

Abstract
Characterization of the genetic regulation of proteins is essential for understanding disease etiology and developing therapies. We identified 10,674 genetic associations for 3,892 plasma proteins to create a cis-anchored gene-protein-disease map of 1,859 connections that highlights strong cross-disease biological convergence. This proteo-genomic map provides a framework to 1) connect etiologically related diseases, 2) provide biological context for new or emerging disorders, and 3) integrate different biological domains to establish mechanisms for known gene-disease links. Our results identify proteo-genomic connections within and between diseases and establish the value of cis-protein variants for annotation of likely causal disease genes at GWAS loci, addressing a major barrier for experimental validation and clinical translation of genetic discoveries.
One Sentence summary:
A genetically anchored map of protein – disease links identifies shared etiologies suggesting therapeutic directions.
Proteins are the central layer of information transfer from the genome to the phenome and recent studies have started to elucidate how natural sequence variation in the human genome impacts on protein concentrations measured from readily available biofluids such as blood (1–6). Investigation of the clinical consequences of these so-called protein-quantitative trait loci (pQTLs) can help to better understand disease mechanisms and provide insights into the shared genetic architecture across diseases within a translational framework that puts humans as the model organism at the center (2, 4). This approach is now pursued at scale by pharmaceutical companies for the discovery of drug targets or repurposing opportunities (7, 8). Earlier studies have started to characterize the genetic architecture of proteins using bespoke panels (3, 6, 9) or larger proteomic platforms (1, 2, 4, 5), and have demonstrated how this can provide insight into the pathogenesis of specific diseases. There has been less attention on: a) providing a framework to assess the protein specificity of genetic variation residing outside (trans) the protein encoding gene, b) understanding the clinical relevance of pQTLs for proteins detected in plasma but known to not be actively secreted (7), c) classifying thousands of proteins based on their genetic architecture as explained by cis variants, specific trans variants, or unspecific trans variants, d) demonstrating the specific utility of pQTLs for the prioritization of candidate genes at established risk loci, and e) systematically mapping shared gene-protein-disease signals to uncover connections among thousands of considered diseases and other phenotypes.
Profiling thousands of proteins circulating in blood at population-scale is currently only possible using large libraries of affinity reagents, namely antibodies or alternatively short oligonucleotides, called aptamers, since gold standard methods such as mass spectrometry lack throughput. We have previously provided a detailed comparison of 871 overlapping proteins measured in 485 individuals (10) using the two most comprehensive platforms, the aptamer-based SomaScan v4 assay and the antibody-based Olink proximity extension assay. We demonstrated that the majority of pQTLs are consistent across platforms (64%), in line with smaller scale efforts (4), but highlighted the need to triangulate pQTLs with gene expression and phenotypic information to derive tangible biological hypotheses. Here we present a genome-proteome-wide association study targeting 4,775 distinct proteins measured from plasma samples of 10,708 generally healthy European-descent individuals who were participants in the Fenland study (Table S1) (11). We identified 10,674 variant – protein associations and developed a framework to systematically identify protein- and pathway-specific pQTLs augmenting current ontology-based classifications in a data-driven manner. We show that half of all pQTLs close to the protein-encoding gene, cis-pQTLs, colocalize with gene expression or splicing QTLs in various tissues allowing us to derive functional insights within tissues by integrating genetics with plasma proteomics. We demonstrate the specific ability of cis-pQTLs to prioritize candidate causal genes at established genetic risk loci. By means of phenome-wide colocalization screens we generate a proteo-genomic map of human health covering 1,859 gene-protein-phenotype triplets providing insights into the shared etiology across diseases and the identification of pathophysiological pathways through cross-domain integration.
RESULTS
Genetic associations for protein targets
We performed a genome-proteome-wide association analysis by testing 10.2 million genotyped or imputed autosomal and X-chromosomal genetic variants with minor allele frequency (MAF) >1% among 10,708 participants in the Fenland study targeting 4,775 distinct proteins (12). We identified 2,584 genomic regions (1,543 within ±500 kb of the protein-encoding genes, cis) associated with at least one of 3,892 protein targets at p<1.004×10−11. 1,097 of these regions covered variants that have not been reported to be associated with plasma proteins so far (1–6, 9) (r2<0.1), of which 64% (867 out of 1,356 pQTLs) available in (4) replicated (p<0.05, directionally consistent). Further, 61% (488 out of 797, Table S2) of pQTLs replicated using the complementary Olink technique (12), with a higher proportion of replication for variants in cis (81.2%) compared to trans (44.2%). Most regions (79.3%, n=2,050) were associated with a single protein target, but we observed pleiotropy (≥2 protein targets) at the remaining regions, including association with up to five (16.1%, n=418), 6–20 (3.4%, n=88), or 21–50 (0.7%, n=19) associated protein targets, and substantial pleiotropy at eight regions (CFH, ARF4-ARHGEF3, C4A-CFB, BCHE, VTN, CFD, ABO, GCKR) associated with 59 to 1,539 protein targets (Fig. 1). The 194 pleiotropic regions harboring a cis-pQTL identified master regulators of the plasma proteome, including glycosyltransferases such as the histo-blood group ABO system transferase (ABO), key metabolic enzymes like glucokinase regulatory protein (GCKR), or lipid mediators such as apolipoprotein E, establishing a network-like structure of the circulating proteome (1).
Fig. 1. Regional sentinel genetic variants associated (p<1.004×10−11) with at least one protein target in up to 10,708 participants from the Fenland Study.
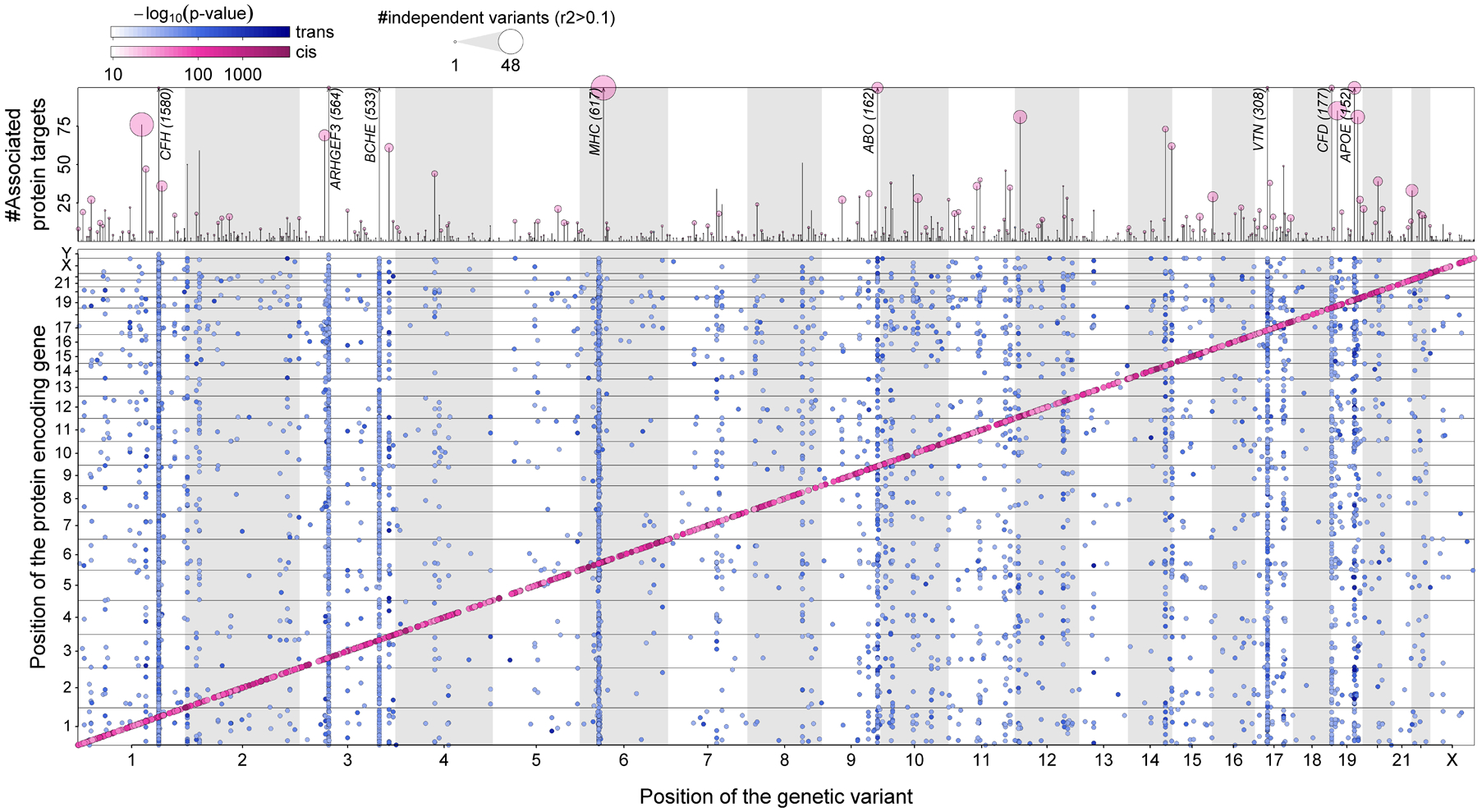
The lower panel maps the genomic locations of the genetic variants against the genomic locations of the protein-encoding genes. Genetic variants close to the protein-encoding gene (±500 kb) are highlighted in pink (cis-pQTLs) and all others are shown in blue (trans-pQTLs). Darker shades indicate more significant p-values. The upper panel shows the number of associated protein targets for each genomic region (vertical line), with circles above representing the number of approximately independent genetic variants (r2<0.1), such that larger circles indicate more genetic variants in the region.
Out of the 3,892 protein targets, 26.8% (n=1,046) had pQTLs in both cis and trans, 13.4% (n=523) in cis only, and 59.6% (n=2,323) in trans only, among a total of 8,328 sentinel variant-protein target associations (Fig. 1 and Tables S2 and S3). We identified another 2,346 secondary pQTLs at those loci using an adapted stepwise conditional analysis (median: 1, range: 1 – 13) indicating widespread allelic heterogeneity in cis (68.8%) and trans (31.2%). The majority of the 5,442 distinct variants were located in introns (~44%) or were in high LD (r2>0.6) with a missense variant (~21%), with similar distributions across cis- and trans-pQTLs (Fig. S1). We observed 663 cis-pQTLs with direct consequences for the structure of the protein target (protein-altering variants, PAVs), including important substructures, such as disulfide bonds (4.2%), α-helices (3.1%), and β-strands (2.6%) (Fig. S1). Such variants are predicted to affect correct folding of protein targets, including diminished secretion or reduced half-life in the bloodstream, rather than expression of the protein-encoding gene (13). For example, we observed an enrichment of PAVs among actively secreted proteins (14) (39.6% vs 33.7%, p=0.04, X2-test) possibly indicating modulation of common posttranslational modifications, such as glycosylation.
An integrated classification system for pathway-specific pQTLs
We integrated a data-driven protein network with ontology mapping (GO terms, Fig. 2A–B and S2) to distinguish pathway-specific pQTLs from those exerting effects on multiple unrelated targets (12, 15). We successfully assigned 40.8% (n=1,790 in cis, n=423 in trans) of the 5,442 genetic variants as protein-specific and 5.9% (n=236 in cis, n=86 in trans) as pathway-specific based on converging evidence from the network and ontology mapping, and another 16.5% (n=498 in cis, n=402 in trans) to be likely pathway-specific based on either source. In total, 1,802 protein targets had at least one (likely) specific pQTL in cis (n=1,385) or trans (n=417). We classified 648 variants that would have been missed by ontology mapping as protein community-specific through our data-driven network approach. One example is rs738408 (PNPLA3), a non-alcoholic fatty liver disease variant (16) which was associated with 22 out of 70 aptamers from the same protein community (Fig. 2C). PNPLA3 encodes patatin-like phospholipase domain-containing protein 3 (PNPLA3), and rs738408 tags the missense variant rs738409 (I148M) rendering PNPLA3 resistant to ubiquitylation-mediated degradation and resulting in subsequent accumulation on hepatic lipid droplets causing fatty liver disease (17). The associated protein targets included multiple metabolic and detoxification enzymes highly expressed in the liver, such as alcohol dehydrogenases, arginosuccinate lyase, bile salt sulfotransferase, or aminoacylase-1. Our results support the hypothesis that those might only appear in plasma of otherwise healthy individuals as a result of lipid overload-induced lysis of hepatocytes. The putative liver damage-specific effect, anchored on the PNPLA3 trans-pQTL, makes those protein targets potential biomarker candidates compared to tissue unspecific proteins currently used to identify fatty liver disease or liver injury in the clinic (18).
Fig. 2. Classification of protein quantitative trait loci (pQTLs, cis and trans) and subsequent partition of the explained variance in plasma abundances of protein targets.
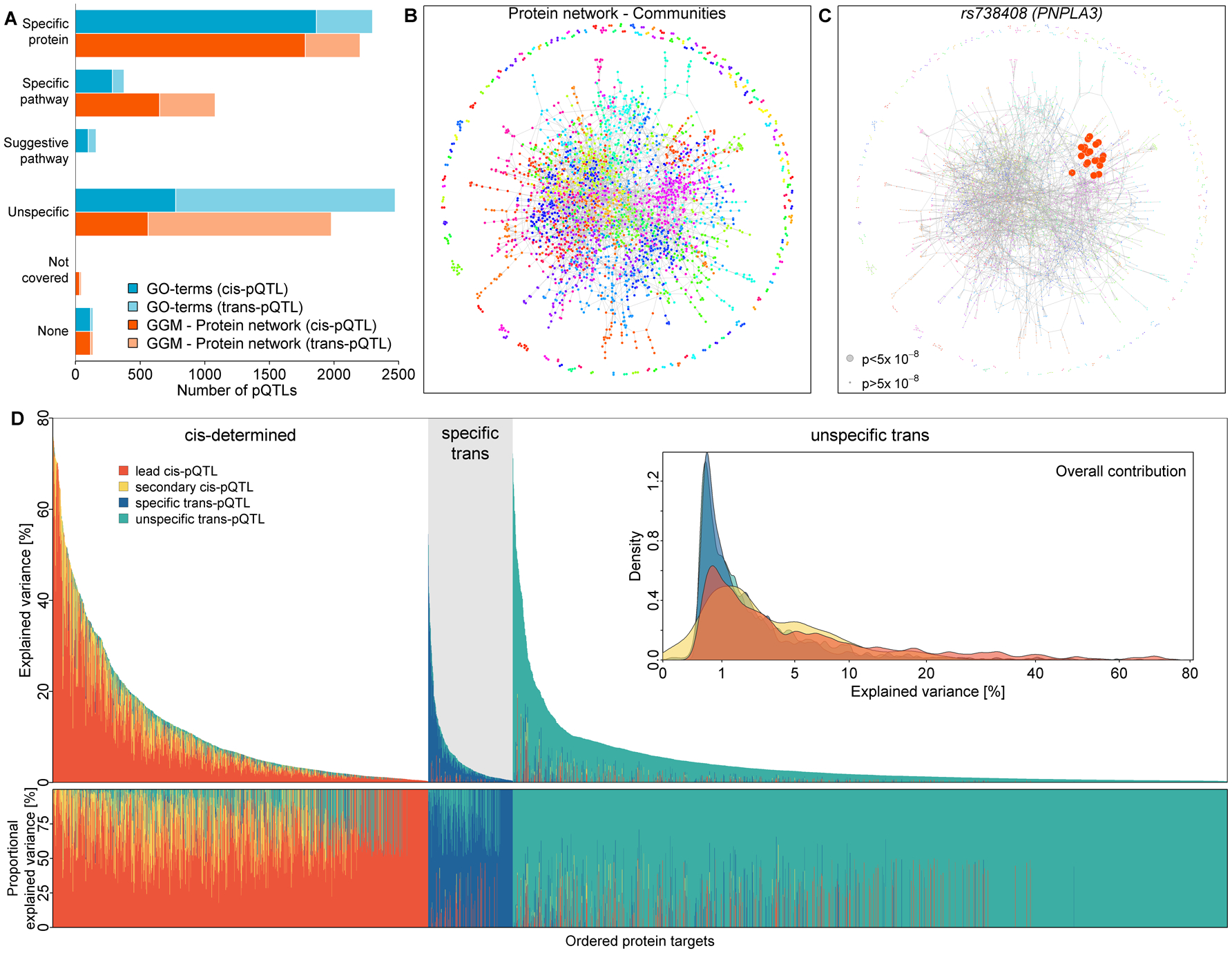
A) Bar chart of pQTL classification based on GO term mapping (blue) or community mapping in a protein network derived by Gaussian graphical modeling (GGM; orange) of associated protein targets. Darker shades indicate cis-pQTLs and lighter colors trans-pQTLs. B) Data-driven protein network colored according to 191 identified protein communities. C) a community-specific pQTL (PNPLA3) that was not captured by GO term mapping. Gene annotation as reported in the Materials and Methods. D) Absolute (upper panel) and relative (lower panel) explained variance in plasma abundances of protein targets by identified pQTLs. Coloring indicates contribution of the lead cis-pQTL (orange), secondary cis-pQTLs (yellow), protein- or pathway-specific trans-pQTLs (blue), and unspecific trans-pQTLs (green). Protein targets have been grouped by underlying genetic architecture as: mostly explained by cis-pQTLs (‘cis-determined’), mostly explained by specific trans-pQTLs (‘specific trans’), and mostly explained by unspecific trans-pQTLs (‘unspecific trans’). The inset displays the overall distribution of explained variance by each of the four categories. The variance explained was computed using linear regression models. A graphical display of effect size distributions can be found in Fig. S3.
Contribution of cis and trans genetic architecture
We observed three major categories of protein targets based on the contribution of genetic variation to plasma concentrations (Fig. 2D and S3, and Table S3). For about a third (n=1,249) of the protein targets, genetic variance was mainly (>50% of genetic variance) explained by one or more cis-pQTLs, whereas for 7.2% (n=282) protein- or pathway-specific trans-pQTLs accounted for most of the genetic variation, leaving two-thirds (n=2,361) mainly explained by unspecific trans-pQTLs (12). Overall, we observed a median genetic contribution of 2.7% (IQR: 1.0% - 7.6%) reaching values above 70% for proteins like vitronectin (rs704, MAF=47.3%) or sialic acid-binding Ig-like lectin 9 (rs2075803, MAF=44.1%) which were often driven by only a single common cis-pQTL. PAVs, affecting the binding epitope of the protein target, are the likely explanation for such strong and isolated genetic effects. While more than two-thirds of the protein targets with at least one cis-pQTL were unrelated to PAVs, we provide evidence that 158 (32.9%) of the protein targets linked to a PAV (r2>0.6) shared a genetic signal with at least one disease or risk factor (see below). This suggests that conformation and possibly function of the protein target, rather than plasma abundance of the protein target, might be more relevant as mediators of downstream phenotypic consequences and that aptamers are able to detect such probably dysfunctional proteins.
Our approach to identify protein-/pathway-specific trans-pQTLs allowed us to uncover biologically relevant information, which was otherwise hidden by strong and unspecific trans-pQTLs that possibly interfere with the measurement technique rather than the biology of the protein target. For example, rs704, a missense variant within VTN associated with a higher fraction of single chain vitronectin with altered binding properties (19, 20), explained 72% of the variance in MICOS complex subunit MIC10 (MOS1), far outperforming the contribution of the specific trans-pQTL rs398041972 (0.7%). rs398041972 resides about 1 Mb upstream of TMEM11, encoding transmembrane protein 11, a physical interaction partner of MOS1 as part of the MICOS complex (21). In general, we observed that the median contribution of specific trans-pQTLs to the variance in plasma concentrations was 1.1% (IQR: 0.6%−2.6%) across 687 protein targets, reaching values as high as 38.3% for catenin β−1 via two trans-pQTLs (rs1392446 and rs35024584) within the same region for which we prioritized CDH6 as a candidate causal gene. CDH6 encodes cadherin 6, which physically interacts with catenin β−1 (22). We systematically tested for an enrichment of putative protein interaction partners among the 20 closest genes at each specific trans locus and observed a 1.53-fold enrichment (Chi-square test p-value=1.8×10−10) of first- and second-degree neighbors from the STRING network (23), highlighting the ability of our classification system to identify biologically meaningful trans-pQTLs.
Shared genetic architecture with gene expression and splicing
We integrated plasma pQTL results with both gene expression and splicing QTL data (eQTL and sQTL, respectively) from the GTEx version 8 release (24) using statistical colocalization (posterior probability (PP) > 80%) for all 1,584 protein targets with at least one cis-pQTL (12). There was strong evidence that half (50.1%) of these had a shared signal with gene expression in at least one and a median of 4.5 tissues (IQR: 2–12; Fig. 3A), vastly expanding previous knowledge of gene expression contribution across tissues (4, 9). The majority of cis-pQTLs (n=584, 73.4%) showed plasma protein and gene expression effects in the same direction in all tissues (Fig. 3A), but 26.6% (n=212) showed evidence of at least one pair with opposite effects, including 108 where the protein effect was opposite to the direction observed for gene expression across all tissues with evidence for colocalization. For example, the A-allele of the lead cis-pQTL rs2295621 for immunoglobulin superfamily member 8 (IGSF8) was inversely associated with plasma abundance of the protein target (beta=−0.19, p<1.65×10−32) but positively associated with expression of the corresponding mRNA across 33 tissues (Table S4). Uncoupling of gene and protein expression, even within the same cell, is a frequently described phenomenon, and possible mechanisms include differential translation, protein degradation, contextual confounders, such as time and developmental state, or protein-level buffering (25). For 145 protein targets, we identified strong evidence of a tissue-specific contribution to plasma abundances based on a single tissue strongly outweighing all others (Fig. 3A and Table S4). These included known tissue-specific examples such as Vitamin K-dependent protein C in liver tissue, but also less obvious ones, such as hepatitis A virus cellular receptor 1 (or TIM-1), an entry receptor for multiple human viruses, for which the cis-pQTL and cis-eQTL specifically colocalized in tissue from the transverse colon. To maximize power for the most closely aligned tissue compartment, whole blood, we integrated gene expression data from the eQTLGen consortium (26), which confirmed 140 cis-eQTL/pQTL pairs and revealed another 38 cis-eQTL/pQTL pairs not seen in the GTEx resource, including immune cell-specific mediators of the inflammatory response such as leukocyte immunoglobulin-like receptor subfamily A member 3 (Table S4).
Fig. 3. Integration of gene and splicing quantitative trait loci (eQTLs and sQTLs).
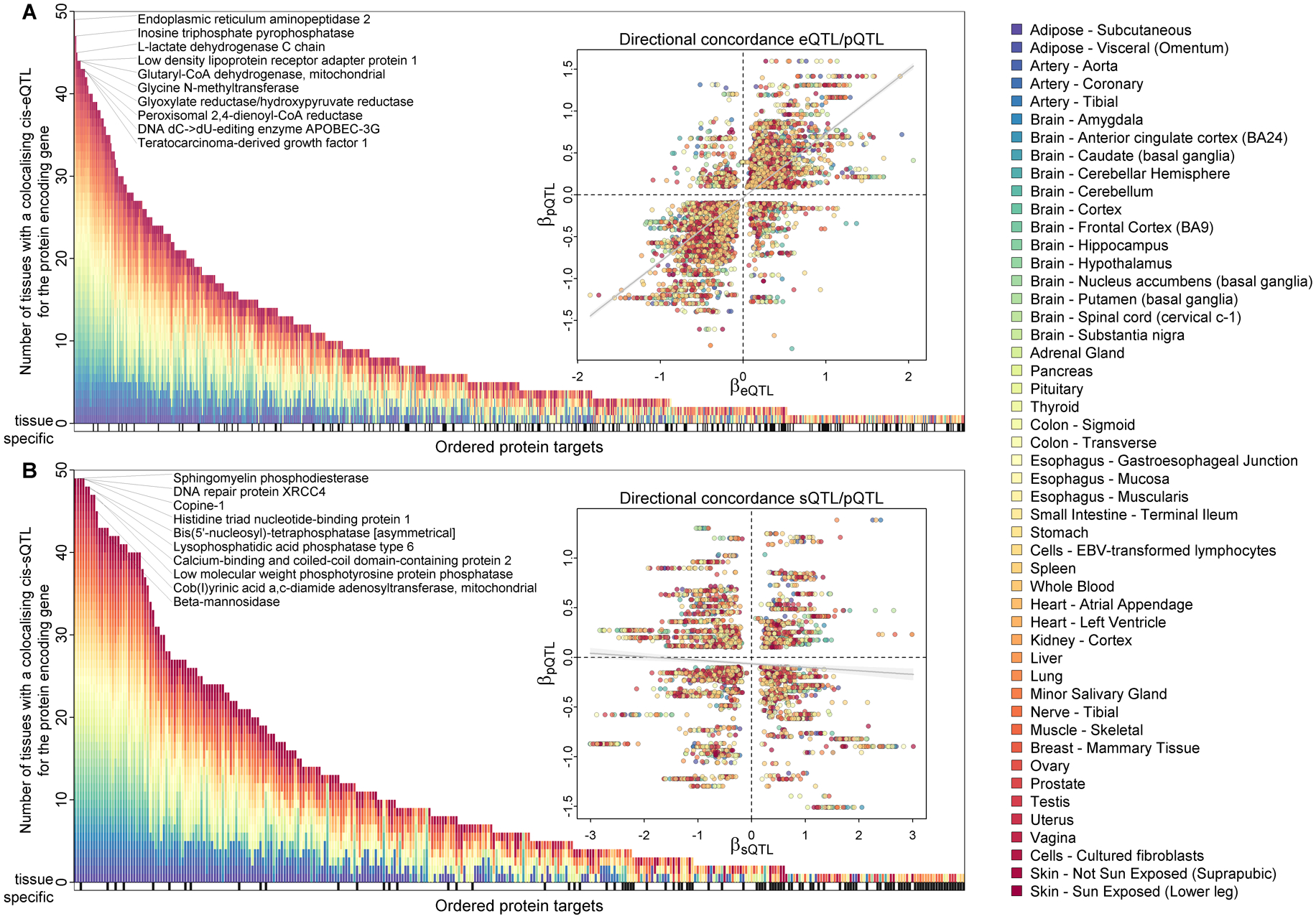
A) Protein targets ordered by the number of tissues for which at least one of the cis-pQTLs was also a cis-eQTL as determined by statistical colocalization (posterior probability >80% for a shared signal). Protein targets for which the eQTL showed evidence for a tissue-specific effect are indicated by black vertical lines underneath. B) Same as A) but considering cis-sQTLs.
To obtain insights beyond the average readout across all transcript species, we examined alternative splicing as a source of protein target variation (12). One-fifth (20.1%) of cis-signals were shared with a cis-sQTL in at least one tissue (median: 6 tissues, IQR: 2–15) (Fig. 3B), and 84 of these were not seen with eQTL data, suggesting that the pQTL-relevant transcript isoform was masked from the bulk of assayed transcripts. In contrast to the eQTL colocalization, we did not observe an overall pattern of aligning effect directions (Fig. 3B). This might be best explained by the intron-usage quantification of splicing events within GTEx version 8, which does not allow straightforward mapping of the eventually transcribed isoforms, and the expression of an alternative protein isoform with less affinity to the SOMAmer reagent. The latter may have accounted for the 90 protein target examples where the colocalizing cis-sQTL explained more than 10% of the variance in plasma concentrations (Table S4) and emphasizes the ability of splicing QTLs to determine the underlying sources of variation in plasma abundances of protein targets. In summary, our results demonstrate that proteins measured in plasma can be used as proxies for tissue processes when anchored on a shared genetic variation with tissue-specific gene expression or alternative splicing data.
cis-pQTLs enable identification of candidate causal genes at GWAS loci
We used the inherent biological specificity of cis-pQTLs to systematically identify candidate causal genes for genome-wide significant variants reported in the GWAS catalog (p<5×10−8; download: 25/01/2021) by assessing 558 cis-regions for which the pQTL was in strong LD (r2>0.8) with at least one variant for 537 collated traits and diseases (Fig. 4 and Table S5) (12). For a quarter of these (24.6%), we annotated a gene different from the reported or mapped gene, and for another 79 cis-regions (14.2%), our predicted causal gene was reported as part of a longer list of potential causal genes.
Fig. 4. Causal gene assignment for associations reported in the GWAS catalog using identified cis-pQTLs.
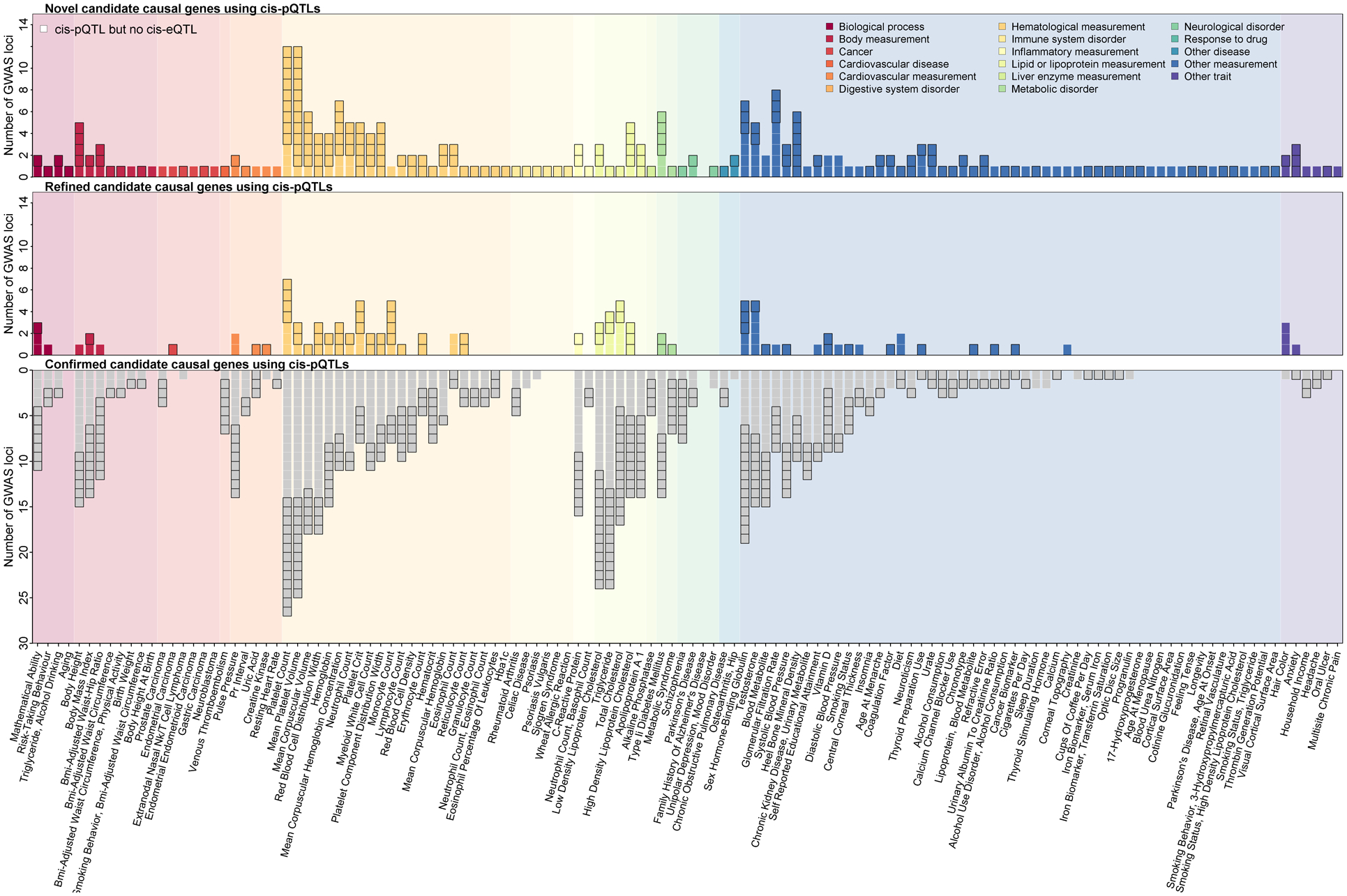
Each panel displays the number of loci that have been reported in the GWAS catalog for a curated phenotype and were identified as protein quantitative trait in close proximity (±500 kb) to the protein-encoding gene (cis-pQTL) in the current study. Mapping of GWAS loci and cis-pQTLs was done using the LD between the reported variants (r2>0.8). The upper panel displays the number of GWAS loci for which cis-pQTLs provided candidate causal genes. The middle panel displays the number of GWAS loci for which cis-pQTLs refined the list of candidate causal genes at the locus. The lower panel displays the number of GWAS loci with confirmative evidence from cis-pQTLs for already assigned candidate causal genes. Examples where gene prioritization was facilitated through pQTL but not gene expression QTL (eQTL) evidence are highlighted by a border around the box. Colors represent broad trait categories.
Among the genes we identified are candidates with strong biological plausibility, such as AGRP, encoding Agouti-related protein, a neuropeptide involved in appetite regulation (27), suggesting a possible mechanism for measures of body fat distribution associated at this locus. Another example was NSF, encoding N-ethylmaleimide-sensitive factor (NSF), which may be involved in the fusion of vesicles with membranes, enabling the release of neurotransmitters into the extracellular space (28); a locus that was previously identified for Parkinson’s disease (Table S5).
We further assigned PRSS8 as a candidate causal gene at the KAT8 locus for Alzheimer’s disease (AD), supported by strong LD (r2=0.96) and a high posterior probability of a shared genetic signal (98%) between the lead cis-pQTL (rs368991827, MAF=27.8%) and the common KAT8 intronic variant (rs59735493) that has been reported for AD (Fig. S4). PRSS8 codes for prostasin, and we estimated a 13% reduction in AD risk (odds ratio: 0.87; 95%-CI: 0.82–0.91, p=3.8×10−8) for each 1 s.d. higher normalized plasma abundance of prostasin. The locus has been identified by multiple GWAS efforts (29), yet prioritization strategies have failed to provide conclusive evidence for a causal gene (30). Prostasin is a serine protease highly expressed in epithelial tissue, which regulates sodium channels (31) and represses TLR4-mediated inflammation in human and mouse models of inflammatory bowel disease (32), a mechanism which might also be relevant to TLR4-mediated neuroinflammation in AD (33).
We observed multiple examples in which our cis-pQTL mapping identified biologically plausible candidate genes that were not implicated by cis-eQTL mapping (Fig. 4). For example, we assigned RSPO1 as a candidate causal gene at the eQTL-supported CDCA8 locus for endometrial cancer (34). The intergenic variant rs113998067 is the lead signal for endometrial cancer and was a secondary cis-pQTL for R-spondin-1, encoded by RSPO1. Statistical colocalization confirmed a highly likely shared signal (PP=98.2%) (Fig. S5). Accordingly, we estimated a 91% increased risk for endometrial cancer per 1 s.d. higher plasma abundance of R-spondin-1 (odds ratio: 1.91, 95%-CI: (1.52–2.41), p-value=3.6×10−8). R-spondin-1 is a secreted activator protein which acts as an agonist for the canonical Wnt signaling pathway (35), playing a regulatory role as an adult stem cell growth factor. Work in mouse models (36), however, suggests that R-spondin-1 upregulates estrogen receptor alpha independent of Wnt/β-catenin signaling and might therefore amplify estrogen-mediated endometrial cancer risk (36). We note that the effect estimate for rs113998067 did not differ by sex (p=0.12), and knockout models in male and female mice have shown abnormal development of testis and ovary, respectively (37, 38), possibly indicating a wider impact on diseases of reproductive tissues.
A map of proteo-genomic connections across the phenome
We systematically assessed sharedness of gene-protein-disease triplets through phenome-wide colocalization of cis-pQTL regions (12) to identify and create a genetically anchored map of proteins involved in the etiology of common complex diseases, which could represent potential druggable targets. We identified 1,859 gene-protein-trait triplets (network edges, Fig. 5 and S6) comprising 412 protein targets and 506 curated phenotypes (Fig. S7 and Table S6). The mapping of these shared gene-protein-phenome connections highlights a large number of insights, as discussed below, while confirming previously established connections for known pleiotropic loci (for example GCKR (n=197 traits), alpha-1-antitrypsin (n=79 traits), or apolipoprotein A-V (n=64 traits)) and established disease genes (for example roto-oncogene tyrosine protein kinase receptor RET (RET) and Hirschsprung’s disease (39) or C-C motif chemokine 21 (CCL21) and rheumatoid arthritis (40)).
Fig. 5. Network representation of phenome-wide colocalization analysis for protein-encoding loci.
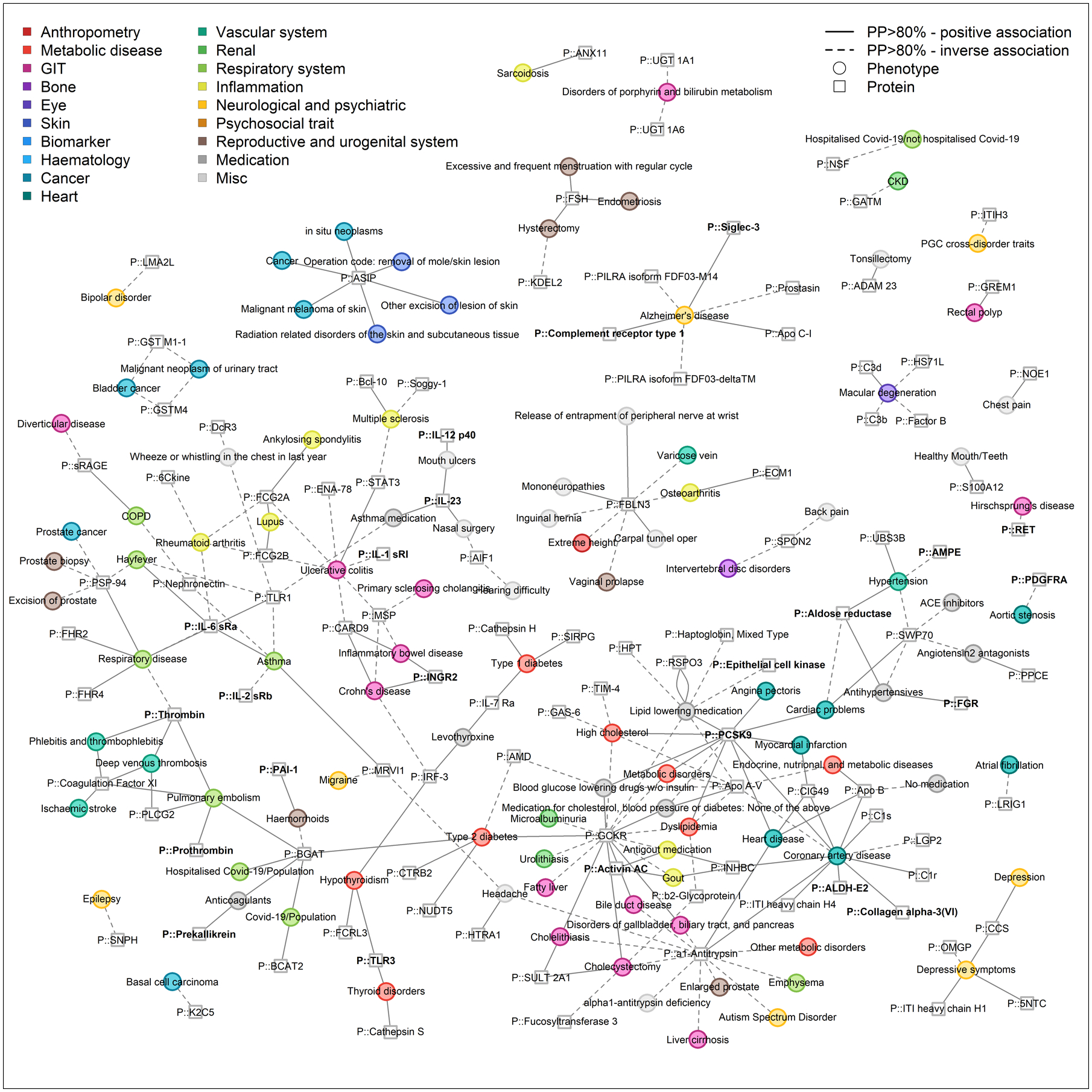
This figure is restricted to connections between proteins and binary endpoints, mainly diseases, to increase visibility and show shared etiology. Only protein targets and phenotypes with at least one connection are included. Effect directions are indicated by the line type (solid = higher protein abundance, increased risk, dashed = higher protein abundance, reduced risk). Colors indicate categories of phenotypes. The entire network is composed of 412 protein targets (squares) and 506 phenotypes (circles) as nodes, which are connected (n=1,859 edges) if there is evidence of a shared genetic signal (posterior probability >80%) and is shown in Fig. S6. An interactive version of the figure can be found at www.omicscience.org/apps/pgwas.
The map highlights ten diseases for which we identified five or more colocalizing cis-pQTLs, including coronary artery disease (n=12), hyperlipidemia indicated by lipid-lowering medication (n=8), ulcerative colitis (n=7), Alzheimer’s disease (n=6), and type 2 diabetes (n=5). Statistical power was greatest for the detection of shared genetic architecture for traits for which measures were available in the largest number of people, in line with a median of 2 colocalizing cis-pQTLs (IQR: 2 – 4, maximum 32 for mean platelet volume) for blood cell parameters and biomarkers available in large-scale biobanks. For 104 out of 191 curated phenotypes with at least 3 colocalizing protein targets, we observed significant enrichment of pathways (false discovery rate (q-value) < 5%; Table S7). These reflected known biology of the corresponding clinical entities, such as ‘wound healing’ for platelet count, ‘skeletal system development’ for height, ‘cholesterol metabolism’ for coronary artery disease, or ‘response to virus’ for Crohn’s disease, as well as yet less understood ones such as ‘toll-like receptor signaling’ for hypothyroidism, for which two of the genes (IRF3 and TLR3) have already been shown to confer virus-induced disease onset in mouse models (41).
The proteo-genomic map provides a new framework to 1) connect etiologically related diseases, 2) provide biological context for new or emerging disorders, such as COVID-19, and 3) integrate information from different biological domains to establish mechanisms for known gene-disease links. For each of these scenarios, we provide selected examples to highlight the scientific opportunities arising from this map below and on the related open resource platform (www.omicscience.org).
Potential candidate genes for COVID-19 outcomes
We integrated GWAS summary statistics in our map for four different outcome definitions related to COVID-19, ranging from susceptibility to COVID-19 to severe cases requiring hospitalization (42). These GWAS differed substantially in the number of included cases (5,101 – 38,984), and we observed that results were sensitive to the choice of outcome. We replicated the previously reported candidate genes ABO and OAS1 (43) (Fig. S8), both of which showed consistent evidence across these different outcome definitions. For ABO, the lead cis-pQTL (rs576125, MAF=33.5%) also colocalized with pulmonary embolism (Fig. 5), a common complication of severe COVID-19 (44), potentially attributable to altered abundances of proteins involved in the coagulation cascade (15). We further observed suggestive evidence for NSF (for the risk of COVID-19 hospitalization) and BCAT2 (for severe COVID-19), each of which shared a genetic signal with only one of these four outcomes, and therefore require external validation of their possible role in COVID-19 or associated pathologies.
Integrating multiple OMICs layers elucidates a disease mechanism for gallstones
We identified a signal at SULT2A1, a known gallstone locus (45), to be shared between bile salt sulfotransferase (SULT2A1) and the risk of cholelithiasis (odds ratio per 1 s.d. higher normalized protein abundance: 2.12, 95%-CI: 1.66 – 2.70, p-value=2.1×10−37) as well as cholecystectomy (odds ratio: 2.09, 95%-CI: 1.86 – 2.34, p-value=7.8×10−38). Multi-trait colocalization (46) further identified that the signal was also shared with mRNA expression of SULT2A1 in the liver, plasma concentrations of multiple sulfated steroids (47), including sulfate conjugates of androgen and pregnenolone metabolites, and bile acids. The high posterior probability (PP=99%) was largely explained (63%) by rs212100, a variant in high LD (r2 = 0.90) with the lead cis-pQTL at this locus (Fig. 6A and Fig. S9). The consistent positive effect directions across all physiological entities, and in particular sulfated steroids and primary bile acid metabolites, suggest higher SULT2A1 activity as the mode of action. The concurrent inverse association with lower plasma concentrations of the secondary bile acid glycholithocholate indicates diminished formation of lithocholic acid, an essential detergent to solubilize fats, including cholesterol (48). Our vertical integration of diverse biology entities points to a supersaturated bile that promotes cholesterol crystallization and gallstone formation as a causal mechanism at a locus for which the mode of action has only been vaguely hypothesized (45).
Fig. 6. Selected phenotypic examples from the proteogenomic map.
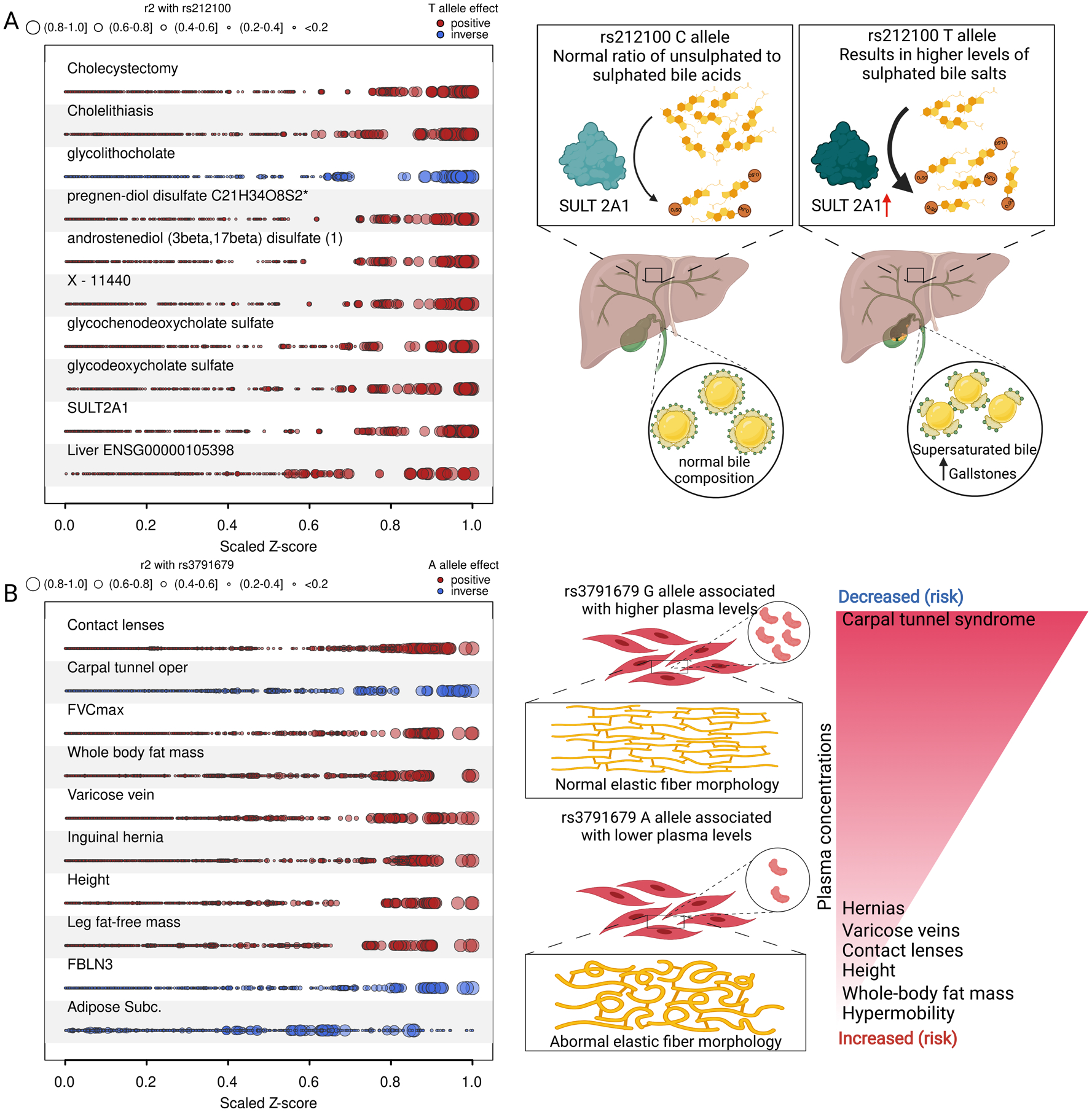
A) Plot visualizing convergence of genetic variants at the SULT2A1 locus in relation to the LD with the candidate gene variant identified by multi-trait colocalization. Z-scores from GWAS for each annotated trait have been scaled by the absolute maximum, and dot size is proportional to the LD (r2). Colors indicate the direction of effect aligned to the risk-increasing allele (red – positive, blue - inverse). The scheme on the right depicts the suggested mode of action by which higher SULT2A1 activity translates to higher risk of gallstones. B) Same as A, but for diseases and other phenotypes colocalizing at the EFEMP1 locus. The scheme on the right depicts a proposed mechanism by which altered secretion of FBLN3 leads to the observed phenotypes. Stacked regional association plots for A and B can be found in Figs. S9 and S10.
Convergence of soft tissue disorders through FBLN3
A protein target connected to a very large number (n=37) of diseases and other phenotypes was FBLN3 (extracellular matrix glycoprotein encoded by EFEMP1), which showed gene-protein convergence of diverse connective tissue disorders as well as gene expression of EFEMP1 in subcutaneous adipose tissue, with high confidence in the lead cis-pQTL (rs3791679, MAF=23.4%) being the causal variant in multi-trait colocalization (Fig. 6B and Fig. S10). The common A-allele of rs3791679 was associated with lower plasma abundance of FBLN3 and increased risk for a range of connective or soft tissue abnormalities, including hernias, varicose veins, vaginal prolapse, and hypermobility, several of which have previously been reported in individual GWAS but have not been connected (49–54). This spectrum of human clinical features suggests that lower plasma levels of A-allele carriers results in altered elastic fiber morphology and/or lower content, in line with evidence from Efemp1 knock-out mice that display abnormal elastic fiber morphology, develop different types of hernias, and pelvic organ prolapse (55). FBLN3 is part of the extracellular matrix and widely expressed but its function is incompletely understood (56). We provide insights about its role in the etiology of a large number of connective tissue disorders, including a potential explanation for the established link between carpal tunnel syndrome and shorter stature (51). Mutations in EFEMP1 cause a rare eye disease called Doyne honeycomb retinal dystrophy (DHRD) (57), characterized by visual disturbances and drusenoid deposits due to accumulating intracellular FBLN3. We observed sharedness of the signal at this protein locus with vision-related phenotypes, including use of contact lenses (myopia) and decreased optic disc area, a risk factor for open-angle glaucoma (50), with lower protein concentrations associated with greater risk, as also observed in patients with DHRD.
Differential effect sizes of cis-pQTLs by sex and age
We systematically tested differences in the genetic associations of all protein targets included in the proteogenomic map (N=412) by age or sex. We identified a total of 14 protein targets that showed evidence for significant (p<5.9×10−5) effect modification of the cis-pQTL by sex (N=10) or age (N=8), including four common to both (Table S8). This included biologically plausible candidates, such as annexin II, where the cis-pQTL showed a stronger effect in women, albeit with a strong significant effect in either sex (women: beta=−0.86, p-value<1.7×10−467; men: beta=−0.64, p-value<2.5×10−231). This finding is in line with evidence of isoform expression of the protein-encoding gene ANXA2 in male and female reproductive tissues, including prostate (PP=81.9%) and vagina (PP=87.4%) and a possible role of the locus in puberty timing (58, 59).
We noted that most of the identified cis-pQTLs showed age- and sex-differential and not dimorphic effects (60) and were linked to missense variation (inhibin C, vitronectin, Siglec 9, GCKR, SOD3, CPA4, and PILRA) or alternative splicing events (annexin II, BGAT, and CO8G) with very strong overall effects, enabling the detection of even small effect differences between strata more easily (61). In general, our results are concordant with the few sex-specific effects of molecular QTLs reported so far (62, 63) and show that systematic efforts for both molecular QTLs and disease GWAS are needed to better understand the mechanisms underlying such differences. Crucially, investigating the relevance of these genetic differences for phenotypic expression depends on the availability of sex-specific GWAS results across the human phenome.
Druggable targets and repurposing opportunities
We systematically identified druggable proteins in the proteo-genomic map by linking the protein-encoding gene to the druggable genome (64) and identified 60 protein targets linked to at least one phenotype, including 22 protein targets linked to a disease (Table S9). We replicate established examples, such as the IL-6 receptor for rheumatoid arthritis or thrombin for deep venous thrombosis (Fig. 5). We also identified 31 candidates with potential repurposing opportunities for 1 to 8 diseases (for a total of 32 different indications), following a search and prioritization strategy in Open Targets (65).
Webserver
To enable customized and in-depth exploration of high-priority protein targets, that is, those with at least one cis-pQTL, we created an interactive online resource (www.omiscience.org/apps/pgwas). The webserver provides intuitive representations of genetic findings and enables the look-up of summary statistics for individual SNPs, genes, and whole genomic regions across all protein targets. To interactively assess specificity and identify pleotropic cis-pQTLs that present strong trans-like association profiles, we generated an interactive heatmap of genetic associations of all cis-pQTLs across all high-priority candidate proteins. We further provide detailed annotations of the protein targets, including links to external databases, such as UniProt or Reactome, information on currently available drugs, characterization of associated SNPs, as well as results from our colocalization analysis with eQTLs, sQTLs, diseases, and other phenotypes. An interactive version of the proteo-genomic map allows a deep dive into proteins or phenotypes of particular interest to explore cross-disease connections within subnetworks.
DISCUSSION
The promise of proteomic technologies and their integration with genomic data lies in their application to rare and common human diseases. While previous studies started to exploit the phenotypic consequences of pQTLs, they have mainly focused on identifying and describing the genetic architecture of proteins measured by specific platforms (1–6, 9). We performed a systematic integration of the phenome and created a proteo-genomic map of human health that identifies many potential causal disease genes and highlights genetically driven connections across diverse human conditions. The traditional classification of diseases relies on the aggregation of symptoms commonly presenting together and, with the exception of Mendelian disorders, is rarely based on shared etiology (66). Our network anchors the convergence of diseases in their shared genetic etiology, as shown for FBLN3, providing mechanistic understanding and a starting point for the identification of treatment strategies targeting underlying genetic causes.
Uncertainty in assigning causal genes and variants remains a major limitation for experimental validation and clinical translation of results from the plethora of hypothesis-free genetic association studies. We show how cis-pQTLs identify causal candidate genes at established disease risk loci, including COVID-19, providing immediate hypotheses for experimental follow-up for a large number of disease genes.
The uncertain specificity of genetic variation affecting protein content outside of the protein-encoding region, trans-pQTLs, restricts the discovery of de novo biological insights in protein regulation and instrumentation of such variants for genetic prediction, such as with polygenic scores. We show how data-driven network clustering augments ontology-based classification approaches and identifies biologically plausible examples, such as for PNLPA3 and a community of liver-derived protein targets.
Genetic variation found for proteins circulating in blood raises the question of transferability to disease-relevant tissue processes. We demonstrate that for about half of the protein targets with a cis-pQTL, this can be linked to gene expression in various tissues and provide examples, such as for SULT2A1, that illustrate how multi-domain integration can identify tissue-specific mechanism. In its most simple form such cis-pQTLs determine the basal rate of protein production within cells and are more or less constantly released into plasma due to natural cell turnover (67). Integration of genetic information allowed us to separate out such enclosed effects from other mechanisms leading to higher cell turn over or leakage, such as for SULT2A1 and the liver-specific effect of the PNPLA3 variant. While this provides a strategy to point to relevant tissues, overlapping data for tissue-specific gene and protein expression is required to quantify the contribution of various tissues to the plasma proteome.
To accelerate use and translational potential of our findings, we generated an open access interactive web resource that enables the scientific community to easily and rapidly capitalize on these results for future research across clinical specialties. We demonstrate for multiple examples how this resource can be used to put gene-phenotype findings into a systems biological context.
While our study is distinguished by its comprehensive discovery and characterization of pQTLs in cis and trans along with a systematic integration of the phenome, it does have limitations. Firstly, the nature of the technology used to measure protein concentrations is designed to maximize discovery by generating a large library of affinity reagents, which rely on a preserved shape of the target protein and hence might miss genetic effects specific to a particular isoform of the protein (10). The semi-quantitative nature of the assay makes risk estimates based on Mendelian randomization studies challenging. A thorough discussion of assay differences can be found in our previous work (10), and we observed consistent cis-pQTLs for the highlighted examples, including RSPO1, SULT2A1, and FBLN3, as measured with Olink. Secondly, our study cohort consisted of predominantly healthy middle-aged participants of European-descent and replication of our results in ethnically diverse populations is warranted, in particular for the discovery of drug targets. Further work would also be required to investigate possible modifying effects of phenotypic characteristics on gene – protein associations, such as by sex, age, or behavioral factors. Thirdly, our study concentrated on the common spectrum of variation in the genome. Investigation of rare variation is likely to identify pQTLs with larger effect sizes and possibly more severe phenotypic consequences. Finally, our proteo-genomic map is limited to publicly available GWAS summary statistics and inclusion of further data for additional phenotypes, in particular cancers, and understudied diseases, will provide additional insights.
MATERIALS and METHODS
Detailed materials and methods are provided in the supplementary materials (12). We performed a genome-proteome-wide association study among 10,708 participants of European-descent in the Fenland study (Table S1) on 10.2 million genetic variants and plasma abundances of 4,775 distinct protein targets measured in plasma using established workflows (15). Protein targets were measured using the SomaScan v4 assay employing 4,979 single-stranded oligonucleotides (aptamers) with specific binding affinities to 4,775 unique protein targets (68, 69). We used the term ‘protein target’ to refer to proteins targeted by at least one aptamer. We define significant genetic variant – protein target associations (pQTLs) at a stringent Bonferroni-threshold (p<1.004×10−11) and performed approximate conditional analysis to detect secondary signals for each genomic region identified by distance-based clumping of association statistics. We defined cross-aptamer regions using a combined approach of multi-trait colocalization (46) and LD-clumping. We classified pQTLs as protein- or pathway-specific by assessing pQTL-specificity across the entire proteome (p<5×10−8) while testing whether associated protein targets were captured by a common GO term or a protein community in a data-driven protein network. We computed the variance explained in plasma abundances of protein targets by cis- (within ±500kb of the protein-encoding gene) or trans-pQTLs according to different specificity categories using linear regression models. We used statistical colocalization (70) to test for a shared genetic signal between expression or alternative splicing of the protein-encoding gene and the cis-pQTL in one out of at least 49 tissues of the GTEx v8 project (24). We systematically cross-referenced established genetic risk loci for common complex diseases and phenotypes with pQTLs by identifying cis-pQTLs or strong proxies (r2>0.8) in the GWAS catalog (https://www.ebi.ac.uk/gwas/). We finally performed phenome-wide colocalization screens at 1,548 protein-encoding loci using publicly available (71) as well as in-house curated genome-wide association statistics for thousands of phenotypes. We applied stringent priors and conservative filters to derive high confidence protein – phenotype links. We used basic functions of R (v.3.6.0), the R package igraph, and the BioRender web application (https://biorender.com/) to create figures. The Fenland study was approved by the National Health Service (NHS) Health Research Authority Research Ethics Committee (NRES Committee – East of England Cambridge Central, ref. 04/Q0108/19), and all participants provided written informed consent.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to all Fenland volunteers and to the General Practitioners and practice staff for assistance with recruitment. We thank the Fenland Study Investigators, Fenland Study Co-ordination team and the Epidemiology Field, Data and Laboratory teams. Proteomic measurements were supported and governed by a collaboration agreement between the University of Cambridge and SomaLogic. This research has been conducted using the UK Biobank Resource (application no. 20361 and 44448).
FUNDING
The Fenland Study (10.22025/2017.10.101.00001) is funded by the Medical Research Council (MC_UU_12015/1). We further acknowledge support for genomics from the Medical Research Council (MC_PC_13046). ERG is supported by the National Institutes of Health (NIH) Awards R35HG010718, R01HG011138, R01GM140287, and NIH/NIA AG068026. MAW, MA, and GK are supported by grants from the National Institute on Aging (NIA): U01 AG061359, RF1 AG057452, and RF1 AG059093). JCZ is supported by a 4-year Wellcome Trust PhD Studentship and the Cambridge Trust; MK is supported by a Gates Fellowship; CL, EW, MP, JL, EO, IS, NK, and NJW are funded by the Medical Research Council (MC_UU_00006/1 - Aetiology and Mechanisms). This work was supported in part by the UKRI/NIHR Strategic Priorities Award in Multimorbidity Research for the Multimorbidity Mechanism and Therapeutics Research Collaborative (MR/V033867/1).
Footnotes
COMPETING INTERESTS
RAS and AC are current employees and/or stockholders of GlaxoSmithKline. ERG receives an honorarium from the journal Circulation Research of the American Heart Association as a member of the Editorial Board. SOR has received remuneration for consultancy services provided to Pfizer Inc, Astra Zeneca, ERX Pharmaceuticals, GSK, Third Rock Ventures and LG Life Sciences. All other authors declare that they have no competing interests.
DATA and MATERIALS AVAILABILITY
Data from the Fenland cohort can be requested by bona fide researchers for specified scientific purposes via the study website (https://www.mrc-epid.cam.ac.uk/research/studies/fenland/information-for-researchers/). Data will either be shared through an institutional data sharing agreement or arrangements will be made for analyses to be conducted remotely without the necessity for data transfer. Summary statistics can be obtained from www.omicscience.org/apps/pgwas. Publicly available summary statistics for look-up and colocalisation of pQTLs were obtained from https://gwas.mrcieu.ac.uk/ and https://www.ebi.ac.uk/gwas/. Associated code and scripts for the analysis is available on GitHub (https://github.com/MRC-Epid/pGWAS_discovery) and has been permanently archived using Zenodo (12).
REFERENCES AND NOTES
- 1.Emilsson V, Ilkov M, Lamb JR, Finkel N, Gudmundsson EF, Pitts R, Hoover H, Gudmundsdottir V, Horman SR, Aspelund T, Shu L, Trifonov V, Sigurdsson S, Manolescu A, Zhu J, Olafsson Ö, Jakobsdottir J, Lesley SA, To J, Zhang J, Harris TB, Launer LJ, Zhang B, Eiriksdottir G, Yang X, Orth AP, Jennings LL, Gudnason V, Co-regulatory networks of human serum proteins link genetics to disease. Science 361, 769–773 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suhre K, Arnold M, Bhagwat AM, Cotton RJ, Engelke R, Raffler J, Sarwath H, Thareja G, Wahl A, Delisle RK, Gold L, Pezer M, Lauc G, Selim MAED, Mook-Kanamori DO, Al-Dous EK, Mohamoud YA, Malek J, Strauch K, Grallert H, Peters A, Kastenmüller G, Gieger C, Graumann J, Connecting genetic risk to disease end points through the human blood plasma proteome. Nat. Commun 8 (2017), doi: 10.1038/ncomms14357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Folkersen L, Fauman E, Sabater-Lleal M, Strawbridge RJ, Frånberg M, Sennblad B, Baldassarre D, Veglia F, Humphries SE, Rauramaa R, de Faire U, Smit AJ, Giral P, Kurl S, Mannarino E, Enroth S, Johansson Å, Enroth SB, Gustafsson S, Lind L, Lindgren C, Morris AP, Giedraitis V, Silveira A, Franco-Cereceda A, Tremoli E, IMPROVE study group, Gyllensten U, Ingelsson E, Brunak S, Eriksson P, Ziemek D, Hamsten A, Mälarstig A, Mapping of 79 loci for 83 plasma protein biomarkers in cardiovascular disease. PLoS Genet 13, e1006706 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, Burgess S, Jiang T, Paige E, Surendran P, Oliver-Williams C, Kamat MA, Prins BP, Wilcox SK, Zimmerman ES, Chi A, Bansal N, Spain SL, Wood AM, Morrell NW, Bradley JR, Janjic N, Roberts DJ, Ouwehand WH, Todd JA, Soranzo N, Suhre K, Paul DS, Fox CS, Plenge RM, Danesh J, Runz H, Butterworth AS, Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao C, Chen G, Song C, Keefe J, Mendelson M, Huan T, Sun BB, Laser A, Maranville JC, Wu H, Ho JE, Courchesne P, Lyass A, Larson MG, Gieger C, Graumann J, Johnson AD, Danesh J, Runz H, Hwang S-JJ, Liu C, Butterworth AS, Suhre K, Levy D, Genome‐wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat. Commun 9, 3268 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilly A, Park Y-C, Png G, Barysenka A, Fischer I, Bjørnland T, Southam L, Suveges D, Neumeyer S, Rayner NW, Tsafantakis E, Karaleftheri M, Dedoussis G, Zeggini E, Whole-genome sequencing analysis of the cardiometabolic proteome. Nat. Commun 11, 6336 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suhre K, McCarthy MI, Schwenk JM, Genetics meets proteomics: perspectives for large population-based studies. Nat. Rev. Genet, 1–19 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Zheng J, Haberland V, Baird D, Walker V, Haycock PC, Hurle MR, Gutteridge A, Erola P, Liu Y, Luo S, Robinson J, Richardson TG, Staley JR, Elsworth B, Burgess S, Sun BB, Danesh J, Runz H, Maranville JC, Martin HM, Yarmolinsky J, Laurin C, Holmes MV, Liu JZ, Estrada K, Santos R, McCarthy L, Waterworth D, Nelson MR, Smith GD, Butterworth AS, Hemani G, Scott RA, Gaunt TR, Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat. Genet 52, 1122–1131 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Folkersen L, Gustafsson S, Wang Q, Hansen DH, Hedman ÅK, Schork A, Page K, Zhernakova DV, Wu Y, Peters J, Eriksson N, Bergen SE, Boutin TS, Bretherick AD, Enroth S, Kalnapenkis A, Gådin JR, Suur BE, Chen Y, Matic L, Gale JD, Lee J, Zhang W, Quazi A, Ala-Korpela M, Choi SH, Claringbould A, Danesh J, Davey Smith G, de Masi F, Elmståhl S, Engström G, Fauman E, Fernandez C, Franke L, Franks PW, Giedraitis V, Haley C, Hamsten A, Ingason A, Johansson Å, Joshi PK, Lind L, Lindgren CM, Lubitz S, Palmer T, Macdonald-Dunlop E, Magnusson M, Melander O, Michaelsson K, Morris AP, Mägi R, Nagle MW, Nilsson PM, Nilsson J, Orho-Melander M, Polasek O, Prins B, Pålsson E, Qi T, Sjögren M, Sundström J, Surendran P, Võsa U, Werge T, Wernersson R, Westra HJ, Yang J, Zhernakova A, Ärnlöv J, Fu J, Smith JG, Esko T, Hayward C, Gyllensten U, Landen M, Siegbahn A, Wilson JF, Wallentin L, Butterworth AS, Holmes MV, Ingelsson E, Mälarstig A, Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nat. Metab 2, 1135–1148 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pietzner M, Wheeler E, Carrasco-Zanini J, Kerrison ND, Oerton E, Koprulu M, Luan J, Hingorani AD, Williams SA, Wareham NJ, Langenberg C, bioRxiv, in press, doi: 10.1101/2021.03.18.435919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindsay T, Westgate K, Wijndaele K, Hollidge S, Kerrison N, Forouhi N, Griffin S, Wareham N, Brage S, Descriptive epidemiology of physical activity energy expenditure in UK adults (The Fenland study). Int. J. Behav. Nutr. Phys. Act 16, 126 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Associated code is available on GitHub. Extended Material and Methods are avialable as Supplemental Material (2021), doi: 10.5281/zenodo.5385532. [DOI] [Google Scholar]
- 13.Narayan M, Disulfide bonds: protein folding and subcellular protein trafficking. FEBS J 279, 2272–82 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Uhlén M, Karlsson MJ, Hober A, Svensson A, Scheffel J, Kotol D, Zhong W, Tebani A, Strandberg L, Edfors F, Sjöstedt E, Mulder J, Mardinoglu A, Berling A, Ekblad S, Dannemeyer M, Kanje S, Rockberg J, Lundqvist M, Malm M, Volk A, Nilsson P, Månberg A, Dodig-crnkovic T, Pin E, Zwahlen M, Oksvold P, Von Feilitzen K, Häussler RS, Hong M, Lindskog C, Ponten F, Katona B, Vuu J, Lindström E, Nielsen J, Robinson J, Ayoglu B, Mahdessian D, Sullivan D, Thul P, Danielsson F, Stadler C, Lundberg E, Bergström G, Gummesson A, Voldborg BG, Tegel H, Hober S, Forsström B, Schwenk JM, Fagerberg L, Sivertsson Å, The human secretome 0274, 1–9 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Pietzner M, Wheeler E, Carrasco-Zanini J, Raffler J, Kerrison ND, Oerton E, Auyeung VPW, Luan J, Finan C, Casas JP, Ostroff R, Williams SA, Kastenmüller G, Ralser M, Gamazon ER, Wareham NJ, Hingorani AD, Langenberg C, Genetic architecture of host proteins involved in SARS-CoV-2 infection. Nat. Commun 11, 6397 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eslam M, Valenti L, Romeo S, Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol 68, 268–279 (2018). [DOI] [PubMed] [Google Scholar]
- 17.BasuRay S, Wang Y, Smagris E, Cohen JC, Hobbs HH, Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc. Natl. Acad. Sci. U. S. A 116, 9521–9526 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newsome PN, Cramb R, Davison SM, Dillon JF, Foulerton M, Godfrey EM, Hall R, Harrower U, Hudson M, Langford A, Mackie A, Mitchell-Thain R, Sennett K, Sheron NC, Verne J, Walmsley M, Yeoman A, Guidelines on the management of abnormal liver blood tests. Gut 67, 6–19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tollefsen DM, Weigel CJ, Kabeer MH, The presence of methionine or threonine at position 381 in vitronectin is correlated with proteolytic cleavage at arginine 379. J. Biol. Chem 265, 9778–9781 (1990). [PubMed] [Google Scholar]
- 20.Leavesley DI, Kashyap AS, Croll T, Sivaramakrishnan M, Shokoohmand A, Hollier BG, Upton Z, Vitronectin--master controller or micromanager? IUBMB Life 65, 807–18 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Guarani V, McNeill EM, Paulo JA, Huttlin EL, Fröhlich F, Gygi SP, Van Vactor D, Harper JW, QIL1 is a novel mitochondrial protein required for MICOS complex stability and cristae morphology. Elife 4 (2015), doi: 10.7554/eLife.06265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maguire PB, Donlon T, Parsons M, Wynne K, Dillon E, Ní Áinle F, Szklanna PB, Proteomic Analysis Reveals a Strong Association of β-Catenin With Cadherin Adherens Junctions in Resting Human Platelets. Proteomics 18 (2018), doi: 10.1002/pmic.201700419. [DOI] [PubMed] [Google Scholar]
- 23.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, von Mering C, STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47, D607–D613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.GTEx Consortium, The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buccitelli C, Selbach M, mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet 21, 630–644 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Võsa U, Claringbould A, Westra H-J, Jan Bonder M, Deelen P, Zeng B, Kirsten H, Saha A, Kreuzhuber R, Kasela S, Pervjakova N, Alvaes I, Fave M-J, Agbessi M, Christiansen M, Jansen R, Seppälä I, Tong L, Teumer A, Schramm K, Hemani G, Verlouw J, Yaghootkar H, Sönmez R, Brown A, Kukushkina V, Kalnapenkis A, Rüeger S, Porcu E, Kronberg-Guzman J, Kettunen J, Powell J, Lee B, Zhang F, Arindrarto W, Beutner F, Consortium B, Brugge H, Consortium Q, Dmitreva J, Elansary M, Fairfax BP, Georges M, Heijmans BT, Kähönen M, Kim Y, Knight JC, Kovacs P, Krohn K, Li S, Loeffler M, Marigorta UM, Mei H, Momozawa Y, Müller-Nurasyid M, Nauck M, Nivard M, Penninx B, Pritchard J, Raitakari O, Rotzchke O, Slagboom EP, DA Stehouwer C, Stumvoll M, Sullivan P, Thiery J, Tönjes A, van Dongen J, van Iterson M, Veldink J, Völker U, Wijmenga C, Swertz M, Andiappan A, Montgomery GW, Ripatti S, Perola M, Kutalik Z, Awadalla P, Milani L, Ouwehand W, Downes K, Stegle O, Battle A, Yang J, Visscher PM, Scholz M, Gibson G, Esko T, Franke L, Bonder MJ, Deelen P, Zeng B, Kirsten H, Saha A, Kreuzhuber R, Kasela S, Pervjakova N, Alvaes I, Fave M-J, Agbessi M, Christiansen M, Jansen R, Seppälä I, Tong L, Teumer A, Schramm K, Hemani G, Verlouw J, Yaghootkar H, Sönmez R, Brown A, Kukushkina V, Kalnapenkis A, Rüeger S, Porcu E, Kronberg-Guzman J, Kettunen J, Powell J, Lee B, Zhang F, Arindrarto W, Beutner F, Brugge H, Dmitreva J, Elansary M, Fairfax BP, Georges M, Heijmans BT, Kähönen M, Kim Y, Knight JC, Kovacs P, Krohn K, Li S, Loeffler M, Marigorta UM, Mei H, Momozawa Y, Müller-Nurasyid M, Nauck M, Nivard M, Penninx B, Pritchard J, Raitakari O, Rotzchke O, Slagboom EP, DA Stehouwer C, Stumvoll M, Sullivan P, Hoen P, Thiery J, Tönjes A, van Dongen J, van Iterson M, Veldink J, Völker U, Wijmenga C, Swertz M, Andiappan A, Montgomery GW, Ripatti S, Perola M, Kutalik Z, Dermitzakis E, Bergmann S, Frayling T, van Meurs J, Prokisch H, Ahsan H, Pierce B, Lehtimäki T, Boomsma D, Psaty B, Gharib S, Awadalla P, Milani L, Ouwehand W, Downes K, Stegle O, Battle A, Yang J, Visscher PM, Scholz M, Gibson G, Esko T, Franke L, Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv 18, 447367 (2018). [Google Scholar]
- 27.Sternson SM, Atasoy D, Agouti-related protein neuron circuits that regulate appetite. Neuroendocrinology 100, 95–102 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Baker RW, Hughson FM, Chaperoning SNARE assembly and disassembly. Nat. Rev. Mol. Cell Biol 17, 465–79 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hägg S, Athanasiu L, Voyle N, Proitsi P, Witoelar A, Stringer S, Aarsland D, Almdahl IS, Andersen F, Bergh S, Bettella F, Bjornsson S, Brækhus A, Bråthen G, de Leeuw C, Desikan RS, Djurovic S, Dumitrescu L, Fladby T, Hohman TJ, Jonsson PV, Kiddle SJ, Rongve A, Saltvedt I, Sando SB, Selbæk G, Shoai M, Skene NG, Snaedal J, Stordal E, Ulstein ID, Wang Y, White LR, Hardy J, Hjerling-Leffler J, Sullivan PF, van der Flier WM, Dobson R, Davis LK, Stefansson H, Stefansson K, Pedersen NL, Ripke S, Andreassen OA, Posthuma D, Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet 51, 404–413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwartzentruber J, Cooper S, Liu JZ, Barrio-Hernandez I, Bello E, Kumasaka N, Young AMH, Franklin RJM, Johnson T, Estrada K, Gaffney DJ, Beltrao P, Bassett A, Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat. Genet (2021), doi: 10.1038/s41588-020-00776-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aggarwal S, Dabla PK, Arora S, Prostasin: An Epithelial Sodium Channel Regulator. J. biomarkers 2013, 179864 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugitani Y, Nishida A, Inatomi O, Ohno M, Imai T, Kawahara M, Kitamura K, Andoh A, Sodium absorption stimulator prostasin (PRSS8) has an anti-inflammatory effect via downregulation of TLR4 signaling in inflammatory bowel disease. J. Gastroenterol 55, 408–417 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Calvo-Rodriguez M, García-Rodríguez C, Villalobos C, Núñez L, Role of Toll Like Receptor 4 in Alzheimer’s Disease. Front. Immunol 11, 1588 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Mara TA, Glubb DM, Amant F, Annibali D, Ashton K, Attia J, Auer PL, Beckmann MW, Black A, Bolla MK, Brauch H, Brenner H, Brinton L, Buchanan DD, Burwinkel B, Chang-Claude J, Chanock SJ, Chen C, Chen MM, Cheng THT, Clarke CL, Clendenning M, Cook LS, Couch FJ, Cox A, Crous-Bous M, Czene K, Day F, Dennis J, Depreeuw J, Doherty JA, Dörk T, Dowdy SC, Dürst M, Ekici AB, Fasching PA, Fridley BL, Friedenreich CM, Fritschi L, Fung J, García-Closas M, Gaudet MM, Giles GG, Goode EL, Gorman M, Haiman CA, Hall P, Hankison SE, Healey CS, Hein A, Hillemanns P, Hodgson S, Hoivik EA, Holliday EG, Hopper JL, Hunter DJ, Jones A, Krakstad C, Kristensen VN, Lambrechts D, Le Marchand L, Liang X, Lindblom A, Lissowska J, Long J, Lu L, Magliocco AM, Martin L, McEvoy M, Meindl A, Michailidou K, Milne RL, Mints M, Montgomery GW, Nassir R, Olsson H, Orlow I, Otton G, Palles C, Perry JRB, Peto J, Pooler L, Prescott J, Proietto T, Rebbeck TR, Risch HA, Rogers PAW, Rübner M, Runnebaum I, Sacerdote C, Sarto GE, Schumacher F, Scott RJ, Setiawan VW, Shah M, Sheng X, Shu X-O, Southey MC, Swerdlow AJ, Tham E, Trovik J, Turman C, Tyrer JP, Vachon C, VanDen Berg D, Vanderstichele A, Wang Z, Webb PM, Wentzensen N, Werner HMJ, Winham SJ, Wolk A, Xia L, Xiang Y-B, Yang HP, Yu H, Zheng W, Pharoah PDP, Dunning AM, Kraft P, De Vivo I, Tomlinson I, Easton DF, Spurdle AB, Thompson DJ, Identification of nine new susceptibility loci for endometrial cancer. Nat. Commun 9, 3166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Binnerts ME, Kim K-A, Bright JM, Patel SM, Tran K, Zhou M, Leung JM, Liu Y, Lomas WE, Dixon M, Hazell SA, Wagle M, Nie W-S, Tomasevic N, Williams J, Zhan X, Levy MD, Funk WD, Abo A, R-Spondin1 regulates Wnt signaling by inhibiting internalization of LRP6. Proc. Natl. Acad. Sci. U. S. A 104, 14700–5 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geng A, Wu T, Cai C, Song W, Wang J, Yu QC, Zeng YA, A novel function of R-spondin1 in regulating estrogen receptor expression independent of Wnt/β-catenin signaling. Elife 9 (2020), doi: 10.7554/eLife.56434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chassot A-A, Bradford ST, Auguste A, Gregoire EP, Pailhoux E, de Rooij DG, Schedl A, Chaboissier M-C, WNT4 and RSPO1 together are required for cell proliferation in the early mouse gonad. Development 139, 4461–72 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Chassot A-A, Ranc F, Gregoire EP, Roepers-Gajadien HL, Taketo MM, Camerino G, de Rooij DG, Schedl A, Chaboissier M-C, Activation of beta-catenin signaling by Rspo1 controls differentiation of the mammalian ovary. Hum. Mol. Genet 17, 1264–77 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Edery P, Lyonnet S, Mulligan LM, Pelet A, Dow E, Abel L, Holder S, Nihoul-Fékété C, Ponder BA, Munnich A, Mutations of the RET proto-oncogene in Hirschsprung’s disease. Nature 367, 378–80 (1994). [DOI] [PubMed] [Google Scholar]
- 40.Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, Li Y, Kurreeman FAS, Zhernakova A, Hinks A, Guiducci C, Chen R, Alfredsson L, Amos CI, Ardlie KG, Barton A, Bowes J, Brouwer E, Burtt NP, Catanese JJ, Coblyn J, Coenen MJH, Costenbader KH, Criswell LA, Crusius JBA, Cui J, De Bakker PIW, De Jager PL, Ding B, Emery P, Flynn E, Harrison P, Hocking LJ, Huizinga TWJ, Kastner DL, Ke X, Lee AT, Liu X, Martin P, Morgan AW, Padyukov L, Posthumus MD, Radstake TRDJ, Reid DM, Seielstad M, Seldin MF, Shadick NA, Steer S, Tak PP, Thomson W, Van Der Helm-Van Mil AHM, Van Der Horst-Bruinsma IE, Van Der Schoot CE, Van Riel PLCM, Weinblatt ME, Wilson AG, Wolbink GJ, Wordsworth BP, Wijmenga C, Karlson EW, Toes REM, De Vries N, Begovich AB, Worthington J, Siminovitch KA, Gregersen PK, Klareskog L, Plenge RM, Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet 42, 508–514 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harii N, Lewis CJ, Vasko V, McCall K, Benavides-Peralta U, Sun X, Ringel MD, Saji M, Giuliani C, Napolitano G, Goetz DJ, Kohn LD, Thyrocytes express a functional toll-like receptor 3: overexpression can be induced by viral infection and reversed by phenylmethimazole and is associated with Hashimoto’s autoimmune thyroiditis. Mol. Endocrinol 19, 1231–50 (2005). [DOI] [PubMed] [Google Scholar]
- 42.C.−19 H. G. Initiative, Mapping the human genetic architecture of COVID-19. Nature (2021), doi: 10.1038/s41586-021-03767-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou S, Butler-Laporte G, Nakanishi T, Morrison DR, Afilalo J, Afilalo M, Laurent L, Pietzner M, Kerrison N, Zhao K, Brunet-Ratnasingham E, Henry D, Kimchi N, Afrasiabi Z, Rezk N, Bouab M, Petitjean L, Guzman C, Xue X, Tselios C, Vulesevic B, Adeleye O, Abdullah T, Almamlouk N, Chen Y, Chassé M, Durand M, Paterson C, Normark J, Frithiof R, Lipcsey M, Hultström M, Greenwood CMT, Zeberg H, Langenberg C, Thysell E, Pollak M, Mooser V, Forgetta V, Kaufmann DE, Richards JB, A Neanderthal OAS1 isoform protects individuals of European ancestry against COVID-19 susceptibility and severity. Nat. Med 27, 659–667 (2021). [DOI] [PubMed] [Google Scholar]
- 44.Whyte MB, Kelly PA, Gonzalez E, Arya R, Roberts LN, Pulmonary embolism in hospitalised patients with COVID-19. Thromb. Res 195, 95–99 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Joshi AD, Andersson C, Buch S, Stender S, Noordam R, Weng L-C, Weeke PE, Auer PL, Boehm B, Chen C, Choi H, Curhan G, Denny JC, De Vivo I, Eicher JD, Ellinghaus D, Folsom AR, Fuchs C, Gala M, Haessler J, Hofman A, Hu F, Hunter DJ, Janssen HLA, Kang JH, Kooperberg C, Kraft P, Kratzer W, Lieb W, Lutsey PL, Darwish Murad S, Nordestgaard BG, Pasquale LR, Reiner AP, Ridker PM, Rimm E, Rose LM, Shaffer CM, Schafmayer C, Tamimi RM, Uitterlinden AG, Völker U, Völzke H, Wakabayashi Y, Wiggs JL, Zhu J, Roden DM, Stricker BH, Tang W, Teumer A, Hampe J, Tybjærg-Hansen A, Chasman DI, Chan AT, Johnson AD, Four Susceptibility Loci for Gallstone Disease Identified in a Meta-analysis of Genome-Wide Association Studies. Gastroenterology 151, 351–363.e28 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Foley CN, Staley JR, Breen PG, Sun BB, Kirk PDW, Burgess S, Howson JMM, A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat. Commun 12, 764 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shin S-YY, Fauman EB, Petersen A-KK, Krumsiek J, Santos R, Huang J, Arnold M, Erte I, Forgetta V, Yang T-PP, Walter K, Menni C, Chen L, Vasquez L, Valdes AM, Hyde CL, Wang V, Ziemek D, Roberts P, Xi L, Grundberg E, Waldenberger M, Richards JB, Mohney RP, Milburn MV, John SL, Trimmer J, Theis FJ, Overington JP, Suhre K, Brosnan MJ, Gieger C, Kastenmüller G, Spector TD, Soranzo N, Soranzo N, An atlas of genetic influences on human blood metabolites. Nat. Genet 46, 543–550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lammert F, Gurusamy K, Ko CW, Miquel J-F, Méndez-Sánchez N, Portincasa P, van Erpecum KJ, van Laarhoven CJ, Wang DQ-H, Gallstones. Nat. Rev. Dis. Prim 2, 16024 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, Chu AY, Estrada K, Luan J, Kutalik Z, Amin N, Buchkovich ML, Croteau-Chonka DC, Day FR, Duan Y, Fall T, Fehrmann R, Ferreira T, Jackson AU, Karjalainen J, Lo KS, Locke AE, Mägi R, Mihailov E, Porcu E, Randall JC, Scherag A, Vinkhuyzen AAE, Westra H-J, Winkler TW, Workalemahu T, Zhao JH, Absher D, Albrecht E, Anderson D, Baron J, Beekman M, Demirkan A, Ehret GB, Feenstra B, Feitosa MF, Fischer K, Fraser RM, Goel A, Gong J, Justice AE, Kanoni S, Kleber ME, Kristiansson K, Lim U, Lotay V, Lui JC, Mangino M, Mateo Leach I, Medina-Gomez C, Nalls MA, Nyholt DR, Palmer CD, Pasko D, Pechlivanis S, Prokopenko I, Ried JS, Ripke S, Shungin D, Stancáková A, Strawbridge RJ, Sung YJ, Tanaka T, Teumer A, Trompet S, van der Laan SW, van Setten J, V Van Vliet-Ostaptchouk J, Wang Z, Yengo L, Zhang W, Afzal U, Arnlöv J, Arscott GM, Bandinelli S, Barrett A, Bellis C, Bennett AJ, Berne C, Blüher M, Bolton JL, Böttcher Y, Boyd HA, Bruinenberg M, Buckley BM, Buyske S, Caspersen IH, Chines PS, Clarke R, Claudi-Boehm S, Cooper M, Daw EW, De Jong PA, Deelen J, Delgado G, Denny JC, Dhonukshe-Rutten R, Dimitriou M, Doney ASF, Dörr M, Eklund N, Eury E, Folkersen L, Garcia ME, Geller F, Giedraitis V, Go AS, Grallert H, Grammer TB, Gräßler J, Grönberg H, de Groot LCPGM, Groves CJ, Haessler J, Hall P, Haller T, Hallmans G, Hannemann A, Hartman CA, Hassinen M, Hayward C, Heard-Costa NL, Helmer Q, Hemani G, Henders AK, Hillege HL, Hlatky MA, Hoffmann W, Hoffmann P, Holmen O, Houwing-Duistermaat JJ, Illig T, Isaacs A, James AL, Jeff J, Johansen B, Johansson Å, Jolley J, Juliusdottir T, Junttila J, Kho AN, Kinnunen L, Klopp N, Kocher T, Kratzer W, Lichtner P, Lind L, Lindström J, Lobbens S, Lorentzon M, Lu Y, Lyssenko V, Magnusson PKE, Mahajan A, Maillard M, McArdle WL, McKenzie CA, McLachlan S, McLaren PJ, Menni C, Merger S, Milani L, Moayyeri A, Monda KL, Morken MA, Müller G, Müller-Nurasyid M, Musk AW, Narisu N, Nauck M, Nolte IM, Nöthen MM, Oozageer L, Pilz S, Rayner NW, Renstrom F, Robertson NR, Rose LM, Roussel R, Sanna S, Scharnagl H, Scholtens S, Schumacher FR, Schunkert H, Scott RA, Sehmi J, Seufferlein T, Shi J, Silventoinen K, Smit JH, Smith AV, Smolonska J, V Stanton A, Stirrups K, Stott DJ, Stringham HM, Sundström J, Swertz MA, Syvänen A-C, Tayo BO, Thorleifsson G, Tyrer JP, van Dijk S, van Schoor NM, van der Velde N, van Heemst D, van Oort FVA, Vermeulen SH, Verweij N, Vonk JM, Waite LL, Waldenberger M, Wennauer R, Wilkens LR, Willenborg C, Wilsgaard T, Wojczynski MK, Wong A, Wright AF, Zhang Q, Arveiler D, Bakker SJL, Beilby J, Bergman RN, Bergmann S, Biffar R, Blangero J, Boomsma DI, Bornstein SR, Bovet P, Brambilla P, Brown MJ, Campbell H, Caulfield MJ, Chakravarti A, Collins R, Collins FS, Crawford DC, Cupples LA, Danesh J, de Faire U, den Ruijter HM, Erbel R, Erdmann J, Eriksson JG, Farrall M, Ferrannini E, Ferrières J, Ford I, Forouhi NG, Forrester T, Gansevoort RT, V Gejman P, Gieger C, Golay A, Gottesman O, Gudnason V, Gyllensten U, Haas DW, Hall AS, Harris TB, Hattersley AT, Heath AC, Hengstenberg C, Hicks AA, Hindorff LA, Hingorani AD, Hofman A, Hovingh GK, Humphries SE, Hunt SC, Hypponen E, Jacobs KB, Jarvelin M-R, Jousilahti P, Jula AM, Kaprio J, Kastelein JJP, Kayser M, Kee F, Keinanen-Kiukaanniemi SM, Kiemeney LA, Kooner JS, Kooperberg C, Koskinen S, Kovacs P, Kraja AT, Kumari M, Kuusisto J, Lakka TA, Langenberg C, Le Marchand L, Lehtimäki T, Lupoli S, Madden PAF, Männistö S, Manunta P, Marette A, Matise TC, McKnight B, Meitinger T, Moll FL, Montgomery GW, Morris AD, Morris AP, Murray JC, Nelis M, Ohlsson C, Oldehinkel AJ, Ong KK, Ouwehand WH, Pasterkamp G, Peters A, Pramstaller PP, Price JF, Qi L, Raitakari OT, Rankinen T, Rao DC, Rice TK, Ritchie M, Rudan I, Salomaa V, Samani NJ, Saramies J, Sarzynski MA, Schwarz PEH, Sebert S, Sever P, Shuldiner AR, Sinisalo J, Steinthorsdottir V, Stolk RP, Tardif J-C, Tönjes A, Tremblay A, Tremoli E, Virtamo J, Vohl M-C, Electronic Medical Records and Genomics (eMEMERGEGE) Consortium, MIGen Consortium, PAGEGE Consortium, LifeLines Cohort Study, Amouyel P, Asselbergs FW, Assimes TL, Bochud M, Boehm BO, Boerwinkle E, Bottinger EP, Bouchard C, Cauchi S, Chambers JC, Chanock SJ, Cooper RS, de Bakker PIW, Dedoussis G, Ferrucci L, Franks PW, Froguel P, Groop LC, Haiman CA, Hamsten A, Hayes MG, Hui J, Hunter DJ, Hveem K, Jukema JW, Kaplan RC, Kivimaki M, Kuh D, Laakso M, Liu Y, Martin NG, März W, Melbye M, Moebus S, Munroe PB, Njølstad I, Oostra BA, Palmer CNA, Pedersen NL, Perola M, Pérusse L, Peters U, Powell JE, Power C, Quertermous T, Rauramaa R, Reinmaa E, Ridker PM, Rivadeneira F, Rotter JI, Saaristo TE, Saleheen D, Schlessinger D, Slagboom PE, Snieder H, Spector TD, Strauch K, Stumvoll M, Tuomilehto J, Uusitupa M, van der Harst P, Völzke H, Walker M, Wareham NJ, Watkins H, Wichmann H-E, Wilson JF, Zanen P, Deloukas P, Heid IM, Lindgren CM, Mohlke KL, Speliotes EK, Thorsteinsdottir U, Barroso I, Fox CS, North KE, Strachan DP, Beckmann JS, Berndt SI, Boehnke M, Borecki IB, McCarthy MI, Metspalu A, Stefansson K, Uitterlinden AG, van Duijn CM, Franke L, Willer CJ, Price AL, Lettre G, Loos RJF, Weedon MN, Ingelsson E, O’Connell JR, Abecasis GR, Chasman DI, Goddard ME, Visscher PM, Hirschhorn JN, Frayling TM, Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet 46, 1173–86 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Springelkamp H, Iglesias AI, Mishra A, Höhn R, Wojciechowski R, Khawaja AP, Nag A, Wang YX, Wang JJ, Cuellar-Partida G, Gibson J, Bailey JNC, Vithana EN, Gharahkhani P, Boutin T, Ramdas WD, Zeller T, Luben RN, Yonova-Doing E, Viswanathan AC, Yazar S, Cree AJ, Haines JL, Koh JY, Souzeau E, Wilson JF, Amin N, Müller C, Venturini C, Kearns LS, Kang JH, NEIGHBORHOOD Consortium, Tham YC, Zhou T, van Leeuwen EM, Nickels S, Sanfilippo P, Liao J, van der Linde H, Zhao W, van Koolwijk LME, Zheng L, Rivadeneira F, Baskaran M, van der Lee SJ, Perera S, de Jong PTVM, Oostra BA, Uitterlinden AG, Fan Q, Hofman A, Tai E-S, Vingerling JR, Sim X, Wolfs RCW, Teo YY, Lemij HG, Khor CC, Willemsen R, Lackner KJ, Aung T, Jansonius NM, Montgomery G, Wild PS, Young TL, Burdon KP, Hysi PG, Pasquale LR, Wong TY, Klaver CCW, Hewitt AW, Jonas JB, Mitchell P, Lotery AJ, Foster PJ, Vitart V, Pfeiffer N, Craig JE, Mackey DA, Hammond CJ, Wiggs JL, Cheng C-Y, van Duijn CM, MacGregor S, New insights into the genetics of primary open-angle glaucoma based on meta-analyses of intraocular pressure and optic disc characteristics. Hum. Mol. Genet 26, 438–453 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wiberg A, Ng M, Schmid AB, Smillie RW, Baskozos G, V Holmes M, Künnapuu K, Mägi R, Bennett DL, Furniss D, A genome-wide association analysis identifies 16 novel susceptibility loci for carpal tunnel syndrome. Nat. Commun 10, 1030 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jorgenson E, Makki N, Shen L, Chen DC, Tian C, Eckalbar WL, Hinds D, Ahituv N, Avins A, A genome-wide association study identifies four novel susceptibility loci underlying inguinal hernia. Nat. Commun 6, 10130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shrine N, Guyatt AL, Erzurumluoglu AM, Jackson VE, Hobbs BD, Melbourne CA, Batini C, Fawcett KA, Song K, Sakornsakolpat P, Li X, Boxall R, Reeve NF, Obeidat M, Zhao JH, Wielscher M, Weiss S, Kentistou KA, Cook JP, Sun BB, Zhou J, Hui J, Karrasch S, Imboden M, Harris SE, Marten J, Enroth S, Kerr SM, Surakka I, Vitart V, Lehtimäki T, Allen RJ, Bakke PS, Beaty TH, Bleecker ER, Bossé Y, Brandsma CA, Chen Z, Crapo JD, Danesh J, DeMeo DL, Dudbridge F, Ewert R, Gieger C, Gulsvik A, Hansell AL, Hao K, Hoffman JD, Hokanson JE, Homuth G, Joshi PK, Joubert P, Langenberg C, Li X, Li L, Lin K, Lind L, Locantore N, Luan J, Mahajan A, Maranville JC, Murray A, Nickle DC, Packer R, Parker MM, Paynton ML, Porteous DJ, Prokopenko D, Qiao D, Rawal R, Runz H, Sayers I, Sin DD, Smith BH, Soler Artigas M, Sparrow D, Tal-Singer R, Timmers PRHJ, Van den Berge M, Whittaker JC, Woodruff PG, Yerges-Armstrong LM, Troyanskaya OG, Raitakari OT, Kähönen M, Polašek O, Gyllensten U, Rudan I, Deary IJ, Probst-Hensch NM, Schulz H, James AL, Wilson JF, Stubbe B, Zeggini E, Jarvelin MR, Wareham N, Silverman EK, Hayward C, Morris AP, Butterworth AS, Scott RA, Walters RG, Meyers DA, Cho MH, Strachan DP, Hall IP, Tobin MD, Wain LV, New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat. Genet 51, 481–493 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pickrell JK, Berisa T, Liu JZ, Ségurel L, Tung JY, Hinds DA, Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet 48, 709–717 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McLaughlin PJ, Bakall B, Choi J, Liu Z, Sasaki T, Davis EC, Marmorstein AD, Marmorstein LY, Lack of fibulin-3 causes early aging and herniation, but not macular degeneration in mice. Hum. Mol. Genet 16, 3059–70 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Livingstone I, Uversky VN, Furniss D, Wiberg A, The Pathophysiological Significance of Fibulin-3. Biomolecules 10 (2020), doi: 10.3390/biom10091294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marmorstein LY, Munier FL, Arsenijevic Y, Schorderet DF, McLaughlin PJ, Chung D, Traboulsi E, Marmorstein AD, Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration. Proc. Natl. Acad. Sci. U. S. A 99, 13067–72 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hollis B, Day FR, Busch AS, Thompson DJ, Soares ALG, Timmers PRHJ, Kwong A, Easton DF, Joshi PK, Timpson NJ, PRACTICAL Consortium, 23andMe Research Team, Ong KK, Perry JRB, Genomic analysis of male puberty timing highlights shared genetic basis with hair colour and lifespan. Nat. Commun 11, 1536 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Day FR, Thompson DJ, Helgason H, Chasman DI, Finucane H, Sulem P, Ruth KS, Whalen S, Sarkar AK, Albrecht E, Altmaier E, Amini M, Barbieri CM, Boutin T, Campbell A, Demerath E, Giri A, He C, Hottenga JJ, Karlsson R, Kolcic I, Loh P-R, Lunetta KL, Mangino M, Marco B, McMahon G, Medland SE, Nolte IM, Noordam R, Nutile T, Paternoster L, Perjakova N, Porcu E, Rose LM, Schraut KE, V Segrè A, V Smith A, Stolk L, Teumer A, Andrulis IL, Bandinelli S, Beckmann MW, Benitez J, Bergmann S, Bochud M, Boerwinkle E, Bojesen SE, Bolla MK, Brand JS, Brauch H, Brenner H, Broer L, Brüning T, Buring JE, Campbell H, Catamo E, Chanock S, Chenevix-Trench G, Corre T, Couch FJ, Cousminer DL, Cox A, Crisponi L, Czene K, Davey Smith G, de Geus EJCN, de Mutsert R, De Vivo I, Dennis J, Devilee P, dos-Santos-Silva I, Dunning AM, Eriksson JG, Fasching PA, Fernández-Rhodes L, Ferrucci L, Flesch-Janys D, Franke L, Gabrielson M, Gandin I, Giles GG, Grallert H, Gudbjartsson DF, Guénel P, Hall P, Hallberg E, Hamann U, Harris TB, Hartman CA, Heiss G, Hooning MJ, Hopper JL, Hu F, Hunter DJ, Arfan Ikram M, Kyung Im H, Järvelin M-R, Joshi PK, Karasik D, Kellis M, Kutalik Z, LaChance G, Lambrechts D, Langenberg C, Launer LJ, E Laven JS, Lenarduzzi S, Li J, Lind PA, Lindstrom S, Liu Y, Luan an, Mägi R, Mannermaa A, Mbarek H, McCarthy MI, Meisinger C, Meitinger T, Menni C, Metspalu A, Michailidou K, Milani L, Milne RL, Montgomery GW, Mulligan AM, Nalls MA, Navarro P, Nevanlinna H, Nyholt DR, Oldehinkel AJ, O TA, Padmanabhan S, Palotie A, Pedersen N, Peters A, Peto J, Pharoah PDP, Pouta A, Radice P, Rahman I, Ring SM, Robino A, Rosendaal FR, Rudan I, Rueedi R, Ruggiero D, Sala CF, Schmidt MK, Scott RA, Shah M, Sorice R, Southey MC, Sovio U, Stampfer M, Steri M, Strauch K, Tanaka T, Tikkanen E, Timpson NJ, Traglia M, Truong T, Tyrer JP, Uitterlinden AG, Velez Edwards DR, Vitart V, Völker U, Vollenweider P, Wang Q, Widen E, Willems van Dijk K, Willemsen G, Winqvist R, R Wolffenbuttel BH, Hua Zhao J, Zoledziewska M, Zygmunt M, Alizadeh BZ, Boomsma DI, Ciullo M, Cucca F, Esko T, Franceschini N, Gieger C, Gudnason V, Hayward C, Kraft P, Lawlor DA, E Magnusson PK, Martin NG, Mook-Kanamori DO, Nohr EA, Polasek O, Porteous D, Price AL, Ridker PM, Snieder H, Spector TD, Stöckl D, Toniolo D, Ulivi S, Visser JA, Völzke H, Wareham NJ, Wilson JF, Spurdle AB, Thorsteindottir U, Pollard KS, Easton DF, Tung JY, Chang-Claude J, Hinds D, Murray A, Murabito JM, Stefansson K, Ong KK, B Perry JR, Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat. Publ. Gr 49 (2017), doi: 10.1038/ng.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khramtsova EA, Davis LK, Stranger BE, The role of sex in the genomics of human complex traits. Nat. Rev. Genet 20, 173–190 (2019). [DOI] [PubMed] [Google Scholar]
- 61.Aschard H, A perspective on interaction effects in genetic association studies. Genet. Epidemiol 40, 678–688 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oliva M, Muñoz-Aguirre M, Kim-Hellmuth S, Wucher V, Gewirtz ADH, Cotter DJ, Parsana P, Kasela S, Balliu B, Viñuela A, Castel SE, Mohammadi P, Aguet F, Zou Y, Khramtsova EA, Skol AD, Garrido-Martín D, Reverter F, Brown A, Evans P, Gamazon ER, Payne A, Bonazzola R, Barbeira AN, Hamel AR, Martinez-Perez A, Soria JM, GTEx Consortium, Pierce BL, Stephens M, Eskin E, Dermitzakis ET, V Segrè A, Im HK, Engelhardt BE, Ardlie KG, Montgomery SB, Battle AJ, Lappalainen T, Guigó R, Stranger BE, The impact of sex on gene expression across human tissues. Science 369 (2020), doi: 10.1126/science.aba3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mittelstrass K, Ried JS, Yu Z, Krumsiek J, Gieger C, Discovery of Sexual Dimorphisms in Metabolic and Genetic Biomarkers. PLoS Genet 7, 1002215 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Finan C, Gaulton A, Kruger FA, Lumbers RT, Shah T, Engmann J, Galver L, Kelley R, Karlsson A, Santos R, Overington JP, Hingorani AD, Casas JP, The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med 9 (2017), doi: 10.1126/scitranslmed.aag1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ochoa D, Hercules A, Carmona M, Suveges D, Gonzalez-Uriarte A, Malangone C, Miranda A, Fumis L, Carvalho-Silva D, Spitzer M, Baker J, Ferrer J, Raies A, Razuvayevskaya O, Faulconbridge A, Petsalaki E, Mutowo P, Machlitt-Northen S, Peat G, McAuley E, Ong CK, Mountjoy E, Ghoussaini M, Pierleoni A, Papa E, Pignatelli M, Koscielny G, Karim M, Schwartzentruber J, Hulcoop DG, Dunham I, McDonagh EM, Open Targets Platform: supporting systematic drug-target identification and prioritisation. Nucleic Acids Res 49, D1302–D1310 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kola I, Bell J, A call to reform the taxonomy of human disease. Nat. Rev. Drug Discov 10, 641–2 (2011). [DOI] [PubMed] [Google Scholar]
- 67.Sender R, Milo R, The distribution of cellular turnover in the human body. Nat. Med 27, 45–48 (2021). [DOI] [PubMed] [Google Scholar]
- 68.Williams SA, Kivimaki M, Langenberg C, Hingorani AD, Casas JP, Bouchard C, Jonasson C, Sarzynski MA, Shipley MJ, Alexander L, Ash J, Bauer T, Chadwick J, Datta G, DeLisle RK, Hagar Y, Hinterberg M, Ostroff R, Weiss S, Ganz P, Wareham NJ, Plasma protein patterns as comprehensive indicators of health. Nat. Med 25, 1851–1857 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, Carter J, Dalby AB, Eaton BE, Fitzwater T, Flather D, Forbes A, Foreman T, Fowler C, Gawande B, Goss M, Gunn M, Gupta S, Halladay D, Heil J, Heilig J, Hicke B, Husar G, Janjic N, Jarvis T, Jennings S, Katilius E, Keeney TR, Kim N, Koch TH, Kraemer S, Kroiss L, Le N, Levine D, Lindsey W, Lollo B, Mayfield W, Mehan M, Mehler R, Nelson SK, Nelson M, Nieuwlandt D, Nikrad M, Ochsner U, Ostroff RM, Otis M, Parker T, Pietrasiewicz S, Resnicow DI, Rohloff J, Sanders G, Sattin S, Schneider D, Singer B, Stanton M, Sterkel A, Stewart A, Stratford S, Vaught JD, Vrkljan M, Walker JJ, Watrobka M, Waugh S, Weiss A, Wilcox SK, Wolfson A, Wolk SK, Zhang C, Zichi D, Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One 5 (2010), doi: 10.1371/journal.pone.0015004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V, Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10, e1004383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Elsworth B, Lyon M, Alexander T, Liu Y, Matthews P, Hallett J, Bates P, Palmer T, Haberland V, Smith GD, Zheng J, Haycock P, Gaunt TR, Hemani G, bioRxiv, in press, doi: 10.1101/2020.08.10.244293. [DOI] [Google Scholar]
- 72.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, Luo Y, Sidore C, Kwong A, Timpson N, Koskinen S, Vrieze S, Scott LJ, Zhang H, Mahajan A, Veldink J, Peters U, Pato C, Van Duijn CM, Gillies CE, Gandin I, Mezzavilla M, Gilly A, Cocca M, Traglia M, Angius A, Barrett JC, Boomsma D, Branham K, Breen G, Brummett CM, Busonero F, Campbell H, Chan A, Chen S, Chew E, Collins FS, Corbin LJ, Smith GD, Dedoussis G, Dorr M, Farmaki AE, Ferrucci L, Forer L, Fraser RM, Gabriel S, Levy S, Groop L, Harrison T, Hattersley A, Holmen OL, Hveem K, Kretzler M, Lee JC, McGue M, Meitinger T, Melzer D, Min JL, Mohlke KL, Vincent JB, Nauck M, Nickerson D, Palotie A, Pato M, Pirastu N, McInnis M, Richards JB, Sala C, Salomaa V, Schlessinger D, Schoenherr S, Slagboom PE, Small K, Spector T, Stambolian D, Tuke M, Tuomilehto J, Van Den Berg LH, Van Rheenen W, Volker U, Wijmenga C, Toniolo D, Zeggini E, Gasparini P, Sampson MG, Wilson JF, Frayling T, De Bakker PIW, Swertz MA, McCarroll S, Kooperberg C, Dekker A, Altshuler D, Willer C, Iacono W, Ripatti S, Soranzo N, Walter K, Swaroop A, Cucca F, Anderson CA, Myers RM, Boehnke M, McCarthy MI, Durbin R, Abecasis G, Marchini J, A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet 48, 1279–1283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang J, Howie B, McCarthy S, Memari Y, Walter K, Min JL, Danecek P, Malerba G, Trabetti E, Zheng HF, Gambaro G, Richards JB, Durbin R, Timpson NJ, Marchini J, Soranzo N, Al Turki S, Amuzu A, Anderson CA, Anney R, Antony D, Artigas MS, Ayub M, Bala S, Barrett JC, Barroso I, Beales P, Benn M, Bentham J, Bhattacharya S, Birney E, Blackwood D, Bobrow M, Bochukova E, Bolton PF, Bounds R, Boustred C, Breen G, Calissano M, Carss K, Casas JP, Chambers JC, Charlton R, Chatterjee K, Chen L, Ciampi A, Cirak S, Clapham P, Clement G, Coates G, Cocca M, Collier DA, Cosgrove C, Cox T, Craddock N, Crooks L, Curran S, Curtis D, Daly A, Day INM, Day-Williams A, Dedoussis G, Down T, Du Y, Van Duijn CM, Dunham I, Edkins S, Ekong R, Ellis P, Evans DM, Farooqi IS, Fitzpatrick DR, Flicek P, Floyd J, Foley AR, Franklin CS, Futema M, Gallagher L, Gasparini P, Gaunt TR, Geihs M, Geschwind D, Greenwood C, Griffin H, Grozeva D, Guo X, Guo X, Gurling H, Hart D, Hendricks AE, Holmans P, Huang L, Hubbard T, Humphries SE, Hurles ME, Hysi P, Iotchkova V, Isaacs A, Jackson DK, Jamshidi Y, Johnson J, Joyce C, Karczewski KJ, Kaye J, Keane T, Kemp JP, Kennedy K, Kent A, Keogh J, Khawaja F, Kleber ME, Van Kogelenberg M, Kolb-Kokocinski A, Kooner JS, Lachance G, Langenberg C, Langford C, Lawson D, Lee I, Van Leeuwen EM, Lek M, Li R, Li Y, Liang J, Lin H, Liu R, Lönnqvist J, Lopes LR, Lopes M, Luan J, MacArthur DG, Mangino M, Marenne G, März W, Maslen J, Matchan A, Mathieson I, McGuffin P, McIntosh AM, McKechanie AG, McQuillin A, Metrustry S, Migone N, Mitchison HM, Moayyeri A, Morris J, Morris R, Muddyman D, Muntoni F, Nordestgaard BG, Northstone K, O’Donovan MC, O’Rahilly S, Onoufriadis A, Oualkacha K, Owen MJ, Palotie A, Panoutsopoulou K, Parker V, Parr JR, Paternoster L, Paunio T, Payne F, Payne SJ, Perry JRB, Pietilainen O, Plagnol V, Pollitt RC, Povey S, Quail MA, Quaye L, Raymond L, Rehnström K, Ridout CK, Ring S, Ritchie GRS, Roberts N, Robinson RL, Savage DB, Scambler P, Schiffels S, Schmidts M, Schoenmakers N, Scott RH, Scott RA, Semple RK, Serra E, Sharp SI, Shaw A, Shihab HA, Shin SY, Skuse D, Small KS, Smee C, Smith GD, Southam L, Spasic-Boskovic O, Spector TD, St. Clair D, St. Pourcain B, Stalker J, Stevens E, Sun J, Surdulescu G, Suvisaari J, Syrris P, Tachmazidou I, Taylor R, Tian J, Tobin MD, Toniolo D, Traglia M, Tybjaerg-Hansen A, Valdes AM, Vandersteen AM, Varbo A, Vijayarangakannan P, Visscher PM, Wain LV, Walters JTR, Wang G, Wang J, Wang Y, Ward K, Wheeler E, Whincup P, Whyte T, Williams HJ, Williamson KA, Wilson C, Wilson SG, Wong K, Xu CJ, Yang J, Zaza G, Zeggini E, Zhang F, Zhang P, Zhang W, Improved imputation of low-frequency and rare variants using the UK10K haplotype reference panel. Nat. Commun 6, 1–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, Cortes A, Welsh S, Young A, Effingham M, McVean G, Leslie S, Allen N, Donnelly P, Marchini J, The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Willer CJ, Li Y, Abecasis GR, METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang J, Lee SH, Goddard ME, Visscher PM, GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet 88, 76–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F, The Ensembl Variant Effect Predictor. Genome Biol 17, 1–14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Berisa T, Pickrell JK, Approximately independent linkage disequilibrium blocks in human populations. Bioinformatics 32, 283–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, Tan VY, Yarmolinsky J, Shihab HA, Timpson NJ, Evans DM, Relton C, Martin RM, Davey Smith G, Gaunt TR, Haycock PC, The MR-Base platform supports systematic causal inference across the human phenome. Elife 7 (2018), doi: 10.7554/eLife.34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lotta LA, Pietzner M, Stewart ID, Wittemans LBL, Li C, Bonelli R, Raffler J, Biggs EK, Oliver-Williams C, Auyeung VPW, Luan J, Wheeler E, Paige E, Surendran P, Michelotti GA, Scott RA, Burgess S, Zuber V, Sanderson E, Koulman A, Imamura F, Forouhi NG, Khaw K-T, MacTel Consortium, Griffin JL, Wood AM, Kastenmüller G, Danesh J, Butterworth AS, Gribble FM, Reimann F, Bahlo M, Fauman E, Wareham NJ, Langenberg C, A cross-platform approach identifies genetic regulators of human metabolism and health. Nat. Genet 53, 54–64 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yu G, Wang LG, Han Y, He QY, ClusterProfiler: An R package for comparing biological themes among gene clusters. Omi. A J. Integr. Biol 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data from the Fenland cohort can be requested by bona fide researchers for specified scientific purposes via the study website (https://www.mrc-epid.cam.ac.uk/research/studies/fenland/information-for-researchers/). Data will either be shared through an institutional data sharing agreement or arrangements will be made for analyses to be conducted remotely without the necessity for data transfer. Summary statistics can be obtained from www.omicscience.org/apps/pgwas. Publicly available summary statistics for look-up and colocalisation of pQTLs were obtained from https://gwas.mrcieu.ac.uk/ and https://www.ebi.ac.uk/gwas/. Associated code and scripts for the analysis is available on GitHub (https://github.com/MRC-Epid/pGWAS_discovery) and has been permanently archived using Zenodo (12).