

Abstract
Triggering receptors expressed on myeloid cells (TREMs) encompass a family of cell-surface receptors chiefly expressed by granulocytes, monocytes and tissue macrophages. These receptors have been implicated in inflammation, neurodegenerative diseases, bone remodelling, metabolic syndrome, atherosclerosis and cancer. Here, I review the structure, ligands, signalling modes and functions of TREMs in humans and mice and discuss the challenges that remain in understanding TREM biology.
Subject terms: Immunology, Innate immunity
In this Review, Marco Colonna provides a comprehensive summary of the triggering receptor expressed on myeloid cells (TREM) family of receptors. TREMs are important for modulating signalling in myeloid cells and have now been implicated in many different disease settings, including inflammatory diseases, autoimmunity, neurodegeneration and cancer.
Introduction
Myeloid lineage cells have crucial functions in immune responses as well as in the development, homeostasis and remodelling of all tissues; they recognize soluble mediators in the tissue microenvironment and surface ligands expressed on neighbouring cells through multiple receptors. Pattern recognition receptors, such as Toll-like receptors (TLRs), NOD-like receptors and scavenger receptors, signal the presence of molecular patterns that portend microbial invasion or tissue damage. Other receptors perceive the state of the organism and help to set thresholds for cellular responses to each molecular pattern encountered, ultimately controlling the amplitude and duration of the response. By integrating the responses of all receptors, myeloid cells can generate a carefully graded response to a specific set of conditions.
Triggering receptors expressed on myeloid cells (TREMs) are a family of cell-surface receptors expressed broadly on myeloid cells. The first TREM identified, TREM1, was characterized as an amplifier of the immune response that potentiates granulocyte and monocyte responses to microbial products1. The TREM family has since been extended to include proteins expressed on granulocytes, monocytes and tissue macrophages, as well as on macrophage and dendritic cell (DC) lines. Generally, TREMs appear to function primarily as modulators that define the threshold and duration of myeloid cell responses. TREMs have been found to have both positive and negative functions in regulating the activation and differentiation of myeloid cells and have been implicated in multiple conditions and diseases.
In this Review, I detail the genomic organization, structure and signalling mechanisms of TREMs, and extensively discuss the functions of TREM1 and TREM2, which are the best-characterized TREMs. I briefly review our knowledge of other, less-investigated TREM family members. Finally, I summarize the challenges that remain in understanding TREM biology and harnessing our knowledge for therapeutic intervention in human diseases.
The TREM family of receptors
TREMs are evolutionary conserved
TREMs encompass a family of related immunoglobulin superfamily cell-surface receptors with a single extracellular V-type immunoglobulin domain that sits on a stalk in the extracellular region (Fig. 1). Human and mouse TREMs are encoded by gene clusters on human chromosome 6p21.1 and mouse chromosome 17C, respectively (Fig. 2). The human cluster includes NCR2 (encoding NKp44), TREM1, TREML4 (encoding TREM-like 4), TREML2, TREM2 and TREML1. The mouse cluster includes Trem5, Trem4, Trem1, Trem3, Treml4, Treml2, Treml6, Trem2 and Treml1 (gene order from centromere to telomere). TREMs are conserved from teleost fish to humans2–4. However, their numbers and signalling motifs vary across species (Fig. 2), suggesting that TREMs evolved through duplication, followed by selection of variants by yet unknown factors, perhaps including infectious agents. The closest relatives of TREMs include the CD300 family members (also known as CMRF or IREM receptors in humans5–8 and CLM receptors in mice9) and the polymeric immunoglobulin receptor. Other structurally related but evolutionary distant receptors include VISTA and TIM3. Part of the TREM gene cluster, NCR2, encodes the NKp44 receptor and is expressed by natural killer cells, type 1 and type 3 innate lymphoid cells, rather than by myeloid cells. The unique biology of the NKp44 receptor is not examined here, as it has been reviewed elsewhere10.
Fig. 1. General structure of the TREM molecules.
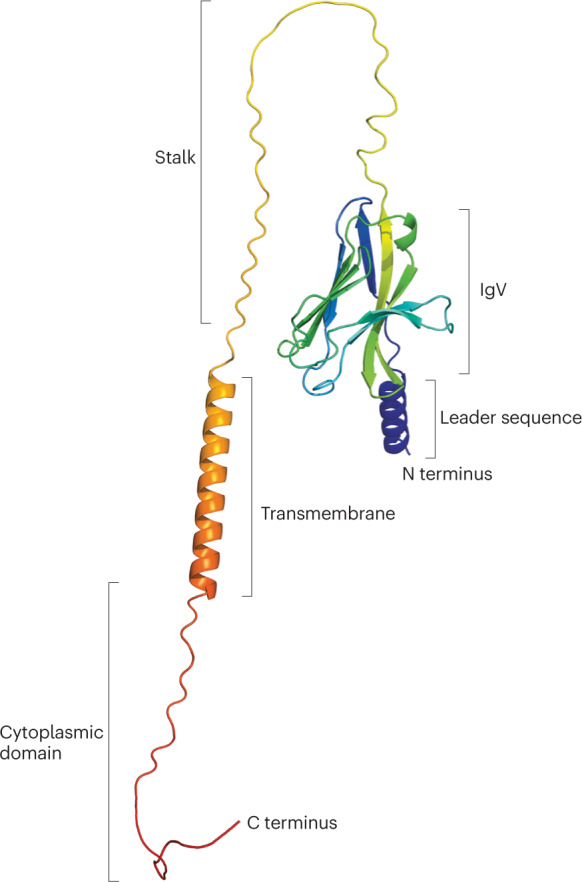
A three-dimensional model of the structure of the triggering receptors expressed on myeloid cells (TREMs), based on the three-dimensional TREM2 structure predicted by AlphaFold219. The extracellular region of the TREMs consists of an IgV fold on a long stalk. The N terminus leader sequence and the C terminus transmembrane and cytoplasmic domains are also depicted.
Fig. 2. Human and mouse TREM gene clusters.

The triggering receptor expressed on myeloid cell (TREM) gene clusters are located on human chromosome 6p21.1 and mouse chromosome 17C. Both clusters include genes encoding TREM1, TREM2, TREML1, TREML2 and TREML4. The human cluster includes NCR2, which encodes NKp44, a receptor expressed on natural killer cells and group 3 innate lymphoid cells. The mouse cluster contains additional genes encoding TREM3, TREM4, TREM5 and TREML6. Genes highlighted in green associate with DNAX-activating protein 12 kDa (DAP12), although TREM2 also associates with DAP10. TREML6 (gene indicated in red) is a bona fide inhibitory receptor with cytoplasmic immunoreceptor tyrosine-based inhibitory motifs. TREML1 (gene highlighted in yellow) contains a cytoplasmic motif similar to an immunoreceptor tyrosine-based inhibitory motif but delivers activating signals. The signalling properties of TREML2 (gene highlighted in grey) remain unknown. Arrows indicate the orientation of the gene. For additional details, see ref. 2.
TREM signalling as part of an integrated network
Most TREMs have a short cytoplasmic domain that lacks obvious signalling motifs. However, a positively charged amino acid present in the transmembrane domain enables pairing with one or more transmembrane adapters that contain a negatively charged residue in the transmembrane domain and a signalling motif in the cytoplasmic domain. The most prominent adapter is DNAX-activating protein of 12 kDa (DAP12), encoded by TYROBP, which contains a cytoplasmic immunoreceptor tyrosine-based activation motif (ITAM)11 (Fig. 3). In response to TREM receptor ligation, SRC kinases phosphorylate ITAM tyrosines that form a docking site for the protein tyrosine kinase SYK, which instigates activation of scaffolding proteins and downstream signalling molecules. These include phosphatidylinositol 3-kinase (PI3K), linker for activation of T cells family member 2 (LAT2), phospholipase Cγ2, guanine exchange factor VAV family members and the E3 ubiquitin ligase CBL. PI3K triggers the kinase AKT, which activates mTOR and inactivates glycogen synthase kinase 3β (GSK3β). Activation of the mTOR pathway supports protein synthesis and energy metabolism12. Inactivation of GSK3β stabilizes β-catenin, thereby promoting cell survival and proliferation13. Phospholipase Cγ2 degrades phosphatidylinositol 4,5-biphosphate into inositol 1,4,5-triphosphate and diacylglycerol, which induce intracellular Ca2+ mobilization and NF-κB activation, respectively. VAV family members promote actin cytoskeleton rearrangement, whereas CBL acts as a negative regulator; LAT2 is a hub for other signalling molecules but has also been reported as a negative signalling regulator14. Although the ITAM signalling pathway in T cells has been shown to activate transcription factors such as AP-1, NFAT and NF-κB, which drive gene expression, transcription factors downstream of the TREM–DAP12 pathway have not been identified, with the exception of early growth response 2 (EGRF2)15, and thus represent an important area of future research.
Fig. 3. TREM activating signals through DAP12 and DAP10.

Engagement of activating triggering receptors expressed on myeloid cells (TREMs) by specific ligands triggers phosphorylation of the immunoreceptor tyrosine-based activation motifs (ITAMs) located in the cytosolic domain of homodimeric DNAX-activating protein 12 kDa (DAP12) by an SRC kinase family member. The phosphorylated ITAMs recruit the protein tyrosine kinase SYK, which is also phosphorylated and activated by the SRC kinase. SYK activates multiple downstream targets and scaffolds, including phosphatidylinositol 3-kinase (PI3K) comprising the p110 and p85 subunits (part a), the adapter linker for activation of T cells family member 2 (LAT2) (parts b,c) and β2 integrins (part d). In part a, PI3K activates AKT, which in turn activates mechanistic target of rapamycin (mTOR) signalling. AKT also phosphorylates glycogen synthase kinase 3β (GSK3β), resulting in GSK3β inactivation, stabilization of β-catenin and cell cycling. In part b, LAT2 phosphotyrosines form docking sites for phospholipase Cγ2 (PLCγ2), which degrades phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) into inositol 1,4,5-triphosphate (Ins(1,4,5)P3) and diacylglycerol (DAG) to elicit Ca2+ mobilization and NF-κB activation, respectively. In part c, LAT2 phosphotyrosines also form docking sites for the adaptors GRB2 and SLP76. These adaptors initiate the mitogen-activated protein kinase (MAPK) pathway and recruit guanine exchange factors of the VAV family, which promote rearrangement of the actin cytoskeleton. In part d, β2 integrins require SYK signals to acquire an open and active conformation that can bind to ligands. TREM2 and perhaps other DNAX-activating protein 12 kDa (DAP12)-associated TREMs also recruit homodimeric DAP10, which contains an YXNM motif that directly recruits the p85 subunit of PI3K and the adapter GRB2, activating the pathways highlighted earlier. Of note, signalling pathways presented here are generalized and may vary depending on the TREM, the cell type in which the receptor is expressed and the physiological or pathological context. PtdIns(3,4,5)P3, phosphatidylinositol 3,4,5-trisphosphate.
DAP12 signalling intermingles with other signalling pathways. DAP12 and SYK activation are required for β2 integrin signalling16,17 (Fig. 3). Conversely, DAP12 modulates TLR signalling, particularly activation of extracellular signal-regulated kinase (ERK)18,19 (Fig. 4). One study demonstrated that the DAP12 ITAM binds to the phosphotyrosine-binding domain of downstream of kinase 3 (DOK3). In turn, DOK3 recruits the adapters GRB2 and SOS1 that are required to initiate ERK signalling, sequestering them from TLRs20. DAP12 and DOK3 can also recruit the inositol phosphatase SHIP1, which can also cross-regulate intracellular signalling pathways. In addition to DAP12, TREMs can associate with another transmembrane adapter resembling DAP12, known as DAP10, encoded by HCST21,22 (Fig. 3). Like DAP12, DAP10 contains a negatively charged residue in the transmembrane domain that enables binding to TREMs. However, DAP10 lacks a canonical cytoplasmic ITAM and instead contains a cytoplasmic YXNM motif, similar to that identified in the T cell co-stimulatory molecules CD28 and ICOS. This motif can directly recruit the p85 subunit of PI3K11 and GRB2 (refs. 23,24). Overall, the integration of DAP12 and DAP10 signalling with other signal transduction pathways creates a network that facilitates regulation of cellular responses to extracellular stimuli. Of note, TREM-mediated signals highlighted here and their functional outcomes may vary depending on the individual TREM molecule, the cell type in which TREMs are expressed and the physiological and pathological contexts in which TREM signals occur.
Fig. 4. TREM-modulating signals through DAP12.

DNAX-activating protein 12 kDa (DAP12) can deliver inhibitory signals that modulate Toll-like receptor (TLR) signalling. In part a, DAP12 immunoreceptor tyrosine-based activation motifs (ITAMs) bind the phosphotyrosine-binding domain of downstream of kinase 3 (DOK3), which in turn recruits the adapters GRB2 and SOS, subtracting them from TLRs and thereby interfering with the capacity of TLRs to initiate extracellular signal-regulated kinase (ERK) signalling. In part b, DAP12 and DOK3 can also recruit the inositol phosphatase SHIP1, which degrades phosphatidylinositol 3,4,5-trisphosphate (PtdIns(3,4,5)P3) to phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2), regulating the recruitment and activation of intracellular signalling molecules tethered to the membrane through PtdIns(3,4,5)P3.
Biology of TREM1
TREM1: an amplifier of inflammation in pathogenic infections
TREM1 is expressed on neutrophils, monocytes and some tissue macrophages, such as alveolar macrophages. Initial studies linked TREM1 to microbial infections: TREM1 was found to be upregulated in response to bacterial lipopolysaccharide (LPS) and other microbial products; immunohistochemistry revealed strong expression in the inflammatory infiltrates of tissues affected by bacterial infections25,26. Of note, the ligands of TREM1 are still poorly understood (Box 1). Although antibody-mediated stimulation of TREM1 alone induced modest cellular activation and cytokine secretion, combined stimulation with anti-TREM1 antibody and ligands of TLRs25,26 or NOD-like receptors27 resulted in synergistic cytokine secretion, suggesting that TREM1 is an amplifier of the inflammatory response in the context of microbial infections. In vivo studies using mouse models of sepsis induced by LPS, polymicrobial abdominal infection or Pseudomonas aeruginosa corroborated this function. Modulation of TREM1 signalling in these models using soluble decoys — for instance, the extracellular domain of TREM1 fused with IgG-Fc (TREM1-Fc) or the synthetic peptide LP17, which mimics a highly conserved domain of TREM1 — attenuated blood levels of inflammatory cytokines and improved survival25,28,29. These results were further supported by studies in DAP12-deficient mice, which found that abrogating DAP12 signalling attenuated the inflammatory response to septic infection30. Together, these studies suggested that careful modulation of TREM1 signalling may be beneficial in infections and sepsis by attenuating excessive inflammation without markedly affecting pathogen elimination by neutrophils and monocytes. However, complete abrogation of TREM1 signalling does impair control of infection, at least in the case of certain pathogens. P. aeruginosa-induced pneumonia in mice lacking TREM1 was associated with elevated production of local and systemic cytokines along with higher mortality than seen in control mice31. TREM1 deficiency impaired transepithelial migration of neutrophils into the airspace, preventing neutrophils from reaching the site of infection and killing bacteria. A follow-up study showed that TREM1 enhances neutrophil chemotaxis by promoting NOX-dependent superoxide production32. TREM1-deficient mice were more susceptible to pneumonia induced by Streptococcus pneumoniae33, whereas antibody-mediated TREM1 activation improved host defence34. In mice infected with Klebsiella pneumoniae, TREM1 deficiency enhanced bacterial translocation in the small intestine, increasing the generation of liver abscesses and mortality35, whereas the control of infections with Leishmania major, Legionella pneumoniae and influenza virus was unaffected in TREM1-deficient mice36. In conclusion, TREM1-mediated amplification of inflammation may have variable consequences on host defence, ranging from effective pathogen control to exaggerated and detrimental inflammation, depending on the context. These variable effects impact the potential for therapeutic manipulation of TREM1.
Box 1 TREM1 ligands.
Despite considerable progress in the functional characterization of triggering receptor expressed on myeloid cells 1 (TREM1), TREM1 ligands remain incompletely defined. Two studies have implicated peptidoglycan recognition protein 1 as a ligand for TREM1, either as a complex with bacterially derived peptidoglycan220 or as an N-terminal peptide221. Another study has shown that TREM1 binds to the extracellular cold-inducible RNA-binding protein eCIRP, an endogenous molecule that can be released by damaged tissues222. Whether these molecules or other ligands activate TREM1 in mice or in humans is poorly understood. Two independent crystallographic structures of TREM1 have been solved. In one structure, TREM1 forms a unique head-to-tail dimer in solution and has two potential ligand-binding surfaces on the basis of surface conformation and electrostatic potential223. In another structure, TREM1 exists as a monomer224. However, no structure of TREM1 in a complex with any of the reported ligands has been solved. Identification of TREM1 ligands and their molecular interaction with TREM1 is an important goal for the field.
Soluble TREM1 is a biomarker and a potential treatment in sepsis
The extracellular portion of TREM1 is produced and released as soluble TREM1 (sTREM1). A major source of sTREM1 is the translation of a TREM1 mRNA splice variant lacking the sequence encoding the transmembrane and cytoplasmic regions of TREM1 (refs. 37,38). However, proteolytic cleavage of surface-expressed TREM1 by matrix metalloproteases may also contribute to sTREM1 production39,40. sTREM1 was detected in vivo in the plasma of mice challenged with LPS or subjected to polymicrobial abdominal infection28. Clinical studies reported detection of sTREM1 in serum and bronchioalveolar lavage fluid of patients with pneumonia41. sTREM1 in the bronchioalveolar lavage fluid was more accurate than any other clinical or laboratory finding in predicting the presence of bacterial or fungal infection. Further studies in sepsis have corroborated that increased sTREM1 plasma concentrations correlate with mortality, but are insufficient to predict mortality alone42. sTREM1 was also elevated in the plasma of patients with cystic fibrosis and correlated with levels of proteases43. Very recently, sTREM1 plasma concentrations were found to be significantly elevated in patients with COVID-19 compared with healthy controls44. sTREM1 together with IL-6 concentrations was an accurate predictor for intubation, mortality and oxygen requirement in patients with COVID-19 in intensive care units45. In addition to the use of sTREM1 as a prognostic biomarker, research has focused on sTREM1 as a treatment in sepsis. In a recent phase IIa clinical trial, patients with septic shock were treated with nangibotide (also known as LR12), a 12-amino acid peptide that acts as a ligand-trapping molecule, modulating the TREM1-mediated amplification of inflammatory-response46. The study has demonstrated LR12 safety and revealed trends towards clinically relevant benefits. A phase IIb clinical trial in patients with septic shock is currently underway to further explore this effect (NCT04429334, ClinicalTrials.gov).
TREM1 sustains non-infectious inflammation
Beyond infections, TREM1 has been implicated in many sterile inflammatory processes. TREM1 potentiated neutrophil-mediated inflammation in gout, which is induced by deposition of monosodium urate monohydrate crystals47. Overexpression of the TREM1-Fc decoy in the liver by adenoviral gene transfer tempered hepatic granulomatous inflammation elicited by zymosan48. TREM1 deficiency was beneficial in a mouse model of chronic liver injury and fibrosis induced by carbon tetrachloride administration. In this model, TREM1 was highly upregulated on Kupffer cells, circulating monocytes and monocyte-derived macrophages; lack of TREM1 restricted Kupffer cell activation, infiltration of inflammatory cells, liver injury and fibrogenesis49. TREM1 also amplifies inflammatory responses in cardiovascular and cerebrovascular diseases; TREM1 was expressed in advanced human atheromas and myeloid cells infiltrating vascular lesions in mouse models of dyslipidaemia and atherosclerosis50. Genetic deletion of TREM1 substantially attenuated diet-induced atherogenesis by moderating pro-atherogenic cytokine production and foamy cell formation induced by lipids. TREM1 expression was also upregulated in ischaemic myocardium after infarction in mice and humans51. Genetic deletion of TREM1 or pharmacological blockade using the LP17 peptide attenuated myocardial inflammation, neutrophil recruitment, release of CCL2 and, consequently, monocyte mobilization to the heart, ultimately resulting in improved left ventricular function and survival. Moreover, TREM1 induced monocyte recruitment and inflammation in a mouse model of angiotensin II-induced abdominal aortic aneurysm52. During stroke, the initial cerebral ischaemia is followed by secondary injury owing to immune responses to ischaemic brain components. TREM1 was shown to be induced, not only in the brain but also in the gut, following stroke and to amplify inflammatory responses to endogenous brain components as well as bacterial components derived from intestinal inflammation and leakage53. Overall, these studies suggest that targeting TREM1 may be helpful in limiting sterile inflammation, particularly in cardiac ischaemia and stroke in which this event can be particularly harmful. Beyond inflammation, a study proposed a role for TREM1 signals in the repair of kidney tubules after acute kidney injury54, extending the potential impact of TREM1 in human pathology.
TREM1 is a pathogenic factor in autoimmunity
In the setting of non-infectious inflammation, TREM1 has been implicated in autoimmune diseases, such as inflammatory bowel disease, rheumatoid arthritis, psoriasis, nephritis and systemic lupus erythematosus. Patients with inflammatory bowel disease had more TREM1+ macrophages in the gut lamina propria55 and higher serum concentrations of sTREM1 than did healthy controls, regardless of whether the disease was active or inactive56,57. In mouse models of colitis induced by T cell transfer or dextran sulfate sodium, disease, inflammatory infiltrates and expression of pro-inflammatory cytokines were markedly attenuated in Trem1–/– mice36. In comparison to healthy controls, patients with rheumatoid arthritis had relatively high plasma levels of sTREM1 that correlated with disease activity58. Moreover, TREM1 blockade using a recombinant adenovirus encoding TREM1-Fc or the LP17 peptide significantly ameliorated joint inflammation in mice with collagen-induced arthritis59. Similarly, blockade of TREM1 assuaged a model of nephritis induced by administration of an anti-glomerular basement membrane antibody60. Increased expression of TREM1 in the skin and plasma sTREM1 has been reported in psoriasis61; however, TREM1 deficiency had no functional impact in models of psoriasiform dermatitis, contact dermatitis or epidermolysis bullosa62. Finally, although patients with systemic lupus erythematosus had higher circulating levels of sTREM1 than did healthy controls63, TREM1 deficiency counterintuitively promoted autoimmunity in the Faslpr mouse model; in this case, autoimmunity was associated with enhanced DC production of BAFF64, suggesting a novel function of TREM1 in modulating BAFF expression through an as-yet undefined mechanism.
TREM1 contributes to cancer inflammation
Myeloid cells infiltrating the tumour microenvironment, such as tumour-associated macrophages (TAMs) and granulocytes, are key components of the inflammatory infiltrate of many solid human tumours that promote angiogenesis, immunosuppression, cancer progression and resistance to therapy65,66. Growing evidence suggests that TREM1+ myeloid cells contribute to tumour stroma inflammation and tumour progression. In humans, high levels of TREM1 and/or its soluble form have been associated with poorer survival in several solid malignancies, such as hepatocellular carcinoma67 and non-small-cell lung cancer68. TREM1 deficiency in the diethylnitrosamine-induced model of hepatocellular carcinogenesis attenuated Kupffer cell activation, curbing liver injury and ultimately tumour development69. In an orthotopic liver cancer model, hypoxia induced TREM1+ TAMs, which caused dysfunction of CD8+ T cells, tumour progression and resistance to checkpoint blockade. In this model, TREM1 signalling was inhibited with the nonapeptide GF9 that blocks the interaction of TREM1 with DAP12 without affecting ligand binding. This treatment reversed immunosuppression and resistance to the checkpoint inhibitor anti-PDL1 (ref. 70). The GF9 peptide also attenuated tumour growth and prolonged survival in a model of pancreatic cancer71.
Although TREM1 is mainly expressed in granulocytes and macrophages, it has been shown that TREM1 is induced in tumour-associated DCs through activation of the nuclear receptors RXR, RAR, LXR and VDR72. In patients with cancer, TREM1 was found in DCs from pleural effusions and ascites, which contain the LXR ligands such as oxysterols and the RXR ligand such as 9-cis retinoic acid; moreover, in vitro exposure of monocyte-derived DCs to tumour-conditioned fluids induced the expression of TREM1 together with LXR and RAR target genes. TREM1 engagement enhanced DC release of TNF and IL-1β, suggesting an active role of TREM1+ DCs in inflammation-driven cancer. Accordingly, in bone marrow chimeric mice, B6 mice reconstituted with Trem1–/– bone marrow controlled Lewis lung carcinoma better than B6 mice reconstituted with wild type bone marrow. Together, these studies suggest that TREM1 may be both a potential biomarker and a therapeutic target in human oncology. An anti-TREM1 antibody is currently in a phase 1a–1b FIH study in combination with pembrolizumab in patients with advanced solid tumours (NCT04682431, ClinicalTrials.gov), with the goal of reprogramming TAMs towards an immunostimulatory phenotype.
Biology of TREM2
TREM2 is a lipid-binding receptor
TREM2 is an activating receptor associated with DAP12 and DAP10. It is expressed by tissue macrophages, both on the cell surface and in intracellular pools. TREM2+ macrophages include microglia in the central nervous system, osteoclasts in the bone and macrophage subsets in the liver, adipose tissue, skin, gut and tumours. TREM2 binds to phospholipids and sulfatides73, enabling recognition of apoptotic cells that expose phospholipids, and lipoproteins such as high-density lipoprotein and low-density lipoprotein74. Among lipoproteins, much attention has been focused on high-density lipoprotein containing apolipoprotein E (APOE)75,76, which is particularly abundant in the brain. TREM2 also binds to microbial lipids, such as certain bacterial LPS77 and non-glycosylated mycolic acids of mycobacteria78. The structural basis for lipid binding remains unclear. An initial characterization of the TREM2 three-dimensional structure identified cationic and hydrophobic regions in the immunoglobulin domain of TREM2 that were predicted to bind to the polar head and the acyl chains of phospholipids, respectively79. However, another TREM2 three-dimensional structure together with phosphatidylserine found that TREM2 forms trimers of dimers with a phospholipid-binding site of the interface of each dimer80.
TREM2 has also been reported to bind to proteins with a strong tendency to aggregate and accumulate in neurodegenerative diseases. TREM2 binds to neurotoxic β-amyloid (Aβ) peptides, which form amyloid plaques in Alzheimer disease (AD)81–83. As Aβ binds to many receptors with various binding affinities, it will be important to elucidate the structural basis of this interaction and how different receptors contribute to cellular responses to Aβ. A recent study also showed TREM2 binding to TDP-43, a nuclear protein that becomes mislocalized to the cytoplasm, forming inclusion bodies implicated in frontotemporal dementia and amyotrophic lateral sclerosis84. Whether TREM2 interacts in vivo with neuronal intracellular inclusion requires further investigations. Additional TREM2-binding partners have been reported. One study has proposed that TREM2 binds to galectin-3, a β-galactoside-binding lectin broadly expressed in the human body85. However, it should be noted that galectin-3 binds to numerous surface proteins decorated with glycans. Endogenous TREM2 ligands have also been hypothesized to exist on astrocytoma cell lines77 and bone marrow-derived macrophages86, but have never been identified. Finally, TREM2 has been proposed to form a complex in cis with plexin-A1, which binds to semaphorin 6D for axon guidance, bone remodelling and cardiac morphogenesis. In these circumstances, TREM2 may function as a co-receptor for plexin-A1 (ref. 87). Overall, the TREM2–lipid interaction is quite established, whereas other interactions may require more substantial validation through structural and functional studies.
TREM2 impacts microglia and osteoclast functions
TREM2 was originally identified and characterized in vitro in human DCs generated from blood monocytes cultured in vitro with GM–CSF and IL-4 (ref. 88) and a mouse macrophage cell line89. The first insights into the function of TREM2 in natura came from studies of a rare disease known as Nasu–Hakola disease (NHD). NHD is an autosomal recessive disorder that causes joint swelling and recurring bone fractures during adolescence, followed by an early-onset frontal-type dementia that ultimately causes death before the fifth decade90. Pathological features include multiple bone cysts and a sclerosing leukoencephalopathy characterized by substantial loss of myelin and gliosis. Genetic studies have shown that NHD is caused by homozygous loss-of-function mutations in either TYROBP (encoding DAP12) or TREM2 genes91,92. Similar TREM2 mutations can also cause frontotemporal dementia with no bone pathology93,94. These studies suggested an essential function of TREM2 in osteoclasts and microglia, the macrophages of the bone and brain, respectively.
Accordingly, in vitro studies have shown that monocytes and bone marrow cells derived from patients with NHD or TREM2-deficient or DAP12-deficient mice were unable to generate multinucleated osteoclasts that resorb bone95–99. Manipulation of TREM2 expression also affected multiple functions of cultured microglia: TREM2 overexpression enhanced chemokine receptor expression, cell migration and phagocytosis; conversely, attenuation of endogenous TREM2 expression hindered phagocytosis of apoptotic cells but augmented transcription of inflammatory cytokines100. However, these in vitro studies have provided limited insight into how TREM2 impacts such a wide range of macrophage functions. Moreover, mice lacking either TREM2 or DAP12 did not reproduce the strong clinical features of NHD. DAP12-deficient and TREM2-deficient mice showed low-to-mild osteopetrosis in vivo rather than developing the bone cysts seen in patients with NHD97,98,101. In addition, neuropathology in DAP12-deficient and TREM2-deficient mice was much milder than in NHD sclerosing leukoencephalopathy: only moderate myelin reduction in certain brain regions and slight impairment of neuronal synapse functions and behaviour were observed97,98. Why mouse models do not recapitulate NHD pathology remains unclear.
Microglia require TREM2 to restrain Alzheimer disease pathology
In 2013, heterozygous hypomorphic variants of TREM2, chiefly TREM2R47H, have been found to increase risk for developing AD102,103. Thus, the TREM2–DAP12 pathway and its impact in microglia biology gained much broader attention. AD is the leading cause of late-onset dementia among the elderly, affecting millions of individuals worldwide104. The pathology of AD is quite different from that of NHD: it initiates with the formation of brain extracellular aggregates of Aβ peptides. Aβ deposits elicit hyperphosphorylation of neuronal tau, which forms intracellular cytotoxic aggregates that cause neuron and synaptic loss, ultimately leading to cognitive impairment. Familial early-onset AD is linked to gene variants that facilitate the processing of amyloid precursor protein into Aβ105. However, the risk of sporadic late-onset AD is associated with multiple gene variants, among which TREM2R47H and others have pinpointed the involvement of genes expressed in microglia. The AD-associated TREM2R47H variant is unable to effectively bind to phospholipid ligands73–76. The structural basis of impaired ligand recognition is unclear. In one structural study of TREM2, R47H and other AD-associated variants, such as R62H, affected the cationic surface predicted to bind to the polar head of phospholipids79. However, another structure of TREM2R47H indicated that the R47H mutation may induce conformational changes that indirectly affect a distinct phospholipid-binding site generated by TREM2 oligomerization80. Studies involving mouse models of Aβ accumulation and in patients with AD have demonstrated that the hypofunctional TREM2R47H variant impairs the ability of microglia to encase Aβ plaques, facilitating spreading and neurotoxicity of Aβ plaques as well as progression of AD106,107. A similar phenotype was observed in AD models in mice that lacked TREM2 (refs. 73,108,109) or that were grafted with human microglia lacking TREM2 (ref. 110). Conversely, overexpression of TREM2 protected from Aβ pathology111.
Characterization of the microglial transcriptome in TREM2-deficient mice during accumulation of Aβ plaques demonstrated that TREM2 is required to sustain microglia acquisition of an Aβ-responsive programme widely known as disease-associated microglia107,112,113. This programme entails the expression of TREM2, integrins (Cd11c), pro-inflammatory chemokines (Ccl4), extracellular matrix proteins (Spp1) and genes directing lipid metabolism (Lpl), cholesterol efflux (ApoE) and lysosomal function (Ctsb, Ctsd and Cts7) (Table 1). In parallel with disease-associated microglia, Aβ induces additional distinct microglial responses delineated by an interferon-response profile, MHC class II expression114 and proliferation, all of which are also TREM2-dependent115,116.
Table 1.
Signatures of TREM2+ macrophages in human diseases and mouse models
| Species | Organ, tissue or cell type | Gene signature associated with TREM2+ macrophages | Refs. | |||||
|---|---|---|---|---|---|---|---|---|
| Lipid and cholesterol metabolism genes | Tetraspanin genes | Phagocytic genes | Galectin genes | Osteopontin | Other genes | |||
| Mouse | Microglia | ApoE, Lpl | Cd9 | Cst7, Ctsd | – | Spp1 | Clec7a, Cd11c, Tyrobp, C1q | 112 |
| Adipose tissue from animals on a high-fat diet | Lpl, Lipa, Cd36, Fabp4, Fabp5 | Cd9 | Ctsb | Lgals1, Lgals3 | – | Ctsl | 159 | |
| Atherosclerotic lesions | Fabp4, Abcg1 | Cd9 | Ctsd, Ctsb, Ctsz | Lgals3 | Spp1 | Hvcn1 | 162,163 | |
| Liver in nonalcoholic steatohepatitis or nonalcoholic fatty liver disease | – | Cd9, Cd63 | – | – | Spp1 | Gpnmb | 165,166 | |
| Sarcoma tumours | – | Cd9, Cd63 | – | – | – | Arg1, Vgefa, Cx3cr1, Mrc1, Clec4d | 175,176 | |
| Human | Liver, cirrhosis | – | CD9 | – | LGALS3 | SPP1 | C1QC, IL1B, CCR2, TNFSF12, CXCL8, PDGFB, VEGFA | 164 |
| Skin, acne | APOE, LPL | – | CTSB | LGALS3 | SPP1 | CD68, GPNMB, TYROBP, CCL18, IL18, S100A8 | 173 | |
| Breast tumours | APOE, FABP5 | CD9 | – | – | SPP1 | FN1, C1QA, C1QB, C1QC | 182–184,186 | |
| Non-small-cell lung carcinoma tumours | APOE, APOC1 | – | – | – | SPP1 | MARCO, C1QA, C1QB, C1QC, CD163 | 178–181 | |
| Hepatocellular carcinoma tumours | FABP5 | – | – | – | SPP1 | CD163, FOLR2, PDL3, NUPR1 | 192,218 | |
TREM2, triggering receptor expressed on myeloid cells 2.
Why does the microglial response to Aβ require TREM2? The ability of the TREM2–DAP12–SYK complex to elicit PI3K–AKT signalling is crucial. In turn, AKT activates the mTOR signalling pathway, which sustains synthesis of proteins and production of energy that microglia require to cope with and respond to Aβ accumulation12,22. Moreover, AKT phosphorylates and inactivates GSK3β, enabling β-catenin-induced proliferation of microglia22,117. Finally, AKT supports cell survival. These studies provide a unifying mechanism for the wide-ranging microglial functions that have been attributed to TREM2, including phagocytosis, migration, cytokine secretion, as well as lipid and cholesterol metabolism118.
One outstanding question is what binds to and stimulates TREM2 in AD. TREM2 binds to phospholipids and sulfatides; hence, apoptotic cells, lipidated APOE and myelin debris are all potential candidates. In addition, TREM2 can bind to Aβ, stimulating intracellular activating signals and Aβ phagocytosis. A few reports have suggested that TREM2 may also recognize non-lipidated APOE119, which is a component of Aβ plaques that enhances their compactness120. In turn, TREM2-activated microglia produce APOE107,112,113, which binds to plaques, generating an autocrine circuit that sustains the microglial response to Aβ plaques121–123. Supporting this notion, in APOE-deficient mice, microglia fail to cluster around amyloid plaques, resulting in reduced Aβ plaque compaction and increased neurite dystrophy as observed in TREM2-deficient mice124. However, it should be noted that APOE has a complex function in lipid metabolism that may impact AD pathogenesis independently of TREM2 activation. Moreover, APOE is encoded by three polymorphic alleles, APOE2, APOE3 and APOE4, among which the APOE4 allele increases the risk of AD compared with the most prevalent APOE3 allele, whereas the APOE2 allele reduces risk125. Whether TREM2 binds to all APOE isoforms with similar affinity75,76 or has higher affinity for APOE4 (ref. 110) is unclear. Thus, future studies should determine whether the interaction with TREM2 contributes to the known effect of APOE4 on disease risk.
Although TREM2 supports the microglial response to Aβ plaques, the impact of TREM2 in regulating intracellular tau pathology in mouse models is a matter of debate. Mice lacking TREM2 or carrying the hypofunctional R47H variant of TREM2 exhibited less severe pathology in the P301S model of taupathy, suggesting that TREM2 activation may be detrimental at late disease stages when tau pathology is prevalent126,127. However, TREM2 deficiency aggravated pathology in several models, namely, in humanized tau mice that lack the endogenous tau128, in Aβ plaque-bearing mice injected with human tau prepared from patients with AD129 and in mice that develop Aβ plaques along with tauopathy pathology and neurodegeneration130. Regardless of the reasons underlying these discrepancies, given the established importance of the TREM2 pathway in eliciting microglial responses to Aβ, perhaps the most crucial question is whether we can harness TREM2 for AD therapy. Three studies have shown that an agonistic anti-TREM2 antibody injected in the circulation can cross the blood–brain barrier, reach the brain parenchyma and activate microglia in mouse models of AD115,116,131. Importantly, the first phase II clinical study evaluating the efficacy and safety of an anti-TREM2 antibody in slowing AD progression is currently ongoing (NCT04592874, ClinicalTrials.gov). Results of this trial will be essential to gauge the suitability of TREM2 as a therapeutic target in AD and perhaps in other neurodegenerative diseases.
TREM2 sustains microglial processing of myelin
Microglia contribute to myelination remodelling in the central nervous system during development and homeostasis; in response to pathology that causes myelin damage, microglia clear myelin debris, allowing replacement of damaged myelin with newly formed myelin and they neutralize toxicity of oxidized lipid by-products132,133. TREM2 binds to myelin lipids, phagocytoses them and delivers intracellular signals that induce metabolic programmes of lipid catabolism and cholesterol efflux. The impact of TREM2 in this process has been clearly demonstrated in a model of demyelination induced by cuprizone, a toxic substance that causes death of oligodendrocytes and massive demyelination in the corpus callosum. A marked defect in the ability of microglia to clear myelin debris in TREM2-deficient mice treated with cuprizone was evident, resulting in delayed remyelination134,135. Furthermore, TREM2 deficiency impaired microglial expression of genes controlling lipid catabolism and cholesterol secretion134,135. This defect caused microglial accumulation of lipid droplets and biased their composition towards the accumulation of cholesteryl ester and their oxidized derivatives118. Consistent with these results, TREM2-deficient microglia were also unable to cope with excessive myelin debris generated in another model of demyelination induced by local injection of lysolecithin136. Overall, these reports support the possibility of harnessing TREM2 for treating demyelinating diseases. An encouraging observation in this direction is that the administration of an agonistic TREM2 antibody promoted myelin clearance and remyelination in the cuprizone model137.
Soluble TREM2 as a predictor of Alzheimer disease pathology
A remarkable feature of TREM2 is that the extracellular domain is shed by proteases such as ADAM10 and ADAM17 at the H157–S158 bond within the stalk region, leading to the generation of sTREM2 (refs. 138–140). This phenomenon explains early reports showing that TREM2 expression is rapidly lost when myeloid cells are incubated with either interferon-γ (IFNγ) or LPS, which activate proteases18,88. However, TREM2 surface expression is also regulated in part by ligand-induced internalization. Indeed, antibodies to the TREM2 extracellular immunoglobulin domain that do not affect the putative ligand-binding sites were shown to induce TREM2 internalization upon engaging the target, blocking TREM2 cleavage141. A further source of sTREM2 may be provided by an alternatively spliced transcript that lacks the transmembrane domain142. The biological significance of TREM2 shedding is being actively investigated. Mutations of H157 that block shedding augment TREM2 cell-surface expression and signalling143. Moreover, sTREM2 may have its own functions: it may scavenge TREM2 ligands, further curtailing TREM2 activation; it may have a prosurvival effect on other cells, perhaps by interacting with a yet unknown receptor144,145.
The impact of TREM2 shedding in AD has been actively investigated. A rare human TREM2 H157Y variant that sheds more rapidly is a risk factor for AD in a Chinese cohort146, suggesting that shedding is detrimental in AD. However, stabilization of TREM2 surface expression in mice accelerated Aβ pathology143. Moreover, variants of the tetraspanin MS4A associated with reduced AD risk were correlated with increased levels of sTREM2 in the cerebrospinal fluid (CSF), suggesting a protective role of sTREM2 (refs. 147,148). A beneficial effect on Aβ pathology has also been reported after brain injection of sTREM2 (ref. 149). As sTREM2 can bind to amyloid aggregates106, it may contribute to compacting amyloid plaques, reducing diffuse neurotoxic Aβ plaques that damage adjacent neurons. Regardless of its function in disease progression, sTREM2 can be a useful marker of AD pathology and cognitive decline. CSF levels of sTREM2 in patients with AD versus healthy controls positively correlated with the levels of the neurodegenerative markers Aβ42 and total levels of tau and phosphorylated tau, indicating co-occurrence of microglial activation with AD pathology150–153. Moreover, longitudinal studies of patients with AD showed that a higher ratio of sTREM2 versus phosphorylated tau in the CSF predicts slower cognitive decline; in addition, high CSF concentrations of sTREM2 were associated with attenuated amyloid and tau positron-emission tomography154,155. Thus, higher CSF levels of sTREM2 in patients with AD indicate longitudinal reduction of disease progression. Of note, administration of an anti-TREM2 antibody in humans for AD therapy caused a dose-dependent decrease in sTREM2 in the CSF115, suggesting that sTREM2 can also be a useful biomarker in AD therapy to trace target engagement. Finally, an increase in sTREM2 levels in the CSF can indicate microglial activation in other neurodegenerative and neuroinflammatory conditions such as multiple sclerosis156.
TREM2 involvement in metabolic disorders and atherosclerosis
Beyond neurological disorders, TREM2 has been implicated in the pathogenesis of other diseases, such as metabolic syndrome. People who are obese tend to develop a metabolic syndrome encompassing insulin resistance and various risk factors for cardiovascular disease157. Metabolic syndrome is in part caused by chronic low-grade inflammation resulting from immune responses to lipid accumulation in the adipose tissue158. A study in mice with insulin resistance induced by a high-fat diet revealed an accumulation of TREM2+ macrophages, called lipid-associated macrophages (LAMs), which formed crown-like structures around hypertrophic adipocytes159. Mainly derived from blood monocytes, LAMs acquired distinctive expression of Cd9, Cd63 and Gpmnb and exhibited a functional programme for lipid catabolism (Table 1). Genetic ablation of Trem2 inhibited the LAM programme and aggravated adipocyte hypertrophy159,160. However, the metabolic consequences of TREM2 deficiency in this model of obesity remain unclear. One study reported body fat accumulation, hypercholesterolaemia and insulin resistance159, whereas another study showed no effect on these parameters160. Further studies are necessary to resolve this conundrum.
A complex interaction between macrophages and lipid accumulation also occurs in atherosclerosis. Lipid accumulation in the arterial walls is paralleled by the recruitment of macrophages that phagocytose lipids through their scavenger receptors and become lipid-laden cells, called foamy cells, which form a major component of atherosclerotic lesions161. In mouse models of atherosclerosis, foamy macrophages express high levels of TREM2 as part of a transcriptional programme similar to that observed in LAMs162. This transcriptional profile is distinguished by high expression of CD9, markers of lipid loading and cholesterol efflux (Fabp4 and Abcg1), lysosomal cathepsins (Ctsd), galectin-3 (Lgals3) and Spp1 (ref. 163) (Table 1). Conversely, expression of inflammatory-response genes is low. Similar findings were made in human macrophages from atherosclerosis lesions. Whether TREM2 directly contributes to capture or lipid metabolism in foamy macrophages has not been tested.
TREM2+ macrophages have been implicated in liver cirrhosis, a form of progressive scarring of the liver secondary to various conditions, such as hepatitis and chronic alcoholism. A population of scar-associated TREM2+CD9+ macrophages with profibrogenic function differentiates from circulating monocytes and expands in human liver fibrosis164 (Table 1). TREM2+ macrophages have also been identified in mouse models of liver diseases caused by diets excessively rich in fats and calories, such as nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. A nonalcoholic steatohepatitis diet caused remodelling of Kupffer cell gene expression programmes, resulting in TREM2 and CD9 expression, a scar-associated phenotype and cell death165 (Table 1). Dead Kupffer cells were replaced by monocyte-derived macrophages that acquired convergent programmes, including TREM2 expression165–167. In a mouse model of nonalcoholic fatty liver disease characterized by steatohepatitis and fibrosis, TREM2 deficiency facilitated macrophage release of exosomes containing a microRNA affecting mitochondria homeostasis in hepatocytes168. Further functional investigation in TREM2-deficient mice will be required to establish the impact of TREM2 in various liver diseases and its suitability as a therapeutic target. As sTREM2 levels were found to be increased in the serum in human and mouse nonalcoholic steatohepatitis169, it will also be important to validate specificity and sensitivity of sTREM2 as a diagnostic or prognostic marker.
Of note, LAM, foamy macrophages and liver TREM2+ macrophages share the expression of several genes controlling lipid and cholesterol metabolism (Table 1). Some of these genes are also expressed in microglia in models of AD. This shared gene signature may be underpinned by common regulatory circuits utilizing transcription factors, such as the nuclear receptors LXR and RXR, which are known to control fatty acid metabolism and cholesterol efflux170. These regulatory circuits may also induce TREM2 to enhance lipid sensing, together with phagocytic receptors, such as AXL and MERTK, which are capable of engulfing apoptotic cells and lipidated particles171.
A TREM2–lipid connection in skin macrophages
Acne is a human skin disease characterized by excessive production of lipids, particularly squalene, secreted as sebum by the pilosebaceous unit. Increased sebum secretion, along with growth of the commensal Cutibacterium acnes and obstruction of the hair follicle, causes inflammation and clinical manifestations of acne172. Multipronged analysis of acne lesions revealed TREM2+ macrophages adjacent to hair follicles and sebaceous glands173. TREM2+ macrophages were close to the epithelium lining the hair follicle and sebaceous glands and expressed a lipid metabolic programme similar to that found in foamy macrophages of atherosclerotic lesions (Table 1). Furthermore, the addition of squalene to monocyte-derived macrophages induced TREM2 expression but inhibited phagocytosis of C. acnes, suggesting that these macrophages may contribute to the pathogenesis of acne. TREM2+ macrophages were also found within the hair follicles of mice. These macrophages produce oncostatin M, which maintains hair follicle stem cells in a state of quiescence, inhibiting hair growth174. How TREM2 contributes to the functions of skin TREM2+ macrophages is currently unknown.
TREM2 in cancer
TREM2 was recently found to be highly expressed on TAMs in various types of human cancer and mouse models and was implicated in TAM immunosuppressive functions. One study found that TREM2 was expressed in TAMs that infiltrate a wide variety of human tumours and three mouse models of sarcoma, colorectal cancer and breast cancer175; TREM2 deletion or blockade with a monoclonal antibody in these models constrained tumour growth, enhanced antitumour CD8+ T cell responses and modified the TAM landscape, altering the balance between immunosuppressive macrophages (CX3CR1+ or CD206+) and immunostimulatory macrophages (CXCL9+CD83+ or iNOS+)175. TREM2 deletion or functional blockade also synergized with anti-PD1 treatment in promoting antitumour CD8+ T cells, tumour rejection and reshaping of TAMs175. Another study characterizing immune cell subsets in a sarcoma model by single-cell RNA sequencing coupled with intracellular protein activity also identified an immunosuppressive TREM2+ macrophage population expressing Arg1, Gpnmb, Il7r, Cd63, Vegfa, Hilpda, Hmox1 and Clec4d176 (Table 1). Trem2 deletion in this model curbed tumour growth by facilitating cytotoxic T cell and natural killer cell responses, accompanied by waning of PD1-expressing and TIM3-expressing dysfunctional T cells176. Another study delineated the relationship of TREM2+ macrophages and T cell composition in human ovarian cancer as well as additional different tumours by single-cell RNA sequencing of CD45+ infiltrates177. Further analysis of thousands of tumours from The Cancer Genome Atlas and the Genotype Tissue-Expression Project confirmed broad TREM2 expression. TREM2+ TAMs were particularly enriched in ovarian cancer, and TREM2 expression correlated with the severity of the disease stage and worsened recurrence-free survival. Depletion of TREM2+ TAMs with an anti-TREM2-specific antibody curtailed tumour growth in an orthotopic ovarian cancer model as well as in models of colon and breast cancer; a combination of anti-TREM2 and anti-PD1 effectively controlled growth of an anti-PD1-resistant colon cancer model, although neither alone curbed tumour growth177. On the basis of this study, a TREM2-depleting antibody is currently being tested as a single agent or in combination with pembrolizumab for tumour therapy (NCT04691375, ClinicalTrials.gov).
In humans, TREM2-expressing TAMs have been extensively found in single-cell immune cell atlases of early and late metastatic non-small-cell lung cancer178–181. In these tumours, TREM2+ TAMs mainly derived from blood monocytes and expressed molecules associated with lipid metabolism, immunosuppression and complement activation; profuse TREM2+ TAM infiltration correlated with disease progression181. TREM2 has also been extensively studied in human breast cancer182–186. TREM2+ TAMs showed a transcriptome profile similar to that found in lung cancer, derived mainly from blood monocytes and were associated with immunosuppression and negative prognosis. Of note, TREM2 and folate receptor-β (FOLR2) expression were mutually exclusive, defining immunosuppressive and immunostimulatory TAMs, respectively184. Histological analyses further showed a different spatial distribution of these subsets: although FORL2+ TAMs were mostly present in the tumour stroma, TREM2+ TAMs were found in both stroma and tumour nests, particularly along the invasive margin, suggesting that TREM2+ TAMs may be directly imprinted by tumour cells. TREM2+ TAMs inversely correlated with responsiveness to checkpoint blockade immunotherapy. Colorectal carcinoma and its liver metastases were also shown to harbour a heterogenous population of TAMs that included one subset expressing both TREM2 and the complement component C1QC187. TREM2+ TAMs were also found in melanoma188, clear cell renal carcinoma189 and pancreatic ductal adenocarcinoma190 and shown to associate with immunosuppression and disease recurrence. Single-cell transcriptomics of 15 human parenchymal brain metastases identified metastasis-associated macrophages expressing TREM2, APOE and C1QB+ (ref. 191).
Studies on the impact of TREM2+ TAMs in hepatocellular carcinoma are divergent. One report corroborated that these immunosuppressive macrophages inversely correlated with survival192. However, another report showed that mice deficient in TREM2 developed more diethylnitrosamine-induced liver tumours along with exacerbated liver damage, inflammation and oxidative stress193. Overall, TREM2+ TAMs generally enforce immunosuppression and support tumour growth, although their impact may vary depending on the type and pathogenesis of the tumour.
A function for TREM2 in tissue repair versus inflammation
Although not expressed in peripheral blood monocytes, TREM2 is induced by CSF1, CSF2 and IL-4. This explains why TREM2 was detected in human monocyte-derived DCs cultured in CSF2 and IL-4 but not in primary DCs88. IL-4 inducibility also explains why TREM2 was detected in alternatively activated macrophages in vitro18 as well as in lung and lymph node myeloid cells during allergic airway inflammation194. The induction of TREM2 expression by CSF1 and IL-4 supports the idea that TREM2 facilitates tissue remodelling and repair, rather than inflammation. Consistent with this notion, it has been shown that TREM2 modulates macrophage responses to TLRs. Genetic deletion or downregulation of TREM2 in bone marrow-derived or peritoneal macrophages enhanced their capacity to produce inflammatory cytokines, such as TNF and IL-6, in response to various TLR agonists18,86. TREM2 deficiency completely accounted for a similar increase in cytokine production observed in DAP12-deficient mice upon stimulation with TLR ligands18,86. As noted earlier, biochemical studies have demonstrated that DAP12 inhibits ERK activation by TLRs by recruiting DOK3, which sequesters the adapters SOS and GRB2 that are required for initiating the RAS–MEK–ERK signalling pathway20 (Fig. 3). However, as TREM2 has also been reported to recruit the inhibitory phosphatase SHIP1 (ref. 21), TREM2 may attenuate inflammation also through additional pathways. Finally, TREM2 has been reported to facilitate wound repair of colonic mucosa195 but exacerbate colonic inflammation in two mouse models of colitis196. As both models are dependent on gut microbiota, these seemingly contrasting observations suggest a complex interaction between TREM2+ myeloid cells in the gut mucosa, intestinal microbiota and their products, which needs to be explored in detail.
Other TREM family members
TREML4
TREML4 is a DAP12-associated receptor expressed on a broad range of myeloid cells. It was initially found in mouse CD8α+ DCs and spleen macrophages197 and shown to mediate phagocytosis of dead cells197 and to present antigens that artificially bound TREML4 through conjugation with an anti-TREML4 mAb198. Subsequently, an unbiased in vitro genome-scale screen found that TREML4 amplifies TLR7 signalling in macrophages199. As TLR7 detects single-stranded viral RNAs, TREML4-deficient macrophages failed to produce type I interferons in response to influenza virus. Conversely, TREML4 deficiency moderated the production of inflammatory cytokines and autoantibodies in the MRL–lpr mouse model of autoimmunity. Another unbiased in vivo genome-scale screen in a mouse model of polymicrobial sepsis identified TREML4 as a regulator of neutrophil survival and function200. Deletion of Treml4 improved survival and phagocytic function of mature neutrophils, paralleled by attenuated release of extracellular traps, free radical generation and associated organ damage; this combination ultimately provided protection from cytokine storm and mortality during polymicrobial sepsis, as well as secondary infection by P. aeruginosa. In humans, TREML4 has been linked to macrophage involvement in coronary artery calcification, a feature of atherosclerosis that strongly predicts risk for future cardiovascular events201. TREML4 expression was upregulated in the peripheral blood of patients with coronary artery calcification and linked to a single-nucleotide polymorphism that increases the relative risk of coronary artery calcification. Moreover, TREML4 was expressed in CD68+ macrophages surrounding the necrotic core of coronary plaques complicated by calcification in atherosclerotic lesions.
TREML1
TREML1 is unique within the TREM cluster in terms of expression, signalling and function. TREML1 is exclusively expressed in the α-granules of megakaryocytes and platelets202 under the control of RUNX1 (ref. 203). Upon activation, TREML1 translocates to the cell surface of platelets and binds to fibrinogen204, facilitating platelet aggregation and protecting against haemorrhage205. TREML1 was also found to promote platelet–neutrophil interactions and neutrophil accumulation in the lung during acute lung injury204, as well as platelet–monocyte aggregate formation in patients with tuberculosis206. TREML1 may sustain platelet activation at least in part by facilitating outside-in signalling of β3 integrin207. The modalities of these signals are poorly understood. TREML1 does not associate with DAP12 but has an extended cytoplasmic domain containing two tyrosine motifs resembling immunoreceptor tyrosine-based inhibitory motifs202. One motif was shown to recruit the protein tyrosine phosphatase SHP2 (ref. 208), which can mediate either activation or inhibition depending on context. In the case of TREML1, recruitment of SHP2 was associated with intracellular calcium release. Further biochemical and signalling studies are warranted to elucidate this pathway.
Like TREM1 and TREM2, TREML1 is shed from the cell surface, generating a soluble form of TREML1 detectable in the serum of humans and mice. Plasma levels of sTREML1 correlated with the presence of disseminated intravascular coagulation in patients with sepsis, providing a sensitive marker of platelet activation204. Moreover, sTREML1 regulated inflammation in a mouse model of sepsis by tempering platelet–neutrophil crosstalk209 and alleviated pulmonary haemorrhage during acute lung injury204, suggesting the potential therapeutic relevance of TREML1 decoys in these conditions. The crystal structure of the extracellular domain of TREML1 has been solved210, corroborating the presence of a classical immunoglobulin fold containing a region analogous to complementarity determining region 3, which may function as a ligand-binding site.
TREML2
TREML2 transcripts were initially found in human and mouse B cells211, although how TREML2 functions in B cells is not yet known. TREML2 neither associates with DAP12 nor contains canonical signalling motifs in the cytoplasmic domain, although potential endocytosis and SH3 binding motifs have been noted. TREML2 is also expressed in mouse neutrophils as well as in resident peritoneal and lung macrophages, but not in monocytes, suggesting that its expression is associated with terminal differentiation of monocyte-derived macrophages. In response to inflammatory mediators, neutrophils but not B cells strongly upregulate TREML2 expression211. In humans, TREML2 is expressed by B cells, monocytes and neutrophils. TREML2 was detected in primary granules in neutrophils and was retained in the plasma membrane following granule exocytosis, rather than being expelled on neutrophil extracellular traps212. A missense variant in TREML2 was reported to be associated with reduced AD risk213,214. Given that the patterns of TREML2 expression are unrelated to microglia and other brain macrophages, these reports may reflect a linkage disequilibrium between TREML2 and TREM2 variants within the TREM gene cluster.
TREM family members expressed solely in mice
TREM3, TREM4, TREM5 and TREML6 are only encoded in mouse2,215. TREML6 is the only TREM family member that contains a canonical immunoreceptor tyrosine-based inhibitory motif in the cytoplasmic tail, which is indicative of an inhibitory function, but this has not been confirmed experimentally. TREM3, TREM4 and TREM5 contain positively charged amino acids in their transmembrane domains and associate with DAP12 (refs. 2,215). TREM4 is uniquely expressed in plasmacytoid DCs, a subset of DCs specialized in the production of IFNα216. Similar to TREM2 (ref. 87) (discussed earlier), TREM4 was found to associate with plexin-A1, the receptor for semaphorin 6D, on the plasmacytoid DC cell surface. Limited TLR signals induced TREM4 expression and, together with semaphorin 6D, triggered IFNα production. The ability of plasmacytoid DCs to produce IFNα was markedly weakened by each of the following: diminished expression of TREM4, inhibited formation of the TREM4–plexin-A complex and lack of DAP12 (ref. 216). TREM3 was identified from a murine macrophage library215. Although transcripts for TREM3 have been detected in murine macrophage and myeloid cell lines, protein expression has not been reported and a potential activating function of TREM3 has been shown only in an artificially transfected natural killer cell line. TREM5 has not been studied.
Concluding remarks
Although much has been achieved in understanding the biology of TREM1 and TREM2 since their identification, many questions remain to be addressed. Given the multiple ligands reported for TREM1 and TREM2, future biochemical and structural studies should define their relative affinities, the three-dimensional details of the interaction between these ligands and their cognate receptors, and their functional impact. The signalling mediators downstream of TREM1 and TREM2 have been extensively defined in the cytosol, but not much is known about how they impact transcription factors and chromatin accessibility. Most in vivo studies have been performed in knockout mice. However, TREM molecules differ between mice and humans. For example, the Trem3 gene closely linked and homologous to Trem1 is expressed in mice but is not found in humans. Thus, it will be important to establish whether the presence of additional and/or non-homologous TREM molecules in mice may impact disease models; perhaps, generating new mouse models that more closely recapitulate human TREMs, such as transgenic or humanized mice, would be appropriate. TREM1-based therapies have been designed and tested for sepsis and cancer, as have TREM2-based therapies for neurodegeneration and cancer (Table 2). It is predictable that these approaches will further develop through a new generation of TREM agonists and antagonists that may impact ligand binding or delivery of intracellular signals through DAP12 and DAP10 adapters. Modified TREM2 agonists with increased brain availability can be developed to increase efficacy and reduce potential activation of peripheral macrophages217. Moreover, indications for TREM-based treatments may extend to other diseases, such as metabolic and cardiovascular disorders (Table 2). Methods for detecting sTREM1 and sTREM2 in human biological fluids will further enable diagnosis and prognosis of several diseases as well as the identification of patients who may specifically benefit from TREM-based therapies. Other TREM family molecules, particularly TREML4 and TREML1, may also be considered potential therapeutic targets in sepsis or other inflammatory diseases (Table 2). This will require further studies of their ligands, signalling properties, interaction with other pathways and impact in various mouse models of infection, inflammation and cancer.
Table 2.
Potential therapeutic indications of TREM-based therapies
| TREM target | Goal | Indications | Therapeutics | ClinicalTrials.gov ID |
|---|---|---|---|---|
| TREM1 | Activation | Tumoursa, synergy with immune checkpoint therapya, liver disease, stroke, cardiovascular disease | Agonistic TREM1 antibodies, small TREM1 agonists | NCT04682431 |
| TREM1 | Inhibition | Sepsisa, tumours, liver inflammation | Ligand-trapping peptide (LR12), TREM1-Fc decoy, signalling-blocking peptide (GF9) | NCT04429334 |
| TREML1 | Inhibition | Sepsis | Ligand-trapping peptide, blocking antibodies | – |
| TREM2 | Activation | Alzheimer diseasea, metabolic disease | Agonistic antibodies, small molecules | NCT04592874 |
| TREM2 | Inhibition | Tumoursa, synergy with immune checkpoint therapya | TREM2-blocking antibodies, small-molecule TREM2 inhibitors, antibodies depleting TREM2+ tumour-associated macrophages | NCT04691375 |
aIndications in which triggering receptor expressed on myeloid cells (TREM)-based therapeutics are currently being tested in clinical trials.
Acknowledgements
The author thanks S. Gilfillan, J. Klesney-Tait, M. Molgora, Y. Chen, M. B. Humphrey, D. McVicar and M. Cella for critical reading. This work is supported by the US National Institutes of Health (RF1 AG051485, R21 AG059176, R01 CA262684 and RF1 AG059082).
Glossary
- Genotype Tissue-Expression Project
A data resource and tissue bank established by the US National Institutes of Health.
- Polymeric immunoglobulin receptor
A transmembrane protein expressed by mucosal epithelial cells that transports polymeric IgA, IgM and immune complexes from the basolateral to the apical surface of the epithelium.
- The Cancer Genome Atlas
A project aiming to catalogue the genetic mutations responsible for cancer by using genome sequencing and bioinformatics.
- V-type immunoglobulin domain
An immunoglobulin-like domain resembling the antibody variable domain.
Peer review
Peer review information
Nature Reviews Immunology thanks G. Bu, M. Yuka and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Competing interests
M.C. is a member of the Vigil Scientific Advisory Board and NGM Bio, is consultant for Cell Signalling Technology and has received research grants from Vigil, NGM Bio and Ono during the conduct of the study. In addition, M.C. has a patent related to TREM2 pending.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bouchon A, Dietrich J, Colonna M. Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J. Immunol. 2000;164:4991–4995. doi: 10.4049/jimmunol.164.10.4991. [DOI] [PubMed] [Google Scholar]
- 2.Kasamatsu J, et al. Double-stranded RNA analog and type I interferon regulate expression of Trem paired receptors in murine myeloid cells. BMC Immunol. 2016;17:9. doi: 10.1186/s12865-016-0147-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stet RJ, et al. Novel immunoglobulin-like transcripts in teleost fish encode polymorphic receptors with cytoplasmic ITAM or ITIM and a new structural Ig domain similar to the natural cytotoxicity receptor NKp44. Immunogenetics. 2005;57:77–89. doi: 10.1007/s00251-005-0771-9. [DOI] [PubMed] [Google Scholar]
- 4.Viertlboeck BC, Schmitt R, Gobel TW. The chicken immunoregulatory receptor families SIRP, TREM, and CMRF35/CD300L. Immunogenetics. 2006;58:180–190. doi: 10.1007/s00251-006-0091-8. [DOI] [PubMed] [Google Scholar]
- 5.Clark GJ, Green BJ, Hart DN. The CMRF-35H gene structure predicts for an independently expressed member of an ITIM/ITAM pair of molecules localized to human chromosome 17. Tissue Antigens. 2000;55:101–109. doi: 10.1034/j.1399-0039.2000.550201.x. [DOI] [PubMed] [Google Scholar]
- 6.Green BJ, Clark GJ, Hart DN. The CMRF-35 mAb recognizes a second leukocyte membrane molecule with a domain similar to the poly Ig receptor. Int. Immunol. 1998;10:891–899. doi: 10.1093/intimm/10.7.891. [DOI] [PubMed] [Google Scholar]
- 7.Jackson DG, Hart DN, Starling G, Bell JI. Molecular cloning of a novel member of the immunoglobulin gene superfamily homologous to the polymeric immunoglobulin receptor. Eur. J. Immunol. 1992;22:1157–1163. doi: 10.1002/eji.1830220508. [DOI] [PubMed] [Google Scholar]
- 8.Aguilar H, et al. Molecular characterization of a novel immune receptor restricted to the monocytic lineage. J. Immunol. 2004;173:6703–6711. doi: 10.4049/jimmunol.173.11.6703. [DOI] [PubMed] [Google Scholar]
- 9.Chung DH, et al. CMRF-35-like molecule-1, a novel mouse myeloid receptor, can inhibit osteoclast formation. J. Immunol. 2003;171:6541–6548. doi: 10.4049/jimmunol.171.12.6541. [DOI] [PubMed] [Google Scholar]
- 10.Barrow AD, Martin CJ, Colonna M. The natural cytotoxicity receptors in health and disease. Front. Immunol. 2019;10:909. doi: 10.3389/fimmu.2019.00909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lanier LL. DAP10- and DAP12-associated receptors in innate immunity. Immunol. Rev. 2009;227:150–160. doi: 10.1111/j.1600-065X.2008.00720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ulland TK, et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell. 2017;170:649–663. doi: 10.1016/j.cell.2017.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otero K, et al. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and β-catenin. Nat. Immunol. 2009;10:734–743. doi: 10.1038/ni.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tessarz AS, et al. Non-T cell activation linker (NTAL) negatively regulates TREM-1/DAP12-induced inflammatory cytokine production in myeloid cells. J. Immunol. 2007;178:1991–1999. doi: 10.4049/jimmunol.178.4.1991. [DOI] [PubMed] [Google Scholar]
- 15.Yuan Z, et al. Triggering receptor expressed on myeloid cells 1 (TREM-1)-mediated Bcl-2 induction prolongs macrophage survival. J. Biol. Chem. 2014;289:15118–15129. doi: 10.1074/jbc.M113.536490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mocsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA. Syk is required for integrin signaling in neutrophils. Immunity. 2002;16:547–558. doi: 10.1016/S1074-7613(02)00303-5. [DOI] [PubMed] [Google Scholar]
- 17.Mocsai A, et al. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat. Immunol. 2006;7:1326–1333. doi: 10.1038/ni1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turnbull IR, et al. Cutting edge: TREM-2 attenuates macrophage activation. J. Immunol. 2006;177:3520–3524. doi: 10.4049/jimmunol.177.6.3520. [DOI] [PubMed] [Google Scholar]
- 19.Hamerman JA, Tchao NK, Lowell CA, Lanier LL. Enhanced Toll-like receptor responses in the absence of signaling adaptor DAP12. Nat. Immunol. 2005;6:579–586. doi: 10.1038/ni1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng Q, Long CL, Malhotra S, Humphrey MB. A physical interaction between the adaptor proteins DOK3 and DAP12 is required to inhibit lipopolysaccharide signaling in macrophages. Sci. Signal. 2013;6:ra72. doi: 10.1126/scisignal.2003801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peng Q, et al. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci. Signal. 2010;3:ra38. doi: 10.1126/scisignal.2000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang S, et al. TREM2 drives microglia response to amyloid-β via SYK-dependent and -independent pathways. Cell. 2022;185:4153–4169. doi: 10.1016/j.cell.2022.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang C, et al. Cutting edge: KAP10, a novel transmembrane adapter protein genetically linked to DAP12 but with unique signaling properties. J. Immunol. 1999;163:4651–4654. doi: 10.4049/jimmunol.163.9.4651. [DOI] [PubMed] [Google Scholar]
- 24.Upshaw JL, et al. NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat. Immunol. 2006;7:524–532. doi: 10.1038/ni1325. [DOI] [PubMed] [Google Scholar]
- 25.Bouchon A, Facchetti F, Weigand MA, Colonna M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature. 2001;410:1103–1107. doi: 10.1038/35074114. [DOI] [PubMed] [Google Scholar]
- 26.Bleharski JR, et al. A role for triggering receptor expressed on myeloid cells-1 in host defense during the early-induced and adaptive phases of the immune response. J. Immunol. 2003;170:3812–3818. doi: 10.4049/jimmunol.170.7.3812. [DOI] [PubMed] [Google Scholar]
- 27.Netea MG, et al. Triggering receptor expressed on myeloid cells-1 (TREM-1) amplifies the signals induced by the NACHT-LRR (NLR) pattern recognition receptors. J. Leukoc. Biol. 2006;80:1454–1461. doi: 10.1189/jlb.1205758. [DOI] [PubMed] [Google Scholar]
- 28.Gibot S, et al. A soluble form of the triggering receptor expressed on myeloid cells-1 modulates the inflammatory response in murine sepsis. J. Exp. Med. 2004;200:1419–1426. doi: 10.1084/jem.20040708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang F, et al. Blocking TREM-1 signaling prolongs survival of mice with Pseudomonas aeruginosa induced sepsis. Cell Immunol. 2012;272:251–258. doi: 10.1016/j.cellimm.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 30.Turnbull IR, et al. DAP12 (KARAP) amplifies inflammation and increases mortality from endotoxemia and septic peritonitis. J. Exp. Med. 2005;202:363–369. doi: 10.1084/jem.20050986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klesney-Tait J, et al. Transepithelial migration of neutrophils into the lung requires TREM-1. J. Clin. Invest. 2013;123:138–149. doi: 10.1172/JCI64181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baruah S, et al. TREM-1 regulates neutrophil chemotaxis by promoting NOX-dependent superoxide production. J. Leukoc. Biol. 2019;105:1195–1207. doi: 10.1002/JLB.3VMA0918-375R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hommes TJ, et al. Triggering receptor expressed on myeloid cells-1 (TREM-1) improves host defence in pneumococcal pneumonia. J. Pathol. 2014;233:357–367. doi: 10.1002/path.4361. [DOI] [PubMed] [Google Scholar]
- 34.Lagler H, et al. TREM-1 activation alters the dynamics of pulmonary IRAK-M expression in vivo and improves host defense during pneumococcal pneumonia. J. Immunol. 2009;183:2027–2036. doi: 10.4049/jimmunol.0803862. [DOI] [PubMed] [Google Scholar]
- 35.Lin YT, et al. TREM-1 promotes survival during Klebsiella pneumoniae liver abscess in mice. Infect. Immun. 2014;82:1335–1342. doi: 10.1128/IAI.01347-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weber B, et al. TREM-1 deficiency can attenuate disease severity without affecting pathogen clearance. PLoS Pathog. 2014;10:e1003900. doi: 10.1371/journal.ppat.1003900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gingras MC, Lapillonne H, Margolin JF. TREM-1, MDL-1, and DAP12 expression is associated with a mature stage of myeloid development. Mol. Immunol. 2002;38:817–824. doi: 10.1016/S0161-5890(02)00004-4. [DOI] [PubMed] [Google Scholar]
- 38.Baruah S, et al. Identification of a novel splice variant isoform of TREM-1 in human neutrophil granules. J. Immunol. 2015;195:5725–5731. doi: 10.4049/jimmunol.1402713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Begum NA, et al. Mycobacterium bovis BCG cell wall-specific differentially expressed genes identified by differential display and cDNA subtraction in human macrophages. Infect. Immun. 2004;72:937–948. doi: 10.1128/IAI.72.2.937-948.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomez-Pina V, et al. Metalloproteinases shed TREM-1 ectodomain from lipopolysaccharide-stimulated human monocytes. J. Immunol. 2007;179:4065–4073. doi: 10.4049/jimmunol.179.6.4065. [DOI] [PubMed] [Google Scholar]
- 41.Gibot S, et al. Soluble triggering receptor expressed on myeloid cells and the diagnosis of pneumonia. N. Engl. J. Med. 2004;350:451–458. doi: 10.1056/NEJMoa031544. [DOI] [PubMed] [Google Scholar]
- 42.Su L, Liu D, Chai W, Liu D, Long Y. Role of sTREM-1 in predicting mortality of infection: a systematic review and meta-analysis. BMJ Open. 2016;6:e010314. doi: 10.1136/bmjopen-2015-010314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Forrester DL, Barr HL, Fogarty A, Knox A. sTREM-1 is elevated in cystic fibrosis and correlates with proteases. Pediatr. Pulmonol. 2017;52:467–471. doi: 10.1002/ppul.23650. [DOI] [PubMed] [Google Scholar]
- 44.de Nooijer AH, et al. Increased sTREM-1 plasma concentrations are associated with poor clinical outcomes in patients with COVID-19. Biosci. Rep. 2021;41:BSR20210940. doi: 10.1042/BSR20210940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Singer M, et al. COVID-19 risk stratification algorithms based on sTREM-1 and IL-6 in emergency department. J. Allergy Clin. Immunol. 2021;147:99–106. doi: 10.1016/j.jaci.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Francois B, et al. Nangibotide in patients with septic shock: a phase 2a randomized controlled clinical trial. Intensive Care Med. 2020;46:1425–1437. doi: 10.1007/s00134-020-06109-z. [DOI] [PubMed] [Google Scholar]
- 47.Murakami Y, et al. Induction of triggering receptor expressed on myeloid cells 1 in murine resident peritoneal macrophages by monosodium urate monohydrate crystals. Arthritis Rheum. 2006;54:455–462. doi: 10.1002/art.21633. [DOI] [PubMed] [Google Scholar]
- 48.Nochi H, et al. Modulation of hepatic granulomatous responses by transgene expression of DAP12 or TREM-1-Ig molecules. Am. J. Pathol. 2003;162:1191–1201. doi: 10.1016/S0002-9440(10)63915-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nguyen-Lefebvre AT, et al. The innate immune receptor TREM-1 promotes liver injury and fibrosis. J. Clin. Invest. 2018;128:4870–4883. doi: 10.1172/JCI98156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zysset D, et al. TREM-1 links dyslipidemia to inflammation and lipid deposition in atherosclerosis. Nat. Commun. 2016;7:13151. doi: 10.1038/ncomms13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boufenzer A, et al. TREM-1 mediates inflammatory injury and cardiac remodeling following myocardial infarction. Circ. Res. 2015;116:1772–1782. doi: 10.1161/CIRCRESAHA.116.305628. [DOI] [PubMed] [Google Scholar]
- 52.Vandestienne M, et al. TREM-1 orchestrates angiotensin II-induced monocyte trafficking and promotes experimental abdominal aortic aneurysm. J. Clin. Invest. 2021;131:e142468. doi: 10.1172/JCI142468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Q, et al. Peripheral TREM1 responses to brain and intestinal immunogens amplify stroke severity. Nat. Immunol. 2019;20:1023–1034. doi: 10.1038/s41590-019-0421-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tammaro A, et al. TREM1/3 deficiency impairs tissue repair after acute kidney injury and mitochondrial metabolic flexibility in tubular epithelial cells. Front. Immunol. 2019;10:1469. doi: 10.3389/fimmu.2019.01469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schenk M, Bouchon A, Seibold F, Mueller C. TREM-1-expressing intestinal macrophages crucially amplify chronic inflammation in experimental colitis and inflammatory bowel diseases. J. Clin. Invest. 2007;117:3097–3106. doi: 10.1172/JCI30602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Park JJ, et al. Correlation of serum-soluble triggering receptor expressed on myeloid cells-1 with clinical disease activity in inflammatory bowel disease. Dig. Dis. Sci. 2009;54:1525–1531. doi: 10.1007/s10620-008-0514-5. [DOI] [PubMed] [Google Scholar]
- 57.Tzivras M, et al. Role of soluble triggering receptor expressed on myeloid cells in inflammatory bowel disease. World J. Gastroenterol. 2006;12:3416–3419. doi: 10.3748/wjg.v12.i21.3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi ST, Kang EJ, Ha YJ, Song JS. Levels of plasma-soluble triggering receptor expressed on myeloid cells-1 (sTREM-1) are correlated with disease activity in rheumatoid arthritis. J. Rheumatol. 2012;39:933–938. doi: 10.3899/jrheum.111218. [DOI] [PubMed] [Google Scholar]
- 59.Murakami Y, et al. Intervention of an inflammation amplifier, triggering receptor expressed on myeloid cells 1, for treatment of autoimmune arthritis. Arthritis Rheum. 2009;60:1615–1623. doi: 10.1002/art.24554. [DOI] [PubMed] [Google Scholar]
- 60.Du Y, Wu T, Zhou XJ, Davis LS, Mohan C. Blockade of CD354 (TREM-1) ameliorates anti-GBM-induced nephritis. Inflammation. 2016;39:1169–1176. doi: 10.1007/s10753-016-0351-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hyder LA, et al. TREM-1 as a potential therapeutic target in psoriasis. J. Invest. Dermatol. 2013;133:1742–1751. doi: 10.1038/jid.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Drager S, et al. Increased TREM-1 expression in inflamed skin has no functional impact on the pathogenesis of cutaneous disorders. J. Dermatol. Sci. 2017;88:152–155. doi: 10.1016/j.jdermsci.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 63.Bassyouni IH, et al. Clinical association of a soluble triggering receptor expressed on myeloid cells-1 (sTREM-1) in patients with systemic lupus erythematosus. Immunol. Invest. 2017;46:38–47. doi: 10.1080/08820139.2016.1211140. [DOI] [PubMed] [Google Scholar]
- 64.Liu CJ, et al. Triggering receptor expressed on myeloid cells-1 (TREM-1) deficiency augments BAFF production to promote lupus progression. J. Autoimmun. 2017;78:92–100. doi: 10.1016/j.jaut.2016.12.010. [DOI] [PubMed] [Google Scholar]
- 65.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017;14:399–416. doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lopez-Yrigoyen M, Cassetta L, Pollard JW. Macrophage targeting in cancer. Ann. N. Y. Acad. Sci. 2021;1499:18–41. doi: 10.1111/nyas.14377. [DOI] [PubMed] [Google Scholar]
- 67.Liao R, et al. Expression of TREM-1 in hepatic stellate cells and prognostic value in hepatitis B-related hepatocellular carcinoma. Cancer Sci. 2012;103:984–992. doi: 10.1111/j.1349-7006.2012.02273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ho CC, et al. TREM-1 expression in tumor-associated macrophages and clinical outcome in lung cancer. Am. J. Respir. Crit. Care Med. 2008;177:763–770. doi: 10.1164/rccm.200704-641OC. [DOI] [PubMed] [Google Scholar]
- 69.Wu J, et al. The proinflammatory myeloid cell receptor TREM-1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res. 2012;72:3977–3986. doi: 10.1158/0008-5472.CAN-12-0938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu Q, et al. Blocking triggering receptor expressed on myeloid cells-1-positive tumor-associated macrophages induced by hypoxia reverses immunosuppression and anti-programmed cell death ligand 1 resistance in liver cancer. Hepatology. 2019;70:198–214. doi: 10.1002/hep.30593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shen ZT, Sigalov AB. Novel TREM-1 inhibitors attenuate tumor growth and prolong survival in experimental pancreatic cancer. Mol. Pharm. 2017;14:4572–4582. doi: 10.1021/acs.molpharmaceut.7b00711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fontana R, et al. Nuclear receptor ligands induce TREM-1 expression on dendritic cells: analysis of their role in tumors. Oncoimmunology. 2019;8:1554967. doi: 10.1080/2162402X.2018.1554967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Y, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160:1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Song W, et al. Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement. 2017;13:381–387. doi: 10.1016/j.jalz.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Atagi Y, et al. Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2) J. Biol. Chem. 2015;290:26043–26050. doi: 10.1074/jbc.M115.679043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bailey CC, DeVaux LB, Farzan M. The triggering receptor expressed on myeloid cells 2 binds apolipoprotein E. J. Biol. Chem. 2015;290:26033–26042. doi: 10.1074/jbc.M115.677286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Daws MR, et al. Pattern recognition by TREM-2: binding of anionic ligands. J. Immunol. 2003;171:594–599. doi: 10.4049/jimmunol.171.2.594. [DOI] [PubMed] [Google Scholar]
- 78.Iizasa E, et al. TREM2 is a receptor for non-glycosylated mycolic acids of mycobacteria that limits anti-mycobacterial macrophage activation. Nat. Commun. 2021;12:2299. doi: 10.1038/s41467-021-22620-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kober DL, et al. Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. eLife. 2016;5:e20391. doi: 10.7554/eLife.20391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sudom A, et al. Molecular basis for the loss-of-function effects of the Alzheimer’s disease-associated R47H variant of the immune receptor TREM2. J. Biol. Chem. 2018;293:12634–12646. doi: 10.1074/jbc.RA118.002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao Y, et al. TREM2 is a receptor for beta-amyloid that mediates microglial function. Neuron. 2018;97:1023–1031. doi: 10.1016/j.neuron.2018.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lessard CB, et al. High-affinity interactions and signal transduction between Aβ oligomers and TREM2. EMBO Mol. Med. 2018;10:e9027. doi: 10.15252/emmm.201809027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhong L, et al. Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2) Mol. Neurodegener. 2018;13:15. doi: 10.1186/s13024-018-0247-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xie M, et al. TREM2 interacts with TDP-43 and mediates microglial neuroprotection against TDP-43-related neurodegeneration. Nat. Neurosci. 2022;25:26–38. doi: 10.1038/s41593-021-00975-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Boza-Serrano A, et al. Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. 2019;138:251–273. doi: 10.1007/s00401-019-02013-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hamerman JA, et al. Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J. Immunol. 2006;177:2051–2055. doi: 10.4049/jimmunol.177.4.2051. [DOI] [PubMed] [Google Scholar]
- 87.Takegahara N, et al. Plexin-A1 and its interaction with DAP12 in immune responses and bone homeostasis. Nat. Cell. Biol. 2006;8:615–622. doi: 10.1038/ncb1416. [DOI] [PubMed] [Google Scholar]
- 88.Bouchon A, Hernandez-Munain C, Cella M, Colonna M. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J. Exp. Med. 2001;194:1111–1122. doi: 10.1084/jem.194.8.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Daws MR, Lanier LL, Seaman WE, Ryan JC. Cloning and characterization of a novel mouse myeloid DAP12-associated receptor family. Eur. J. Immunol. 2001;31:783–791. doi: 10.1002/1521-4141(200103)31:3<783::AID-IMMU783>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 90.Paloneva J, et al. CNS manifestations of Nasu–Hakola disease: a frontal dementia with bone cysts. Neurology. 2001;56:1552–1558. doi: 10.1212/WNL.56.11.1552. [DOI] [PubMed] [Google Scholar]
- 91.Paloneva J, et al. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat. Genet. 2000;25:357–361. doi: 10.1038/77153. [DOI] [PubMed] [Google Scholar]
- 92.Paloneva J, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 2002;71:656–662. doi: 10.1086/342259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chouery E, et al. Mutations in TREM2 lead to pure early-onset dementia without bone cysts. Hum. Mutat. 2008;29:E194–E204. doi: 10.1002/humu.20836. [DOI] [PubMed] [Google Scholar]
- 94.Guerreiro RJ, et al. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 2013;70:78–84. doi: 10.1001/jamaneurol.2013.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Paloneva J, et al. DAP12/TREM2 deficiency results in impaired osteoclast differentiation and osteoporotic features. J. Exp. Med. 2003;198:669–675. doi: 10.1084/jem.20030027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cella M, et al. Impaired differentiation of osteoclasts in TREM-2-deficient individuals. J. Exp. Med. 2003;198:645–651. doi: 10.1084/jem.20022220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kaifu T, et al. Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J. Clin. Invest. 2003;111:323–332. doi: 10.1172/JCI16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nataf S, et al. Brain and bone damage in KARAP/DAP12 loss-of-function mice correlate with alterations in microglia and osteoclast lineages. Am. J. Pathol. 2005;166:275–286. doi: 10.1016/S0002-9440(10)62251-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Humphrey MB, et al. The signaling adapter protein DAP12 regulates multinucleation during osteoclast development. J. Bone Miner. Res. 2004;19:224–234. doi: 10.1359/JBMR.0301234. [DOI] [PubMed] [Google Scholar]
- 100.Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005;201:647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Otero K, et al. TREM2 and β-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis. J. Immunol. 2012;188:2612–2621. doi: 10.4049/jimmunol.1102836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Guerreiro R, et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jonsson T, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179:312–339. doi: 10.1016/j.cell.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Griciuc A, Tanzi RE. The role of innate immune genes in Alzheimer’s disease. Curr. Opin. Neurol. 2021;34:228–236. doi: 10.1097/WCO.0000000000000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Song WM, et al. Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J. Exp. Med. 2018;215:745–760. doi: 10.1084/jem.20171529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhou Y, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020;26:131–142. doi: 10.1038/s41591-019-0695-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jay TR, et al. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci. 2017;37:637–647. doi: 10.1523/JNEUROSCI.2110-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Griciuc A, et al. TREM2 acts downstream of CD33 in modulating microglial pathology in Alzheimer’s disease. Neuron. 2019;103:820–835. doi: 10.1016/j.neuron.2019.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McQuade A, et al. Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat. Commun. 2020;11:5370. doi: 10.1038/s41467-020-19227-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lee CYD, et al. Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron. 2018;97:1032–1048. doi: 10.1016/j.neuron.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Keren-Shaul H, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169:1276–1290.e17. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 113.Krasemann S, et al. The TREM2–APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566–581. doi: 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mathys H, et al. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 2017;21:366–380. doi: 10.1016/j.celrep.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang S, et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020;217:e20200785. doi: 10.1084/jem.20200785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ellwanger DC, et al. Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer’s disease. Proc. Natl Acad. Sci. USA. 2021;118:e2017742118. doi: 10.1073/pnas.2017742118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ennerfelt H, et al. SYK coordinates neuroprotective microglial responses in neurodegenerative disease. Cell. 2022;185:4135–4152. doi: 10.1016/j.cell.2022.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nugent AA, et al. TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron. 2020;105:837–854. doi: 10.1016/j.neuron.2019.12.007. [DOI] [PubMed] [Google Scholar]
- 119.Kober DL, et al. Functional insights from biophysical study of TREM2 interactions with apoE and Aβ1–42. Alzheimers Dement. 2020 doi: 10.1002/alz.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liao F, et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Invest. 2018;128:2144–2155. doi: 10.1172/JCI96429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Parhizkar S, et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019;22:191–204. doi: 10.1038/s41593-018-0296-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. 2016;91:328–340. doi: 10.1016/j.neuron.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 123.Nguyen AT, et al. APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol. 2020;140:477–493. doi: 10.1007/s00401-020-02200-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ulrich JD, et al. ApoE facilitates the microglial response to amyloid plaque pathology. J. Exp. Med. 2018;215:1047–1058. doi: 10.1084/jem.20171265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen Y, Strickland MR, Soranno A, Holtzman DM. Apolipoprotein E: structural insights and links to Alzheimer disease pathogenesis. Neuron. 2021;109:205–221. doi: 10.1016/j.neuron.2020.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Leyns CEG, et al. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl Acad. Sci. USA. 2017;114:11524–11529. doi: 10.1073/pnas.1710311114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gratuze M, et al. Impact of TREM2R47H variant on tau pathology-induced gliosis and neurodegeneration. J. Clin. Invest. 2020;130:4954–4968. doi: 10.1172/JCI138179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bemiller SM, et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol. Neurodegener. 2017;12:74. doi: 10.1186/s13024-017-0216-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Leyns, C, et al. TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci. 2019;22:1217–1222. doi: 10.1038/s41593-019-0433-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee SH, et al. Trem2 restrains the enhancement of tau accumulation and neurodegeneration by β-amyloid pathology. Neuron. 2021;109:1283–1301. doi: 10.1016/j.neuron.2021.02.010. [DOI] [PubMed] [Google Scholar]
- 131.Schlepckow K, et al. Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol. Med. 2020;12:e11227. doi: 10.15252/emmm.201911227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Santos EN, Fields RD. Regulation of myelination by microglia. Sci. Adv. 2021;7:eabk1131. doi: 10.1126/sciadv.abk1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dong Y, et al. Oxidized phosphatidylcholines found in multiple sclerosis lesions mediate neurodegeneration and are neutralized by microglia. Nat. Neurosci. 2021;24:489–503. doi: 10.1038/s41593-021-00801-z. [DOI] [PubMed] [Google Scholar]
- 134.Poliani PL, et al. TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Invest. 2015;125:2161–2170. doi: 10.1172/JCI77983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cantoni C, et al. TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol. 2015;129:429–447. doi: 10.1007/s00401-015-1388-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gouna G, et al. TREM2-dependent lipid droplet biogenesis in phagocytes is required for remyelination. J. Exp. Med. 2021;218:e20210227. doi: 10.1084/jem.20210227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cignarella, F, et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 2020;140:513–534. doi: 10.1007/s00401-020-02193-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Thornton P, et al. TREM2 shedding by cleavage at the H157–S158 bond is accelerated for the Alzheimer’s disease-associated H157Y variant. EMBO Mol. Med. 2017;9:1366–1378. doi: 10.15252/emmm.201707673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Feuerbach D, et al. ADAM17 is the main sheddase for the generation of human triggering receptor expressed in myeloid cells (hTREM2) ectodomain and cleaves TREM2 after histidine 157. Neurosci. Lett. 2017;660:109–114. doi: 10.1016/j.neulet.2017.09.034. [DOI] [PubMed] [Google Scholar]
- 140.Schlepckow K, et al. An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol. Med. 2017;9:1356–1365. doi: 10.15252/emmm.201707672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Szykowska A, et al. Selection and structural characterization of anti-TREM2 scFvs that reduce levels of shed ectodomain. Structure. 2021;29:1241–1252.e5. doi: 10.1016/j.str.2021.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Del-Aguila JL, et al. TREM2 brain transcript-specific studies in AD and TREM2 mutation carriers. Mol. Neurodegener. 2019;14:18. doi: 10.1186/s13024-019-0319-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dhandapani R, et al. Sustained Trem2 stabilization accelerates microglia heterogeneity and Aβ pathology in a mouse model of Alzheimer’s disease. Cell Rep. 2022;39:110883. doi: 10.1016/j.celrep.2022.110883. [DOI] [PubMed] [Google Scholar]
- 144.Zhong L, et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017;214:597–607. doi: 10.1084/jem.20160844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wu K, et al. TREM-2 promotes macrophage survival and lung disease after respiratory viral infection. J. Exp. Med. 2015;212:681–697. doi: 10.1084/jem.20141732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jiang T, et al. A rare coding variant in TREM2 increases risk for Alzheimer’s disease in Han Chinese. Neurobiol. Aging. 2016;42:217.e1–213.e3. doi: 10.1016/j.neurobiolaging.2016.02.023. [DOI] [PubMed] [Google Scholar]
- 147.Deming Y, et al. The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci. Transl. Med. 2019;11:eaau2291. doi: 10.1126/scitranslmed.aau2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Piccio L, et al. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016;131:925–933. doi: 10.1007/s00401-016-1533-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhong L, et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat. Commun. 2019;10:1365. doi: 10.1038/s41467-019-09118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Henjum K, et al. Cerebrospinal fluid soluble TREM2 in aging and Alzheimer’s disease. Alzheimers Res. Ther. 2016;8:17. doi: 10.1186/s13195-016-0182-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Heslegrave A, et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegener. 2016;11:3. doi: 10.1186/s13024-016-0071-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Suarez-Calvet M, et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med. 2016;8:466–476. doi: 10.15252/emmm.201506123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pascoal TA, et al. Microglial activation and tau propagate jointly across Braak stages. Nat. Med. 2021;27:1592–1599. doi: 10.1038/s41591-021-01456-w. [DOI] [PubMed] [Google Scholar]
- 154.Morenas-Rodriguez E, et al. Soluble TREM2 in CSF and its association with other biomarkers and cognition in autosomal-dominant Alzheimer’s disease: a longitudinal observational study. Lancet Neurol. 2022;21:329–341. doi: 10.1016/S1474-4422(22)00027-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ewers, M. et al. Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci. Transl. Med. 11, (2019). [DOI] [PMC free article] [PubMed]
- 156.Piccio L, et al. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain. 2008;131:3081–3091. doi: 10.1093/brain/awn217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 158.DiSpirito JR, Mathis D. Immunological contributions to adipose tissue homeostasis. Semin. Immunol. 2015;27:315–321. doi: 10.1016/j.smim.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jaitin DA, et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell. 2019;178:686–698. doi: 10.1016/j.cell.2019.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Winn NC, Wolf EM, Garcia JN, Hasty AH. Exon 2-mediated deletion of Trem2 does not worsen metabolic function in diet-induced obese mice. J. Physiol. 2022;600:4485–4501. doi: 10.1113/JP283684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat. Immunol. 2011;12:204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 162.Cochain C, et al. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ. Res. 2018;122:1661–1674. doi: 10.1161/CIRCRESAHA.117.312509. [DOI] [PubMed] [Google Scholar]
- 163.Zernecke A, et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ. Res. 2020;127:402–426. doi: 10.1161/CIRCRESAHA.120.316903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ramachandran P, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 2019;575:512–518. doi: 10.1038/s41586-019-1631-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Seidman JS, et al. Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity. 2020;52:1057–1074. doi: 10.1016/j.immuni.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Daemen S, et al. Dynamic shifts in the composition of resident and recruited macrophages influence tissue remodeling in NASH. Cell Rep. 2021;34:108626. doi: 10.1016/j.celrep.2020.108626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Xiong X, et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol. Cell. 2019;75:644–660. doi: 10.1016/j.molcel.2019.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hou J, et al. TREM2 sustains macrophage–hepatocyte metabolic coordination in nonalcoholic fatty liver disease and sepsis. J. Clin. Invest. 2021;131:e135197. doi: 10.1172/JCI135197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hendrikx T, et al. Soluble TREM2 levels reflect the recruitment and expansion of TREM2+ macrophages that localize to fibrotic areas and limit NASH. J. Hepatol. 2022;77:1373–1385. doi: 10.1016/j.jhep.2022.06.004. [DOI] [PubMed] [Google Scholar]
- 170.Spann NJ, et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell. 2012;151:138–152. doi: 10.1016/j.cell.2012.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Savage JC, et al. Nuclear receptors license phagocytosis by trem2+ myeloid cells in mouse models of Alzheimer’s disease. J. Neurosci. 2015;35:6532–6543. doi: 10.1523/JNEUROSCI.4586-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Leyden JJ. New understandings of the pathogenesis of acne. J. Am. Acad. Dermatol. 1995;32:S15–S25. doi: 10.1016/0190-9622(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 173.Do TH, et al. TREM2 macrophages induced by human lipids drive inflammation in acne lesions. Sci. Immunol. 2022;7:eabo2787. doi: 10.1126/sciimmunol.abo2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang ECE, Dai Z, Ferrante AW, Drake CG, Christiano AM. A subset of TREM2+ dermal macrophages secretes oncostatin m to maintain hair follicle stem cell quiescence and inhibit hair growth. Cell Stem Cell. 2019;24:654–669. doi: 10.1016/j.stem.2019.01.011. [DOI] [PubMed] [Google Scholar]
- 175.Molgora M, et al. TREM2 modulation remodels the tumor myeloid landscape enhancing anti-PD-1 immunotherapy. Cell. 2020;182:886–900 e817. doi: 10.1016/j.cell.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Katzenelenbogen Y, et al. Coupled scRNA-seq and intracellular protein activity reveal an immunosuppressive role of TREM2 in cancer. Cell. 2020;182:872–885. doi: 10.1016/j.cell.2020.06.032. [DOI] [PubMed] [Google Scholar]
- 177.Binnewies M, et al. Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep. 2021;37:109844. doi: 10.1016/j.celrep.2021.109844. [DOI] [PubMed] [Google Scholar]
- 178.Lavin Y, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell. 2017;169:750–765. doi: 10.1016/j.cell.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Maynard A, et al. Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell. 2020;182:1232–1251. doi: 10.1016/j.cell.2020.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Leader AM, et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell. 2021;39:1594–1609. doi: 10.1016/j.ccell.2021.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhang H, et al. Immunosuppressive TREM2+ macrophages are associated with undesirable prognosis and responses to anti-PD-1 immunotherapy in non-small cell lung cancer. Cancer Immunol. Immunother. 2022;71:2511–2522. doi: 10.1007/s00262-022-03173-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wu SZ, et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 2021;53:1334–1347. doi: 10.1038/s41588-021-00911-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Azizi E, et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018;174:1293–1308. doi: 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Nalio Ramos R, et al. Tissue-resident FOLR2+ macrophages associate with CD8+ T cell infiltration in human breast cancer. Cell. 2022;185:1189–1207. doi: 10.1016/j.cell.2022.02.021. [DOI] [PubMed] [Google Scholar]
- 185.Zhang Y, et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell. 2021;39:1578–1593. doi: 10.1016/j.ccell.2021.09.010. [DOI] [PubMed] [Google Scholar]
- 186.Timperi E, et al. Lipid-associated macrophages are induced by cancer-associated fibroblasts and mediate immune suppression in breast cancer. Cancer Res. 2022;82:3291–3306. doi: 10.1158/0008-5472.CAN-22-1427. [DOI] [PubMed] [Google Scholar]
- 187.Liu Y, et al. Immune phenotypic linkage between colorectal cancer and liver metastasis. Cancer Cell. 2022;40:424–437. doi: 10.1016/j.ccell.2022.02.013. [DOI] [PubMed] [Google Scholar]
- 188.Xiong D, Wang Y, You M. A gene expression signature of TREM2hi macrophages and γδ T cells predicts immunotherapy response. Nat. Commun. 2020;11:5084. doi: 10.1038/s41467-020-18546-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Obradovic A, et al. Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell. 2021;184:2988–3005. doi: 10.1016/j.cell.2021.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kemp SB, et al. Pancreatic cancer is marked by complement-high blood monocytes and tumor-associated macrophages. Life Sci. Alliance. 2021;4:e202000935. doi: 10.26508/lsa.202000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Gonzalez H, et al. Cellular architecture of human brain metastases. Cell. 2022;185:729–745. doi: 10.1016/j.cell.2021.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhou L, et al. Integrated analysis highlights the immunosuppressive role of TREM2+ macrophages in hepatocellular carcinoma. Front. Immunol. 2022;13:848367. doi: 10.3389/fimmu.2022.848367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Esparza-Baquer A, et al. TREM-2 defends the liver against hepatocellular carcinoma through multifactorial protective mechanisms. Gut. 2021;70:1345–1361. doi: 10.1136/gutjnl-2019-319227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hall SC, Agrawal DK. Increased TREM-2 expression on the subsets of CD11c+ cells in the lungs and lymph nodes during allergic airway inflammation. Sci. Rep. 2017;7:11853. doi: 10.1038/s41598-017-12330-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Seno H, et al. Efficient colonic mucosal wound repair requires Trem2 signaling. Proc. Natl Acad. Sci. USA. 2009;106:256–261. doi: 10.1073/pnas.0803343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Correale C, et al. Bacterial sensor triggering receptor expressed on myeloid cells-2 regulates the mucosal inflammatory response. Gastroenterology. 2013;144:346–356. doi: 10.1053/j.gastro.2012.10.040. [DOI] [PubMed] [Google Scholar]
- 197.Hemmi H, et al. A new triggering receptor expressed on myeloid cells (Trem) family member, Trem-like 4, binds to dead cells and is a DNAX activation protein 12-linked marker for subsets of mouse macrophages and dendritic cells. J. Immunol. 2009;182:1278–1286. doi: 10.4049/jimmunol.182.3.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hemmi H, et al. Treml4, an Ig superfamily member, mediates presentation of several antigens to T cells in vivo, including protective immunity to HER2 protein. J. Immunol. 2012;188:1147–1155. doi: 10.4049/jimmunol.1102541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ramirez-Ortiz ZG, et al. The receptor TREML4 amplifies TLR7-mediated signaling during antiviral responses and autoimmunity. Nat. Immunol. 2015;16:495–504. doi: 10.1038/ni.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Nedeva C, et al. TREML4 receptor regulates inflammation and innate immune cell death during polymicrobial sepsis. Nat. Immunol. 2020;21:1585–1596. doi: 10.1038/s41590-020-0789-z. [DOI] [PubMed] [Google Scholar]
- 201.Sen SK, et al. Integrative DNA, RNA, and protein evidence connects TREML4 to coronary artery calcification. Am. J. Hum. Genet. 2014;95:66–76. doi: 10.1016/j.ajhg.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Washington AV, et al. A TREM family member, TLT-1, is found exclusively in the α-granules of megakaryocytes and platelets. Blood. 2004;104:1042–1047. doi: 10.1182/blood-2004-01-0315. [DOI] [PubMed] [Google Scholar]
- 203.Glembotsky AC, et al. Downregulation of TREM-like transcript-1 and collagen receptor α2 subunit, two novel RUNX1-targets, contributes to platelet dysfunction in familial platelet disorder with predisposition to acute myelogenous leukemia. Haematologica. 2019;104:1244–1255. doi: 10.3324/haematol.2018.188904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Morales-Ortiz J, et al. Platelet-derived TLT-1 is a prognostic indicator in ALI/ARDS and prevents tissue damage in the lungs in a mouse model. Blood. 2018;132:2495–2505. doi: 10.1182/blood-2018-03-841593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Washington AV, et al. TREM-like transcript-1 protects against inflammation-associated hemorrhage by facilitating platelet aggregation in mice and humans. J. Clin. Invest. 2009;119:1489–1501. doi: 10.1172/JCI36175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Wang M, et al. TLT-1 promotes platelet–monocyte aggregate formation to induce IL-10-producing B cells in tuberculosis. J. Immunol. 2022;208:1642–1651. doi: 10.4049/jimmunol.2001218. [DOI] [PubMed] [Google Scholar]
- 207.Jessica MO, et al. Tlt-1-controls early thrombus formation and stability by facilitating Aiibb3 outside-in signaling in mice. Int. J. Adv. Res. 2018;6:1143–1149. doi: 10.21474/IJAR01/7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Barrow AD, et al. Cutting edge: TREM-like transcript-1, a platelet immunoreceptor tyrosine-based inhibition motif encoding costimulatory immunoreceptor that enhances, rather than inhibits, calcium signaling via SHP-2. J. Immunol. 2004;172:5838–5842. doi: 10.4049/jimmunol.172.10.5838. [DOI] [PubMed] [Google Scholar]
- 209.Derive M, et al. Soluble TREM-like transcript-1 regulates leukocyte activation and controls microbial sepsis. J. Immunol. 2012;188:5585–5592. doi: 10.4049/jimmunol.1102674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gattis JL, et al. The structure of the extracellular domain of triggering receptor expressed on myeloid cells like transcript-1 and evidence for a naturally occurring soluble fragment. J. Biol. Chem. 2006;281:13396–13403. doi: 10.1074/jbc.M600489200. [DOI] [PubMed] [Google Scholar]
- 211.King RG, Herrin BR, Justement LB. Trem-like transcript 2 is expressed on cells of the myeloid/granuloid and B lymphoid lineage and is up-regulated in response to inflammation. J. Immunol. 2006;176:6012–6021. doi: 10.4049/jimmunol.176.10.6012. [DOI] [PubMed] [Google Scholar]
- 212.Thomas KA, King RG, Sestero CM, Justement LB. TREM-like transcript 2 is stored in human neutrophil primary granules and is up-regulated in response to inflammatory mediators. J. Leukoc. Biol. 2016;100:177–184. doi: 10.1189/jlb.3AB1115-507R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Benitez BA, et al. Missense variant in TREML2 protects against Alzheimer’s disease. Neurobiol. Aging. 2014;35:1510.e19–1510.26. doi: 10.1016/j.neurobiolaging.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zheng H, et al. Opposing roles of the triggering receptor expressed on myeloid cells 2 and triggering receptor expressed on myeloid cells-like transcript 2 in microglia activation. Neurobiol. Aging. 2016;42:132–141. doi: 10.1016/j.neurobiolaging.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Chung DH, Seaman WE, Daws MR. Characterization of TREM-3, an activating receptor on mouse macrophages: definition of a family of single Ig domain receptors on mouse chromosome 17. Eur. J. Immunol. 2002;32:59–66. doi: 10.1002/1521-4141(200201)32:1<59::AID-IMMU59>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 216.Watarai H, et al. PDC-TREM, a plasmacytoid dendritic cell-specific receptor, is responsible for augmented production of type I interferon. Proc. Natl Acad. Sci. USA. 2008;105:2993–2998. doi: 10.1073/pnas.0710351105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Zhao, P, et al. A tetravalent TREM2 agonistic antibody reduced amyloid pathology in a mouse model of Alzheimer’s disease. Sci. Transl. Med. 2022;7:14. doi: 10.1126/scitranslmed.abq0095. [DOI] [PubMed] [Google Scholar]
- 218.Sharma A, et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell. 2020;183:377–394. doi: 10.1016/j.cell.2020.08.040. [DOI] [PubMed] [Google Scholar]
- 219.Jumper, J, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Read CB, et al. Cutting Edge: identification of neutrophil PGLYRP1 as a ligand for TREM-1. J. Immunol. 2015;194:1417–1421. doi: 10.4049/jimmunol.1402303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Sharapova TN, Ivanova OK, Romanova EA, Sashchenko LP, Yashin DV. N-terminal peptide of PGLYRP1/Tag7 is a novel ligand for TREM-1 receptor. Int. J. Mol. Sci. 2022;23:5752. doi: 10.3390/ijms23105752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Denning NL, et al. Extracellular CIRP as an endogenous TREM-1 ligand to fuel inflammation in sepsis. JCI Insight. 2020;5:e134172. doi: 10.1172/jci.insight.134172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Radaev S, Kattah M, Rostro B, Colonna M, Sun PD. Crystal structure of the human myeloid cell activating receptor TREM-1. Structure. 2003;11:1527–1535. doi: 10.1016/j.str.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 224.Kelker MS, et al. Crystal structure of human triggering receptor expressed on myeloid cells 1 (TREM-1) at 1.47 Å. J. Mol. Biol. 2004;342:1237–1248. doi: 10.1016/j.jmb.2004.07.089. [DOI] [PubMed] [Google Scholar]