

Abstract
Background:
Neutrophilic asthma is associated with disease severity and corticosteroid insensitivity. Novel therapies are required to manage this life-threatening asthma phenotype. Programmed cell death protein-1 (PD-1) is a key homeostatic modulator of the immune response for T cell effector functions.
Objective:
Our aim here was to investigate the role of PD-1 in the regulation of acute neutrophilic inflammation in a murine model of airway hyperreactivity (AHR).
Methods:
House dust mite was used to induce and compare neutrophilic AHR in wild-type and PD-1 knockout mice. Then the therapeutic potential of a human PD-1 agonist was tested in a humanized mouse model in which the PD-1 extracellular domain is entirely humanized. Single-cell RNA sequencing and flow cytometry were mainly used to investigate molecular and cellular mechanisms.
Results:
PD-1 was highly induced on pulmonary T cells in our inflammatory model. PD-1 deficiency was associated with an increased neutrophilic AHR and high recruitment of inflammatory cells to the lungs. Consistently, PD-1 agonist treatment dampened AHR, decreased neutrophil recruitment, and modulated cytokine production in a humanized PD-1 mouse model. Mechanistically, we demonstrated at the transcriptional and protein levels that the inhibitory effect of PD-1 agonist is associated with the reprogramming of pulmonary effector T cells that showed decreased number and activation.
Conclusion:
PD-1 agonist treatment is efficient in dampening neutrophilic AHR and lung inflammation in a preclinical humanized mouse model.
Keywords: Neutrophilic asthma, PD-1 agonist, T cells, lung inflammation, humanized mice
Capsule summary:
This study demonstrates the first proof of concept supporting the beneficial utilization of PD-1 agonist in neutrophilic asthma via specific targeting and reprogramming of activated T cells in the lungs.
Introduction
Asthma is a chronic inflammatory disease of the airways associated with respiratory symptoms ranging from coughing to shortness of breath during acute exacerbations. Asthma is a heterogeneous disease that consists of many different phenotypes including eosinophilic asthma, neutrophilic asthma, exercise-induced, occupational, and obesity-related asthma, each associated with different immunopathology(1–3). Although available treatments including inhaled corticosteroids and long-acting β2- agonists are effective to manage asthma symptoms and avoid serious outcomes, asthma could affect patients’ daily life if not treated appropriately. Additionally, approximately 10 to 15 percent of patients develop a severe steroid-refractory phenotype associated with a higher risk of hospitalization and intubation(2,4,5). Neutrophilic asthma is the most common phenotype among adult patients associated with high disease severity and therefore requires more aggressive therapies and often the patients do not respond to the treatment. Neutrophil-driven inflammation is characterized by high neutrophil recruitment and is associated with airway hyperreactivity (AHR) and pathological tissue remodeling(6–9). Given the serious outcomes, novel therapeutic targets are required to develop a more specific and effective treatment against severe forms of asthma.
Immune checkpoints constitute a group of gatekeeper receptors that determine the intensity of an immune response. Programmed cell death protein-1 (PD-1) is one of the principal inhibitory checkpoints that mainly suppresses T cell activation. PD-L1 and PD-L2, are the two known ligands that interact with PD-1 allowing the induction of intracellular signaling pathways through the immunoreceptor tyrosine-based inhibitory motif in the cytoplasmic domain(10–12). Although PD-1 has received considerable attention for its role in T cell exhaustion, PD-1 has diverse roles in regulating host immunity including regulation of T cell activation, T cell priming, and effector functions at the early stages of T cell response(13–15). Despite the compelling promise, agonist antibodies targeting PD-1 in autoimmune diseases still require further steps in clinical trials. This is mainly due to the complexity of immune agonist development that should carefully take into consideration many factors including affinity, the choice of the isotype, and toxicity(16,17).
The role of immune checkpoints in the immunopathology of asthma has been an underrepresented area of investigation during the last decades when compared to cancer and autoimmune diseases. At the therapeutic level, the development of alternative therapies for neutrophilic asthma mainly focuses on interleukin antagonists to the detriment of other targets. However, strategies targeting neutrophil chemotaxis have failed to improve lung functions and to decrease inflammation, as is the example for interleukin- (IL-)17 receptor and CXCR2 antagonists(18,19). In contrast, strong evidence supports considering immune checkpoints like PD-1 as new potential targets. Data from asthma patients have demonstrated the expression of PD-1 on CD4+ T lymphocytes and established a negative correlation between PD-1 expression and immune activation(20–22). Moreover, PDCD1 polymorphism was associated with an increased risk of allergic bronchial asthma development in certain populations(23,24). In murine preclinical models, different studies have implicated the PD-1 axis in the regulation of allergic asthma (25–27). Recent works by our lab and others have demonstrated the efficacy of PD-1 agonists in modulating T cell and type-2 innate lymphoid cell (ILC2) effector functions(28–30). However, the effect of PD-1 agonistic activation in steroid-refractory asthma has yet to be explored.
Herein, we investigated the role of PD-1 in an established mouse model of steroid-resistant neutrophilic asthma(31). Interestingly, we have demonstrated that neutrophilic lung inflammation is associated with PD-1 induction mainly on T helper cells, and PD-1 deficiency led to exacerbated AHR and lung inflammation. The induction and the protective role of PD-1 in this inflammatory context make it a promising target. To validate this hypothesis, we induced neutrophil-driven lung inflammation in a preclinical PD-1 humanized mouse model and tested the therapeutic potential of a novel human PD-1 agonist. Strikingly, agonist-treated mice showed controlled AHR, inflammatory cell recruitment, and cytokine production, as compared to the control group. Moreover, single-cell RNA sequencing (scRNAseq) and flow cytometry data underlined the capacity of our PD-1 agonist to downregulate effector T cells (Teff) in the lungs. Altogether, this study provides new insights regarding a PD-1-targeted innovative approach for the treatment of neutrophilic asthma.
Methods
Mice
Wild-type (WT) BALB/cByJ mice were purchased from Jackson Laboratory (Bar Harbor, ME). PD-1-knockout (KO) BALB/c mice were generated in the Sharpe laboratory(32). Humanized PD-1 mice on a C57BL/6 background were purchased from GenOway (Lyon, France) and distributed by Charles River laboratories. Eight to ten-week-old aged and sexed-matched mice were used in the study. All experimentation protocols were approved by the USC Institutional Animal Care and Use Committee and conducted in accordance with the principles of the Declaration of Helsinki.
Induction of acute neutrophilic lung inflammation
A well-established model of mice acute neutrophilic asthma was used with some modifications(31). Mice were sensitized via a subcutaneous (s.c.) tail base injection of house dust mite (HDM) mixed in Complete Freund’s Adjuvant (CFA) (1:1 v/v, 200 μg of HDM). After 13 days, mice were challenged with one intranasal (i.n.) dose of HDM (100 μg). Control mice only received PBS. In some experiments, mice were treated via the intraperitoneal route (i.p.) with Dexamethasone (1mg/Kg, Sigma Aldrich). On day 14, lung function was evaluated by direct measurement of lung resistance and dynamic compliance (cDyn) in restrained, tracheostomized, and mechanically ventilated mice using the FinePointe RC system (Buxco Research Systems) under general anesthesia(33–35). Mice were sequentially challenged with aerosolized PBS (baseline), followed by increasing doses of methacholine ranging from 5 to 40 mg.mL−1. Maximum lung resistance and minimum compliance values were recorded during a 3 min period after each methacholine challenge. AHR data were analyzed by repeated measurements of a general linear model. When indicated, hPD-1 mice received 2 doses of PD-1 agonist (Janssen Pharmaceuticals) or the corresponding isotype: intraperitoneal injection of 500 μg on day 13 and intravenous (i.v.) injection of 250 μg on day 14. For these experiments, AHR was assessed on day 15. The PD-1 agonist has been fully characterized and tested in vivo and in vitro(28,29). Briefly, antibodies were generated in mice that were immunized with the extracellular domain of human PD-1 conjugated to fragment crystallizable region (Fc). B cells were isolated from the spleen to generate hybridomas. The hybridomas were screened by ELISA for binding to recombinant PD -1 (isotype IgG2a). The selected antibody binds to human PD-1 with an equilibrium dissociation constant (KD) of 5 × 10−8 M; an association constant (ka) of about 3 × 104 l.Ms−1 and a dissociation constant (kd) of about 3 × 10−3 l.s−1.
Tissue processing and flow cytometry
After measurements of AHR, lungs were injected with 3 mL of ice-cold PBS to collect bronchoalveolar lavage (BAL) cells. Lung tissue was then cut into small pieces and incubated in type IV collagenase (1.6 mg.ml−1; Worthington Biochemicals) at 37°C for 60 minutes(36). Single-cell suspensions were obtained by passing the lung tissue digest through a 70 μm cell strainer. Spleens were also processed through a 70 μm cell strainer using the flat end of syringe plunger. Red blood cells were lysed in all cell suspensions using RBC Lysis Buffer from BioLegend. The following antibodies were used to characterize different immune populations: BV421 anti-human PD-1 (EH12.2H7), APC-Cy7 anti-mouse CD3 (145-2C11), APC anti-mouse CD4 (GK1.5), BV510 anti-mouse CD25 (PC61), PE-Cy7 anti-mouse CD44 (IM7), BV650 anti-mouse CD45 (30-F11), APC anti-mouse Siglec F (S17007L), BV421 anti-mouse PD-1 (29F.1A12), PercP-Cy5.5 anti-mouse CD11c (N418), PE anti-mouse CD64 (X54-5/7.1), BV785 anti-mouse Ly6G (1A8), APC-Cy7 anti-mouse MHC II (M5/114.15.2), PE-Cy7 anti-mouse PD-L2 (TY25), BV711 anti-mouse PD-L1 (10F.9G2), PE/Dazzle anti-mouse/human CD11b (M1/70), FITC anti-mouse CD19 (6D5) (all from BioLegend); FITC anti-mouse CD8a (53-6.7), PerCP-Cy5.5 anti-mouse CD62L (MEL-14) (both from eBioscience); BV510 anti-mouse CD3 (M1/69) (from BD Biosciences) and PE anti-mouse FOXP3 (FJK-16s) (from Invitrogen). Intranuclear staining was performed using the FOXP3 Transcription Factor Staining Kit (ThermoFisher Scientific). CountBright Absolute Count Beads were used to count lung immune cells (Invitrogen). The acquisition was performed on BD FACSCanto II and FACSARIA III system (BD Biosciences) using the BD FACSDiva software v8.0.1. Data were analyzed with FlowJo software (TreeStar) version 10. To prepare tissue lysates, lungs were mechanically homogenized in 500 μL of Triton X-100 lysis buffer (0.5% Triton X-100, 150 mmol/L NaCl, 15 mmol/L Tris, 1 mmol/L CaCl2, and 1 mmol/L MgCl2), as previously described(37). The lysates were then centrifuged for 20 minutes at 10,000g to collect the supernatants.
Cytokine measurement
The levels of IFN-γ, MCP-1, TNF-α, CXCL-10, IL-4, IL-5, IL-6, IL-10, IL-13, and GM-CSF were measured in the BAL or in lung lysates using Legendplex multiplex kits (BioLegend) and data were analyzed via the LEGENDplex data analysis software v8.0. IL-17A and G-CSF were quantified in the BAL and in lung lysates using the ELISA MAX Deluxe Set Mouse IL-17A kit (BioLegend) and the G-CSF (CSF3) Mouse ELISA Kit (Invitrogen), respectively.
scRNA-Seq
Sorted live CD45+ cells (viability ≥95%) were stained using TotalSeq™-A antibodies and cell hashing reagents. Three samples from each group were mixed using the cell hashing technique (~50,000 cells/group). Each sample was initially stained with a monoclonal antibody conjugated with a short DNA oligo carrying a sequence barcode and tagging samples in the final suspension (TotalSeq-A0301 anti-mouse Hashtag 1 Antibody, TotalSeq-A0302 anti-mouse Hashtag 2 Antibody, TotalSeq-A0303 anti-mouse Hashtag 3 Antibody). After cell labeling, 10x Genomics single cell 3’ v3 assay (Chromium Next GEM Single Cell 3ʹReagent Kits v3.1), Antibody derived tag (ADT) and mRNA library preparation was performed according to the manufacturer’s instructions (BioLegend and 10X Genomics). The pooled library was loaded and sequenced on an Illumina HiSeq X Ten System (150bp × 8bp × 150bp). Single-cell sequencing data were processed and analyzed using Partek® Flow® software (v10.0.21.1116), where read pairs were first trimmed, according to the 10x Genomics Chromium Single Cell 3’ v3 prep kit used, to extract barcode and UMI information. Trimmed reads were then aligned to the MM10 genome using STAR v2.7.3a aligner, UMIs deduplicated, barcodes filtered, and then quantified against the Ensembl v98 transcripts annotation. Single-cell QA/QC was performed filtering out cells with alignment counts 600–15000, detected features 500–4000, mitochondrial counts 0–10, and minimum ribosomal reads percent > 0.13. The average total count after filtering was approximately 15,000 cells per sample. Read counts were normalized by first calculating counts per million (CPM), adding 1, then performing a log2 transformation, and genes not expressed in 99.9% of cells were excluded. The normalized count tables were exported from Partek Flow and used for annotation of cell classifications using SingleR, after which the resultant cell annotations were re-imported back into Partek Flow. PCA and tSNE was applied to reduce the dimensionality of the single cell normalized count data, and Partek Flow’s Data Viewer visualization tools were subsequently used to generate the various tSNE, bubble heatmap, gene expression, and dot plots used in the figures. Gene ontology (GO) enrichment analysis was performed using Enrichr(38). scRNA-seq data from this study have been deposited in Genbank with the primary accession code GSE210349.
Histology
When indicated, lungs were collected for histology and stored in paraformaldehyde 4% buffered in PBS. Lungs were embedded in paraffin, cut into 4-μm sections, and stained with hematoxylin and eosin (H&E) or alcian blue/periodic acid–13chiff (AB-PAS) according to standard protocols. Images were captured on a Keyence BZ-X700 using a 20x objective.
Statistical analysis
A two-tailed student t-test for unpaired data was applied for comparisons between 2 groups. Data were analyzed with Prism Software (GraphPad Software Inc.). Error bars represent standard error of the mean. P value < 0.05 was considered to denote statistical significance (*P < .05, **P < .01, ***P < .001, n.s. non-significant).
Results
PD-1 is expressed and induced on T cells in acute neutrophilic lung inflammation
The study of PD-1 in autoimmune and allergic diseases is recently gaining more interest for therapeutic purposes. We have recently explored the protective role of PD-1 in eosinophilic asthma mouse models induced by either the alarmin IL-33 or the fungal allergen Alternaria alternata(28). However, the induction of PD-1 in a mouse model of neutrophilic lung inflammation is yet to be investigated. For this purpose, we used here an established mouse model of acute neutrophilic asthma resistant to corticosteroids (Figure E1) and induced by HDM extracts(31), a common clinically relevant allergen. On day 0, BALB/c wild-type (WT) mice received a subcutaneous injection of HDM (200μg) mixed with CFA, followed by an intranasal injection of HDM (100μg) after 13 days (Fig. 1A). We first characterized the model by measuring lung resistance and inflammatory cell recruitment. On day 14, lung function was assessed before euthanasia by direct measurement of lung resistance in anesthetized tracheostomized mice using the FinePointe RC system (Buxco Research Systems), followed by BAL and lung collection. HDM-treated mice displayed 2 to 3-fold higher lung resistance in response to increasing concentrations of methacholine as compared to PBS control mice (Fig. 1B), suggesting the development of AHR. Additionally, immune CD45+ cells were highly recruited to the airway lumen as revealed by flow cytometry analysis in the BAL (Fig. 1C). Although these cells included alveolar macrophages, recruited T and B lymphocytes as well as eosinophils, neutrophils were the major recruited cells and represented about 80% of the recruited immune cells in the BAL (Fig. 1D–G, Figure E2A). To assess the role of PD-1 in this model, we identified the principal PD-1+ cells in the lungs of control and HDM-treated mice. T cells constituted around 70% of PD-1+ cells, followed by B cells that represented about 15%, while innate cells including macrophages, DCs, and neutrophils were weakly represented (0.5 to 2.5%). Interestingly, the percentage of T cells, macrophages, and DCs among PD-1+ cells increased in HDM-treated mice (Fig. 1H, Figure E2B). As T cells represented the major population, we further assessed the induction of PD-1 in the total CD3+ population. As shown in Figure 1I, PD-1 expression was highly induced on lung T cells in response to HDM and the percentage of PD-1+ T cells significantly increased from 10 to approximately 30%. Within this positive population, CD4+ and CD8+ T cells represented around 80% and 20% respectively (Fig. 1J), suggesting that PD-1 induction mainly concerns T helper cells. Consistently, FOXP3+ regulatory T cells (Treg) and CD62L+ cells each represented around 15% of total PD-1+ CD4 T cells in HDM-challenged mice, while FOXP3− CD62L− cells represented around 70% (Fig. 1K). Interestingly, HDM challenge did not affect the percentage of Treg cells among total PD-1+ CD4 T cells, while the percentage of CD62L− significantly increased to the detriment of CD62L+ cells that mainly represent naive T cells. These results indicated therefore that PD-1 is mainly expressed and induced on CD4+ Teff cells in neutrophilic lung inflammation.
Figure 1: Neutrophilic inflammation is associated with PD-1 induction in lung immune cells.
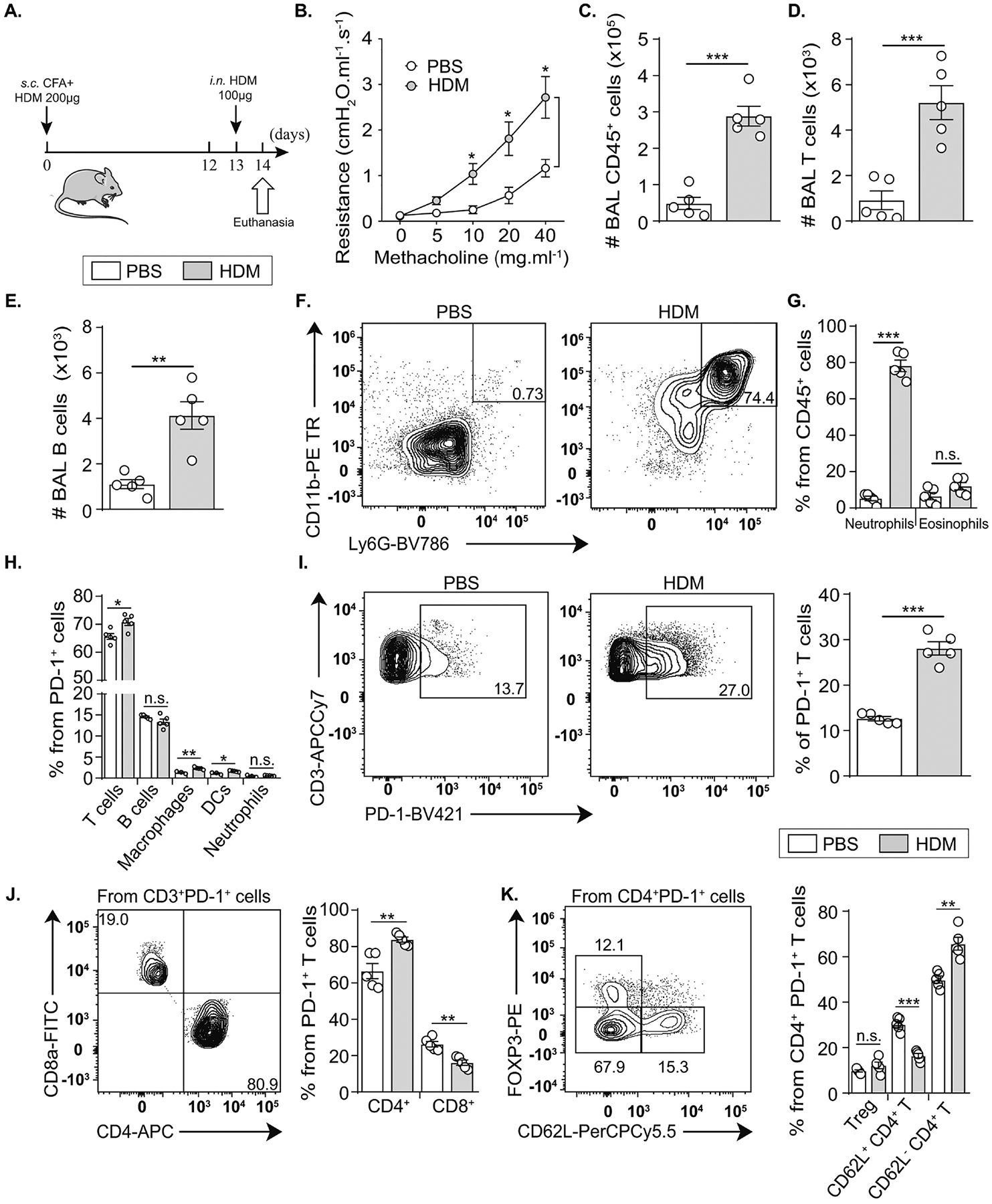
(A) BALB/cByJ mice (WT) mice were sensitized via subcutaneous (s.c.) tail base injection of HDM (200 μg) mixed in CFA (1:1 v/v). After 13 days, mice were intranasally (i.n.) challenged with HDM (100 μg). On day 14, lung resistance was measured before euthanasia, BAL collection and lung processing.
(B) Variations in lung resistance measured in restrained ventilated mice.
(C) Total number of CD45+ cells, (D) CD3+ T lymphocytes and (E) B lymphocytes.
(F) Representative flow cytometry plots of neutrophils and (G) the quantification of lung neutrophils and eosinophils presented as the percentage among CD45+ cells.
(H) Composition of PD-1+ cells in the lung suspensions.
(I) Representative flow cytometry plots of PD-1 induction on CD3+ CD45+ cells and the corresponding quantification presented as the percentage of PD-1+ T cells in the lungs.
(J) Comparison between the percentage of CD4+ and CD8+ T cells expressing PD-1 in the lungs of HDM-treated mice within PD-1+ T cells.
(K) Composition of PD-1+ CD4+ T cells according to FOXP3 and CD62L markers.
Data are representative of at least 2 independent experiments and are presented as means ± SEM (two-tailed Student’s t-test; n=5).
Lack of PD-1 induces severe neutrophilic lung inflammation
To elucidate the capacity of PD-1 to regulate lung neutrophilic inflammation, we introduced a PD-1-knockout (KO) mouse on BALB/c background. WT and PD-1 KO mice were exposed to the experimental protocol detailed in Figure 1A. On day 14, lung function was assessed by direct measurement of lung resistance and cDyn in anesthetized tracheostomized WT and PD-1 KO mice. Interestingly, PD-1 KO mice developed a higher lung resistance and lower cDyn as compared to WT mice (Fig. 2A, B). This indicates that PD-1 deficiency is associated with an increased AHR. We also collected the BAL to investigate the role of PD-1 in the regulation of inflammatory cell recruitment to the lungs. In agreement with AHR results, PD-1 KO mice had more CD45+ cells in the BAL than WT mice, mainly due to a significantly increased recruitment of neutrophils and T cells (Fig. 2C–E). Moreover, the cell count of B cells, eosinophils, and alveolar macrophages indicated a non-significant trend in the BAL from PD-1 KO mice (Fig. 2F–H; Figure E2A). Of note, no statistical differences were observed between WT and PD-1 KO mice receiving PBS. Cytokines in the BAL were also quantified to assess the activation level of recruited inflammatory cells. We detected significantly higher production of several pro-inflammatory mediators including tumor necrosis factor-alpha (TNF-α), IL-17A, interferon-gamma (IFN-γ), and IL-6 in the BAL of PD-1 KO mice when compared to WT mice (Fig. 2I–L; Figure E3). Also, a more potent secretion of three neutrophil recruiting mediators: monocyte chemoattractant protein-1 (MCP-1), CXCL-10, and granulocyte colony-stimulating factor (G-CSF) was detected in PD-1 KO mice (Fig. 2M–O). In parallel, IL-4 levels showed a non-significant trend toward reduced production in PD-1 KO mice (Fig. 2P), suggesting a potential inhibitory effect of this cytokine on neutrophil recruitment and function in our model(39). Histological analysis also revealed an increased thickness of airway epithelium as well as increased inflammatory cell recruitment in the lung sections from PD-1 KO mice as compared to WT mice (Fig. 2Q). Altogether, these results demonstrated that PD-1 downregulates AHR and controls neutrophilic lung inflammation.
Figure 2: PD-1 deficiency increases neutrophilic AHR and lung inflammation.
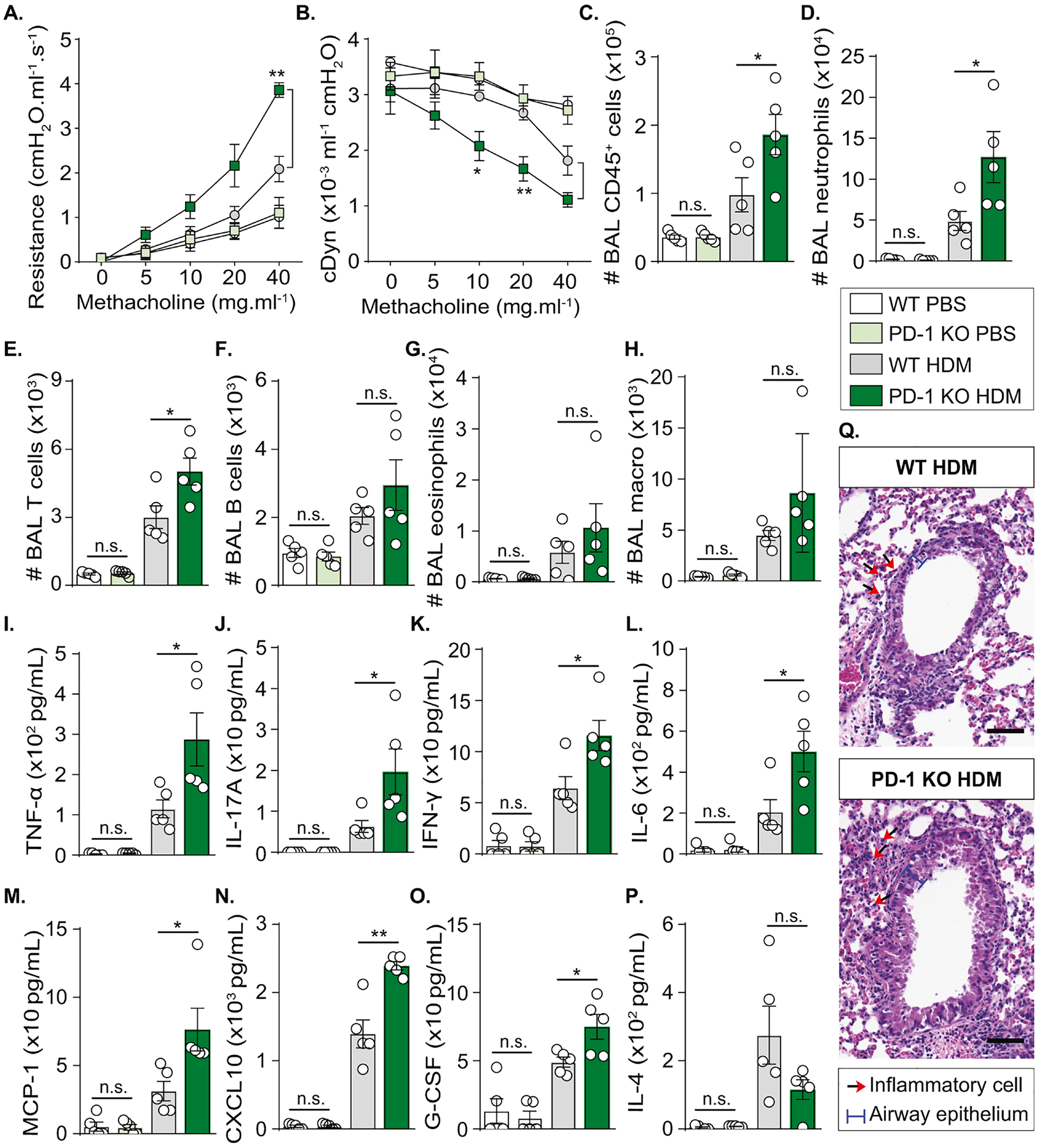
WT and PD-1 KO mice were sensitized via s.c. tail base injection of HDM (200 μg) mixed in CFA (1:1 v/v). After 13 days, mice were i.n. challenged with HDM (100 μg). Control mice received PBS only. On day 14, lung functions were measured before euthanasia and BAL collection.
(A) Lung resistance and (B) dynamic compliance measured in tracheostomized mechanically ventilated mice exposed to increasing concentrations of methacholine.
(C) Absolute count of CD45+ cells, (D) neutrophils, (E) T cells, (F) B cells, (G) eosinophils, and (H) alveolar macrophages quantified in the BAL using the count precision beads in flow cytometry analysis.
(I) Levels of TNF-α, (J) IL-17A, (K) IFN-γ, (L) IL-6, (M) MCP-1, (N) CXCL-10, (O) G-CSF, and (P) IL-4 quantified in the BAL.
(Q) Hematoxylin and eosin (H&E) staining of lung sections (scale bar = 20 μm).
Data are representative of at least 2 independent experiments and are presented as means ± SEM (two-tailed Student’s t-test; n=5).
PD-1 is induced in humanized PD-1 mice during neutrophilic lung inflammation
Since our data suggested a protective role for PD-1 in neutrophilic lung inflammation, we wanted to elucidate the benefits of targeting PD-1 in a translational approach. Human PD-1 agonists were recently developed, fully characterized, and successfully tested in a humanized mice model(28,29). To elucidate the therapeutic potential of a human PD-1 agonist in acute neutrophilic asthma model, we used a preclinical humanized PD-1 (hPD-1) mouse in which the mouse extracellular domain of PD-1 was replaced by the human equivalent (Fig. 3A). Before applying a treatment strategy, it was first crucial to confirm that the genetic manipulation of Pdcd1 gene did not affect the expression and induction of PD-1 in our experimental inflammatory context (Fig. 3B). An anti-human PD-1 antibody was used in flow cytometry analysis and revealed similar basal expression of PD-1 in the lungs of hPD-1 mice, as compared to the level of murine PD-1 expression in WT mice. The induction of PD-1 in acute neutrophilic inflammation was also comparable, revealing an increase from 1 to 2–2.5% among total CD45+ cells (Fig. 3C, D; Figure E4). As T lymphocytes represented the major PD-1+ population, we checked the induction of PD-1 on T cells in lungs and spleens of hPD-1 mice. As expected, human PD-1 was significantly induced on lung T cells following HDM challenge (Fig. 3E, F), while the expression remained unchanged in the spleen (Fig. 3G). This suggests that a systemic therapy targeting PD-1 may specifically affect lung T cells with minor effects on other organs. To investigate other aspects related to PD-1 axis engagement, we also assessed the expression of PD-1 ligands, PD-L1 and PD-L2, on CD45+ cells. Only PD-L1 was expressed on immune cells at a steady state. However, HDM challenge increased the percentage of PD-L1+ PD-L2− population from 30 to 40% and induced the expression of PD-L2 resulting in a new PD-L1 PD-L2 double positive population (about 7%) (Fig. 3H–J). Taken together, these results supported the use of hPD-1 mouse as a relevant preclinical model for neutrophilic asthma and paved the way to investigate the therapeutic potential of a human PD-1 agonist in this inflammatory context.
Figure 3: PD-1 axis is comparably induced in humanized PD-1 mice and WT mice following HDM challenge.
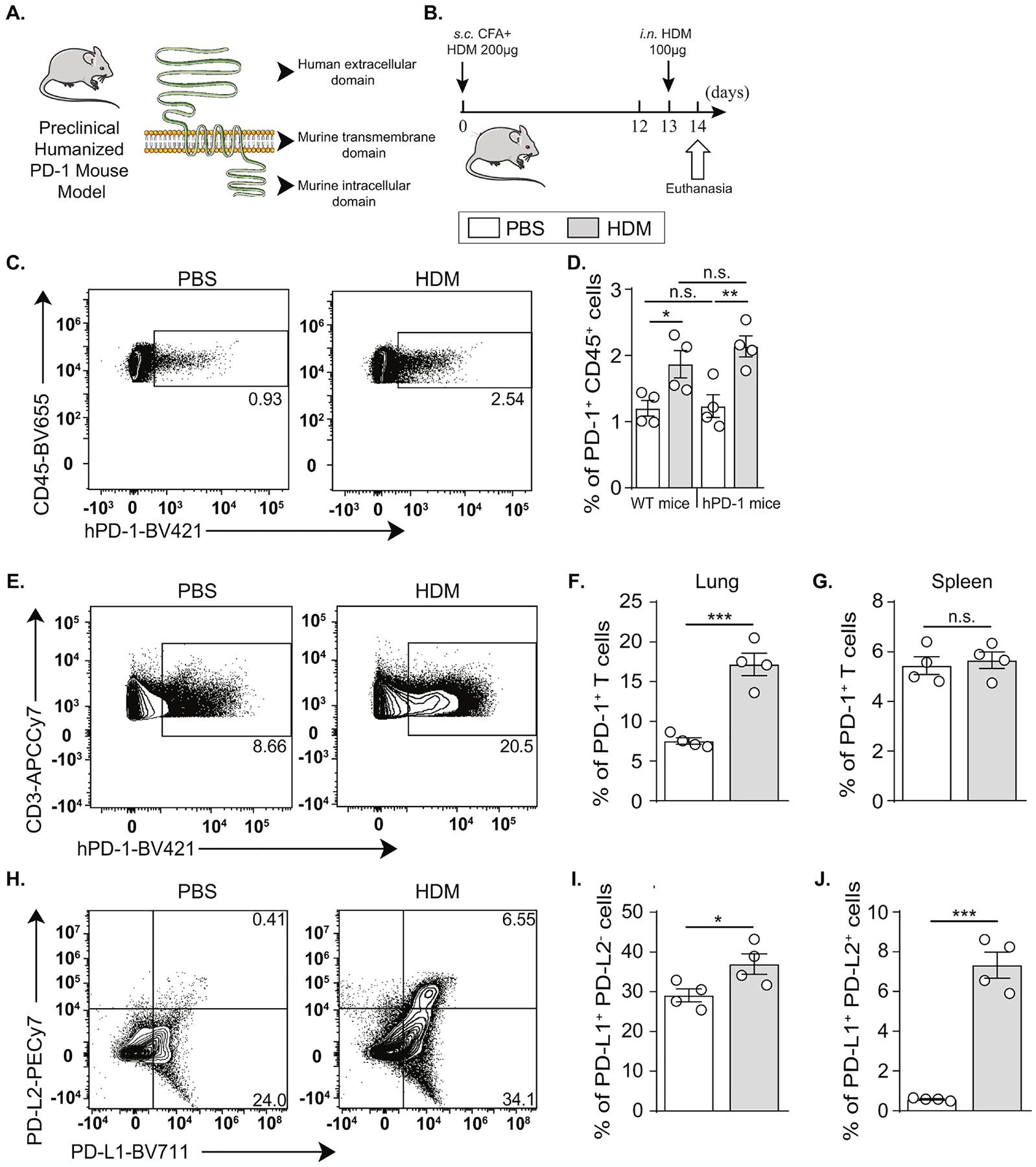
(A) Representation of extracellular, transmembrane, and intracellular domains of PD-1 in hPD-1 mouse.
(B)hPD-1 mice were sensitized via s.c. tail base injection of HDM (200 μg) mixed in CFA (1:1 v/v). After 13 days, mice were i.n. challenged with HDM (100 μg). On day 14, lung and spleen processing followed euthanasia.
(C) Representative flow cytometry plots of human PD-1 induction on CD45+ cells from hPD-1 mice.
(D) PD-1+ CD45+ cell percentage in the lungs of WT and hPD-1 mice following HDM challenge.
(E) Representative flow cytometry plots of human PD-1 induction on CD3+ cells and (F) the corresponding quantification presented as the percentage of PD-1+ T cells in the lungs of hPD-1 mice.
(G) Percentage of PD-1+ T cells in the spleens of hPD-1 mice.
(H) Representative flow cytometry plots of PD-L1 and PD-L2 induction on CD45+ cells and
(I) the corresponding quantification presented as the percentage of PD-L1+ PD-L2− cells and (J) PD-L1+ PD-L2+ cells in the lungs of hPD-1 mice.
Data are representative of at least 2 independent experiments and are presented as means ± SEM (two-tailed Student’s t-test; n=4).
PD-1 agonist treatment dampens AHR and ameliorates neutrophilic lung inflammation
First, we studied the impact of the PD-1 agonist on lung function and inflammatory burden, in order to investigate its therapeutic potential. hPD-1 mice received the PD-1 agonist or the corresponding isotype control along with HDM challenge as shown in Figure 4A, while naïve mice only received PBS and were not challenged or treated. Assessment of lung resistance on day 15 in anesthetized tracheostomized mice revealed that mice treated with PD-1 agonist have a decreased lung resistance in comparison with isotype-treated mice (Fig. 4B). Consistently, BAL analysis showed a significant decrease in CD45+ cell count (Fig. 4C). In particular, PD-1 agonist treatment decreased neutrophil, T cell, and alveolar macrophage count significantly, but not eosinophils (Fig. 4D–G). We also measured several cytokines in the BAL and observed a marked decrease in IFN-γ, IL-6, and TNF-α levels (Fig. 4H–J). The levels of Th2 cytokines including IL-4, IL-5, and IL-13 were as well decreased in the BAL of PD-1 agonist-treated mice as compared to isotype-treated mice (Figure E5A–C). Importantly, the secretion of IL-17, known as a potent inducer of neutrophil recruitment, was significantly reduced in the BAL and lung lysates of PD-1 agonist-treated mice (Fig. 4K; Figure E5D), in association with a significantly reduced production of MCP-1 and CXCL-10 (Fig. 4L, M). However, PD-1 agonist treatment did not affect the production of G-CSF in the BAL and in lung lysates, nor the production of granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-10 (Fig. 4N; Figure E5E–G). Last, histological analysis revealed the capacity of PD-1 agonist to decrease airway epithelial thickness, inflammatory cell recruitment, and mucous production (Fig. 4O–Q). Altogether, these data indicated that PD-1 agonist treatment can successfully downregulate acute neutrophilic lung inflammation.
Figure 4: PD-1 agonist downregulates AHR and controls neutrophilic lung inflammation.
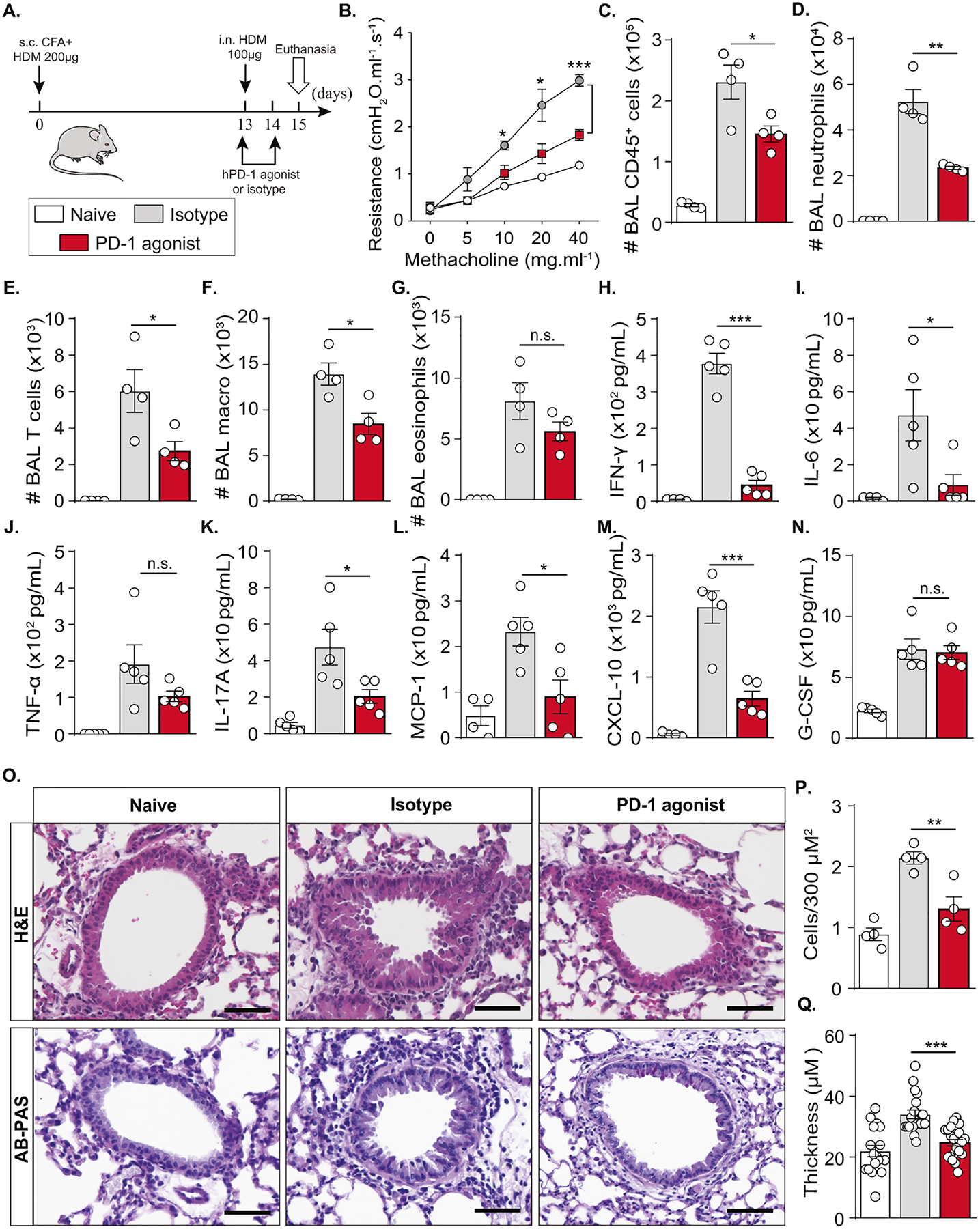
(A) Humanized PD-1 mice were sensitized via s.c. tail base injection of HDM (200 μg) mixed in CFA (1:1 v/v). After 13 days, mice were i.n. challenged with HDM (100 μg) and received 500 μg of PD-1 agonist via the intraperitoneal route or the corresponding isotype, while the dose was reduced to 250 μg via the intravenous route on day 14. Naïve mice were not sensitized, challenged, or treated. On day 15, lung function was measured. BAL and lungs were collected after euthanasia.
(B) Lung resistance measured in tracheostomized ventilated mice.
(C) Absolute count of CD45+ cells, (D) neutrophils, (E) T cells, (F) alveolar macrophages, and (G) eosinophils quantified in the BAL.
(H) Levels of IFN-γ, (I) IL-6, (J) TNF-α, (K) IL-17A, (L) MCP-1,(M) CXCL-10, and (N) G-CSF quantified in the BAL.
(O) Representative images of H&E and AB-PAS-stained histology sections (scale bar = 50 μm) with the corresponding quantifications of (P) recruited immune cells and (Q) airway epithelial thickness.
Data are representative of at least 2 independent experiments and are presented as means ± SEM (two-tailed Student’s t-test; n=4–5). Mouse image provided with permission from Servier Medical Art.
PD-1 agonist induces a dynamic modulation of lung immune cell transcriptome
We used single-cell RNA sequencing (scRNAseq) to better understand the effect of PD-1 agonist on immune responses in neutrophilic asthma(40,41). To obtain a high-dimensional analysis of the immune landscape following PD-1 agonist treatment, we performed scRNAseq using the 10× Genomics platform and analyzed the data using Partek® Flow® software, v10.0.21.1116. We compared the transcriptional profile of sorted live CD45+ cells from HDM-challenged mice, treated with PD-1 agonist or the corresponding isotype (Fig. 4A). The t-SNE plots in Figure 5A showed main immune cells that were annotated using SingleR(42). Interestingly, these t-SNE plots showed a considerable rearrangement in different immune populations, notably T and B cells. Moreover, the bubble heatmap in Figure 5B showed the effect of PD-1 agonist treatment on different cytokine axes in the identified immune populations. In particular, PD-1 agonist treatment was associated with a significant decrease in the expression of Cxcl5 in macrophages, Cxcl10 and Cxcl11 in neutrophils, macrophages and monocytes, and Il17rc in pro B cells and NKT cells. To investigate the direct effect of the agonist on PD-1+ lung cells, we generated t-SNE plots showing the distribution of Pdcd1+ cells (Fig. 5C). The comparison between isotype- and PD-1 agonist- treated groups revealed the lack of a sub-population that highly expresses Pdcd1 while other Pdcd1+ cell clusters were slightly or unaffected. This was confirmed in a quantitative graph showing the loss of a small population of cells that highly express Pdcd1>10 (Fig. 5D). Altogether, these transcriptomic data indicated that PD-1 agonist treatment does not systemically deplete PD-1+ cells but directly or indirectly shapes the transcriptomic signature of inflammatory cells that are implicated in the pathogenesis of neutrophilic lung inflammation.
Figure 5: scRNAseq analysis reveals the impact of PD-1 agonist treatment on the lung immune landscape.
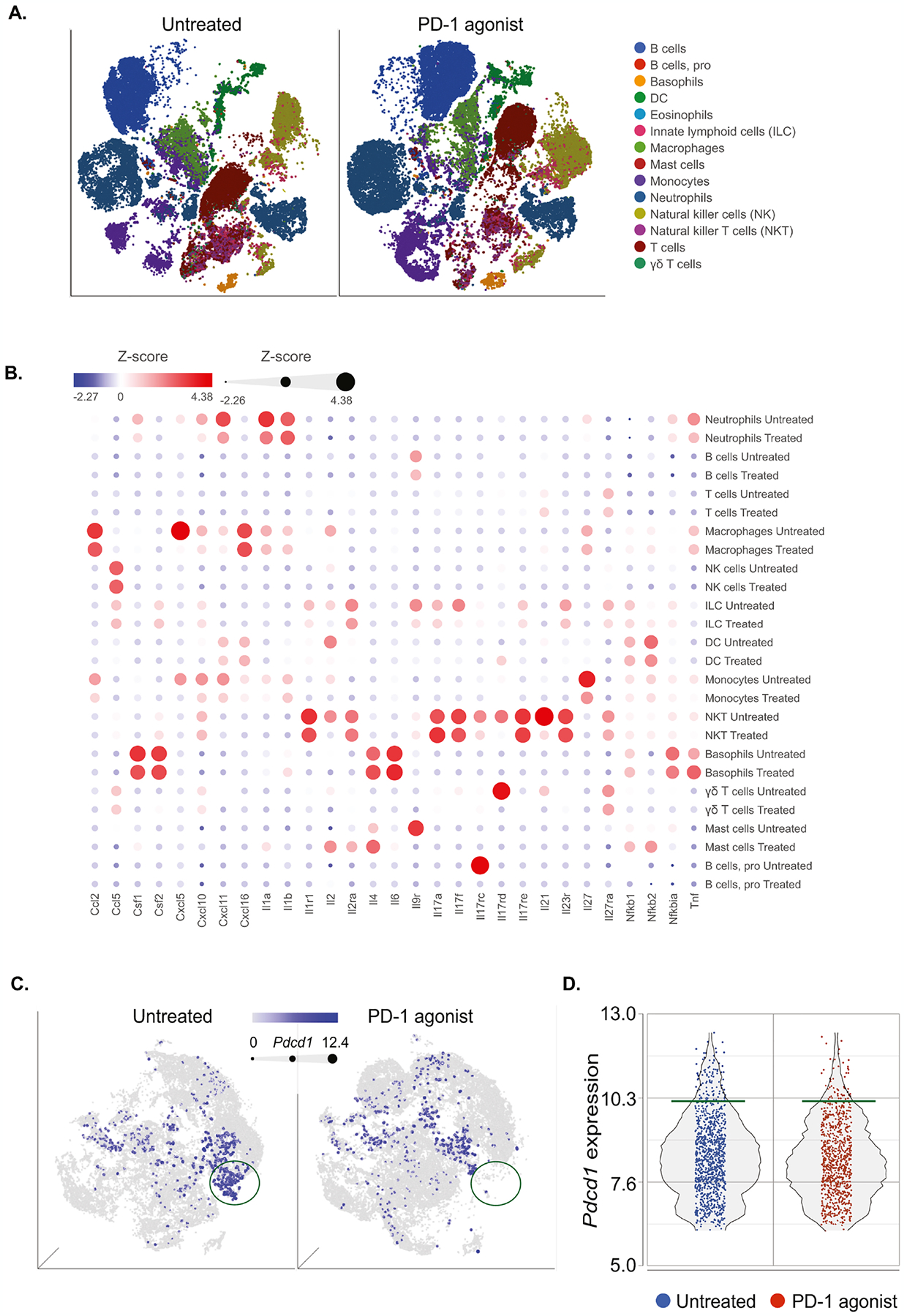
Single-cell RNA-seq was performed on sorted CD45+ cell suspensions pooled from 3 lungs per group (isotype- versus PD-1 agonist- treated mice). Samples were analyzed using Partek Flow genomic analysis software.
(A) tSNE plot (2D graph) revealing the effect of PD-1 agonist on main immune cell populations annotated using SingleR.
(B) Bubble heatmap showing the expression of relevant genes implicated in cytokine pathways.
(C) tSNE plot (3D graph) of CD45+ cells colored by Pdcd1 expression. The intensity of the blue color, as well as the dot size, are dependent on the level of Pdcd1 expression. Green circles represent a cluster of Pdcd1+ high cells.
(D) Dot plot showing the intensity of Pdcd1 expression in total CD45+ lung cells.
PD-1 agonist targets and reprograms Teff cells in the lungs
Having demonstrated that PD-1 is mainly expressed on CD4+T cells, we wanted to better understand the effect of PD-1 agonist on the distribution of main subsets within this immune population. In agreement with the BAL cellular analysis, we first demonstrated that PD-1 agonist treatment significantly decreases the number of CD45+ cells in the lungs and particularly reduces the number of neutrophils (Fig. 6A, B; Figure E6). Consistently, the count of T cells gated as CD3+ cells significantly decreased in the lungs of PD-1 agonist-treated mice and reached similar levels as naïve mice (Fig. 6C). To better understand the molecular mechanisms associated with this remarkable decrease in T cell number, we performed a gene ontology (GO) enrichment analysis on our scRNAseq data. Interestingly, pathways related to IFN responses, TNF-α signaling via NF-κB as well as IL-6/JAK/STAT3 and IL-2/STAT5 signaling pathways were considerably affected, suggesting an important impact of PD-1 agonist treatment on T cell effector functions (Fig. 6D).
Figure 6: PD-1 agonist treatment downregulates the effector phenotype of lung T cells.
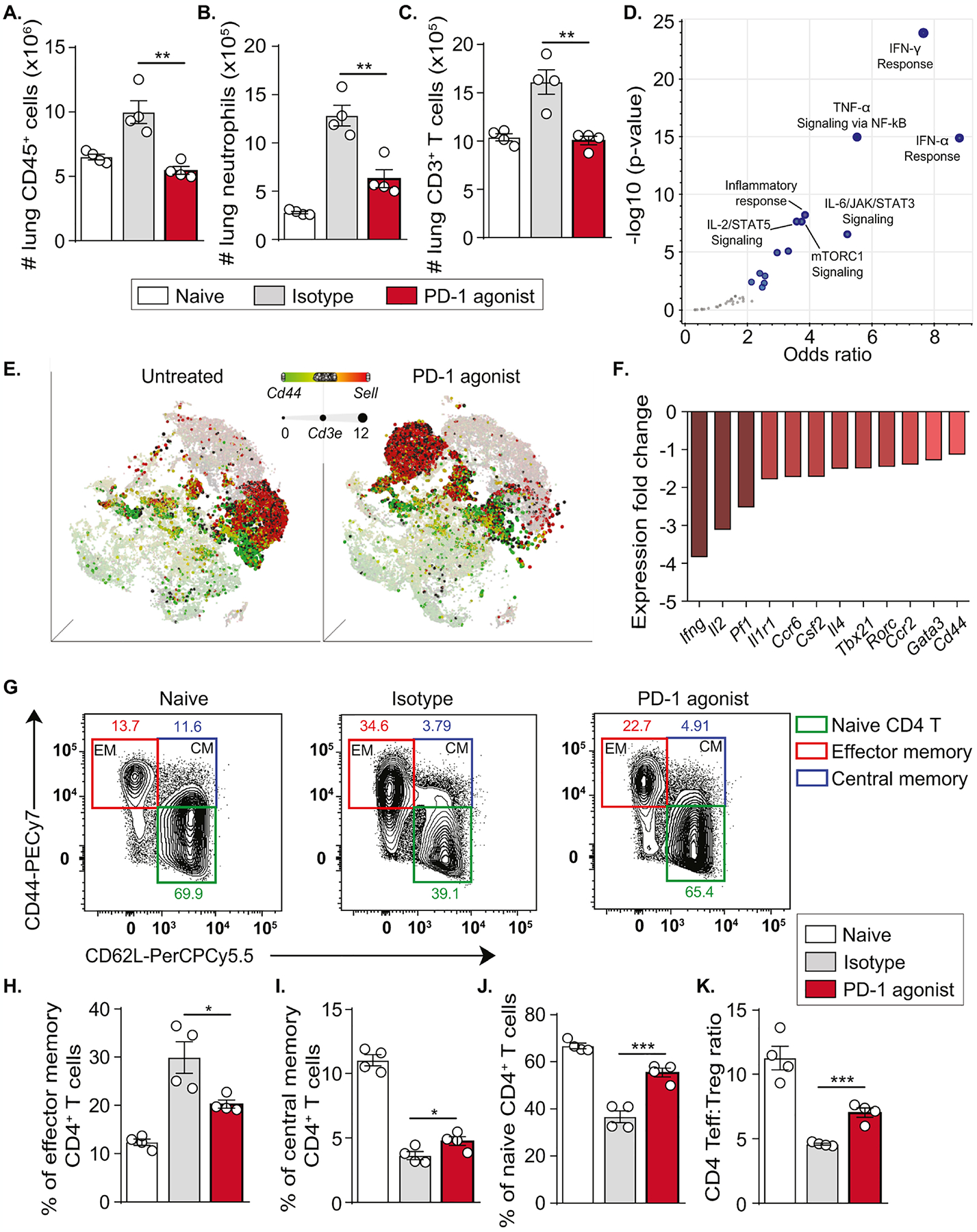
Following the induction of neutrophilic lung inflammation, humanized PD-1 mice received two injections of the PD-1 agonist or the corresponding isotype, as previously described. On day 15, lungs were washed and collected after euthanasia.
(A) Absolute count of CD45+ cells, (B) neutrophils, (C) CD3+ T cells.
(D) Volcano plot showing significantly affected pathways in lung immune cells from PD-1 agonist-treated mice as compared to isotype-treated mice (MSigDB_Hallmark_2020 gene set).
(E) tSNE plot representation based on the expression of Cd44 and Sell genes in lung immune cells. The size of dots is dependent on the level of Cd3e expression.
(F) Expression of genes related to the T effector phenotype expressed as fold change (PD-1 agonist vs isotype group).
(G) Representative flow cytometry plots showing the gating strategy for the identification of CD4+ T cells subsets according to CD44 and CD62L expression.
(H) Percentages of effector memory CD4+ T cells, (I) central memory CD4+ T cells, (J) naïve CD4+ T cells, and (K) Teff:Treg ratio in the lungs.
Data are representative of at least 2 independent experiments and are presented as means ± SEM (two-tailed Student’s t-test; n=4).
In mice, CD44 and CD62L (L-selectin) could be used to classify T cells into naïve, effector memory, and central memory subsets. We further analyzed the expression of the genes Cd44 and Sell, encoding CD44 and CD62L respectively, within Cd3e+ cells. Interestingly, the t-SNE plot revealed a major translocation of Sell+ cells in response to PD-1 agonist treatment, suggesting a remapping of naïve T cell transcriptional signature. Moreover, an important fraction of the Cd44+ T cell population that colocalized Pdcd1 high cells was missing in the lungs of PD-1 agonist-treated mice as compared to isotype-treated mice (Fig. 6E). Consistently, CD44+ CD4+ T cells displayed a significantly induced expression of PD-1 as compared to CD44− CD4+ T cells (Figure E7A–C). We next focused on the effector phenotype of CD4+ T cells that were shown to represent the majority of PD-1+ T cells (Fig. 1J). Interestingly, genes coding for key cytokines in CD4+ T cells including IFN-γ, IL-2, and IL-4, as well as genes coding for main regulatory transcription factors such as GATA-3, T-bet, ROR-γt and ROR-α were downregulated in PD-1 agonist-treated group in comparison with the isotype-treated group (Fig. 6F). This also supports previous GO analysis and suggested a downregulation of the identified inflammatory pathways in response to PD-1 agonist treatment. To confirm our findings at the protein level and assess the effect of PD-1 agonist on T cell subset distribution, we used multiparametric flow cytometry. Effector memory (EM) CD4+ T cells, central memory (CM) CD4+ T cells, and naïve CD4+ T cells were gated based on the expression of CD44 and CD62L (Fig. 6G). PD-1 agonist treatment decreased the percentage of EM T cells compared to the isotype control and slightly increased the percentage of CM T cells (Fig. 6H, I). Consistently, the percentage of naïve CD4+ T cells was significantly increased in PD-1 agonist treated group similar to the levels of the naive unchallenged group (Fig. 6J). Last, we evaluated the effect of PD-1 agonism on the Teff:Treg ratio which provides a unique parameter to evaluate immunomodulation. Total effector CD4+ T cells were gated as CD4+ CD25− FOXP3− cells while Treg cells were gated as CD4+ CD25+ FOXP3+ cells (Figure E7D). Interestingly, isotype-treated mice display a skewed Teff:Treg ratio while PD-1 agonist treatment significantly increases this ratio indicating a relevant shift in the proinflammatory/tolerogenic balance. Of note, the distribution of T cell subsets was not affected in the spleen, indicating a local effect for PD-1 agonist treatment (Figure E8). Taken together, these data provided solid evidence that agonistic activation of PD-1 shapes the balance of Teff cell activation in neutrophilic lung inflammation.
Discussion
During the last decade, many studies have investigated the role of PD-1 axis in the homeostasis of the immune system. These studies provided a strong rationale for targeting and modulating PD-1 axis in several immune disorders. Here, we characterized for the first time PD-1 implication in a clinically relevant mouse model of neutrophilic AHR that mimics the immunopathological features of human neutrophilic asthma. We have demonstrated that PD-1 is mainly inducible on CD4+ T cells while the lack of PD-1 leads to a significant exacerbation of AHR and lung inflammation. Based on a scRNAseq approach, our data in hPD-1 mice support the preclinical therapeutic potential of a PD-1 agonist in neutrophilic asthma.
The role of PD-1 in different asthma phenotypes is not yet completely clear. A few studies have suggested an impairment of the PD-1 axis in human asthma. In particular, correlations between low PD-1 expression on Teff cells and imbalanced immune responses were established in asthmatic patients(20,22). Among other key inhibitory checkpoints including CTLA-4 and LAG-3, the unique capacity of PD-1 to control allergen-specific human T cells was recently demonstrated(43). In parallel, we and others have previously demonstrated the effect of PD-1 axis impairment in murine models of allergic asthma(44,45,27). We have also recently described the inhibitory role of PD-1 and its capacity to regulate eosinophilic response in ILC2-dependent asthma models(28). Since asthma is a heterogenous disease, we wanted here to investigate the protective role of PD-1 in a more severe phenotype associated with neutrophilic inflammation(31). Interestingly, we first demonstrate in a mouse model of acute neutrophilic asthma that PD-1 is highly expressed and inducible on T lymphocytes, in particular on effector CD4+ T cells. In parallel, Treg cells represent a small percentage of PD-1+ CD4+ T cells that does not increase in our neutrophilic asthma model. At the functional level, we report using a PD-1 KO mouse that PD-1 deficiency leads to exacerbated AHR and inflammatory cell recruitment, notably high neutrophil infiltration. This exacerbated neutrophilic response was associated with high levels of key inflammatory cytokines including TNF-α, IL-6, IL-17 and IFN-γ(46,47), as well as high production of MCP-1, G-CSF, and CXCL-10 chemokines, known to recruit neutrophils to the lungs(48–51). These data support the notion that targeting PD-1 axis could be a promising strategy in severe and steroid resistant forms of asthma.
Although strong rationale supports the inhibitory role of PD-1 in different disorders related to impaired immune tolerance, the development of agonistic therapies is limited and clinical trials have not yet reached advanced phases(17). A few studies have recently demonstrated the downregulatory effect of PD-1 agonism on the immune response, notably on T cell activation(30,52,53). In parallel, new human PD-1 agonist antibodies were designed, fully characterized, and successfully underwent early phases of clinical trials in graft-versus-host disease(29). In this regard, we have recently tested one of these human PD-1 agonists using a humanized model of ILC2-dependent asthma, in which alymphoid mice were reconstituted with human ILC2s and treated with the selected agonist(28). Promising results provided supportive evidence for the use of this agonist against neutrophilic inflammation in relevant preclinical models. To achieve this goal, we used a fully immunocompetent humanized mouse that expresses an entirely humanized PD-1 extracellular domain. Importantly, we validated the use of this mouse in our inflammatory context and showed induced expression of PD-1 and PD-1 ligands upon exposure to the clinically relevant allergen, HDM. This study provides therefore a robust translational approach to test a well characterized human PD-1 agonist in a preclinical fully immunocompetent humanized mouse.
Management of neutrophilic asthma is a challenging task since patients become unresponsive to high doses of corticosteroids and to standard therapies(6,54). In particular, neutrophils are resistant to corticosteroids that exert limited downregulatory effects on steroid-sensitive cells, notably CD4+ T lymphocytes(55,56). Although several monoclonal antibodies targeting key interleukin axes were approved during the last decade to treat eosinophilic asthma, clinical trials investigating the benefits of these immunotherapies in neutrophilic asthma are yet to be conclusive. Several antibodies have already failed to provide evidence of effectiveness against severe asthma, including IL-6 and IL-17R antagonists(18,47,57). Therefore, a novel strategy targeting a potent inhibitory checkpoint like PD-1 could be of great interest. Based on relevant preclinical approach and cutting-edge techniques, this study reveals a significant therapeutic effect of PD-1 agonist against neutrophilic inflammation in asthma. Interestingly, PD-1 agonist treatment dampened AHR and resulted in a 2-to-3-fold decrease in the number of neutrophils in the BAL and lungs, as well as the number of macrophages and T cells. This was associated with a remarkable decrease in the production of key cytokines such as IFN-γ, IL-17, and CXLC-10, confirmed at both the transcriptional and the protein levels. Consistently, several studies have highlighted a prominent role of IL-17(58,59) and IFN-γ–CXCL-10 axis in steroid-resistant asthma in mouse models and human patients(54,57,60,61). In particular, increased IFN-γ response is linked to the disruption of airway epithelium and exacerbated AHR in severe type-2 low asthma (62,63). This suggests that a PD-1 agonist could directly reprogram Teff cells and disrupt fundamental crosstalk between lung inflammatory cells, leading to the inhibition of neutrophil recruitment.
The PD-1 axis is dominantly involved in the regulation of the crosstalk between T cells and antigen-presenting cells within immunological synapses(10,64). Although airway neutrophilia is the main feature of neutrophilic asthma, T cells and antigen-presenting cells have a central role in the pathogenesis of steroid-resistant asthma and shape neutrophil chemotactic recruitment(65,66). In our inflammatory model, PD-1 is mainly expressed on T cells and slightly on antigen-presenting cells, while neutrophils do not express PD-1, as expected. Therefore, PD-1 agonist treatment principally targets activated subsets of effector Th1/Th17 cells that promote neutrophil recruitment but also activated Th2 cells as revealed by cytokine profiling. However, Treg cells are not negatively affected, since PD-1 agonist treatment ameliorates the Teff:Treg ratio and does not decrease the levels of IL-10 in the lungs. Nonetheless, PD-1 agonist treatment failed in our experimental setup to decrease the levels of G-CSF, known to be mainly secreted in the lungs by epithelial cells(49), but not T cells. Altogether, the therapeutic activity of PD-1 agonist is tightly related to the reprogramming of activated Teff cells independently from their polarization.
It is known that the choice of isotype is a critical step in immune agonist development and defines the implication of antibody-dependent cellular cytotoxicity (ADCC) activity in the antibody’s mechanism of action. Unlike autoimmunity and allergies, depleting isotypes have a major drawback in cancer allowing the loss of the targeted T cells, while the treatment aims to promote their activation and proliferation(67). Indeed, the most promising agonist antibody targeting inducible T cell costimulator (ICOS) in cancer, has non-depleting properties(68). The PD-1 agonist that we used was not described as a non-depleting antibody, and our data do not show a systemic depletion of PD-1+ cells in the lungs. This suggests selective elimination of highly activated Teff cells in the lungs and reprogramming of T cell subsets based on the enrichment of naïve T cells, with no significant alteration of Treg cells and T cell repertoire in other organs. This decline in Teff cell number could be the key event leading to a state of recovery in neutrophilic asthma. Since T cell activation represents a common feature uniting different asthma types, our PD-1-based strategy could alleviate the challenging therapeutic task associated with asthma heterogeneity and the clustering of clinical phenotypes.
In conclusion, this study highlights the potential of PD-1 axis to inhibit neutrophilic lung inflammation in asthma via T cell reprogramming and suggests PD-1 agonist as a novel therapy in steroid-refractory asthma. Although the rationale is strong, immune agonist development is very challenging and requires relevant models for preclinical assessment of effectiveness and safety. Our experimental study is based on solid approaches for the evaluation of the therapeutic potential of PD-1 agonists in neutrophil-mediated allergic asthma and therefore provides an experimental model for other preclinical studies. Taken together, this study supports the notion of considering PD-1 agonistic activation as a new opportunity to achieve tremendous clinical goals in severe forms of asthma.
Supplementary Material
Clinical implication:
Our findings provide new insight into the utilization of PD-1 agonist as an effective therapy against a severe and refractory asthma phenotype that lacks efficient and targeted therapies.
Acknowledgments
We are grateful to USC Libraries Bioinformatics Service for assisting with data analysis, in particular Dr. Yibu Chen, Dr Yong-Hwee E Loh, and Meng Li. The bioinformatics software and computing resources used in the analysis are funded by the USC Office of Research and the Norris Medical Library. We thank Dr. Long Hung and Dr. Xiangming Ding from Single-Cell, Sequencing, and CyTOF Core (SC2) at Children’s Hospital Los Angeles for the technical support.
This article was financially supported by National Institutes of Health Public Health Service grants R01 HL144790, R01 HL151493, R01 AI145813, R01 HL151769 and R01 HL159804 (O.A.) and P01 AI56299 (A.H.S.).
Conflict of interest:
A.H.S. has patents/pending royalties on the PD-1 pathway from Roche and Novartis. P.S. is employee of Janssen R&D. O.A. receives grant support from the NIH and Janssen Pharmaceuticals. The rest of the authors declare that they have no relevant conflicts of interest.
Abbreviations
- AHR
Airway hyperreactivity
- BAL
Bronchoalveolar lavage
- CFA
Complete Freund’s adjuvant
- CM
Central memory
- EM
Effector memory
- G-CSF
Granulocyte colony-stimulating factor
- HDM
House dust mite
- i.n.
Intranasally
- IFN-γ
Interferon-gamma
- IL-
Interleukin-
- ILC2
Type-2 innate lymphoid cell
- KO
Knockout
- MCP-1
Monocyte chemoattractant protein-1
- PD-1
Programmed cell death protein-1
- s.c.
Subcutaneous
- scRNAseq
Single-cell RNA sequencing
- Teff
Effector T cells
- TNF-α
Tumor necrosis factor-alpha
- Treg
Regulatory T cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shilovskiy IP, Nikolskii AA, Kurbacheva OM, Khaitov MR. Modern View of Neutrophilic Asthma Molecular Mechanisms and Therapy. Biochem Biokhimiia. 2020. Aug;85(8):854–68. [DOI] [PubMed] [Google Scholar]
- 2.Copperbelt University, Syabbalo N. Clinical Features And Management Of Neutrophilic Asthma. Pulm Med Respir Res. 2020. Aug 19;6(1):1–15. [Google Scholar]
- 3.Papi A, Brightling C, Pedersen SE, Reddel HK. Asthma. The Lancet. 2018. Feb 24;391(10122):783–800. [DOI] [PubMed] [Google Scholar]
- 4.Pike KC, Levy ML, Moreiras J, Fleming L. Managing problematic severe asthma: beyond the guidelines. Arch Dis Child. 2018. Apr;103(4):392–7. [DOI] [PubMed] [Google Scholar]
- 5.Castillo JR, Peters SP, Busse WW. Asthma Exacerbations: Pathogenesis, Prevention, and Treatment. J Allergy Clin Immunol Pract. 2017. Aug;5(4):918–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nair P, Surette MG, Virchow JC. Neutrophilic asthma: misconception or misnomer? Lancet Respir Med. 2021. May 1;9(5):441–3. [DOI] [PubMed] [Google Scholar]
- 7.Nair P, Prabhavalkar KS. Neutrophilic Asthma and Potentially Related Target Therapies. Curr Drug Targets. 2020;21(4):374–88. [DOI] [PubMed] [Google Scholar]
- 8.Radermecker C, Louis R, Bureau F, Marichal T. Role of neutrophils in allergic asthma. Curr Opin Immunol. 2018. Oct;54:28–34. [DOI] [PubMed] [Google Scholar]
- 9.Shi B, Li W, Hao Y, Dong H, Cao W, Guo J, et al. Characteristics of inflammatory phenotypes among patients with asthma: relationships of blood count parameters with sputum cellular phenotypes. Allergy Asthma Clin Immunol. 2021. May 11;17(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. 2018. Mar;18(3):153–67. [DOI] [PubMed] [Google Scholar]
- 11.Pauken KE, Torchia JA, Chaudhri A, Sharpe AH, Freeman GJ. Emerging concepts in PD1 checkpoint biology. Semin Immunol. 2021. Feb 1;52:101480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong Y, Sun Q, Zhang X. PD-1 and its ligands are important immune checkpoints in cancer. Oncotarget. 2016. Dec 10;8(2):2171–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahn E, Araki K, Hashimoto M, Li W, Riley JL, Cheung J, et al. Role of PD-1 during effector CD8 T cell differentiation. Proc Natl Acad Sci. 2018. May 1;115(18):4749–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He X, Xu C. PD-1: A Driver or Passenger of T Cell Exhaustion? Mol Cell. 2020. Mar 5;77(5):930–1. [DOI] [PubMed] [Google Scholar]
- 15.Shimizu K, Sugiura D, Okazaki I mi, Maruhashi T, Takegami Y, Cheng C, et al. PD-1 Imposes Qualitative Control of Cellular Transcriptomes in Response to T Cell Activation. Mol Cell. 2020. Mar 5;77(5):937–950.e6. [DOI] [PubMed] [Google Scholar]
- 16.Garber K Immune agonist antibodies face critical test. Nat Rev Drug Discov. 2019. Dec 11;19(1):3–5. [DOI] [PubMed] [Google Scholar]
- 17.Grebinoski S, Vignali DA. Inhibitory receptor agonists: the future of autoimmune disease therapeutics? Curr Opin Immunol. 2020. Dec 1;67:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Busse WW, Holgate S, Kerwin E, Chon Y, Feng J, Lin J, et al. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med. 2013. Dec 1;188(11):1294–302. [DOI] [PubMed] [Google Scholar]
- 19.Nair P, Gaga M, Zervas E, Alagha K, Hargreave FE, O’Byrne PM, et al. Safety and efficacy of a CXCR2 antagonist in patients with severe asthma and sputum neutrophils: a randomized, placebo-controlled clinical trial. Clin Exp Allergy J Br Soc Allergy Clin Immunol. 2012. Jul;42(7):1097–103. [DOI] [PubMed] [Google Scholar]
- 20.Bratke K, Fritz L, Nokodian F, Geißler K, Garbe K, Lommatzsch M, et al. Differential regulation of PD-1 and its ligands in allergic asthma. Clin Exp Allergy J Br Soc Allergy Clin Immunol. 2017. Nov;47(11):1417–25. [DOI] [PubMed] [Google Scholar]
- 21.Mosayebian A, Koohini Z, Hossein-Nataj H, Abediankenari S, Abedi S, Asgarian-Omran H. Elevated Expression of Tim-3 and PD-1 Immune Checkpoint Receptors on T-CD4+ Lymphocytes of Patients with Asthma. Iran J Allergy Asthma Immunol. 2018. Dec 2;17(6):517–25. [PubMed] [Google Scholar]
- 22.Xi X, Liu JM, Guo JY. Correlation of PD-1/PD-L1 Signaling Pathway with Treg/Th17 Imbalance from Asthmatic Children. Int Arch Allergy Immunol. 2018;176(3–4):255–67. [DOI] [PubMed] [Google Scholar]
- 23.Dmitrieva-Zdorova EV, Gabaeva MV, Seregin YA, Bodoev NV, Voronko OE. PDCD1 PD-1.3 polymorphism and allergic bronchial asthma in Russian and Buryat patients. J Asthma Off J Assoc Care Asthma. 2017. Jan 2;54(1):46–52. [DOI] [PubMed] [Google Scholar]
- 24.James ES, Harney S, Wordsworth BP, Cookson WOCM, Davis SJ, Moffatt MF. PDCD1: a tissue-specific susceptibility locus for inherited inflammatory disorders. Genes Immun. 2005. Aug;6(5):430–7. [DOI] [PubMed] [Google Scholar]
- 25.Lewkowich I, Lajoie S, Stoffers S, Suzuki Y, Richgels P, Dienger K, et al. PD-L2 modulates asthma severity by directly decreasing dendritic cell IL-12 production. Mucosal Immunol. 2013. Jul;6(4):728–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh AK, Stock P, Akbari O. Role of PD-L1 and PD-L2 in allergic diseases and asthma. Allergy. 2011. Feb;66(2):155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akbari O, Stock P, Singh A, Lombardi V, Lee WL, Freeman G, et al. PD-L1 and PD-L2 modulate airway inflammation and iNKT-cell-dependent airway hyperreactivity in opposing directions. Mucosal Immunol. 2010. Jan;3(1):81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Helou DG, Shafiei-Jahani P, Lo R, Howard E, Hurrell BP, Galle-Treger L, et al. PD-1 pathway regulates ILC2 metabolism and PD-1 agonist treatment ameliorates airway hyperreactivity. Nat Commun. 2020. Aug 10;11(1):3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Q, Cole S, Duffy K, Gardner D, Guo Y, Hamel D, et al. Antibodies That Specifically Bind PD-1 and Methods of Use. 800/850 Ridgeview Drive, Horsham, Pennsylvania 19044; WO 2018/226580 A2. [Google Scholar]
- 30.Bryan CM, Rocklin GJ, Bick MJ, Ford A, Majri-Morrison S, Kroll AV, et al. Computational design of a synthetic PD-1 agonist. Proc Natl Acad Sci. 2021; 118(29). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ouyang S, Liu C, Xiao J, Chen X, Lui AC, Li X. Targeting IL-17A/glucocorticoid synergy to CSF3 expression in neutrophilic airway diseases. JCI Insight. 2020; 5(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keir ME, Freeman GJ, Sharpe AH. PD-1 Regulates Self-Reactive CD8+ T Cell Responses to Antigen in Lymph Nodes and Tissues. J Immunol. 2007. Oct 15;179(8):5064–70. [DOI] [PubMed] [Google Scholar]
- 33.Shafiei-Jahani P, Helou DG, Hurrell BP, Howard E, Quach C, Painter JD, et al. CD200–CD200R immune checkpoint engagement regulates ILC2 effector function and ameliorates lung inflammation in asthma. Nat Commun. 2021. May 5;12(1):2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Howard E, Lewis G, Galle-Treger L, Hurrell BP, Helou DG, Shafiei-Jahani P, et al. IL-10 production by ILC2s requires Blimp-1 and cMaf, modulates cellular metabolism and ameliorates airway hyperreactivity. J Allergy Clin Immunol. 2020. Sep 6; [DOI] [PubMed] [Google Scholar]
- 35.Hurrell BP, Galle-Treger L, Jahani PS, Howard E, Helou DG, Banie H, et al. TNFR2 Signaling Enhances ILC2 Survival, Function, and Induction of Airway Hyperreactivity. Cell Rep. 2019. Dec 24;29(13):4509–4524.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Helou DG, Shafiei-Jahani P, Hurrell BP, Painter JD, Quach C, Howard E, et al. LAIR-1 acts as an immune checkpoint on activated ILC2s and regulates the induction of airway hyperreactivity. J Allergy Clin Immunol. 2022; 149(1), pp.223–236.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kerzerho J, Maazi H, Speak AO, Szely N, Lombardi V, Khoo B, et al. Programmed cell death ligand 2 regulates TH9 differentiation and induction of chronic airway hyperreactivity. J Allergy Clin Immunol. 2013. Apr;131(4):1048–1057.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, et al. Gene Set Knowledge Discovery with Enrichr. Curr Protoc. 2021;1(3):e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woytschak J, Keller N, Krieg C, Impellizzieri D, Thompson RW, Wynn TA, et al. Type 2 Interleukin-4 Receptor Signaling in Neutrophils Antagonizes Their Expansion and Migration during Infection and Inflammation. Immunity. 2016. Jul 19;45(1):172–84. [DOI] [PubMed] [Google Scholar]
- 40.Wang L, Netto KG, Zhou L, Liu X, Wang M, Zhang G, et al. Single-cell transcriptomic analysis reveals the immune landscape of lung in steroid-resistant asthma exacerbation. Proc Natl Acad Sci. 2021; 118(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Wang H, Sokulsky L, Liu S, Yang R, Liu X, et al. Single-cell transcriptomic analysis reveals key immune cell phenotypes in the lungs of patients with asthma exacerbation. J Allergy Clin Immunol. 2021. Mar 1;147(3):941–54. [DOI] [PubMed] [Google Scholar]
- 42.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019. Feb;20(2):163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosskopf S, Jahn-Schmid B, Schmetterer KG, Zlabinger GJ, Steinberger P. PD-1 has a unique capacity to inhibit allergen-specific human CD4 + T cell responses. Sci Rep. 2018. Sep 10;8(1):13543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McAlees JW, Lajoie S, Dienger K, Sproles AA, Richgels PK, Yang Y, et al. Differential control of CD4+ T cell subsets by the PD-1/PD-L1 axis in allergic asthma. Eur J Immunol. 2015. Apr;45(4):1019–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oflazoglu E, Swart DA, Anders-Bartholo P, Jessup HK, Norment AM, Lawrence WA, et al. Paradoxical role of programmed death-1 ligand 2 in Th2 immune responses in vitro and in a mouse asthma model in vivo. Eur J Immunol. 2004. Dec;34(12):3326–36. [DOI] [PubMed] [Google Scholar]
- 46.Quoc QL, Choi Y, Thi Bich TC, Yang EM, Shin YS, Park HS. S100A9 in adult asthmatic patients: a biomarker for neutrophilic asthma. Exp Mol Med. 2021. Jul;53(7):1170–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gubernatorova EO, Namakanova OA, Gorshkova EkaterinaA, Medvedovskaya AD, Nedospasov SA, Drutskaya MS. Novel Anti-Cytokine Strategies for Prevention and Treatment of Respiratory Allergic Diseases. Front Immunol. 2021;12:1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Metzemaekers M, Gouwy M, Proost P. Neutrophil chemoattractant receptors in health and disease: double-edged swords. Cell Mol Immunol. 2020. May;17(5):433–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim YM, Kim H, Lee S, Kim S, Lee JU, Choi Y, et al. Airway G-CSF identifies neutrophilic inflammation and contributes to asthma progression. Eur Respir J. 2019; 55(2), p.1900827. [DOI] [PubMed] [Google Scholar]
- 50.Jin L, Ghimire L, Paudel S, Cai S, Rangasamy T, Jeyaseelan S. MCP-1 plays a critical role in neutrophil function and pyroptosis during Carbapenemf-Resistant Klebsiella Pneumoniae. J Immunol. 2018. May 1;200(1 Supplement):46.17–46.17. [Google Scholar]
- 51.Ichikawa A, Kuba K, Morita M, Chida S, Tezuka H, Hara H, et al. CXCL10-CXCR3 Enhances the Development of Neutrophil-mediated Fulminant Lung Injury of Viral and Nonviral Origin. Am J Respir Crit Care Med. 2013. Jan 1;187(1):65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Banerjee I, Edwards L, Halvey P, Alioto S, Cluckley D, Mitchell C, et al. Ab0034 Pd-1 Agonism Inhibits Activation of Plasmacytoid Dendritic Cells. Ann Rheum Dis. 2021. Jun 1;80(Suppl 1):1050–1050. [Google Scholar]
- 53.Curnock AP, Bossi G, Kumaran J, Bawden LJ, Figueiredo R, Tawar R, et al. Cell-targeted PD-1 agonists that mimic PD-L1 are potent T cell inhibitors. JCI Insight. 2021. Oct 22;6(20):e152468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ray A, Kolls JK. Neutrophilic Inflammation in Asthma and Association with Disease Severity. Trends Immunol. 2017. Dec;38(12):942–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crisford H, Sapey E, Rogers GB, Taylor S, Nagakumar P, Lokwani R, et al. Neutrophils in asthma: the good, the bad and the bacteria. Thorax. 2021. Aug 1;76(8):835–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brooks CR, Van Dalen CJ, Harding E, Hermans IF, Douwes J. Effects of treatment changes on asthma phenotype prevalence and airway neutrophil function. BMC Pulm Med. 2017. Dec 4;17(1):169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kalchiem-Dekel O, Yao X, Levine SJ. Meeting the Challenge of Identifying New Treatments for Type 2-Low Neutrophilic Asthma. Chest. 2020. Jan;157(1):26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wei Q, Liao J, Jiang M, Liu J, Liang X, Nong G. Relationship between Th17-mediated immunity and airway inflammation in childhood neutrophilic asthma. Allergy Asthma Clin Immunol. 2021. Jan 6;17(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rahmawati SF, te Velde M, Kerstjens HAM, Dömling ASS, Groves MR, Gosens R. Pharmacological Rationale for Targeting IL-17 in Asthma. Front Allergy. 2021; Frontiers in Allergy, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu C, Zhang X, Xiang Y, Qu X, Liu H, Liu C, et al. Role of epithelial chemokines in the pathogenesis of airway inflammation in asthma (Review). Mol Med Rep. 2018. May 1;17(5):6935–41. [DOI] [PubMed] [Google Scholar]
- 61.Gauthier M, Chakraborty K, Oriss TB, Raundhal M, Das S, Chen J, et al. Severe asthma in humans and mouse model suggests a CXCL10 signature underlies corticosteroid-resistant Th1 bias. JCI Insight. 2(13):e94580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Branchett WJ, Stölting H, Oliver RA, Walker SA, Puttur F, Gregory LG, et al. A T cell–myeloid IL-10 axis regulates pathogenic IFN-γ–dependent immunity in a mouse model of type 2–low asthma. J Allergy Clin Immunol. 2020. Feb 1;145(2):666–678.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Raundhal M, Morse C, Khare A, Oriss TB, Milosevic J, Trudeau J, et al. High IFN-γ and low SLPI mark severe asthma in mice and humans. J Clin Invest. 2015. Aug 3;125(8):3037–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 pathway. Sci Adv. 6(38):eabd2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leόn B T Cells in Allergic Asthma: Key Players Beyond the Th2 Pathway. Curr Allergy Asthma Rep. 2017. Jul;17(7):43. [DOI] [PubMed] [Google Scholar]
- 66.Nabe T Steroid-Resistant Asthma and Neutrophils. Biol Pharm Bull. 2020;43(1):31–5. [DOI] [PubMed] [Google Scholar]
- 67.Garber K Biochemistry: A radical treatment. Nature. 2012. Sep 27;489(7417):S4–6. [DOI] [PubMed] [Google Scholar]
- 68.Angevin E, Groenland SL, Lim AML, Martin-Liberal J, Moreno V, Trigo JM, et al. Updated analysis of the inducible T-cell co-stimulatory receptor (ICOS) agonist, GSK3359609 (GSK609), combination with pembrolizumab (PE) in patients (pts) with anti-PD-1/L1 treatment-naïve head and neck squamous cell carcinoma (HNSCC). J Clin Oncol. 2020. May 20;38(15_suppl):6517–6517. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.