

Abstract
Aspirin-exacerbated respiratory disease (AERD) has fascinated and frustrated specialists in allergy/immunology, pulmonology, and otorhinolaryngology for decades. It generally develops in previously healthy young adults and is unremitting and challenging to treat. The classical triad of asthma, nasal polyposis, and pathognomonic respiratory reactions to aspirin and other cyclooxygenase (COX)-1 inhibitors is accompanied by high levels of mast cell activation, cysteinyl leukotriene (cysLT) production, platelet activation, and severe type 2 respiratory inflammation (T2I). The “unbraking” of mast cell activation and further cysLT generation induced by COX-1 inhibition reflect an idiosyncratic dependency on COX-1-derived products, likely prostaglandin E2, to maintain a tenuous homeostasis. While cysLTs are clear disease effectors, little else was known about their cellular sources and targets, and the contributions from other mediators and T2I effector cells to disease pathophysiology were unknown until recently. The applications of targeted biological therapies, single cell genomics, and transgenic animal approaches have substantially advanced our understanding of AERD pathogenesis and treatment and have also revealed disease heterogeneity. This review covers novel insights into the immunopathogenesis of AERD from each of these lines of research, including the roles of lipid mediators, effector cell populations, and inflammatory cytokines, discusses unanswered questions regarding cause and pathogenesis, and considers potential future therapeutic options.
Keywords: AERD, CRSwNP, Prostaglandin E2, Leukotriene E4, mast cells, mechanisms
Introduction.
Over the last few years, new insights into the mechanisms of immune dysregulation that drive aspirin-exacerbated respiratory disease (AERD) have come from a variety of study types, including clinical trials using targeted biologics and therapeutics, investigations of mouse models of respiratory inflammation, and the application of novel technologies like single-cell RNA sequencing. Some of these recent advances in our understanding of AERD pathogenesis have provided insights into the mechanism of action by which our current therapies provide therapeutic benefit, and others have provided the rationale for the development of new therapies directed against selected targets. Though there is much more yet to be learned, there is now considerable knowledge of the involvement of particular lipid mediators, respiratory effector cells, antibody overproduction, and cytokine-driven inflammation in AERD.
Lipid mediators.
Once regarded principally as regulators of smooth muscle tone and vascular permeability, arachidonic acid-derived lipid mediators are now well known to regulate features of innate and adaptive immunity. Long-recognized disturbances in lipid mediator production and receptor function associated with AERD now can be considered in the greater context of the dysregulated type 2 inflammation (T2I) that underlies the disease.
Prostaglandin (PG)E2.
PGE2 is the most ubiquitous COX product, being generated in some quantity by most cell types, although the most prominent sources are macrophages, epithelial cells, and fibroblasts. Either constitutive COX-1 or inducible COX-2 can drive PGE2 production by converting arachidonic acid to PGH2, which then serves as a substrate for conversion to PGE2 by one of three different PGE2 synthase (PGES) enzymes (1). PGE2 stabilizes and prevents the activation of mast cells (2) and group 2 innate lymphoid cells (ILC2s)(3), and prevents the formation of cysLTs by inhibiting the translocation of 5-lipoxygenase (5-LO) (4). Several studies link AERD to diminished production (or increased catabolism) of PGE2 in the respiratory tract (5, 6), associated with diminished expression and function of COX-2 and/or microsomal (m)PGES-1, the dominant PGES isoform that is upregulated with inflammation (7)(Fig. 1). Additionally, expression of the PGE2-selective EP2 receptor by hematopoietic cells (8, 9) and fibroblasts (7, 10) is reduced in AERD. Since COX-1 is exquisitely sensitive to inhibition by aspirin, a chronic reduction in COX-2/mPGES-1-derived PGE2 could account for aspirin-induced reactions in AERD, by removing a critical COX-1-dependent “braking” function. Consistent with this hypothesis, inhaled PGE2 not only blocks changes in lung function in response to aspirin challenges in AERD, but also blocks the characteristic aspirin-induced increase in urinary leukotriene (LT)E4 (11). Mice lacking mPGES-1 (Ptges−/− mice) or EP2 receptors (Ptger2−/− mice) display higher levels of airway T2I, mast cell activation, ILC2 expansion, and cysLT production than do wild-type controls after intranasal instillation with dust mite antigens, and display AERD-like physiologic reactions to aspirin (12). While the causes of impairments in the COX-2/mPGES-1 system and EP2 expression are unknown, it is likely that they contribute to the T2I, mast cell activation, and overproduction of cysLTs that characterize AERD.
Figure 1. Summary of the pathogenesis of the chronic inflammation in the respiratory tissue in AERD.
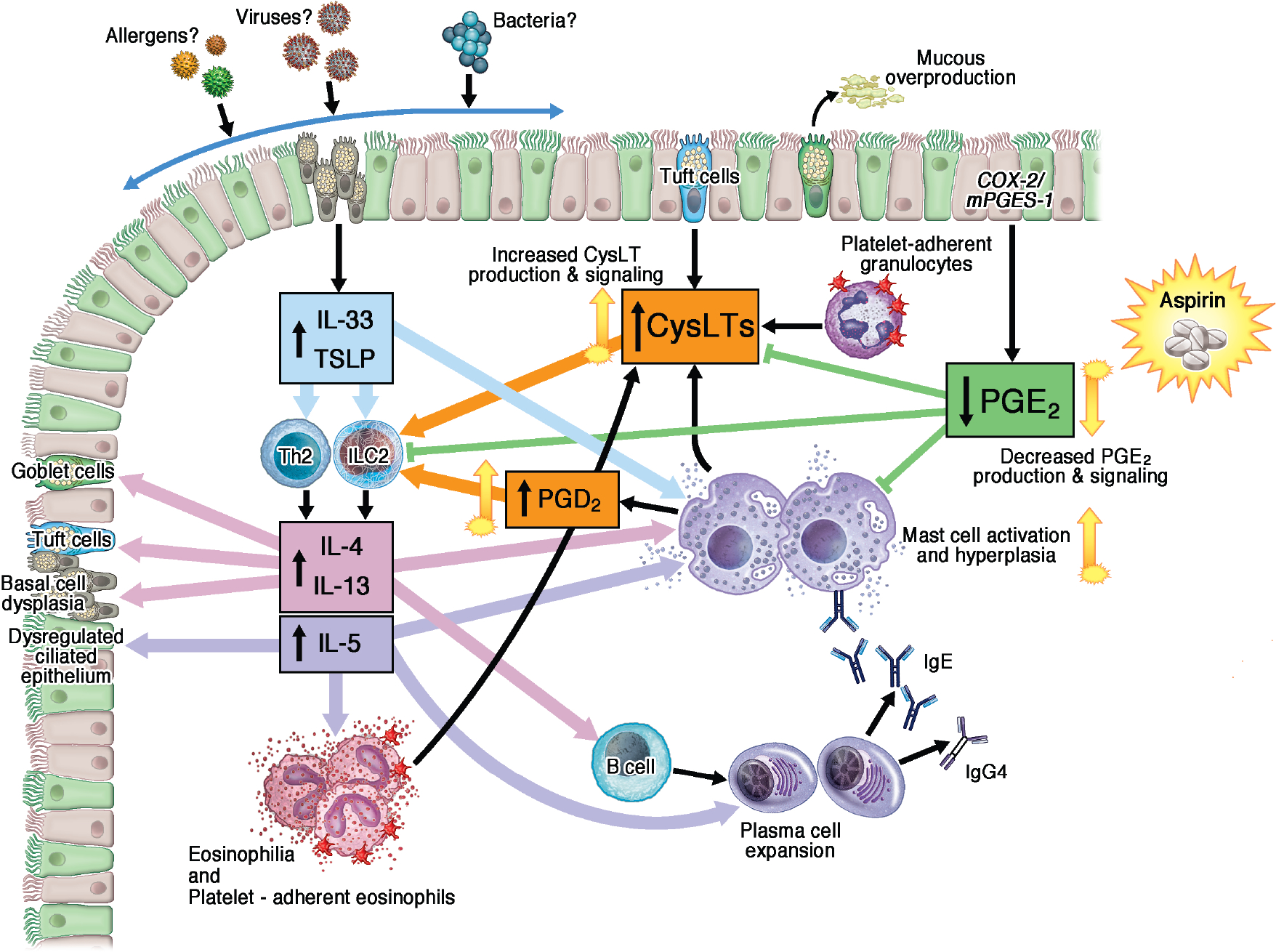
IL-33 and TSLP derived from dysplastic epithelial basal cells activate Th2 cells and ILC2s to produce IL4, IL-13, and IL-5, as well as activating mast cells. Those cytokines in turn activate and recruit B cells, plasma cells, mast cells and eosinophils and also feedback to target the epithelium. Proinflammatory lipids CysLTs and PGD2 are overproduced and act in concert with IL-33 to activate ILC2s, the anti-inflammatory lipid, PGE2, is underproduced, contributing to ILC2 expansion and unchecked mast cell activation.
ILC, innate lymphoid cell; CysLT, cysteinyl leukotriene; PG, prostaglandin; TSLP, thymic stromal lymphopoietin
cysLTs.
CysLTs form from conversion of arachidonic acid to LTA4 by 5-LO, which is then conjugated to reduced glutathione by LTC4 synthase (LTC4S) to form LTC4. Mast cells, basophils, eosinophils, epithelial brush cells, and platelet-adherent granulocytes all have the capacity to generate LTC4, which is converted extracellularly to the potent bronchoconstrictor LTD4 (13) and its metabolite LTE4. Urinary levels of LTE4, a marker of systemic cysLT production, are characteristically elevated in patients with AERD relative to aspirin-tolerant controls, and increase further during NSAID-induced reactions (14). Additionally, patients with AERD are highly sensitive to bronchoconstriction induced by inhaled LTE4 (15). Treatment with zileuton, which inhibits 5-LO and decreases production of leukotrienes, attenuates the sinonasal symptoms (16), lung function changes (17), and gastrointestinal and cutaneous symptoms that occur during aspirin-induced reactions in AERD. Additionally, a post hoc analysis of the Phase 3 studies of zileuton for the treatment of asthma revealed that the subgroup of asthmatic patients with comorbid aspirin sensitivity experienced a 22.8% increase in FEV1 from baseline, which was significantly greater than that in the aspirin-tolerant subgroup (18). These findings verify the importance of cysLTs as drivers of both the ongoing airway disease and the acute aspirin-induced reactions in AERD. Moreover, both overproduction and end-organ hyperresponsiveness likely contribute to the cysLT-driven features of AERD.
Animal studies suggest that cysLTs play a much broader role than previously appreciated in driving the exaggerated features of T2I in AERD. All three cysLTs have potent and distinct bioactivities in lung T2I, displaying differential preferences for their receptors, CysLT2R, CysLT1R and CysLT3R/GPR99, respectively(19–21). As is the case for humans with AERD (16, 17), treatment of Ptges−/− mice (which display AERD-like reactions to inhaled lysine aspirin and selective hyperresponsiveness to LTE4) with the 5-LO inhibitor zileuton blocks the aspirin-induced changes in both lung function and mast cell activation (12). Genetic deletion of LTC4S attenuates the eosinophilic lung inflammation, ILC2 expansion, and upregulation of IL-33 that occurs in Ptges−/− mice treated with intranasal dust mite (20). Notably, respective deletions of CysLT1R, CysLT2R, or CysLT3R also reduce lung eosinophilia and ILC2 expansion in these AERD-like mice, but only CysLT2R deletion inhibits the upregulation of IL-33, limiting the expansion of IL-33+ epithelial cells (20). Moreover, LTE4 acts at CysLT3R to drive the expansion of brush/tuft cells (22), a unique population of epithelial chemosensory cells that are the dominant source of IL-25 in the lung and gut mucosa (22, 23), and are also capable of generating both LTC4 and prostaglandin D2 (PGD2) (24, 25) Additionally, LTC4 and LTD4 activate and elicit proliferation of mouse lung ILC2s by engaging CysLT1R (26)(Fig. 1), and synergize with IL-33 and IL-25 in this process (27, 28). Collectively, these studies suggest that cysLTs and their receptors function in a network that amplifies T2I through synergistic actions on ILC2s and epithelial-derived cytokines. The high levels of cysLTs in AERD, the substantial therapeutic effects of zileuton, and the robust features of respiratory inflammation in this disease suggest that this putative network may be especially important in driving disease features. Further, many of these leukotriene-dependent features may depend on cysLT receptors that are not targeted by the currently available drugs zafirlukast and montelukast, which only block CysLT1R. This may explain the comparatively modest clinical effects of CysLT1R blockade in AERD relative to the more robust effects of zileuton (29).
PGD2.
PGD2 is the dominant COX product of mast cells and tuft/brush cells and is also generated in lesser amounts by eosinophils, T helper type 2 (Th2) cells (30), and ILC2s(31). The levels of urinary PGD2 metabolites in patients with AERD exceed those in aspirin-tolerant controls, and often paradoxically increase in parallel with LTE4 during reactions to aspirin (32). Interestingly, a subset of patients with AERD display both especially high baseline levels of urinary PGD2 metabolites and dramatic further increases during aspirin reactions, which is usually associated with their development of a pruritic macular rash and abdominal pain (33). PGD2 induces chemotaxis of Th2 cells (34) and ILC2s (35) and elicits their production of cytokines (35, 36), and also elicits eosinophil and basophil chemotaxis (34). These actions all depend on the chemoattractant receptor-homologous molecule expressed on T-helper type 2 cells (CRTH2), one of two known PGD2 receptors. In a 12-week placebo-controlled study of 43 patients with CRSwNP, 27 of whom had AERD, treatment with the selective CRTH2 antagonist AZD1981 had no effect on nasal polyp score, measures of olfaction, or measures of sinus disease symptoms compared with placebo (37). Further, in a 16-week study of the CRTH2 inhibitor fevipiprant for the treatment of CRSwNP, fevipiprant did not significantly improve nasal polyp score, nasal congestion, measures of olfaction, or measures of sinus disease symptoms compared with placebo, though there was only one patient with AERD was randomized to the treatment group(38). Neither study assessed baseline systemic or respiratory tract PGD2 levels in the patient population, so it is not known whether these were the most appropriate target populations(39). Though these results do not completely exclude a role for PGD2 in CRSwNP in general or AERD in particular, they do suggest that signaling through the PGD2-CRTH2 pathway is unlikely to be a major contributor to the ongoing respiratory inflammation in CRSwNP/AERD. Whether CRTH2 blockade might attenuate features of reactions to aspirin or benefit the “PGD2-high” subgroup remains to be determined.
Effector cell-related features.
Severe T2I of both the upper and lower respiratory tract is typical of AERD, and accounts for the clinical severity of the asthma and CRSwNP in the syndrome. Recent clinical trials and the application of molecular tools have challenged dogma regarding the contributions of particular cell types, while revealing previously unrecognized additional effectors.
Tissue eosinophilia.
Eosinophilia is a prominent histologic feature of CRSwNP and asthma, especially in AERD (40, 41). Although eosinophils are regarded as drivers of tissue dysfunction and remodeling, recent studies prompt a reassessment of these assumptions in AERD. Dexpramipexole is a small orally available molecule that depletes blood and respiratory tissue eosinophils. In an open-label study of 16 patients with CRSwNP, several of whom had AERD, dexpramipexole induced a 97% reduction from baseline in nasal polyp tissue eosinophilia. Nonetheless, there was no significant reduction in nasal polyp score, nor any significant improvement in measures of olfaction or measures of sinus disease symptoms (42). These ‘negative’ findings bring the pathogenetic role of tissue eosinophils into question. Notably, while polyp neutrophils were not affected by dexpramipexole, polyp mast numbers in the biopsies actually increased significantly over the treatment period, consistent with a possible compensatory role for mast cells disease drivers. The lack of effects on symptoms and polyp size induced by eosinophil depletion with dexpramipexole contrasts with the modest therapeutic efficacy of anti-IL-5 treatments, such as benralizumab, mepolizumab, or reslizumab, suggesting that IL-5 acts on cells other than eosinophils to support features of T2I.
Platelet activation.
Compared with aspirin-tolerant asthmatic controls, the serum of patients with AERD contains high levels of platelet activation markers (43). Additionally, nasal fluid from patients with AERD has higher levels of platelet-derived microparticles than does nasal fluid from aspirin-tolerant controls with CRSwNP (44). During inflammatory responses, activated platelets form complexes with circulating leukocytes. Platelet-adherent leukocytes become primed for adhesion and activation. Moreover, platelets strongly express LTC4S, and adherent platelets convert granulocyte-derived LTA4 to LTC4 through a transcellular pathway (45). The frequencies of platelet-adherent granulocytes in the blood of patients with AERD exceed those in the blood of aspirin tolerant-controls (43, 46), and correlate with the levels of urinary LTE4, suggesting that transcellular platelet-derived cysLTs contribute significantly to systemic cysLTs generation. A placebo-controlled cross-over trial of prasugrel, which inhibits platelet activation by blocking the P2Y12 receptor, was undertaken with 40 AERD patients to determine whether prasugrel would inhibit platelet activation and attenuate the severity of respiratory symptoms induced during aspirin challenge. While prasugrel produced no benefit for the group as a whole, a subgroup analysis revealed 5 patients for whom prasugrel pretreatment completely blocked all reaction symptoms. Those 5 responders had higher baseline indices of platelet activation and less evidence of aspirin-induced mast cell activation, suggesting a contribution from P2Y12 signaling and platelets in this subset (47). Because prasugrel did not decrease the frequency of platelet-leukocyte aggregates, it is likely that the P2Y12 pathway does not account for the aberrant platelet phenotype of AERD.
Mast cell hyperplasia and activation.
Several lines of evidence indicate that mast cells play a central role in AERD. Mast cells accumulate in the nasal and bronchial mucosa of patients with AERD at higher concentrations than in aspirin-tolerant controls (41, 48). The levels of mast cell products in the urine and nasal fluid from patients with AERD are elevated compared with aspirin-tolerant controls, as are the levels of mast cell-derived microparticles in their nasal fluids (44), indicative of steady-state mast cell activation. Mast cell products increase further during reactions to aspirin (32, 33, 49), likely reflecting the depletion of PGE2. Treatment with cromolyn, a mast cell stabilizing drug, rapidly increases baseline lung function (50) and reduces sputum eosinophil counts (51) in subjects with AERD, suggesting that persistent mast cell activation drives several steady-state features of AERD. Moreover, cromolyn pretreatment also blocks changes in lung function during aspirin challenges, and blocks the characteristic increase in urinary LTE4 (52). A recent study demonstrated a high frequency of proliferating (Ki67+) mast cells in AERD sinonasal tissues compared with aspirin-tolerant controls (48) and single cell RNA-sequencing revealed a high degree of activation in nasal polyp mast cells, with enrichment for both IgE- and IL-33-driven transcriptional signatures. Moreover, treatment of human cord blood mast cells with IL-4 ex vivo elicited a transcriptional signature that closely mirrored the intraepithelial population of mast cells that is the most richly expanded population in AERD tissues. These studies indicate that both innate and adaptive immune pathways drive mast cell activation in AERD, and that proliferation is the principal mechanism responsible for the marked mast cell hyperplasia observed in this disease.
ILC2 recruitment.
ILC2s are tissue resident poised effector cells capable of generating large quantities of IL-5, IL-13 and other cytokines in response to both innate cytokines (IL-33, thymic stromal lymphopoietin (TSLP), IL-25) and lipid mediators. Nasal aspirin challenges in AERD elicit a reduction in circulating ILC2s, accompanied by an increase in ILC2s in the nasal tissue (53), which parallels the peaks of urinary LTE4 and PGD2 metabolites. Given that PGD2 and LTE4 cooperatively activate human ILC2s and elicit their chemotaxis via CRTH2 and CysLT1R, respectively (35, 54), it is possible that their recruitment in AERD is driven by the release of lipid mediators from the respiratory tract. This may also result in incremental tissue eosinophilia, as studies have noted a similar reduction in blood eosinophil counts and influx of nasal eosinophil counts associated with aspirin challenges (47, 55), suggesting that ILC2s and eosinophils may be recruited by overlapping mechanisms. Interestingly, the antibody-mediated depletion of ILC2s markedly inhibited all physiologic and biochemical features of aspirin-induced reactions in a mouse model (20), suggesting that products of ILC2s may be essential to the clinical phenotype.
Plasma cell expansion.
While the majority of mechanistic studies in AERD focus on innate effectors, the adaptive immune system also likely plays an important role. Plasma cells are found in nasal polyp tissues. A transcriptionally distinct population of plasma cells was identified within AERD nasal polyps, which was notable for strong expression of the alpha subunit of the IL-5 receptor (IL-5Rα)(56). Flow cytometry confirmed the presence of IL-5Rα+ plasma cells at higher frequencies in nasal polyps from AERD patients than in those from aspirin-tolerant CRSwNP. These plasma cells also expressed IGHG4 and IGHE, encoding the heavy chains of IgG4 and IgE, respectively. Interestingly, the levels of both IgG4 and IgE in sinonasal tissue from patients with AERD far exceeded those in aspirin-tolerant CRSwNP, but there was no relationship between the nasal and serum levels of these antibodies. Further, nasal IgE levels were highest and IgG4 levels the lowest in patients whose nasal polyps regrew the most quickly after surgery. This study highlights a potentially unique role for plasma cells, locally generated antibodies, and IL-5Rα in AERD pathophysiology, although the immune stimuli driving these responses and the role of IL-5 in plasma cell ontogeny remain to be determined.
Epithelial dysplasia.
Epithelial cells have emerged as major contributors to the pathogenesis of T2I. A recent study used single cell RNA-sequencing to define the functional and transcriptional differences between epithelial cells from CRSsNP and CRSwNP tissue (57). Although the study was underpowered to detect differences between AERD and aspirin-tolerant CRSwNP, several key findings emerged with respect to the epithelium. First, epithelial basal cells, the principal cellular site of IL33 and TSLP expression, were markedly expanded in polyp tissue compared to non-polypoid sinus tissue. Second, the expanded basal cells bore a strong imprint of IL-4 receptor alpha (IL-4Rα)-driven signature transcripts (e.g., ALOX15, POSTN, CCL26). Third, analyses of developmental trajectories and chromatin architecture revealed that polyp epithelial basal cells display evidence of “blocked” differentiation, characterized by persistent stem-ness and loss of normal development into secretory cells, mediated by chromatin remodeling. A subsequent study revealed that ALOX15 is specifically upregulated in AERD nasal polyps relative to that in aspirin-tolerant CRSwNP (58), and that its expression correlates with that of 15-hydroxyprostaglandin dehydrogenase (PGDH) by mast cells. 15-hydroperoxyeicosatertraenoic acid (15-HETE), the product of ALOX15, is converted by PGDH into 15-oxo-ETE, a potent eosinophil chemoattractant, suggesting a mechanism by which epithelial-mast cell cross talk may facilitate tissue eosinophilia in AERD. Moreover, esterified 15-HETE was recently shown to regulate ferroptosis (59), a form of programmed cell death in epithelial cells that is driven by lipid peroxidation (60). Thus, nasal polyposis in general, and AERD in particular, may involve disturbances in epithelial homeostasis, redox balance, and regenerative capacity, while driving T2I through production of IL-33 and TSLP. The therapeutic efficacy of targeted anti-IL-4Rα antibodies, such as dupilumab, as treatments for CRSwNP and AERD could reflect a partial restoration of these functions (61).
Lessons learned from targeted therapeutics.
The advent of biologics targeting cytokines and antibodies involved in T2I, and the investigation of high-dose aspirin as a treatment regimen, has not only provided therapeutic breakthroughs for patients with AERD, but has also provided the opportunity to test existing hypotheses regarding the pathogenesis of the disease. As a result, clinical trials have yielded important and, in some instances, unexpected information regarding the causal mechanisms of AERD.
IgE.
Although AERD is not thought to be driven by classical allergen-induced pathways, some studies do support a potentially significant role for IgE in its pathogenesis. In an open-label study, treatment of AERD patients with omalizumab, a monoclonal anti-IgE antibody, dramatically decreased urinary levels of both LTE4 and PGD2 metabolites (62) and induced improvement in baseline symptoms. Subsequently, a placebo-controlled trial of omalizumab pretreatment followed by aspirin challenge in AERD showed that 5 of the 7 patients randomized to omalizumab then had no respiratory reaction during aspirin challenge (63). These findings support a role for IgE, likely derived locally from plasma cells in the respiratory tract (56), in driving both the steady-state eicosanoid formation and the mast cell-dependent clinical reactions to aspirin, and are consistent with the enrichment for IgE-driven activation transcripts in nasal polyp mast cells (48). The mechanisms by which IgE can induce a chronically activated mast cell population, and the antigen specificity of the locally produced IgE remain to be determined. As the entry criteria for the omalizumab studies included at least one positive skin test to environmental allergens, it is unclear whether the efficacy of anti-IgE is limited to patients with AERD who are also atopic, or whether locally produced respiratory IgE of unknown specificity drives significant mast cell activation and pathology.
IL-5.
IL-5, produced by activated Th2 cells, ILCs, and mast cells, is elevated in the respiratory tissues of patients with AERD relative to controls and is required for the development of tissue eosinophilia. Neutralization of IL-5 with the humanized antibody mepolizumab reduces blood and tissue eosinophilia in patients with severe asthma and CRSwNP and improves symptomatic control over placebo. Open label studies reveal that mepolizumab provides moderate symptomatic benefit for patients with AERD (64, 65), but does not consistently block NSAID-induced reactions (66). Notably, single cell RNA-sequencing of nasal polyps has revealed that several non-eosinophil cell types strongly express IL5RA transcript, including mast cells, ciliated epithelial cells, and plasma cells. These findings indicate that, contrary to dogma, the actions of IL-5 are not limited to eosinophils. Inhibition of IL-5 signaling on these other IL-5Rα+ cell types could account for the therapeutic benefit provided by mepolizumab, considering the lack of benefit that dexpramipexole afforded despite its complete eosinophil depletion in CRSwNP and AERD (42). A recent case-control study of 18 AERD patients on mepolizumab and 18 matched AERD patients not treated with mepolizumab revealed that mepolizumab decreased production of inflammatory eicosanoids, especially PGD2, and upregulated tight junction-associated nasal epithelial cell transcripts, potentially reflecting the actions of IL-5 on mast cells and epithelial cells, respectively (65). However, the function of the IL-5Rα and the role of IL-5 signaling on these non-eosinophil airway cells still remains to be determined.
IL-4/IL-13.
IL-4 and IL-13 are critical to the pathogenesis of T2I, signal through a shared common receptor subunit, IL-4Rα, and are elevated in the nasal fluid of AERD patients compared to controls(67). RNA-sequencing has revealed that mast cells and Th2 cells are the principal sources of IL-13 in the nasal polyps (57). Treatment with dupilumab, a humanized antibody against IL-4Rα, leads to robust improvements in upper and lower respiratory tract symptoms in AERD patients, including improved sense of smell, reduced nasal polyp size, and improved lung function (68–70). Patients with AERD generally report onset of clinical improvement with dupilumab within the first month of initiating treatment (71). Additionally, perioperative initiation of dupilumab decreases the rate of polyp regrowth after endoscopic sinus surgery (72). An open label study of 22 AERD patients treated with dupilumab for three months showed a rapid and sustained improvement in both upper and lower airway symptoms and sense of smell. Concomitantly, dupilumab reduced nasal fluid albumin levels, suggesting an improvement in epithelial/endothelial barrier function, and decreased serum and nasal fluid IgE levels. Additionally, dupilumab markedly decreased nasal and urinary levels of LTE4, while increasing nasal fluid concentrations of PGE2. Since IL-4 and IL-13 suppress the expressions of COX-2, mPGES-1, and EP2 receptors by human bronchial epithelial cells ex vivo (73), it is tempting to speculate that IL-4Rα blockade could at least partially restore PGE2-dependent homeostasis of the cysLT pathway. Consistent with this hypothesis, treatment with dupilumab can increase aspirin tolerance and often completely prevent aspirin-induced reactions in AERD (74). Given the broad distribution of IL-4Rα expression, the efficacy of dupilumab in AERD likely reflects its ability to block IL-4Rα on multiple relevant cell types, including mast cells, granulocytes, epithelial cells, endothelial cells, Th2 cells, and B/plasma cells(71). Whether its effects are disease modifying or dependent on indefinite IL-4Rα inhibition remains to be determined.
Epithelial cytokines.
IL-25, IL-33, and TSLP are alarmin-type cytokines that are produced and released by epithelial barrier cells in response to injury or activation. These cytokines act directly on ILC2s, mast cells, and Th2 cells to drive T2I. Sinonasal tissues from patients with AERD contain high concentrations of TSLP (32) and IL-33 (75), both of which are expressed principally by epithelial basal cells and (in the case of IL-33) endothelial cells (57). Platelets, which contain pre-formed IL-33 (76), are another potential source in polyp tissue. Nasal polyp mast cells are strongly enriched for IL-33-driven activation signature genes (48) and blockade of IL-33 dramatically attenuates the aspirin-induced AERD-like responses of Ptges−/− mice (75). Nasal polyp levels of TSLP mRNA correlate with mast cell-related transcripts and with urinary PGD2 metabolites, and TSLP stimulates human mast cells to generate PGD2 ex vivo (32). Tezepelumab, an anti-TSLP humanized monoclonal antibody (77), was recently FDA approved for the treatment of severe asthma, and antibodies against IL-33 and its receptor are currently under investigation (78, 79). Clinical trials are necessary to verify the roles of TSLP and IL-33 in the pathogenesis of AERD.
Aspirin desensitization.
Following a reaction to aspirin or any COX-1-active NSAID, patients with AERD are transiently refractory to the effects of aspirin and can be desensitized and escalate to a daily dose of up to 650 mg twice daily. For about 60–70% of patients with AERD, this daily aspirin dose significantly improves symptoms of sinonasal dysfunction and asthma, and can reduce the rate of polyp regrowth (80). Despite these clinical benefits, treatment with high-dose aspirin does not diminish the characteristic biomarkers of inflammation. In a prospective study of 40 individuals with AERD who underwent desensitization, treatment with high-dose aspirin for 8 weeks resulted in substantially increased urinary concentrations of LTE4 (concomitantly with significantly reduced urinary levels of PGE2 metabolites), along with increases in serum tryptase (indicative of mast cell activation and/or mast cell burden), higher blood eosinophil counts, and increased exhaled nitric oxide concentrations, which may be suggestive of increased airway eosinophilia (81). Notably, none of these parameters changed in aspirin-tolerant controls. These observations indicate that the mechanism(s) responsible for the clinical benefit of high-dose aspirin are likely at least partly due to a loss of end-organ reactivity to cysLTs, as reported previously (15, 82). The depletion of PGE2, unique to AERD, may be consistent with the lack of COX-2/mPGES-1 function in their respiratory tract (83) and could account for the observed increases in tryptase, LTE4, and nitric oxide.
Remaining unknowns and hypotheses.
Although advances have been made in our understanding of the immune mechanisms that underlie AERD, many unknowns still remain (Table I). For example, there are only two known potential risk factors for the development of the disease. Prolonged tobacco smoke exposure may be one risk, as a patient exposed to environmental tobacco smoke during both childhood and adulthood has an odds ratio of 5.09 for developing AERD (95% CI, 2.75–9.43)(84). However, a study of adults in Japan revealed that smoking cessation 1–4 years prior was positively associated with the development of AERD compared with the development of aspirin-tolerant asthma (85). Female sex is another risk factor, identified as such as early as 1968 when Samter and Beer described the first large cohort of patients with AERD, 57% of whom were female(86). More recent work has confirmed this female predominance, and has shown that on average, women develop the disease a few years earlier than men, with 6.8% of female AERD patients reporting the onset of their disease prior to 18 years of age [REF Bensko, once Epub online available]. The reasons for these sex-based differences are still unknown, although several plausible mechanisms are reported. For example, androgens suppress 5-LO translocation in granulocytes (87), and thus granulocytes from females generate significantly more leukotrienes than do those from males. Androgens also suppress the proliferation and activation of ILC2s (88). Finally, COX-2 expression and PGE2 production are both significantly lower in women than in men (89, 90). Each of these sex differences are potentially relevant to the unique pathophysiology of AERD. However, aside from female sex, and possibly tobacco smoke exposure, other risk factors that predispose to the development of AERD remain unknown.
Table I.
Knowns and unknowns in the causative immune mechanisms in AERD.
| Known or Suspected | Unknown | |
|---|---|---|
| Risk factors for the development of AERD | ||
| Female sex | Why is there a female predominance, and why do women tend to develop the disease earlier than men? | |
| Tobacco smoke exposure | What is the mechanism by which tobacco may increase risk of developing AERD? | |
| Age | Why is AERD so uncommon in children? | |
| Inciting trigger for AERD | ||
| Respiratory infection | Is there a universal bacterial or viral infection that precipitates the initial immune dysregulation? | |
| Autoimmunity | Is there a predisposition for the development of autoantibodies in patients with AERD? | |
| Causes of ongoing inflammation and chronic olfactory dysfunction | ||
| CysLTs and mast cell activation | What drives the chronic overproduction of cysLTs and mast cell activation? | |
| Epithelial dysfunction | Is the epithelial barrier dysregulation a primary cause of the inflammation in AERD, or a result of it? | |
| Elevations of inflammatory cytokines and decreased levels of PGE2 in the respiratory tissue | What are the cellular sources of these type 2 inflammatory cytokines, and what are their effects on their respective effector cell targets? Do inflammatory cytokines or low levels of PGE2 directly cause olfactory dysfunction? |
|
| Mechanisms of COX-1 inhibitor-induced reactions | ||
| Decreased tissue levels of PGE2 and EP2/4 receptors | What drives the deficiency in respiratory PGE2 production or signaling? | |
| Further increase in cysLTs during reactions | Which cells drive the COX-1 inhibitor-induced rise in cysLTs? | |
| Variability in reaction severity and development of extra-pulmonary symptoms | Is AERD a local respiratory disease or a systemic disease that can involve the GI tract and skin as well? | |
| Mechanisms of benefit of high-dose aspirin | ||
| ~60% of AERD patients derive benefit from high-dose aspirin (delay of polyp regrowth) | Why is daily aspirin not beneficial for all AERD patients, and what biomarkers predict who will derive benefit? | |
| Aspirin doses of ≥325mg can provide benefit | Why is such a high dose of aspirin needed for the respiratory benefit? | |
| Other non-aspirin NSAIDs do not provide respiratory benefit | Why do other COX-1 inhibitors provide no respiratory benefit? | |
The inciting trigger for the development of AERD is also unknown. For most patients, the disease begins quite suddenly, often described as though it was “a cold that never went away”. Decades ago, Szczeklik proposed the hypothesis that AERD may be initiated by an upper respiratory viral infection (91). While both viral (92) and bacterial pathogens, particularly Staphylococcus aureus (93, 94), have been implicated in the pathogenesis of CRSwNP, none have proven causality, and none are specific to AERD. Nonetheless, it seems plausible that infection-induced damage to the respiratory epithelium could result in exposure of the self-antigens and allow for the development of an autoimmune process. Several studies have found the presence of class-switched autoantibodies within CRSwNP tissue, with measurable autoantibody levels against a number of self-antigens, including against nuclear antigens (anti-dsDNA), B2-microglobulin, and collagen (95–97). However, whether these autoantibodies play a causative role in AERD is not known.
The cause of the severe chronic inflammation in AERD remains unknown. Further, nasal polyps in these patients have frustratingly high rates of post-operative recurrence (98), without known causes of polyp recurrence. The adult onset of AERD, along with its minimal heritability, and intransient nature are consistent with a contribution from epigenetic changes, perhaps as a result of a prior insult. In particular, epigenetic modifications may impact the control of lipid mediator biosynthesis that is fundamental to AERD, which could in turn contribute to the chronic inflammation and polyp regrowth. One study reported hypomethylation of PGDS and ALOX5AP in whole nasal polyp tissue from subjects with AERD relative to aspirin-tolerant controls, which could affect the production of PGD2 and cysLTs, and hypermethylation of PTGES, which could affect the synthesis of PGE2 (99). Additionally, PTGER2 expression by polyp fibroblasts from patients with AERD controlled by histone acetylation, and an epigenetically mediated reduction of EP2 expression could contribute to the PGE2 resistance observed in the disease (10). Still, the nature of the insult driving the development of these epigenetic changes is unknown. The profound olfactory dysfunction, often leaving patients with decades of complete anosmia, is an additional source of frustration, without clear causes. One recent study of 22 patients with AERD found that baseline severity of smell loss was correlated with lower nasal PGE2 levels and treatment-induced improvement in olfaction related to increases in nasal PGE2 (71). Nonetheless, a direct immunologic link between PGE2 and olfactory function is elusive. Elevations in mucous levels of IL-2, IL-5, IL-6, IL-10, and IL-13 have also been shown to associate with loss of smell (100). However, the induction of an acute transient systemic inflammation with an experimental endotoxemia model, which does trigger a rise in some inflammatory cytokines, was not associated with a reduction in smell (101), suggesting that the cytokine associations may be correlative but not causative.
Conclusions.
Our understanding of the immune mechanisms that underlie AERD has been considerably advanced in the last decade, through investigations into the mechanistic changes induced by targeted therapies, the application of single cell genomics of the respiratory tissue, and the preclinical study of interventions afforded by the availability of transgenic animal models. The pathogenic roles of both mast cell activation and epithelial dysregulation have been further elucidated, along with the particular hypersensitivity to cysLTs and reliance on PGE2 that is evident in AERD. The importance of type 2 cytokines as drivers of the chronic disease has also been clearly established. However, many questions remain in regard to the initial disease triggers and the causes of the persistent inflammation. As further investigations into the immunopathogenesis of AERD reveal additional clues, there may be future therapeutic options that have yet to be explored.
Funding:
This work was supported by NIH/NIAID U19 AI095219, R37AI052353, R01AI136041, R01AI078908, and by generous contributions from the Vinik and Kaye Families.
Abbreviations:
- AERD
Aspirin-exacerbated respiratory disease
- COX
Cyclooxygenase
- T2I
Type 2 inflammation
- CysLTs
Cysteinyl leukotrienes
- PG
Prostaglandins
- ILC2
Group 2 innate lymphoid cells
- 5-LO
5-lipoxygenase
- Th2
T helper type 2
- CRTH2
chemoattractant receptor-homologous molecule expressed on T-helper type 2 cells
- CRSwNP
Chronic rhinosinusitis with nasal polyps
- CRSsNP
Chronic rhinosinusitis without nasal polyps
- TSLP
thymic stromal lymphopoietin
Footnotes
Conflict of Interest: TM Laidlaw has served on scientific advisory boards for GlaxoSmithKline and Sanofi-Genzyme, Novartis, and Regeneron. JA Boyce has served on the advisory boards of Sanofi-Genzyme, Siolta Therapeutics, and Third Harmonic Bio.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Inada M, Matsumoto C, Uematsu S, Akira S, Miyaura C. Membrane-bound prostaglandin E synthase-1-mediated prostaglandin E2 production by osteoblast plays a critical role in lipopolysaccharide-induced bone loss associated with inflammation. J Immunol. 2006;177(3):1879–85. [DOI] [PubMed] [Google Scholar]
- 2.Kay LJ, Yeo WW, Peachell PT. Prostaglandin E2 activates EP2 receptors to inhibit human lung mast cell degranulation. Br J Pharmacol. 2006;147(7):707–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maric J, Ravindran A, Mazzurana L, Bjorklund AK, Van Acker A, Rao A, et al. Prostaglandin E2 suppresses human group 2 innate lymphoid cell function. J Allergy Clin Immunol. 2018;141(5):1761–73 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brock TG. Regulating leukotriene synthesis: the role of nuclear 5-lipoxygenase. J Cell Biochem. 2005;96(6):1203–11. [DOI] [PubMed] [Google Scholar]
- 5.Yoshimura T, Yoshikawa M, Otori N, Haruna S, Moriyama H. Correlation between the prostaglandin D(2)/E(2) ratio in nasal polyps and the recalcitrant pathophysiology of chronic rhinosinusitis associated with bronchial asthma. Allergol Int. 2008;57(4):429–36. [DOI] [PubMed] [Google Scholar]
- 6.Roca-Ferrer J, Perez-Gonzalez M, Garcia-Garcia FJ, Pereda J, Pujols L, Alobid I, et al. Low Prostaglandin E(2) and Cyclooxygenase Expression in Nasal Mucosa Fibroblasts of Aspirin-Intolerant Asthmatics. Respirology. 2013. [DOI] [PubMed] [Google Scholar]
- 7.Machado-Carvalho L, Martin M, Torres R, Gabasa M, Alobid I, Mullol J, et al. Low E-prostanoid 2 receptor levels and deficient induction of the IL-1beta/IL-1 type I receptor/COX-2 pathway: Vicious circle in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2016;137(1):99–107. [DOI] [PubMed] [Google Scholar]
- 8.Corrigan CJ, Napoli RL, Meng Q, Fang C, Wu H, Tochiki K, et al. Reduced expression of the prostaglandin E2 receptor E-prostanoid 2 on bronchial mucosal leukocytes in patients with aspirin-sensitive asthma. J Allergy Clin Immunol. 2012;129(6):1636–46. [DOI] [PubMed] [Google Scholar]
- 9.Ying S, Meng Q, Scadding G, Parikh A, Corrigan CJ, Lee TH. Aspirin-sensitive rhinosinusitis is associated with reduced E-prostanoid 2 receptor expression on nasal mucosal inflammatory cells. J Allergy Clin Immunol. 2006;117(2):312–8. [DOI] [PubMed] [Google Scholar]
- 10.Cahill KN, Raby BA, Zhou X, Guo F, Thibault D, Baccarelli A, et al. Impaired E Prostanoid2 Expression and Resistance to Prostaglandin E2 in Nasal Polyp Fibroblasts from Subjects with Aspirin-Exacerbated Respiratory Disease. Am J Respir Cell Mol Biol. 2016;54(1):34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sestini P, Armetti L, Gambaro G, Pieroni MG, Refini RM, Sala A, et al. Inhaled PGE2 prevents aspirin-induced bronchoconstriction and urinary LTE4 excretion in aspirin-sensitive asthma. Am J Respir Crit Care Med. 1996;153(2):572–5. [DOI] [PubMed] [Google Scholar]
- 12.Liu T, Laidlaw TM, Katz HR, Boyce JA. Prostaglandin E2 deficiency causes a phenotype of aspirin sensitivity that depends on platelets and cysteinyl leukotrienes. Proc Natl Acad Sci U S A. 2013;110(42):16987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weiss JW, Drazen JM, McFadden ER Jr., Weller P, Corey EJ, Lewis RA, et al. Airway constriction in normal humans produced by inhalation of leukotriene D. Potency, time course, and effect of aspirin therapy. JAMA. 1983;249(20):2814–7. [PubMed] [Google Scholar]
- 14.Christie PE, Tagari P, Ford-Hutchinson AW, Charlesson S, Chee P, Arm JP, et al. Urinary leukotriene E4 concentrations increase after aspirin challenge in aspirin-sensitive asthmatic subjects. Am Rev Respir Dis. 1991;143(5 Pt 1):1025–9. [DOI] [PubMed] [Google Scholar]
- 15.Arm JP, O’Hickey SP, Spur BW, Lee TH. Airway responsiveness to histamine and leukotriene E4 in subjects with aspirin-induced asthma. Am Rev Respir Dis. 1989;140(1):148–53. [DOI] [PubMed] [Google Scholar]
- 16.Fischer AR, Rosenberg MA, Lilly CM, Callery JC, Rubin P, Cohn J, et al. Direct evidence for a role of the mast cell in the nasal response to aspirin in aspirin-sensitive asthma. J Allergy Clin Immunol. 1994;94(6 Pt 1):1046–56. [DOI] [PubMed] [Google Scholar]
- 17.Israel E, Fischer AR, Rosenberg MA, Lilly CM, Callery JC, Shapiro J, et al. The pivotal role of 5-lipoxygenase products in the reaction of aspirin-sensitive asthmatics to aspirin. Am Rev Respir Dis. 1993;148(6 Pt 1):1447–51. [DOI] [PubMed] [Google Scholar]
- 18.Laidlaw TM, Fuentes DJ, Wang Y. Efficacy of Zileuton in Patients with Asthma and History of Aspirin Sensitivity: A Retrospective Analysis of Data from Two Phase 3 Studies. AAAAI: J Allergy Clin Immunol; 2017. p. AB384. [Google Scholar]
- 19.Bankova LG, Lai J, Yoshimoto E, Boyce JA, Austen KF, Kanaoka Y, et al. Leukotriene E4 elicits respiratory epithelial cell mucin release through the G-protein-coupled receptor, GPR99. Proc Natl Acad Sci U S A. 2016;113(22):6242–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu T, Barrett NA, Kanaoka Y, Yoshimoto E, Garofalo D, Cirka H, et al. Type 2 Cysteinyl Leukotriene Receptors Drive IL-33-Dependent Type 2 Immunopathology and Aspirin Sensitivity. J Immunol. 2018;200(3):915–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu T, Barrett NA, Kanaoka Y, Buchheit K, Laidlaw TM, Garofalo D, et al. Cysteinyl leukotriene receptor 2 drives lung immunopathology through a platelet and high mobility box 1-dependent mechanism. Mucosal Immunol. 2019;12(3):679–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bankova LG, Dwyer DF, Yoshimoto E, Ualiyeva S, McGinty JW, Raff H, et al. The cysteinyl leukotriene 3 receptor regulates expansion of IL-25-producing airway brush cells leading to type 2 inflammation. Sci Immunol. 2018;3(28). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.von MJ, Ji M, Liang HE, Locksley RM. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature. 2016;529(7585):221–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ualiyeva S, Lemire E, Aviles EC, Wong C, Boyd AA, Lai J, et al. Tuft cell-produced cysteinyl leukotrienes and IL-25 synergistically initiate lung type 2 inflammation. Sci Immunol. 2021;6(66):eabj0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ualiyeva S, Hallen N, Kanaoka Y, Ledderose C, Matsumoto I, Junger WG, et al. Airway brush cells generate cysteinyl leukotrienes through the ATP sensor P2Y2. Science Immunology. 2020;5(43):eaax7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, Broide DH. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J Allergy Clin Immunol. 2013;132(1):205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.von Moltke J, O’Leary CE, Barrett NA, Kanaoka Y, Austen KF, Locksley RM. Leukotrienes provide an NFAT-dependent signal that synergizes with IL-33 to activate ILC2s. J Exp Med. 2017;214(1):27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGinty JW, Ting H-A, Billipp TE, Nadjsombati MS, Khan DM, Barrett NA, et al. Tuft-Cell-Derived Leukotrienes Drive Rapid Anti-helminth Immunity in the Small Intestine but Are Dispensable for Anti-protist Immunity. Immunity. 2020;52(3):528–41.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stevenson DD, Simon RA, Mathison DA, Christiansen SC. Montelukast is only partially effective in inhibiting aspirin responses in aspirin-sensitive asthmatics. Ann Allergy Asthma Immunol. 2000;85(6 Pt 1):477–82. [DOI] [PubMed] [Google Scholar]
- 30.Mitson-Salazar A, Prussin C. Pathogenic Effector Th2 Cells in Allergic Eosinophilic Inflammatory Disease. Front Med (Lausanne). 2017;4:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maric J, Ravindran A, Mazzurana L, Van Acker A, Rao A, Kokkinou E, et al. Cytokine-induced endogenous production of prostaglandin D2 is essential for human group 2 innate lymphoid cell activation. J Allergy Clin Immunol. 2019;143(6):2202–14 e5. [DOI] [PubMed] [Google Scholar]
- 32.Buchheit KM, Cahill KN, Katz HR, Murphy KC, Feng C, Lee-Sarwar K, et al. Thymic stromal lymphopoietin controls prostaglandin D2 generation in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2016;137(5):1566–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cahill KN, Bensko JC, Boyce JA, Laidlaw TM. Prostaglandin D2: A dominant mediator of aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2015;135(1):245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med. 2001;193(2):255–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xue L, Salimi M, Panse I, Mjosberg JM, McKenzie AN, Spits H, et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol. 2014;133(4):1184–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xue L, Gyles SL, Wettey FR, Gazi L, Townsend E, Hunter MG, et al. Prostaglandin D2 causes preferential induction of proinflammatory Th2 cytokine production through an action on chemoattractant receptor-like molecule expressed on Th2 cells. J Immunol. 2005;175(10):6531–6. [DOI] [PubMed] [Google Scholar]
- 37.Price CPE, Guo A, Stevens WW, Cousens L, Vu TT, Suh LA, et al. Efficacy of an oral CRTH2 antagonist (AZD1981) in the treatment of chronic rhinosinusitis with nasal polyps in adults: a randomized controlled clinical trial. Clin Exp Allergy. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gevaert P, Bachert C, Maspero JF, Cuevas M, Steele D, Acharya S, et al. Phase 3b randomized controlled trial of fevipiprant in patients with nasal polyposis with asthma (THUNDER). J Allergy Clin Immunol. 2022;149(5):1675–82 e3. [DOI] [PubMed] [Google Scholar]
- 39.Cahill KN. Fevipiprant in CRSwNP and comorbid asthma: Wrong target population or wrong PGD2 receptor? J Allergy Clin Immunol. 2022;149(5):1587–9. [DOI] [PubMed] [Google Scholar]
- 40.Cowburn AS, Sladek K, Soja J, Adamek L, Nizankowska E, Szczeklik A, et al. Overexpression of leukotriene C4 synthase in bronchial biopsies from patients with aspirin-intolerant asthma. J Clin Invest. 1998;101(4):834–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nasser SM, Pfister R, Christie PE, Sousa AR, Barker J, Schmitz-Schumann M, et al. Inflammatory cell populations in bronchial biopsies from aspirin-sensitive asthmatic subjects. Am J Respir Crit Care Med. 1996;153(1):90–6. [DOI] [PubMed] [Google Scholar]
- 42.Laidlaw TM, Prussin C, Panettieri RA, Lee S, Ferguson BJ, Adappa ND, et al. Dexpramipexole depletes blood and tissue eosinophils in nasal polyps with no change in polyp size. Laryngoscope. 2019;129(2):E61–E6. [DOI] [PubMed] [Google Scholar]
- 43.Mitsui C, Kajiwara K, Hayashi H, Ito J, Mita H, Ono E, et al. Platelet activation markers overexpressed specifically in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2015. [DOI] [PubMed] [Google Scholar]
- 44.Takahashi T, Kato A, Berdnikovs S, Stevens WW, Suh LA, Norton JE, et al. Microparticles in nasal lavage fluids in chronic rhinosinusitis: Potential biomarkers for diagnosis of aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2017;140(3):720–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maclouf J, Murphy RC, Henson PM. Transcellular biosynthesis of sulfidopeptide leukotrienes during receptor-mediated stimulation of human neutrophil/platelet mixtures. Blood. 1990;76(9):1838–44. [PubMed] [Google Scholar]
- 46.Laidlaw TM, Kidder MS, Bhattacharyya N, Xing W, Shen S, Milne GL, et al. Cysteinyl leukotriene overproduction in aspirin-exacerbated respiratory disease is driven by platelet-adherent leukocytes. Blood. 2012;119(16):3790–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laidlaw TM, Cahill KN, Cardet JC, Murphy K, Cui J, Dioneda B, et al. A trial of type 12 purinergic (P2Y12) receptor inhibition with prasugrel identifies a potentially distinct endotype of patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dwyer DF, Ordovas-Montanes J, Allon SJ, Buchheit KM, Vukovic M, Derakhshan T, et al. Human airway mast cells proliferate and acquire distinct inflammation-driven phenotypes during type 2 inflammation. Sci Immunol. 2021;6(56). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bochenek G, Nagraba K, Nizankowska E, Szczeklik A. A controlled study of 9alpha,11beta-PGF2 (a prostaglandin D2 metabolite) in plasma and urine of patients with bronchial asthma and healthy controls after aspirin challenge. J Allergy Clin Immunol. 2003;111(4):743–9. [DOI] [PubMed] [Google Scholar]
- 50.Imokawa S, Sato A, Taniguchi M, Toyoshima M, Nakazawa K, Hayakawa H, et al. [Sodium cromoglycate nebulized solution has an acute bronchodilative effect in patients with aspirin-intolerant asthma (AIA)]. Arerugi = [Allergy]. 1992;41(10):1515–20. [PubMed] [Google Scholar]
- 51.Amayasu H, Nakabayashi M, Akahori K, Ishizaki Y, Shoji T, Nakagawa H, et al. Cromolyn sodium suppresses eosinophilic inflammation in patients with aspirin-intolerant asthma. Ann Allergy Asthma Immunol. 2001;87(2):146–50. [DOI] [PubMed] [Google Scholar]
- 52.Yoshida S, Amayasu H, Sakamoto H, Onuma K, Shoji T, Nakagawa H, et al. Cromolyn sodium prevents bronchoconstriction and urinary LTE4 excretion in aspirin-induced asthma. Ann Allergy Asthma Immunol. 1998;80(2):171–6. [DOI] [PubMed] [Google Scholar]
- 53.Eastman JJ, Cavagnero KJ, Deconde AS, Kim AS, Karta MR, Broide DH, et al. Group 2 innate lymphoid cells are recruited to the nasal mucosa in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2017;140(1):101–8 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salimi M, Stoger L, Liu W, Go S, Pavord I, Klenerman P, et al. Cysteinyl leukotriene E4 activates human group 2 innate lymphoid cells and enhances the effect of prostaglandin D2 and epithelial cytokines. J Allergy Clin Immunol. 2017;140(4):1090–100 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kupczyk M, Kurmanowska Z, Kuprys-Lipinska I, Bochenska-Marciniak M, Kuna P. Mediators of inflammation in nasal lavage from aspirin intolerant patients after aspirin challenge. Respir Med. 2010;104(10):1404–9. [DOI] [PubMed] [Google Scholar]
- 56.Buchheit KM, Dwyer DF, Ordovas-Montanes J, Katz HR, Lewis E, Vukovic M, et al. IL-5Ralpha marks nasal polyp IgG4 and IgE-expressing cells in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ordovas-Montanes J, Dwyer DF, Nyquist SK, Buchheit KM, Vukovic M, Deb C, et al. Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature. 2018;560(7720):649–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stevens WW, Staudacher AG, Hulse KE, Carter RG, Winter DR, Kato A, et al. Activation of the 15-lipoxygenase pathway in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell. 2017;171(3):628–41 e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nagasaki T, Schuyler AJ, Zhao J, Samovich SN, Yamada K, Deng Y, et al. 15LO1 dictates glutathione redox changes in asthmatic airway epithelium to worsen type 2 inflammation. J Clin Invest. 2022;132(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laidlaw TM, Buchheit KM. Biologics in chronic rhinosinusitis with nasal polyposis. Ann Allergy Asthma Immunol. 2020;124(4):326–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hayashi H, Mitsui C, Nakatani E, Fukutomi Y, Kajiwara K, Watai K, et al. Omalizumab reduces cysteinyl leukotriene and 9alpha,11beta-prostaglandin F2 overproduction in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2016;137(5):1585–7.e4. [DOI] [PubMed] [Google Scholar]
- 63.Lang DM, Aronica MA, Maierson ES, Wang XF, Vasas DC, Hazen SL. Omalizumab can inhibit respiratory reaction during aspirin desensitization. Ann Allergy Asthma Immunol. 2018;121(1):98–104. [DOI] [PubMed] [Google Scholar]
- 64.Tuttle KL, Buchheit KM, Laidlaw TM, Cahill KN. A retrospective analysis of mepolizumab in subjects with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol Pract. 2018;6(3):1045–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Buchheit KM, Lewis E, Gakpo D, Hacker J, Sohail A, Taliaferro F, et al. Mepolizumab targets multiple immune cells in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2021;148(2):574–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martin H, Barrett NA, Laidlaw T. Mepolizumab does not prevent all aspirin-induced reactions in patients with aspirin-exacerbated respiratory disease: A case series. J Allergy Clin Immunol Pract. 2021;9(3):1384–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Scott WC, Cahill KN, Milne GL, Li P, Sheng Q, Huang LC, et al. Inflammatory heterogeneity in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2021;147(4):1318–28 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bachert C, Mannent L, Naclerio RM, Mullol J, Ferguson BJ, Gevaert P, et al. Effect of Subcutaneous Dupilumab on Nasal Polyp Burden in Patients With Chronic Sinusitis and Nasal Polyposis: A Randomized Clinical Trial. JAMA. 2016;315(5):469–79. [DOI] [PubMed] [Google Scholar]
- 69.Laidlaw TM, Mullol J, Fan C, Zhang D, Amin N, Khan A, et al. Dupilumab improves nasal polyp burden and asthma control in patients with CRSwNP and AERD. J Allergy Clin Immunol Pract. 2019;7(7):2462–5 e1. [DOI] [PubMed] [Google Scholar]
- 70.Bavaro N, Gakpo D, Mittal A, Bensko JC, Laidlaw TM, Buchheit KM. Efficacy of dupilumab in patients with aspirin-exacerbated respiratory disease and previous inadequate response to anti-IL-5 or anti-IL-5Ralpha in a real-world setting. J Allergy Clin Immunol Pract. 2021;9(7):2910–2 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buchheit KM, Sohail A, Hacker J, Maurer R, Gakpo D, Bensko JC, et al. Rapid and sustained effect of dupilumab on clinical and mechanistic outcomes in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Patel P, Bensko JC, Bhattacharyya N, Laidlaw TM, Buchheit KM. Dupilumab as an adjunct to surgery in patients with aspirin-exacerbated respiratory disease. Ann Allergy Asthma Immunol. 2022;128(3):326–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Trudeau J, Hu H, Chibana K, Chu HW, Westcott JY, Wenzel SE. Selective downregulation of prostaglandin E2-related pathways by the Th2 cytokine IL-13. J Allergy Clin Immunol. 2006;117(6):1446–54. [DOI] [PubMed] [Google Scholar]
- 74.Mustafa SS, Vadamalai K. Dupilumab increases aspirin tolerance in aspirin-exacerbated respiratory disease. Ann Allergy Asthma Immunol. 2021;126(6):738–9. [DOI] [PubMed] [Google Scholar]
- 75.Liu T, Kanaoka Y, Barrett NA, Feng C, Garofalo D, Lai J, et al. Aspirin-Exacerbated Respiratory Disease Involves a Cysteinyl Leukotriene-Driven IL-33-Mediated Mast Cell Activation Pathway. J Immunol. 2015;195(8):3537–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu T, Barrett NA, Kanaoka Y, Buchheit K, Laidlaw TM, Garofalo D, et al. Cysteinyl leukotriene receptor 2 drives lung immunopathology through a platelet and high mobility box 1-dependent mechanism. Mucosal immunology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Menzies-Gow A, Corren J, Bourdin A, Chupp G, Israel E, Wechsler ME, et al. Tezepelumab in Adults and Adolescents with Severe, Uncontrolled Asthma. N Engl J Med. 2021;384(19):1800–9. [DOI] [PubMed] [Google Scholar]
- 78.Kelsen SG, Agache IO, Soong W, Israel E, Chupp GL, Cheung DS, et al. Astegolimab (anti-ST2) efficacy and safety in adults with severe asthma: A randomized clinical trial. J Allergy Clin Immunol. 2021;148(3):790–8. [DOI] [PubMed] [Google Scholar]
- 79.Wechsler ME, Ruddy MK, Pavord ID, Israel E, Rabe KF, Ford LB, et al. Efficacy and Safety of Itepekimab in Patients with Moderate-to-Severe Asthma. New England Journal of Medicine. 2021;385(18):1656–68. [DOI] [PubMed] [Google Scholar]
- 80.Berges-Gimeno MP, Simon RA, Stevenson DD. Long-term treatment with aspirin desensitization in asthmatic patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2003;111(1):180–6. [DOI] [PubMed] [Google Scholar]
- 81.Cahill KN, Cui J, Kothari P, Murphy K, Raby BA, Singer J, et al. Unique Effect of Aspirin Therapy on Biomarkers in Aspirin-exacerbated Respiratory Disease. A Prospective Trial. Am J Respir Crit Care Med. 2019;200(6):704–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sousa AR, Parikh A, Scadding G, Corrigan CJ, Lee TH. Leukotriene-receptor expression on nasal mucosal inflammatory cells in aspirin-sensitive rhinosinusitis. N Engl J Med. 2002;347(19):1493–9. [DOI] [PubMed] [Google Scholar]
- 83.Picado C, Fernandez-Morata JC, Juan M, Roca-Ferrer J, Fuentes M, Xaubet A, et al. Cyclooxygenase-2 mRNA is downexpressed in nasal polyps from aspirin-sensitive asthmatics. Am J Respir Crit Care Med. 1999;160(1):291–6. [DOI] [PubMed] [Google Scholar]
- 84.Chang JE, Ding D, Martin-Lazaro J, White A, Stevenson DD. Smoking, environmental tobacco smoke, and aspirin-exacerbated respiratory disease. Ann Allergy Asthma Immunol. 2012;108(1):14–9. [DOI] [PubMed] [Google Scholar]
- 85.Hayashi H, Fukutomi Y, Mitsui C, Nakatani E, Watai K, Kamide Y, et al. Smoking Cessation as a Possible Risk Factor for the Development of Aspirin-Exacerbated Respiratory Disease in Smokers. J Allergy Clin Immunol Pract. 2018;6(1):116–25 e3. [DOI] [PubMed] [Google Scholar]
- 86.Samter M, Beers RF, Jr. Intolerance to aspirin. Clinical studies and consideration of its pathogenesis. Ann Intern Med. 1968;68(5):975–83. [DOI] [PubMed] [Google Scholar]
- 87.Pace S, Pergola C, Dehm F, Rossi A, Gerstmeier J, Troisi F, et al. Androgen-mediated sex bias impairs efficiency of leukotriene biosynthesis inhibitors in males. J Clin Invest. 2017;127(8):3167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Laffont S, Blanquart E, Savignac M, Cenac C, Laverny G, Metzger D, et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J Exp Med. 2017;214(6):1581–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pace S, Rossi A, Krauth V, Dehm F, Troisi F, Bilancia R, et al. Sex differences in prostaglandin biosynthesis in neutrophils during acute inflammation. Sci Rep. 2017;7(1):3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kolmert J, Gomez C, Balgoma D, Sjodin M, Bood J, Konradsen JR, et al. Urinary Leukotriene E4 and Prostaglandin D2 Metabolites Increase in Adult and Childhood Severe Asthma Characterized by Type-2 Inflammation. Am J Respir Crit Care Med. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Szczeklik A Aspirin-induced asthma as a viral disease. Clin Allergy. 1988;18(1):15–20. [DOI] [PubMed] [Google Scholar]
- 92.Zaravinos A, Bizakis J, Spandidos DA. Prevalence of human papilloma virus and human herpes virus types 1–7 in human nasal polyposis. J Med Virol. 2009;81(9):1613–9. [DOI] [PubMed] [Google Scholar]
- 93.Sachse F, Becker K, von Eiff C, Metze D, Rudack C. Staphylococcus aureus invades the epithelium in nasal polyposis and induces IL-6 in nasal epithelial cells in vitro. Allergy. 2010;65(11):1430–7. [DOI] [PubMed] [Google Scholar]
- 94.Bachert C, Gevaert P, Holtappels G, Johansson SG, van Cauwenberge P. Total and specific IgE in nasal polyps is related to local eosinophilic inflammation. J Allergy Clin Immunol. 2001;107(4):607–14. [DOI] [PubMed] [Google Scholar]
- 95.Tan BK, Li QZ, Suh L, Kato A, Conley DB, Chandra RK, et al. Evidence for intranasal antinuclear autoantibodies in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2011;128(6):1198–206 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kato A, Peters A, Suh L, Carter R, Harris KE, Chandra R, et al. Evidence of a role for B cell-activating factor of the TNF family in the pathogenesis of chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2008;121(6):1385–92, 92 e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jeffe JS, Seshadri S, Hamill KJ, Huang JH, Carter R, Suh L, et al. A role for anti-BP180 autoantibodies in chronic rhinosinusitis. Laryngoscope. 2013;123(9):2104–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mendelsohn D, Jeremic G, Wright ED, Rotenberg BW. Revision rates after endoscopic sinus surgery: a recurrence analysis. Ann Otol Rhinol Laryngol. 2011;120(3):162–6. [DOI] [PubMed] [Google Scholar]
- 99.Cheong HS, Park SM, Kim MO, Park JS, Lee JY, Byun JY, et al. Genome-wide methylation profile of nasal polyps: relation to aspirin hypersensitivity in asthmatics. Allergy. 2011;66(5):637–44. [DOI] [PubMed] [Google Scholar]
- 100.Wu J, Chandra RK, Li P, Hull BP, Turner JH. Olfactory and middle meatal cytokine levels correlate with olfactory function in chronic rhinosinusitis. Laryngoscope. 2018;128(9):E304–E10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tognetti A, Sarolidou G, Lasselin J, Lekander M, Olsson MJ, Lundstrom JN. Acute Systemic Experimental Inflammation Does Not Reduce Human Odor Identification Performance. Chem Senses. 2021;46. [DOI] [PMC free article] [PubMed] [Google Scholar]