

Abstract
Background and Aims
Diploid and polyploid Urochloa (including Brachiaria, Panicum and Megathyrsus species) C4 tropical forage grasses originating from Africa are important for food security and the environment, often being planted in marginal lands worldwide. We aimed to characterize the nature of their genomes, the repetitive DNA and the genome composition of polyploids, leading to a model of the evolutionary pathways within the group including many apomictic species.
Methods
Some 362 forage grass accessions from international germplasm collections were studied, and ploidy was determined using an optimized flow cytometry method. Whole-genome survey sequencing and molecular cytogenetic analysis were used to identify chromosomes and genomes in Urochloa accessions belonging to the ‘brizantha’ and ‘humidicola’ agamic complexes and U. maxima.
Key Results
Genome structures are complex and variable, with multiple ploidies and genome compositions within the species, and no clear geographical patterns. Sequence analysis of nine diploid and polyploid accessions enabled identification of abundant genome-specific repetitive DNA motifs. In situ hybridization with a combination of repetitive DNA and genomic DNA probes identified evolutionary divergence and allowed us to discriminate the different genomes present in polyploids.
Conclusions
We suggest a new coherent nomenclature for the genomes present. We develop a model of evolution at the whole-genome level in diploid and polyploid accessions showing processes of grass evolution. We support the retention of narrow species concepts for Urochloa brizantha, U. decumbens and U. ruziziensis, and do not consider diploids and polyploids of single species as cytotypes. The results and model will be valuable in making rational choices of parents for new hybrids, assist in use of the germplasm for breeding and selection of Urochloa with improved sustainability and agronomic potential, and assist in measuring and conserving biodiversity in grasslands.
Keywords: Polyploidy, apomixis, repetitive DNA motifs, genome-specific sequences, evolution, Brachiaria, tropical forage grasses
Introduction
Most arable crops have well-understood evolution and domestication processes, and the genetic diversity of their wild relatives is being exploited in breeding new varieties (Vaughan et al., 2007). Native grasslands include high biodiversity that can be threatened by expansion of cultivated areas, while forage grasses occupy half the world’s agricultural land. Genomic knowledge is being increasingly applied to breeding the temperate Lolium–Festuca (ryegrass) complex (Velmurugan et al., 2016), and there are a number of genetic selection and breeding programmes for tropical and sub-tropical forage (e.g. Worthington and Miles, 2015) but applications of omics-based technologies (Ishitani et al., 2004) remain limited. The tropical forage grasses include clusters of species with various ploidies, and many reproduce through apomixis, but their genomic composition and diversity in general remain poorly characterized. The rational choice of parents for making crosses in breeding programmes, however, requires the knowledge of genome composition and ploidy. The integration of sequencing, molecular cytogenetic and bioinformatic tools allows the identification of genomes which come together in polyploids (Soltis et al., 2013). Many crop species with polyploid members, including Brassica (Alix et al., 2008) and the Brassicaceae (Cheng et al., 2013), Avena (Tomaszewska and Kosina, 2018, 2021; Liu et al., 2019) and particularly the tribe Triticeae (Hordeae) (Linde-Laursen et al., 1997) have well-established genome designations (as single letters) to describe the ancestral genomes in auto- and allo-polyploids (amphiploids). Resolution of genome relationships in the wheat group has mainly assisted with extensive use of the germplasm pool in breeding (Feldman and Sears, 1981; Ali et al., 2016; Rasheed et al., 2018). Although it has proved difficult to identify conclusively the genomes present in Urochloa tropical forage grasses, some suggestions can be made based on a range of evidence (Corrêa et al., 2020). Here, we aim to establish genome differences between diploids, and the genome composition in polyploids using advanced bioinformatic analysis of whole genome sequencing data to assist with genome nomenclature.
The pantropical grass genus Urochloa includes species previously classified under Brachiaria, Megathyrsus, and some Eriochloa and Panicum (Webster, 1987; González and Morton, 2005; Kellogg, 2015) and is a member of the Panicoideae tribe Paniceae, subtribe Melinidinae, comprising an estimated 150 annual and perennial grasses centred in sub-Saharan Africa (Kellogg, 2015; Soreng et al., 2017). Joint missions in the early 1980s conducted by CGIAR (Consultative Group on International Agricultural Research) centres, CIAT (Centro Internacional de Agricultura Tropical) and ILRI (International Livestock Research Institute) in several African countries collected wild species mostly as live plant cuttings or ramets. These activities built a global grass collection with 700 accessions of Urochloa species representing a highly diverse gene pool for breeding and systematic studies (Keller-Grein et al., 1996). Valuable traits of Urochloa include biomass yield, physiological tolerance to low-fertility acid soils of the tropics (Arroyave et al., 2011), digestibility and energy content (Hanley et al., 2020), insect tolerance (particularly to neotropical spittlebugs; Miles et al., 2006) and disease resistance (Valério et al., 1996; Alvarez et al., 2014; Hernandez et al., 2017). However, undesirable traits are also present, such as allelopathy (leaving bare soil; Kato-Noguchi et al., 2014), cold-susceptibility (hybrid Mulato II: Pizarro et al., 2013) and invasiveness (Durigan et al., 2007 in the Brazilian Cerrado; Urochloa panicoides is on the US Federal Noxious Weed List https://www.aphis.usda.gov/plant_health/plant_pest_info/weeds/downloads/weedlist.pdf; accessed on 16 February 2021). These Urochloa grass collections have huge potential for sustainable improvement as well as conservation of grasslands, including pastures, rangelands, savannah, prairie, cerrado, and roadsides and verges, with various degrees of management of grazing. Breeding or trial programmes based in Colombia, Brazil, Thailand, Zimbabwe, Ethiopia, South Africa and Australia have led to the development of over a dozen cultivars (do Valle and Savidan, 1996; Singh et al., 2010) and Urochloa is now the most widely planted forage grass in South America occupying 60 million hectares of grasslands in the tropical savannah ecoregion of Brazil (Gracindo et al., 2014).
The extent of the monophyletic Urochloa lineage, encompassing most species previously placed in the genus Brachiaria on morphological grounds, is now established (Webster, 1987; Salariato et al., 2010, 2012). However, understanding of the genetic and genomic relationships within the diploid and polyploid species within the genus is limited. The species-level taxonomy within Urochloa established in African floras (Hutchinson and Dalziel, 1972; Clayton and Renvoize, 1982; Clayton, 1989) has not been fully maintained by recent floristic work (Sosef, 2016). Some Urochloa species have been arranged in agamic (apomictic) complexes: U. brizantha, U. decumbens and U. ruziziensis were classified into the ‘brizantha’ complex, and U. humidicola together with U. dictyoneura were assigned to the ‘humidicola’ complex (Lutts et al., 1991; Renvoize and Maass, 1993). Urochloa maxima was previously assigned to Megathyrsus and Panicum. These species complexes have long been recognized as productive forages (Keller-Grein et al., 1996). Some Urochloa species reproduce sexually, but others with apomictic or mixed reproduction allow odd levels of ploidy and contribute to increased intraspecific variability, making classification difficult. Some species are only known in the wild as diploids, but chromosome numbers of U. ruziziensis (Timbó et al., 2014) and diploid U. brizantha (Pinheiro et al., 2000) have been doubled in the laboratory to enable crossing with tetraploid apomictic species (Risso-Pascotto et al., 2005; de Souza-Kaneshima et al., 2010; Felismino et al., 2010). The most common basic chromosome number is x = 9 (de Wet, 1986; Bernini and Marin-Morales, 2001), but x = 8, x = 7 (Basappa et al., 1987) and x = 6 (Risso-Pascotto et al., 2006; Boldrini et al., 2009b; Worthington et al., 2019) have been reported, making the genus Urochloa complex.
Characterization of the genome composition and diversity of Urochloa germplasm, phenotypes and ploidy is required for its effective use by researchers, breeders and farmers. Both whole genome sequencing and RNA-sequencing (RNAseq) (Higgins et al., 2021) reveals unique repetitive and single-copy sequences present only in one genome and enables recognition and designation of diploid genomes and their relationships, and characterization of the genome composition in polyploids. Despite the agronomic importance of Urochloa, and the need to make crosses for breeding, genomes are not clearly defined (Boldrini et al., 2009a), although some ploidy measurements have been made (Penteado et al., 2010; Jungmann et al., 2010). The use of transposable element probes against Urochloa chromosomes indicates that many species are allopolyploid with differentiation in their transposable element composition (Santos et al., 2015). Allopolyploidy is also shown by genetic analysis in apomicts (Worthington et al., 2016) and genomic in situ hybridization (Corrêa et al., 2020).
Here, we aimed to define the evolution and relationships of forage species in the tropical genus Urochloa, and understand evolutionary processes in polyploid, apomictic groups, and the diversification of abundant repetitive DNA sequences in their genomes. We measured ploidy in most of the Urochloa germplasm collection at CIAT (Colombia). We then aimed to use genomic and molecular cytogenetic approaches to identify repetitive DNA motifs and identify genome-specific sequences, to characterize the genomes present in the polyploid accessions (genomic composition), and to develop a model of evolution at the whole-genome level in diploid and polyploid accessions in the tropical forage grass group.
MATERIALS AND METHODS
Plant material
Studies were carried out on 362 accessions of Urochloa and related species (17 species, one synthetic hybrid, three unidentified accessions) focusing on material available on request for research and to breeders from CIAT and USDA (United States Department of Agriculture, USA) germplasm collections (Supplementary Data Table S1). Three notable unidentified accessions were included in our study: because of their unique characteristics they are already used in breeding programmes and the genomic composition must be analysed, along with our target species belonging to ‘brizantha’ and ‘humidicola’ agamic complexes and U. maxima, all having a huge potential for sustainable grazing and pasture management.
For diploids and polyploids, we use the narrow species concepts of Clayton and Renvoize (1982) and Clayton (1989) rather than the broader concepts of Sosef (2016) for ease of communication regarding the diverse genetic variants within U. decumbens and U. ruziziensis. Synonymy was updated and reconciled (POWO, 2019). For accessions from the CIAT and USDA germplasm collections, RNAseq data show that 111 lines (sampled from the 362 analysed here) are genetically distinct (Higgins et al., 2021), supporting continued validity of correlation of collection locality with accession number, and a commendation to the CIAT germplasm resource collection group who maintained true lines through violent conflict, not allowing a small number of vigorous and robust lines to dominate the collection.
Fresh leaf material from apomictic and sexual plants was collected in the field in Colombia and trial plots grown at CIAT, and dried in silica gel according to the protocol presented by Tomaszewska et al. (2021). The leaf samples were then used to isolate nuclei for flow cytometry and extract whole genomic DNA. Seed samples for chromosome preparation and further cytogenetic studies were provided by CIAT (Colombia) and USDA (USA) (Table 1). Only one diploid species was used for chromosome preparation (U. ruziziensis). The reason for this was the lack of available diploid U. brizantha and U. decumbens seeds. Those diploid accessions supplied to us gave contradictory results, suggesting possible wrong assignment to species, and thus were excluded from further analysis. The use of polyploid species from the maintained collection for analysis of genomic composition yielded reliable results.
Table 1.
List of accessions used in cytological studies, their chromosome numbers and ploidy levels
| Species | Accession number | Donor | Number of chromosomes | Base number |
|---|---|---|---|---|
| Urochloa sp. | PI 6576531 | USDA | 2n = 4x = 32 | 8 |
| Urochloa sp. | PI 5085712 | USDA | 2n = 4x = 36 | 9 |
| Urochloa sp. | PI 5085703 | USDA | 2n = 5x = 45 | 9 |
| U. brizantha (A.Rich.) R.D.Webster | PI 226049 | USDA | 2n = 6x = 54 | 9 |
| U. brizantha (A.Rich.) R.D.Webster | PI 292187 | USDA | 2n = 4x = 36 | 9 |
| U. brizantha (A.Rich.) R.D.Webster | PI 2107244 | USDA | 2n = 4x = 36 | 9 |
| U. decumbens (Stapf) R.D.Webster | 6370 | CIAT | 2n = 4x = 36 | 9 |
| U. decumbens (Stapf) R.D.Webster | 664 | CIAT | 2n = 4x = 36 | 9 |
| U. humidicola (Rendle) Morrone & Zuloaga | 16867 | CIAT | 2n = 8x+2/9x-4 = 50 | 6 |
| U. humidicola (Rendle) Morrone & Zuloaga | 26151 | CIAT | 2n = 6x = 36 | 6 |
| U. ruziziensis (R.Germ. & C.M.Evrard) Crins | 6419 | CIAT | 2n = 2x = 18 | 9 |
| U. maxima (Jacq.) R.D.Webster | PI 284156 | USDA | 2n = 4x = 32 | 8 |
| U. maxima (Jacq.) R.D.Webster | 6171 | CIAT | 2n = 4x = 32 | 8 |
| U. maxima (Jacq.) R.D.Webster | 16004 | CIAT | 2n = 4x = 32 | 8 |
1 Urochloa sp. PI 657653 was received by USDA as Panicum miliaceum, re-identified at first as Panicum sumatrense, and finally described as Urochloa sp. We failed to determine the species identity of PI 657653, which has spikelets like Urochloa adspersa but leaves like U. decumbens, so does not match any species and may be of hybrid origin, and hence the difficulties in its taxonomic identification.
2 Urochloa sp. PI 508571 was not identified.
3 Urochloa sp. PI 508570 morphologically corresponds to Urochloa panicoides.
4Accession PI 210724 previously assigned to Urochloa decumbens has been re-identified as U. brizantha.
Ploidy determination
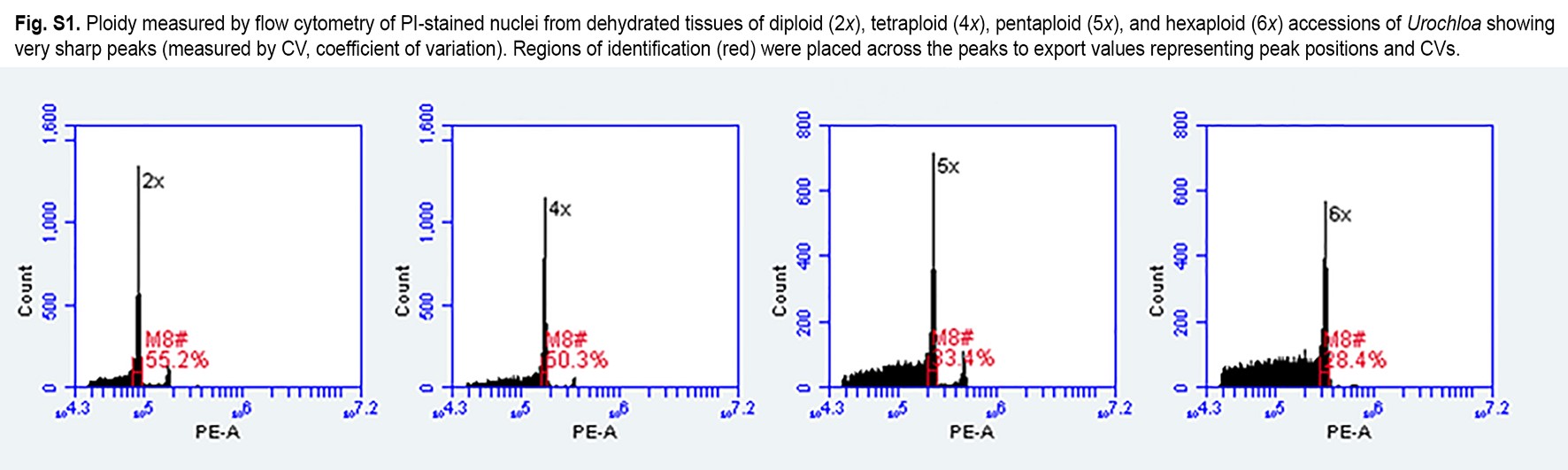
Flow cytometry was conducted to establish ploidy levels of 362 accessions of Urochloa and related species (355 accessions from the CIAT germplasm collection, and seven accessions from the USDA germplasm collection; Supplementary Data Table S1). Cell nuclei from dehydrated leaf tissues were isolated mechanically, using the method described by Doležel et al. (2007) with some modifications following Tomaszewska et al. (2021). Approximately 500 mg of tissue was chopped with razor blade in a Petri dish containing 1 mL lysis buffer (0.1 m Tris, 2.5 mm MgCl2 x 6H2O, 85 mm NaCl, 0.1 % Triton X-100; pH = 7.0) supplemented with 15 mm β-mercaptoethanol and 1 % PVP-40 to reduce negative effects of cytosolic and phenolic compounds. The nuclear suspension was recovered and filtered through a 50-µm nylon mesh (CellTrics, Partec) to remove cell fragments and large debris, and then stained with 50 µg mL−1 propidium iodide (PI), supplemented with 50 µg mL−1 RNase to prevent staining of double-stranded RNA. Samples were incubated on ice and analysed within 10 min in an Accuri C6 Flow Cytometer (Becton Dickinson at the Flow Cytometry Facility, University of Leicester), equipped with a 20-mW laser illumination operating at 488 nm. The results were acquired using the CFlow Plus software. The software was set up according to Galbraith and Lambert (2012). The flow cytometry measurements were standardized following the methods described by Tomaszewska et al. (2021). Ploidy levels of Urochloa were estimated by comparing the relative fluorescence values of the peak positions of PI-stained nuclei (FL) of target samples to that of an external standard, following the protocol presented by Tomaszewska et al. (2021). The coefficient of variation (CV) of the G0/G1 peak was evaluated in each sample to estimate nuclei integrity and variation in DNA staining.
DNA extraction and sequencing
Genomic DNA was extracted from fresh and dried leaves with the standard cetyltrimethylammonium bromide (CTAB)-based method (Doyle and Doyle, 1990). Whole genomic DNA from nine Urochloa accessions of various ploidies (Supplementary Data Table S2) was sequenced commercially (Novogene) with Illumina HiSeq 2× 150-bp paired-end reads (~12 Gb) (with a mean coverage of ~13×). Apomictic lines used by the breeders were selected for sequencing. Our aim was to generate universal probes that can be used on multiple accessions, not just sexual reproducing individuals that produce seeds (as used for chromosome preparations).
Project data have been deposited at the National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov/sra/) under BioProject PRJNA771228.
Identification and analysis of repetitive DNA sequences
Whole genome sequencing data were used to discover the most abundant repeats, and establish genomic compositions of Urochloa accessions of different ploidy levels. The whole genome shotgun sequence from U. ruziziensis cultivar CIAT 26162 (deposited in SRA under accession PRJNA437375; Worthington et al., 2021) was used as a reference genome. Highly abundant repetitive DNA sequences were extracted as high-frequency 50-mers using the program Jellyfish v.2.2.6 (Marçais and Kingsford, 2011). Similarity-based clustering, repeat identification and classification of a subset of raw reads were performed using RepeatExplorer (RE; Novak et al., 2013) and TAREAN (Novak et al., 2017). All potential specific sequences extracted as 50-mer repeats or clusters were mapped to the reference genome (U. ruziziensis,Worthington et al., 2021) and paired reads from nine sequenced genomes (Supplementary Data Table S2) using the program Geneious (Kearse et al., 2012). The 50-mer repeats, contigs and clusters were analysed by BLAST searches against the NCBI database to check for repeat identification (Sayers et al., 2019). The polymerase chain reaction (PCR) primer pairs were designed using Primer3 (Rozen and Skaletsky, 1999), and are listed in Supplementary Data Tables S3 and S4.
Probes used for in situ hybridization
Four different types of probes were used for fluorescence in situ hybridization (FISH):
Two ribosomal DNA sequences: pTa71 (Gerlach and Bedbrook, 1979), which contains a 9-kb EcoRI fragment of Triticum aestivum L. consisting of the 18S–5.8S–25S rRNA genes and the transcribed and non-transcribed intergenic spacer regions; and pTa794 (Gerlach and Dyer, 1980), which contains part of the T. aestivum 5S rRNA gene and spacer sequences.
Whole genomic DNA extracted from six diploid species (Table 2).
Conserved regions found by k-mer and RepeatExplorer analysis, and amplified in a standard PCR using newly designed genome-specific primers synthesized commercially (Sigma-Aldrich; Supplementary Data Tables S3 and S4).
Newly designed synthetic 50-mer oligonucleotide probes (Supplementary Data Table S3) synthesized commercially (Sigma-Aldrich).
Table 2.
List of genomic DNA and genome-specific probes used for in situ hybridization
| Name of probe | Description |
|---|---|
| gDNA_Ubriz1 | Genomic DNA extracted from U. brizantha, CIAT 16341 (2n = 2x = 18) |
| gDNA_Udec1 | Genomic DNA extracted from U. decumbens, CIAT 26305 (2n = 2x = 18) |
| gDNA_Udec2 | Genomic DNA extracted from U. decumbens, CIAT 6133a (2n = 2x = 18) |
| gDNA_Uruz1 | Genomic DNA extracted from U. ruziziensis, CIAT 26348 (2n = 2x = 18) |
| gDNA_Umax1 | Genomic DNA extracted from U. maxima, CIAT 16049 (2n = 2x = 16) |
| gDNA_Umax2 | Genomic DNA extracted from U. maxima, CIAT 6898 (2n = 2x = 16) |
| Uruz-spec1 | Sequence found in U. ruziziensis, CIAT 26162 (2n = 2x = 18) by k-mer analysis |
| Udec2x-spec1 | Sequence found in U. decumbens, CIAT 26305 (2n = 2x = 18) by RepeatExplorer |
| Udec2x-spec3 | Sequence found in U. decumbens, CIAT 26305 (2n = 2x = 18) by RepeatExplorer |
| Udec2x-spec6 | Sequence found in U. decumbens, CIAT 26305 (2n = 2x = 18) by RepeatExplorer |
| Udec4x-spec3 | Sequence found in U. decumbens, CIAT 664 (2n = 4x = 36) by k-mer analysis |
| Ubriz-spec2 | Sequence found in U. brizantha, CIAT 26032 (2n = 4x = 36), 26745 (2n = 4x = 36), 16292 (2n = 4x = 36) and 16290 (2n = 5x = 45) by k-mer analysis |
| Ubriz-spec3 | Sequence found in U. brizantha, CIAT 26032 (2n = 4x = 36), 26745 (2n = 4x = 36), 16292 (2n = 4x = 36) and 16290 (2n = 5x = 45) by k-mer analysis |
| Uhum-spec1 | Sequence found in U. humidicola, CIAT 26155 (2n = 6x = 36) by k-mer analysis |
| Uhum-spec3 | Sequence found in U. humidicola, CIAT 26155 (2n = 6x = 36) by k-mer analysis |
| Uhum-spec7 | Sequence found in U. humidicola, CIAT 26155 (2n = 6x = 36) by k-mer analysis |
| Uhum-spec12 | Sequence found in U. humidicola, CIAT 26155 (2n = 6x = 36) by RepeatExplorer analysis |
Probes from groups 1–3 were labelled with digoxigenin-11-dUTP or biotin-11-dUTP (Roche) using a BioPrime Array CGH, and then purified using a BioPrime Purification Module (Invitrogen), according to the manufacturers’ instructions. Fluorescent nucleotides were incorporated during commercial synthesis for probes from group 4.
Chromosome preparation
Chromosome preparation was carried out according to Schwarzacher and Heslop-Harrison (2000). The root-tips were collected from plants cultivated in a glasshouse, treated with α-bromonaphthalene at 4 °C for 6 h to accumulate metaphases, and fixed in 3 : 1 ethanol/acetic acid. Fixed root-tips were washed in enzyme buffer (10 mm citric acid/sodium citrate) for 15 min, digested in enzyme solution: 20 U mL–1 cellulase (Sigma C1184), 10 U mL–1 ‘Onozuka’ RS cellulase (RPI C32400), and 20 U mL–1 pectinase (Sigma P4716 from Aspergillus niger; solution in 40 % glycerol) in 10 mm enzyme buffer, and squashed in 60 % acetic acid. Cover-slips were removed after freezing with dry ice. Slides were air-dried and used for in situ hybridization within 3 months.
In situ hybridization procedure
FISH was carried out using the method described by Schwarzacher and Heslop-Harrison (2000), with minor modifications as described below. The hybridization mixture consisted of 50 % deionized formamide, 10 % dextran sulphate, 1 % sodium dodecyl sulphate, 2× SSC, probe(s) (2 ng μL−1 each) and 200 ng μL−1 salmon sperm DNA. Additional use of genomic DNA extracted from diploid species, as a blocker, did not give different in situ hybridization results. The hybridization mixture and the chromosome slides were denatured together in a hybridization oven for 7 min at 75 °C. Hybridization was performed at 37 °C overnight (for amplified conserved regions and genomic DNA probes) or 2 days (for 50-mer oligonucleotide probes). Post-hybridization washes were carried out at 42 °C: in 2× SSC for 2 min, in 0.1× SSC (for 50-mer oligonucleotide probes and amplified conserved regions) or 20 % formamide in 0.1× SSC (for genomic DNA probes) for 6 min, and 2× SSC for 20 min. Hybridization sites were detected with streptavidin conjugated to Alexa 594 (Life Technologies-Molecular Probes) and antidigoxigenin conjugated to fluorescein isothiocyanate (FITC; Roche Diagnostics). Slides were then counterstained with DAPI. Mounted slides were examined with a Nikon Eclipse 80i epifluorescence microscope, and photographs were taken using a DS-QiMc monochromatic camera, and NIS-Elements v.2.34 software (Nikon) and assembled in Photoshop (Adobe) using only software functions affecting the whole image.
RESULTS
Taxonomic identification and ploidy measurement
We studied 362 accessions of Urochloa and related genera (summary in Table 3), and verified these taxonomically using live plants in CIAT, Cali, Colombia and reference herbarium specimens at the Royal Botanic Gardens, Kew, UK, linked where available with collection localities in Africa, morphological traits and cultivar status (data collected from CIAT GenBank, Genesys database and archived reports; Supplementary Data Table S1).
Table 3.
Number of analysed accessions and their distribution in the various levels of ploidy
| Species | 2n = 2x | 2n = 4x | 2n = 5x | 2n = 6x | 2n = 7x | 2n = 8x + 2 or 2n = 9x − 4 | 2n = 9x |
|---|---|---|---|---|---|---|---|
| Andropogon gayanus | 1 | ||||||
| Panicum coloratum | 1 | ||||||
| Paspalum dilatatum | 1 | ||||||
| Pennisetum polystachion | 1 | ||||||
| Pennisetum purpureum | 1 | ||||||
| Urochloa arrecta | 1 | ||||||
| Urochloa brizantha | 6 | 59 | 25 | 2 | |||
| Urochloa decumbens | 18 | 26 | 1 | ||||
| Urochloa dictyoneura | 1 | ||||||
| Urochloa dura | 1 | ||||||
| Urochloa humidicola | 16 | 33 | 1 | 3 | |||
| Urochloa jubata | 1 | 1 | |||||
| Urochloa maxima | 25 | 104 | |||||
| Urochloa nigropedata | 1 | ||||||
| Urochloa plantaginea | 1 | ||||||
| Urochloa platynota | 1 | ||||||
| Urochloa ruziziensis | 26 | ||||||
| Urochloa hybrid | 1 | ||||||
| Urochloa sp. 1 | 1 | ||||||
| Urochloa sp. 2 | 1 | ||||||
| Urochloa sp. 3 | 1 |
The ploidy of studied accessions was measured (Supplementary Data Fig. S1) using flow cytometry of fluorescently stained nuclei from dried leaf materials with an optimized method (Tomaszewska et al., 2021) achieving a CV (coefficient of variation of the G0/G1 peak) of typically 2–5 %. Ploidy levels of 2x, 4x, 5x and 6x for the ‘brizantha’ agamic complex, 6x, 7x and 9x for the ‘humidicola’ agamic complex, and 2x and 4x for U. maxima were found (Table 3). Some accessions differed from published values (Supplementary Data Table S1). Urochloa ruziziensis was only found as a diploid (2n = 2x = 18), while other species, such as U. humidicola, were found only as polyploids.
Urochloa brizantha and U. maxima are widespread in sub-Saharan Africa, the range of U. decumbens and U. ruziziensis is restricted to the area of Lake Victoria, and U. humidicola occurs from Nigeria eastwards to southern Ethiopia and southwards to South Africa. No correlation between the level of ploidy of the examined accessions of U. brizantha, U. decumbens, U. ruziziensis and U. humidicola with the area of their original East African collection sites was evident (Fig. 1). A mixture of species and ploidy levels was found at most collection sites; 4x and 5x accessions were predominant for U. brizantha, and 6x and 7x for U. humidicola (see also Table 3).
Fig. 1.

The natural distribution ranges of Urochloa (diagonal shading) and geographical origin of the accessions studied here (symbols). For relevant species, multiple ploidies were found at each collection location. (A) Urochloa brizantha is marked in blue, U. decumbens in green and U. ruziziensis in pink. (B) Urochloa humidicola is marked in orange. (C) Spikelet morphology. Diploid accessions are shown as squares, tetraploid as empty diamonds, pentaploid as upright crosses, hexaploid as triangles, heptaploid as diagonal crosses, octoploid as circles and nonaploid as filled diamonds. Natural distribution ranges are from wcsp.science.kew.org. Photograph of U. humidicola by Russell Cumming.
Number of chromosomes and rDNA sites
The studied accessions were euploid with basic chromosome numbers of x = 6 for U. humidicola, x = 8 for U. maxima, and x = 9 for U. brizantha, U. decumbens and U. ruziziensis (Table 1) with the exception of one aneuploid accession of U. humidicola CIAT 16867 with 2n = 8x + 2 or 9x − 4 = 50. Unidentified accessions had basic chromosome numbers of x = 8 and x = 9. FISH with a wheat 45S rDNA (18S–5.8S–25S; probe pTa71) and 5S rDNA (probe pTa794) (Fig. 2; details on number of rDNA sites in figure legend) showed, typically, one pair of major 45S rDNA sites per diploid chromosome complement, and two pairs of 5S rDNA sites on different chromosomes in species belonging to the ‘brizantha’ complex (Fig. 2A–E). Differences in the number and position of rDNA sites were not observed between studied accessions belonging to the ‘brizantha’ agamic complex. Urochloa humidicola had one pair of chromosomes showing both 45S and 5S rDNA signals and two pairs of chromosomes with only 45S rDNA signals (Fig. 2G, H). Two studied accessions of U. humidicola differed in number of 5S rDNA sites. In U. maxima, one pair of 45S rDNA sites and one pair of 5S rDNA sites per diploid chromosome complement were observed (Fig. 2I–K). The pattern of rDNA sites in unidentified accessions PI 657653 and PI 508570 did not correspond to the other polyploids studied here (Fig. 2L–N).
Fig. 2.

Localization of ribosomal 5S (red) and 18S–5.8S–25S (green) DNA sites on metaphase chromosomes of Urochloa species by fluorescence in situ hybridization. (A) Urochloa ruziziensis (2x; CIAT 6419); one pair of chromosomes with large 18S–5.8S–25S sites detected on the satellites, and two pairs of chromosomes with 5S sites observed in the interstitial regions. (B) Urochloa decumbens (4x; CIAT 664); 18S–5.8S–25S sites on satellites of two chromosome pairs, and 5S observed in the interstitial regions of three chromosome pairs; one pair of chromosomes showed much stronger 5S rDNA signals, as did diploid U. ruziziensis. (C) Urochloa decumbens (4x; CIAT 6370); same number and position of rDNA signals as in U. decumbens CIAT 664 (B). (D) Urochloa brizantha (4x; PI 210724); same number and position of rDNA signals as in tetraploid U. decumbens CIAT 664 (B) and CIAT 6370 (C). (E) Urochloa brizantha (6x; PI 226049); three pairs of chromosomes with 18S–5.8S–25S rDNA sites and five pairs of chromosomes with 5S rDNA sites. (F) Urochloa brizantha (4x; PI 292187); same number and position of rDNA signals as in tetraploid U. decumbens CIAT 664 (B) and CIAT 6370 (C). (G) Urochloa humidicola (6x; CIAT 26151); three pairs of chromosomes showed 18S–5.8S–25S rDNA signals on their satellites, and one pair was different, having additional 5S rDNA sites; another three pairs of chromosomes had 5S rDNA signals. (H) Urochloa humidicola (8x + 2 or 9x − 4; CIAT 16867); three pairs of chromosomes showed 18S–5.8S–25S rDNA signals, and one pair had additional 5S rDNA sites; another four pairs of chromosomes had 5S rDNA signals. (I) Urochloa maxima (4x; CIAT 6171); two pairs of chromosomes with large 18S–5.8S–25S rDNA signals, and two pairs of chromosomes with 5S rDNA signals detected at the pericentromeric regions. (J) Urochloa maxima (4x; CIAT 16004); same number and position of rDNA signals as in U. maxima CIAT 6171 (I). (K) Urochloa maxima (4x; PI284156); same number and position of rDNA signals as in CIAT 6171 (I) and CIAT 16004 (J). (L) Urochloa sp. (4x; PI 657653); one pair of chromosomes with 18S–5.8S–25S rDNA signals and two pairs of chromosomes with 5S rDNA, which does not correspond to the pattern of rDNA signals in other tetraploids studied here. (M) Urochloa sp. (4x; PI 508571); same number of rDNA signals as in PI 657653 (L). (N) Urochloa sp. (5x; PI 508570); two pairs of chromosomes showing 18S–5.8S–25S rDNA and two pairs of chromosomes with 5S rDNA signals. Scale bars = 5µm.
Repetitive DNA motifs identified by k-mer and graph-based clustering of DNA sequence reads
The most abundant 50-mer repeats were extracted from 2 Gb of whole genome sequence reads from each of the ten accessions (our whole genome sequencing data from nine accessions listed in Supplementary Data Table S2 along with published whole genome shotgun sequence from U. ruziziensis, Worthington et al., 2021). Those with sequence homology to rDNA, sequencing primers or chloroplast genomes, or with extreme GC ratio were omitted from further analysis as well as the telomeric repeat, (TTTAGGG)n, that represented about 0.1 % of the reads. The genome proportion of the remaining abundant 50-mers from reads or from assembled contigs of abundant 50-mers showed that most motifs were on the whole present with similar abundance in most Urochloa accessions (Supplementary Data Table S3; BLASTN search in Supplementary Data Table S5). Only a few motifs showed differences in abundance, indicating only limited species or genome-specific repeats are present. Comparison of abundant 50-mer motifs from the three diploid species, U. ruziziensis CIAT 26162, U. decumbens CIAT 26305 and U. maxima CIAT 16049, did not reveal any U. decumbens-specific repeats. However, some repeats showed different abundance: repeat 1392_24 was much more abundant in U. ruziziensis (1.18 %, Supplementary Data Table S3), and repeat 1101_42 represents 0.43 % of the diploid U. maxima genome (2n = 2x = 16), and has very low homology to U. brizantha and U. humidicola accessions. Repeat 1162_31 represents 1.72 % of the diploid U. maxima genome, and is also highly abundant in genomes of two out of five polyploid accessions of U. brizantha, representing slightly above 1 % of their genomes.
Since the whole genome sequence of diploid U. brizantha was not available, two k-mer strategies were used to find sequences potentially specific for the U. brizantha genome. In the first, the abundant 50-mer motifs from four polyploid accessions of U. brizantha were mapped to each other. Sequences that occurred in all four accessions were then de novo assembled. Contig 5 (Supplementary Data Fig. S2) with the highest genome proportion in U. brizantha accessions, but no or very low genome proportion in diploid U. decumbens and U. ruziziensis, was a candidate motif specific to the genome of U. brizantha. In the second strategy, we tested our hypothesis that tetraploid U. decumbens is an allopolyploid with the genomic composition XXYZ (where X, Y and Z represent genomes to be determined). We have not ruled out such a possibility because synthetic multi-generation hybrids involving U. ruziziensis, U. decumbens and U. brizantha are known (Supplementary Data Table S1), and such crosses could take place in nature. We mapped abundant unassembled 50-mer motifs from tetraploid U. decumbens CIAT 664, to 50-mer datasets from diploid U. decumbens, U. ruziziensis and U. maxima. The differentially abundant sequence 1771_76 (>100× the genome proportion in tetraploid U. decumbens and four polyploid accessions of U. brizantha compared to the diploids where it represented <0.01 % of the genome; Supplementary Data Table S3) was a candidate repeat specific to genome Z.
The 50-mer sequence dataset from U. humidicola (6x) was mapped to highly abundant 50-mers from the three diploid species and identified four motifs unique to U. humidicola: 5899, 1014, 1015_2 and 7000 (Supplementary Data Table S3). Similarly, to find abundant repetitive motifs in 4x Urochloa sp. (PI 657653, unidentified accession, potentially of hybrid origin), two 50-mer sequences were extracted, 1134_5 and 1644_4, representing 0.33 and 0.2 % of the genome, respectively, with abundance below 0.01 % in the diploids (U. decumbens, U. maxima, and already published U. ruziziensis, Worthington et al., 2021).
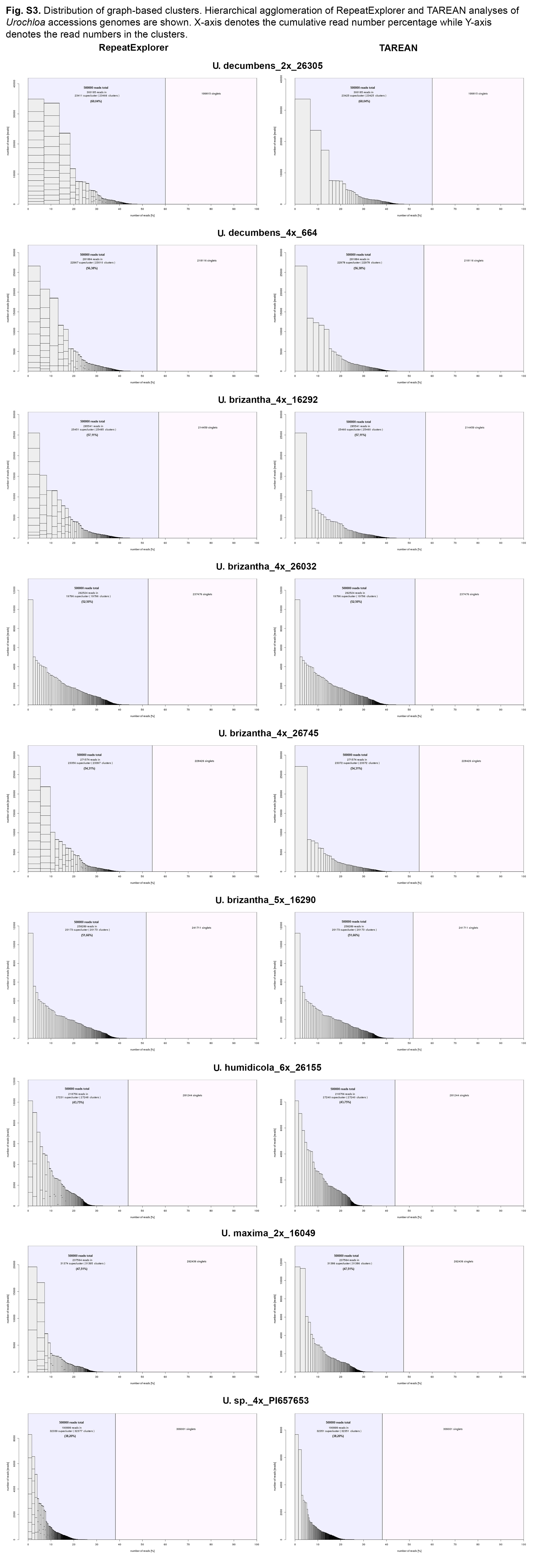
For graph-based sequence clustering and characterization of repeats, 2-Gb subsets of raw sequence from each of the nine Urochloa accessions were analysed using RepeatExplorer and TAREAN (Novak et al., 2013, 2017). Generally, 38.2–60.0 % of reads were assigned into clusters of related sequence reads (Supplementary Data Fig. S3; Supplementary Data Tables S6-S9). As with k-mers, sequences showing high homology to rDNA or chloroplast genomes, and extreme GC ratio were omitted from further analysis, and the final list of putative genome-specific sequences was created by comparing genome abundance between accessions (Supplementary Data Table S4). The number of raw reads with high homology to the most abundant clusters/RE motifs in each one of the sequence datasets were then counted for ten whole genome sequence reads. Those clusters showing a high proportion in one diploid genome are candidate genome-specific sequences (Supplementary Data Table S4), and some were selected for testing as probes by chromosomal in situ hybridization (see below and Table 2).
Transposable elements were recognized in each of the nine sequenced genomes (Fig. 3; Supplementary Data Table S10; for U. ruziziensis see Worthington et al., 2021). The automated annotation provided by RepeatExplorer will omit, or give incorrect, identification of some elements based on homology to known sequences; therefore, regardless of annotation, any elements differing in abundance between accessions were candidates for use as probes for in situ hybridization to distinguish genomes. Thus, sequences with differential abundance identified were the Bianca retrotransposon in U. brizantha polyploids, the highly abundant Tekay retrotransposon in diploid U. decumbens, the retrotransposon CRM in 4× PI 657653, and the long interspersed elements (LINE) in U. humidicola (arrows in Fig. 3). The Tork retrotransposon was found in some U. brizantha, suggesting differences in genome structure between accessions.
Fig. 3.

Relative abundance (log) of DNA sequence classes in Urochloa species and hybrids from whole-genome sequence reads. Automated repeat identification of graph-based clustering (RepeatExplorer, TAREAN) and abundant k-mer motifs were classified by nucleotide domain hits and database BLASTN searches (Supplementary Data Tables S7 and S9). Arrows indicate some motifs with differential abundance between accessions. Bars below 1 (y-axis) indicate abundance <0.01 % of genome.
Chromosomal organization and genome specificity of repetitive DNA sequences
Total genomic DNAs (gDNA; Table 2) from diploid species of U. brizantha (Ubriz), U. decumbens (Udec), U. ruziziensis (Uruz) and U. maxima (Umax) were used as probes for genomic in situ hybridization (GISH) on 14 accessions of Urochloa diploids and polyploids (Table 1). The results are given in Table 4, and example micrographs are shown in Fig. 4 (giving details regarding probe combinations and observed signals in the legend). In summary, probes gDNA_Uruz and gDNA_Udec showed signals in broadly pericentromeric regions rather than painting whole chromosomes, and the differential hybridization conditions (hybridization stringency 72 and 85 %; using only salmon sperm DNA or together with gDNA extracted from diploid species as an additional block of cross-hybridization of common sequence motifs) did not affect GISH results. Different strengths of signals in pericentromeric position of chromosomes in polyploids belonging the to ‘brizantha’ complex indicated that these species might be allopolyploids (Fig. 4C–J). Further investigation using genome-specific probes showed that polyploids from the ‘brizantha’ agamic complex are allopolyploids, but the signal strengths of gDNA probes were not sufficient to recognize genomes (see the last paragraph of this section, and the legend to Fig. 5). The simultaneous use of probes gDNA_Uruz and gDNA_Udec against chromosomes of U. humidicola showed differential dispersed signals on many chromosomes, indicating the differences between diploid U. ruziziensis and U. decumbens genomes (Fig. 4K). The gDNA_Ubriz probe hybridized to rDNA sites of different species, but not to pericentromeric regions of chromosomes (Fig. 4I; Table 4). The gDNA_Umax probe showed very strong pericentromeric signals on all 32 chromosomes of tetraploid U. maxima, in addition to terminal and subterminal regions (Fig. 4L). Urochloa accessions not assigned to species are clearly allopolyploids (Fig. 4N, O; Table 4). Ultimately, our GISH results were difficult to interpret, and thus there was a need to develop specific probes to gain the much-needed genome specificity.
Table 4.
Description of genomic in situ hybridization results
| Accession | Genomic DNA probes | |||
|---|---|---|---|---|
| gDNA_Ubriz | gDNA_Udec | gDNA_Uruz | gDNA_Umax | |
| U. ruziziensis CIAT 6419 | No signal | Signals in pericentromeric position of 18 chromosomes; same position of signals as gDNA_Uruz probe | Signals in pericentromeric position of 18 chromosomes; same position of signals as gDNA_Udec probe | No signal |
| U. decumbens PI 210724 | Four 45S rDNA signals | Strong signals in pericentromeric position of 18 chromosomes, and weak signals in pericentromeric position of remaining 18 chromosomes; same position of signals as gDNA_Uruz probe | Strong signals in pericentromeric position of 18 chromosomes, and weak signals in pericentromeric position of remaining 18 chromosomes; same position of signals as gDNA_Udec probe | Four 45S rDNA signals |
| U. decumbens CIAT 664 | Four 45S rDNA signals | Strong signals in pericentromeric position of 18 chromosomes, and weak signals in pericentromeric position of remaining 18 chromosomes; same position of signals as gDNA_Uruz probe | Strong signals in pericentromeric position of 18 chromosomes, and weak signals in pericentromeric position of remaining 18 chromosomes; same position of signals as gDNA_Udec probe | Four 45S rDNA signals |
| U. decumbens CIAT 6370 | Four 45S rDNA signals | Strong signals in pericentromeric position of 18 chromosomes, and weak signals in pericentromeric position of remaining 18 chromosomes; same position of signals as gDNA_Uruz probe | Strong signals in pericentromeric position of 18 chromosomes, and weak signals in pericentromeric position of remaining 18 chromosomes; same position of signals as gDNA_Udec probe | Four 45S rDNA signals |
| U. brizantha PI 292187 | Four 45S rDNA signals | Nine chromosomes painted; 27 chromosomes show pericentromeric signals; same position of signals as gDNA_Uruz probe | Nine chromosomes painted; 27 chromosomes show pericentromeric signals; same position of signals as gDNA_Udec probe | Four 45S rDNA signals |
| U. brizantha PI 226049 | Six 45S rDNA signals | Eighteen strong, 18 weaker and 18 weak signals in pericentromeric position of chromosomes | Eighteen strong, 18 weaker and 18 weak signals in pericentromeric position of chromosomes | Six 45S rDNA signals |
| U. humidicola CIAT 26151 | Six 45S rDNA signals | Interspersed signals along chromosomes | Interspersed signals along chromosomes | Six 45S rDNA signals |
| U. humidicola CIAT 16867 | Six 45S rDNA signals | Interspersed signals along chromosomes | Interspersed signals along chromosomes | Six 45S rDNA signals |
| U. maxima CIAT 6171 | Four 45S rDNA signals | Four 45S rDNA signals | Four 45S rDNA signals | Thirty-two chromosomes show signals in telomeric–subtelomeric and pericentromeric position |
| U. maxima CIAT 16004 | Four 45S rDNA signals | Four 45S rDNA signals | Four 45S rDNA signals | Thirty-two chromosomes show signals in telomeric–subtelomeric and pericentromeric position |
| U. maxima PI 284156 | Four 45S rDNA signals | Four 45S rDNA signals | Four 45S rDNA signals | Thirty-two chromosomes show signals in telomeric-subtelomeric and pericentromeric position |
| Urochloa sp. PI 657653 | Four 45S rDNA signals, weak signals in centromeres | Four 45S rDNA signals | Four 45S rDNA signals | Signals along 16 chromosomes |
| Urochloa sp. PI 508571 | No signal | Signals in pericentromeric position of 18 chromosomes; same position of signals as gDNA_Uruz probe | Signals in pericentromeric position of 18 chromosomes; same position of signals as gDNA_Udec probe | No signal |
| Urochloa sp. PI 508570 | Signals along 18 chromosomes; some chromosomes show gDNA_Ubriz and gDNA_Umax signals | Four 45S rDNA signals | No signal | Signals along 27 chromosomes |
Fig. 4.

Localization of labelled whole genomic DNA (gDNA) from diploid species used as probes for in situ hybridization on metaphase chromosomes (fluorescing blue). (A) Eighteen chromosomes of U. ruziziensis (2x, CIAT 6419) showing strong signals of gDNA_Uruz1 probe (red) in pericentromeric regions of chromosomes and some terminal signals on one or both arms of several chromosomes. (B) Urochloa ruziziensis (2x, CIAT 6419) metaphase showing strong signals of gDNA_Udec1 probe (green) in eight pairs of chromosomes (satellites of two chromosomes remain unlabelled; red arrowheads), and weak signals in centromeres of two chromosomes (yellow arrowheads). (C) Metaphase of U. decumbens (4x, CIAT 6370) with 18 strong and 18 weak signals of gDNA_Udec2 probe (green), and four red signals of rDNA after hybridization with gDNA_Umax1 probe. (D) Metaphase of U. decumbens (4x, CIAT 6370) showing 18 strong and 18 weak signals of gDNA_Udec1 probe (green). (E) Same metaphase as in D, showing signals of gDNA_Uruz1 probe (red) in the same position as gDNA_Udec1 probe, which confirms the similarity of the genomes of these accessions. (F) Chromosomes of U. brizantha (4x, PI 292187) with red signals of gDNA_Uruz1 probe: nine very strong and 27 weak. (G) Chromosomes of U. brizantha (4x, PI 292187) with green signals of gDNA_Udec1 probe: nine very strong and 27 weak; gDNA_Umax1 probe shows four red signals (rDNA): one signal in chromosome with strong gDNA_Udec1 signal (yellow arrowheads) and three signals in chromosomes with weak gDNA_Udec1 signals (red arrowheads). (H) Metaphase of U. brizantha (4x, PI 292187) with some rDNA and dispersed signals of gDNA_Umax2 probe (green); gDNA_Ubriz1 probe did not show signals. (I) Metaphase of U. brizantha (6x, PI 226049) with gDNA_Uruz1 probe signals (green) in centromeres: 18 strong, 18 weaker and 18 weak; gDNA_Ubriz1 probe (red) gave four red signals (rDNA). (J) Metaphase of U. brizantha (6x, PI 226049) with gDNA_Udec1 probe signals (green) in centromeres: 18 strong, 18 weaker and 18 weak. (K) Metaphase of U. humidicola (8x + 2 or 9x − 4, CIAT 16867) showing dispersed signals of gDNA_Uruz1 (red) and gDNA_Udec2 (green) probes along chromosomes; many of these signals did not overlap. (L) Metaphase of U. maxima (4x, CIAT 6171) with pericentromeric and telomeric signals of gDNA_Umax1 probe (red). (M) Chromosomes of U. decumbens (4x, CIAT 664) with four rDNA (red arrowheads) and very weak dispersed signals in centromeres after hybridization with gDNA_Umax1 probe (red). (N) Metaphase of Urochloa sp. (5x, PI 508570) showing 18 chromosomes with dispersed signals of gDNA_Ubriz1 probe (red) and 27 chromosomes with dispersed signals of gDNA_Umax1 probe (green). (O) Metaphase of Urochloa sp. (4x, PI 508571) showing signals of gDNA_Uruz1 probe (red) and gDNA_Udec1 probe (green) in 18 chromosomes; the 18 remaining chromosomes showed no signals. Scale bars = 5 µm.
Fig. 5.

Localization of abundant repetitive sequences on chromosomes of different species belonging to the ‘brizantha’ complex. Probes are described in Supplementary Data Table S3. Chromosomes (right) were arranged by chromosomal distribution of FISH signals and chromosome length. (A) Eighteen strong signals of Uruz-spec1 probe (green) at pericentromeric regions of U. ruziziensis (2x, CIAT 6419) chromosomes. (B) Uruz-spec1_50mer labelled 18 chromosomes of U. decumbens (4x, CIAT 664; green). (C) Ubriz-spec2 probe showed strong signals in terminal regions of nine chromosomes of U. decumbens (4x, CIAT 664). Udec2x-spec3 probe showed strong signals in pericentromeric positions of chromosomes, even those with terminal Ubriz-spec2 signals. Two chromosomes exhibited very strong green fluorescence (white arrows). Some signals were more dispersed along chromosomes, and the 18 chromosomes without Ubriz-spec2 signal had weak Udec2x-spec3 signals. (D) Metaphase of U. decumbens (4x, CIAT 664); Ubriz-spec3 showed a similar pattern of signals as Ubriz-spec2 in C. Udec2x-spec6 showed only nine chromosomes with weak signals. (E) Metaphase of U. decumbens (4x, CIAT 6370); Ubriz-spec2 probe produced seven signals in the terminal position of chromosomes; one chromosome with (yellow arrow) and one chromosome without Ubriz-spec2 signals (white arrow) showed strong signals in pericentromeric and subtelomeric positions of Udec2x-spec1 probe. (F) Metaphase of U. brizantha (4x, PI 210724); 12 signals of Ubriz-spec2 probe at terminal regions of chromosomes. (G) Metaphase of U. brizantha (4x, PI 292187); same number and position of Ubriz-spec2 signals as in F where two of the 12 signals were weaker (white arrows). Thirty chromosomes showed strong to weak Udec2x-spec1 signals, while the other six had very weak or no signals. (H) Urochloa brizantha (4x, PI 292187); gDNA-Udec probe gave strong signals on some chromosomes with Ubriz-spec3 signals and those without Ubriz-spec3 signals. (I) Urochloa brizantha (6x, PI 226049); Ubriz-spec2 and Udec2x-spec1 probes differentiated chromosomes into five types: nine chromosomes with Ubriz-spec2 signals (group I), 11 chromosomes with Ubriz-spec2 and Udec2x-spec1 signals (group II), 11 chromosomes with strong Udec2x-spec1 signals (group III), 14 chromosomes with very weak Udec2x-spec1 signals (group IV), and nine chromosomes without any signals (group V). In group II, there was a pair of chromosomes showing the same pattern of signals (white arrows), although it seems that another chromosome from this group (yellow arrow) had the same strong pericentromeric signal of Udec2x-spec1 probe as another chromosome from group III (yellow arrow). (J) Metaphase of U. brizantha (6x, PI 226049) showing nine chromosomes with very strong pericentromeric signals of Uruz-spec1 probe. (K) Metaphase of U. brizantha (4x, PI 292187); Ubriz-spec3 probe gave 12 signals at the terminal position of chromosomes. One very strong signal of Udec4x-spec3 detected at the terminal region of one chromosome (white arrow). (L) Metaphase of U. decumbens (4x, CIAT 664) showing four terminal signals of Udec4x-spec3 probe: one strong on chromosome without Ubriz-spec3 signals, and three weak on chromosomes showing Ubriz-spec3 signals (white arrows). Scale bars = 5 µm.
Probes designed from highly abundant sequences recognized by k-mer (Supplementary Data Table S3), and RepeatExplorer and TAREAN (Supplementary Data Table S4) analyses were used, mostly in differential pairs, for in situ hybridization to localize repeats on Urochloa chromosomes, and distinguish genomes in polyploids (see Fig. 5 for ‘brizantha’ and Fig. 6 for ‘humidicola’ complexes; signal summary in Supplementary Data Tables S3 and S4: chromosomes were grouped by signal location and intensity). Overall, in situ hybridization strength correlated with in silico analysis (percentage of sequence in the genomes), now showing the genome and chromosomal distribution of the probes and enabling discrimination of the genome of origin of most chromosomes in the polyploid accessions. The Uruz-specific probe perfectly labelled 18 chromosomes belonging to the diploid U. ruziziensis, allowing us to also recognize this genome in polyploids. All putative Ubriz-specific probes designed using two strategies gave the same number and position of signals; and some chromosomes shared both Ubriz- and Udec-specific signals. Uhum-specific probes enabled recognition of all genomes which come together in hexaploid U. humidicola. Detailed descriptions of the probes and hybridization results are given in the extended legend (Figs 5 and 6) and probe description in Supplementary Data Tables S3 and S4. A summary of the in situ hybridization results is presented in Fig. 7, showing the possible genome composition of the studied accessions.
Fig. 6.

Localization of abundant repetitive sequences on chromosomes of U. humidicola accessions. Probes are described in Supplementary Data Table S3; Chromosomes (right) were arranged by chromosomal distribution of FISH signals and chromosome length. (A) Uhum-spec1 and Uhum-spec3 probes differentiated chromosomes of U. humidicola (6x, CIAT 26151) into four types: eight chromosomes with terminal Uhum-spec1 signals (group I), eight chromosomes with Uhum-spec1 and Uhum-spec3 signals (group II), eight chromosomes with Uhum-spec3 signals (group III), and 12 chromosomes without any signals (group IV). Two chromosomes belonging to group III differed from the other six: one of them had two additional signals of Uhum-spec1 and Uhum-spec3 probes (white arrow), while the other had only one additional signal of Uhum-spec1 probe (yellow arrow). (B) Urochloa humidicola (6x, CIAT 26151) showed signals of Uhum-spec12 probe at pericentromeric and intercalary position of 12 chromosomes. The intensity and distribution of these signals indicated the presence of six pairs of chromosomes. In particular, one pair of shorter chromosomes exhibited very strong pericentromeric signals of Uhum-spec12 (white arrows). (C) Urochloa humidicola (8x + 2 or 9x − 4, CIAT 16867); Uhum-spec7 signals were dispersed along chromosomes, some of which were more intensive, but it is difficult to deduce if there was any specific pattern of their distribution (high stringency conditions). Uhum-spec1 probe showed signals on 26 chromosomes, but four chromosomes were different, showing additional signals: two chromosomes had extra Uhum-spec1 signals on the opposite arms (white arrows), one chromosome showed doubled Uhum-spec1 signal (yellow arrow), and one chromosome had strong terminal Uhum-spec7 signal (green arrow). (D) Chromosomes of U. humidicola (8x + 2 or 9x − 4, CIAT 16867); the low stringency conditions, allowing hybridization between DNAs sharing 72 % sequence identity, revealed eight additional weak signals of Uhum-spec1 probe. Three chromosomes had Uhum-spec1 signals on both arms (white arrows), and one chromosome showed signals on one arm (yellow arrow). Scale bars = 5 µm.
Fig. 7.

Summary of in situ hybridization results with gDNA and genome-specific probes. Inferred genomic composition (coloured blocks) and chromosome numbers (shown) of studied accessions belonging to the ‘brizantha’ (left) and ‘humidicola’ (middle) complexes, and U. maxima (right). White blocks with numbers: undetermined or diverged genome. Probe names are given in Table 2. Details of the probes are given in Supplementary Data Tables S3 and S9.
Discussion
Through our analysis of repetitive DNA sequences using unassembled raw-reads, molecular cytogenetic and flow cytometry tools, we were able to define the nature and similarity between the Urochloa species and genomes available internationally in germplasm resource collections. By identifying repetitive sequences that were unique to the different genomes present in the species, and identifying distinct genomes in the polyploids, we revealed the genome composition of polyploids and the nature of evolutionary changes in the primary DNA sequence of repetitive motifs and changes in their abundance. Together with growth habit and morphological data, evaluation of the Urochloa material confirmed the challenges in defining the genetic relationships of the accessions used in forage breeding. Analyses of data including collection localities in Africa, morphological traits and cultivar status, together with ploidy levels and sequencing data are critical for understanding biodiversity in the wild, and using diverse genebank material in breeding.
Ploidy and geographical origin
All the species analysed here are native to sub-Saharan Africa (Renvoize and Maass, 1993). Information regarding the collection sites, most from the international 1984/85 expeditions representing the majority of germplasm in Colombia and Brazil, and reintroductions within Africa (Wassie et al., 2018), allowed us to correlate geographical distribution and ploidy levels as determined by flow cytometry. For Urochloa species with multiple ploidies, representatives of all ploidies were found in each geographical region, indicating co-occurrence, no major niche specialization, and the opportunity for hybridization and introgression, including segmental allopolyploidy. This is not uncommon for species with multiple ploidies. In wild Tripleurospermum inodorum (Asteraceae) in central Europe, for example, Čertner et al. (2017) studying the spatio-temporal patterns of ploidy coexistence found tetraploid cytotypes alone in about half or more of the populations, diploids in about 10 % of populations, with the remaining populations being a mixture of ploidies. Natural selection may produce polyploids and hybrids with strong geographical signals (Hagl et al., 2021; Alix et al., 2017). Even in species with no significant ecological differences between cytotypes (e.g. in Aster amellus), no mixing of ploidies is seen even in contact zones (Mandáková and Münzbergová, 2006). Deliberate or accidental roadside or forage introductions (likely to be over-represented in the genebank material sampled here) may introduce different ploidies, although our accessions are genetically different (Hanley et al., 2020; Higgins et al., 2021). Polyploids are often argued to have a competitive advantage over diploids (Alix et al., 2017) and production of polyploid seeds and individuals by diploids is widespread, although subsequent establishment of whole polyploid populations and their expansion can be hindered by insufficient seed production (Levin, 2021). Thus, it is not surprising that multiple ploidy levels (2x–9x) in many collection areas, including new polyploids and fertile 3x hybrids, were found and suggests co-existence of the various ploidy levels in both U. brizantha and U. humidicola.
Chromosome and genome differentiation in Urochloa polyploids
The karyotypes of three Urochloa species belonging to the ‘brizantha’ complex show little differences, having chromosomes similar in size and morphology (Bernini and Marin-Morales, 2001; Nielen et al., 2010). Physical mapping of 5S and 18S–5.8S–25S rDNA locations provides a chromosome marker, but the mostly similar patterns in the ‘brizantha’ complex did not assist in identification of genome composition (see Fig. 2; Akiyama et al., 2010; Nielen et al., 2010; Santos et al., 2015; Nani et al., 2018). In the three accessions with desirable agronomic characteristics that could not be assigned to species based on morphology, the number of rDNA sites did not correspond to ploidy, with only one pair of 45S rDNA sites in the two tetraploids (two pairs of sites expected), and four 45S sites in a pentaploid (expectation five), suggesting a more complex origin involving processes such as karyotype reorganization, aneuploidy or segmental allopolyploidy and introgression.
Using two diploid total genomic, gDNA, probes (Uruz and Udec) to chromosomes of three species belonging to the ‘brizantha’ complex, in situ hybridization results showed very small differences in hybridization patterns between groups of chromosomes (candidate genomes), with strong signals in centromeres consistent with Corrêa et al. (2020). However, the genome-specific motifs identified in sequence data (see below) suggested that some chromosomes sharing similar pericentromeric signals actually belong to different genomes. Polyploid U. humidicola showed dispersed signals of gDNA probes along all chromosomes, making it impossible to discriminate genomes. GISH indicated that tetraploid U. maxima is autopolyploid, which is in contrast to the other polyploids in Urochloa that have been identified as allopolyploids. The autopolyploid origin of U. maxima and its facultative apomixis type of reproduction have been proved by different authors (Toledo-Silva et al., 2013; Lara et al., 2019), meaning that gDNA probes here showing both terminal and pericentromeric signals are informative.
Santos et al. (2015) revealed some differentiation of candidate genome-specific Ty3-gypsy retrotransposons in pericentromeric regions of Urochloa chromosomes. Urochloa contrasts with another Poaceae, Avena, where GISH can characterize individual genomes (Katsiotis et al., 2000; Tomaszewska and Kosina, 2021), ‘painting’ most of the chromosomal lengths. The Urochloa results indicate that bulk repetitive sequences present in the gDNA probes have diverged only slightly in sequence and copy number during speciation of the diploid U. brizantha ancestors combined in polyploids, showing only weak genome specificity (Corrêa et al., 2020). Centromeres of plants are often composed of abundant tandemly repeated sequences and sometimes centromere-specific retrotransposon families (e.g. the CR family in grasses; Miller et al., 1998; Presting et al., 1998; Heslop-Harrison and Schwarzacher, 2011). The centromere-specific distribution pattern of signals of genomic (this study; Corrêa et al., 2020) and transposable element (Santos et al., 2015) probes in Urochloa may be due to the retrotransposons being clustered in centromeres and thus generating strong signals, whereas copies located along chromosome arms are dispersed (Miller et al., 1998).
While GISH did not differentiate Urochloa genomes, bioinformatic analysis of unassembled raw DNA sequences identified short sequence motifs that showed differential abundance among accessions. In situ hybridization of the various motifs to metaphase chromosomes confirmed the differential abundance and enabled identification of the genomes present in polyploids, leading to a model of Urochloa evolution (see below). All the sequences were present on multiple chromosomes, showing both amplification and dispersion or homogenization of the motifs after speciation from a common ancestral Urochloa genome, and each sequence had a characteristic proximal, distal or more dispersed chromosomal location. However, in contrast to a parallel analysis in Avena species (Liu et al., 2019), no major DNA satellite or tandem repeats giving chromosomal bands were revealed in Urochloa. Triticeae species with much larger genomes and chromosomes have many tandem repeats, including simple sequence motifs, that are tribe-, genus- or species-specific and have been widely used to identify chromosomes (along with total genomic DNA; e.g. Ali et al., 2016; Patokar et al., 2016). More generally, in a wide range of species, repetitive sequences have been identified as a key component of evolutionary mechanisms and karyotypic differentiation, playing an important role in speciation (Heslop-Harrison and Schwarzacher, 2011; Mehrotra and Goyal, 2014). Comparison of GISH, and the sequences and chromosomal distribution of repetitive sequences identified by cloning or sequence analysis, suggests considerable differences in repetitive sequence evolution between taxonomic ‘groups’ (family, tribe or genus). It is evident that each group has distinctive rules for chromosome and repetitive sequence evolution, but these are not easily transferrable as models between species groups.
Taxonomy and the genomic composition of Urochloa polyploids
Species concepts for many of the genebank accessions of Urochloa (including Brachiaria, and other species which have previously been placed in the genera Megathyrsus, Eriochloa and Panicum) have been problematic, not least because of the range of ploidies, apomixis, vegetative propagated lines, intermediate morphological traits and growth habits, and the presence of hybrids occurring in the wild or as landraces selected by forage grass breeders and farmers. Our results support the maintenance of distinct species for U. ruziziensis, U. brizantha, U. decumbens, U. humidicola and U. maxima (chromosomal organization in Figs 4–6; relationship models in Figs 7 and 8). We accept the species concepts for diploids (Clayton and Renvoize, 1982; Clayton, 1989), and do not consider allopolyploids as cytotypes.
Fig. 8.

A model for the evolutionary origin of Urochloa species in the ‘brizantha’ (left) and ‘humidicola’ (middle) complexes, and in U. maxima (right), built from this study and published data: genome sizes and ploidy (Supplementary Data Table S1); repetitive DNA sequences from whole genome sequence analysis (k-mer counts and graph-based clustering; Supplementary Data Tables S3 and S4); in situ hybridization with defined repeat probes (Figs 5 and 6) and genomic DNA (Fig. 4; and Corrêa et al., 2020); karyotype analyses (Corrêa et al., 2020); meiotic behaviour (Risso-Pascotto et al., 2005; Mendes-Bonato et al., 2007; Fuzinatto et al., 2007); chloroplast genome (Pessoa-Filho et al., 2017); hybrid occurrence (Table 1; Mendes et al., 2006; Vigna et al., 2016; Risso-Pascotto et al., 2005); CIAT breeding programmes (Renvoize and Maass, 1993; Miles et al., 1996); and reported apomixis (Roche et al., 2001). The three line types show evolutionary sequence divergence (solid line), and hybridization events involving haploid, n (dotted line), or unreduced, 2n (dash-dotted line), gametes from different genomes (designated in Fig. 7). White blocks: putative species/hybrids.
Following the genome labelling system adopted across the Triticeae (Hordeae) tribe (Linde-Laursen et al., 1997) or in Brassica (Cheng et al., 2013; Alix et al., 2008), the level of genomic differentiation as found here by extensive sequence and chromosomal analysis is high enough that we propose designating basic genomes in Urochloa using the upper-case letters R, B and D for the ‘brizantha’ complex, rather than the superscript designations Bb, Bd and Br (Corrêa et al., 2020) for U. brizantha, U. decumbens and U. ruziziensis, which would indicate a much closer relationship of the three genomes than we think is present. Similarly, we suggest use of A and B or even C (for ‘humidicola’ complex), and M for U. maxima (Figs 7 and 8). More limited differentiation allows us to suggest use of superscript designations, referring to modified basic genomes, for less-well differentiated genomes including Ba and Bb. Figure 7 illustrates the chromosome and genome composition of the accessions studied here. Urochloa ruziziensis was diploid; U. brizantha with multiple polyploid levels shows a variation of chromosomes and genomes, as does U. decumbens. An important question to be answered is whether allopolyploid species should be considered separate species or not, since their genomic composition indicates that they are of hybrid origin and their parental species are known?
Our analysis supported the genome composition of hexaploid U. humidicola (based on meiotic behaviour, Vigna et al., 2016; and transposable elements, Santos et al., 2015) as including A and B genomes (and probably the C genome in higher ploidy levels). Ty1-gypsy Tat probe (Santos et al., 2015) and Uhum-spec12 (Fig. 6B) are good markers for the A genome. The B genome is more variable in showing three types of chromosomes.
Evolutionary model for Urochloa species
Three substantive models (Fig. 8) to explain the evolution of Urochloa polyploids in the ‘brizantha’ and ‘humidicola’ agamic complexes, and U. maxima were generated from multiple lines of evidence. Renvoize and Maass (1993) suggested that diploid U. decumbens evolved from U. brizantha: the natural range of U. decumbens covers the area of a candidate ancestral U. brizantha form or variety (e.g. U. brizantha var. latifolium Oliver or U. brizantha var. angustifolia Stent & Rattray) with lanceolate hairy leaves and a decumbent habit. We found genome-specific repetitive sequences in U. decumbens, but all of them were shared with U. brizantha, supporting the order of evolutionary branching. These data contradict Basappa et al.’s (1987) suggestion that U. decumbens is a natural hybrid between U. brizantha and U. ruziziensis, and confirmation of this hypothesis would be meiotic abnormalities found in U. decumbens. We support this hypothesis for tetraploid U. decumbens, but not the diploid accession we studied. Our results were inconclusive for the hexaploid U. brizantha accessions (see Fig. 7). Pessoa-Filho et al. (2017) found that tetraploid U. brizantha and U. decumbens show high similarity of their plastid sequences and low number of single nucleotide polymorphisms, which may suggest that a single polyploidization event took place to establish both the tetraploid U. brizantha and U. decumbens: namely a potential fertilization of a tetraploid U. brizantha BD gamete and an unreduced RR gamete of a diploid U. ruziziensis.
Risso-Pascotto et al. (2006) suggested that hexaploid U. brizantha probably resulted from ‘chromosome doubling of a triploid derived from species that did not display the same behaviour for spindle organization’. Triploid hybrids were found in nature (Timbó et al., 2014), and may originate from crosses between diploid U. ruziziensis and tetraploid U. decumbens or U. brizantha. Thus, a hexaploid species would be created by crossing two different triploids rather than doubling of genomes of a triploid hybrid. This suggestion arises from the presence of only one R genome in the hexaploid U. brizantha, as indicated by our in situ hybridization analysis (see Figs 4, 5 and 7). We also suggest, based on our in situ hybridization and repetitive sequence composition in hexaploid U. brizantha, that there are at least two cytotypes/varieties of diploid U. brizantha. Another possibility is that the genomes of the hexaploid U. brizantha have undergone structural changes after polyploidization, and therefore some chromosomes show signals of both U. brizantha- and U decumbens-specific probes, and some only show U. brizantha-specific signals. This hypothesis can be supported by Bernini and Marin-Morales (2001) and Nielen et al. (2010), who showed differences in karyotypes of diploid and tetraploid U. brizantha accessions.
The most likely evolution of species belonging to the ‘humidicola’ complex is much more difficult to propose, because all accessions are polyploid and there is no suggestion as to which diploid species may be considered ancestral. There are three known levels of ploidy in this species: hexaploid, heptaploid and nonaploid (Boldrini et al., 2009a; Jungmann et al., 2010; Vigna et al., 2016; we also had an inconclusive accession 2n = 8x + 2 or 9x − 4 = 50). Our analysis of the genomic composition of the hexaploid species matches with meiotic analyses conducted by Boldrini et al. (2009b) and Vigna et al. (2016), and the model of evolution of species belonging to the ‘humidicola’ complex is supported by in situ hybridization with genome-specific probes (see Fig. 6). The B genome includes chromosomes showing three different types of signals, which may suggest that U. humidicola has gone through several rounds of polyploidization. Broader analysis of genome composition of species belonging to the ‘humidicola’ agamic complex, including different accessions of U. humidicola and U. dictyoneura, would be desirable to understand the process of speciation, especially as tetraploid accessions with 2n = 4x = 24 are known (Boldrini et al., 2010) and could have contributed to the evolution of U. humidicola, which shows odd ploidy levels.
Our in situ hybridization studies gave evidence for potential introgression within Urochloa. Some polyploid lines (U. brizantha and U. humidicola) here have chromosome pairs that are different from others within their genome (see Figs 5G, J–L and 6A, C, D), resembling segmental allopolyploidy (Mendes-Bonato et al., 2002) or disomic introgression lines. Frequent introgression seems to occur in wheat (Cheng et al., 2019) and oat polyploids, and in breeding, whole chromosomes, chromosome arms or segments may be substituted. An example is Triticale, which may have not the expected seven chromosome pairs of each genome but 14 A, 12 B, two D and 14 R chromosomes (Neves et al., 1997). Some hybrid species are diploid or reduce chromosome numbers so they are not clearly tetraploid – Petunia hybrida is 2n = 14, like its ancestors (Bombarely et al., 2016), with a mixture of ancestral genomes, while the octaploid Nicotiana cell fusion hybrid (4x + 4x) has lost a few chromosomes (Patel et al., 2011).
Conclusions
Genome composition and evolution are complex in Urochola tropical forage grasses. Grasslands are not only a major source of food production but also provide environmental services: water, soil preservation, carbon capture, etc., often in more biodiverse regions, where identification of species and their relationships will assist in grass conservation. Despite their lower economic value, breeding and exploitation of biodiversity is required within the group (whether using sequence data or a genetic map, for example as in Lolium, Tomaszewski et al., 2012). Like wheat and Brassica crops, wild relatives contribute to the current pool of diversity used in Urochloa tropical forage grass improvement, with additional complexities from apomixis. Knowledge of genome relationships and polyploid genome composition gives opportunities for rational and systematic use of accessions in forage improvement programmes (superdomestication: Vaughan et al., 2007). Complementing our study showing the diversification of genomes and repetitive DNA, a parallel study (Hanley et al., 2020) found high levels of genetic diversity in 20 genes related to forage quality in 104 of the accessions studied here.
Our study was focused on accessions available from international germplasm collections to breeders and researchers. As Keller-Grein et al. (1996) correctly pointed out, further collecting of the Urochloa species in Africa would be worthwhile to enrich the germplasm collection with new accessions, finding further useful characteristics that can be exploited, and to better understand its complicated evolution, adding to the analysis here. For legal regulations regarding biosecurity restrictions (diseases and invasive species) and Plant Breeders Rights and germplasm ownership, it is necessary to have an accepted name for every species, and our identification of genomes and genome composition in Urochloa polyploids presents the necessary framework.
Breeding programmes often work with a single ploidy because directed crosses among parents with different ploidies are challenging. We suggest that Urochloa species are all part of a common gene pool, and any hybrid combination might be possible and become a successful forage variety, noxious weed or disease host. The current breeding programmes at CIAT manage tetraploid interspecific crosses within the ‘brizantha’ agamic complex, hexaploid crosses within the ‘humidicola’ agamic complex and tetraploid intraspecific crosses of U. maxima. The choice of appropriate strategies to generate hybrids requires knowledge of ploidy provided by our research, supported by the model of evolution and diversification of the species.
SUPPLEMENTARY DATA
Supplementary data are available online at https://academic.oup.com/aob and consist of the following. Fig. S1. Ploidy measured by flow cytometry of PI-stained nuclei from dehydrated tissues of diploid, tetraploid, pentaploid and hexaploid accessions of Urochloa showing very sharp peaks. Fig. S2. Contig 5 as a candidate motif specific to the U. brizantha genome. Fig. S3. Distribution of graph-based clusters. Table S1. List of accessions used in the study, their ploidy levels, growth habits and geographical distribution. Table S2. Summary of sequencing data quality. Table S3. Potential genome-specific 50-mer sequences, their genome proportion, and description of probes and in situ hybridization signals. Table S4. Potential genome-specific repeats and their genome proportion. Table S5. BLASTN search of highly abundant potential genome-specific 50-mers. Table S6. NCBI BLASTN results of clusters found using RepeatExplorer. Table S7. RepeatExplorer characterization of selected repeat clusters of Urochloa accessions. Table S8. NCBI BLASTN results of clusters found using TAREAN. Table S9. TAREAN characterization of selected repeat clusters of Urochloa accessions. Table S10. Repetitive DNA composition of Urochloa genomes.
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We would like to thank Dr Jennifer Hincks from The Centre for Core Biotechnology Services, Flow Cytometry Facility at University of Leicester, for assistance with developing our flow cytometry protocol. We are grateful to the USDA-Germplasm Resources Information Network (GRIN) for their generous provision of seeds. Germplasm held in the CIAT collections is available on request (http://genebank.ciat.cgiar.org) and we thank colleagues there for assistance with access to reports of collection expeditions and trials which are linked to the accessions from the germplasm collections. The authors declare no conflict of interest.
Contributor Information
Paulina Tomaszewska, Department of Genetics and Genome Biology, University of Leicester, Leicester, UK.
Maria S Vorontsova, Royal Botanic Gardens, Kew, Richmond, Surrey, UK.
Stephen A Renvoize, Royal Botanic Gardens, Kew, Richmond, Surrey, UK.
Sarah Z Ficinski, Royal Botanic Gardens, Kew, Richmond, Surrey, UK.
Joseph Tohme, International Center for Tropical Agriculture (CIAT), A.A. 6713, Cali, Colombia.
Trude Schwarzacher, Department of Genetics and Genome Biology, University of Leicester, Leicester, UK; Key Laboratory of Plant Resources Conservation and Sustainable Utilization/Guangdong Provincial Key Laboratory of Applied Botany, South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, China.
Valheria Castiblanco, International Center for Tropical Agriculture (CIAT), A.A. 6713, Cali, Colombia.
José J de Vega, Earlham Institute, Norwich Research Park, Norwich, UK.
Rowan A C Mitchell, Rothamsted Research, Harpenden, Hertfordshire, UK.
J S (Pat) Heslop-Harrison, Department of Genetics and Genome Biology, University of Leicester, Leicester, UK; Key Laboratory of Plant Resources Conservation and Sustainable Utilization/Guangdong Provincial Key Laboratory of Applied Botany, South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, China.
FUNDING
This work was supported under the RCUK-CIAT Newton-Caldas Initiative ‘Exploiting biodiversity in Brachiaria and Panicum tropical forage grasses using genetics to improve livelihoods and sustainability’, with funding from UK’s Official Development Assistance Newton Fund awarded by UK Biotechnology and Biological Sciences Research Council (BB/R022828/1). P.T. has received further support (polyploidy and chromosome evolution) from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreements No. 844564 and No. 101006417.
LITERATURE CITED
- Akiyama Y, Yamada-Akiyama H, Ebina M. 2010. Morphological diversity of chromosomes bearing ribosomal DNA loci in Brachiaria species. Grassland Science 56: 217–223. [Google Scholar]
- Ali N, Heslop-Harrison JS, Ahmad H, Graybosch RA, Hein GL, Schwarzacher T. 2016. Introgression of chromosome segments from multiple alien species in wheat breeding lines with wheat streak mosaic virus resistance. Heredity 117: 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alix K, Gérard PR, Schwarzacher T, Heslop-Harrison JS. 2017. Polyploidy and interspecific hybridization: partners for adaptation, speciation and evolution in plants. Annals of Botany 120: 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alix K, Joets J, Ryder CD, et al. 2008. The CACTA transposon Bot1 played a major role in Brassica genome divergence and gene proliferation. The Plant Journal 56: 1030–1044. [DOI] [PubMed] [Google Scholar]
- Alvarez E, Latorre M, Bonilla X, Miles JW. 2014. Assessing the resistance of Brachiaria hybrids to pathogenic Rhizoctonia. Plant Disease 98: 306–310. [DOI] [PubMed] [Google Scholar]
- Arroyave C, Barceló J, Poschenrieder C, Tolrà R. 2011. Aluminium-induced changes in root epidermal cell patterning, a distinctive feature of hyperresistance to Al in Brachiaria decumbens. Journal of Inorganic Biochemistry 105: 1477–1483. [DOI] [PubMed] [Google Scholar]
- Basappa GP, Muniyamma M, Chinnappa CC. 1987. An investigation of chromosome numbers in the genus Brachiaria (Poaceae: Paniceae) in relation to morphology and taxonomy. Canadian Journal of Botany 65: 2297–2309. [Google Scholar]
- Bernini C, Marin-Morales MA. 2001. Karyotype analysis in Brachiaria (Poaceae) species. Cytobios 104: 157–171. [PubMed] [Google Scholar]
- Boldrini KR, Micheletti PL, Gallo PH, Mendes-Bonato AB, Pagliarini MS, Valle CB. 2009a. Origin of a polyploid accession of Brachiaria humidicola (Poaceae; Panicoideae: Paniceae). Genetics and Molecular Research 8: 888–895. [DOI] [PubMed] [Google Scholar]
- Boldrini KR, Pagliarini MS, Valle CB. 2009b. Meiotic behavior of a nonaploid accession endorses x = 6 for Brachiaria humidicola (Poaceae). Genetics and Molecular Research 8: 1444–1450. [DOI] [PubMed] [Google Scholar]
- Boldrini KR, Pagliarini MS, Valle CB. 2010. Evidence of natural hybridization in Brachiaria humidicola (Rendle) Schweick. (Poaceae: Panicoideae: Paniceae). Journal of Genetics 89: 91–94. [DOI] [PubMed] [Google Scholar]
- Bombarely A, Moser M, Amrad A, et al. 2016. Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nature Plants 16074. doi: 10.1038/NPLANTS.2016.74. [DOI] [PubMed] [Google Scholar]
- Čertner M, Fenclová E, Kúr P, et al. 2017. Evolutionary dynamics of mixed-ploidy populations in an annual herb: dispersal, local persistence and recurrent origins of polyploids. Annals of Botany 120: 303–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Liu J, Wen J, et al. 2019. Frequent intra- and inter-species introgression shapes the landscape of genetic variation in bread wheat. Genome Biology 20: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F, Mandáková T, Wu J, Xie Q, Lysak MA, Wang X. 2013. Deciphering the diploid ancestral genome of the mesohexaploid Brassica rapa. The Plant Cell 25: 1541–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton WD. 1989. Gramineae. In: Launert E, Pope GV, eds. Flora Zambesiaca 10 (3). Kew: Royal Botanic Gardens. [Google Scholar]
- Clayton WD, Renvoize SA. 1982. Flora of tropical East Africa. Gramineae. Part 3. Rotterdam: AA Balkema. [Google Scholar]
- Corrêa CTR, Bonetti NGZ, Barrios SCL, do Valle CB, Torres GA, Techio VH. 2020. GISH-based comparative genomic analysis in Urochloa P. Beauv. Molecular Biology Reports 47: 887–896. [DOI] [PubMed] [Google Scholar]
- Doležel J, Greilhuber J, Suda J. 2007. Estimation of nuclear DNA content in plants using flow cytometry. Nature Protocols 2: 2233–2244. [DOI] [PubMed] [Google Scholar]
- Doyle JJ, Doyle JL. 1990. A rapid total DNA preparation procedure for fresh plant tissue. Focus 12: 13–15. [Google Scholar]
- Durigan G, de Siqueira MF, Franco GADC. 2007. Threats to the Cerrado remnants of the state of São Paulo, Brasil. Scientia Agricola 64: 355–363. [Google Scholar]
- Feldman M, Sears ER. 1981. The wild gene resources of wheat. Scientific American 244: 102–112. [Google Scholar]
- Felismino MF, Pagliarini MS, do Valle CB. 2010. Meiotic behavior of interspecific hybrids between artificially tetraploidized sexual Brachiaria ruziziensis and tetraploid apomictic B. brizantha (Poaceae). Scientia Agricola (Piracicaba, Braz.) 67: 191–197. [Google Scholar]
- Fuzinatto VA, Pagliarini MS, Valle CB. 2007. Microsporogenesis in sexual Brachiaria hybrids (Poaceae). Genetics and Molecular Research 6: 1107–1117. [PubMed] [Google Scholar]
- Galbraith DW, Lambert GM. 2012. Using the BD AccuriTM C6 cytometer for rapid and accurate analysis of the nuclear DNA contents of flowering plants. BD Bioscience 1–10. [Google Scholar]
- Gerlach WL, Bedbrook JR. 1979. Cloning and characterization of ribosomal RNA genes from wheat and barley. Nucleic Acids Research 7: 1869–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach WL, Dyer TA. 1980. Sequence organization of the repeating units in the nucleus of wheat which contain 5S rRNA genes. Nucleic Acids Research 8: 4851–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracindo CV, Louvandini H, Riet-Correa F, Barbosa-Ferreira M, de Castro MB. 2014. Performance of sheep grazing in pastures of Brachiaria decumbens, Brachiaria brizantha, Panicum maximum, and Andropogon gayanus with different protodioscin concentrations. Tropical Animal Health and Production 46: 733–737. [DOI] [PubMed] [Google Scholar]
- Hagl PA, Gargiulo R, Fay MF, et al. 2021. Geographical structure of genetic diversity in Loudetia simplex (Poaceae) in Madagascar and South Africa. Botanical Journal of the Linnean Society. [Google Scholar]
- Hanley SJ, Pellny TK, de Vega JJ, et al. 2020. Allele mining in diverse accessions of Urochloa and Megathyrsus sp. tropical grasses to improve forage quality and reduce environmental impact. bioRxiv preprint 10.1101/2020.12.09.418087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez LM, Sotelo G, Bonilla X, Alvarez E, John W. 2017. Phenotyping Brachiaria genotypes to assess Rhizoctonia resistance by comparing three inoculum types. Plant Disease 101: 1–30. [DOI] [PubMed] [Google Scholar]
- Heslop-Harrison JS, Schwarzacher T. 2011. Organisation of the plant genome in chromosomes. Plant Journal 66: 18–33. [DOI] [PubMed] [Google Scholar]
- Higgins J, Tomaszewska P, Pellny TK, et al. 2021. The five Urochloa spp. used in development of tropical forage cultivars originate from defined subpopulations with differentiated gene pools. bioRxiv 10.1101/2021.07.21.453213 [DOI] [Google Scholar]
- Hutchinson J, Dalziel JM. 1972. Flora of West Tropical Africa: Vol. 3, part 2 Juncaceae-Gramineae. London: Crown Agents. [Google Scholar]
- Ishitani M, Rao I, Wenzl P, Beebe S, Tohme J. 2004. Integration of genomics approach with traditional breeding towards improving abiotic stress adaptation: drought and aluminum toxicity as case study. Field Crops Research 90: 35–45. [Google Scholar]
- Jungmann L, Vigna BBZ, Boldrini KR, et al. 2010. Genetic diversity and population structure analysis of the tropical pasture grass Brachiaria humidicola based on microsatellites, cytogenetics, morphological traits, and geographical origin. Genome 53: 698–709. [DOI] [PubMed] [Google Scholar]
- Kato-Noguchi H, Kobayashi A, Ohno O, Kimura F, Fujii Y, Suenaga K. 2014. Phytotoxic substances with allelopathic activity may be central to the strong invasive potential of Brachiaria brizantha. Journal of Plant Physiology 171: 525–530. [DOI] [PubMed] [Google Scholar]
- Katsiotis A, Loukas M, Heslop-Harrison JS. 2000. Repetitive DNA, genome and species relationships in Avena and Arrhenatherum (Poaceae). Annals of Botany 86: 1135–1142. [Google Scholar]
- Kearse M, Moir R, Wilson A, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller-Grein G, Maass BL, Hanson J. 1996. Natural variation in Brachiaria and existing germplasm collections. In: Miles JW, Maass BL, do Valle CB, eds. Brachiaria: biology, agronomy, and improvement. Cali, Columbia: Centro Internacional de Agricultura Tropical (CIAT); Campo Grande, BR: Empresa Brasileira de Pesquisa Agropecuaria (EMBRAPA), Centro Nacional de Pesquisa de Gado de Corte (CNPGC), 16–39. [Google Scholar]
- Kellogg EA. 2015. Flowering Plants. Monocots: Poaceae. In: Kubitzki K, ed. The families and genera of vascular plants, Vol. 13. Heidelberg, Germany: Springer. [Google Scholar]
- Lara LAC, Santos MF, Jank L, et al. 2019. Genomic selection with allele dosage in Panicum maximum Jacq. G3 9: 2463–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin DA. 2021. Propagule pressure and the establishment of polyploid populations (Viewpoint). Annals of Botany 127: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linde-Laursen I, Heslop-Harrison JS, Shepherd KW, Taketa S. 1997. The barley genome and its relationship with the wheat genomes. A survey with an internationally agreed recommendation for barley chromosome nomenclature. Hereditas 126: 1–16. [Google Scholar]
- Liu Q, Li X, Zhou X, et al. 2019. The repetitive DNA landscape in Avena (Poaceae): chromosome and genome evolution defined by major repeat classes in whole-genome sequence reads. BMC Plant Biology 19: 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutts S, Ndikumana J, Louant BP. 1991. Fertility of Brachiaria ruziziensis in interspecific crosses with Brachiaria decumbens and Brachiaria brizantha: meiotic behaviour, pollen viability and seed set. Euphytica 57: 267–274. [Google Scholar]
- Mandáková T, Münzbergová Z. 2006. Distribution and ecology of cytotypes of the Aster amellus aggregates in the Czech Republic. Annals of Botany 98: 845–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais G, Kingsford C. 2011. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27: 764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrotra S, Goyal V. 2014. Repetitive sequences in plant nuclear DNA: types, distribution, evolution and function. Genomics, Proteomics & Bioinformatics 12: 164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes DV, Boldrini KR, Mendes-Bonato AB, Pagliarini MS, Do Valle CB. 2006. Cytological evidence of natural hybridization in Brachiaria brizantha Stapf (Gramineae). Biological Journal of the Linnean Society 150: 441–446. [Google Scholar]
- Mendes-Bonato AB, Gargione R, Filho J, Pagliarini MS, Do Valle CB, Penteado MI. 2002. Unusual cytological patterns of microsporogenesis in Brachiaria decumbens: abnormalities in spindle and defective cytokinesis causing precocious cellularization. Cell Biology International 26: 641–646. [DOI] [PubMed] [Google Scholar]
- Mendes-Bonato AB, Pagliarini MS, do Valle CB. 2007. Meiotic arrest compromises pollen fertility in an interspecific hybrid between Brachiaria ruziziensis x Brachiaria decumbens (Poaceae: Paniceae). Brazilian Archives of Biology and Technology 50: 831–837. [Google Scholar]
- Miles JW, Cardona C, Sotelo G. 2006. Recurrent selection in a synthetic Brachiaria grass population improves resistance to three spittlebug species. Crop Science 46: 1088–1093. [Google Scholar]
- Miles JW, Maass BL, do Valle CB, eds.1996. Brachiaria: Biology, agronomy, and improvement. Campo Grande, BR: Internacional de Agricultura Tropical (CIAT); Cali, Columbia:Empresa Brasileira de Pesquisa Agropecuaria (EMBRAPA), Centro Nacional de Pesquisa de Gado de Corte (CNPGC), 288. [Google Scholar]
- Miller JT, Dong F, Jackson SA, Song J, Jiang J. 1998. Retrotransposon-related DNA sequences in the centromeres of grass chromosomes. Genetics 150: 1615–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nani TF, Schnable JC, Washburn JD, et al. 2018. Location of low copy genes in chromosomes of Brachiaria spp. Molecular Biology Reports 45: 109–118. [DOI] [PubMed] [Google Scholar]
- Neves N, Silva M, Heslop-Harrison JS, Viegas W. 1997. Nucleolar dominance in triticales: control by unlinked genes. Chromosome Research 5: 125–131. [DOI] [PubMed] [Google Scholar]
- Nielen S, Almeida LM, Carneiro VTC, Araujo ACG. 2010. Physical mapping of rDNA genes corroborates allopolyploid origin in apomictic Brachiaria brizantha. Sexual Plant Reproduction 23: 45–51. [DOI] [PubMed] [Google Scholar]
- Novak P, Neumann P, Pech J, Steinhais J, Macas J. 2013. RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next generation sequence reads. Bioinformatics 29: 792–793. [DOI] [PubMed] [Google Scholar]
- Novak P, Robledillo LA, Koblizkova A, Vrbova I, Neumann P, Macas J. 2017. TAREAN: a computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acid Research 45: e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D, Power JB, Anthony P, Badakshi F, Heslop-Harrsion JS, Davey MR. 2011. Somatic hybrid plants of Nicotiana × sanderae (+) N. debneyi with fungal resistance to Peronospora tabacina. Annals of Botany 108: 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patokar C, Sepsi A, Schwarzacher T, Kishii M, Heslop-Harrison JS. 2016. Molecular cytogenetic characterization of novel wheat–Thinopyrum bessarabicum recombinant lines carrying intercalary translocations. Chromosoma 125: 163–172. [DOI] [PubMed] [Google Scholar]
- Penteado MIO, Santos ACM, Rodrigues IF, do Valle CB, Seixas MAC, Esteves A. 2010. Determinação de ploidia e avaliação da quantidade de DNA total em diferentes espécies do gênero Brachiaria. Campo Grande: Embrapa Gado de Corte. (Embrapa Gado de Corte. Boletim de Pesquisa, 11). [Google Scholar]
- Pessoa-Filho M, Martins AM, Ferreira ME. 2017. Molecular dating of phylogenetic divergence between Urochloa species based on complete chloroplast genomes. BMC Genomics 18: 516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro AA, Pozzobon MT, do Valle CB, Penteado MIO, Carneiro VTC. 2000. Duplication of the chromosome number of diploid Brachiaria brizantha plants using colchicine. Plant Cell Reports 19: 274–278. [DOI] [PubMed] [Google Scholar]
- Pizarro EA, Hare MD, Mutimura M, Changjun B. 2013. Brachiaria hybrids: potential, forage use and seed yield. Tropical Grasslands - ForrajesTropicales 1: 31–35. [Google Scholar]
- POWO. 2019. Plants of the World Online. Kew: Facilitated by the Royal Botanic Gardens. http://www.plantsoftheworldonline.org/ Retrieved 1 December 2020. [Google Scholar]
- Presting GG, Malysheva L, Fuchs J, Schubert I. 1998. ATY3/GYPSY retrotransposon‐like sequence localizes to the centromeric regions of cereal chromosomes. Plant Journal 16: 721–728. [DOI] [PubMed] [Google Scholar]
- Rasheed A, Mujeeb-Kazi A, Ogbonnaya FC, He Z, Rajaram S. 2018. Wheat genetic resources in the post-genomics era: promise and challenges. Annals of Botany 121: 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renvoize SA, Maass B. 1993. Brachiaria. A report to CIAT, Colombia, on the species and specimens held in the germplasm collection. Kew: Royal Botanic Gardens. [Google Scholar]
- Risso-Pascotto C, Pagliarini MS, Valle CB. 2005. Meiotic behavior in interspecific hybrids between Brachiaria ruziziensis and Brachiaria brizantha (Poaceae). Euphytica 145: 155–159. [Google Scholar]
- Risso-Pascotto C, Pagliarini MS, Valle CB. 2006. A new basic chromosome number for the genus Brachiaria (Trin.) Griseb. (Poaceae: Panicoideae: Paniceae). Genetic Resources and Crop Evolution 53: 7–10. [Google Scholar]
- Roche D, Hanna WW, Ozias-Akins. 2001. Is supernumerary chromatin involved in gametophytic apomixis of polyploid plants? Sexual Plant Reproduction 13: 343–349. [Google Scholar]
- Rozen S, Skaletsky H. 1999. Primer3 on the WWW for general users and for biologist programmers. Bioinformatics Methods and Protocols 132: 365–386. [DOI] [PubMed] [Google Scholar]
- Salariato DL, Morrone O, Zuloaga FO. 2012. Mayariochloa, a new monotypic genus segregated from Scutachne (Poaceae, Panicoideae, Paniceae). Systematic Botany 37: 105–116. [Google Scholar]
- Salariato DL, Zuloaga FO, Giussani LM, Morrone O. 2010. Molecular phylogeny of the subtribe Melinidinae (Poaceae: Panicoideae: Paniceae) and evolutionary trends in the homogenization of inflorescences. Molecular Phylogenetics and Evolution 56: 355–369. [DOI] [PubMed] [Google Scholar]
- Santos FC, Guyot R, do Valle CB, et al. 2015. Chromosomal distribution and evolution of abundant retrotransposons in plants: gypsy elements in diploid and polyploid Brachiaria forage grasses. Chromosome Research 23: 571–582. [DOI] [PubMed] [Google Scholar]
- Sayers EW, Agarwala R, Bolton EE, et al. 2019. Database resources of the National Center for Biotechnology Information. Nucleic Acids Research 47: D23–D28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzacher T, Heslop-Harrison JS. 2000. Practical in situ hybridization. Oxford, UK: Bios. [Google Scholar]
- Singh AK, Singh L, Kumar C, Kumar P, Dimree A. 2010. Para grass hybrid (Brachiaria sp.) – a potential forage for India. Environment & Ecology 28: 1602–1606. [Google Scholar]
- Soltis DE, Gitzendanner MA, Stull G, et al. 2013. The potential of genomics in plant systematics. Taxon 62: 886–898. [Google Scholar]
- Soreng RJ, Peterson PM, Romaschenko K, et al. 2017. A worldwide phylogenetic classification of the Poaceae (Gramineae) II: an update and a comparison of two 2015 classifications. Journal of Systematics and Evolution 55: 259–290. [Google Scholar]
- Sosef MSM. 2016. Taxonomic novelties in central African grasses (Poaceae), Paniceae 1. Plant Ecology and Evolution 149: 356–365. [Google Scholar]
- de Souza-Kaneshima AM, Simioni C, Felismino MF, et al. 2010. Meiotic behaviour in the first interspecific hybrids between Brachiaria brizantha and Brachiaria decumbens. Plant Breeding 129: 186–191. [Google Scholar]
- Timbó ALO, Pereira RC, Sobrinho FS, Davide C. 2014. Nuclear DNA content and chromosome number in Brachiaria spp. Revista Ciência Agronômica 45: 62–67. [Google Scholar]
- Toledo-Silva G, Cardoso-Silva CB, Jank L, Souza AP. 2013. De novo transcriptome assembly for the tropical grass Panicum maximum Jacq. PLoS One 8: e70781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaszewska P, Kosina R. 2018. Instability of endosperm development in amphiploids and their parental species in the genus Avena L. Plant Cell Reports 37: 1145–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaszewska P, Kosina R. 2021. Cytogenetic events in the endosperm of amphiploid Avena magna x A. longiglumis. Journal of Plant Research 134: 1047–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaszewska P, Pellny TK, Hernández LM, et al. 2021. Flow cytometry-based determination of ploidy from dried leaf specimens in genomically complex collections of the tropical forage grass Urochloa s. l. Genes 12: 957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaszewski C, Byrne SL, Foito A, et al. 2012. Genetic linkage mapping in an F2 perennial ryegrass population using DArT markers. Plant Breeding 131: 345–349. [Google Scholar]
- Torres González AM, Morton CM. 2005. Molecular and morphological phylogenetic analysis of Brachiaria and Urochloa (Poaceae). Molecular Phylogenetics and Evolution 37: 36–44. [DOI] [PubMed] [Google Scholar]
- Valério JR, Lapointe SL, Kelemu S, Fernandes CD, Morales FJ. 1996. Pests and diseases of Brachiaria species. In: Miles JW, Maass BL, do Valle CB, eds. Brachiaria: biology, agronomy, and improvement. Campo Grande, BR: Centro Internacional de Agricultura Tropical (CIAT); Cali, Columbia:Empresa Brasileira de Pesquisa Agropecuaria (EMBRAPA), Centro Nacional de Pesquisa de Gado de Corte (CNPGC),87–105. [Google Scholar]
- Vaughan DA, Balazs E, Heslop-Harrison JS. 2007. From crop domestication to super-domestication. Annals of Botany 100: 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- do Valle CB, Savidan YH. 1996. Genetics, cytogenetics, and reproductive biology of Brachiaria. In: Miles JW, Maass BL, do Valle CB, eds. Brachiaria: biology, agronomy, and improvement. Campo Grande, BR: Centro Internacional de Agricultura Tropical (CIAT); Cali, Columbia@: Empresa Brasileira de Pesquisa Agropecuaria (EMBRAPA), Centro Nacional de Pesquisa de Gado de Corte (CNPGC),147–163. [Google Scholar]
- Velmurugan J, Mollison E, Barth S, et al. 2016. An ultra-high density genetic linkage map of perennial ryegrass (Lolium perenne) using genotyping by sequencing (GBS) based on a reference shotgun genome assembly. Annals of Botany 118: 71–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigna BBZ, Santos JCS, Jungmann L, et al. 2016. Evidence of allopolyploidy in Urochloa humidicola based on cytological analysis and genetic linkage mapping. PLoS One 11: e0153764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassie WA, Tsegay BA, Wolde AT, Limeneh BA. 2018. Evaluation of morphological characteristics, yield and nutritive value of Brachiaria grass ecotypes in northwestern Ethiopia. Agriculture & Food Security 7: 89. [Google Scholar]
- Webster RD. 1987. The Australian Paniceae (Poaceae). Berlin and Stuttgart: J. Cramer. [Google Scholar]
- de Wet JMJ. 1986. Hybridization and polyploidy in the Poaceae. In: Soderstrom TR, Hilu TR, Campbell WH, Barkworth ME, eds. Grass systematics and evolution. Washington, DC, USA: Smithsonian Institution Press, 179–187. [Google Scholar]
- Worthington M, Ebina M, Yamanaka N, et al. 2019. Translocation of a parthenogenesis gene candidate to an alternate carrier chromosome in apomictic Brachiaria humidicola. BMC Genomics 20: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthington M, Heffelfinger C, Bernal D, et al. 2016. A parthenogenesis gene candidate and evidence for segmental allopolyploidyan in apomictic Brachiaria decumbens. Genetics 203: 1117–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthington ML, Miles JW. 2015. Reciprocal full-sib recurrent selection and tools for accelerating genetic gain in apomictic Brachiaria. In: Budak H, Spangenberg G, eds. Molecular breeding of forage and Turf. Cham: Springer, 19–30. [Google Scholar]
- Worthington M, Perez JG, Mussurova S, et al. 2021. A new genome allows the identification of genes associated with natural variation in aluminium tolerance in Brachiaria grasses. Journal of Experimental Botany 72: 302–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.