rickettsia, as determined by immunofluorescence microscopy (75). As described above (see “Listeria” and “Shigella”), VASP normally localizes to focal contacts and regions of dynamic actin rearrangements. It binds proline-rich motifs, such as FPPPP of Listeria ActA. Profilin, an actin monomer-binding protein that can deliver actin monomers to sites of actin assembly in the cell, binds to proline-rich motifs in VASP that have the consensus (G/A/L/S)PPPPP. Other proteins that localize to the Rickettsia actin tails are the focal adhesion proteins vinculin and filamin (75). As describe above (see “Shigella”), the amino-terminal head fragment of vinculin has been proposed to serve as a linker between the Shigella actin assembly protein IcsA and VASP.
Clearly, colocalization of a protein to the site of actin assembly does not necessarily indicate a function for it in the process, and additional studies are required to directly test the role of each. Proteins that do not localize to the Rickettsia actin tail and therefore are unlikely to be required for Rickettsia actin assembly include the actin cross-linking protein α-actinin, the focal adhesion proteins ezrin and paxillin, the actin side-binding protein tropomyosin, and, as mentioned above, the Arp2/3 complex (58, 75).
Summary
Several members of the Rickettsia family utilize actin-based motility to move within and spread between cells. Rickettsia cells move approximately 2.5-fold more slowly than do Listeria, Shigella, and vaccinia virus. Actin tails formed by Rickettsia are distinctly different in appearance from those formed by these other microbes. In particular, the actin filaments within the Rickettsia tails are significantly longer and are arranged in parallel, rather than being cross-linked at acute angles. These observations, in conjunction with the lack of detectable Arp2/3 complex within the tail, suggest that the molecular mechanism of actin tail assembly by Rickettsia may differ from that of assembly by Listeria, Shigella, and vaccinia virus. However, given the lack of genetic systems for manipulating Rickettsia, relatively little is known at the molecular level.
CONCLUSIONS AND FUTURE DIRECTIONS
Listeria spp., Shigella spp., vaccinia virus, and Rickettsia spp. all assemble actin tails to move through the cytoplasm of mammalian cells. Listeria, Shigella, and vaccinia virus utilize existing cellular pathways of actin assembly. The molecular mechanism of actin assembly by Rickettsia is unknown but may well also involve preexisting cellular pathways. Listeria, Shigella, and vaccinia virus make use of the Cdc42 pathway (Fig. 3), in which WASP family members activate the actin assembly activity of the Arp2/3 complex. In this pathway, Cdc42 and PI(4,5)P2 activate WASP family members by inducing them to reverse intramolecular bonds and unfold. The activated WASP family member then activates the Arp2/3 complex, which nucleates, polymerizes, and cross-links actin. Arp2/3 assembly of actin is normally associated with reorganization of the cellular cytoskeleton.
Remarkably, Listeria, Shigella, and vaccinia virus each enter into this pathway of actin assembly at a distinct step (Fig. 27). The Listeria protein that mediates actin assembly, ActA, mimics WASP family members. Like WASP family members, ActA activates Arp2/3 complex-mediated actin assembly and delivers monomeric actin to the actin assembly machinery. The Shigella protein that mediates actin assembly, IcsA, has been proposed to mimic Cdc42. Like Cdc42, IcsA binds and probably activates the WASP family member N-WASP. Activated N-WASP in turn activates Arp2/3 complex-mediated actin assembly. The vaccinia virus protein that is the principal, if not the only, mediator of vaccinia virus actin assembly, A36R, interacts with the cellular actin machinery. A36R binding to the adapter protein Nck is dependent on its prior tyrosine phosphorylation by cellular Src kinases. Nck binds WIP, and WIP binds and activates WASP family members. However, binding of Nck and WIP to vaccinia virus is dependent on the presence of a WASP family member, suggesting that Nck, WIP, and the WASP family member may exist as a preformed complex and that Nck and WIP binding may not be upstream of WASP family member binding. The activated WASP family member in turn activates Arp2/3 complex-mediated actin assembly. In these ways, Listeria, Shigella, and vaccinia virus appear to have convergently evolved mechanisms to utilize the cellular Cdc42 pathway of actin assembly, which is highly conserved and ancient. Of note, this pathway is also used by Salmonella during entry into mammalian cells (25).
FIG. 27.

Comparison of the models of actin assembly by Listeria, Shigella, and vaccinia virus. Diagrams of molecules that interact directly or indirectly with the microbial proteins that mediate actin assembly are shown. (A) Listeria ActA; (B) Shigella IcsA; (C) vaccinia virus A36R. A, acidic domain; AB, actin-binding domain; P, profilin; Ph, phosphorylation. N-WASP abbreviations are as in the legend to Fig. 20.
Much about the molecular mechanisms of actin assembly by these organisms remains to be characterized. While motility mediated by Listeria ActA and Shigella IcsA has been partially reconstituted using purified proteins, it appears that the full array of proteins involved in the process may not yet be known. Similarly, the full array of proteins involved in vaccinia virus motility does not appear to have been identified yet. Further, the biochemical, molecular, and structural aspects of interactions among these proteins have been described for few of the involved molecules. Finally, for Rickettsia, very little is known about either the bacterial or the cellular proteins required for the process. Nevertheless, in the past few years an enormous amount has been learned about microbial actin assembly. Moreover, since these microbes utilize existing cellular pathways, in addition to increasing our understanding of microbial actin-based motility, the study of these systems has already yielded insights into the molecular mechanisms of actin reorganization and cell motility of mammalian cells and will certainly continue to do so.
ACKNOWLEDGMENTS
I thank S. B. Snapper, P. Lauer, J. A. Theriot, and D. A. Portnoy for their willingness to provide data prior to their publication and C. Taggart for administrative support.
ADDENDUM IN PROOF
The structure of the Arp2/3 complex has been solved (N. Volkmann et al., Science 293:2456–2459).
REFERENCES
- 1.Ahern-Djamali S M, Comer A R, Bachmann C, Kastenmeier A S, Reddy S K, Beckerle M C, Walter U, Hoffmann F M. Mutations in Drosophila Enabled and rescue by human vasodilator-stimulated phosphoprotein (VASP) indicate important functional roles for Ena/VASP homology domain 1 (EVH1) and EVH2 domains. Mol Biol Cell. 1998;9:2157–2171. doi: 10.1091/mbc.9.8.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amann K J, Pollard T D. The Arp2/3 complex nucleates actin filament branches from the sides of pre-existing filaments. Nat Cell Biol. 2001;3:306–310. doi: 10.1038/35060104. [DOI] [PubMed] [Google Scholar]
- 3.Anton I M, Lu W, Mayer R J, Ramesh N, Geha R S. The Wiskott-Aldrich syndrome protein-interacting protein (WIP) binds to the adaptor protein Nck. J Biol Chem. 1998;273:20992–20995. doi: 10.1074/jbc.273.33.20992. [DOI] [PubMed] [Google Scholar]
- 4.Bachmann C, Fischer L, Walter U, Reinhard M. The EVH2 domain of the vasodilator-stimulated phosphoprotein mediates tetramerization, F-actin binding and actin bundle formation. J Biol Chem. 1999;274:23549–23557. doi: 10.1074/jbc.274.33.23549. [DOI] [PubMed] [Google Scholar]
- 5.Bailly M, Ichetovkin I, Grant W, Zebda N, Machesky L M, Segall J E, Condeelis J. The F-actin side binding activity of the Arp2/3 complex is essential for actin nucleation and lamellipod extension. Curr Biol. 2001;11:620–625. doi: 10.1016/s0960-9822(01)00152-x. [DOI] [PubMed] [Google Scholar]
- 6.Balasubramanian M K, Feoktistiva A, McCollum D, Gould K L. Fission yeast Sop2p: a novel and evolutionarily conserved protein that interacts with Arp3p and modulates profilin function. EMBO J. 1996;15:6426–6437. [PMC free article] [PubMed] [Google Scholar]
- 7.Ball L J, Kuhne R, Hoffmann B, Hafner A, Schmieder P, Volkmer-Engert R, Hof M, Wahl M, Schneider-Mergener J, Walter U, Oschkinat H, Jarchau T. Dual epitope recognition by the VASP EVH1 domain modulates polyproline ligand specificity and binding affinity. EMBO J. 2000;19:4903–4914. doi: 10.1093/emboj/19.18.4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bear J E, Krause M, Gertler F B. Regulating cellular actin assembly. Curr Opin Cell Biol. 2001;13:158–166. doi: 10.1016/s0955-0674(00)00193-9. [DOI] [PubMed] [Google Scholar]
- 9.Bear J E, Loureiro J J, Libova I, Fassler R, Wehland J, Gertler F B. Negative regulation of fibroblast motility by Ena/VASP proteins. Cell. 2000;101:717–728. doi: 10.1016/s0092-8674(00)80884-3. [DOI] [PubMed] [Google Scholar]
- 10.Bernardini M L, Mounier J, d'Hauteville H, Coquis-Rondon M, Sansonetti P J. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci USA. 1989;86:3867–3871. doi: 10.1073/pnas.86.10.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bishop A L, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348:241–255. [PMC free article] [PubMed] [Google Scholar]
- 12.Blanchoin L, Amann K J, Higgs H N, Marchand J-B, Kaiser D A, Pollard T D. Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASP/Scar proteins. Nature. 2000;404:1007–1011. doi: 10.1038/35010008. [DOI] [PubMed] [Google Scholar]
- 13.Blanchoin L, Pollard T D. Interaction of actin monomers with Acanthamoeba actophorin (ADF/cofilin) and profilin. J Biol Chem. 1998;273:25106–25111. doi: 10.1074/jbc.273.39.25106. [DOI] [PubMed] [Google Scholar]
- 14.Blanchoin L, Pollard T D, Mullins R D. Interactions of ADF/cofilin, Arp2/3 complex, capping protein and profilin in remodeling of branched actin filament networks. Curr Biol. 2000;10:1273–1282. doi: 10.1016/s0960-9822(00)00749-1. [DOI] [PubMed] [Google Scholar]
- 15.Brindle N P J, Holt M R, Davies J E, Price C J, Critchley D R. The focal adhesion vasodilator-stimulated phosphoprotein (VASP) binds to the proline-rich domain in vinculin. Biochem J. 1996;318:753–757. doi: 10.1042/bj3180753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brundage R A, Smith G A, Camilli A, Theriot J A, Portnoy D A. Expression and phosphorylation of the Listeria monocytogenes ActA protein in mammalian cells. Proc Natl Acad Sci USA. 1993;90:11890–11894. doi: 10.1073/pnas.90.24.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burridge K, Feramisco J R. Microinjection and localization of a 130K protein in living fibroblasts: a relationship to actin and fibronectin. Cell. 1980;19:587–595. doi: 10.1016/s0092-8674(80)80035-3. [DOI] [PubMed] [Google Scholar]
- 18.Cameron L A, Footer M J, Oudenaarden A v, Theriot J A. Motility of ActA protein-coated microspheres driven by actin polymerization. Proc Natl Acad Sci USA. 1999;96:4908–4913. doi: 10.1073/pnas.96.9.4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cameron L A, Giardini P A, Soo F S, Theriot J A. Secrets of actin-based motility revealed by a bacterial pathogen. Nat Rev Mol Cell Biol. 2000;1:110–119. doi: 10.1038/35040061. [DOI] [PubMed] [Google Scholar]
- 20.Cameron L A, Svitkina T A, Vignjevic D, Theriot J A, Borisy G G. Dendritic organization of actin comet tails. Curr Biol. 2001;11:130–135. doi: 10.1016/s0960-9822(01)00022-7. [DOI] [PubMed] [Google Scholar]
- 21.Carlier M-F, Laurent V, Santolini J, Melki R, Didry D, Xia G-X, Hong Y, Chua N-H, Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol. 1997;136:1307–1323. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chakraborty T, Ebel F, Domann E, Niebuhr K, Gerstel B, Pistor S, Temm-Grove C J, Jockusch B M, Reinhard M, Walter U, Wehland J. A focal adhesion factor directly linking intracellularly motile Listeria monocytogenes and Listeria ivanovii to the actin-based cytoskeleton of mammalian cells. EMBO J. 1995;14:1314–1321. doi: 10.1002/j.1460-2075.1995.tb07117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan A Y, Bailly M, Zebda N, Segall J E, Condeelis J S. Role of cofilin in epidermal growth factor-stimulated actin polymerization and lamellipod protrusion. J Cell Biol. 2000;148:531–542. doi: 10.1083/jcb.148.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Charles M, Perez M, Kobil J H, Goldberg M B. Polar targeting of Shigella virulence factor IcsA in Enterobacteriacae and Vibrio. Proc Natl Acad Sci USA. 2001;98:9871–9876. doi: 10.1073/pnas.171310498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L M, Hobbie S, Galan J E. Requirement of CDC42 for Salmonella-induced cytoskeletal and nuclear responses. Science. 1996;274:2115–2118. doi: 10.1126/science.274.5295.2115. [DOI] [PubMed] [Google Scholar]
- 26.Chou M, Hanafusa N. A novel ligand for SH3 domains. J Biol Chem. 1995;270:7359–7364. doi: 10.1074/jbc.270.13.7359. [DOI] [PubMed] [Google Scholar]
- 27.Chou M M, Fajardo J E, Hanafusa H. The SH2 and SH3 containing Nck protein transforms mammalian fibroblasts in the absence of elevated phosphotyrosine levels. Mol Cell Biol. 1992;12:5834–5842. doi: 10.1128/mcb.12.12.5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cicchetti G, Maurer P, Wagener P, Kocks C. Actin and phosphoinositide binding by the ActA protein of the bacterial pathogen Listeria monocytogenes. J Biol Chem. 1999;274:33616–33626. doi: 10.1074/jbc.274.47.33616. [DOI] [PubMed] [Google Scholar]
- 29.Condeelis J. How is actin polymerization nucleated in vivo. Trends Cell Biol. 2001;11:288–293. doi: 10.1016/s0962-8924(01)02008-6. [DOI] [PubMed] [Google Scholar]
- 30.Cooper J A, Walker S B, Pollard T D. Pyrene actin: documentation of the validity of a sensitive assay for actin polymerization. J Muscle Res Cell Motil. 1983;4:253–262. doi: 10.1007/BF00712034. [DOI] [PubMed] [Google Scholar]
- 31.Cossart P. Actin-based motility of pathogens: the Arp2/3 complex is a central player. Cell Microbiol. 2000;2:195–205. doi: 10.1046/j.1462-5822.2000.00053.x. [DOI] [PubMed] [Google Scholar]
- 32.Cudmore S, Cossart P, Griffiths G, Way M. Actin-based motility of vaccinia virus. Nature. 1995;378:636–638. doi: 10.1038/378636a0. [DOI] [PubMed] [Google Scholar]
- 33.Cudmore S, Reckmann I, Griffiths G, Way M. Vaccinia virus: a model system for actin-membrane interactions. J Cell Sci. 1996;109:1739–1747. doi: 10.1242/jcs.109.7.1739. [DOI] [PubMed] [Google Scholar]
- 34.Dabiri G A, Sanger J M, Portnoy D A, Southwick F S. Listeria monocytogenes moves rapidly through the host-cell cytoplasm by inducing directional actin assembly. Proc Natl Acad Sci USA. 1990;87:6068–6072. doi: 10.1073/pnas.87.16.6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Derry J M, Ochs H D, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell. 1994;78:635–644. doi: 10.1016/0092-8674(94)90528-2. [DOI] [PubMed] [Google Scholar]
- 36.Devriendt K, Kim A S, Mathijs G, Frints S G, Schwartz M, Van Der Oord J J, Verhoef G E, Boogaerts M A, Fryns J P, You D, Rosen M K, Vandenberghe P. Constituitively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet. 2001;27:313–317. doi: 10.1038/85886. [DOI] [PubMed] [Google Scholar]
- 37.Domann E, Wehland J, Rohde M, Pistor S, Hartl M, Goebel W, Leimeister-Wachter M, Wuenscher M, Chakraborty T. A novel bacterial virulence gene in Listeria monocytogenes required for host cell microfilament interaction with homology to the proline-rich region of vinculin. EMBO J. 1992;11:1981–1990. doi: 10.1002/j.1460-2075.1992.tb05252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Donnelly S F, Pocklington M J, Pallotta D, Orr E. A proline-rich protein, verprolin, involved in cytoskeletal organization and cellular growth in theyeast Saccharomyces cerevisiae. Mol Microbiol. 1993;10:585–596. doi: 10.1111/j.1365-2958.1993.tb00930.x. [DOI] [PubMed] [Google Scholar]
- 39.Egile C, Loisel T P, Laurent V, Li R, Pantaloni D, Sansonetti P J, Carlier M-F. Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J Cell Biol. 1999;146:1319–1332. doi: 10.1083/jcb.146.6.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reference deleted.
- 41.Ermekova K S, Zambrano N, Linn H, Minopoli, Gertler F, Russo T, Sudol M. The WW domain of neural protein FE65 interacts with proline-rich motifs in Mena, the mammalian homolog of Drosophila enabled. J Biol Chem. 1997;272:32869–32877. doi: 10.1074/jbc.272.52.32869. [DOI] [PubMed] [Google Scholar]
- 42.Federov A A, Federov E, Gertler F, Almo S C. Structure of EVH1, a novel proline-rich ligand-binding module involved in cytoskeletal dynamics and neural function. Nat Struct Biol. 1999;6:661–665. doi: 10.1038/10717. [DOI] [PubMed] [Google Scholar]
- 43.Freitag N E, Jacobs K E. Examination of Listeria monocytogenes intracellular gene expression by using the green fluorescent protein of Aequorea victoria. Infect Immun. 1999;67:1844–1852. doi: 10.1128/iai.67.4.1844-1852.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frischknecht F, Cudmore S, Moreau V, Reckmann I, Rottger S, Way M. Tyrosine phosphorylation is required for actin-based motility of vaccinia but not Listeria or Shigella. Curr Biol. 1999;9:89–92. doi: 10.1016/s0960-9822(99)80020-7. [DOI] [PubMed] [Google Scholar]
- 45.Frischknecht F, Moreau V, Rottger S, Gonfioni S, Reckmann I, Superti-Furga G, Way M. Actin-based motility of vaccinia mimics receptor tyrosine kinase signalling. Nature. 1999;401:926–929. doi: 10.1038/44860. [DOI] [PubMed] [Google Scholar]
- 46.Frischknecht F, Way M. Surfing pathogens and the lessons learned for actin polymerization. Trends Cell Biol. 2001;11:30–38. doi: 10.1016/s0962-8924(00)01871-7. [DOI] [PubMed] [Google Scholar]
- 47.Geiger B. A 130K protein from chicken gizzard: its localization at the termini of microfilament bendles in cultured chicken cells. Cell. 1979;18:193–205. doi: 10.1016/0092-8674(79)90368-4. [DOI] [PubMed] [Google Scholar]
- 48.Geiger B, Dutton A H, Tokuyasu K T, Singer S J. Immunoelectron microscope studies of membrane-microfilament interactions: distributions of α-actinin, tropomyosin, and vinculin in intestinal epithelial brush border and chicken gizzard smooth muscle cells. J Cell Biol. 1981;91:614–628. doi: 10.1083/jcb.91.3.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geiger B, Tokuyasu K T, Dutton A H, Singer S J. Vinculin, an intracellular protein localized at specialized sites where microfilament bundles terminate at cell membranes. Proc Natl Acad Sci USA. 1980;77:4127–4131. doi: 10.1073/pnas.77.7.4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gerstel B, Grobe L, Pistor S, Chakraborty T, Wehland J. The ActA polypeptides of Listeria ivanovii and Listeria monocytogenes harbor related binding sites for host microfilament proteins. Infect Immun. 1996;64:1929–1936. doi: 10.1128/iai.64.6.1929-1936.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gertler F B, Comer A R, Juang J-L, Ahern S M, Clark M J, Liebl E C, Hoffmann F M. enabled, a dosage-sensitive suppressor of mutations in the Drosophila Abl tyrosine kinase, encodes an Abl substrate with SH3 domain-binding properties. Genes Dev. 1995;9:521–533. doi: 10.1101/gad.9.5.521. [DOI] [PubMed] [Google Scholar]
- 52.Gertler F B, Niebuhr K, Reinhard M, Wehland J, Soriano P. Mena, a relative of VASP and Drosophila Enabled, is implicated in the control of microfilament dynamics. Cell. 1996;87:227–239. doi: 10.1016/s0092-8674(00)81341-0. [DOI] [PubMed] [Google Scholar]
- 53.Giuliano K A, Khatib F A, Hayden S M, Daoud E W, Adams M E, Amorese D A, Bernstein B W, Bamburg J R. Properties of purified actin depolymerizing factor from chick brain. Biochemistry. 1988;27:8931–8938. doi: 10.1021/bi00425a009. [DOI] [PubMed] [Google Scholar]
- 54.Goldberg M B. Shigella actin-based motility in the absence of vinculin. Cell Motil Cytoskeleton. 1997;37:44–53. doi: 10.1002/(SICI)1097-0169(1997)37:1<44::AID-CM5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 55.Goldberg M B, Barzu O, Parsot C, Sansonetti P J. Unipolar localization and ATPase activity of IcsA, a Shigella flexneri protein involved in intracellular movement. J Bacteriol. 1993;175:2189–2196. doi: 10.1128/jb.175.8.2189-2196.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goldberg M B, Theriot J A. Shigella flexneri surface protein IcsA is sufficient to direct actin-based motility. Proc Natl Acad Sci USA. 1995;92:6572–6576. doi: 10.1073/pnas.92.14.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gouin E, Dehoux P, Mengaud J, Kocks C, Cossart P. iactA of Listeria ivanovii, although distantly related to Listeria monotytogenes actA, restores actin tail formation in an L. monocytogenes actA mutant. Infect Immun. 1995;63:2729–2737. doi: 10.1128/iai.63.7.2729-2737.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gouin E, Gantelet H, Egile C, Lasa I, Ohayon H, Villiers V, Gounon P, Sansonetti P J, Cossart P. A comparative study of the actin-based motilities of the pathogenic bacteria Listeria monocytogenes, Shigella flexneri, and Rickettsia conorii. J Cell Sci. 1999;112:1697–1708. doi: 10.1242/jcs.112.11.1697. [DOI] [PubMed] [Google Scholar]
- 59.Heinzen R A, Grieshaber S S, Kirk L S V, Devin C J. Dynamics of actin-based movement by Rickettsia rickettsii in Vero cells. Infect Immun. 1999;67:4201–4207. doi: 10.1128/iai.67.8.4201-4207.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heinzen R A, Hayes S F, Peacock M G, Hackstadt T. Directional actin polymerization associated with spotted fever group Rickettsia infection of Vero cells. Infect Immun. 1993;61:1926–1935. doi: 10.1128/iai.61.5.1926-1935.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Henderson I R, Navarro-Garcia F, Nataro J P. The great escape: structure and function of the autotransporter proteins. Trends Microbiol. 1998;6:370–378. doi: 10.1016/s0966-842x(98)01318-3. [DOI] [PubMed] [Google Scholar]
- 62.Higgs H N, Pollard T D. Regulation of actin filament network formation through Arp2/3 complex. Annu Rev Biochem. 2001;70:649–676. doi: 10.1146/annurev.biochem.70.1.649. [DOI] [PubMed] [Google Scholar]
- 63.Higgs H N, Pollard T D. Regulation of actin polymerization by Arp2/3 complex and WASp/Scar proteins. J Biol Chem. 1999;274:32531–32534. doi: 10.1074/jbc.274.46.32531. [DOI] [PubMed] [Google Scholar]
- 64.Hiller G, Jungwirth C, Weber K. Fluorescence microscopic analysis of the life cycle of vaccinia virus in chick embryo fibroblasts. Virus-cytoskeleton interactions. Exp Cell Res. 1981;132:81–87. doi: 10.1016/0014-4827(81)90085-9. [DOI] [PubMed] [Google Scholar]
- 65.Hiller G, Weber K. A phosphorylated basic vaccinia virion polypeptide of molecular weight 11,000 is exposed on the surface of mature particles and interacts with actin-containing cytoskeletal elements. J Virol. 1982;44:647–657. doi: 10.1128/jvi.44.2.647-657.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hiller G, Weber K, Schneider L, Parajsz C, Jungwirth C. Interaction of assembled progeny pox viruses with the cellular cytoskeleton. Virology. 1979;98:142–153. doi: 10.1016/0042-6822(79)90533-6. [DOI] [PubMed] [Google Scholar]
- 67.Ichetovkin I, Han J, Pang K M, Knecht D A, Condeelis J S. Actin filaments are severed by both native and recombinant Dictyostelium cofilin but to different extents. Cell Motil Cytoskeleton. 2000;45:293–306. doi: 10.1002/(SICI)1097-0169(200004)45:4<293::AID-CM5>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 68.Johnson R P, Craig S W. F-actin binding site masked by the intramolecular association of vinculin head and tail domains. Nature. 1995;373:261–264. doi: 10.1038/373261a0. [DOI] [PubMed] [Google Scholar]
- 69.Johnson R P, Craig S W. An intramolecular association between the head and tail domains of vinculin modulates talin binding. J Biol Chem. 1994;269:12611–12619. [PubMed] [Google Scholar]
- 70.Kadurugamuwa J L, Rohde M, Wehland L, Timmis K N. Intercellular spread of Shigella flexneri through a monolayer mediated by membraneous protrusions and associated with reorganization of the cytoskeletal protein vinculin. Infect Immun. 1991;59:3463–3471. doi: 10.1128/iai.59.10.3463-3471.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kaibuchi K, Kuroda S, Amano M. Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu Rev Biochem. 1999;68:459–486. doi: 10.1146/annurev.biochem.68.1.459. [DOI] [PubMed] [Google Scholar]
- 72.Karuansagar I, Krohne G, Goebel W. Listeria ivanovii is capable of cell-to-cell spread involving actin polymerization. Infect Immun. 1993;61:162–169. doi: 10.1128/iai.61.1.162-169.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kelleher J F, Atkinson S J, Pollard T D. Sequences, structural models, and cellular localization of the actin-related proteins Arp2 and Arp3 from Acanthamoeba. J Cell Biol. 1995;131:385–397. doi: 10.1083/jcb.131.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim A S, Kakalis L T, Abdul-Manan N, Liu G A, Rosen M K. Autoinhibition and activation mechanisms of the Wiskott-Aldrich syndrome protein. Nature. 2000;404:151–158. doi: 10.1038/35004513. [DOI] [PubMed] [Google Scholar]
- 75.Kirk L S V, Hayes S F, Heinzen R A. Ultrastructure of Rickettsia rickettsii actin tails and localization of cytoskeletal proteins. Infect Immun. 2000;68:4706–4713. doi: 10.1128/iai.68.8.4706-4713.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell. 1992;68:521–531. doi: 10.1016/0092-8674(92)90188-i. [DOI] [PubMed] [Google Scholar]
- 77.Kocks C, Hellio R, Gounon P, Ohayon H, Cossart P. Polarized distribution of Listeria monocytogenes surface protein ActA at the site of directional actin assembly. J Cell Sci. 1993;105:699–710. doi: 10.1242/jcs.105.3.699. [DOI] [PubMed] [Google Scholar]
- 78.Kocks C, Marchand J-B, Gouin E, d'Hauteville H, Sansonetti P J, Carlier M-F, Cossart P. The unrelated surface proteins ActA of Listeria monocytogenes and IcsA of Shigella flexneri are sufficient to confer actin-based motility on Listeria innocua and Escherichia coli, respectively. Mol Microbiol. 1995;18:413–423. doi: 10.1111/j.1365-2958.1995.mmi_18030413.x. [DOI] [PubMed] [Google Scholar]
- 79.Kotloff K L, Noriega F, Losonsky G A, Sztein M B, Wasserman S S, Nataro J P, Levine M M. Safety, immunogenicity, and transmissibility in humans of CVD 1203, a live oral Shigella flexneri 2a vaccine candidate attenuated by deletions in aroA and virG. Infect Immun. 1996;64:4542–4548. doi: 10.1128/iai.64.11.4542-4548.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kouyama T, Mihashi K. Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labelled F-actin. Local structure change of actin protome both on polymerization and on binding of heavy meromysin. Eur J Biochem. 1981;114:33–38. [PubMed] [Google Scholar]
- 81.Kozma R, Ahmed S, Best A, Lim L. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol Biol Cell. 1995;15:325–331. doi: 10.1128/mcb.15.4.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kreft J, Dumbsky M, Theiss S. The actin-polymerization protein from Listeria ivanovii is a large repeat protein which shows only limited amino acid sequence homology to ActA from Listeria monocytogenes. FEMS Microbiol Lett. 1995;126:113–121. doi: 10.1111/j.1574-6968.1995.tb07403.x. [DOI] [PubMed] [Google Scholar]
- 83.Krempien U, Schneider L, Hiller G, Weber K, Katz E, Jungwirth C. Conditions for pox virus-specific microvilli formation studied during synchronous virus asembly. Virology. 1981;113:556–564. doi: 10.1016/0042-6822(81)90183-5. [DOI] [PubMed] [Google Scholar]
- 84.Laine R O, Zeile W, Kang F, Purich D L, Southwick F S. Vinculin proteolysis unmasks an ActA homolog for actin-based Shigella motility. J Cell Biol. 1997;138:1255–1264. doi: 10.1083/jcb.138.6.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lasa I, David V, Gouin E, Marchand J-B, Cossart P. The amino-terminal part of ActA is critical for the actin-based motility of Listeria monocytogenes; the central proline-rich region acts as a stimulator. Mol Microbiol. 1995;18:425–436. doi: 10.1111/j.1365-2958.1995.mmi_18030425.x. [DOI] [PubMed] [Google Scholar]
- 86.Lasa I, Gouin E, Goethals M, Vancompernolle K, David V, Vandekerckhove J, Cossart P. Identification of two regions in the N-terminal domain of ActA involved in the actin comet tail formation by Listeria monocytogenes. EMBO J. 1997;16:1531–1540. doi: 10.1093/emboj/16.7.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86a.Lauer, P., J. A. Theriot, J. Skoble, M. D. Welch, and D. A. Portnoy. Systematic mutational analysis of the amino-terminal domain of the Listeria monocytogenes ActA protein reveals novel functions in actin-based motility. Mol. Microbiol., in press. [DOI] [PubMed]
- 87.Laurent V, Loisel T P, Harbeck B, Wehman A, Grobe L, Jockusch B M, Wehland J, Gertler F B, Carlier M-F. Role of proteins of the Ena/VASP family in actin-based motility of Listeria monocytogenes. J Cell Biol. 1999;144:1245–1258. doi: 10.1083/jcb.144.6.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lees-Miller J P, Henry G, Helfman D M. Identification of act2, an essential gene in the fission yeast Schizosaccharomyces pombe that encodes a protein related to actin. Proc Natl Acad Sci USA. 1992;89:80–83. doi: 10.1073/pnas.89.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lett M-C, Sasakawa C, Okada N, Sakai T, Makino S, Yamada M, Komatsu K, Yoshikawa M. virG, a plasmid-coded virulence gene of Shigella flexneri: identification of the virG protein and determination of the complete coding sequence. J Bacteriol. 1989;171:353–359. doi: 10.1128/jb.171.1.353-359.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li R. Bee1, a yeast protein with homology to Wiskott-Aldrich syndrome protein, is critical for the assembly of cortical actin cytoskeleton. J Cell Biol. 1997;136:649–658. doi: 10.1083/jcb.136.3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Loisel T P, Boujemaa R, Pantaloni D, Carlier M-F. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature. 1999;401:613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- 92.Lorenzo M M, Galindo I, Griffiths G, Blasco R. Intrecellular localization of vaccinia virus extracellular enveloped virus envelope proteins individually expressed using a Semliki Forest virus replicon. J Virol. 2000;74:10535–10550. doi: 10.1128/jvi.74.22.10535-10550.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Machesky L M, Atkinson S J, Ampe C, Vandekerckhove J, Pollard T D. Purification of a cortical complex containing two unconventional actins from Acanthamoeba by affinity chromatography on profilin-agarose. J Cell Biol. 1994;127:107–115. doi: 10.1083/jcb.127.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Machesky L M, Gould K L. The Arp2/3 complex: a multifunctional actin organizer. Curr Opin Cell Biol. 1999;11:117–121. doi: 10.1016/s0955-0674(99)80014-3. [DOI] [PubMed] [Google Scholar]
- 95.Machesky L M, Insall R H. Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr Biol. 1998;8:1347–1356. doi: 10.1016/s0960-9822(98)00015-3. [DOI] [PubMed] [Google Scholar]
- 96.Machesky L M, Mullins R D, Higgs H N, Kaiser D A, Blanchoin L, May R C, Hall M E, Pollard T D. Scar, a WASp-related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc Natl Acad Sci USA. 1999;96:3739–3744. doi: 10.1073/pnas.96.7.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Machesky L M, Reeves E, Wientjes F, Mattheyse F J, Grogan A, Totty N F, Burlingame A L, Hsuan J J, Segal A W. Mammalian actin-related protein 2/3 complex localizes to regions of lammelipodia protrusion and is composed of evolutionarily conserved proteins. Biochem J. 1997;328:105–112. doi: 10.1042/bj3280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Makino S, Sasakawa C, Kamata K, Kurata T, Yoshikawa M. A genetic determinant required for continuous reinfection of adjacent cells on large plasmid in S. flexneri 2a. Cell. 1986;46:551–555. doi: 10.1016/0092-8674(86)90880-9. [DOI] [PubMed] [Google Scholar]
- 99.Marchand J-B, Moreau P, Paoletti A, Cossart P, Carlier M-F, Pantaloni D. Actin-based movement of Listeria monocytogenes: actin assembly results from the local maintenance of uncapped filament barbed ends at the bacterium surface. J Cell Biol. 1995;130:331–343. doi: 10.1083/jcb.130.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Marchand J B, Kaiser D A, Pollard T D, Higgs H N. Interaction of WASP/Scar proteins with actin and vertebrate Arp2/3 complex. Nat Cell Biol. 2001;3:76–82. doi: 10.1038/35050590. [DOI] [PubMed] [Google Scholar]
- 101.May R C, Hall M E, Higgs H N, Pollard T D, Chakraborty T, Wehland J, Machesky L M, Sechi A S. The Arp2/3 complex is essential for the actin-based motility of Listeria monocytogenes. Curr Biol. 1999;9:759–762. doi: 10.1016/s0960-9822(99)80337-6. [DOI] [PubMed] [Google Scholar]
- 102.McCarty J H. The Nck SH2/SH3 adaptor protein: a regulator of multiple intracellular signal transduction events. Bioessays. 1998;20:913–921. doi: 10.1002/(SICI)1521-1878(199811)20:11<913::AID-BIES6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 103.McCollum D, Feoktistova A, Morphew M, Balasubramanian M, Gould K L. The Schizosaccharomyces pombe actin-related protein, Arp3, is a component of the cortical actin cytoskeleton and interacts with profilin. EMBO J. 1996;15:6438–6446. [PMC free article] [PubMed] [Google Scholar]
- 104.Miki H, Miura K, Takenawa T. N-WASP, a novel actin-depolymerizing protein, regulates the cortical cytoskeleton rearrangement in a PIP2-dependent manner downstream of tyrosine kinases. EMBO J. 1996;15:5326–5335. [PMC free article] [PubMed] [Google Scholar]
- 105.Miki H, Sasaki T, Takai Y, Takenawa T. Induction of filopodia formation by a WASP-related actin-depolymerizing protein N-WASP. Nature. 1998;391:93–96. doi: 10.1038/34208. [DOI] [PubMed] [Google Scholar]
- 106.Miki H, Suetsugu S, Takenawa T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 1998;17:6932–6941. doi: 10.1093/emboj/17.23.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Miki H, Takenawa T. Direct binding of the verprolin-homology domain in N-WASP to actin is essential for cytoskeletal reorganization. Biochem Biophys Res Commun. 1998;243:73–78. doi: 10.1006/bbrc.1997.8064. [DOI] [PubMed] [Google Scholar]
- 108.Mimuro H, Suzuki T, Suetsugu S, Miki H, Takenawa T, Sasakawa C. Profilin is required for sustaining efficient intra- and intercellular spreading of Shigella flexneri. J Biol Chem. 2000;275:28893–28901. doi: 10.1074/jbc.M003882200. [DOI] [PubMed] [Google Scholar]
- 109.Mogilner A, Oster G. Cell motility driven by actin polymerization. Biophys J. 1996;71:3030–3045. doi: 10.1016/S0006-3495(96)79496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Moors M A, Levitt B, Youngman P, Portnoy D A. Expression of listeriolysin O and ActA by intracellular and extracellular Listeria monocytogenes. Infect Immun. 1999;67:131–139. doi: 10.1128/iai.67.1.131-139.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Moreau V, Frischknecht F, Reckmann I, Vincentelli R, Rabut G, Stewart D, Way M. A complex of N-WASP and WIP integrates signalling cascades that lead to actin polymerization. Nat Cell Biol. 2000;2:441–448. doi: 10.1038/35017080. [DOI] [PubMed] [Google Scholar]
- 112.Moreau V, Galan J M, Devilliers G, Haguenauer-Tsapis R, Winsor B. The yeast actin-related protein Arp2p is required for the internalization step of endocytosis. Mol Biol Cell. 1997;8:1361–1375. doi: 10.1091/mbc.8.7.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Moreau V, Madania A, Martin R P, Winsor B. The Saccharomyces cerevisiae actin-related protein Arp2 is involved in the actin cytoskeleton. J Cell Biol. 1996;134:117–132. doi: 10.1083/jcb.134.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mourrain P, Lasa I, Gautreau A, Gouin E, Pugsley A, Cossart P. ActA is a dimer. Proc Natl Acad Sci USA. 1997;94:10034–10039. doi: 10.1073/pnas.94.19.10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mullins R D, Heuser J A, Pollard T D. The interaction of Arp2/3 complex with actin: nucleation, high-affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci USA. 1998;95:6181–6186. doi: 10.1073/pnas.95.11.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mullins R D, Kelleher J F, Xu J, Pollard T D. Arp2/3 complex from Acanthamoeba binds profilin and cross-links actin filaments. Mol Biol Cell. 1998;9:841–852. doi: 10.1091/mbc.9.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mullins R D, Pollard T D. Structure and function of the Arp2/3 complex. Curr Opin Struct Biol. 1999;9:244–249. doi: 10.1016/s0959-440x(99)80034-7. [DOI] [PubMed] [Google Scholar]
- 118.Mullins R D, Stafford W F, Pollard T D. Structure, subunit topology, and actin-binding activity of the Arp2/3 complex from Acanthamoeba. J Cell Biol. 1997;136:331–343. doi: 10.1083/jcb.136.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Neibuhr K, Ebel F, Frank R, Reinhard M, Domann E, Carl U D, Walter U, Gertler F B, Wehland J, Chakraborty T. A novel proline-rich motif present in ActA of Listeria monocytogenes and cytoskeletal proteins is the ligand for the EVH1 domain, a protein module present in the Ena/VASP family. EMBO J. 1997;16:5433–5444. doi: 10.1093/emboj/16.17.5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Niebuhr K, Chakraborty T, Rohde M, Gazlig T, Jansen B, Kollner P, Wehland J. Localization of the ActA polypeptide of Listeria monocytogenes in infected tissue culture cell lines: ActA is not associated with actin comets. Infect Immun. 1993;61:2793–2802. doi: 10.1128/iai.61.7.2793-2802.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nollau R R, Ho H-Y H, Kirschner M W, Mayer B J. Nck and phosphatidylinositol 4,5-bisphosphate synergistically activate actin polymerization through the N-WASP-Arp2/3 pathway. J Biol Chem. 2001;276:26448–26452. doi: 10.1074/jbc.M103856200. [DOI] [PubMed] [Google Scholar]
- 122.Ogawa H, Nakamura A, Nakaya R. Cinemicrographic study of tissue cell cultures infected with Shigella flexneri. Jpn J Med Sci Biol. 1968;21:259–273. doi: 10.7883/yoken1952.21.259. [DOI] [PubMed] [Google Scholar]
- 123.Pal T, Newland J W, Tall B D, Formal S B, Hale T L. Intracellular spread of Shigella flexneri associated with the kcpA locus and a 140-kilodalton protein. Infect Immun. 1989;57:477–486. doi: 10.1128/iai.57.2.477-486.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pantaloni D, Boujemaa R, Didry D, Gounon P, Carlier M-F. The Arp2/3 complex branches filament barbed ends: functional antagonism with capping proteins. Nat Cell Biol. 2000;2:385–391. doi: 10.1038/35017011. [DOI] [PubMed] [Google Scholar]
- 125.Pantaloni D, LeClainche C, Carlier M-F. Mechanism of actin-based motility. Science. 2001;292:1502–1506. doi: 10.1126/science.1059975. [DOI] [PubMed] [Google Scholar]
- 126.Pistor S, Chakraborty T, Niebuhr K, Domann E, Wehland J. The ActA protein of Listeria monocytogenes acts as a nucleator inducing reorganization of the actin cytoskeleton. EMBO J. 1994;13:758–763. doi: 10.1002/j.1460-2075.1994.tb06318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pistor S, Chakraborty T, Walter U, Wehland J. The bacterial actin nucleator protein ActA of Listeria monocytogenes contains multiple binding sites for host microfilament proteins. Curr Biol. 1995;5:517–525. doi: 10.1016/s0960-9822(95)00104-7. [DOI] [PubMed] [Google Scholar]
- 128.Pistor S, Grobe L, Sechi A S, Domann E, Gerstel B, Machesky L M, Chakraborty T, Wehland J. Mutations of arginine residues within the 146-KKRRK-150 motif of the ActA protein of Listeria monocytogenes abolish intracellular motility by interfering with the recruitment of the Arp2/3 complex. J Cell Sci. 2000;113:3277–3287. doi: 10.1242/jcs.113.18.3277. [DOI] [PubMed] [Google Scholar]
- 129.Ploubidou A, Way M. Viral transport and the cytoskeleton. Curr Opin Cell Biol. 2001;13:97–105. doi: 10.1016/S0955-0674(00)00180-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Prehoda K E, Lee D J, Lim W A. Structure of the Enabled/VASP homology 1 domain-peptide complex: a key component in the spatial control of actin assembly. Cell. 1999;97:471–480. doi: 10.1016/s0092-8674(00)80757-6. [DOI] [PubMed] [Google Scholar]
- 131.Prehoda K E, Scott J A, Mullins R D, Lim W A. Integration of multiple signals through cooperative regulation of the N-WASP-Arp2/3 complex. Science. 2000;290:801–806. doi: 10.1126/science.290.5492.801. [DOI] [PubMed] [Google Scholar]
- 132.Purich D L, Southwick F S. ABM-1 and ABM-2 homology sequences: consensus docking sites for actin-based motility defined by oligoproline regions in Listeria ActA surface protein and human VASP. Biochem Biophys Res Commun. 1997;231:686–691. doi: 10.1006/bbrc.1997.6158. [DOI] [PubMed] [Google Scholar]
- 133.Quilliam L A, Lambert Q T, Mickelson-Young L A, Westwick J K, Sparks A B, Kay B K, Jenkins N A, Gilbert D A, Copeland N G, Der C J. Isolation of a NCK-associated kinase, PRK2, an SH3-binding protein and potential effector of Rho protein signaling. J Biol Chem. 1996;271:28772–28776. doi: 10.1074/jbc.271.46.28772. [DOI] [PubMed] [Google Scholar]
- 134.Ramesh N, Anton I E, Hartwig J H, Geha R S. WIP, a protein associated with Wiskott-Aldrich syndrome protein, induces actin polymerization and redistribution in lymphoid cells. Proc Natl Acad Sci USA. 1997;94:14671–14676. doi: 10.1073/pnas.94.26.14671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ramesh N, Anton I M, Martinez-Quiles N, Geha R S. Waltzing with WASP. Trends Cell Biol. 1999;9:15–19. doi: 10.1016/s0962-8924(98)01411-1. [DOI] [PubMed] [Google Scholar]
- 136.Reinhard M, Giehl K, Abel K, Haffner C, Jarchau T, Hoppe V, Jockusch B M, Walter U. The proline-rich focal adhesion and microfilament protein VASP is a ligand for profilins. EMBO J. 1995;14:1583–1589. doi: 10.1002/j.1460-2075.1995.tb07146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Reinhard M, Halbrugge M, Scheer U, Wiegand C, Jockusch B M, Walter U. A 46/50 kDa phosphoprotein VASP purified from human platelets is a novel protein associated with actin filaments and focal contacts. EMBO J. 1992;11:2063–2070. doi: 10.1002/j.1460-2075.1992.tb05264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Reinhard M, Jouvenal K, Tripier D, Walter U. Identification, purification and characterization of a zyxin-related protein which binds the focal adhesion and microfilament protein VASP. Proc Natl Acad Sci USA. 1995;92:7956–7960. doi: 10.1073/pnas.92.17.7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Reinhard M, Rudiger M, Jockusch B M, Walter U. VASP interaction with vinculin: a recurring theme of interactions with proline-rich motifs. FEBS Lett. 1996;399:103–107. doi: 10.1016/s0014-5793(96)01295-1. [DOI] [PubMed] [Google Scholar]
- 140.Ridley A J, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 141.Ridley A J, Paterson H F, Johnston C L, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 142.Rivero-Lezcano O M, Marcilla A, Sameshima J H, Robbins K C. Wiskott-Aldrich syndrome protein physically associates with Nck through Src3 homology domains. Mol Cell Biol. 1995;15:5725–5731. doi: 10.1128/mcb.15.10.5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Robbins J R, Monack D, McCallum S J, Vegas A, Pham E, Goldberg M B, Theriot J A. The making of a gradient: IcsA (VirG) polarity in Shigella flexneri. Mol Microbiol. 2001;41:861–872. doi: 10.1046/j.1365-2958.2001.02552.x. [DOI] [PubMed] [Google Scholar]
- 144.Rohatgi R, Ho H H, Kirschner M W. Mechanism of N-WASP activation by CDC42 and phosphatidylinositol 4,5 bisphosphate. J Cell Biol. 2000;150:1299–1309. doi: 10.1083/jcb.150.6.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, Kirschner M W. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–231. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- 146.Roper R L, Wolffe E L, Weisberg A, Moss B. The envelope protein encoded by the A33R gene is required for formation of actin-containing microvilli and efficient cell-to-cell apread of vaccinia virus. J Virol. 1998;72:4192–4204. doi: 10.1128/jvi.72.5.4192-4204.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rottger S, Frischknecht F, Reckmann I, Smith G L, Way M. Interactions between vaccinia virus IEV membrane proteins and their roles in IEV assembly and actin tail formation. J Virol. 1999;73:2863–2875. doi: 10.1128/jvi.73.4.2863-2875.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sanderson C M, Frischknecht F, Way M, Hollinshead M, Smith G L. Roles of vaccinia virus EEV-specific proteins in intracellular actin tail formation and low pH-induced cell-cell fusion. J Gen Virol. 1998;79:1415–1425. doi: 10.1099/0022-1317-79-6-1415. [DOI] [PubMed] [Google Scholar]
- 149.Sansonetti P J, Arondel J, Fontaine A, d'Hauteville H, Bernardini M L. OmpB (osmo-regulation) and icsA (cell-to-cell spread) mutants of Shigella flexneri: vaccine candidates and probes to study the pathogenesis of shigellosis. Vaccine. 1991;9:416–422. doi: 10.1016/0264-410x(91)90128-s. [DOI] [PubMed] [Google Scholar]
- 150.Savoy D N, Billadeau D D, Leibson P J. Cutting edge: WIP, a binding partner for Wiskott-Aldrich syndrome protein, cooperates with Vav in the regulation of T cell activation. J Immunol. 2000;164:2866–2870. doi: 10.4049/jimmunol.164.6.2866. [DOI] [PubMed] [Google Scholar]
- 151.Schaechter M, Bozeman F M, Smadel J E. Study on growth of rickettsiae. II. Morphologic observations of living rickettsiae in tissue culture cells. Virology. 1957;3:160–172. doi: 10.1016/0042-6822(57)90030-2. [DOI] [PubMed] [Google Scholar]
- 152.Schafer D A, Jennings P B, Cooper J A. Dynamics of capping protein and actin assembly in vitro: uncapping barbed ends by polyphosphoinositides. J Cell Biol. 1996;135:169–179. doi: 10.1083/jcb.135.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Schlaepfer D D, Broome M A, Hunter T. Fibronectin-stimulated signaling from a focal adhesion kinase–c-src complex: involvement of the Grb2, p130cas, and Nck adapter proteins, Mol. Cell Biol. 1997;17:1702–1713. doi: 10.1128/mcb.17.3.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Schutt C E, Myslik J C, Rozycki M D, Goonesekere N C, Lindberg U. The structure of crystalline profilin-beta-actin. Nature. 1993;365:810–816. doi: 10.1038/365810a0. [DOI] [PubMed] [Google Scholar]
- 155.Schwob E, Martin R P. New yeast actin-like gene required late in the cell cycle. Nature. 1992;355:179–182. doi: 10.1038/355179a0. [DOI] [PubMed] [Google Scholar]
- 156.Sechi A S, Wehland J, Small J V. The isolated comet tail pseudopodium of Listeria monocytogenes: a tail of two actin filament populations, long and axial and short and random. J Cell Biol. 1997;137:155–167. doi: 10.1083/jcb.137.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.She H, Rockow S, Tang J, Nishimura R, Skolnik E V, Chen M, Margolis B, Lei W. Wiskott-Aldrich syndrome protein is associated with the adaptor protein Grb2 and the epidermal growth factor in living cells. Mol Biol Cell. 1997;8:1709–1721. doi: 10.1091/mbc.8.9.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shere K D, Sallustio S, Manessis A, D'Aversa T G, Goldberg M B. Disruption of IcsP, the major Shigella protease that cleaves IcsA, accelerates actin-based motility. Mol Microbiol. 1997;25:451–462. doi: 10.1046/j.1365-2958.1997.4681827.x. [DOI] [PubMed] [Google Scholar]
- 159.Skoble J, Portnoy D A, Welch M D. Three regions within ActA promote Ap2/3 complex-mediated actin nucleation and Listeria monocytogenes motility. J Cell Biol. 2000;150:527–537. doi: 10.1083/jcb.150.3.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Smith G A, Portnoy D A. How the Listeria monocytogenes ActA protein converts actin polymerization into a motile force. Trends Microbiol. 1997;5:272–276. doi: 10.1016/S0966-842X(97)01048-2. [DOI] [PubMed] [Google Scholar]
- 161.Smith G A, Portnoy D A, Theriot J A. Asymmetric distribution of the Listeria monocytogenes ActA protein is required and sufficient to direct actin-based motility. Mol Microbiol. 1995;17:945–951. doi: 10.1111/j.1365-2958.1995.mmi_17050945.x. [DOI] [PubMed] [Google Scholar]
- 162.Smith G A, Theriot J A, Portnoy D A. The tandem repeat domain in the Listeria monocytogenes ActA protein controls the rate of actin-based motility, the percentage of moving bacteria, and the localization of vasodilator-stimulated phosphoprotein and profilin. J Cell Biol. 1996;135:647–660. doi: 10.1083/jcb.135.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Snapper S B, Rosen F S. The Wiskott-Aldrich syndrome protein (WASP): roles in signalling and cytoskeletal organization. Annu Rev Immunol. 1999;17:905–929. doi: 10.1146/annurev.immunol.17.1.905. [DOI] [PubMed] [Google Scholar]
- 163a.Snapper S B, Takeshima F, Anton I, Liu C-H, Thomas S, Nguyen D, Dudley D, Fraser H, Putich D, Lopez-Ilasaca M, Klein C, Davidson L, Bronson R, Mulligan R C, Southwick F, Geha R, Goldberg M B, Rosen F S, Hartwig J H, Alt F H. N-WASP deficiency reveals distinct pathways for cell surface projections and microbial actin-based motility. Nat Cell Biol. 2001;3:897–904. doi: 10.1038/ncb1001-897. [DOI] [PubMed] [Google Scholar]
- 164.Southwick F S, Adamson E D, Purich D L. Shigella actin-based motility in the presence of truncated vinculin. Cell Motil Cytoskeleton. 2000;45:272–278. doi: 10.1002/(SICI)1097-0169(200004)45:4<272::AID-CM3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 165.Southwick F S, Purich D L. Arrest of Listeria movement in host cells by a bacterial ActA analogue: implications for actin-based motility. Proc Natl Acad Sci USA. 1994;91:5168–5172. doi: 10.1073/pnas.91.11.5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Steffen P, Schafer D A, David V, Gouin E, Cooper J A, Cossart P. Listeria monocytogenes ActA protein interacts with phosphstidylinositol 4,5-bisphosphate in vitro. Cell Motil Cytoskeleton. 2000;45:58–66. doi: 10.1002/(SICI)1097-0169(200001)45:1<58::AID-CM6>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 167.Steinhauer J, Agha R, Pham T, Varga A W, Goldberg M B. The unipolar Shigella surface protein IcsA is directly targeted to the old pole: IcsP cleavage of IcsA occurs over the entire bacterial surface. Mol Microbiol. 1999;32:367–378. doi: 10.1046/j.1365-2958.1999.01356.x. [DOI] [PubMed] [Google Scholar]
- 168.Stewart D M, Tian L, Nelson L D. Mutations that cause Wiskott-Aldrich syndrome impair the interaction of Wiskott-Aldrich syndrome protein (WASP) with WASP interacting protein. J Immunol. 1999;162:5019–5024. [PubMed] [Google Scholar]
- 169.Suetsugu S, Miki H, Takenawa T. The essential role of profilin in the assembly of actin for microspike formation. EMBO J. 1998;17:6516–6526. doi: 10.1093/emboj/17.22.6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Suzuki T, Miki H, Takenawa T, Sasakawa C. Neural Wiskott-Aldrich syndrome protein is implicated in the actin-based motility of Shigella flexneri. EMBO J. 1998;17:2767–2776. doi: 10.1093/emboj/17.10.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Suzuki T, Miki H M, Takenawa T, Sasaki T, Nakanishi H, Takai Y, Sasakawa C. Rho family GTPase Cdc42 is essential for the actin-based motility of Shigella in mammalian cells. J Exp Med. 2000;191:1905–1920. doi: 10.1084/jem.191.11.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Suzuki T, Saga S, Sasakawa C. Functional analysis of Shigella VirG domains essential for interaction with vinculin and actin-based motility. J Biol Chem. 1996;271:21878–21885. doi: 10.1074/jbc.271.36.21878. [DOI] [PubMed] [Google Scholar]
- 173.Svitkina T M, Borisy G G. Arp2/3 complex and actin depolymerizing factor/cofilin in dentritic organization and treadmilling of actin filament array in lamellipodia. J Cell Biol. 1999;145:1009–1026. doi: 10.1083/jcb.145.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Theriot J A, Mitchison T J. Actin microfilament dynamics in locomoting cells. Nature. 1991;352:126–131. doi: 10.1038/352126a0. [DOI] [PubMed] [Google Scholar]
- 175.Theriot J A, Mitchison T J, Tilney L G, Portnoy D A. The rate of actin-based motility of intracellular Listeria monocytogenes equals the rate of actin polymerization. Nature. 1992;357:257–260. doi: 10.1038/357257a0. [DOI] [PubMed] [Google Scholar]
- 176.Theriot J A, Rosenblatt J, Portnoy D A, Goldschmidt-Clermont P J, Mitchison T J. Involvement of profilin in the actin-based motility of L. monocytogenes in cells and in cell-free extracts. Cell. 1994;76:505–517. doi: 10.1016/0092-8674(94)90114-7. [DOI] [PubMed] [Google Scholar]
- 177.Tilney L G, Connelly P S, Portnoy D A. Actin filament nucleation by the bacterial pathogen, Listeria monocytogenes. J Cell Biol. 1990;111:2979–2988. doi: 10.1083/jcb.111.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tilney L G, DeRosier D J, Tilney M S. How Listeria exploits host cell actin to form its own cytoskeleton. I. Formation of a tail and how that tail might be involved in movement. J Cell Biol. 1992;118:71–81. doi: 10.1083/jcb.118.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tilney L G, DeRosier D J, Weber A, Tilney M S. How Listeria exploits host cell actin to form its own cytoskeleton. II. Nucleation, actin filament polarity, filament assembly, and evidence for a pointed end capper. J Cell Biol. 1992;118:83–93. doi: 10.1083/jcb.118.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Tilney L G, Portnoy D A. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J Cell Biol. 1989;109:1597–1608. doi: 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Vaduva G, Martin N C, Hooper A K. Actin-binding verprolin is a polarity development protein required for the morphogenesis and function of the yeast actin cytoskeleton. J Cell Biol. 1997;139:1821–1833. doi: 10.1083/jcb.139.7.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Vaduva G, Martinez-Quiles N, Anton I M, Martin N C, Geha R S, Hooper A K, Ramesh N. The human WASP-interacting protein, WIP, activates the cell polarity pathway in yeast. J Biol Chem. 1999;274:17103–17108. doi: 10.1074/jbc.274.24.17103. [DOI] [PubMed] [Google Scholar]
- 182a.van Eijl H, Hollinshead M, Smith G L. The vaccinia virus A36R protein is a type Ib membrane protein present on intracellular but not extracellular enveloped virus particles. Virology. 2000;271:26–36. doi: 10.1006/viro.2000.0260. [DOI] [PubMed] [Google Scholar]
- 183.VanTroys M, Dewitte D, Goethals M, Carlier M F, Vandekerckhove J, Ampe C. The actin binding site of thymosin beta 4 mapped by mutational analysis. EMBO J. 1996;15:201–210. [PMC free article] [PubMed] [Google Scholar]
- 184.Wear M A, Schafer D A, Cooper J A. Actin dynamics: assembly and disassembly of actin networks. Curr Biol. 2000;10:R891–R895. doi: 10.1016/s0960-9822(00)00845-9. [DOI] [PubMed] [Google Scholar]
- 185.Welch M D, DePace A H, Verma S, Iwamatsu A, Mitchison T J. The human Arp2/3 complex is composed of evolutionarily conserved subunits and is localized to cellular regions of dynamic actin filament assembly. J Cell Biol. 1997;138:375–384. doi: 10.1083/jcb.138.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Welch M D, Iwamatsu A, Mitchison T J. Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. Nature. 1997;385:265–269. doi: 10.1038/385265a0. [DOI] [PubMed] [Google Scholar]
- 187.Welch M D, Rosenblatt J, Skoble J, Portnoy D A, Mitchison T J. Interaction of human Arp2/3 complex and the Listeria monocytogenes ActA protein in actin filament elongation. Science. 1998;281:105–108. doi: 10.1126/science.281.5373.105. [DOI] [PubMed] [Google Scholar]
- 188.Winter D, Lechler T, Li R. Activation of the yeast Arp2/3 complex by Bee1p, a WASP-family protein. Curr Biol. 1999;9:501–504. doi: 10.1016/s0960-9822(99)80218-8. [DOI] [PubMed] [Google Scholar]
- 189.Winter D, Podtelejnikov A V, Mann M, Li R. The complex containing actin-related proteins Arp2 and Arp3 is required for the motlity and integrity of yeast actin patches. Curr Biol. 1997;7:519–529. doi: 10.1016/s0960-9822(06)00223-5. [DOI] [PubMed] [Google Scholar]
- 190.Winter D C, Choe E Y, Li R. Genetic dissection of the budding yeast Arp2/3 complex: a comparison of the in vivo and structural roles of individual subunits. Proc Natl Acad Sci USA. 1999;96:7288–7293. doi: 10.1073/pnas.96.13.7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Wolffe E J, Weisberg A S, Moss B. Role for the vaccinia virus A36R outer envelope protein in the formation of virus-tipped actin-containing microvilli and cell-to-cell spread. Virology. 1998;25:20–26. doi: 10.1006/viro.1998.9103. [DOI] [PubMed] [Google Scholar]
- 192.Wolffe E J, Weisberg A S, Moss B. The vaccinia virus A33R protein provides a chaperone function for viral membrane localization and tyrosine phosphorylation of the A36R protein. J Virol. 2001;75:303–310. doi: 10.1128/JVI.75.1.303-310.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yang C, Huang M, DeBiasio J, Pring M, Joyce M, Miki H, Takenawa T, Zigmond S H. Profilin enhances Cdc42-induced nucleation of actin polymerization. J Cell Biol. 2000;150:1001–1012. doi: 10.1083/jcb.150.5.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Yarar D, To W, Abo A, Welch M D. The Wiskott-Aldrich syndrome protein directs actin-based motility by stimulating actin nucleation with the Arp2/3 complex. Curr Biol. 1999;9:555–558. doi: 10.1016/s0960-9822(99)80243-7. [DOI] [PubMed] [Google Scholar]
- 195.Zalevsky J, Grigirova I, Mullins R D. Activation of the Arp2/3 complex by the Listeria ActA protein. ActA binds two actin monomers and three subunits of the Arp2/3 complex. J Biol Chem. 2001;276:3468–3475. doi: 10.1074/jbc.M006407200. [DOI] [PubMed] [Google Scholar]
- 196.Zebda N, Bernard O, Bailly M, Welti S, Lawrence D S, Condeelis J S. Phosphorylation of ADF/cofilin abolishes EGF-induced actin nucleation at the leading edge and subsequent lamellipod extension. J Cell Biol. 2000;151:1119–1127. doi: 10.1083/jcb.151.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zeile W L, Purich D L, Southwick F S. Recognition of two classes of oligoproline sequences in profilin-mediated acceleration of actin-based Shigella motility. J Cell Biol. 1996;133:49–59. doi: 10.1083/jcb.133.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zhang W-H, Wilcock D, Smith G L. Vaccinia virus F12L protein is required for actin tail formation, normal plaque size, and virulence. J Virol. 2000;74:11654–11662. doi: 10.1128/jvi.74.24.11654-11662.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Zigmond S H. How WASP regulates actin polymerization. J Cell Biol. 2000;150:F117–F120. doi: 10.1083/jcb.150.6.f117. [DOI] [PMC free article] [PubMed] [Google Scholar]