

Abstract
Antiestrogen medication is the only chemoprevention currently available for women at a high risk of developing breast cancer; however, antiestrogen therapy requires years to achieve efficacy and has adverse side effects. Therefore, it is important to develop an efficacious chemoprevention strategy that requires only a short-course of treatment. PIK3CA is commonly activated in breast atypical hyperplasia, the known precancerous precursor of breast cancer. Targeting PI3K signaling in these precancerous lesions may offer a new strategy for chemoprevention. Here, we first established a mouse model that mimics the progression from precancerous lesions to breast cancer. Next, we demonstrated that a short-course prophylactic treatment with the clinically approved PI3K inhibitor alpelisib slowed early lesion expansion and prevented cancer formation in this model. Furthermore, we showed that alpelisib suppressed ex vivo expansion of patient-derived atypical hyperplasia. Together, these data indicate that the progression of precancerous breast lesions heavily depends on the PI3K signaling, and that prophylactic targeting of PI3K activity can prevent breast cancer.
Keywords: breast cancer, chemoprevention, mouse model, PIK3CA, alpelisib
Introduction
Preventing breast cancer in high-risk patients is an effective strategy for reducing the burden of breast cancer. Current chemoprevention relies on 3–5 years of endocrine therapy and can prevent approximately 50% of estrogen receptor-positive (ER+) breast cancer (1). However, adherence is low due to the lengthy treatment time required to achieve prevention, and side effects are common, including hot flashes, increased uterine cancer risk and blood clots (1). In addition, no significant protection is provided against the more aggressive ER-negative breast cancer. A chemoprevention strategy that does not require years of treatment is needed. Lessons learned from the biology of breast tumorigenesis and tumor treatment that target phosphatidylinositol-3-kinase (PI3K) signaling point to a promising strategy in cancer prevention. Activating mutations in PIK3CA, which encodes the catalytic subunit of PI3K, are detected in up to 70% of proliferative benign lesions (2). PIK3CA mutations also exist in 35.7% of breast cancers, including 42% of ER+ breast cancers, 31% of HER2+ breast cancers, and 16% of triple-negative breast cancers (TNBCs, which lack ER, progesterone receptor, and HER2) (3). PIK3CA H1047R is a hotspot mutation accounting for 35% of identified PIK3CA mutations in breast tumors. Knocking in or overexpressing PIK3CA with the H1047R mutation (PIK3CAH1047R) in murine mammary glands resulted in tumors with a long latency (4–6), signifying its role in driving the tumorigenic process.
The high prevalence of genetic alterations in PIK3CA in breast and other cancers has stimulated the development of PI3K inhibitors. PI3K inhibitors targeting PIK3CA together with other family members (PIK3CB, PIK3CD, and PIK3CG) often cause multiple adverse side effects, while PI3K isoform-selective inhibitors demonstrate better tolerance (7,8). Alpelisib is the first FDA-approved PIK3CA-selective inhibitor and degrader for treating ER+ breast cancer harboring PIK3CA mutations (9,10). The higher frequency of PIK3CA mutations in early breast lesions provides rationale for targeting the PI3K pathway as a chemopreventive strategy for all subtypes of breast cancer. Wang et al. recently demonstrated that the experimental pan-PI3K inhibitor GDC-0941 can delay the appearance of murine mammary tumors initiated by transgenic expression of HER2 (11). However, it remains unknown whether prophylactic treatment targeting PI3K prevents human breast cancer, in particular the predominant HER2-negative breast tumors comprising ~75–80% of all breast cancer cases.
There are no mouse models in which precancerous human breast cells reliably progress to invasive carcinoma. In this study, we established a clinically relevant mouse model that mimics human breast tumorigenesis by up-the-teat injection of the immortalized human mammary epithelial cell line MCF10A transduced with PIK3CAH1047R. We demonstrated that alpelisib treatment drastically delayed breast tumor formation. We also showed that alpelisib suppressed the growth of both PIK3CA-mutated and -WT patient-derived atypia cultures in vitro. Together, these preclinical data indicate that short-term alpelisib treatment is an efficient chemoprevention strategy that should be tested in clinical trials.
Materials and Methods
Cell lines
The immortalized human mammary epithelial cell line MCF10A (RRID:CVCL_0598; undocumented source) was grown in DMEM/F12 medium supplemented with 5% horse serum, epidermal growth factor (20 ng/ml), insulin (10 ng/ml), cholera toxin (100 ng/ml), hydrocortisone (500 ng/ml), and 1% penicillin-streptomycin. Human primary breast cells with hyperplasia were obtained from the Behbod lab following IRB protocol. Cells from female donors in 2017 arrived in single cell suspension and were cultured in EpiCult™-C human medium (STEMCELL Technologies, Vancouver, Canada) supplemented with 10 μM Forskolin (Cayman Chemical, MI). HEK293T cells (RRID:CVCL_0063; undocumented source) were maintained in DMEM medium supplemented with 10% heat-inactivated FBS and 1% penicillin-streptomycin. All cell experiments were performed within 10 passages after thawing. In particular, experiments utilizing primary cells were performed within 5 passages. MCF10A was authenticated by ATCC using STR profiling. HEK293T cells and human primary cells were not authenticated. In addition, all cell lines were routinely tested and confirmed negative for mycoplasma contamination by Lonza™ Mycoalert™ Mycoplasma Detection Kit (Lonza, TX, USA).
Virus production
FUCGW-PIK3CAH1047R expression plasmid was generated as previously published (6). For virus production, HEK293T cells were transfected with expression plasmid and helper plasmids using jetPRIME transfection reagents per user’s manual (Polyplus, France). Lentivirus was concentrated by ultracentrifugation, and titration was determined by flow cytometry. MCF10A cells were transduced by virus (M.O.I.=10) and enriched by fluorescence-activated cell sorting (FACS) to generate MCF10A-vector and MCF10A-PIK3CAH1047R cell lines.
Chemicals
Alpelisib (BYL719) (Chemietek, IN, USA) stock solution was made at 20 mM in DMSO for in vitro assays. For oral gavage, alpelisib was formulated in 5% DMSO, 50% PEG300 (90878, Sigma, USA), 5% Tween 80 (P4780, Sigma, USA), and 40% distilled water. Fulvestrant (Selleck Chemicals, TX, USA) was made as 1 mM stock in alcohol for in vitro assays. For subcutaneous injection, fulvestrant was formulated in 5% DMSO and 95% corn oil.
Cell viability assay
MCF10A cells were seeded at 2,000 cells per well in 96-well plate format, and then treated with drug or matched vehicle 1 day post-seeding. Human primary UDH or ADH cells were seeded at 5,000 cells per well in 96-well plate format, and then treated with alpelisib (1, 2, 5, 10 μM, Chemietek, USA) or vehicle 1 day post-seeding. The cell viability was determined 3 days post treatment using the colorimetric cell counting kit-8 (CCK8) following the manufacturer’s instructions (ApexBio, Houston, TX, USA). The measurement was read at absorbance at 450 nm after 3 hours of incubation with CCK reagent.
Digital PCR workflow
DNA was extracted from each sample using the DNeasy kit (Qiagen, Germany) after which 10 μL at 20 ng/μL was submitted to the LSSBC Molecular Diagnostics Laboratory (LSSBC-MDL) at BCM for analysis via droplet digital PCR (ddPCR). The assay was performed using the QX200 AutoDG Droplet Digital PCR System (Bio-Rad, California, US) per manufacturer’s protocol. Following droplet generation, the plate was transferred to a Thermo Cycler (C1000 Touch, Biorad) and conditions were set following Biorad’s standard recommendations. Each PCR reaction was performed on 50 ng of purified input genomic DNA. To monitor for potential contamination, no template controls were used throughout. Each sample was digested with Haelll restriction enzyme prior to droplet generation. Biorad assays (probes) were selected for each target of interest: FAM and HEX-labeled probes (PIK3CA p.E545K, dHsaMDS986963652; PIK3CA p.E542K, dHsaMDV2010073; PIK3CA p.H1047L, dHsaMDV2010123; and PIK3CA p.H1047R, dHsaMDV2010077; Bio-Rad). All preparation and assays were performed in a dedicated pre- and post-PCR rooms. All ddPCR assays included positive and negative controls. Samples were read on the Bio-Rad QX200 droplet reader and analyzed with QX Manager™ software (v.1.1) to obtain fractional abundance values for each sample assay.
Animal study
All animals were handled according to the protocol AN-2834 as approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine (Houston, TX). Mammary glands of 8~12-week old female NSG mice (RRID:IMSR_JAX:021885) were implanted with 30,000 cells per gland in 10 μL DMEM/F12 basal medium. The growth of implanted cells was monitored by bioluminescence using IVIS and initiation of tumor was scored at 2.0 mm in diameter by palpation. Tumors were harvested and subjected to full body organ survey for bioluminescence signals when reaching 2.0 cm in diameter or at the ethical endpoint. The primary tumors were fixed in 4% paraformaldehyde or snap frozen for subsequent applications. In the short-term alpelisib treatment scheme, mice were randomized and alpelisib was administered via a once-daily oral gavage at 50 mg/kg/day. A 12-day period was based on 5-day on, 2-day off, and 5-day on schedule, repeated after a week break. The bioluminescence signal was read on day 0 (pre) and the last day of drug administration (post).
To establish lung lesions, 8-week-old female NSG mice received 1,000,000 cells of MCF10A-PIK3CAH1047R cells via tail vein injection, and tumor growth was monitored by bioluminescence using IVIS weekly. Mice were randomized for drug treatment when the overall signal in lung reached the initial baseline. Fulvestrant at 200 mg/kg or vehicle was administered by subcutaneous (s.c.) injection weekly.
Protein extraction and Western blotting analysis
Cells were harvested in RIPA buffer with a Xpert duo inhibitor cocktail (GenDEPOT, TX, USA). Total protein of cell lysates was quantified by BCA™ Protein Assay Kit (Pierce Biotechnology, IL, USA) and equal amounts of protein (30 μg) were resolved by SDS-PAGE with 10% gel and transferred to PVDF membranes (GE Life Sciences, USA). Blots were incubated overnight at 4°C with rabbit anti-phospho-AKT (Ser473), diluted 1:1,000 (Cell Signaling Technology Cat# 3787, RRID:AB_331170), rabbit anti-AKT, diluted 1:1,000 (Cell Signaling Technology Cat# 9272, RRID:AB_329827), rabbit anti-phospho-4E-BP1 (Thr 37/46), diluted 1:1,000 (Cell Signaling Technology Cat# 2855, RRID:AB_560835), rabbit anti- 4E-BP1, diluted 1:1,000 (Cell Signaling Technology Cat# 9644, RRID:AB_2097841), rabbit anti-phospho-S6, diluted 1:1,000 (Cell Signaling Technology Cat# 2211, RRID:AB_331679), mouse anti-S6, diluted 1:1,000 (Santa Cruz Biotechnology, Santa Cruz, USA, Cat# sc-74576, RRID:AB_2181030), mouse anti-β-actin antibody, diluted 1:5,000 (Sigma-Aldrich Cat# A5441, RRID:AB_476744), and then with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (Sigma). Protein signals were detected using the SuperSignal West Pico Stable Peroxide Solution (Thermo Scientific, USA) and captured by ChemiDoc Imager (Biorad).
Immunostaining
Tissue specimens were fixed, paraffin-embedded, and sectioned to 3 μm in thickness. The sections were then heated in a 37 °C incubator overnight, dewaxed with xylene, and dehydrated with gradient alcohol. Next, the slides received 3% peroxidase treatment for 10 min, underwent antigen retrieval in 0.01 M citric acid buffer with pressure cooker, cooled at room temperature, and then rinsed with PBS (pH 7.4). The sections were blocked with normal goat serum and incubated with diluted antibodies against KU80 (1:800, Cell Signaling Technology Cat# 2180, RRID:AB_2218736), ERα (1:200, Leica Biosystems Cat# NCL-L-ER-6F11, RRID:AB_563706), PR (1:1600, Agilent Cat# M3568, RRID:AB_2252608), HER2 (1:100, Lab Vision Cat# RM-9103-S, RRID:AB_149908), phospho-AKT (Ser473) (1:50, Cell Signaling Technology Cat# 3787, RRID:AB_331170), and ERα (SP1) (1:200, Thermo Fisher Scientific Cat# MA5-14501, RRID:AB_10981779). The sections were counterstained with hematoxylin, dehydrated with gradient alcohol, cleared with xylene, and mounted for observation and imaging using the Olympus image analysis software CellSens standard (Model U-MDOB). In parallel, tumor slides were counterstained by Hematoxylin and Eosin and submitted to the pathologist for a blinded scoring at the tumor grade.
Statistical analysis
All graphs were generated and analyzed using GraphPad Prism 9.0 software (GraphPad, San Diego, California, USA). Kaplan-Meier survival analysis for tumor latency and ethical endpoint survival was analyzed by log-rank (Mantel-Cox) test. All data sets were presented as mean ± S.D. unless otherwise indicated. Comparisons between two groups or multiple groups were performed by two-tailed Student’s t-test or one-way ANOVA, respectively. To examine the influence of two independent parameters on multiple groups, two-way ANOVA was used. Statistically significant differences were indicated as follows: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. Statistical details of experiments can be found in the figure legends.
Data Availability
The data generated in this study are available upon request from the corresponding author.
Results
Intraductal delivery of MCF10A-PIK3CAH1047R leads to ER+ ductal carcinoma in mice
The PIK3CA hotspot mutation H1047R (PIK3CAH1047R) is known to activate PI3K signaling for oncogenic transformation in culture and for driving mammary tumor formation in mice (4–6,12–15). Previous attempts to drive tumorigenesis of the immortalized human breast epithelial line MCF10A by overexpressing PIK3CAH1047R and grafting the resulting cells into the mouse fat-pad have not been very successful (12). Whether MCF10A- PIK3CAH1047R cells can form tumors with a more supportive microenvironment has not been reported. The mammary intraductal (MIND) model (16–18) provides a native niche for precancerous breast cells to progress. To test whether MCF10A-PIK3CAH1047R cells are able to progress to tumors in the ductal microenvironment, we injected intraductally via the teat 30,000 luciferase-labelled MCF10A-PIK3CAH1047R cells or MCF10A-vector control cells into mammary glands of 12-week-old NSG virgin female mice. We then tracked lesion growth and the progression to tumors by bioluminescence imaging and palpation (Fig. 1A). MCF10A-PIK3CAH1047R cells expanded and established detectable lesions within 9 weeks, but MCF10A-vector cells did not (Fig. 1B, 1C, 1D). We confirmed by H&E the presence of early lesions from MCF10A-PIK3CAH1047R cells at 6 weeks after cell implantation (Fig. 1D). These lesions advanced to palpable tumors with a medium tumor latency of 22.85 weeks (Fig. 1E, N=47) and a median ethical endpoint survival of 51.1 weeks (Fig. 1F, N=34). H&E staining showed that 9 out of 10 (90%) MCF10A-PIK3CAH1047R tumors were high-grade adenocarcinoma with secretory features (Fig. 2A). The deposition of oil droplets was confirmed by Oil Red O (ORO) staining, and the presence of glycogen was confirmed by Periodic acid–Schiff (PAS) staining (Fig. S1). Immunohistochemistry (IHC) staining against the human biomarker KU80 confirmed the human origin of these tumors (Fig. 2B). Mammary tumor profiling by IHC showed that these tumors were ER-positive, PR-negative, and HER2-negative (Fig. 2B). The percentage of ER-positive cells in tumors ranged from 5 to 20%, and the ER expression was validated of human origin (Fig. S2).
Figure 1. PIK3CAH1047R confers tumorigenicity to MCF10A cells in vivo.
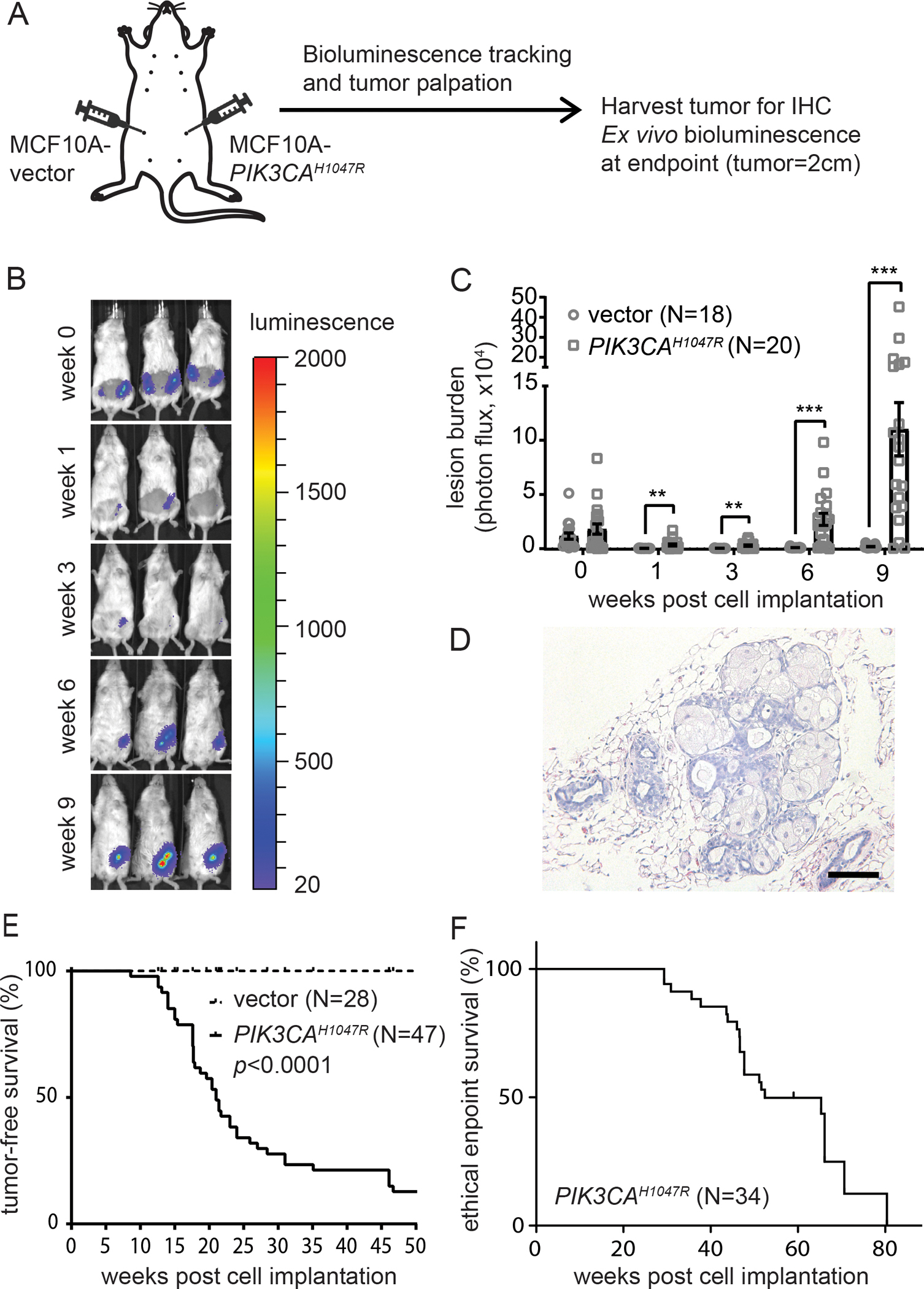
A, Diagram of the mouse intraductal model for MCF10A-PIK3CAH1047R cells. Each #4 mammary glands were intraductally implanted with 30,000 cells. One side is MCF10A-vector and the other side is MCF10A-PIK3CAH1047R. B, Representative mouse images depict the IVIS signals over 9 weeks post cell implantation. C, Quantification of bioluminescence signals by IVIS over time. D, H&E of PIK3CAH1047R-initiated early lesions at 6 weeks post cell implantation. Scale = 100 μm. E, Tumor-free survival. F, Ethical endpoint survival. N of each group is as indicated. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Figure 2. PIK3CAH1047R drives MCF10A cells to form mammary tumors and metastasize to multiple organs.

A, H&E of PIK3CAH1047R-initiated mammary tumors. Left, high-grade (9/10). Right, low-grade (1/10). Scale = 100 μm. B, Immunohistochemical staining for the human biomarker KU80, ERα, PR, and HER2. Insets are positive control. Scale = 100 μm. C, Ex vivo bioluminescence image of the indicated organs at the ethical endpoint. D, Quantification of bioluminescence signals in the indicated organs.
To investigate the metastatic potential of MCF10A-PIK3CAH1047R tumors, we euthanized the mice at the ethical endpoint (tumor size = 2.0 cm), and with ex vivo bioluminescence we determined whether disseminated tumor cells were present in different organs (N=11) including the bones, lungs, kidneys, spleen, livers, ovaries, and brain (Fig. 2C, 2D). We found spontaneous metastases in multiple organs, but predominantly in the lungs and bones. Among 11 mice, 9 (81.8%) exhibited bone metastasis and 5 (45.5%) exhibited lung metastasis, while 4 (36.4%) presented both bone and lung metastases. Thus, we have established a MCF10A-PIK3CAH1047R MIND model for testing chemoprevention against human breast cancer.
Alpelisib inhibits the growth of precancerous cells in vitro
To study whether the growth of precancerous human breast cells depends on sustained PI3K signaling, we first subjected MCF10A-vector cells and MCF10A-PIK3CAH1047R cells in culture dishes to the PI3K inhibitor alpelisib, at doses ranging from 0 to 10 μM. Without alpelisib treatment, MCF10A-PIK3CAH1047R cells proliferated 52.8% more than MCF10A-vector cells did at 72 hours post plating (Fig. 3A). Upon alpelisib treatment, both MCF10A-PIK3CAH1047R cells and MCF10A-vector cells showed dose-dependent inhibition, but MCF10A-PIK3CAH1047R cells were inhibited to a significantly greater extent (Fig. 3B). In line with the finding, alpelisib effectively deactivated PI3K signaling components including AKT, 4E-BP1, and S6 (Fig. 3C). These data indicate that alpelisib is effective in suppressing PI3K signaling and arresting the growth of PIK3CA-mutated precancerous cells while showing a therapeutic window where normal cells may be spared. Given MCF10A-PIK3CAH1047R cells generated ER-positive tumors, we also tested whether fulvestrant, an ER degrader, can inhibit the growth of MCF10A-PIK3CAH1047R cells, but we did not observe any inhibitory effect on cell growth in the absence or presence of alpelisib (Fig. S3A). Similarly, fulvestrant treatment of mice did not inhibit the growth of MCF10A-PIK3CAH1047R lesions in lungs (Fig. S3B). Therefore, for the remainder of this study, we did not further pursue endocrine treatment or consider combination treatment.
Figure 3. Alpelisib inhibits the growth of precancerous human breast cells in vitro.

A, Cell viability of MCF10A-PIK3CAH1047R cells at 3 days post plating measured by CCK8 reagents. Data was normalized to MCF10A-vector. B, Alpelisib dose response curves of MCF10A-PIK3CAH1047R cells and MCF10A-vector cells. Cell viability was measured 3 days post alpelisib treatment, and data was normalized to vehicle treatment of the respective cell line. C, Western blotting for the indicated proteins after alpelisib treatment at 1 μM for 3 hours. D, Dose response curves of human UDH and ADH cells. Cell viability was measured 3 days post alpelisib treatment, and data was normalized to vehicle control of respective cell line. Orange line, PIK3CA MUT cell line; black line, PIK3CA WT cell line. Data points are mean ± S.D. Two-way ANOVA. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
To validate the finding of alpelisib’s cancer preventive effects, we established short-term cultures of human proliferative lesions from human breast, including one case of usual ductal hyperplasia (UDH), and three cases of atypical ductal hyperplasia (ADH). UDH is a non-cancerous lesion of the breast, while ADH is a precancerous lesion linked to a higher breast cancer risk (19). Using droplet digital PCR (ddPCR) to determine PIK3CA mutant status in these primary cultures, we found that one (ADH#SL082317) carried two hot spot PIK3CA mutations (H1047R and H1047L; in approximately 8% cells for each), while the UDH culture and two other ADH cultures were WT (Fig. S4). We next tested the response of these three cultures to alpelisib. We found that they all showed a dose-dependent response, as did MCF10A-PIK3CAH1047R cells (Fig. 3D). At least two of three ADH lines responded significantly better than the UDH line, and the PIK3CA-mutated line trended towards a better response (although not statistically significant). These data suggest that alpelisib is efficacious in inhibiting the expansion of precancerous human breast cells, particularly those with PIK3CA mutations.
Short-term alpelisib prevents MCF10A-PIK3CAH1047R initiated lesions from progressing to cancer
We next tested the cancer prevention efficacy of alpelisib in this MCF10A-PIK3CAH1047R MIND model (Fig. 4). 35 NSG mice were intraductally injected with 30,000 MCF10A-PIK3CAH1047R cells. Six weeks later, these mice were randomized to vehicle vs. alpelisib for two cycles of treatment with a one-week break between the two cycles. Each cycle consists of once daily oral gavage (50 mg/kg)(20,21) for 5 days on, 2 days off, and another 5 days on. Prolonged use of alpelisib is known to have side effects in breast cancer patients that must be carefully monitored (22). In our scheme of short-term prophylactic treatment, alpelisib did not affect body weight, confirming that 2 cycles of alpelisib administration were well tolerated in mice (Fig. S5), consistent with our previous chemoprevention studies using short-term treatment to moderate side effects (6,23,24). We determined the impact of treatment on precancerous lesion burden by comparing the bioluminescence signals in the mammary glands on day 0 and the final day of treatment. The lesion burden increased by 16.26-fold in the vehicle group, but only by 2.5-fold in the alpelisib group (p = 0.0003) (Fig. 4B, 4C), a drastic reduction. As expected, pAKT levels were reduced n in these lesions (Fig. 4D). These data demonstrate that prophylactic alpelisib can block the expansion of precancerous early lesions in vivo.
Figure 4. Short-term prophylactic alpelisib prevents MCF10A-PIK3CAH1047R precancerous lesions from progressing to tumors in mice.

A, Diagram of prophylactic alpelisib treatment in the MCF10A-PIK3CAH1047R MIND model. 30,000 MCF10A-PIK3CAH1047R cells were intraductally injected into one #4 mammary gland. After 6 weeks, mice were randomized to receive alpelisib (50 mg/kg) or vehicle treatments for 2 cycles (5-day on, 2-day off, and 5-day on, then repeat after 1 week). Lesion burden was determined by bioluminescence signals on day 0 (pre) and day 31 (post). B, Representative bioluminescence images of mice pre and post alpelisib treatment. C, Quantification of bioluminescence signals. Two-way ANOVA (p = 0.0003). D, Immunostaining for pAKT (S473) in MCF10A-PIK3CAH1047R early lesions 12 hours post 2 cycles of alpelisib or vehicle treatment. Scale = 100 μm. E, Tumor-free survival. Log-rank (Mantel-Cox) test (p < 0.0001). F, Ethical endpoint survival. Log-rank (Mantel-Cox) test (p = 0.03).
To determine whether this short-term alpelisib prevents these precancerous lesions from progressing to cancer, we palpated both cohorts of mice weekly for tumor appearance. Prophylactic alpelisib treatment increased the median tumor latency from 14.3 weeks to 20.9 weeks (p < 0.0001; Fig. 4E). Prophylactic Alpelisib also extended the median ethical endpoint survival from 37.7 weeks to 46.35 weeks (p = 0.03; Fig. 4F). Prophylactic alpelisib treatment did not affect eventual metastasis burden in lungs or in bones when assayed at the same ethical endpoint (primary tumor size = 2.0 cm; Fig. S6). Together, these data demonstrate that a short course of alpelisib treatment can prevent human breast cancer and improve overall survival.
Discussion
There is increasing evidence that targeted PI3K inhibition benefits patients with PIK3CA-mutated tumors (9,10), but it is still unclear whether prophylactic blockade of PI3K signaling prevents human breast cancer due to the lack of in vivo models to study human breast tumorigenesis. The MCF10A cell line is well-characterized and provides a model to study how PIK3CA mutations contribute to the transition from normal epithelia to tumorigenic cells (12–14). A supportive microenvironment is a key determinant in tumor phenotype and behavior. MCF10A-PIK3CAH1047R cells failed to induce tumor formation when injected to the fat pad (12). Unlike the adipose tissue that relates poorly to where the breast lesions arise, milk ducts provide the best niche. Sflomos et al., reported that the ductal microenvironment allows a higher engraftment rate (25). In this study, we successfully established a MCF10A-PIK3CAH1047R in vivo MIND model which can develop breast lesions and tumors with metastatic potential through intraductally injecting as few as 30,000 cells. Recent reports have demonstrated that mutant PIK3CA stimulated metabolic reprogramming, including de novo fatty acid biosynthesis and increased glutamine metabolism (12,26). In accordance with these findings, MCF10A- PIK3CAH1047R tumors displayed enriched lipid and glycogen content.
Endocrine therapy can reduce the risk of ER+ breast cancer, but does not protect against other breast cancer subtypes (1). A chemoprevention strategy that covers a wider spectrum of breast lesions regardless of ER status is urgently needed. Current evidence points to potential efficacy in targeting PI3K pathways in human breast cancer prevention. Several lines of in vivo evidence clearly demonstrated the inhibiting PI3K activity exerts anti-tumor efficacy by decreasing cell proliferation and increasing cell death in mouse and human cancer xenograft models of breast cancer (11,20,21). Wang et al. reported that the GDC-0941, a PI3K inhibitor, can significantly delay mammary tumor initiation in MMTV-neu mice (11), and they found evidence of potential enhanced immune surveillance as a result of GDC-0941. As our NSG mice lack T, B and NK cells, our chemopreventive efficacy in vivo is likely cell-autonomous in agreement with our in vitro data. But whether an intact immune system may affect chemoprevention efficacy of alpelisib remains to be tested.
Since alpelisib prevented expansion of both PIK3CA-mutated and WT ADH cells in culture, prophylactic alpelisib may prevent cancer from even PIK3CA WT precancerous lesions. This is plausible as besides mutations, PI3K signaling can be activated by a variety of ways including receptor tyrosine kinase activation and PTEN loss in breast tumorigenesis. Of course, these in vitro ADH data need to be validated using in vivo ADH MIND models when they are developed for chemoprevention studies.
We observed excellent chemoprevention efficacy after two cycles of alpelisib (50 mg/kg) treatment over one month. This short-course treatment did not cause significant adverse effects, but future preclinical experiments should test additional treatment schedules including intermittent treatment to achieve high prevention efficacy and low side effects. Alpelisib should also be tested in combination with short-term endocrine treatment to determine whether they together can effectively prevent breast tumor formation, probably in ADH MIND models and rat models since rats readily development estrogen-dependent tumors unlike mice (27)(28–31)(32,33). Finding new chemoprevention strategies is very important since current endocrine-based chemoprevention requires multiple years of continuous treatment, can cause significant side effects and suffer low adherence rates (1).
In conclusion, we demonstrated that alpelisib inhibits precancerous breast cell expansion in culture. Importantly, we established a MIND model of precancerous human lesions that can progress to invasive breast carcinoma with multi-organ metastases. Using this model, we demonstrate that a short-term alpelisib treatment can slow precancerous cell expansion and prevent breast tumor formation. These preclinical data indicate that a short-course alpelisib treatment may prevent breast cancer in high-risk women and should be tested in clinical trials.
Supplementary Material
Prevention Relevance Statement:
PI3K protein is abnormally high in breast precancerous lesions. This preclinical study demonstrates that the FDA-approved anti-PI3K inhibitor alpelisib can prevent breast cancer and thus warrant future clinical trials in high-risk women.
Acknowledgements
This work was financially supported by an NIH-T32 grant (#1T32CA203690-01A1 to A.T. Ku under S.A.W. Fuqua), an NIH R01 grant (R01 CA271498 to Y. Li), and DOD-CDMRP grants (BC191649 and BC191646 to Y. Li). This project was also supported by the Brest Center Pathology Core as part of the SPORE program (#P50 CA186784), and Cytometry and Cell Sorting Core with fundings from the CPRIT (CPRIT-RP180672) and the NIH (P30 CA125123 and S10 RR024574), with the expert assistance of J.M. Sederstrom, and resources from the Dan L. Duncan Cancer Center (P30CA125123).
Footnotes
Conflict of interest statement: The authors declare no potential conflicts of interest.
References
- 1.Britt KL, Cuzick J, Phillips KA. Key steps for effective breast cancer prevention. Nature reviews Cancer 2020;20(8):417–36 doi 10.1038/s41568-020-0266-x. [DOI] [PubMed] [Google Scholar]
- 2.Ang DC, Warrick AL, Shilling A, Beadling C, Corless CL, Troxell ML. Frequent phosphatidylinositol-3-kinase mutations in proliferative breast lesions. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc 2014;27(5):740–50 doi 10.1038/modpathol.2013.197. [DOI] [PubMed] [Google Scholar]
- 3.Martinez-Saez O, Chic N, Pascual T, Adamo B, Vidal M, Gonzalez-Farre B, et al. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast cancer research : BCR 2020;22(1):45 doi 10.1186/s13058-020-01284-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koren S, Reavie L, Couto JP, De Silva D, Stadler MB, Roloff T, et al. PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature 2015;525(7567):114–8 doi 10.1038/nature14669. [DOI] [PubMed] [Google Scholar]
- 5.Tikoo A, Roh V, Montgomery KG, Ivetac I, Waring P, Pelzer R, et al. Physiological levels of Pik3ca(H1047R) mutation in the mouse mammary gland results in ductal hyperplasia and formation of ERalpha-positive tumors. PLoS One 2012;7(5):e36924 doi 10.1371/journal.pone.0036924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young A, Bu W, Jiang W, Ku A, Kapali J, Dhamne S, et al. Targeting the Pro-survival Protein BCL-2 to Prevent Breast Cancer. Cancer prevention research 2022;15(1):3–10 doi 10.1158/1940-6207.CAPR-21-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ellis H, Ma CX. PI3K Inhibitors in Breast Cancer Therapy. Current oncology reports 2019;21(12):110 doi 10.1007/s11912-019-0846-7. [DOI] [PubMed] [Google Scholar]
- 8.Vanhaesebroeck B, Perry MWD, Brown JR, Andre F, Okkenhaug K. PI3K inhibitors are finally coming of age. Nature reviews Drug discovery 2021;20(10):741–69 doi 10.1038/s41573-021-00209-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narayan P, Prowell TM, Gao JJ, Fernandes LL, Li E, Jiang X, et al. FDA Approval Summary: Alpelisib Plus Fulvestrant for Patients with HR-positive, HER2-negative, PIK3CA-mutated, Advanced or Metastatic Breast Cancer. Clin Cancer Res 2021;27(7):1842–9 doi 10.1158/1078-0432.CCR-20-3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner S, Chia S, Kanakamedala H, Hsu WC, Park J, Chandiwana D, et al. Effectiveness of Alpelisib + Fulvestrant Compared with Real-World Standard Treatment Among Patients with HR+, HER2-, PIK3CA-Mutated Breast Cancer. The oncologist 2021;26(7):e1133–e42 doi 10.1002/onco.13804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Zhang Y, Xiao Y, Yuan X, Li P, Wang X, et al. Boosting immune surveillance by low-dose PI3K inhibitor facilitates early intervention of breast cancer. American journal of cancer research 2021;11(5):2005–24. [PMC free article] [PubMed] [Google Scholar]
- 12.Lau CE, Tredwell GD, Ellis JK, Lam EW, Keun HC. Metabolomic characterisation of the effects of oncogenic PIK3CA transformation in a breast epithelial cell line. Scientific reports 2017;7:46079 doi 10.1038/srep46079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bessette DC, Tilch E, Seidens T, Quinn MC, Wiegmans AP, Shi W, et al. Using the MCF10A/MCF10CA1a Breast Cancer Progression Cell Line Model to Investigate the Effect of Active, Mutant Forms of EGFR in Breast Cancer Development and Treatment Using Gefitinib. PLoS One 2015;10(5):e0125232 doi 10.1371/journal.pone.0125232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dawson PJ, Wolman SR, Tait L, Heppner GH, Miller FR. MCF10AT: a model for the evolution of cancer from proliferative breast disease. The American journal of pathology 1996;148(1):313–9. [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H, Liu G, Dziubinski M, Yang Z, Ethier SP, Wu G. Comprehensive analysis of oncogenic effects of PIK3CA mutations in human mammary epithelial cells. Breast cancer research and treatment 2008;112(2):217–27 doi 10.1007/s10549-007-9847-6. [DOI] [PubMed] [Google Scholar]
- 16.Valdez KE, Fan F, Smith W, Allred DC, Medina D, Behbod F. Human primary ductal carcinoma in situ (DCIS) subtype-specific pathology is preserved in a mouse intraductal (MIND) xenograft model. J Pathol 2011;225(4):565–73 doi 10.1002/path.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hong Y, Limback D, Elsarraj HS, Harper H, Haines H, Hansford H, et al. Mouse-INtraDuctal (MIND): an in vivo model for studying the underlying mechanisms of DCIS malignancy. J Pathol 2022;256(2):186–201 doi 10.1002/path.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Behbod F, Kittrell FS, Lamarca H, Edwards D, Kerbawy S, Heestand JC, et al. An intraductal human-in-mouse transplantation model mimics the subtypes of ductal carcinoma in situ. Breast cancer research : BCR 2009;11(5):R66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartmann LC, Radisky DC, Frost MH, Santen RJ, Vierkant RA, Benetti LL, et al. Understanding the premalignant potential of atypical hyperplasia through its natural history: a longitudinal cohort study. Cancer prevention research 2014;7(2):211–7 doi 10.1158/1940-6207.CAPR-13-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tasian SK, Teachey DT, Li Y, Shen F, Harvey RC, Chen IM, et al. Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood 2017;129(2):177–87 doi 10.1182/blood-2016-05-707653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elkabets M, Vora S, Juric D, Morse N, Mino-Kenudson M, Muranen T, et al. mTORC1 inhibition is required for sensitivity to PI3K p110alpha inhibitors in PIK3CA-mutant breast cancer. Science translational medicine 2013;5(196):196ra99 doi 10.1126/scitranslmed.3005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller J, Armgardt E, Svoboda A. The efficacy and safety of alpelisib in breast cancer: A real-world analysis. Journal of oncology pharmacy practice : official publication of the International Society of Oncology Pharmacy Practitioners 2022:10781552221096413 doi 10.1177/10781552221096413. [DOI] [PubMed] [Google Scholar]
- 23.Haricharan S, Dong J, Hein S, Reddy JP, Du Z, Toneff M, et al. Mechanism and preclinical prevention of increased breast cancer risk caused by pregnancy. eLife 2013;2(0):e00996 doi 10.7554/eLife.00996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnston AN, Bu W, Hein S, Garcia S, Camacho L, Xue L, et al. Hyperprolactinemia-inducing antipsychotics increase breast cancer risk by activating JAK-STAT5 in precancerous lesions. Breast cancer research : BCR 2018;20(1):42 doi 10.1186/s13058-018-0969-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sflomos G, Dormoy V, Metsalu T, Jeitziner R, Battista L, Scabia V, et al. A Preclinical Model for ERalpha-Positive Breast Cancer Points to the Epithelial Microenvironment as Determinant of Luminal Phenotype and Hormone Response. Cancer Cell 2016;29(3):407–22 doi 10.1016/j.ccell.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 26.Koundouros N, Karali E, Tripp A, Valle A, Inglese P, Perry NJS, et al. Metabolic Fingerprinting Links Oncogenic PIK3CA with Enhanced Arachidonic Acid-Derived Eicosanoids. Cell 2020;181(7):1596–611 e27 doi 10.1016/j.cell.2020.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bu W, Li Y. Intraductal Injection of Lentivirus Vectors for Stably Introducing Genes into Rat Mammary Epithelial Cells in Vivo. J Mammary Gland Biol Neoplasia 2020;25(4):389–96 doi 10.1007/s10911-020-09469-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huggins C, Grand LC, Brillantes FP. Mammary cancer induced by a single feeding of polymucular hydrocarbons, and its suppression. Nature 1961;189:204–7 doi 10.1038/189204a0. [DOI] [PubMed] [Google Scholar]
- 29.Huggins C, Yang NC. Induction and extinction of mammary cancer. A striking effect of hydrocarbons permits analysis of mechanisms of causes and cure of breast cancer. Science 1962;137(3526):257–62 doi 10.1126/science.137.3526.257. [DOI] [PubMed] [Google Scholar]
- 30.Huggins C, Briziarelli G, Sutton H, Jr. Rapid induction of mammary carcinoma in the rat and the influence of hormones on the tumors. The Journal of experimental medicine 1959;109(1):25–42 doi 10.1084/jem.109.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang BC, Kennan WS, Yasukawa-Barnes J, Lindstrom MJ, Gould MN. Carcinoma induction following direct in situ transfer of v-Ha-ras into rat mammary epithelial cells using replication-defective retrovirus vectors. Cancer Res 1991;51(10):2642–8. [PubMed] [Google Scholar]
- 32.Wang B, Kennan WS, Yasukawa-Barnes J, Lindstrom MJ, Gould MN. Difference in the response of neu and ras oncogene-induced rat mammary carcinomas to early and late ovariectomy. Cancer Res 1992;52(15):4102–5. [PubMed] [Google Scholar]
- 33.Dischinger PS, Tovar EA, Essenburg CJ, Madaj ZB, Gardner EE, Callaghan ME, et al. NF1 deficiency correlates with estrogen receptor signaling and diminished survival in breast cancer. NPJ breast cancer 2018;4:29 doi 10.1038/s41523-018-0080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available upon request from the corresponding author.