

Abstract
Mismatch repair-deficient (MMRd) cancers have varied responses to immune checkpoint blockade (ICB). We conducted a phase 2 clinical trial of the PD-1 inhibitor pembrolizumab in 24 patients with MMRd endometrial cancer (NCT02899793). Patients with mutational MMRd tumors (6 patients) had higher response rates and longer survival than those with epigenetic MMRd tumors (18 patients). Mutation burden was higher in tumors with mutational MMRd compared to epigenetic MMRd; however, within each category of MMRd, mutation burden was not correlated with ICB response. Pre-treatment JAK1 mutations were not associated with primary resistance to pembrolizumab. Longitudinal single-cell RNA-seq of circulating immune cells revealed contrasting modes of anti-tumor immunity for mutational vs. epigenetic MMRd cancers. Whereas effector CD8+ T cells correlated with regression of mutational MMRd tumors, activated CD16+ NK cells were associated with ICB-responsive epigenetic MMRd tumors. These data highlight the interplay between tumor-intrinsic and extrinsic factors that influence ICB response.
Introduction
Mismatch repair-deficient (MMRd) cancers are characterized by exceptionally high mutational loads, particularly at repetitive microsatellite regions that are prone to replication errors (microsatellite instability-high, MSI-H)(1,2). The high mutation burden in MSI-H/MMRd tumors is thought to result in the generation of ample tumor-specific neoantigens that can subsequently be targeted by the immune system in the setting of immune checkpoint blockade (ICB)(3,4). As the first tissue-agnostic ICB therapy approved by the FDA, the PD-1 checkpoint inhibitor pembrolizumab received accelerated approval in 2017 for the treatment of MSI-H/MMRd solid tumors that have progressed on prior lines of therapy(5–8). However, even among MSI-H/MMRd tumors, there is considerable variability in response rates to ICB, both within and across cancer types. For patients with MSI-H/MMRd colorectal cancer or endometrial cancer, 44% and 57% of patients demonstrated an objective radiographic response to PD-1 ICB, respectively(5). In stark contrast, only 18% of patients with MSI-H/MMRd pancreatic cancer and 0% of patients with brain cancer responded to PD-1 ICB(7). To increase the proportion of patients that derive benefit from ICB, it is crucial to identify the genetic, immunologic, and environmental factors underlying these variations in response.
MSI-H/MMRd status is commonly determined by PCR of microsatellite regions and/or immunohistochemistry (IHC) for key MMR proteins(9), and such assays were utilized in the clinical trials that demonstrated the efficacy of pembrolizumab for MSI-H/MMRd solid tumors. However, neither PCR nor IHC can provide insight into the underlying molecular mechanisms of MMRd. Whereas some tumors exhibit somatic loss-of-function mutations in key MMR genes (hereafter termed mutational MMRd, or mut-MMRd), more than 70% of MSI-H/MMRd endometrial cancer cases(10) instead exhibit epigenetic silencing of the MLH1 gene promoter through DNA methylation (epigenetic MMRd, or epi-MMRd)(9).
We recently conducted a phase 2 trial (NCT02899793) evaluating the efficacy of pembrolizumab in patients with MSI-H/MMRd endometrial cancers (n = 24 evaluable patients)(11). In this trial, we classified patients into two groups based on the putative molecular mechanism driving MMRd (i.e., mut-MMRd or epi-MMRd). We observed a 100% objective response rate (ORR) for mut-MMRd patients, compared to an ORR of 44% for epi-MMRd patients(11). Accordingly, mut-MMRd patients had a more rapid clinical response, as well as significantly longer progression-free survival and overall survival compared to epi-MMRd patients, suggesting important differences in the immunologic response to PD-1 ICB across these two subtypes of MSI-H/MMRd tumors.
We therefore hypothesized that certain immunologic parameters may vary between patients with mut-MMRd or epi-MMRd tumors, providing a potential mechanistic explanation for the variable therapeutic benefit of PD-1 ICB across MSI-H/MMRd endometrial cancers. Here, we report a comprehensive characterization of the tumor genomes and circulating immune responses in the participants of our recent phase 2 clinical trial. We performed single cell RNA-sequencing (scRNA-seq) and matched T cell receptor repertoire sequencing (scTCR-seq) of peripheral blood mononuclear cells (PBMCs) from these patients, before and after treatment with pembrolizumab, along with pre-treatment bulk TCR-seq of tumor-infiltrating lymphocytes (TILs) and tumor exome sequencing. By interrogating this dataset, we uncover two distinct axes of anti-tumor immunity that appear to be differentially engaged by PD-1 ICB in patients with mut-MMRd vs. epi-MMRd tumors.
Results
The molecular mechanism of mismatch repair deficiency impacts tumor mutation burden and response to immunotherapy
Study participants were patients with recurrent MSI-H/MMRd endometrial carcinoma that enrolled in our previously described single-arm, open-label phase 2 clinical trial evaluating the efficacy of pembrolizumab in this population (n = 24 evaluable patients)(11). The molecular mechanism of MMRd was determined by MLH1 promoter methylation analysis, FoundationOne profiling, whole exome sequencing (WES), PCR, and IHC for MMR proteins. Six patients were classified as mut-MMRd and the remaining 18 patients as epi-MMRd (Supplementary Table S1)(11).
Response to pembrolizumab (200 mg of pembrolizumab every 3 weeks for ≤ 2 years) was assessed by RECIST 1.1 criteria(12), defining responders as patients with a ≥ 30% decrease in tumor size (Figure 1A). All mut-MMRd patients demonstrated an objective response to pembrolizumab (6/6, 100%), while 44% of epi-MMRd patients responded (8/18) (Figure 1B–C). As we were interested in understanding the factors that distinguished epi-MMRd responders from non-responders, we subsequently categorized the patients into 3 groups for further analysis: epi-MMRd non-responders (NR; n = 10), epi-MMRd responders (epiR; n = 8), and mut-MMRd responders (mutR; n = 6). Patient ages were similar across the three groups (Supplementary Figure S1A).
Figure 1: The molecular mechanism of MMRd influences mutation burden and response to PD-1 immunotherapy.
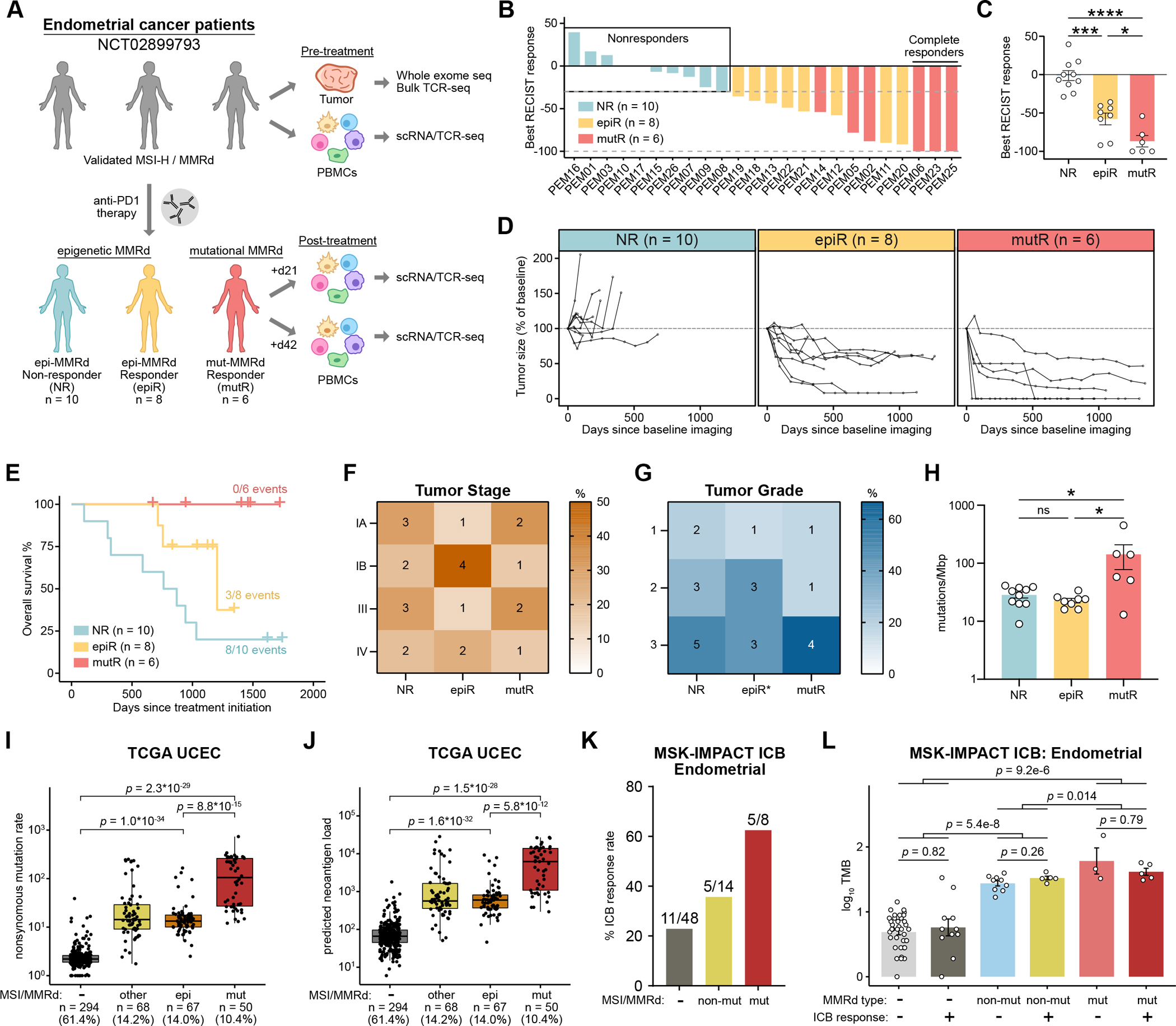
A. Schematic of study design and sample collection strategy. Patients with MSI-H/MMRd endometrial cancer were categorized into three groups based on the molecular mechanism of MMRd, as well as their subsequent response to pembrolizumab: non-responder (NR, n = 10), epigenetic-MMRd responder (epiR, n = 8), and mutational-MMRd responder (mutR, n = 6).
B. The maximum percentage reduction in tumor size from baseline, based on RECIST criteria (n = 24 evaluable patients).
C. Comparison of maximal RECIST response across the three patient groups. Statistical significance was assessed by one-way ANOVA with Tukey’s multiple comparisons testxs.
D. Tumor size over time in each patient, relative to baseline imaging obtained prior to pembrolizumab initiation.
E. Kaplan-Meier overall survival curves, calculated relative to the timepoint of pembrolizumab initiation in each patient.
F-G. Heatmap of tumor stage (F) and grade (G) annotations across groups. Cells are colored by the percentage of patients within each patient category, with the number of patients indicated in each cell. Tumor grade annotation was not available for one epiR patient.
H. Tumor mutation burden across the three patient groups. Statistical significance was assessed by one-way ANOVA with Tukey’s multiple comparisons test.
I-J. Nonsynonymous mutation rate (I) and predicted neoantigen load (J) across the TCGA UCEC cohort, with patients categorized by MSI status and the molecular mechanism of MMRd. Statistical significance was assessed by two-tailed unpaired Mann-Whitney test.
K. Response rates to ICB therapy among endometrial cancer patients in the MSK-IMPACT ICB cohort. Patients are categorized by MSI status and the presence of mutations in canonical MMR genes.
L. Mutation burden across the three groups of endometrial cancer patients in the MSK-IMPACT ICB cohort, further grouped by ICB response. Statistical significance was assessed by two-tailed unpaired Mann-Whitney test.
Longitudinal analysis of tumor sizes revealed that responses to pembrolizumab were sustained for well over a year, with faster response kinetics in mutR patients (Figure 1D). As we previously reported, mut-MMRd patients had longer progression-free survival and overall survival compared to epi-MMRd patients (Figure 1E, Supplementary Figure S1B–C). Tumor stage and grade were comparable across the three groups of patients (Figure 1F–G). Importantly, tumor mutation burden (TMB) was significantly higher in mutR patients compared to NR or epiR patients, while TMB was not significantly different between NR and epiR patients (Figure 1H). Using a set of high-confidence somatic exonic mutations, we predicted the number of MHC-I and MHC-II neoantigens in each sample, revealing a similar trend towards increased neoantigen counts in mutR patients, though not statistically significant (Supplementary Figure S2A–C, Supplementary Table S2).
To corroborate these findings in an independent cohort, we examined the uterine endometrial carcinoma cohort from The Cancer Genome Atlas (TCGA UCEC)(13). 75.4% of the tumors in this cohort were classified as endometrioid carcinomas. We categorized the tumors into four groups: 1) MMR-proficient (MMRp), 2) epi-MMRd, 3) mut-MMRd, and 4) MMRd, not otherwise specified. To define epi-MMRd tumors, we identified tumors that were previously classified as MSI-H and had a methylation beta-value ≥ 0.5 for the MLH1 promoter (Supplementary Figure S2D). To define mut-MMRd samples, we identified tumors that were previously classified as MSI-H(14) and that exhibited mutations in a canonical MMR gene (MLH1, MSH2, MSH6, or PMS2), but did not have MLH1 promoter methylation. As a validation of the beta-value threshold, we confirmed that samples with a methylation beta-value ≥ 0.5 for the MLH1 promoter had significantly decreased expression of MLH1 (Supplementary Figure S2E). There was no association between tumor stage and the four categories of tumors (Supplementary Figure S2F). Comparing across groups, we observed that all MSI-H/MMRd groups had significantly higher TMB than MMRp tumors, as expected (Figure 1I). We further observed that mut-MMRd tumors had significantly higher mutation rates than epi-MMRd tumors, confirming our observations with the trial cohort (Figure 1I). Similarly, all MSI-H/MMRd groups had significantly higher predicted neoantigen load(14) than MMRp tumors (Figure 1J), with mut-MMRd tumors exhibiting the highest numbers of predicted neoantigens. Thus, analysis of an independent endometrial cancer cohort confirmed the relationship between the molecular mechanism of MMRd and TMB.
Of note, the patients profiled in the TCGA UCEC cohort were not treated with ICB therapy. To further investigate whether mut-MMRd tumors have higher ICB response rates than epi-MMRd tumors, we reanalyzed data from the MSK-IMPACT cohort of cancer patients treated with ICB therapy(15–17). Since methylation data were not available for this cohort, we instead categorized the endometrial cancer patients into three groups: 1) MMRp, 2) mut-MMRd, and 3) non-mutational MMRd (non-mut-MMRd). In the MSK-IMPACT cohort, mut-MMRd patients were defined by the combination of MSI-H status and presence of mutations in MLH1, MSH2, MSH6, or PMS2. In contrast, non-mut-MMRd patients were defined by the combination of MSI-H status and the absence of mutations in MLH1, MSH2, MSH6, or PMS2. The classification scheme for non-mut-MMRd tumors in the MSK-IMPACT cohort was thus different from that used to define epi-MMRd in our own cohort or in the TCGA cohort; this approach represented the closest possible surrogate given the available data. Comparing the response rates to ICB therapy across the three groups, we observed that mut-MMRd patients had the highest response rate (5/8, or 63%), while non-mut-MMRd patients and MMRp patients had response rates of 36% and 23%, respectively (Figure 1K). These data further corroborate our finding that mut-MMRd tumors have higher response rates to ICB therapy than non-mutational MMRd tumors, albeit with the limitation that MLH1 promoter methylation data were not available for the MSK-IMPACT cohort.
Given the association between mut-MMRd and higher TMB, one might expect that differences in tumor immunogenicity due to neoantigen load are sufficient to explain the varying ICB responses between mut-MMRd and epi-MMRd patients(4). However, when we further stratified patients by response status, we observed no significant differences in TMB between responders and non-responders within each group (Figure 1L). This finding confirmed our earlier observation that NR and epiR patients had comparable TMB (Figure 1H). Thus, while the molecular mechanism of MMRd is reproducibly associated with TMB, in the setting of elevated neoantigen load (e.g., among MSI-H/MMRd endometrial cancers), factors beyond TMB are crucial in shaping the anti-tumor immune response upon ICB therapy.
Association between specific gene mutations and immunotherapy response
Analysis of computationally-predicted pathogenic and deleterious mutations revealed several recurrently mutated genes across the patient cohort (Figure 2A), including PTEN, ARID1A, RPL22, KMT2B, and KMT2D (Supplementary Table S3). Across the entire cohort, only mutations in ARID1A and CTCF were nominally associated with response to pembrolizumab, with the limitation of the small sample size for this clinical study of phase 2 trial participants (Figure 2B). CTCF encodes a key factor for three-dimensional (3D) chromatin organization(18), and CTCF binding sites are often mutated in cancer(19). We observed that mutations in CTCF were associated with response to pembrolizumab (Figure 2C), suggesting that dysregulated 3D chromatin structure may promote immunotherapy response in this context. On the other hand, ARID1A encodes a subunit of the BAF SWI/SNF chromatin remodeling complex(20), and is frequently mutated across multiple cancer types(21). While ARID1A deficiency has previously been associated with enhanced ICB responsiveness(22), possibly through loss of its interactions with MMR machinery, we found that ARID1A mutations were associated with resistance to pembrolizumab (Figure 2C). This difference may stem from the fact that our current study was restricted to patients with MSI-H/MMRd tumors, as the sensitizing effect of ARID1A deficiency through loss of MMR function would not be relevant in this setting.
Figure 2: JAK1 mutations do not confer primary resistance to PD-1 checkpoint immunotherapy.
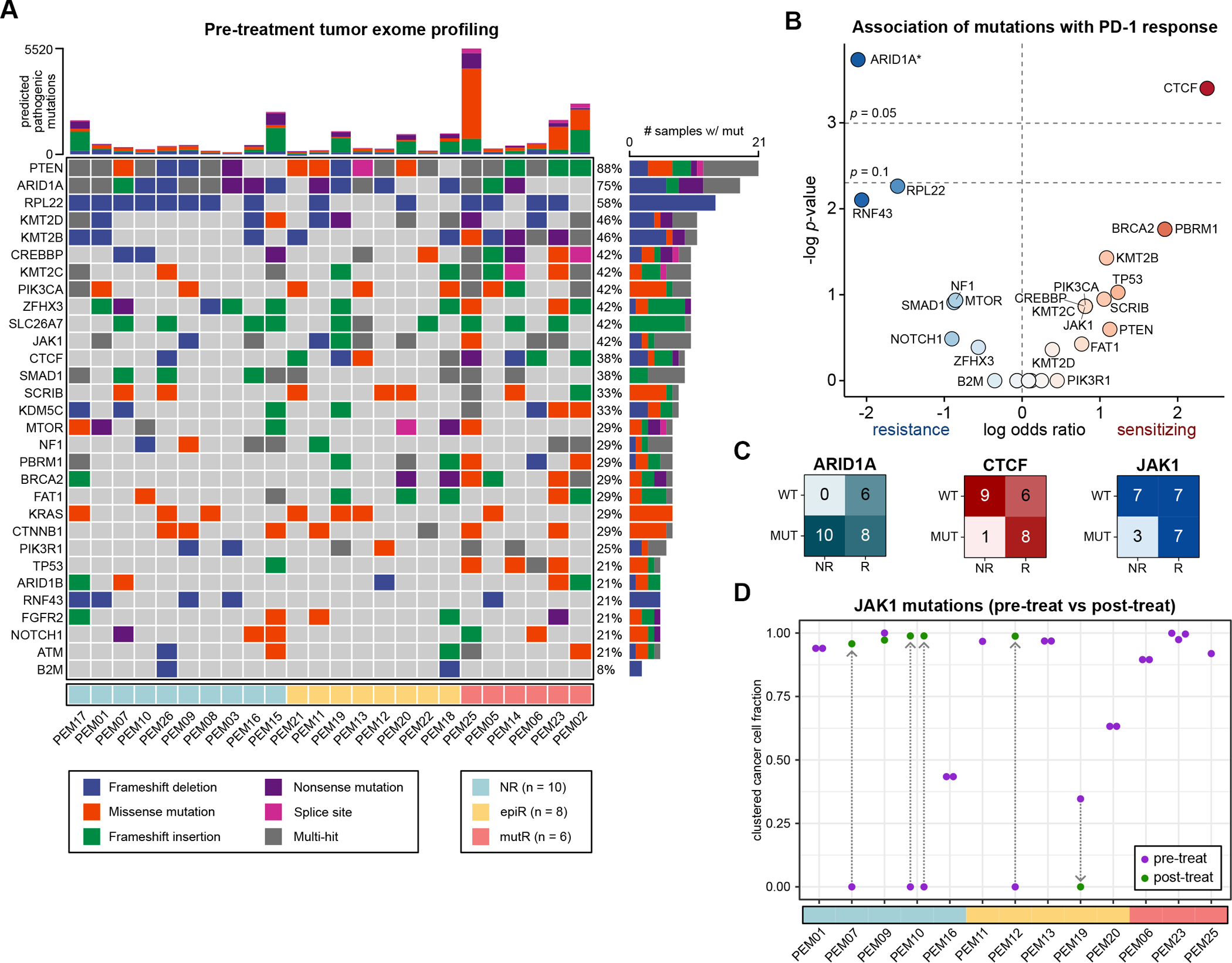
A. Mutation profiles of pre-treatment tumors, filtered for predicted pathogenic or deleterious mutations.
B. Association between individual mutations and response to PD-1 immunotherapy. Data are expressed as log odds ratios. A pseudocount was added for ARID1A, as all patients with non-mutant ARID1A responded to ICB. Statistical significance was assessed by two-tailed Fisher’s exact test.
C. Association of ARID1A, CTCF and JAK1 mutations with PD-1 immunotherapy response.
D. JAK1-mutant cancer cell fractions (CCFs) in JAK1-mutant tumors, after clustering. Each point represents a unique JAK1 variant that was identified in a particular sample. Pre-treatment CCFs are annotated in purple, while post-treatment CCFs are in green, with arrows connecting the same variant across timepoints.
Prior studies across multiple contexts have indicated that JAK1 mutations can protect tumor cells from CD8+ T cell cytotoxicity by disabling IFN-γ pathway signal transduction(23–31). For instance, multiple CRISPR screens using in vitro co-culture systems have identified JAK1 loss as a key mechanism by which tumor cells can evade T cell cytotoxicity(25,32–34). Analogous observations have been described in the clinical setting. For instance, a case series of melanomas with acquired resistance to pembrolizumab demonstrated that new JAK1, JAK2, or B2M mutations can arise following an initial response to ICB(24). Similarly, other case series have described the potential impact of pre-treatment JAK1 mutations on initial ICB responses, concluding that JAK1 loss can also confer primary resistance to ICB(23,35). While these reports are compelling, the relatively small sample sizes of these prior clinical studies are an important limitation. To ascertain whether pre-treatment JAK1 mutations indeed confer resistance to ICB, larger cohorts of JAK1-mutant and JAK1 non-mutant tumors are needed.
Our cohort of MSI-H/MMRd endometrial cancer patients provided an opportunity to explore this question. Of the tumors profiled in this study, 42% (10 of 24) exhibited predicted pathogenic/deleterious mutations in JAK1 prior to treatment (Figure 2A). Analysis of the TCGA Pan-Cancer dataset(36) revealed the highest rates of JAK1 alterations (14%) in the UCEC cohort (Supplementary Figure S3A), further increasing to 30% of MSI-H endometrial cancers(37). Analysis of the AACR-GENIE cohort(38) showed similar findings, with the highest rates of JAK1 mutations (11%) in endometrial cancers (Supplementary Figure S3B). These data indicate that JAK1 mutations are abundant in endometrial cancers, possibly hinting at active immune surveillance even in the absence of ICB therapy.
However, contrary to our expectations, pre-treatment JAK1 mutations were not associated with primary resistance to pembrolizumab in this cohort of MSI-H/MMRd endometrial cancers (Figure 2B). In fact, 70% (7 of 10) of the JAK1-mutant tumors responded to pembrolizumab, compared to 50% (7 of 14) of the non-mutant tumors (Figure 2C). To understand the clonality of these JAK1 mutations, we converted the variant allele frequencies (VAFs) into cancer cell fractions (CCFs). By accounting for tumor cellularity and allele-specific copy number variation, CCFs more accurately represent the clonality of mutations compared to VAFs(39) (Methods). We observed that JAK1-mutant CCFs were consistently above 0.25 in JAK1-mutant tumors, indicating that JAK1 mutations were present in over 25% of the cells in each tumor (Figure 2D, Supplementary Figure S3C–D). Of note, all three of the patients who exhibited a complete response to pembrolizumab (PEM06, PEM23, and PEM25) had JAK1 mutations prior to treatment (Figure 2A). In these patients, JAK1-mutant cells were estimated to comprise a substantial proportion (> 80%) of the total tumor cell population (Figure 2D). If JAK1 mutations indeed conferred to primary resistance to immunotherapy, these JAK1-mutant cells would be expected to drive disease progression while on pembrolizumab. Thus, it is notable that a complete response was observed in patients PEM06, PEM23, and PEM25, even though their tumors were largely comprised of JAK1-mutant cells.
To further understand the relationship between JAK1 alterations and ICB response, we performed WES on 8 epi-MMRd tumors after initiation of pembrolizumab. We observed that three of the tumors (two NR and one epiR) had JAK1 frameshift mutations that were not detected in the pre-treatment samples (Figure 2D, Supplementary Figure S3D). These JAK1 mutations could represent clonal selection in the setting of enhanced anti-tumor immune pressure following pembrolizumab, consistent with acquired resistance to ICB. We speculate that rare JAK1-mutant subclones were already present prior to treatment initiation, but given the limitations of single-site exome sequencing for capturing infrequent subclonal variants in heterogeneous tumors, these JAK1 variants were not detected pre-treatment.
On the other hand, one epiR patient (PEM19) had a JAK1 frameshift mutation (p.M316fs; c.947_948insAC) at baseline that subsequently was not detected on disease progression while on pembrolizumab (Supplementary Figure S3D). This suggests that the pre-treatment JAK1 mutation in patient PEM19 was not under positive selection despite the heightened immune pressure from ICB treatment. Furthermore, it indicates that the pre-treatment JAK1-mutant subclone was unlikely to be responsible for driving disease progression, as an ICB-resistant subclone would be expected to constitute a large enough proportion of the post-treatment tumor to be detectable on WES. With that said, a caveat to this interpretation is that the JAK1 mutation may not have been identified in the post-treatment sample due to tumor heterogeneity and the limitations of single-site exome sequencing. Another possible explanation for the loss of this JAK1-mutant subclone is that an immune response was mounted against a neoantigen encoded by the JAK1 mutation. The top predicted mutant MHC-I epitope at this site showed low predicted affinity (IC50: 5434.3 nM), while the top predicted MHC-II epitope had moderate predicted affinity (354.46 nM). It is therefore possible that tumor cells carrying this JAK1 mutation were eliminated in patient PEM19 following ICB therapy due to a T cell response against this neoepitope.
Taken together, these data indicate that JAK1 mutations may be advantageous for endometrial cancer outgrowth by subverting immune surveillance. This was especially apparent in the case of three patients whose tumors appeared to have acquired JAK1 mutations following pembrolizumab initiation. Interestingly, however, we also found that pre-treatment JAK1 mutations did not necessarily confer primary resistance to pembrolizumab. For instance, all three tumors with a complete response to PD-1 ICB had near-clonal JAK1 mutations at baseline.
Longitudinal single-cell transcriptional profiling of circulating immune cells
We were intrigued that only a fraction of patients with epi-MMRd tumors showed a response to PD-1 immunotherapy, whereas all patients with mut-MMRd tumors had responded (Figure 1B–C). While mut-MMRd tumors had higher TMB than epi-MMRd tumors, TMB alone could not explain the observed variation in ICB response (Figure 1H, 1L). We thus sought to explore the immunological mechanisms driving differential ICB responses across patients. To do so, we sampled PBMCs before and after pembrolizumab treatment (n = 22 patients). In total, we performed paired scRNA-seq and scTCR-seq on over 260,000 PBMCs (n = 52 samples) (Supplementary Figure S4A). We also performed bulk TCR-seq on the pre-treatment TILs to construct a reference library of tumor-reactive TCR sequences.
After standardized data preprocessing, we visualized the scRNA-seq data through uniform manifold approximation and projection (UMAP) (Figure 3A). The cells were classified into cell types by analysis of differentially expressed genes (DEGs) within each cluster (Supplementary Figure S5A). We then further subclustered the T cell populations for a more detailed perspective on the functional status of the circulating T cells in these patients (Figure 3B, Supplementary Figure S5B). We ultimately classified the PBMCs into 21 cell types (Supplementary Table S4). After filtering out samples with fewer than 1000 total cells (n = 3 samples removed) (Supplementary Figure S4A), we compared the relative frequencies of the 21 cell types across timepoints and patient categories. At the level of the 21 cell clusters, we did not observe major systematic differences in cell type frequencies between NR, epiR, and mutR patients (Supplementary Figure S5C). Similarly, there were inconsistent changes in cell type frequencies before and after pembrolizumab treatment, comparing within each category of patients (Supplementary Figure S5C).
Figure 3: Effector CD8+ T cells are enriched in mut-MMRd responders but not in epi-MMRd responders.
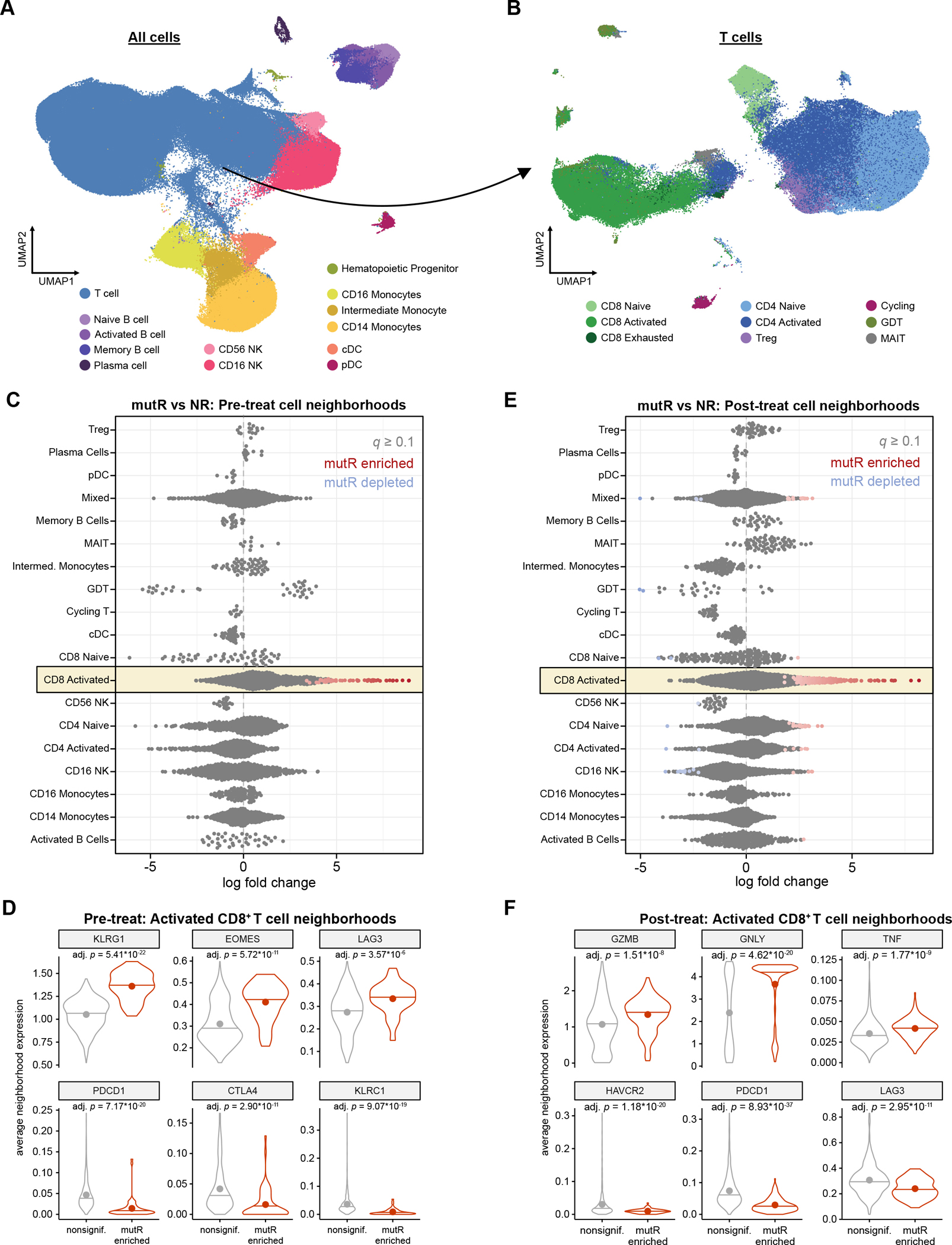
A. UMAP visualization of all PBMCs profiled by scRNA-seq. Data shown are integrated with healthy donor PBMCs for cell annotation and visualization.
B. UMAP visualization of T cells profiled by scRNA-seq, reclustered to identify T cell subsets. Data shown are integrated with healthy donor T cells for cell annotation and visualization.
C. Differential abundance analysis of cell neighborhoods in mutR vs. NR patients, before PD-1 immunotherapy. Each point represents one cell neighborhood. Statistical significance was determined through a generalized linear model with Benjamini-Hochberg multiple hypothesis correction, as implemented in Milo.
D. Violin plots of select differentially expressed genes in activated CD8+ T cell neighborhoods that are enriched in mutR vs. NR patients, before PD-1 immunotherapy. Horizontal lines indicate the median, while points indicate the mean. Statistical significance was assessed by two-sided Mann-Whitney test, with Benjamini-Hochberg multiple hypothesis correction.
E. Differential abundance analysis of cell neighborhoods in mutR vs. NR patients, after PD-1 immunotherapy. Statistical significance was determined through a generalized linear model with Benjamini-Hochberg multiple hypothesis correction, as implemented in Milo.
F. Violin plots of select differentially expressed genes in activated CD8+ T cell neighborhoods that are enriched in mutR vs. NR patients, after PD-1 immunotherapy. Horizontal lines indicate the median, while points indicate the mean. Statistical significance was assessed by two-sided Mann-Whitney test, with Benjamini-Hochberg multiple hypothesis correction.
Mutational MMRd is associated with effector CD8+ T cell responses
We reasoned that finer-resolution changes in specific cell subsets may be masked when analyzing the relative frequencies of broad cluster-based cell type classifications. To identify subtler shifts in circulating immune cell subpopulations and cell states, we grouped the cells into “neighborhoods” through k-nearest neighbor clustering, as implemented through the Milo analysis framework(40). Differential abundance analysis of cell neighborhoods revealed that prior to treatment, epiR patients did not have significant enrichment for any specific subpopulations compared to NR patients (Supplementary Figure S6A). In contrast, mutR patients exhibited significant enrichment for specific subpopulations of activated CD8+ T cells when compared to NR patients (Figure 3C). DEG analysis showed that the specific CD8+ T cell neighborhoods enriched in mutR patients were defined by high expression of KLRG1, EOMES, and LAG3, along with lower expression of inhibitory receptors PDCD1, CTLA4 and KLRC1 (encoding NKG2A(41)) (Figure 3D). There was no difference in IFNG or TNF expression between enriched and non-enriched CD8+ T cell neighborhoods (Supplementary Figure S6B). In aggregate, this expression signature suggests that a CD8+ T cell effector population is poised for mounting anti-tumor responses in mutR patients.
After treatment with PD-1 ICB, epiR patients again did not show any significant differences in cell neighborhood abundances (Supplementary Figure S6C), whereas mutR patients were enriched for several CD8+ T cell neighborhoods (Figure 3E). The mutR-enriched CD8+ T cell neighborhoods in the post-treatment setting were characterized by elevated expression of several cytolytic effector molecules (GZMB, GNLY, TNF) (Figure 3F) but not IFNG (Supplementary Figure S6D). CD8+ T neighborhoods enriched post-treatment in mutR patients also demonstrated reduced expression of inhibitory receptors such as HAVCR2 (encoding TIM-3), PDCD1, and LAG3. Overall, these data point to the presence of an activated CD8+ T cell population in mutR patients that is associated with anti-tumor responses following PD-1 ICB.
To further characterize the T cell responses in these patients, we next focused on the scTCR-seq repertoires. While there were no statistically significant differences in TCR clonality (by Simpson’s clonality index) among the three categories of patients before or after treatment, there was a trend towards increased TCR clonality in responders, particularly in a subset of mutR patients (Supplementary Figure S6E–F). Using the pre-treatment bulk TCR-seq data collected from TILs, we generated a reference library of tumor-reactive TCR sequences in each patient, which we used to identify putative tumor-reactive clonotypes among the peripheral T cells. With this reference, we quantified the percentage of circulating T cells with TIL-matching TCR sequences in each sample. Prior to treatment, there were no significant differences in the relative proportions of TIL-matched circulating T cells among the different patient groups (Supplementary Figure S6G). In contrast, there was a significant increase in the percentage of TIL-matched circulating T cells in mutR patients compared to NR (Supplementary Figure S6H). Of note, expansion of peripheral T cell populations has previously been associated with tumor infiltration and immunotherapy response across cancer types(42). Collectively, these data revealed a circulating effector CD8+ T cell response in mutR patients that appeared to be further amplified by PD-1 immunotherapy.
Our earlier analysis of the tumor exome sequencing data had revealed that pre-treatment JAK1 alterations were not associated with primary resistance to pembrolizumab (Figure 2B–C). We therefore wondered if patients with JAK1-mutant tumors had different circulating immune responses compared to those with JAK1 non-mutant tumors. We performed Milo analyses comparing JAK1-mutant and JAK1 non-mutant tumors within each patient group (NR, epiR, mutR). This analysis revealed minimal changes in cell neighborhood abundances when comparing by JAK1 mutation status (Supplementary Figure S7A–B), suggesting that patients with JAK1-mutant tumors did not have systematic differences in the composition of the circulating immune response. However, we cannot entirely rule out that immune responses against JAK1-mutant tumors may subtly differ from those against JAK1 non-mutant tumors, as the present analysis is limited by the relatively small size of the trial cohort.
Distinct transcriptional profiles of circulating immune cells in epigenetic MMRd responders
Distinct from mutR patients, epiR patients had similar TMB as NR patients (Figure 1H) and did not exhibit significant enrichment for activated CD8+ T cells (Supplementary Figure S6A–B). These data suggest that the underlying mechanism of MMRd (i.e., mutational or epigenetic) influences the intensity of MSI, potentially leading to differences in the intrinsic characteristics of the associated tumors and the cellular composition of the circulating immune response. But while these factors could explain the high response rates against mut-MMRd tumors, they could not explain why approximately half of the patients with epi-MMRd tumors responded to PD-1 ICB. We therefore hypothesized that transcriptional characteristics of the circulating immune response could distinguish patients with epi-MMRd tumors that respond to PD-1 blockade from those that do not.
We subsequently set out to identify the transcriptional features of T and NK cells in epiR patients compared to NR patients. However, since the number of profiled cells varied widely between samples (Supplementary Figure S4A), conventional scRNA-seq approaches for differential expression such as the Mann-Whitney test (the default in the widely used Seurat analysis package)(43,44) would be weighted towards samples with more cells. In a similar vein, we were concerned that considering each single cell as an independent event would artificially inflate statistical power. We instead used a random sampling-based approach to partition the cells in each sample into “pseudobulk” profiles (see Methods). Specifically, we generated five pseudobulk profiles for each cell type in each sample, then performed DEG analysis using a generalized linear model as implemented in DESeq2(45). This approach equally weights each sample and avoids overcounting single cells as independent events, providing advantages over the Mann-Whitney test when analyzing large multi-subject scRNA-seq datasets.
Comparing responders to non-responders prior to PD-1 immunotherapy, we quantified the number of significant DEGs across T and NK cell populations (Supplementary Figure S8A). Pathway analysis revealed that CD16+ NK cells in epiR patients demonstrated strong upregulation of genes involved in the proteasome, NK cell-mediated cytotoxicity, oxidative phosphorylation, and NOD-like receptor signaling (Supplementary Figure S8B), whereas these changes were not seen in CD16+ NK cells from mutR patients. Across these T or NK populations, the majority of upregulated pathways were identified in CD16+ NK cells. These data indicate that prior to PD-1 immunotherapy initiation, CD16+ NK cells in epiR patients were transcriptionally distinct from those in NR patients, with higher expression of genes that are relevant to anti-tumor immunity.
We observed a similar pattern in the post-treatment setting, comparing epiR patients and mutR patients to non-responders (Supplementary Figure S8C). After pembrolizumab treatment, activated CD8+ T cells in epiR patients uniquely demonstrated upregulation of genes involved in oxidative phosphorylation and the proteasome (Supplementary Figure S8D). In a similar manner, activated CD4+ T cells showed enrichment for NOD-like receptor signaling and fatty acid metabolism in epiR patients but not in mutR patients. On the other hand, the predominant significant pathway-level changes in NK cells after treatment were mutR-specific downregulation events. For instance, CD16+ NK cells and CD56+ NK cells in mutR patients both had significant downregulation of NK cell-mediated cytotoxicity, but these pathway-level alterations were not observed in epiR patients.
We next identified DEGs in a longitudinal manner within each patient group, comparing pre-treatment and post-treatment transcriptional profiles for each cell type (Supplementary Figure S9A). For instance, when comparing pre- and post-treatment samples, activated CD4+ T cells, CD8+ T cells and CD16+ NK cells showed more upregulated and downregulated DEGs in epiR patients than NR or mutR patients. On the pathway level, both naïve and activated CD4+ T cells in epiR patients exhibited upregulation of antigen processing and presentation, along with downregulation of oxidative phosphorylation (Supplementary Figure S9B). CD16+ NK cells in epiR patients uniquely demonstrated upregulation of genes involved in the phagosome, tight junctions, focal adhesion, and leukocyte transendothelial migration.
These analyses suggested differing modes of anti-tumor immunity in epiR patients compared to mutR patients. We thus directly compared the transcriptional profiles of immune cells from epiR and mutR patients, stratifying by pre-treatment and post-treatment timepoints (Figure 4A). Pathway analyses of the resulting DEGs showed relative enrichment of TCR signaling and TNF signaling in activated CD8+ T cells from mutR patients compared to epiR patients (Figure 4B), corroborating our earlier observation that mutR patients are characterized by potent effector CD8+ T cells (Figure 3C–F). Conversely, CD16+ NK cells from epiR patients had higher expression of genes involved in NK cell-mediated cytotoxicity, graft vs. host disease, and NOD-like receptor signaling (Figure 4B). The enrichment of these pathways in CD16+ NK cells suggests that NK cells may play a key role in mounting anti-tumor responses in epiR patients.
Figure 4: Transcriptional features of NK cells in epi-MMRd responders are associated with survival.
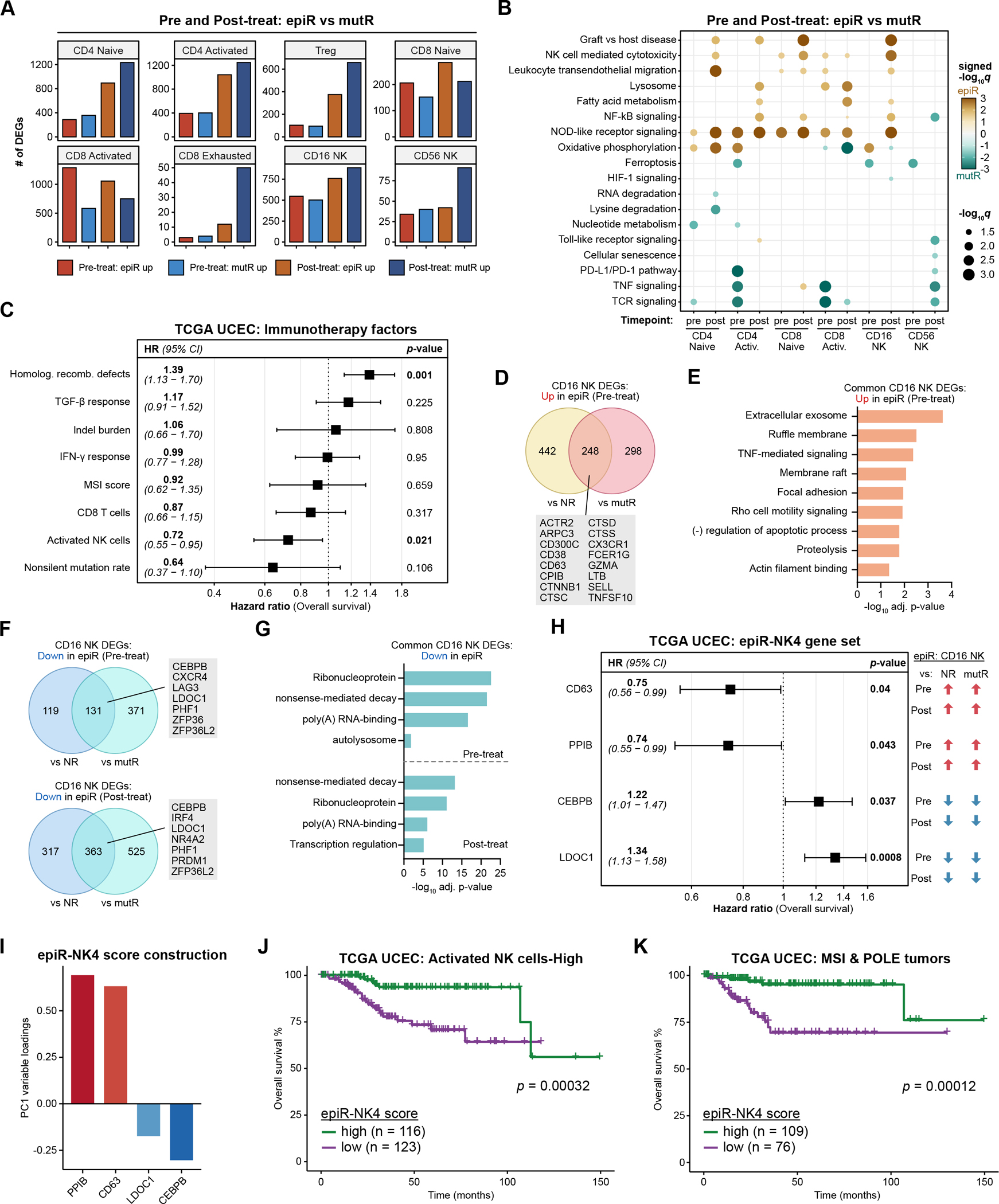
A. The number of DEGs in T and NK cell populations when comparing epiR vs. mutR patients, before or after PD-1 immunotherapy.
B. Dot plots detailing significantly upregulated or downregulated pathways in each of the cell types, comparing epiR patients to mutR patients, before or after PD-1 immunotherapy. Statistical significance was assessed by hypergeometric test with Benjamini-Hochberg multiple hypothesis correction, visualized as signed −log10 q-values.
C. Forest plot detailing the results of a multivariable Cox proportional hazards model for overall survival in the TCGA UCEC cohort, examining several factors previously associated with response or resistance to ICB immunotherapy.
D-E. Venn diagram (D) and gene ontology enrichment analysis (E) of DEGs in CD16+ NK cells that are significantly upregulated in epiR patients compared to NR or mutR patients, before PD-1 immunotherapy. Statistical significance in (E) was assessed by hypergeometric test with Benjamini-Hochberg multiple hypothesis correction.
F-G. Venn diagram (F) and gene ontology enrichment analysis (G) of DEGs in CD16+ NK cells that are significantly downregulated in epiR patients compared to NR or mutR patients, before (upper panel) or after PD-1 immunotherapy (lower panel). Statistical significance in (G) was assessed by hypergeometric test with Benjamini-Hochberg multiple hypothesis correction.
H. Forest plot detailing the results of a multivariable Cox proportional hazards model for overall survival in the TCGA UCEC cohort, examining a set of four genes (epiR-NK4) that were consistently upregulated (CD63, PPIB) or downregulated (CEBPB, LDOC1) in CD16+ NK cells from epiR patients. These four genes were selected through Lasso regression from a total of 111 genes that were concordantly upregulated or downregulated in CD16+ NK cells from epiR patients.
I. Variable loadings for the first principal component (PC1) of the TCGA UCEC dataset, based on expression levels of the epiR-NK4 gene set.
J-K. Kaplan-Meier survival analysis of the TCGA UCEC cohort, subsetted on tumors with a high activated NK cell score (J) or tumors annotated as MSI-H/POLE-hypermutated (K). Patients were stratified by their epiR-NK4 scores, corresponding to PC1 of the epiR-NK4 gene set as in (I), and classifying into high and low groups based on the median score. Statistical significance was assessed by log-rank test.
Collectively, these findings indicate that the transcriptional profiles of T and NK cell populations in epiR patients are distinct from those of NR and mutR patients, both before and after pembrolizumab treatment. While epiR patients did not show significant numerical enrichment of CD8+ T cells over their non-responding epi-MMRd counterparts, transcriptional profiling pointed to heightened functionality of circulating NK cells in epiR patients.
Activated NK cells are associated with survival in endometrial cancer
The DEG analyses pointed to enhanced functionality of NK cells in epiR patients compared to NR patients. Given that mutR patients, but not epiR patients, had increased TMB and activated CD8+ T cell frequencies compared to NR patients (Figure 3C–F), and that T cells from mutR patients had comparatively higher expression of TCR signaling-related genes (Figure 4B), we wondered if NK cells might be responsible for promoting anti-tumor responses in epiR patients. This notion was further supported by the observation that CD16+ NK cells in epiR patients showed higher expression of genes involved in NK cell-mediated cytotoxicity compared to their mutR counterparts (Figure 4B).
To explore this possibility, we first examined the TCGA UCEC cohort of endometrial cancer patients to identify tumor features that correlate with overall survival(13). Although the TCGA UCEC study predates the advent of ICB therapies for this patient population, we reasoned that by understanding the features that associate with survival at baseline (i.e., in the absence of ICB therapy), we could nevertheless identify factors that are clinically relevant for endometrial cancer biology. We constructed a multivariable Cox proportional hazards model examining a number of factors that have previously been associated with response to immunotherapy(14,46): non-silent mutation rate(47), homologous recombination defects(48,49), TGF-B response(50,51), IFN-γ response(52,53), CD8+ T cell abundance(47,53–55), indel burden(56), MSI score(4) (determined by MSIsensor(57)), and NK cell abundance(58,59). In the fully adjusted model, we found that homologous recombination defects were significantly associated with worse survival (HR 1.39, 95% CI 1.13–1.70), while activated NK cells were associated with better survival (HR 0.72, 95% CI 0.55–0.95) (Figure 4C). In contrast, CD8+ T cells were not significantly associated with survival in the multivariable analysis, though there was a trend towards improved survival (HR 0.87, 95% CI 0.66–1.15). Similarly, elevated non-silent mutation rate was associated with improved survival, but did not reach statistical significance (HR 0.64, 95% CI 0.37–1.10).
Transcriptional signatures of CD16+ NK cells in epigenetic MMRd responders are associated with longer survival
The significant association between activated NK cells and overall survival suggests that NK cells might play an important role in immune responses against endometrial cancers. We therefore further interrogated the transcriptional characteristics of NK cells in epiR patients, compared to NR and mutR patients. We specifically focused on the CD16+ NK cell subset since CD16+ NK cells are the predominant NK cell subset in the circulation, have been demonstrated to be more naturally cytotoxic than CD56+ NK cells, and are preferentially recruited from the circulation to sites of inflammation(60,61). In comparing the expression profiles of CD16+ NK cells from epiR patients to those of NR and mutR patients, we identified 248 genes that were commonly upregulated across both comparisons prior to ICB treatment (Figure 4D). These included a number of genes relevant for NK cell functionality, such as ARPC3, CX3CR1, GZMA, LTB, and TNFSF10 (encoding TRAIL)(62,63). Gene ontology analysis showed significant enrichment for categories such as the exosome, ruffle membrane, TNF-mediated signaling, focal adhesion, and Rho cell motility signaling (Figure 4E), indicative of an activated NK cell state.
Similarly, in the post-treatment setting, CD16+ NK cells from epiR patients had 290 commonly upregulated genes in comparison to NR and mutR patients (Supplementary Figure S10A). These included genes that were already upregulated prior to treatment, such as ARPC3, CX3CR1, GZMA, and LTB, as well as post-treatment-specific DEGs, including EOMES, FLT3LG, LTA, and STAT1. Gene ontology analysis similarly revealed enrichment for the exosome and TNF-mediated signaling, along with upregulation of the proteasome, mitochondrion, immune response, and Fc-epsilon receptor signaling (Supplementary Figure S10B).
As for downregulated DEGs, 131 genes (prior to treatment) and 363 genes (after treatment) were commonly downregulated in CD16+ NK cells from epiR patients compared to NR and mutR patients (Figure 4F). Gene ontology analysis of the downregulated DEGs showed enrichment for ribonucleoprotein, nonsense-mediated decay, and poly(A) RNA-binding, both before and after PD-1 ICB (Figure 4G). We noted that ZFP36L2 was among the most strongly downregulated genes in CD16+ NK cells from epiR patients (Supplementary Figure S10C). The ZFP36 family of RNA-binding proteins has previously been shown to translationally repress cytokine production in T cells by binding to 3’-untranslated regions, and loss of ZFP36 family members leads to enhanced antiviral responses(64,65).
Given that activated NK cell abundances were associated with survival in the TCGA UCEC cohort (Figure 4C), we wondered if the gene signatures of CD16+ NK cells in epiR patients could stratify endometrial cancer patients into prognostic subgroups. We constructed a Lasso Cox regression model that included all 111 genes that were concordantly upregulated or downregulated in CD16+ NK cells from epiR patients, taking the intersection of pre-treatment and post-treatment DEGs. We arrived at a final set of 4 genes (termed epiR-NK4) that were each significantly associated with survival in a multivariable analysis and that demonstrated a reciprocal relationship between survival and differential expression in epiR CD16+ NK cells (Figure 4H). The epiR-NK4 gene set consisted of CD63, PPIB, CEBPB, and LDOC1, which were consistently upregulated (CD63 and PPIB) or downregulated (CEBPB and LDOC1) in CD16+ NK cells from epiR patients, both before and after PD-1 treatment. High expression levels of the epiR-upregulated genes CD63 and PPIB were associated with improved survival, whereas high expression levels of the epiR-downregulated genes CEBPB and LDOC1 were associated with worse survival (Figure 4H).
To derive a summative statistic for the epiR-NK4 gene set across the TCGA UCEC cohort, we computed the first principal component (PC1) of the RNA-seq expression values for CD63, PPIB, CEBPB, and LDOC1 (Figure 4I, Supplementary Figure S10D). The PC1 variable loadings were positive for CD63 and PPIB, while negative for CEBPB, and LDOC1; thus, samples with high expression of CD63/PPIB and low expression of CEBPB/LDOC1 would have a high epiR-NK4 score. We evaluated whether the epiR-NK4 score could stratify survival in specific subgroups of interest: 1) tumors with a high abundance of activated NK cells, and 2) tumors with MSI-H/POLE-hypermutated status. In both subgroup analyses, we found that the median epi-NK4 score defined two distinct prognostic categories, with a high epiR-NK4 score being associated with better survival (Figure 4J–K). These findings suggest that the transcriptional signatures of CD16+ NK cells in epiR patients are associated with improved survival in endometrial cancer patients.
Discussion
A substantial proportion of MSI-H/MMRd tumors do not respond to therapy despite their exceptionally high mutational loads(7). We show here that among MSI-H/MMRd endometrial cancers, the molecular mechanism of MMRd is associated with response to PD-1 ICB. As a potential explanation for why tumors with mut-MMRd have more robust responses compared to those with epi-MMRd, we found that the nature of the circulating immune response differs depending on the mechanism of MMRd. For patients with mut-MMRd tumors, the circulating immune repertoire is enriched for a subpopulation of effector CD8+ T cells; for patients with epi-MMRd tumors that respond to PD-1 ICB, the circulating immune response is defined by a highly active CD16+ NK cell population.
We speculate that mut-MMRd tumors and epi-MMRd tumors have distinct tumor-intrinsic features that shape the nature of the circulating immune system and subsequent responses to PD-1 ICB. While mut-MMRd tumors exhibited higher TMB than epi-MMRd tumors, we also found that TMB could not fully explain the distinction between responders and non-responders (Figure 1H, 1L). Of note, prior studies have demonstrated that out of all the nonsynonymous mutations present within a tumor, only a small fraction will be recognized by TILs(66,67). Considering that even a single neoantigen can be sufficient to drive potent anti-tumor immunity(68,69), it is likely that the reported association between TMB and ICB response(16,47) exhibits diminishing returns. Within a cohort of MSI-H/MMRd tumors that all have high neoantigen loads (as in the present study), neoantigen availability is less likely to be the key limiting factor for mounting effective anti-tumor immune responses. Thus, we hypothesize that other tumor-intrinsic differences beyond TMB collectively influence the state of the host immune system, leading to differential responses to PD-1 ICB in mut-MMRd vs. epi-MMRd tumors.
To that end, how might the molecular mechanism of MMRd influence the nature of the circulating anti-tumor immune response? Although answering this question will require further studies in controlled experimental systems, we hypothesize that there are at least two contributing factors.
First, the observed differences between mut-MMRd and epi-MMRd tumors likely stems in part from the rewritable nature of epigenetic alterations. Whereas all downstream progeny of a mut-MMRd tumor clone will carry the same MMR gene mutations, the extent of MMR silencing through MLH1 promoter methylation may vary within an epi-MMRd tumor(70). This heterogeneity in MLH1 silencing can occur at two separate levels: across different cells within a tumor, and also across time, for instance as the microenvironmental state of the tumor evolves(71). To the extent that transient restoration of MLH1 expression (and thus MMR capacity) occurs in epi-MMRd tumors, it is plausible that a fraction of the mutations that had previously accumulated as a result of MLH1 promoter methylation will then be resolved(72,73). An analogous phenomenon was recently described for BRCA1, where BRCA1-methylated tumors could adaptively regain BRCA1 function and acquire resistance to platinum therapies(74). This hypothesis is further supported by the observation that mut-MMRd tumors had significantly higher TMB than epi-MMRd tumors. As a consequence, some of the neoantigens that were being targeted by CD8+ T cells may be lost in a subset of epi-MMRd tumor cells. In this scenario, NK cells may play a larger role in anti-tumor responses, as they can efficiently mount cytotoxic responses in a neoantigen-independent manner.
Second, epi-MMRd tumors may have global alterations in DNA methylation that could have pleiotropic effects on tumor-immune interactions. For instance, epi-MMRd tumors may have altered expression of immunomodulatory factors such as checkpoint molecules, which would thereby affect the state of the circulating immune response. It would be of great interest in future studies to longitudinally profile epi-MMRd tumors over the course of pembrolizumab therapy to investigate these and other potential mechanisms.
In other cancer types such as colorectal cancer, epi-MMRd tumors tend to develop at an older age compared to mut-MMRd tumors(75). Since the functionality of the immune system changes with aging(76), it is possible that some of the differential immune responses we observed in patients with epi-MMRd tumors are related to aging. However, we note that patient age was not significantly different between the epi-MMRd and mut-MMRd groups in our cohort, so the observed changes in immune system composition and gene expression are less likely to be due to age variations. With that said, we cannot rule out that subsequent studies with larger patient cohorts may detect age differences between patients with epi-MMRd vs. mut-MMRd endometrial tumors, which in turn may influence their response to PD-1 immunotherapy(77).
An unexpected finding from our study was that pre-treatment JAK1 mutations were not associated with primary resistance to PD-1 ICB (Figure 2B–C). At the same time, three patients had JAK1 mutations that were only detected after initiating pembrolizumab, a finding that is consistent with prior reports of acquired resistance to ICB through JAK1 alterations(24). We therefore hypothesize that the functional consequences of JAK1 mutations are partly determined by their temporal context, such that pre-existing JAK1 mutations may reshape tumor-immune interactions in a manner distinct from when JAK1 mutations arise following ICB therapy. Interestingly, a recent study using an in vivo CRISPR screening approach demonstrated that pre-treatment JAK-STAT mutations were associated with enhanced ICB sensitivity across multiple murine tumor models(78), in direct contrast to prior in vitro CRISPR screens that utilized co-culture systems(25). Prior studies have also demonstrated in mouse models that loss of tumor-intrinsic IFN-γ signaling can promote anti-tumor immunity through the enhanced activation of CD8+ T cells and NK cells(79–81). Mechanistically, tumors with IFNGR loss promote IFN-γ production by CD8+ T cells, which in turn enhances the activation of cytotoxic NK cells(80,81).
While we had broadly classified the patients as non-responders and responders based on standard RECIST criteria, we were also intrigued that some of the non-responder patients did not experience disease progression for a year or more. These patients with long-term stabilized disease did not demonstrate substantial reductions in tumor size following pembrolizumab; nevertheless, we speculate that ICB treatment may have shifted the tumor-immune set point towards a new equilibrium, with the immune system restraining (but not effectively eliminating) the tumor. Future studies with larger patient cohorts should seek to interrogate the underlying tumor-immune interactions in patients with long-term stable disease.
Given the observational nature of the clinical trial design, the conclusions that can be drawn from the present study are largely correlative in nature. Nevertheless, we believe that a fruitful area for future research will be to investigate the underlying mechanisms driving the two distinct modes of anti-tumor immunity in patients with epi-MMRd vs. mut-MMRd tumors. For instance, a deeper characterization of the tumor-immune microenvironment in epi-MMRd and mut-MMRd tumors during ICB therapy would be highly informative. In addition to uncovering additional tumor-intrinsic features that differentiate the two classes of MMRd tumors, these data could reveal whether the distinguishing features of the circulating immune response we identified here are similarly observed in tumor-infiltrating immune cells.
Taken together, our findings fill an important gap in our understanding of the factors that influence anti-tumor immunity in the setting of substantial neoantigen burden. By identifying the tumor-intrinsic and extrinsic factors that are associated with response to PD-1 immunotherapy in MSI-H/MMRd endometrial cancers, our work illuminates new avenues for biomarker development and therapeutic intervention. Moving forward, it will be of interest to understand precisely how the molecular mechanism of MMRd can mold the anti-tumor immune response.
Methods
Study design and patient population
The trial was approved by the institutional review board of Yale University (HIC: 1605017712). Written informed consent was obtained from all participants. This study was conducted in accordance with the U.S. Common Rule and the Declaration of Helsinki. The study design and patient population have been previously reported in detail(11). In brief, NCT02899793 is an investigator-initiated phase 2 trial evaluating the safety and efficacy of pembrolizumab in patients with recurrent MMRd/MSI-H endometrial cancer, determined by IHC for MMR proteins and by microsatellite PCR. Patients were treated with pembrolizumab 200mg by IV every 3 weeks, until the occurrence of disease progression or adverse effects prohibiting continued therapy. The primary efficacy endpoint was objective response, as determined by RECIST v1.1 criteria. Secondary outcome measures were the duration of progression-free survival and overall survival. This study was partly supported by Merck, which supplied the study drug pembrolizumab but did not influence any aspect of the study design, data collection, analyses, or interpretation.
Classification of tumors by primary mechanism of MMRd
MSI-H/MMRd tumors with MLH1 promoter methylation were classified as epi-MMRd; tumors with somatic MMR gene mutations and without MLH1 promoter methylation were classified as mut-MMRd. In cases where a tumor exhibited both MLH1 promoter methylation and somatic MMR gene mutations, the methylation event was considered the primary driver, as epi-MMRd can drive the subsequent acquisition of MMR gene mutations due to heightened mutagenic potential(75,82). As a confirmatory test in such cases, we leveraged the fact that tumors with epi-MMRd classically exhibit loss of MLH1 and PMS2 on IHC(83,84). Thus, for tumors exhibiting both MLH1 promoter methylation and somatic MMR gene mutations, loss of MLH1 and PMS2 by IHC helped confirm the classification of epi-MMRd. Patient PEM06 was a special case in that the tumor exhibited complete loss of MSH6, MLH1, and PMS2 by IHC, along with MLH1 promoter methylation and biallelic loss-of-function MSH6 mutations. Given these features, we classified patient PEM06 as mut-MMRd despite the presence of MLH1 promoter methylation. Our reasoning was that if MLH1 promoter methylation was instead the initial driving event for MMRd in patient PEM06, and that the MSH6 mutations were randomly acquired later due to the heightened mutagenic potential from epi-MMRd, we would expect MSH6 loss by IHC only in a subset of the tumor cells. The complete loss of MSH6 by IHC in PEM06 is therefore less consistent with epi-MMRd, so we inferred that MSH6 loss was the initial event driving MMRd (hence the mut-MMRd classification).
Analysis of whole exome sequencing data
Genomic DNA was extracted from the tumor samples and captured on the NimbleGen 2.1M human exome array, then sequenced on an Illumina HiSeq 2000. Whole exome sequencing (WES) data were aligned to the hg19 genome (Human G1Kv37 reference with decoy sequences) using BWA-MEM (RRID:SCR_010910)(85), with duplicate filtering performed by Picard (RRID:SCR_006525). The reads were then processed with GATK (RRID:SCR_001876)(86,87) according to the Best Practice workflow. Matched normal WES data was utilized as a reference to call somatic mutations using MuTect2 (RRID:SCR_001876), with DeTiN analysis to improve detection sensitivity(88). Predicted pathogenic/deleterious exonic mutations were defined as frameshift mutations, nonsense mutations, splicing site mutations, or missense mutations that were predicted to be pathogenic by at least two mutation assessors (out of MutationAssessor (RRID:SCR_005762)(89), SIFT (RRID:SCR_012813)(90), and CADD (RRID:SCR_018393)(91)). We refer to these mutations as “predicted pathogenic” throughout, because many of these mutations have not been experimentally validated. The predicted pathogenic/deleterious mutation calls were summarized and visualized using Maftools(92). To assess the association between specific gene mutations and ICB response, we used Fisher’s exact test on the 2×2 contingency matrix between mutation status and ICB response. A pseudocount was added when calculating the log odds ratio for ARID1A, as all patients with non-mutant ARID1A were responsive to therapy. HLA typing was performed on the matched normal WES profiles using HLA-HD(93). Using the high-confidence exonic mutation set above, we predicted potential MHC-I and MHC-II neoantigens in each sample using pVAC-Seq(94).
For detailed analysis of JAK1 mutations, we transformed all exonic variant allele frequencies (VAFs) from MuTect2 into cancer cell fractions (CCFs)(39,95). To do so, we first utilized Sequenza (RRID:SCR_016662)(96) on the aligned .bam files to estimate tumor cellularity and to generate allele-specific copy number segmentation profiles. We then calculated non-clustered CCFs using the standard formula(39), as well as clustered CCF estimates with PyClone-VI(97). For the purpose of visualization, when comparing the CCFs for the same JAK1 variant in pre-treatment vs. post-treatment samples, we assigned a CCF of 0 when the variant was not detected in a particular timepoint.
Validating the association between MMRd mechanisms, TMB, and immunotherapy response
For external validation of our findings regarding the relation between the mechanism of MMRd, TMB, and immunotherapy response, we reanalyzed two independent cohorts of endometrial cancer patients. From the TCGA UCEC study(13), we categorized the patients into four groups based on MSI status and the mechanism of MMR deficiency. MSI/MMR status was based off the TCGA UCEC subtype annotations, as reported in a prior publication(14). Epigenetic MMRd was determined by analysis of methylation levels of the MLH1 promoter, regardless of the presence of MMR gene mutations. Specifically, we utilized the MEXPRESS methylation browser(98) to identify probes in the MLH1 promoter with a correlation coefficient ≤ −0.8 with MLH1 gene expression. The beta-values for these 29 probes were then averaged within each sample to obtain a composite methylation beta-value for the MLH1 promoter. We defined MLH1 promoter methylation with a cutoff of 0.5. Mutational MMRd was defined by the absence of MLH1 promoter methylation and the presence of mutations in any of the canonical MMR genes: MLH1, MSH2, MSH6, or PMS2. All other MSI-H tumors that were not classified as mut-MMRd or epi-MMRd were labeled as MMRd not otherwise-specified (other). Finally, we utilized the non-synonymous mutation rates and predicted neoantigen loads from a prior publication(14) to assess the relation between MMRd mechanism, TMB, and neoantigen load.
From the MSK-IMPACT cohort(15–17), we filtered for endometrial cancer patients and categorized them into three groups based on MSI status and the presence of mutations in canonical MMR genes (MLH1, MSH2, MSH6, or PMS2). Of note, this dataset did not have available annotations of MLH1 promoter methylation status. The three groups were thus 1) MMRp, 2) mut-MMRd, and 3) non-mutational MMRd (non-mut-MMRd). mut-MMRd patients were defined by the combination of MSI-H status and presence of mutations in MLH1, MSH2, MSH6, or PMS2. In contrast, non-mut-MMRd patients were defined by the combination of MSI-H status and the absence of mutations in MLH1, MSH2, MSH6, or PMS2. We then utilized the authors’ annotations to assess the relation between MMRd mechanism, TMB, and ICB response.
The prevalence of JAK1 alterations across cancer types was queried using cBioPortal(99,100), examining JAK1 mutations and homozygous deletions in the TCGA Pan-Cancer Atlas(36) and the AACR-GENIE cohort(38).
scRNA-seq sample collection, data preprocessing and cell annotation
Peripheral blood mononuclear cells were isolated by standard density gradient centrifugation with Ficoll-Paque (GE Healthcare, Uppsala, Sweden) and used to construct scRNA-seq libraries. The Chromium Single-Cell 5’ and Chromium Single Cell V(D)J Reagent Kit (10X Genomics, Pleasanton, USA) was used following the manufacturer’s instructions.
scRNA/scTCR-seq reads were processed using Cell Ranger (RRID:SCR_017344)(101) to generate both raw expression count matrices and TCR sequence information. Healthy donor PBMC scRNA-seq and scTCR-seq control data were obtained from prior publications: Pappalardo et al(102) and Song et al(103). Expression data were loaded into Seurat (RRID:SCR_016341)(43) and prefiltered for cells with ≥ 400 counts, ≥ 150 unique expressed genes, and ≤ 20% mitochondrial gene expression. Cells were not filtered for complexity or doublet related markers. The data were normalized with SCTransform(104), regressing out counts per cell, features per cell, and mitochondrial gene expression. The datasets were then integrated using reciprocal PCA to identify integration anchors before performing cell clustering and UMAP dimensional reduction.
Clusters were annotated using manual analysis of marker gene expression in combination with automatic cell type identification using SingleR(105). Fourteen T cell clusters were subsetted and reprocessed as a separate dataset by rerunning PCA, UMAP, and clustering. After an initial round of analysis, two clusters from the T cell dataset were determined to be composed of cells with uniformly high mitochondrial reads. Further clusters were determined to be differentiated almost entirely by expression of a single TCR variable gene while encompassing many distinct T cell subtypes. To address these issues, we regenerated the dataset by prefiltering out the high mitochondrial read cells and excluding all TCR variable and joining genes from the PCA and clustering analyses.
Differential abundance of cell neighborhoods with Milo
To identify differentially enriched cell neighborhoods in the scRNA-seq dataset, we utilized Milo(40). The single cells were first assigned to partially overlapping neighborhoods based on a k-nearest neighbor graph, with settings of k = 200, d = 50 that were selected based on the distribution of neighborhood sizes, per the authors’ recommendations. After quantifying the number of cells in each neighborhood for each sample, we performed differential abundance testing with a negative binomial generalized linear model as implemented in edgeR(106). Multiple hypothesis correction was performed with a weighted FDR procedure that accounts for spatial overlapping of neighborhoods. We chose a cutoff of spatial FDR (q) < 0.1 for identifying cell neighborhoods that were differentially enriched.
For characterizing the marker genes of differentially enriched cell neighborhoods, we calculated the average expression in each neighborhood for the 5000 most variable genes in the dataset. We then identified statistically significant marker genes by two-tailed unpaired Mann-Whitney test with Benjamini-Hochberg multiple hypothesis correction, comparing enriched and non-enriched cell neighborhoods.
TCR repertoire analysis
We identified high-confidence single cell TCRαβ sequences using the Cell Ranger VDJ pipeline. The data were loaded into CellaRepertorium and restricted to TCRα and TCRβ chains with CDR3s that were high confidence (no N’s), full length, ≥ 6 amino acids long, and productive. Data from bulk TCR-seq of TILs were loaded into Immunarch (10.5281/zenodo.6984421) and filtered for in-frame, coding CDR3s. Since the bulk data contained only TCRβ sequences, TIL matching cells were identified in the PBMC scTCR-seq data by matching the scTCRβ CDR3s to the bulk TIL TCRseq data from the same patient, similar to the approach utilized in a previous study(107). TCR analyses were therefore conducted on TCRβ sequences alone in order to maintain consistency and allow for the identification of tumor-matching TCRs. To determine the clonality of the TCR repertoire in each sample, we calculated Simpson’s clonality index.
Differential expression analysis
Conventional scRNA-seq differential expression analysis operates on the level of single cells, such as the Mann-Whitney test that is commonly used in the Seurat analysis package. However, this approach has a number of important limitations when analyzing large multi-subject datasets: samples with larger cell numbers will dominate the statistical comparisons, and each individual cell will be counted as an entirely independent event, artificially inflating statistical power. To circumvent these issues, we used an alternative approach to differential expression analysis of scRNA-seq data. For each scRNA-seq library, we generated five random samples of each cell type, then aggregated the counts together. In effect, each cell type within a given library was condensed into five replicates. These “pseudobulk” replicates were then analyzed using DESeq2(45), which has been demonstrated to have robust performance on scRNA-seq data despite being developed for bulk RNA-seq analysis(44,108). Differentially expressed genes (DEGs) were defined as those with adjusted p < 0.05. KEGG pathway analysis was performed with clusterProfiler(109) on the set of DEGs, using a hypergeometric test with Benjamini-Hochberg multiple hypothesis correction to evaluate statistical significance. Signed −log10 q-values were calculated by multiplying the −log10 q-values by the directionality of differential expression, such that pathways associated with downregulated DEGs would have a negative −log10 q-value. In the rare cases where a given pathway was associated with both upregulated and downregulated DEGs, we selected the directionality with the strongest statistical significance for visualization.
Survival analysis of TCGA data
We obtained overall survival data(110) from the TCGA UCEC cohort, evaluating the relationship between survival and several factors previously associated with response to immunotherapy. These factors were previously computed across the TCGA dataset in a prior study(14). We constructed a multivariable Cox proportional hazards model with the following variables, all coded as continuous variables: homologous recombination defects, non-silent mutation rate, TGF-β response, IFN-γ response, MSI score (MSIsensor, available on cBioPortal), CD8+ T cell abundance, and activated NK cell abundance. The resulting hazard ratios were visualized as a forest plot.
To evaluate whether the gene signatures of CD16+ NK cells in epiR patients were associated with patient survival, we first compiled a final set of 111 genes that were concordantly upregulated or downregulated in epiR patients compared to NR and mutR patients. These 111 genes thus met several criteria: 1) they were called as a DEG in epiR vs. NR, both pre-treatment and post-treatment, with the same directionality of differential expression; 2) they were called as a DEG in epiR vs. mutR, both pre-treatment and post-treatment, with the same directionality of differential expression; and 3) the directionality of differential expression was shared between the epiR vs. NR and epiR vs. mutR comparisons. The 111 genes were utilized in a Lasso Cox regression to identify a core set of genes associated with survival. Of the five genes retained by the Lasso model, 1 gene was filtered out (PITPNC1), as high expression of this gene was associated with better survival but this gene was downregulated in CD16+ NK cells. The four remaining genes were termed the epiR-NK4 gene set, comprised of CD63, PPIB, CEBPB, and LDOC1. In a multivariable Cox regression model, all four genes were independently associated with survival. To generate a summative score of the epiR-NK4 gene set, we performed PCA on the TCGA dataset, based on the expression of the epiR-NK4 genes. We took the first principal component (PC1) as the epiR-NK4 score and stratified patients into high vs. low groups by the median score. Kaplan-Meier survival curves were then generated with the epiR-NK4 score classifications, and statistical significance was assessed by log-rank test.
Data Availability
Raw and processed scRNA-seq/scTCR-seq data generated in this study are available in the Gene Expression Omnibus (GSE212217). Raw bulk TCR-seq data of TILs were generated by an external core facility (Adaptive Biotechnologies) and are unavailable, but the processed data are available in Zenodo (10.5281/zenodo.6985601) and the Gene Expression Omnibus (GSE212217). Raw exome sequencing data are unavailable due to the provisions of the trial sponsors; the processed exome variant calls and copy number segmentation files are available in Zenodo (10.5281/zenodo.6985601) and Github (https://github.com/rdchow/endo_PD1). The predicted pathogenic/deleterious variant calls are also available in the supplementary tables. Custom scripts used for analysis and additional processed data files are available on Github (https://github.com/rdchow/endo_PD1). Previously published datasets analyzed in this study are described in the Methods.
Supplementary Material
Statement of Significance.
The molecular mechanism of MMRd is associated with response to PD-1 immunotherapy in endometrial carcinoma. Tumors with epigenetic MMRd or mutational MMRd are correlated with NK cell or CD8+ T cell-driven immunity, respectively. Classifying tumors by the mechanism of MMRd may inform clinical decision-making regarding cancer immunotherapy.
Acknowledgements
R.D. Chow is supported by the NIH Medical Scientist Training Program (T32GM136651) and the NIH/NCI (F30CA250249). A. Iwasaki is an Investigator of the Howard Hughes Medical Institute. E. Song is supported by the NIH Medical Scientist Training Program (T32GM136651) and the NIH/NCI (F30CA239444). R.D. Chow and E. Song are also supported by the Paul & Daisy Soros Fellowship for New Americans. A.D. Santin is supported in part by Gilead Sciences Inc., the NIH (U01CA176067-01A1), NCI (P30CA16359), Stand Up To Cancer (Convergence Grant 2.0), the Discovery to Cure Foundation, and the Guido Berlucchi Foundation. The authors also thank Merck-US for its industry support.
Footnotes
Conflict of Interest Statement:
Alessandro D. Santin reports grants or contracts from Immunomedics, Genentech, Puma, Gilead, Synthon, Boehringer-Ingelheim, Tesaro, and Eisai (to his institution) and consulting fees from Merck, Tesaro, and Eisai. The other authors declare no potential conflicts of interest.
References
- 1.Bonneville R, Krook MA, Kautto EA, Miya J, Wing MR, Chen H-Z, et al. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precision Oncology. Wolters Kluwer; 2017;1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hause RJ, Pritchard CC, Shendure J, Salipante SJ. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med. 2016;22:1342–50. [DOI] [PubMed] [Google Scholar]
- 3.Germano G, Lamba S, Rospo G, Barault L, Magrì A, Maione F, et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature. 2017;552:116–20. [DOI] [PubMed] [Google Scholar]
- 4.Mandal R, Samstein RM, Lee K-W, Havel JJ, Wang H, Krishna C, et al. Genetic diversity of tumors with mismatch repair deficiency influences anti–PD-1 immunotherapy response. Science. 2019;364:485–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.André T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. New England Journal of Medicine. Massachusetts Medical Society; 2020;383:2207–18. [DOI] [PubMed] [Google Scholar]
- 6.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord J-P, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. JCO. Wolters Kluwer; 2020;38:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li K, Luo H, Huang L, Luo H, Zhu X. Microsatellite instability: a review of what the oncologist should know. Cancer Cell International. 2020;20:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Post CCB, Stelloo E, Smit, Ruano, Tops, Vermij, et al. Prevalence and Prognosis of Lynch Syndrome and Sporadic Mismatch Repair Deficiency in Endometrial Cancer. J Natl Cancer Inst. 2021;113:1212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bellone S, Roque DM, Siegel ER, Buza N, Hui P, Bonazzoli E, et al. A phase 2 evaluation of pembrolizumab for recurrent Lynch-like versus sporadic endometrial cancers with microsatellite instability. Cancer. 2022;128:1206–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 13.Levine DA. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Yang T-HO, et al. The Immune Landscape of Cancer. Immunity. Elsevier; 2018;48:812–830.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valero C, Lee M, Hoen D, Weiss K, Kelly DW, Adusumilli PS, et al. Pretreatment neutrophil-to-lymphocyte ratio and mutational burden as biomarkers of tumor response to immune checkpoint inhibitors. Nature Communications. Nature Publishing Group; 2021;12:729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samstein RM, Lee C-H, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valero C, Lee M, Hoen D, Wang J, Nadeem Z, Patel N, et al. The association between tumor mutational burden and prognosis is dependent on treatment context. Nat Genet. 2021;53:11–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rowley MJ, Corces VG. Organizational principles of 3D genome architecture. Nat Rev Genet. 2018;19:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katainen R, Dave K, Pitkänen E, Palin K, Kivioja T, Välimäki N, et al. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat Genet Nature Publishing Group; 2015;47:818–21. [DOI] [PubMed] [Google Scholar]
- 20.Mashtalir N, D’Avino AR, Michel BC, Luo J, Pan J, Otto JE, et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell. Elsevier; 2018;175:1272–1288.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu JN, Roberts CWM. ARID1A Mutations in Cancer: Another Epigenetic Tumor Suppressor? Cancer Discov American Association for Cancer Research; 2013;3:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med Nature Publishing Group; 2018;24:556–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. American Association for Cancer Research; 2017;7:188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. New England Journal of Medicine. 2016;375:819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lawson KA, Sousa CM, Zhang X, Kim E, Akthar R, Caumanns JJ, et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature. 2020;586:120–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghoreschi K, Laurence A, O’Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228:273–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547:413–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016;167:397–404.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Müller M, Briscoe J, Laxton C, Guschin D, Ziemiecki A, Silvennoinen O, et al. The protein tyrosine kinase JAK1 complements defects in interferon-α/β and -γ signal transduction. Nature. 1993;366:129–35. [DOI] [PubMed] [Google Scholar]
- 30.Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, et al. Disruption of the Jak1 Gene Demonstrates Obligatory and Nonredundant Roles of the Jaks in Cytokine-Induced Biologic Responses. Cell. 1998;93:373–83. [DOI] [PubMed] [Google Scholar]
- 31.Torrejon DY, Abril-Rodriguez G, Champhekar AS, Tsoi J, Campbell KM, Kalbasi A, et al. Overcoming Genetically Based Resistance Mechanisms to PD-1 Blockade. Cancer Discovery. 2020;10:1140–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, et al. A major chromatin regulator determines resistance of tumor cells to T cell–mediated killing. Science. American Association for the Advancement of Science; 2018;359:770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548:537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kearney CJ, Vervoort SJ, Hogg SJ, Ramsbottom KM, Freeman AJ, Lalaoui N, et al. Tumor immune evasion arises through loss of TNF sensitivity. Science Immunology. 2018;3:eaar3451. [DOI] [PubMed] [Google Scholar]
- 35.Gulhan DC, Garcia E, Lee EK, Lindemann NI, Liu JF, Matulonis UA, et al. Genomic Determinants of De Novo Resistance to Immune Checkpoint Blockade in Mismatch Repair-Deficient Endometrial Cancer. JCO Precis Oncol. 2020;4:492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell. 2018;173:291–304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim T-M, Laird PW, Park PJ. The Landscape of Microsatellite Instability in Colorectal and Endometrial Cancer Genomes. Cell. 2013;155:858–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017;7:818–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tarabichi M, Salcedo A, Deshwar AG, Ni Leathlobhair M, Wintersinger J, Wedge DC, et al. A practical guide to cancer subclonal reconstruction from DNA sequencing. Nat Methods. Nature Publishing Group; 2021;18:144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dann E, Henderson NC, Teichmann SA, Morgan MD, Marioni JC. Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat Biotechnol. 2022;40:245–53. [DOI] [PubMed] [Google Scholar]
- 41.van Montfoort N, Borst L, Korrer MJ, Sluijter M, Marijt KA, Santegoets SJ, et al. NKG2A Blockade Potentiates CD8 T Cell Immunity Induced by Cancer Vaccines. Cell. 2018;175:1744–1755.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen Y-JJ, Chitre AS, et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature. 2020;579:274–8. [DOI] [PubMed] [Google Scholar]
- 43.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soneson C, Robinson MD. Bias, robustness and scalability in single-cell differential expression analysis. Nat Methods. 2018;15:255–61. [DOI] [PubMed] [Google Scholar]
- 45.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell. 2021;184:5309–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Litchfield K, Reading JL, Puttick C, Thakkar K, Abbosh C, Bentham R, et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell. 2021;184:596–614.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nolan E, Savas P, Policheni AN, Darcy PK, Vaillant F, Mintoff CP, et al. Combined immune checkpoint blockade as a therapeutic strategy for BRCA1-mutated breast cancer. Sci Transl Med. 2017;9:eaal4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Principe DR, Park A, Dorman MJ, Kumar S, Viswakarma N, Rubin J, et al. TGFβ Blockade Augments PD-1 Inhibition to Promote T-Cell–Mediated Regression of Pancreatic Cancer. Mol Cancer Ther. American Association for Cancer Research; 2019;18:613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. American Society for Clinical Investigation; 2017;127:2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grasso CS, Tsoi J, Onyshchenko M, Abril-Rodriguez G, Ross-Macdonald P, Wind-Rotolo M, et al. Conserved Interferon-γ Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell. 2020;38:500–515.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity. 2018;49:178–193.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. The Lancet Oncology. Elsevier; 2017;18:1009–21. [DOI] [PubMed] [Google Scholar]
- 57.Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics. 2014;30:1015–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med. 2018;24:1178–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell. 2018;172:1022–1037.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends in Immunology. Elsevier; 2001;22:633–40. [DOI] [PubMed] [Google Scholar]
- 61.Hanna J, Bechtel P, Zhai Y, Youssef F, McLachlan K, Mandelboim O. Novel Insights on Human NK Cells’ Immunological Modalities Revealed by Gene Expression Profiling. The Journal of Immunology. American Association of Immunologists; 2004;173:6547–63. [DOI] [PubMed] [Google Scholar]
- 62.Hamann I, Unterwalder N, Cardona AE, Meisel C, Zipp F, Ransohoff RM, et al. Analyses of phenotypic and functional characteristics of CX3CR1-expressing natural killer cells. Immunology. 2011;133:62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Butler B, Cooper JA. Distinct roles for the actin nucleators Arp2/3 and hDia1 during NK-mediated cytotoxicity. Curr Biol. 2009;19:1886–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Salerno F, Engels S, van den Biggelaar M, van Alphen FPJ, Guislain A, Zhao W, et al. Translational repression of pre-formed cytokine-encoding mRNA prevents chronic activation of memory T cells. Nat Immunol. 2018;19:828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moore MJ, Blachere NE, Fak JJ, Park CY, Sawicka K, Parveen S, et al. ZFP36 RNA-binding proteins restrain T cell activation and anti-viral immunity. eLife. 7:e33057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. American Association for the Advancement of Science; 2015;348:69–74. [DOI] [PubMed] [Google Scholar]
- 67.Tran E, Ahmadzadeh M, Lu Y-C, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. American Association for the Advancement of Science; 2015;350:1387–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tran E, Turcotte S, Gros A, Robbins PF, Lu Y-C, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344:641–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. Nature Publishing Group; 2014;515:577–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Varley KE, Mutch DG, Edmonston TB, Goodfellow PJ, Mitra RD. Intra-tumor heterogeneity of MLH1 promoter methylation revealed by deep single molecule bisulfite sequencing. Nucleic Acids Research. 2009;37:4603–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bindra RS, Glazer PM. Co-repression of mismatch repair gene expression by hypoxia in cancer cells: role of the Myc/Max network. Cancer Lett. 2007;252:93–103. [DOI] [PubMed] [Google Scholar]
- 72.Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, et al. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N’-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 1994;54:4308–12. [PubMed] [Google Scholar]
- 73.Buermeyer AB, Patten CW-V, Baker SM, Liskay RM. The Human MLH1 cDNA Complements DNA Mismatch Repair Defects in Mlh1-deficient Mouse Embryonic Fibroblasts. Cancer Res American Association for Cancer Research; 1999;59:538–41. [PubMed] [Google Scholar]
- 74.Menghi F, Banda K, Kumar P, Straub R, Dobrolecki L, Rodriguez IV, et al. Genomic and epigenomic BRCA alterations predict adaptive resistance and response to platinum-based therapy in patients with triple-negative breast and ovarian carcinomas. Science Translational Medicine. American Association for the Advancement of Science; 2022;14:eabn1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Buchanan DD, Clendenning M, Rosty C, Eriksen SV, Walsh MD, Walters RJ, et al. Tumor testing to identify lynch syndrome in two Australian colorectal cancer cohorts. Journal of Gastroenterology and Hepatology. 2017;32:427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nikolich-Žugich J The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol Nature Publishing Group; 2018;19:10–9. [DOI] [PubMed] [Google Scholar]
- 77.Kugel CH, Douglass SM, Webster MR, Kaur A, Liu Q, Yin X, et al. Age Correlates with Response to Anti-PD1, Reflecting Age-Related Differences in Intratumoral Effector and Regulatory T-Cell Populations. Clin Cancer Res. 2018;24:5347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dubrot J, Du PP, Lane-Reticker SK, Kessler EA, Muscato AJ, Mehta A, et al. In vivo CRISPR screens reveal the landscape of immune evasion pathways across cancer. Nat Immunol Nature Publishing Group; 2022;23:1495–506. [DOI] [PubMed] [Google Scholar]
- 79.Wang X, Tokheim C, Gu SS, Wang B, Tang Q, Li Y, et al. In vivo CRISPR screens identify the E3 ligase Cop1 as a modulator of macrophage infiltration and cancer immunotherapy target. Cell. 2021;184:5357–5374.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Benci JL, Xu B, Qiu Y, Wu T, Dada H, Victor CT-S, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell. 2016;167:1540–1554.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z, et al. Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell. 2019;178:933–948.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shia J, Zhang L, Shike M, Guo M, Stadler Z, Xiong X, et al. Secondary mutation in a coding mononucleotide tract in MSH6 causes loss of immunoexpression of MSH6 in colorectal carcinomas with MLH1/PMS2 deficiency. Mod Pathol. 2013;26:131–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kaneko E, Sato N, Sugawara T, Noto A, Takahashi K, Makino K, et al. MLH1 promoter hypermethylation predicts poorer prognosis in mismatch repair deficiency endometrial carcinomas. J Gynecol Oncol. 2021;32:e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Buchanan DD, Tan YY, Walsh MD, Clendenning M, Metcalf AM, Ferguson K, et al. Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. J Clin Oncol. 2014;32:90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:13033997 [q-bio] [Internet]. 2013. [cited 2017 Aug 8]; Available from: http://arxiv.org/abs/1303.3997 [Google Scholar]
- 86.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Taylor-Weiner A, Stewart C, Giordano T, Miller M, Rosenberg M, Macbeth A, et al. DeTiN: overcoming tumor-in-normal contamination. Nat Methods. 2018;15:531–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Research. 2011;39:e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research. 2019;47:D886–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mayakonda A, Lin D-C, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28:1747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kawaguchi S, Higasa K, Shimizu M, Yamada R, Matsuda F. HLA-HD: An accurate HLA typing algorithm for next-generation sequencing data. Hum Mutat. 2017;38:788–97. [DOI] [PubMed] [Google Scholar]
- 94.Hundal J, Carreno BM, Petti AA, Linette GP, Griffith OL, Mardis ER, et al. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Medicine. 2016;8:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tanner G, Westhead DR, Droop A, Stead LF. Benchmarking pipelines for subclonal deconvolution of bulk tumour sequencing data. Nat Commun Nature Publishing Group; 2021;12:6396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, et al. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol. 2015;26:64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gillis S, Roth A. PyClone-VI: scalable inference of clonal population structures using whole genome data. BMC Bioinformatics. 2020;21:571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Koch A, Jeschke J, Van Criekinge W, van Engeland M, De Meyer T. MEXPRESS update 2019. Nucleic Acids Research. 2019;47:W561–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pappalardo JL, Zhang L, Pecsok MK, Perlman K, Zografou C, Raddassi K, et al. Transcriptomic and clonal characterization of T cells in the human central nervous system. Sci Immunol. 2020;5:eabb8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Song E, Bartley CM, Chow RD, Ngo TT, Jiang R, Zamecnik CR, et al. Divergent and self-reactive immune responses in the CNS of COVID-19 patients with neurological symptoms. Cell Rep Med. 2021;2:100288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biology. 2019;20:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20:163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pauken KE, Shahid O, Lagattuta KA, Mahuron KM, Luber JM, Lowe MM, et al. Single-cell analyses identify circulating anti-tumor CD8 T cells and markers for their enrichment. J Exp Med. 2021;218:e20200920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang T, Li B, Nelson CE, Nabavi S. Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data. BMC Bioinformatics. 2019;20:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. The Innovation. 2021;2:100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell. Elsevier; 2018;173:400–416.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed scRNA-seq/scTCR-seq data generated in this study are available in the Gene Expression Omnibus (GSE212217). Raw bulk TCR-seq data of TILs were generated by an external core facility (Adaptive Biotechnologies) and are unavailable, but the processed data are available in Zenodo (10.5281/zenodo.6985601) and the Gene Expression Omnibus (GSE212217). Raw exome sequencing data are unavailable due to the provisions of the trial sponsors; the processed exome variant calls and copy number segmentation files are available in Zenodo (10.5281/zenodo.6985601) and Github (https://github.com/rdchow/endo_PD1). The predicted pathogenic/deleterious variant calls are also available in the supplementary tables. Custom scripts used for analysis and additional processed data files are available on Github (https://github.com/rdchow/endo_PD1). Previously published datasets analyzed in this study are described in the Methods.