

Abstract
Endocannabinoids [2-arachidonoylglycerol (2-AG) and N-arachidonoylethanolamine (AEA)], endogenously produced arachidonate-based lipids, are anti-inflammatory physiological ligands for two known cannabinoid receptors, CB1 and CB2, yet the molecular and cellular mechanisms underlying their effects after brain injury are poorly defined. In the present study, we hypothesize that traumatic brain injury (TBI)-induced loss of endocannabinoids exaggerates neurovascular injury, compromises brain-cerebrospinal fluid (CSF) barriers (BCB) and causes behavioral dysfunction. Preliminary analysis in human CSF and plasma indicates changes in endocannabinoid levels. This encouraged us to investigate the levels of endocannabinoid-metabolizing enzymes in a mouse model of controlled cortical impact (CCI). Reductions in endocannabinoid (2-AG and AEA) levels in plasma were supported by higher expression of their respective metabolizing enzymes, monoacylglycerol lipase (MAGL), fatty acid amide hydrolase (FAAH), and cyclooxygenase 2 (Cox-2) in the post-TBI mouse brain. Following increased metabolism of endocannabinoids post-TBI, we observed increased expression of CB2R, non-cannabinoid receptor Transient receptor potential vanilloid-1 (TRPV1), aquaporin 4 (AQP4), ionized calcium binding adaptor molecule 1 (IBA1), glial fibrillary acidic protein (GFAP), and acute reduction in cerebral blood flow (CBF). The BCB and pericontusional cortex showed altered endocannabinoid expressions and reduction in ventricular volume. Finally, loss of motor functions and induced anxiety behaviors were observed in these TBI mice. Taken together, our findings suggest endocannabinoids and their metabolizing enzymes play an important role in the brain and BCB integrity and highlight the need for more extensive studies on these mechanisms.
Keywords: Brain-CSF Barriers (BCB), Cerebral Blood Flow (CBF), Endocannabinoid system (ECS), Monoacylglycerol lipase (MAGL), fatty acid amide hydrolase (FAAH), 2-arachidonyl glycerol (2-AG), N-arachidonoylethanolamine (AEA), Choroid plexus (CP), Neurological deficits
1. Introduction
Traumatic brain injury (TBI) is a major health concern in terms of human disability, medical expenses, and lost productivity.1–6 In addition to the immediate mechanical trauma at the time of injury, secondary injury including cerebral edema, impaired cerebral blood flow (CBF), neuronal cell death, blood-brain barrier (BBB), and brain-cerebrospinal fluid (CSF) barrier (BCB) damage, develops within the hours and days after injury and leads to poor neurological outcomes.1–12 Cellular necrosis temporally correlates with immune activation and edema development after TBI.13–17 Under physiological conditions, production and reabsorption of CSF in the ventricular system are in equilibrium. However, TBI-induced inflammation in the ventricular system and restricted outflow of CSF contribute to post-TBI edema, and are manifested by enlarged ventricles and increased intracranial pressure (ICP) in the first few days and weeks after TBI18,19 The elevated ICP due to fluid retention in the brain and BCB cavities can be lethal and needs to be reduced sooner.
The BCB consists of a single layer of cuboidal cells, which lines and surrounds vascular choroid plexus (CP) in the four ventricles of brain. The CP is responsible for producing (CSF), and is the principal constituent of the BCB and its continuation into the arachnoid barrier in the meninges.20–22 The apical epithelial cells hang in ventricles where they secrete and monitor CSF composition.22 Additionally, CPs are comprised of pericytes, endothelial cells, and harbor immune cells including microglia and astrocytes.23–25 CP and respective ventricles are highly vulnerable to trauma-induced physical force and mechanical disruption, which can immediately disintegrate the CP, can alter the composition and regulation of CSF, and can lead to exaggerated edema. For example, the third ventricle (V3) is vulnerable to frontal impact, while the lateral ventricles (VL) are highly sensitive to lateral blows.26–28 Thus, altered ventricular system and elevated ICP post-TBI are physiologically disabling and need to be taken care of immediately.18,19
The endocannabinoid system (ECS) is comprised of two primary endocannabinoid receptors (CB1R and CB2R), bioactive endocannabinoid ligands [2-arachidonoylglycerol (2-AG) and N-arachidonoylethanolamine (AEA)] and their synthesizing/metabolizing enzymes that may be manufactured by multiple cell types within the CNS.29–32 The endocannabinoids balance is maintained by their metabolizing enzymes – monoacylglycerol lipase (MAGL) and fatty acid amide hydrolase (FAAH).31,32 The ECS influences a variety of physiological systems that are dysregulated after TBI, including memory function, appetite, mood, pain sensation, and immune activation.30,33 We and others showed that activation of non-psychoactive CB2R were recently implicated in improved cerebral perfusion via reduction of neurovascular inflammation.29,30,33–35 Thus, cannabinoids directly inhibit inflammation, at least in part via activation of endocannabinoid receptors on immune and non-immune cells.35–39 Enzymatic degradation of 2-AG and AEA leads to formation of arachidonic acid (AA) with MAGL playing a rate-limiting role.35–37 The downstream enzyme cyclooxygenase-2 (Cox-2) acts on AA and yields classic prostaglandins such as PGD2, PGE2 etc. and thus, elevates inflammation after injury.35–37 Increasing scientific effort has revealed the role of the ECS in cellular homeostasis in brain, however, roles of endocannabinoids and its metabolizing enzymes in BCB and CP integrity post-TBI are not fully evaluated. Therefore, we hypothesized that endocannabinoid system dysregulation in the CP mediates neuroinflammation, and leads to progressive neurological deficits after TBI. A translational research approach incorporating patient CSF and plasma was used to elucidate the complex role of endocannabinoids metabolism in the development of secondary neurovascular injury after TBI. Additionally, measurements of CBF, ventricular volume, and behavioral outcomes were assessed to evaluate injury progression in mice after TBI.
2. Methods
2.1. Patient CSF, dura and blood Collection
CSF was obtained from 8 consecutive adult TBI patients on 2–3 days of hospital admission and 6 normal pressure hydrocephalus (NPH) patients requiring CSF diversion in the Department of Neurosurgery at the Medical College of Georgia, Augusta University. NPH patients served as an ideal control population for CSF as these patients exhibit significant edema without a traumatic event. Blood from 7 TBI patients and 9 healthy individuals (Control) served for plasma endocannabinoid analysis. In addition, discarded dura were collected from hemicraniectomy procedure from seven TBI patients. Patient specimens were collected from adults (age 17–83) without regard for age, race, gender, or socioeconomic status. All studies were approved by The Institutional Review Board at Augusta University (Formerly Georgia Regents University) [No. 1361000–1; 933254–8; 1344380–3]. All samples were de-identified and coded by the attending physician prior to transport to the laboratory.
2.2. Animals
This study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23) and was approved by the Committee on Animal Use for Research and Education at Augusta University [No. 2021–1046 and 2017–0838]. Mixed sex CD1 or C57Bl/6J mice (5–6 months old) were housed under a 12-h light/12-h dark cycle at 23 ± 1°C. Food and water were provided ad libitum.
2.3. Controlled cortical impact (CCI)
Mice were anesthetized with isoflurane (2%) and subjected to a sham injury or CCI as detailed previously by our laboratory1,40–42. Mice were placed in a stereotaxic frame and a 3.5-mm craniotomy was made in the right parietal bone midway between the lambda and the bregma with the medial edge 1-mm lateral from the midline, leaving the dura intact. Mice were impacted at 3 m/s with a 85 ms dwell time and 3 mm depression using a 3 mm diameter convex tip (PinPoint PCI3000 Precision Cortical Impactor, Hatteras Instruments, Cary, NC), mimicking a moderate to severe TBI. Craniotomy was sealed with bone wax, skin incision was surgically sutured, and mice were returned into a clean warm cage until recovered. Sham-operated mice received similar procedures but no impact. Body temperature was maintained at 37°C using a small animal temperature controller throughout all procedures (Kopf Instruments, Tujunga, CA, USA). Moderate to severe TBI had a mortality rate of about 15%. Any TBI mouse that died before the terminal end-point in the study was removed from the longitudinal analysis.
2.4. Behavioral tests
2.4.1. Habituation
For behavioral tasks, mice were habituated for thirty minutes in the room and training/testing were done under identical lighting conditions at the same time each day. All behavioral chambers were cleaned with 70% ethanol cleaning solution before and after use.
2.4.2. Assessment of motor coordination by Rota-rod and narrow beam walk
Motor coordination was assessed using a rotarod (Stoelting Co., Wood Dale, IL). Mice were trained to run on rotarod prior to injury, which started moving at a speed of 4 rpm and accelerated to 30 rpm in 4 min. On test day, mice were tested on the rotarod for the length of time each animal maintained balance while walking on top of the drum. Trials were ended when the animal either fell off the rod or clung to the rod as it made one complete rotation.43 Motor coordination also was evaluated on stationary narrow beam (6 mm wide and 1 m long) over 3 consecutive days: 2 days of training prior to injury and 1 day of testing. The time required to traverse the beam was quantified in addition to the number of foot faults.44 Each mouse was tested three-times and the average was recorded. All behavioral analyses were done blindly with regard to injury/sham control.
2.4.3. Open field and Novel object recognition test
Mice were tested in a square box (40 cm × 40 cmx 40 cm) for 10 min and activities were recorded by camera. The activities (time spent in center zone and latency to first entry to center zone) were recorded and analyzed with the software “Ethovision XT” (Noldus Information Technology, Asheville, NC, USA)45. The results were presented as mean ± SEM. The time point studies for open field and Novel object recognition tests followed by MRI were performed by Anymaze (Stoelting Co.) and were analyzed by Anymaze video tracking software as described by Lu et al.46
2.5. Laser speckle contrast imaging (LSCI)
Perimed LSCI was used to image cerebral perfusion and record CBF at days 1, 3 and 21 post-TBI/sham surgery. LSCI is a fast, calibrated and reliable technique that others and we have been using to determine blood flow in the brain and other organs.35,47–50 The brain illuminated with laser light, backscatters light as an interference pattern of bright and dark areas (called as speckle pattern). This pattern depends on blood cells movement, and the level of change in this pattern because of blood flow/blood cells motion is quantified as the local speckle contrast by the LSCI.
To calculate CBF, mouse was anesthetized, and a midline incision was made to expose the skull. The skull then was cleaned with sterile phosphate-buffered saline (PBS) and non-toxic silicone oil was applied to improve imaging quality. Body temperature was maintained at 37 ± 0.5°C. Cerebral perfusion was calculated in regions of interest (ROIs) located between bregma and lambda in each hemisphere using a Perimed PSI system with a 70 mW built-in laser diode for illumination and 1388 × 1038 pixels CCD camera installed 10 cm above the skull (speed 19 Hz, and exposure time 6 ms). Acquired high-resolution images were analyzed for changes in CBF (cerebral perfusion) using vendor supplied PIMSoft software and presented as mean perfusion values.51
2.6. Magnetic resonance imaging (MRI) acquisition and image processing
Naïve mice underwent magnetic resonance imaging (MRI) examination at baseline to make sure there is no underlying pathology. Same mice were reimaged again at 3 days and 24 days post-TBI. Each mouse was anesthetized with isoflurane inhalation (induction 3%; maintenance 1.5–2.0%) in 100% O2 gas and secured within a stereotaxic frame within the dedicated animal holder. Body temperature was maintained at 37°C by warm water circulated in a heating cradle and respiration monitored during the procedure using a physiological monitoring system (SAI, New Jersey, USA). Imaging was done on a 7T/20 small-bore animal scanner (Bruker Biospec, Ettlingen, Germany) at the Augusta University, Augusta, Georgia. Two actively decoupled radio frequency (RF) coils were used: a volume coil of 8.4 cm diameter used as the transmitter and a four-channel mouse phase array surface coil as the receiver.
Data Acquisition and analysis:
The full MRI battery scan time was approximately 1-hour and included weighted, scalar images and data for quantitative mapping as listed below. Given the importance of timing between TBI and MRI measurements made at day 3, the scan order within the battery was identical across all animals and time points. Two-dimensional multi-slice T2-weighted RARE images were collected with TE = 35 ms and TR = 5000 ms for visualization of T2-weighted contrast. For T2-mapping, a two-dimensional multi-slice multi-echo images were collected with TE = 10/20/30/………..150 ms and TR = 3600ms. For T2 mapping, in house Matlab code were used to read in the multi-echo data and to perform fitting of the Carr-Purcell Meibloom Gill (CPMG) equation to determine T2 values at each voxel within the brain volume. Three-dimensional T1-weighted MDEFT (T1W-MDEFT) MRI scans were acquired with TE/TR = 3.5/3000 ms and IR = 1100 ms. T2 -weighted fluid attenuated inversion recovery (FLAIR) was collected using a 2D RARE pulse sequence with TE = 22 ms, TR = 10 s, and IR = 2500 ms. Three-dimensional T2* weighted MRI scans were collected using a multi-gradient echo pulse sequence with TE = 3.5/7.5/10.5/14 ms and TR = 600 ms were collected and the maps averaged across all echoes and visualized to determine the presence and conspicuity of blood products in the parenchyma expected to result from micro hemorrhages. The resultant files were analyzed for edema and ventricular volume with the help of NIH ImageJ software.
2.7. Tissue collection
At terminal time points, mice were deeply anesthetizing with isoflurane (5%). Deeply anesthetized mice were perfused with 30 mL of ice-cold phosphate buffered saline, and brains were carefully removed. A 3-mm coronal brain section centered on the contusion was prepared using an acrylic brain matrix, and cerebral cortices were collected for analysis, as detailed below. For histological and immunohistochemical analysis, brains were perfused with 4% paraformaldehyde.
2.8. Immunostaining and Quantification
Coronal sections (5 μm thick) of mouse brains were obtained from the paraffin embedded tissue blocks, deparaffinized with xylene and alcohol gradients and stained with standardized procedure for immunostaining as reported previously52. Deparaffinized sections after standard preparatory procedures and antigen retrieval were incubated with anti-CB2-AF488 (1:100; Bioss); anti-CB1-AF594(1:100; Bioss Inc.); anti-TRPV1-AF647 (1:100; Novus Biologicals); anti-Iba1 (1: 500; Abcam); anti-MAGL(1:100; Bioss Inc.); anti-FAAH (1:100; Bioss Inc.); anti-Aqp4 (1:100; Santacruz Biotech) and anti-GFAP (1:500; Thermo Fisher) overnight at 4 ° C followed by incubation with an appropriate fluorescent secondary antibody (1:500; Jackson Laboratories, Burlingame, CA) for 1 hour. The sections were mounted with Vectamount permanent mounting media (Vector Laboratories, CA). Dura was embedded in OCT and sectioned at 20 um thickness. Dura sections were stained for Iba1, Aqp4, FAAH, Cox-2 and MAGL. Images were captured by Thermo Fisher EVOS M7000 inverted microscope and analyzed by NIH ImageJ software. The expressing cells were quantified for mean fluorescent intensity (MFI) at five discrete areas of ipsilateral cortex or CP in each group with the help of the imageJ software and repeated five times to quantify the expression of proteins (magnification × 200). Briefly, the images were converted into 16 bit images and threshold were set between 41 and 162 for all dura images. For mice brain images, threshold was set between 39–196 (Iba1/Aqp4/GFAP), 31–196 (CB1/CB2/TRPV1) and 44–190 (Cox-2/FAAH/MAGL). As a result stained area show as white (255 value), while background becomes black (0 value).
2.9. Enzyme-Linked Immunoassay
Endocannabinoids (2-AG and AEA) levels in CSF or plasma were determined by competitive inhibition enzyme immunoassay kits (Cat# CEO443Ge and CEO440Ge respectively; Cloud-Clone Corp, Katy, Tx) as described by manufacturer. Prostaglandin E2 (PGE2) content in CSF/plasma was determined by commercial competitive inhibition enzyme immunoassay kit (cat#KGE004B; R&D Systems, Minneapolis, MN).
2.10. Preparative and analytical flow cytometry
Freshly collected dura tissue was sieved through a cell strainer, followed by centrifugation (1,500 rpm, 5 min) to prepare single-cell suspensions. Cells were incubated with antibodies against cannabinoid markers CB1, CB2 (Bioss Inc.), and TRPV1 (Novus Biologicals Ltd.). Following a PBS wash, cells were fixed and permeabilized using a Fixation/Permeabilization Concentrate (Biolegend, CA, USA), and then incubated with antibodies for intracellular labeling of MAGL, FAAH (Purchased from Bioss Inc.), and Cox-2 (Abcam). After a final wash, cells were analyzed using a 4-color flow cytometer (Acea 4-laser NovoCyte Quanteon, Agilent technologies lnc., USA), and analyzed using FCSexpress™ software (De novo software, CA, USA), as described previously (Baban et al., 2005; Baban et al., 2013). Isotype-matched controls were analyzed to set the appropriate gates for each sample. For each marker, samples were analyzed in duplicate. To minimize false-positive events, the number of double-positive events detected with the isotype controls was subtracted from the number of double-positive cells stained with corresponding antibodies (not isotype control), respectively. Cells expressing a specific marker were reported as a percentage of the number of gated events.
2.11. Statistical Analysis
Two group comparisons were made using a two-tailed t-test followed by Mann-Whitney U computation. Multi-group comparisons were made using a two-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test. Results were expressed as mean ± SEM. A p ≤ 0.05 was considered to be statistically significant.
3. Results
3.1. TBI alters endocannabinoid tone in injured brain
Dura from hemicraniectomy of TBI patients showed presence of cannabinoid receptors (CB1 and CB2) and non-cannabinoid receptors TRPV1 (Fig. 1A–B and supplementary Fig. 1). Among these three receptors, CB1 showed higher number of positive cells in dura (Fig. 1B). The dura cells showed expression of metabolizing enzymes such as FAAH, MAGL, and Cox-2 (Fig. 1C–D and supplementary Fig. 1). MAGL showed higher intensity than the other two enzymes in the dura. In agreement, TBI patient plasma had reduced levels of both 2-AG and AEA (Fig. 1E). Further, reduced amount of 2-AG was observed in human CSF from TBI patient as compared to control NPH CSF (Fig. 1F; p<0.05). Subsequently, high amount of PGE2, a terminal pro-inflammatory product of arachidonic acid metabolism was measured in TBI patient plasma and CSF (p<0.01; Fig. 1E and F). However, elevated levels of AEA (p<0.01) were observed in CSF from TBI patients, suggesting a compensatory mechanism of maintaining endocannabinoid tone in the brain during the early period of recovery.
Fig. 1: Presence of key molecules of the ECS in the dura samples from TBI patients and reduced levels of endocannabinoids in human plasma and CSF.
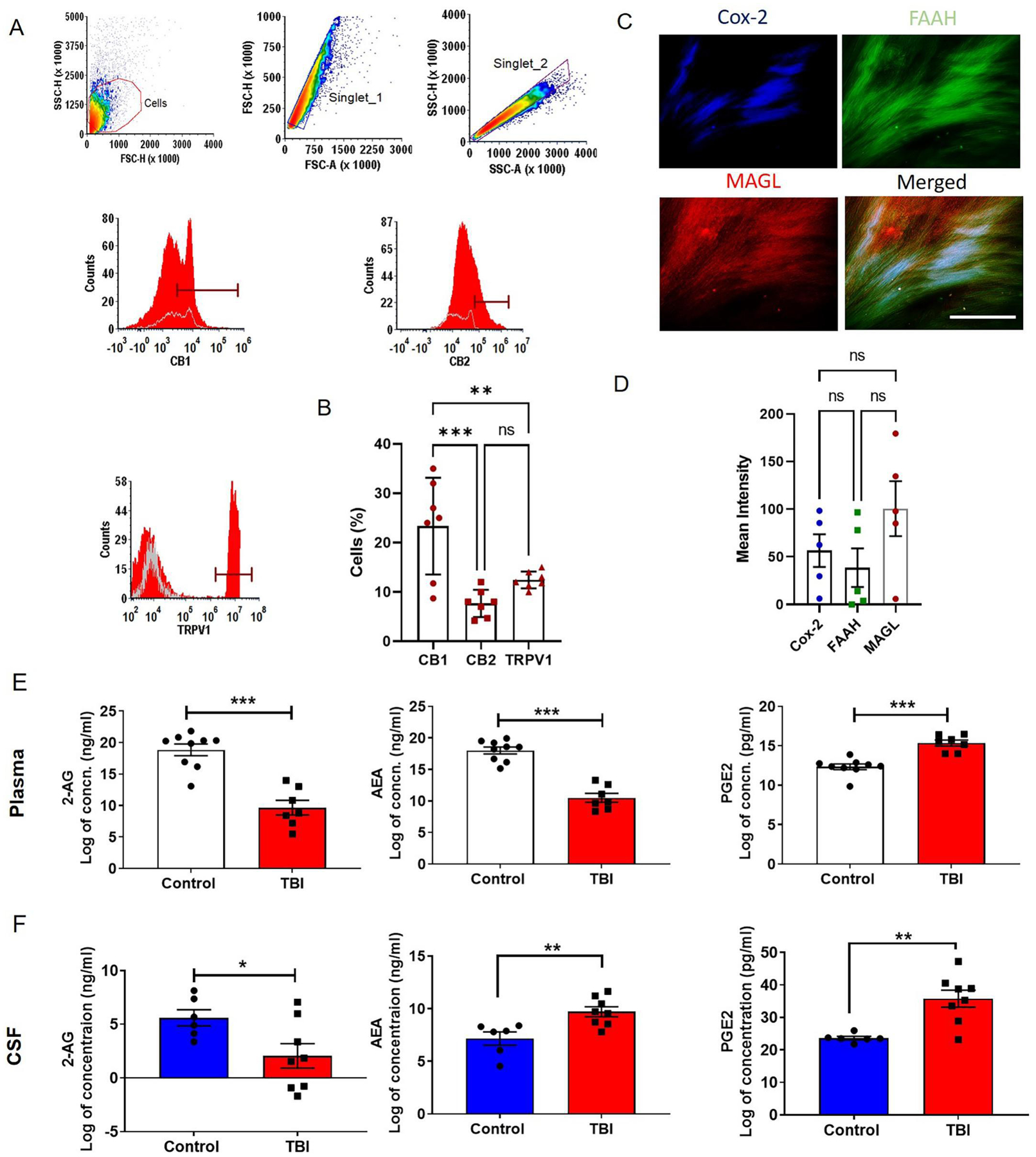
Cannabinoid and non-cannabinoid receptors were studied by flow cytometry. A and B) Our results showed the presence of cannabinoid receptors (CB1 and CB2) and non-cannabinoid receptor TRPV1 in dura from TBI patients (n = 7). The dural tissue was stained for principal cannabinoid metabolizing enzymes FAAH, MAGL and Cox-2 and mean fluorescent intensity was determined (C and D). We observed active metabolizing enzymes in dura (n = 5), suggesting a role for the ECS in brain barriers. The contents of 2-AG, AEA and PGE2 in human plasma and CSF were analyzed with the help of competitive inhibition enzyme immunoassays. Data are expressed as Log of concentrations. (E and F) Reduction in 2-AG contents was observed in TBI patient plasma and CSF. We also observed (E) low content of AEA in TBI patient plasma but, CSF from TBI victims showed a significant (F) increase in AEA level as compared with that from NPH patients. NPH patients’ spinal tap CSFs were used as controls. PGE2, an end product of endocannabinoid-arachidonic acid metabolism, acts as a pro-inflammatory mediator, was found to be elevated in both plasma and CSF (n=6–9). Two groups were compared by Paired t-test followed by Mann-Whitney test. Three groups were compared by One way ANOVA. Results were presented as mean ± SEM. Significance was ascertained as *p<0.05; **p<0.01 and ***p<0.001.
We further analyzed cannabinoid receptor and metabolizing enzyme expression in the post-traumatic brain of mice. The CP and cortex (Fig. 2 and 3) of TBI mice showed high expression of CB1, CB2 and TRPV1 at day 3, while the pericontusional cortex had significantly higher expression of CB1 at day 26 as compared to CB2 and TRPV1, which returned to basal levels by day 26 post-TBI (Fig. 2 E–H). Similarly, we observed elevated MAGL, FAAH and Cox-2 activity in the mouse CP and cortex at 3 days, but not at 26 days post-TBI (Fig. 3). Among these three, Cox-2 was observed to be slightly elevated in the post-TBI mouse brain compared to sham (Fig. 3E and H). Taken together, our data shows that traumatic injury upregulates cannabinoid receptors and metabolizing enzymes acutely which results in decreased endocannabinoids. Overall, these changes result in altered cannabinoid tone in TBI victims. Our findings are consistent with previous reports showing that the endocannabinoid system maintains normal physiology and cellular homeostasis of the brain, and any alteration of this system either by disease or by activators/inhibitors may worsen physiological and functional outcomes.53–55
Fig. 2: Expression of cannabinoid and non-cannabinoid receptors in the CP and cortex in the post-traumatic mouse brain.
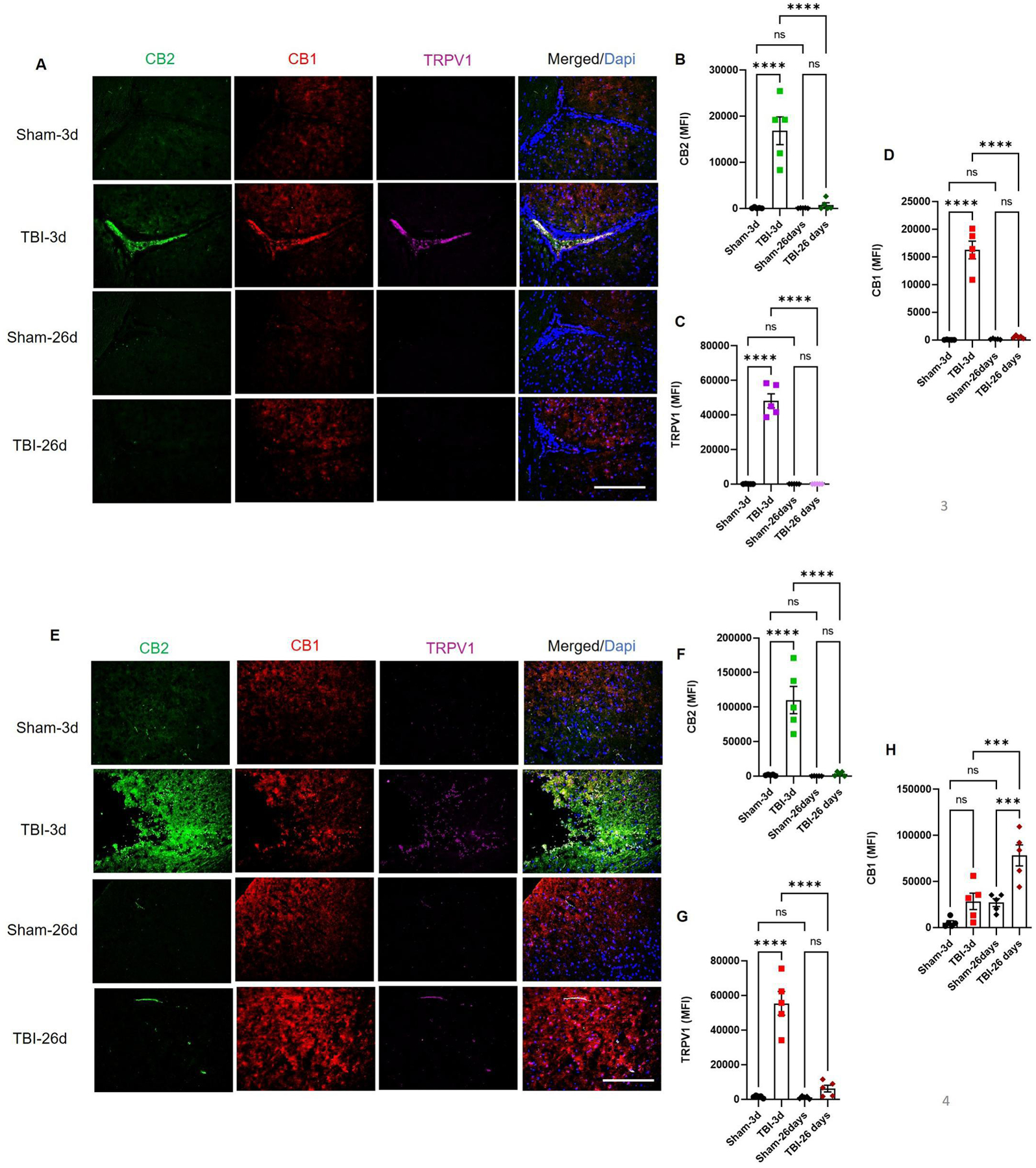
A-D) Fixed frozen mouse brain sections were analyzed by immunohistochemical analysis. Brains were stained for cannabinoid receptors (CB1 and CB2) and non-cannabinoid receptor TRPV1, and were analyzed for their expression up to 26 days post-TBI. CB1, CB2 and TRPV1 showed higher expressions at 3 days and returned to a reduced level at day 26 post-injury (B-D). (E-G) CB2 and TRPV1 showed acute upregulation at day 3 and returned to basal levels at day 26 post-TBI. However, CB1 was found to be high at day 3, but showed significantly increased expression in cortex at day 26 post-TBI (H). Groups were compared by Two-Way ANOVA analysis with Tukey’s post-hoc comparison. Results were calculated as mean fluorescent intensity (MFI) ± SEM. (n=5; ns= not significant; ***p<0.001; ****p<0.0001 vs. sham; Scale bar 125μm).
Fig. 3: The CP and pericontusional cortex are sites of acute endocannabinoid metabolism in the TBI mice.
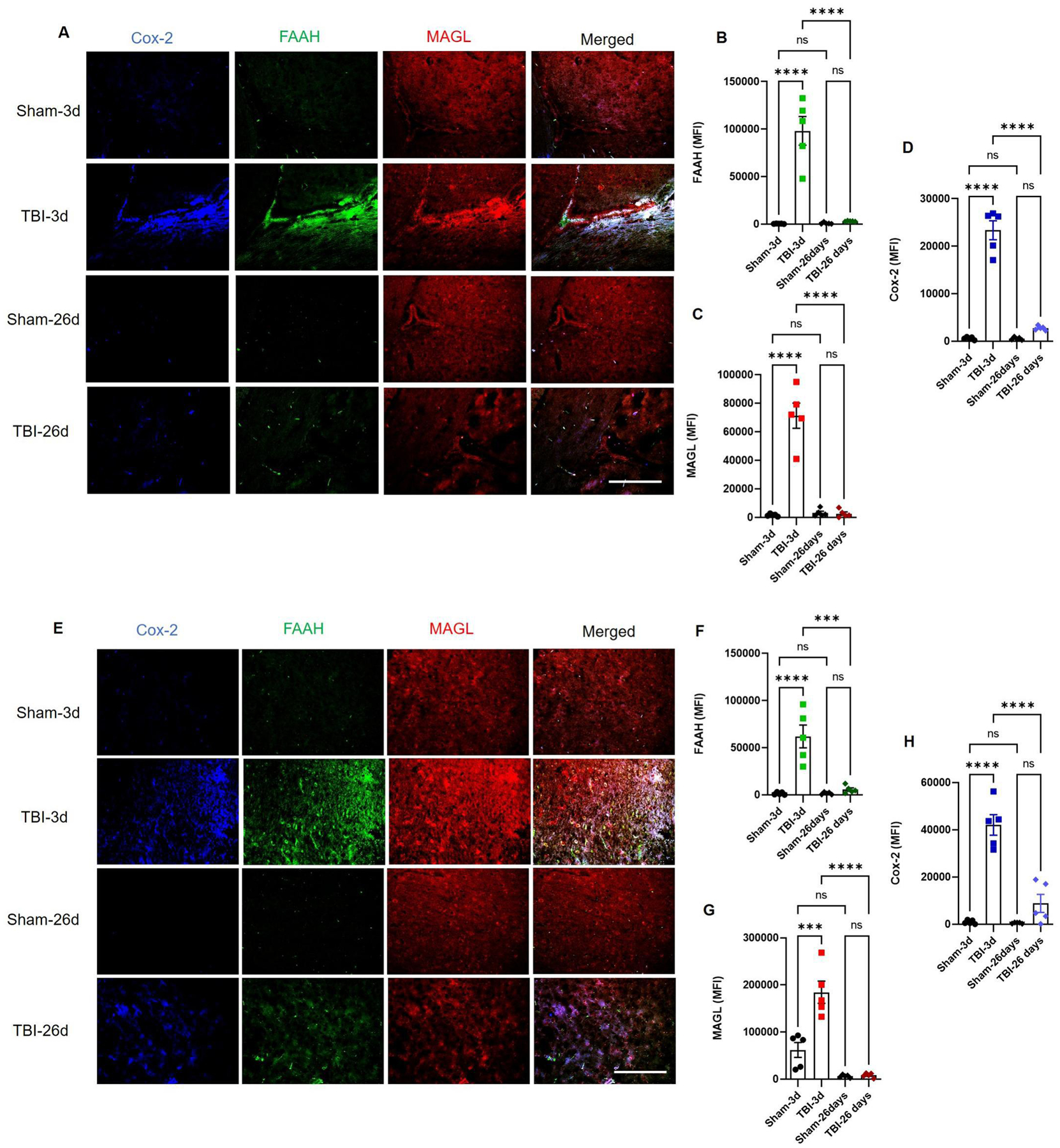
Mouse brain sections were stained for cannabinoid enzymes by immunohistochemistry method. Endocannabinoid metabolizing enzymes FAAH, MAGL and Cox-2 showed higher expressions at 3 days and returned to reduced levels at day 26 post-injury (A-D). As seen at CP, the cortex also shows signs of acute metabolism of 2-AG and AEA as FAAH, MAGL and Cox-2 were highly elevated at day 3 post-TBI with respect to sham (E-H), while only cox-2 remained slightly elevated at day 26th (E, H). Groups were compared by Two-Way ANOVA analysis with Tukey’s post-hoc comparison. Results were calculated as mean fluorescent intensity (MFI) ± SEM. (n=5; ns= not significant; ***p<0.001; ****p<0.0001 vs. sham; Scale bar 125μm).
3.2. Astrogliosis and increased Aqp4 expression mirror changes in the endocannabinoid system in the TBI brain
TBI leads to persistent neuroinflammation, which is often expressed as increased and persistent astrogliosis. The injured brain shows higher expressions of GFAP (a marker of activated astrocyte) and Iba1 (a marker of macrophage/microglia), which reveals extensive damage to the sensory and motor cortex.56–59 Besides neuroinflammtion, TBI leads to edema characterized by increased expression of specific water channels Aqp4 in the hippocampal neurons.60 Interestingly, dura from TBI patients also showed expression of macrophage marker Iba1 and relatively higher water channel Aqp4 (Supplementary Fig. 2A and B). Similarly, TBI mice showed higher expressions of GFAP and Iba1 along the CP and near the contusion area (Fig. 4). The expression of Iba1 remains elevated up to day 26 in both the CP and cortex (Fig. 4A, C, F and H). Cortical GFAP expression remained elevated at day 26 (Fig. 4I and J), but was reduced significantly around the CP at day 26 post-TBI (Fig. 4D and E). However, Aqp4 did not show a significant increase in the pericontusional cortex (Fig. 4G), but showed elevated expression around the CP at day 26 post-TBI (Fig. 4A and B). Our result is in agreement with a previous report showing increased Aqp4 post-TBI was in positive correlation with edema, astrogliosis and neurological impairments.61 Changes in these injury markers mirror the alterations in CB receptors and metabolizing enzymes (Fig. 2 and 3), and thus, indicate the importance of the cannabinoid system in the brain pathology post-TBI. Previously, an increases in Aqp4 and the astrogliosis marker vimentin was observed to be corresponding to increased CB2 in the brain post-TBI61; while chronic deficiency of subcortical and perilesional endocannabinoids mirrored increased astrogliosis and neuroinflammation after TBI.56 Thus astrogliosis and AQP4 may be critical in influencing outcomes following TBI.
Fig. 4: Elevated injury markers in CP and cortex of TBI mice.
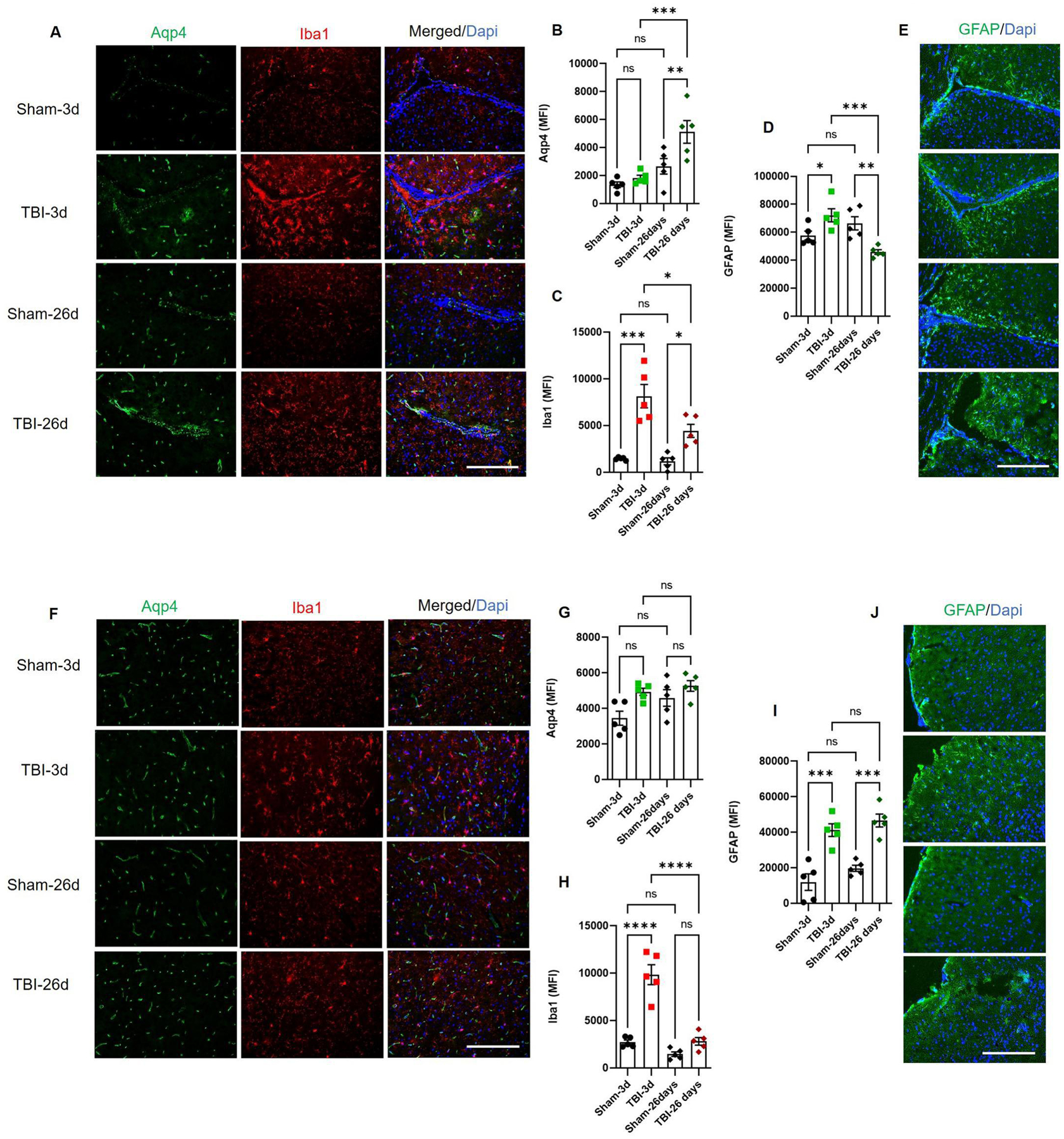
Frozen mouse brain sections were stained for (A-C) Aqp4, Iba1 and (D-E) GFAP. Water channel Aqp4 was significantly elevated in the CP at day 26 post-TBI, while macrophage/microglia marker Iba1and activated astrocyte marker GFAP was elevated at day 3 post-TBI as compared to respective sham. GFAP showed decreased expression in CP at day 26 while, Iba1 remained high at day 26 post-TBI (B-D). Sham mice showed basal expressions of all markers. Traumatic cortex showed differential expressions of Aqp4, Iba1 and GFAP. Water channel Aqp4 was elevated as compared to sham but not significantly at day 3 post-TBI (F and G). Macrophage/microglia marker Iba1 and activated astrocyte marker GFAP were elevated at day 3 post-TBI as compared to respective sham (H and I). However, GFAP sustained higher expression in cortex up to day 26 post-TBI (I and J). Sham mice showed basal expression of all markers. Data were expressed as mean (MFI) ± SEM (n=5). Groups were compared by Two-way ANOVA followed by Tukey’s post hoc test (*p<0.05; **p<0.01; ***p<0.00; ****p<0.00011 vs. Sham; Scale bar 125μm).
3.3. TBI results in reduced CBF and damaged ventricular system in mice
Previously we showed that TBI exerts prominent effects on the neurovascular system. As a result of cortical impact, CBF drops down acutely.35,47 We have examined the CBF in the mouse brain within the period of 1–21 days post-TBI. These TBI mice at all-time points showed significant reductions in CBF as compared to controls (p<0.0001; Fig. 2A–B). TBI mice showed maximum loss of CBF at days 1 and 3 with little recovery at 21 days post-TBI (p<0.05; Fig. 5A–B). Our CBF data showed that neurovascular injury did not resolve even after 3 weeks post-injury. In addition, we performed MRI scans on naïve mice followed by TBI until 24 days. MRI images revealed alterations in ventricular space and edematous tissue at 3 days post-TBI, which sustained for more than 3 weeks (Fig. 5C–D). We observed higher edema at day 3, followed by day 24 post-TBI (Fig. 5C and D). Additionally, ventricles showed the signs of volumetric reduction post-TBI. All ventricles showed reduced volume at day 3 post-TBI as compared to naïve mice (Fig. 5G–H). MRI scans revealed the re-expansion of the third ventricles (V3) and cerebral aqueduct (AQ) to normal levels at 24 days post-TBI. However, right lateral ventricle (Right VL) still showed significantly reduced volume than naïve (Fig. 5H), while the 4th ventricle (V4) showed further reductions in volume (Fig. 5G). The contralateral VL (left VL) did not show any significant changes (Fig. 5H). Our results are in agreement with previous reports showing CP and respective ventricle vulnerability to traumatic injuries.18,19,26–28 TBI can severely affect the integrity and functioning of ventricles and may add to increased edema, astrogliosis and inflammation post-TBI.26–28,62–65
Fig. 5: TBI induces tissue damage, edema and altered ventricular space.
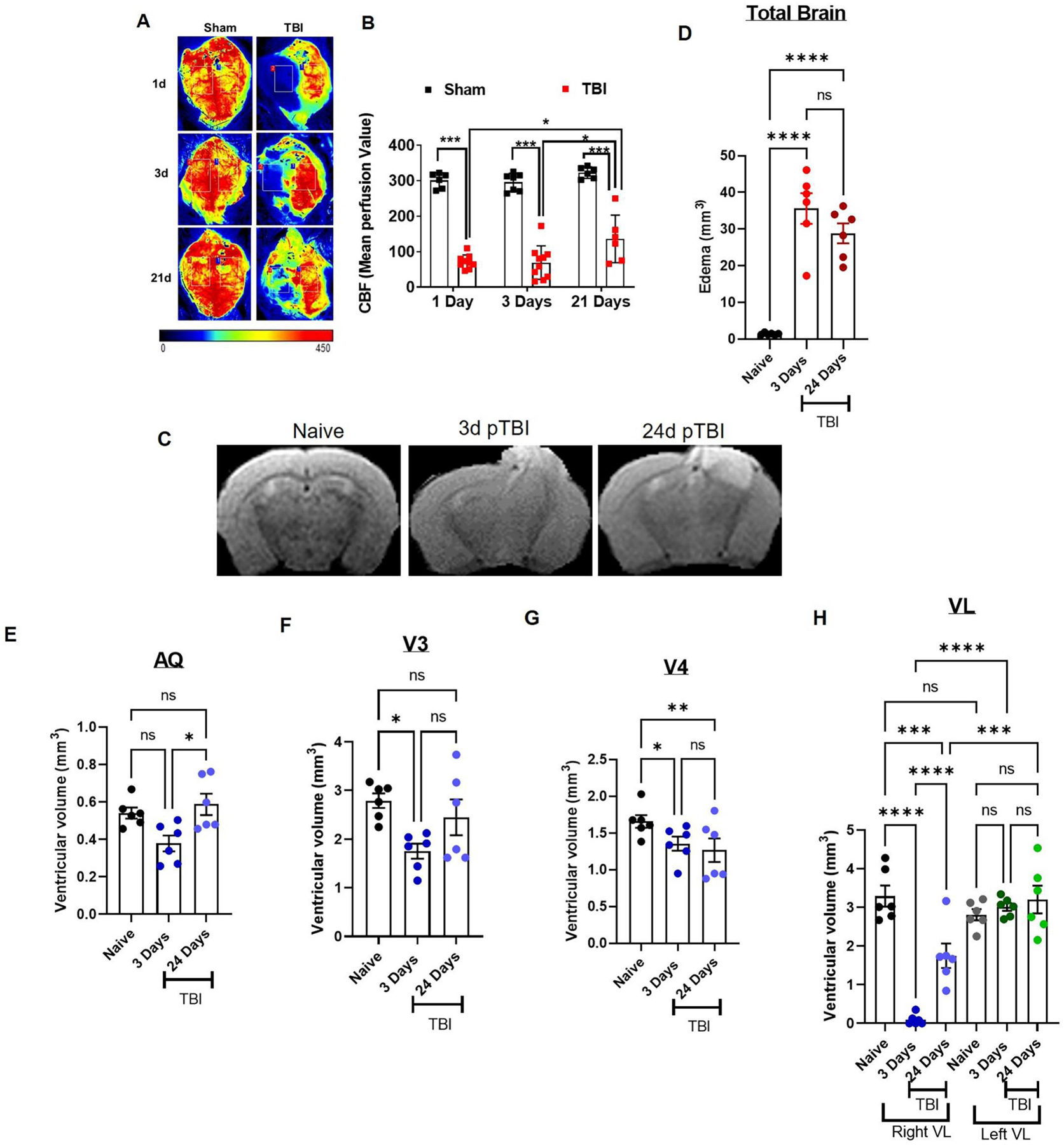
CBF in sham or TBI mice were measured with the help of Perimed LSCI device, while edema and ventricular volume were assessed by MRI. A) Mice were imaged for CBF at day 1, day 3, and day 21 post-TBI or sham surgery using Perimed LSCI. Representative images are provided following Sham or TBI (A). TBI mice showed significant reductions in CBF which persisted up to 21 days post-injury (A, B). Cerebral perfusion was quantified as mean perfusion value and data were presented as mean ± SEM (n=6–10). C-H) Naïve C57Bl/6 mice were imaged by MRI for basal edema and ventricular volume and were introduced to CCI. These mice were rescanned on MRI again on day 3 and day 24 post-TBI longitudinally (n=6). C-D) TBI mice showed higher edema at day 3 which reduced on day 24 post-TBI, but remained significant. Most brain ventricles (AQ, V3 and right VL) showed reduction in volume at day 3 as compared to naïve mice (E, F and H; n=6). However, 4th ventricle (V4) and right VL remained significantly reduced even at day 24 post-TBI (G, H). Left hemispheric VL did not show any reduction in comparison to the basal level. Groups were compared by Two-Way ANOVA analysis with Tukey’s post-hoc comparison. Results were calculated as mean ± SEM. ns= not significant; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001 vs. naïve or sham.
3.4. Behavior deficits correspond with changes in the ventricular system and edema in TBI mice
Finally, those mice who were scanned on MRI for edema and ventricular volume post-TBI, were examined for motor-coordination deficits, anxiety behavior and cognitive impairment. This ventricular alteration and edema was associated with serious motor and behavioral dysfunction (Fig. 6A–E). TBI mice showed motor deficits on rotarod until day 28 post-TBI (Fig. 6C), while grip and motor deficits on hanging wire (Fig. 6A) and stationary narrow beam (NBM) were seen at day 3 post-TBI only (Fig. 6B). These TBI mice also showed signs of anxiety in open field (Fig. 6D) and avoided the open center region in open field test. However, these mice had reduced preference for novel object at day 28, but impairment was not found to be statistically significant as compared to naïve mice at day 28 post-TBI (Fig. 6E). Similarly, others and we have previously shown that TBI mice experience persistent anxiety, motor, cognitive and coordination deficits.47,59 These behavioral deficits are influenced by tissue loss59, endocannabinoid tone35,66–69, and reduced CBF.35,47 Although our results show a strong connection between the alteration in the endocannabinoid system, change in the ventricular system and behavioral deficits, a complementary effect of direct mechanical damage to motor strip or elevated inflammation cannot be ruled out.
Fig. 6: TBI results in neurobehavioral deficits.
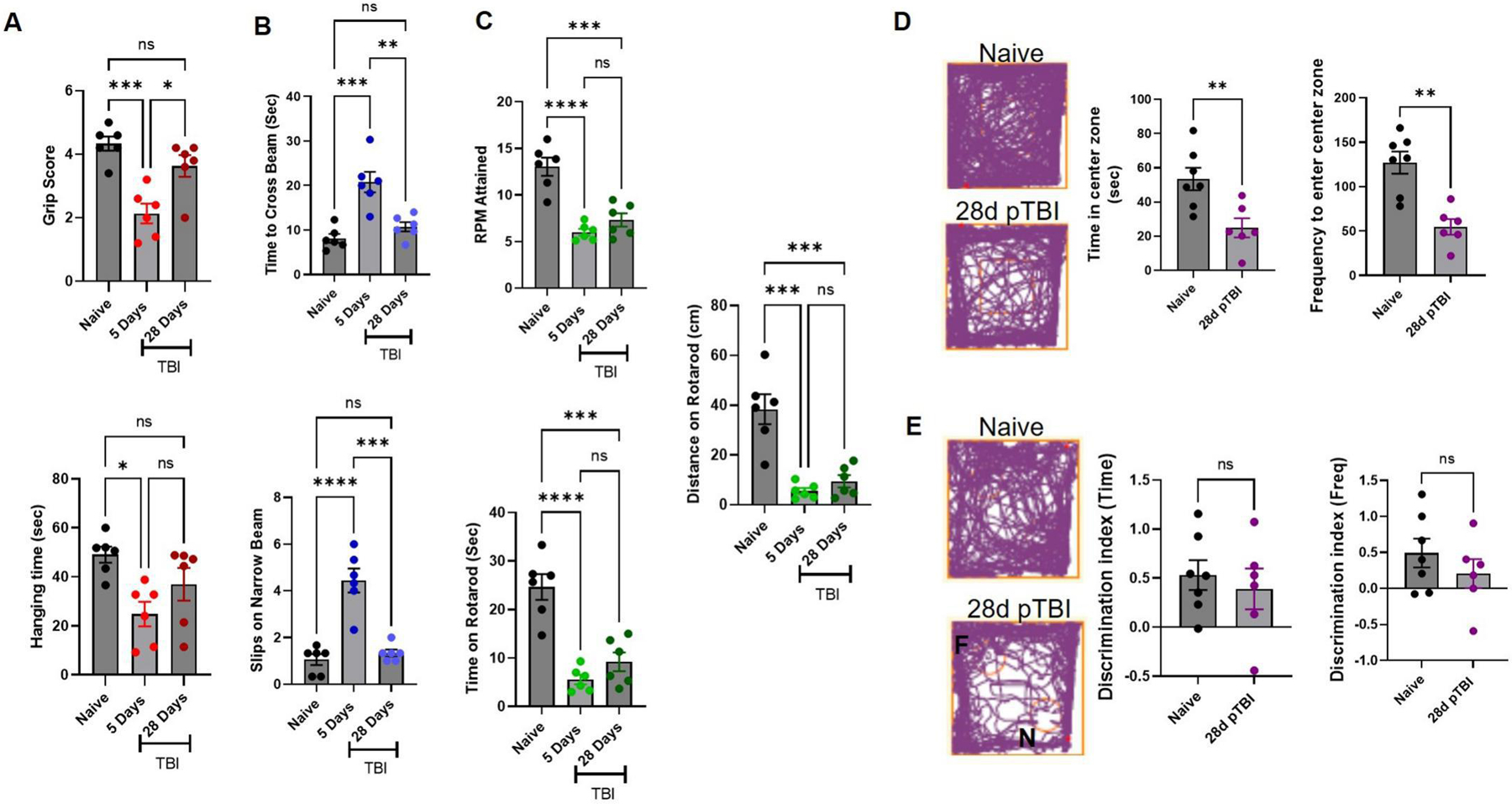
Finally, the same cohort of mice used in Fig. 5, were challenged by a battery of behavioral tests after MRI and longitudinally compared up to day 28 post-TBI (A-E). Mice showed lower grip strength (A) on hanging wire and motor incoordination on stationary beam (B) at day 5 post-injury. (A) TBI mice showed lower scores and less ability to climb on the wire than naïve (baseline) scores. (B) These TBI mice took more time to cross a narrow beam and had higher number of slips than naïve ones. Further, these TBI mice were unable to hold on to an accelerating rotarod and fell off earlier. These mice showed persistent motor impairment on rotarod up to day 28 post-injury (C). At day 28, these mice were further tested for anxiety in open field (D) and for novel object recognition ability (E). Data from open field and novel object recognition test were analyzed by Anymaze software. (D) TBI mice spent more time at the periphery than naïve mice with a lower frequency of visits to the center area, and exhibited higher anxiety than naïve mice. Injured mice also showed reduced preference to explore the novel object, discrimination differences in time spent and number of visits were not observed as significant (E). Groups were compared by Two-Way ANOVA analysis with Tukey’s post-hoc comparison. Results were calculated as mean ± SEM (n=6). ns= not significant; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001 vs. naive.
4. Discussion
TBI is a heterogeneous injury that may persist from a few days to months. Sustained elevations in ICP typically peak within the first days after TBI, while the patient is in the neuro-intensive care unit, and induces tissue hypoperfusion, brain herniation, and a poor clinical prognosis.70,71 Unfortunately, efficacious treatment options to reduce mortality and improve long-term outcomes following TBI are lacking and represent a major unmet clinical need. Preventative measures reduce injury incidence and/or severity, yet one-third of hospitalized TBI patients die from secondary pathological processes that develop during supervised medical care (e.g. within the neurointensive care unit).5,7 In addition to the immediate mechanical traumatic injury at the time of impact secondary injury such as cerebral edema, impaired CBF, BBB damage, ischemic-hypoxia, and neuronal cell death may develop within hours to days and contribute to patient’s poor outcomes.2–7,9–11,72,73 The data presented herein identify the role of endocannabinoid metabolism in the pathology of TBI that possibly cause a coordinated secondary injury response and culminating in the development of neurological deficits. In this report, we demonstrated an important role for the endocannabinoid system in the post-traumatic BCB. In particular, we found prominently reduced 2-AG and AEA in plasma and CSF from TBI patients, mirrored with higher expression of MAGL and FAAH in the BCB and cerebral cortex post-TBI. This reduction in endocannabinoids corresponded with higher levels of PGE2 in plasma and CSF, and elevated Cox-2 in the brain. We further showed that TBI resulted in persistent edema, reduced ventricular volume, decreased cerebral perfusion, and impaired neurological outcomes. Thus, combining all the present results, we suggest an important role for endocannabinoid metabolism in injury progression post-TBI.
The endocannabinoid system is a pleiotropic signaling system involved in the multiple aspects of mammalian physiology and pathology, and controls a wide variety of functions of the normal body like the cardiovascular and neurological systems.74–76 Although several endogenous cannabinoids have been discovered77–79; AEA80 and 2-AG81–83 are the most studied ones. 2-AG is three times more prevalent than AEA in the brain37,84, and has a higher affinity for CB1 and CB2 receptors.85–87 Precursors of these endocannabinoids are present in lipid membranes, which upon demand (typically by injury or by depolarization), are liberated in one or two rapid enzymatic steps and released into the extracellular space in contrast to classical neurotransmitters that are synthesized ahead of time and stored in synaptic vesicles.31,32,88,89 Previously, we have shown that CCI-induced injury causes severe edema35,90 that possibly leads to disrupted vascular perfusion, while selective activation of CB2 minimized edema, protected CBF, and reduced edema.35 In agreement with this, we observed reduced levels of 2-AG, a endogenous ligand to this receptor in CSF and plasma from patients with subsequent high levels of PGE2, an inflammatory prostaglandin downstream to 2-AG in AA metabolism. Our data from human samples are in agreement with the higher expression of MAGL observed in the injured mouse brain, a principal metabolizing enzyme for 2-AG that becomes elevated in response to TBI.
2-AG is an important metabolic intermediate in lipid synthesis and also serves as a major source of AA in prostaglandin synthesis36 and therefore plays an important role in the inflammatory pathways. Distinct sets of enzymes, anabolising or catabolising endocannabinoids, possess distinct anatomical distributions and regulate cannabinoid receptor signaling. Metabolism of AEA occurs predominantly through FAAH91,92 in intracellular membranes of postsynaptic neurons93 in the neocortex, cerebellar cortex, and hippocampus94. Inactivation of 2-AG occurs primarily via MAGL95,96, in presynaptic axon terminals93,95 in the region of the thalamus, hippocampus, cortex, and cerebellum.95 However, FAAH remained highly active for 24–72 h and returned to basal level at 26 days post-TBI. AEA, another endocannabinoid was elevated in the CSF, but was reduced in the plasma from TBI patients. Increased level of AEA in the CSF may be attributed to reduction in acute FAAH activity post-TBI. However, interactions of different immune cells and ruptured BBB can not be ruled out as a cause of increased content of AEA. Although AA is a downstream product of both 2-AG97 and AEA98,99, MAGL represents a rate-limiting biosynthetic enzyme of highly bioactive brain lipid in this pathway and contributes to ~80% eicosanoid in brain.36,100 Given that Cox-2 mediated 2-AG oxidation is considerable in the CNS101,102, Cox-2 can also oxidize AEA to create prostaglandins such as PGE231 and promote inflammatory events.36 PGE2 is produced primarily by immune cells during immune responses, and is dependent upon the activity of Cox-2.103,104 We observed an elevated levels of PGE2 in human plasma and CSF obtained during the acute period post-trauma. This observation was in congruence with elevated Cox-2 in the mouse brain post-TBI. Our result is in agreement with previous reports showing elevated PGE2105–107 and increased Cox-2 activity107,108 up to 7 days post TBI in animal models. Similarly, activation of EP1 receptor by PGE2 mediates Cox-2-induced neurotoxicity.109–111 In a gastric inflammatory lesion model of Wistar rats, upregulation of Cox-2 was accompanied by an increase in PGE2 levels, but not in prostaglandin D2 (PGD2) levels at 6 h and 7 days post-lesion, suggesting PGE2 could be the main product of Cox-2 activation.112
Prolonged pericontusional inflammation is know to occur after TBI.1,47,56,113 This neuroinflammation involves infiltration of peripheral immune cells, and two types of residential glial cells, astrocytes and microglia.57,58 As a result of injury, the expressions of reactive astrocytic marker GFAP58,114 and the microglial/macropahge marker Iba-158,115,116 increase in response to TBI, and is often expressed as astrogliosis. This higher expression of GFAP (activated astrocyte markers) and Iba1, reveals extensive damage to the sensory and motor cortex.56–59 Similarly, we have observed increased expression of GFAP and Iba1 in mouse brain post-TBI. Reactive astrogliosis after TBI is a heterogeneous response that depends and reflects the severity of brain injury and further add to neuroinflammation.57,117 In addition, the water channel protein Aqp4 was also found to be elevated until 26 days post-TBI. Similarly, postmortem brains showed persistent increases in Aqp4 up to 30 days post-TBI.118 This increase in Aqp4 was concomittant to increased GFAP and Iba1 expressions in the TBI brain.118 In addition, a previous report showed that the increase in Aqp4 post-TBI was positively correlated with severity of edema, astrogliosis and neurological impairments. This increase in Aqp4 and the astrogliosis marker vimentin further corresponded to increased CB2 expression in the brain post-TBI.61 Further, previous analysis revealed chronic deficiency of subcortical and perilesional endocananbinoids mirrored increased astrogliosis and neuroinflammation in TBI.56 Therefore, observed astrogliosis in our model could be a reflection of changes in the ECS post-TBI.
The CBF is tightly regulated by multiple processes such as endothelial, metabolic, neural, and myogenic etc. under physiological conditions.30,74–76 However, inflammatory pathways in certain central nervous system pathologies, like TBI, stroke, and vascular dementia greatly affect cerebral blood perfusion.30,74–76 PGE2 derived largely from brain pyramidal cells and from astrocytes to a lesser extent also mediates neurovascular coupling and vasodilation under stimulated condition.119 In the present study, we demonstrated that TBI reduced CBF drastically for up to 3 weeks. Our result is in agreement with previous publications showing brain injuries such as TBI or brain hemorrhages caused reduction in CBF.35,47,48 Alterations in CBF post-TBI are tightly regulated by glial cells and vascular interactions.120,121 Therefore, astrogliosis and inflammatory events may profoundly affect CBF. In line with this, we have observed increased astrogliosis and inflammation in the post-TBI brain, which corresponded to decreased CBF in the injured brain. All major cell types such as, smooth muscle, endothelium, neurons, astrocytes, pericytes, microglia, and leukocytes are involved in cerebrovascular regulation, and are capable of synthesizing endocannabinoids and/or expressing the cannabinoid (CB1 and CB2) receptors and the TRPV1 ion channel. Therefore, the endocannabinoid system may be important in the modulation and regulation of cerebral circulation under physiological and pathophysiological conditions in a very complex manner. However, under stress conditions (e.g., in conscious restrained animals or during hypoxia and hypercapnia) and in certain cerebrovascular pathologies (e.g., subarachnoid hemorrhage, as well as traumatic and ischemic brain injury), cannabinoid receptor activation was shown to induce a reduction of the CBF, likely via inhibition of the electrical and/or metabolic activity of neurons.30,35,74–76
The brain consists of barriers such as -BBB and BCB. These barriers are important components of brain function and immunity, while changes in these barriers in different brain pathologies may cause chronic effects on the functionality of the brain.28,62–64 In the present study, we demonstrated that mechanical trauma causes reduced ventricular volume that leads to altered CSF and water movement, leading to edema. This compromised barriers lead to the infiltration of peripheral immune cells and fluid into brain parenchyma. Our results mirror the previous findings showing mechanical primary injury followed by secondary injury cascade causes biochemical, molecular and inflammatory changes to the BCB. Inflammation, due to accumulation and infiltration of immune cells leads to further distruption of barrier function in the brain.28,64,65 Recently, Yasmin et al. reported that disturbed CSF secretion or absorption by CP may lead to chronic ventricular alterations.63 The group followed MRI findings upto 6 months post fluid percussion injury and observed that ventricular changes were not dependent on cortical lesions, but on CP iron load after injury.63 Similarly, we observed elevated edema and reduced ventricular size at day 3 post- CCI. Edema remains significantly eleveated with necrosis at day 26, while ventricles started expanding after initial reduction. However, V4 and right VL remains significantlty reduced in volume as compared to naïve mice. CPs are present in the lateral, 3rd and 4th ventricles and are comprised of a highly vascular core surrounded by secretary epithelial cells which make up the BCB.28,62 The CPs, particularly those in V3 and VL, are highly sensitive to mechanical cortical trauma, leading to altered fluid regulation and edema. As a result of edema and increased ICP brain tissue suffers from irreversible damage.28,62 Further, elevated expressions of cannabinoid and non-cannabinoid receptors along with key metabolozing enzymes such as MAGL, FAAH, and Cox-2 were observed in CP. Similarly, expression of CB1 and FAAH was observed in epithelial cells of the CP. Further suggesting that the presence of cannabinoid receptors and metabilizing enzymes in the CP may provide evidence of their possible role in regulation of CSF production.122 Moreover, the CP acts as a sink for endocannabinoids or other compounds such as delta9-Tetrahydrocannabinol (delta9-THC) and regulates their distribution in the CSF and brain.123 The TBI mice having altered ventricular shape and volume showed higher anxiety, reduced cognition and loss of motor function. The mechanical injury to the motor cortex and subsequent secondary injury by inflammation could have resulted directly in the observed motor-deficits and anxiety behavior in these TBI mice. In agreement, we and others have previously shown that TBI leads to persistent anxiety, motor, cognitive and coordination deficits.47,59 These behavioral deficits and poor functional outcomes are influenced by tissue loss59,120, neuroinflammation35,47,53, brain barrier damage124,125, endocannabinoid tone35,66–69, and reduced CBF.35,47,126
Research involving CP and cannabinoids is severly lacking. Most of the previous work has focused on the regulation of the BBB by the cannabinoid system post-injury. Notably, the ECS has been found at the BBB. A previous study demonstrated cannabinoids decreased BBB permeability and enhanced membrane integrity in different pathologies.127 A CB2 agonist was also shown to elevate BBB function in people living with HIV.128 Activation of CB2 by agonists exert anti-inflammatory effects either by reducing gliosis129,130 or by polarizing immune cells.35 Similarly, inhibition of 2-AG metabolism by MAGL in a mouse model of BBB distruption by systemic administration of LPS and focal photothrombotic ischemic insult, protected its functional homeostasis.131 Presence of the ECS indicates its important role in BCB integrity and functionality, and may need to be studied in detail. Although the present study highlights the role of endocannabinoid metabolism in the pathogenesis of TBI and chronic neurological deficits, our study has a few limitations. The low number of CSF and plasma samples included in study may be regarded as a limitation of the study. We investigated a murine CCI model of neurotrauma to mimic the moderate to severe injury in humans and delineated the role of metabolizing enzymes MAGL, FAAH, and Cox-2 in the propagation of injury post-TBI. However, the role of other lipid metabolites like various N-acyl amide or glycerol or virodhamine can not be ignored. Secondly, we measured PGE2 and endocannabinoid levels in plasma and CSF; however, other prostaglandins such as PGD2 along with PGE2 may also play an important role in TBI pathology. Examination of other prostaglandins in TBI could be extensive and was out of the scope of the present manuscript; however, we plan to examine this in more detail in a separate future study. Finally, we have followed the study up to 26 days (<1 month) according to study design and resources. It would be interesting to examine the cannabinoid system in the CP chronically up to 3 or 6 months, as TBI is regarded as one of the important factors for neurodegeneration. Inspite of the limitations discussed here, the results from the present study depict an important role for the ECS in brain injury and should be evaluated further for potential use as a drug target. The availability of selective pharmacological inhibitors for FAAH/MAGL has allowed the selective amplification of AEA and 2-AG levels following brain injury as a key strategy to enhance endocannabinoid signaling and to investigate their potential neuroprotective effects.
5. Conclusion
Several lines of evidence suggest a primary function of endocannabinoids, CB1/2, and other components of the ECS in TBI and the degenerative processes. Various investigators using a variety of pre-clinical models have discovered therapeutic intervention strategies modulating the ECS. Furthermore, current approaches to the development of novel therapeutic strategies for neuropathology have focused not only on their neuroprotective properties but also on alleviating symptoms of the injury. These approaches are based on the well-characterized role of cannabinoids that have the ability to control both anti-inflammatory and neuroprotective functions. Our results suggest an important role for cannabinoid receptors, ligands and enzymes in brain-barrier integrity and function. The presence of potentially targetable cannabinoid and non-cannabinoid receptors and enzymes makes the cannabinoid system a preferable target for the treatment of TBI pathology. Promising classes of compounds, such as plant-derived or synthetic cannabinoids are of great therapeutic interest in this regard. However, an improved and detailed characterization of the endocannabinoid system (ligands, enzymes, and receptor populations) will be imperative to understanding its role in TBI pathology.
Supplementary Material
Highlights.
TBI leads to chronic vascular, ventricular, and neurological deficits.
Brain barriers are compromised after traumatic brain injury.
Compromised brain-CSF barrier (BCB) shows altered cannabinoid regulation.
Altered endocannabinoid system (ECS) reduces endocannabinoids (2-AG and AEA).
Restoring cannabinoid homeostasis may promote neurological recovery after TBI.
Acknowledgements
Financial support for this project was provided by a grant from the Augusta University Research Institute and by grants from the National Institutes of Health (R01NS114560, and R03HD094606).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interests
Authors declare that there is no conflict of interest.
REFERENCES
- 1.Braun M et al. Activation of Myeloid TLR4 Mediates T Lymphocyte Polarization after Traumatic Brain Injury. Journal of immunology (Baltimore, Md. : 1950) 198, 3615–3626, doi: 10.4049/jimmunol.1601948 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rubiano AM, Carney N, Chesnut R & Puyana JC Global neurotrauma research challenges and opportunities. Nature 527, S193–197, doi: 10.1038/nature16035 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Fuentes MM, Jimenez N, Apkon SD & Rivara FP Functional outcomes during inpatient rehabilitation for American Indian and Alaska Native children with traumatic brain injury. Journal of pediatric rehabilitation medicine 9, 133–141, doi: 10.3233/prm-160376 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ryan NP et al. Social dysfunction after pediatric traumatic brain injury: A translational perspective. Neuroscience and biobehavioral reviews 64, 196–214, doi: 10.1016/j.neubiorev.2016.02.020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bramlett HM & Dietrich WD Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta neuropathologica 103, 607–614, doi: 10.1007/s00401-001-0510-8 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Glushakova OY, Johnson D & Hayes RL Delayed increases in microvascular pathology after experimental traumatic brain injury are associated with prolonged inflammation, blood-brain barrier disruption, and progressive white matter damage. Journal of neurotrauma 31, 1180–1193, doi: 10.1089/neu.2013.3080 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Bramlett HM & Dietrich WD Progressive damage after brain and spinal cord injury: pathomechanisms and treatment strategies. Progress in brain research 161, 125–141, doi: 10.1016/s0079-6123(06)61009-1 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Bramlett HM & Dietrich WD Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. J Neurotrauma 32, 1834–1848, doi: 10.1089/neu.2014.3352 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bramlett HM, Kraydieh S, Green EJ & Dietrich WD Temporal and regional patterns of axonal damage following traumatic brain injury: a beta-amyloid precursor protein immunocytochemical study in rats. Journal of neuropathology and experimental neurology 56, 1132–1141 (1997). [DOI] [PubMed] [Google Scholar]
- 10.Saul TG & Ducker TB Effect of intracranial pressure monitoring and aggressive treatment on mortality in severe head injury. Journal of neurosurgery 56, 498–503, doi: 10.3171/jns.1982.56.4.0498 (1982). [DOI] [PubMed] [Google Scholar]
- 11.Eisenberg HM et al. Initial CT findings in 753 patients with severe head injury. A report from the NIH Traumatic Coma Data Bank. Journal of neurosurgery 73, 688–698, doi: 10.3171/jns.1990.73.5.0688 (1990). [DOI] [PubMed] [Google Scholar]
- 12.Wu Y, Wu H, Guo X, Pluimer B & Zhao Z Blood-Brain Barrier Dysfunction in Mild Traumatic Brain Injury: Evidence From Preclinical Murine Models. Front Physiol 11, 1030, doi: 10.3389/fphys.2020.01030 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Czigner A et al. Kinetics of the cellular immune response following closed head injury. Acta neurochirurgica 149, 281–289, doi: 10.1007/s00701-006-1095-8 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Hayakata T et al. Changes in CSF S100B and cytokine concentrations in early-phase severe traumatic brain injury. Shock 22, 102–107, doi: 10.1097/01.shk.0000131193.80038.f1 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Katayama Y & Kawamata T Edema fluid accumulation within necrotic brain tissue as a cause of the mass effect of cerebral contusion in head trauma patients. Acta neurochirurgica. Supplement 86, 323–327 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Kawamata T & Katayama Y Surgical management of early massive edema caused by cerebral contusion in head trauma patients. Acta neurochirurgica. Supplement 96, 3–6 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Kawamata T & Katayama Y Cerebral contusion: a role model for lesion progression. Progress in brain research 161, 235–241, doi: 10.1016/s0079-6123(06)61016-9 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Chen K-H et al. Incidence of hydrocephalus in traumatic brain injury: A nationwide population-based cohort study. Medicine 98 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lalou AD et al. Cerebrospinal fluid dynamics in non-acute post-traumatic ventriculomegaly. Fluids and Barriers of the CNS 17, 24, doi: 10.1186/s12987-020-00184-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Solár P, Zamani A, Kubíčková L, Dubový P & Joukal M Choroid plexus and the blood–cerebrospinal fluid barrier in disease. Fluids and Barriers of the CNS 17, 1–29 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liddelow SA Development of the choroid plexus and blood-CSF barrier. Frontiers in neuroscience 9, 32 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghersi-Egea J-F et al. Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta neuropathologica 135, 337–361 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Lun MP, Monuki ES & Lehtinen MK Development and functions of the choroid plexus–cerebrospinal fluid system. Nature Reviews Neuroscience 16, 445–457, doi: 10.1038/nrn3921 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liddelow SA Development of the choroid plexus and blood-CSF barrier. Front Neurosci 9, 32, doi: 10.3389/fnins.2015.00032 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang AC et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 595, 565–571, doi: 10.1038/s41586-021-03710-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Podvin S et al. Esophageal cancer related gene-4 is a choroid plexus-derived injury response gene: evidence for a biphasic response in early and late brain injury. PloS one 6, e24609 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johanson C, Stopa E, Baird A & Sharma H Traumatic brain injury and recovery mechanisms: peptide modulation of periventricular neurogenic regions by the choroid plexus–CSF nexus. Journal of neural transmission 118, 115–133 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bodnar CN, Watson JB, Higgins EK, Quan N & Bachstetter AD Inflammatory Regulation of CNS Barriers After Traumatic Brain Injury: A Tale Directed by Interleukin-1. Frontiers in Immunology 12, doi: 10.3389/fimmu.2021.688254 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amenta PS, Jallo JI, Tuma RF & Elliott MB A cannabinoid type 2 receptor agonist attenuates blood-brain barrier damage and neurodegeneration in a murine model of traumatic brain injury. Journal of neuroscience research 90, 2293–2305, doi: 10.1002/jnr.23114 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Benyo Z, Ruisanchez E, Leszl-Ishiguro M, Sandor P & Pacher P Endocannabinoids in cerebrovascular regulation. American journal of physiology. Heart and circulatory physiology 310, H785–801, doi: 10.1152/ajpheart.00571.2015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu HC & Mackie K An Introduction to the Endogenous Cannabinoid System. Biological psychiatry 79, 516–525, doi: 10.1016/j.biopsych.2015.07.028 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou S & Kumar U Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. International journal of molecular sciences 19, doi: 10.3390/ijms19030833 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buch SJ Cannabinoid receptor 2 activation: a means to prevent monocyte-endothelium engagement. The American journal of pathology 183, 1375–1377, doi: 10.1016/j.ajpath.2013.08.003 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Rom S et al. Selective activation of cannabinoid receptor 2 in leukocytes suppresses their engagement of the brain endothelium and protects the blood-brain barrier. The American journal of pathology 183, 1548–1558, doi: 10.1016/j.ajpath.2013.07.033 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braun M et al. Selective activation of cannabinoid receptor-2 reduces neuroinflammation after traumatic brain injury via alternative macrophage polarization. Brain Behav Immun 68, 224–237, doi: 10.1016/j.bbi.2017.10.021 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nomura DK et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science (New York, N.Y.) 334, 809–813, doi: 10.1126/science.1209200 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schurman LD & Lichtman AH Endocannabinoids: A Promising Impact for Traumatic Brain Injury. Frontiers in pharmacology 8, 69, doi: 10.3389/fphar.2017.00069 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kozela E, Juknat A & Vogel Z Modulation of Astrocyte Activity by Cannabidiol, a Nonpsychoactive Cannabinoid. International journal of molecular sciences 18, doi: 10.3390/ijms18081669 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Puhl S-L Cannabinoid-sensitive receptors in cardiac physiology and ischaemia. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1867, 118462, doi: 10.1016/j.bbamcr.2019.03.009 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Laird MD et al. Curcumin attenuates cerebral edema following traumatic brain injury in mice: a possible role for aquaporin-4? Journal of neurochemistry 113, 637–648, doi: 10.1111/j.1471-4159.2010.06630.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wakade C, Sukumari-Ramesh S, Laird MD, Dhandapani KM & Vender JR Delayed reduction in hippocampal postsynaptic density protein-95 expression temporally correlates with cognitive dysfunction following controlled cortical impact in mice. Journal of neurosurgery 113, 1195–1201, doi: 10.3171/2010.3.JNS091212 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang QG et al. Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS One 7, e34504, doi: 10.1371/journal.pone.0034504 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cernak I, Wing ID, Davidsson J & Plantman S A novel mouse model of penetrating brain injury. Frontiers in neurology 5, 209, doi: 10.3389/fneur.2014.00209 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luong TN, Carlisle HJ, Southwell A & Patterson PH Assessment of motor balance and coordination in mice using the balance beam. Journal of visualized experiments : JoVE, doi: 10.3791/2376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evonuk KS, Prabhu SD, Young ME & DeSilva TM Myocardial ischemia/reperfusion impairs neurogenesis and hippocampal-dependent learning and memory. Brain Behav Immun 61, 266–273, doi: 10.1016/j.bbi.2016.09.001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu Y et al. Neuron-Derived Estrogen Regulates Synaptic Plasticity and Memory. J Neurosci 39, 2792–2809, doi: 10.1523/jneurosci.1970-18.2019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vaibhav K et al. Neutrophil extracellular traps exacerbate neurological deficits after traumatic brain injury. Sci Adv 6, eaax8847, doi: 10.1126/sciadv.aax8847 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vaibhav K et al. Remote ischemic post-conditioning promotes hematoma resolution via AMPK-dependent immune regulation. J Exp Med 215, 2636–2654, doi: 10.1084/jem.20171905 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ren C et al. Limb Ischemic Conditioning Improved Cognitive Deficits via eNOS-Dependent Augmentation of Angiogenesis after Chronic Cerebral Hypoperfusion in Rats. Aging Dis 9, 869–879, doi: 10.14336/ad.2017.1106 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baban B et al. Impact of cannabidiol treatment on regulatory T-17 cells and neutrophil polarization in acute kidney injury. Am J Physiol Renal Physiol 315, F1149–f1158, doi: 10.1152/ajprenal.00112.2018 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Khan MB et al. Remote ischemic postconditioning: harnessing endogenous protection in a murine model of vascular cognitive impairment. Translational stroke research 6, 69–77, doi: 10.1007/s12975-014-0374-6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vaibhav K et al. Piperine suppresses cerebral ischemia-reperfusion-induced inflammation through the repression of COX-2, NOS-2, and NF-kappaB in middle cerebral artery occlusion rat model. Molecular and cellular biochemistry 367, 73–84, doi: 10.1007/s11010-012-1321-z (2012). [DOI] [PubMed] [Google Scholar]
- 53.Jarrahi A et al. Revisiting Traumatic Brain Injury: From Molecular Mechanisms to Therapeutic Interventions. Biomedicines 8, doi: 10.3390/biomedicines8100389 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reddy V et al. Targeting the endocannabinoid system: a predictive, preventive, and personalized medicine-directed approach to the management of brain pathologies. EPMA Journal 11, 217–250, doi: 10.1007/s13167-020-00203-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Howlett AC et al. Endocannabinoid tone versus constitutive activity of cannabinoid receptors. Br J Pharmacol 163, 1329–1343, doi: 10.1111/j.1476-5381.2011.01364.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vogel A et al. Low brain endocannabinoids associated with persistent non-goal directed nighttime hyperactivity after traumatic brain injury in mice. Scientific Reports 10, 14929, doi: 10.1038/s41598-020-71879-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Burda JE, Bernstein AM & Sofroniew MV Astrocyte roles in traumatic brain injury. Experimental neurology 275, 305–315 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mira RG, Lira M & Cerpa W Traumatic Brain Injury: Mechanisms of Glial Response. Front Physiol 12, 740939, doi: 10.3389/fphys.2021.740939 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pöttker B et al. Traumatic brain injury causes long-term behavioral changes related to region-specific increases of cerebral blood flow. Brain structure and function 222, 4005–4021 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Xiong A et al. Aquaporin-4 is a potential drug target for traumatic brain injury via aggravating the severity of brain edema. Burns Trauma 9, tkaa050, doi: 10.1093/burnst/tkaa050 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lopez-Rodriguez AB, Acaz-Fonseca E, Viveros M-P & Garcia-Segura LM Changes in Cannabinoid Receptors, Aquaporin 4 and Vimentin Expression after Traumatic Brain Injury in Adolescent Male Mice. Association with Edema and Neurological Deficit. PLOS ONE 10, e0128782, doi: 10.1371/journal.pone.0128782 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xiang J et al. The choroid plexus as a site of damage in hemorrhagic and ischemic stroke and its role in responding to injury. Fluids and Barriers of the CNS 14, 8, doi: 10.1186/s12987-017-0056-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yasmin A et al. Post-injury ventricular enlargement associates with iron in choroid plexus but not with seizure susceptibility nor lesion atrophy—6-month MRI follow-up after experimental traumatic brain injury. Brain Structure and Function 227, 145–158, doi: 10.1007/s00429-021-02395-5 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaur C, Rathnasamy G & Ling E-A The Choroid Plexus in Healthy and Diseased Brain. Journal of Neuropathology & Experimental Neurology 75, 198–213, doi: 10.1093/jnen/nlv030 (2016). [DOI] [PubMed] [Google Scholar]
- 65.Szmydynger-Chodobska J, Strazielle N, Zink BJ, Ghersi-Egea J-F & Chodobski A The Role of the Choroid Plexus in Neutrophil Invasion after Traumatic Brain Injury. Journal of Cerebral Blood Flow & Metabolism 29, 1503–1516, doi: 10.1038/jcbfm.2009.71 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haller J, Goldberg SR, Pelczer KG, Aliczki M & Panlilio LV The effects of anandamide signaling enhanced by the FAAH inhibitor URB597 on coping styles in rats. Psychopharmacology 230, 353–362 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Griebel G et al. The selective reversible FAAH inhibitor, SSR411298, restores the development of maladaptive behaviors to acute and chronic stress in rodents. Scientific Reports 8, 1–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Panikashvili D et al. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 413, 527–531 (2001). [DOI] [PubMed] [Google Scholar]
- 69.Morena M et al. Enhancing Endocannabinoid Neurotransmission Augments The Efficacy of Extinction Training and Ameliorates Traumatic Stress-Induced Behavioral Alterations in Rats. Neuropsychopharmacology 43, 1284–1296, doi: 10.1038/npp.2017.305 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alberico AM, Ward JD, Choi SC, Marmarou A & Young HF Outcome after severe head injury. Relationship to mass lesions, diffuse injury, and ICP course in pediatric and adult patients. Journal of neurosurgery 67, 648–656, doi: 10.3171/jns.1987.67.5.0648 (1987). [DOI] [PubMed] [Google Scholar]
- 71.Miller JD et al. Significance of intracranial hypertension in severe head injury. Journal of neurosurgery 47, 503–516, doi: 10.3171/jns.1977.47.4.0503 (1977). [DOI] [PubMed] [Google Scholar]
- 72.Braun M et al. Activation of Myeloid TLR4 Mediates T Lymphocyte Polarization after Traumatic Brain Injury. J Immunol, doi: 10.4049/jimmunol.1601948 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bramlett HM & Dietrich WD Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. Journal of neurotrauma, doi: 10.1089/neu.2014.3352 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Iannotti FA, Di Marzo V & Petrosino S Endocannabinoids and endocannabinoid-related mediators: Targets, metabolism and role in neurological disorders. Progress in lipid research 62, 107–128, doi: 10.1016/j.plipres.2016.02.002 (2016). [DOI] [PubMed] [Google Scholar]
- 75.Pertwee RG Endocannabinoids and Their Pharmacological Actions. Handbook of experimental pharmacology 231, 1–37, doi: 10.1007/978-3-319-20825-1_1 (2015). [DOI] [PubMed] [Google Scholar]
- 76.O’Sullivan SE Endocannabinoids and the Cardiovascular System in Health and Disease. Handbook of experimental pharmacology 231, 393–422, doi: 10.1007/978-3-319-20825-1_14 (2015). [DOI] [PubMed] [Google Scholar]
- 77.Chu CJ et al. N-oleoyldopamine, a novel endogenous capsaicin-like lipid that produces hyperalgesia. The Journal of biological chemistry 278, 13633–13639, doi: 10.1074/jbc.M211231200 (2003). [DOI] [PubMed] [Google Scholar]
- 78.Porter AC et al. Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. The Journal of pharmacology and experimental therapeutics 301, 1020–1024 (2002). [DOI] [PubMed] [Google Scholar]
- 79.Heimann AS et al. Hemopressin is an inverse agonist of CB1 cannabinoid receptors. Proceedings of the National Academy of Sciences of the United States of America 104, 20588–20593, doi: 10.1073/pnas.0706980105 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Devane W et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science (New York, N.Y.) 258, 1946–1949, doi: 10.1126/science.1470919 (1992). [DOI] [PubMed] [Google Scholar]
- 81.Mechoulam R et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochemical Pharmacology 50, 83–90, doi: 10.1016/0006-2952(95)00109-D (1995). [DOI] [PubMed] [Google Scholar]
- 82.Mechoulam R & Parker LA The endocannabinoid system and the brain. Annual review of psychology 64, 21–47, doi: 10.1146/annurev-psych-113011-143739 (2013). [DOI] [PubMed] [Google Scholar]
- 83.Sugiura T et al. 2-Arachidonoylgylcerol: A Possible Endogenous Cannabinoid Receptor Ligand in Brain. Biochemical and Biophysical Research Communications 215, 89–97, doi: 10.1006/bbrc.1995.2437 (1995). [DOI] [PubMed] [Google Scholar]
- 84.Bequet F et al. CB1 receptor-mediated control of the release of endocannabinoids (as assessed by microdialysis coupled with LC/MS) in the rat hypothalamus. The European journal of neuroscience 26, 3458–3464, doi: 10.1111/j.1460-9568.2007.05900.x (2007). [DOI] [PubMed] [Google Scholar]
- 85.Pertwee RG & Ross RA Cannabinoid receptors and their ligands. Prostaglandins, leukotrienes, and essential fatty acids 66, 101–121, doi: 10.1054/plef.2001.0341 (2002). [DOI] [PubMed] [Google Scholar]
- 86.Reggio PH Endocannabinoid structure-activity relationships for interaction at the cannabinoid receptors. Prostaglandins, leukotrienes, and essential fatty acids 66, 143–160, doi: 10.1054/plef.2001.0343 (2002). [DOI] [PubMed] [Google Scholar]
- 87.Hillard CJ Biochemistry and pharmacology of the endocannabinoids arachidonylethanolamide and 2-arachidonylglycerol. Prostaglandins & other lipid mediators 61, 3–18 (2000). [DOI] [PubMed] [Google Scholar]
- 88.Gonsiorek W et al. Endocannabinoid 2-arachidonyl glycerol is a full agonist through human type 2 cannabinoid receptor: antagonism by anandamide. Molecular pharmacology 57, 1045–1050 (2000). [PubMed] [Google Scholar]
- 89.Luk T et al. Identification of a potent and highly efficacious, yet slowly desensitizing CB1 cannabinoid receptor agonist. Br J Pharmacol 142, 495–500, doi: 10.1038/sj.bjp.0705792 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Laird MD et al. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia 62, 26–38, doi: 10.1002/glia.22581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cravatt BF et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proceedings of the National Academy of Sciences of the United States of America 98, 9371–9376, doi: 10.1073/pnas.161191698 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cravatt BF et al. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384, 83–87, doi: 10.1038/384083a0 (1996). [DOI] [PubMed] [Google Scholar]
- 93.Gulyas AI et al. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. The European journal of neuroscience 20, 441–458, doi: 10.1111/j.1460-9568.2004.03428.x (2004). [DOI] [PubMed] [Google Scholar]
- 94.Egertova M, Giang DK, Cravatt BF & Elphick MR A new perspective on cannabinoid signalling: complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proceedings. Biological sciences 265, 2081–2085, doi: 10.1098/rspb.1998.0543 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dinh TP et al. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proceedings of the National Academy of Sciences of the United States of America 99, 10819–10824, doi: 10.1073/pnas.152334899 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Blankman JL, Simon GM & Cravatt BF A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chemistry & biology 14, 1347–1356, doi: 10.1016/j.chembiol.2007.11.006 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bell RL, Kennerly DA, Stanford N & Majerus PW Diglyceride lipase: a pathway for arachidonate release from human platelets. Proceedings of the National Academy of Sciences 76, 3238–3241, doi: 10.1073/pnas.76.7.3238 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Deutsch DG & Chin SA Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochemical Pharmacology 46, 791–796, doi: 10.1016/0006-2952(93)90486-G (1993). [DOI] [PubMed] [Google Scholar]
- 99.Deutsch DG et al. Production and physiological actions of anandamide in the vasculature of the rat kidney. The Journal of clinical investigation 100, 1538–1546, doi: 10.1172/jci119677 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nomura DK et al. Monoacylglycerol lipase regulates 2-arachidonoylglycerol action and arachidonic acid levels. Bioorganic & medicinal chemistry letters 18, 5875–5878, doi: 10.1016/j.bmcl.2008.08.007 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim J & Alger BE Inhibition of cyclooxygenase-2 potentiates retrograde endocannabinoid effects in hippocampus. Nature neuroscience 7, 697–698, doi: 10.1038/nn1262 (2004). [DOI] [PubMed] [Google Scholar]
- 102.Straiker A et al. COX-2 and fatty acid amide hydrolase can regulate the time course of depolarization-induced suppression of excitation. Br J Pharmacol 164, 1672–1683, doi: 10.1111/j.1476-5381.2011.01486.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kalinski P Regulation of immune responses by prostaglandin E2. J Immunol 188, 21–28, doi: 10.4049/jimmunol.1101029 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Agard M, Asakrah S & Morici L PGE2 suppression of innate immunity during mucosal bacterial infection. Frontiers in Cellular and Infection Microbiology 3, doi: 10.3389/fcimb.2013.00045 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Moochhala SM et al. Mercaptoethylguanidine inhibition of inducible nitric oxide synthase and cyclooxygenase-2 expressions induced in rats after fluid-percussion brain injury. Journal of Trauma and Acute Care Surgery 59, 448–455 (2005). [DOI] [PubMed] [Google Scholar]
- 106.Gopez JJ et al. Cyclooxygenase-2-specific inhibitor improves functional outcomes, provides neuroprotection, and reduces inflammation in a rat model of traumatic brain injury. Neurosurgery 56, 590–604 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hickey RW et al. Cyclooxygenase-2 Activity Following Traumatic Brain Injury in the Developing Rat. Pediatric Research 62, 271–276, doi: 10.1203/PDR.0b013e3180db2902 (2007). [DOI] [PubMed] [Google Scholar]
- 108.Strauss KI et al. Prolonged cyclooxygenase-2 induction in neurons and glia following traumatic brain injury in the rat. Journal of neurotrauma 17, 695–711 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kawano T, Anrather J, Frys K, Zhou P & Iadecola C Activation of prostaglandin EP1 receptors contributes to COX-2-dependent neurotoxicity. Soc. Neurosci. Abs 30, 600–608 (2004). [Google Scholar]
- 110.Zhou P, Qian L, Anrather J & Iadecola C PGE2 receptor EP1 mediates COX-2-dependent cell death in hippocampal slices. Soc. Neurosci. Abs 30 (2004). [Google Scholar]
- 111.Iadecola C & Gorelick PB The Janus Face of Cyclooxygenase-2 in Ischemic Stroke. Stroke 36, 182–185, doi: 10.1161/01.STR.0000153797.33611.d8 (2005). [DOI] [PubMed] [Google Scholar]
- 112.Motilva V, Alarcón de la Lastra C, Bruseghini L, Manuel Herrerias J & Sánchez-Fidalgo S COX expression and PGE2 and PGD2 production in experimental acute and chronic gastric lesions. International Immunopharmacology 5, 369–379, doi: 10.1016/j.intimp.2004.10.005 (2005). [DOI] [PubMed] [Google Scholar]
- 113.Pernici CD, Kemp BS & Murray TA Time course images of cellular injury and recovery in murine brain with high-resolution GRIN lens system. Scientific reports 9, 1–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Luo J et al. Long-Term Cognitive Impairments and Pathological Alterations in a Mouse Model of Repetitive Mild Traumatic Brain Injury. Frontiers in Neurology 5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mouzon BC et al. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Annals of Neurology 75, 241–254, doi: 10.1002/ana.24064 (2014). [DOI] [PubMed] [Google Scholar]
- 116.Broussard JI et al. Repeated mild traumatic brain injury produces neuroinflammation, anxiety-like behaviour and impaired spatial memory in mice. Brain Injury 32, 113–122, doi: 10.1080/02699052.2017.1380228 (2018). [DOI] [PubMed] [Google Scholar]
- 117.Roth TL et al. Transcranial amelioration of inflammation and cell death after brain injury. Nature 505, 223–228, doi: 10.1038/nature12808 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Neri M et al. Immunohistochemical Evaluation of Aquaporin-4 and its Correlation with CD68, IBA-1, HIF-1α, GFAP, and CD15 Expressions in Fatal Traumatic Brain Injury. Int J Mol Sci 19, doi: 10.3390/ijms19113544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lacroix A et al. COX-2-Derived Prostaglandin E2 Produced by Pyramidal Neurons Contributes to Neurovascular Coupling in the Rodent Cerebral Cortex. The Journal of Neuroscience 35, 11791, doi: 10.1523/JNEUROSCI.0651-15.2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Villapol S, Byrnes KR & Symes AJ Temporal dynamics of cerebral blood flow, cortical damage, apoptosis, astrocyte-vasculature interaction and astrogliosis in the pericontusional region after traumatic brain injury. Front Neurol 5, 82, doi: 10.3389/fneur.2014.00082 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Marina N et al. Astrocytes monitor cerebral perfusion and control systemic circulation to maintain brain blood flow. Nature Communications 11, 131, doi: 10.1038/s41467-019-13956-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ashton JC, Appleton I, Darlington CL & Smith PF Cannabinoid CB1 receptor protein expression in the rat choroid plexus: a possible involvement of cannabinoids in the regulation of cerebrospinal fluid. Neurosci Lett 364, 40–42, doi: 10.1016/j.neulet.2004.04.016 (2004). [DOI] [PubMed] [Google Scholar]
- 123.Agnew WF, Rumbaugh CL & Cheng JT The uptake of delta9-tetrahydrocannabinol in choroid plexus and brain cortex in vitro and in vivo. Brain Res 109, 355–366, doi: 10.1016/0006-8993(76)90535-7 (1976). [DOI] [PubMed] [Google Scholar]
- 124.Sivandzade F, Alqahtani F & Cucullo L Traumatic Brain Injury and Blood-Brain Barrier (BBB): Underlying Pathophysiological Mechanisms and the Influence of Cigarette Smoking as a Premorbid Condition. Int J Mol Sci 21, doi: 10.3390/ijms21082721 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kempuraj D et al. Acute Traumatic Brain Injury-Induced Neuroinflammatory Response and Neurovascular Disorders in the Brain. Neurotox Res 39, 359–368, doi: 10.1007/s12640-020-00288-9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Clark AL et al. Dynamic association between perfusion and white matter integrity across time since injury in Veterans with history of TBI. NeuroImage: Clinical 14, 308–315, doi: 10.1016/j.nicl.2016.12.017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hagan K, Varelas P & Zheng H Endocannabinoid System of the Blood–Brain Barrier: Current Understandings and Therapeutic Potentials. Cannabis and Cannabinoid Research 7, 561–568, doi: 10.1089/can.2021.0101 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ellis RJ et al. Beneficial Effects of Cannabis on Blood–Brain Barrier Function in Human Immunodeficiency Virus. Clinical Infectious Diseases 73, 124–129, doi: 10.1093/cid/ciaa437 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Martín-Moreno AM et al. Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: relevance to Alzheimer’s disease. Mol Pharmacol 79, 964–973, doi: 10.1124/mol.111.071290 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ & Dittel BN Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem 95, 437–445, doi: 10.1111/j.1471-4159.2005.03380.x (2005). [DOI] [PubMed] [Google Scholar]
- 131.Piro JR et al. Inhibition of 2-AG hydrolysis differentially regulates blood brain barrier permeability after injury. Journal of Neuroinflammation 15, 142, doi: 10.1186/s12974-018-1166-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.