

Abstract
Background:
Shearer et al in 2014 articulated well-defined criteria for the diagnosis and classification of severe combined immunodeficiency (SCID) as part of the Primary Immune Deficiency Treatment Consortium’s (PIDTC’s) prospective and retrospective studies of SCID.
Objective:
Because of the advent of newborn screening for SCID and expanded availability of genetic sequencing, revision of the PIDTC 2014 Criteria was needed.
Methods:
We developed and tested updated PIDTC 2022 SCID Definitions by analyzing 379 patients proposed for prospective enrollment into Protocol 6901, focusing on the ability to distinguish patients with various SCID subtypes.
Results:
According to PIDTC 2022 Definitions, 18 of 353 patients eligible per 2014 Criteria were considered not to have SCID, whereas 11 of 26 patients ineligible per 2014 Criteria were determined to have SCID. Of note, very low numbers of autologous T cells (<0.05 × 109/L) characterized typical SCID under the 2022 Definitions. Pathogenic variant(s) in SCID-associated genes was identified in 93% of patients, with 7 genes (IL2RG, RAG1, ADA, IL7R, DCLRE1C, JAK3, and RAG2) accounting for 89% of typical SCID. Three genotypes (RAG1, ADA, and RMRP) accounted for 57% of cases of leaky/atypical SCID; there were 13 other rare genotypes. Patients with leaky/atypical SCID were more likely to be diagnosed at more than age 1 year than those with typical SCID lacking maternal T cells: 20% versus 1% (P < .001). Although repeat testing proved important, an initial CD3 T-cell count of less than 0.05 × 109/L differentiated cases of typical SCID lacking maternal cells from leaky/atypical SCID: 97% versus 7% (P < .001).
Conclusions:
The PIDTC 2022 Definitions describe SCID and its subtypes more precisely than before, facilitating analyses of SCID characteristics and outcomes.
Keywords: Severe combined immunodeficiency, SCID, typical SCID, leaky/atypical SCID, Omenn syndrome, newborn screening
Severe combined immunodeficiency (SCID) is a group of rare genetic disorders that share a common phenotype of low numbers of autologous T lymphocytes with deficient numbers or function of B and/or natural killer cells, causing affected individuals to be at risk for severe and life-threatening infections.1 The immunodeficiency can be so profound that some patients are incapable of rejecting maternal T cells that cross the placenta. Subtypes of SCID are recognized, including (1) SCID with very few or undetectable autologous T cells, often with transplacental maternally engrafted (TME) T cells; (2) SCID with decreased numbers of T cells and no TME; and (3) Omenn syndrome with autoreactive/hyperinflammatory T cells and no TME.2
Historically, there was no universal definition of SCID or its subtypes, which limited multi-institutional studies.3,4 In 2014, Dr William Shearer and members of the Primary Immune Deficiency Treatment Consortium (PIDTC) published a set of criteria developed to facilitate rigorous observational and prospective studies of SCID outcomes following hematopoietic cell transplantation (HCT), gene therapy (GT), or enzyme-replacement therapy (ERT).5 The revised PIDTC 2022 Definitions, published simultaneously with this report, were developed to reflect changes in clinical practice, particularly population-based newborn screening (NBS) for SCID by measuring T-cell receptor excision circles (TRECs) in infant dried blood spots; and genetic sequencing, which has become rapid, inexpensive, and widely available. In this article, we describe the performance of the PIDTC 2022 Definitions, applied to patients proposed for enrollment in a large prospective study of SCID.
METHODS
The PIDTC 2014 Criteria, as reported in Shearer et al,5 were developed by analyzing the diagnostic data of patients with center-designated SCID diagnosed between 2000 and 2009 and enrolled into Protocol 6902 (NCT10346150), a retrospective natural history study of treatment outcomes for SCID, using criteria that could be assessed by the Primary Immune Deficiency community at large.5 Of the 332 patients, 47 (14%) were deemed ineligible due to lack of documented support for a diagnosis of SCID.5 The remainder were divided into stratum A (typical SCID treated by allogeneic HCT, 84%), stratum B (atypical subtypes of SCID, such as leaky SCID, Omenn syndrome, or reticular dysgenesis, all treated by allogeneic HCT, 13%), and stratum C (SCID initially treated by ERT or GT, 3%).
Because of advances in gene mutation analysis and newborn screening, we recently reassessed the PIDTC 2014 Criteria and formulated revisions to develop the PIDTC 2022 Definitions with 3 subtypes of typical SCID, leaky/atypical SCID, and Omenn syndrome (see accompanying article and Tables E1 and E2 in this article’s Online Repository at www.jacionline.org). Changes include (1) modification of the term “leaky” to “leaky/atypical”; (2) setting the T-cell count permissible for typical SCID at less than 0.05 × 109/L (unless maternal T cells are present); (3) recognizing a low (<20%) percentage of naive T cells or presence of oligoclonal T cells as accepted defining features of leaky/atypical SCID; (4) simplifying criteria for Omenn syndrome by scoring the number of supporting features, giving 1 point each for eosinophilia, elevated IgE, abnormal TRECs, lymphadenopathy, hepatosplenomegaly, or oligoclonal T cells; a score of 2 or more is required; (5) eliminating reticular dysgenesis as a separate subtype and assigning these patients to a subtype on the basis of their phenotypes; and (6) excluding all known thymic disorders (only DiGeorge syndrome was excluded in the 2014 Criteria) and cases of idiopathic T-cell lymphopenia. The revised PIDTC 2022 Definitions highlighted the diagnostic value of pathogenic variant(s) in recognized SCID genes, while decreasing reliance on the proliferative response to PHA or other mitogens.
To validate the revised PIDTC 2022 Definitions, we examined baseline clinical and laboratory findings of 379 patients with center-diagnosed SCID between 2010 and 2021 proposed for enrollment into the PIDTC Prospective Protocol 6901 (NCT01186913) and compared eligibility and cohort placement for each potential subject using the PIDTC 2014 Criteria versus the PIDTC 2022 Definitions. Informed consent was obtained by physicians at treating sites, and eligibility data were provided for review. Patients were eligible for Protocol 6901 only if consent was obtained before the start of HCT/GT/ERT. Patients were considered “not SCID” if they did not meet criteria for any of the 3 subtypes of SCID by 2022 Definitions. Of note, the case report forms collected only the percentage of CD3/CD4 T cells that had a CD3/CD4/CD45RO+ memory phenotype (X); therefore, the reported percentage of naive T cells was imputed to be 100-X; this was an imperfect assumption, because a population of CD45RA/RO double-positive cells exists.6
TREC testing was performed either by state laboratories as part of population-wide newborn screening, or in a PIDTC Core Lab, using published methods.7 PHA testing was performed either by incorporation of 3H-thymi-dine (radioactive method) in bulk PBMCs (a mixture of monocytes and lymphocytes), or via flow-cytometric analysis of PBMCs, gating on CD45+ lymphocytes and CD3+ T cells. For the radioactive method, the 3H-thymidine incorporated into newly synthesized DNA in the stimulated and unstimulated cells (background) was expressed as counts per minute and represented as a percentage of proliferating cells of the patient divided by the lower limit of proliferating cells of the reference sample (derived from healthy control data) of the testing laboratory. For the flow-cytometric assay, most laboratories derived the reference cutoff by the 95% CI of the lower 5th percentile of the healthy cohort data, expressed relative to the lower limit of the healthy control cutoff.
Statistical analysis
Demographic and disease-related variables were described with the use of frequencies for categorical variables and medians and ranges for quantitative variables. The association between variables was assessed using Fisher exact test for categorical variables and the Wilcoxon-Mann-Whitney test (for 2 groups) or Kruskal-Wallis (for >2 groups) for continuous variables.
RESULTS
Eligibility status: PIDTC 2014 Criteria versus PIDTC 2022 Definitions
As of September 30, 2021, 379 patients who were reviewed for eligibility on PIDTC 6901 using 2014 Criteria, including 26 patients (7%) determined to be ineligible, were re-reviewed for SCID determination and classification using the 2022 Revised Definitions (Fig 1). The remaining 353 patients eligible per PIDTC 2014 Criteria were assigned into stratum A (typical SCID treated with allogeneic HCT; n = 210, 55%), stratum B (atypical SCID; n = 89, 24%), and stratum C (SCID, either typical or atypical, treated with ERT or GT; n = 58, 14%). On re-review and application of the PIDTC 2022 Definitions, 346 patients were determined to have SCID, of whom 238 (69% of patients with SCID) had typical SCID, 91 (26%) had leaky/atypical SCID, and 17 (5%) had Omenn syndrome.
FIG 1.
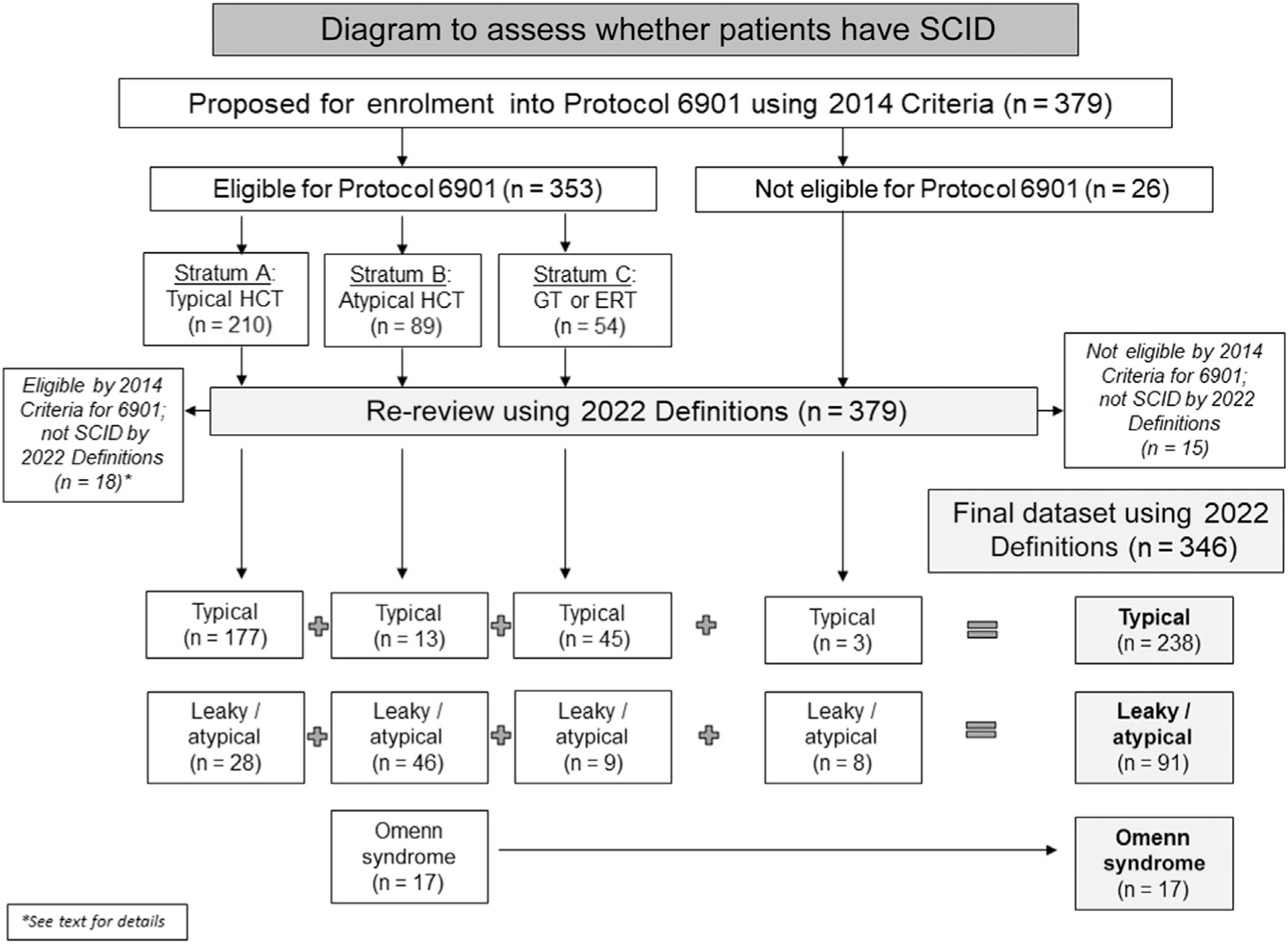
Flow diagram of patients with center-diagnosed SCID proposed for enrollment on PIDTC Protocol 6901. Stratum A: patients with typical SCID planned for allogeneic HCT; stratum B: patients with atypical SCID planned for allogeneic HCT; stratum C: patients with either typical or atypical SCID planned for GT or ERT.
Eighteen patients deemed to be eligible by PIDTC 2014 Criteria were reclassified using the revised 2022 Definitions as not having SCID (see Table E3 in this article’s Online Repository at www.jacionline.org). Of these, 5 patients assigned to stratum A had either no genotype and spontaneous improvement in their T-cell counts consistent with a diagnosis of idiopathic T-cell lymphopenia (n = 2), or thymic dysfunction with pathogenic variants in FOXN1 or FOXI3 (n = 3). Thirteen patients assigned to stratum B by 2014 Criteria were determined not to have SCID per revised 2022 Definitions, due to pathogenic variants in FOXN1 (n = 1), as well as patients with pathogenic variant(s) in ZAP70 (n = 4), IL2RG (n = 2), AK2 (n = 1), or PNP (n = 1), and 4 patients with no established genotype.
Of the 26 ineligible patients by PIDTC 2014 Criteria, 15 were considered not SCID per the revised 2022 Definitions, including 2 with pathogenic variants in ZAP70 and 13 without identified pathogenic gene variants. However, 11 ineligible patients for Protocol 6901 met the PIDTC 2022 Definitions, including 4 for whom consent was obtained after starting treatment (ADA, n = 3; RAG1, n = 1). Five patients (excluded from Protocol 6901 because of proliferation to PHA >30%) met the revised 2022 Definitions for leaky/atypical SCID (1.7% of total), with pathogenic variants in known SCID-associated genes, including RAC2 (n = 1), RAG1 (n = 1), and RMRP (n = 3); 2 patients lacking a genetic diagnosis met the revised 2022 Definitions as leaky/atypical SCID on the basis of low T-cell numbers for age, abnormal TRECs, and low proliferation.
Subtype designation: PIDTC 2014 Criteria versus PIDTC 2022 Definitions
As seen in Fig 1, of the 210 patients considered to have typical SCID according to PIDTC 2014 Criteria, 28 (13%) were reclassified as leaky/atypical SCID per revised 2022 Definitions due to CD3 counts more than 0.05 × 109/L, including patients with pathogenic variant(s) in ADA (n = 2), BCL11B (n = 1), CD3D (n = 2), JAK3 (n = 2), LIG4 (n = 1), MAN2B2 (n = 1), NHEJ1 (n = 1), RAG1 (n = 7), RMRP (n = 5), or TTC7A (n = 1), and unknown (n = 5). Conversely, of the 89 patients considered to have atypical SCID per PIDTC 2014 Criteria (primarily due to lymphocyte proliferation to PHA >10%), 13 (15%) were reclassified per 2022 Definitions as typical SCID, including 10 patients with pathogenic variant(s) in AK2 (n = 4), IL2RG (n = 2), IL7R (n = 1), JAK3 (n = 1), PNP (n = 1), RAG1 (n = 1), and RAG2 (n = 1); 2 of these patients had no identified genotype. All patients assigned to stratum C per 2014 Criteria met the revised 2022 Definitions for either typical (n = 45) or leaky/atypical (n = 9) SCID. The complete genotypic distribution by original 2014 Criteria versus revised 2022 Definitions is presented in Table I.
TABLE I.
Genotypic distribution of patients with SCID per PIDTC 2014 Criteria vs PIDTC 2022 Definitions
| PIDTC 2014 Criteria | PIDTC 2022 Definitions | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | Overall | Stratum A | Stratum B | Stratum C | Not eligible | Overall SCID | Typical SCID | Leaky / atypical SCID | Omenn syndrome | Not SCID |
| IL2RG | 106 | 81 | 9* | 16 | — | 104 | 99 | 5 | — | 2 |
| RAG1 | 58 | 25† | 31* | — | 2 | 58 | 20 | 24 | 14 | — |
| ADA | 41 | 7† | 3 | 28 | 3 | 41 | 26 | 15 | — | — |
| IL7R | 24 | 22 | 2* | — | — | 24 | 23 | — | 1 | — |
| DCLRE1C | 22 | 10 | 2 | 10 | — | 22 | 20 | 2 | — | — |
| JAK3 | 18 | 15† | 3* | — | — | 18 | 14 | 4 | — | — |
| RAG2 | 15 | 10 | 5 | — | — | 15 | 11 | 3 | 1 | — |
| RMRP | 14 | 6† | 5 | — | 3 | 14 | 1 | 13 | — | — |
| CD3D | 8 | 8† | — | — | — | 8 | 6 | 2 | — | — |
| AK2 | 6 | — | 6* | — | — | 5 | 4 | — | 1 | 1 |
| PNP | 4 | — | 4* | — | — | 3 | 1 | 2 | — | 1 |
| MSN | 2 | 1 | 1 | — | — | 2 | 1 | 1 | — | — |
| LIG4 | 2 | 1† | 1 | — | — | 2 | — | 2 | — | — |
| NHEJ1 | 2 | 1† | 1 | — | — | 2 | — | 2 | — | — |
| BCL11B | 1 | 1† | — | — | — | 1 | — | 1 | — | — |
| MAN2B | 1 | 1† | — | — | — | 1 | — | 1 | — | — |
| TTC7A | 1 | 1† | — | — | — | 1 | — | 1 | — | — |
| RAC2 | 1 | — | — | — | 1 | 1 | — | 1 | — | — |
| ZAP70 | 4 | — | 4 | — | — | — | — | — | — | 4 |
| FOXN1 | 3 | 2 | 1 | — | — | — | — | — | — | 3 |
| FOXI3 | 1 | 1 | — | — | — | — | — | — | — | 1 |
| Unknown | 30 | 15† | 11* | — | 2 | 24 | 12 | 12 | — | 6 |
| Total | 364‡ | 210 | 89 | 54 | 11 | 346 | 238 | 91 | 17 | 18 |
Stratum A: Typical SCID undergoing allogeneic HCT; stratum B: atypical SCID (leaky or Omenn or reticular dysgenesis) undergoing allogeneic HCT; stratum C: typical or atypical SCID undergoing autologous GT or ERT.
Twelve patients (IL2RG, n = 2; RAG1, n = 1; IL7R, n = 1, JAK3, n = 1, AK2, n = 4; PNP, n = 1; unknown, n = 2) were moved from stratum B to typical SCID per PIDTC 2022 Criteria.
Twenty-eight patients (RAG1, n = 7; ADA, n = 2; JAK3, n = 2; RMRP, n = 5; CD3D, n = 2; LIG4, n = 1; NHEJ1, n = 1; BCL11B, n = 1; MAN2B, n = 1; ORAI1, n = 1; TTC7A, n = 1; unknown, n = 4) were changed from stratum A to atypical/leaky SCID per PIDTC 2022 Criteria.
Of the 379 total patients, 15 who were not eligible per PIDTC 2014 Criteria and remained “not SCID” per PIDTC 2022 Criteria are not included in this table.
The PIDTC 2022 Definitions classified more patients with pathogenic variants in RAG1, RMRP, and certain rare genotypes as leaky/atypical SCID compared with the PIDTC 2014 Criteria. With the PIDTC 2022 Definitions, just 7 genotypes (IL2RG, RAG1, ADA, IL7R, DCLRE1C, JAK3, and RAG2) comprised 89% of typical SCID. Leaky/atypical SCID was much more genetically heterogeneous with 2 genotypes (RAG1, ADA, and RMRP), representing 57% of cases, and 13 other rare genotypes identified. According to the PIDTC 2022 Definitions, 5% (17 of 346) of all cases of patients with SCID had Omenn syndrome, comprising 16% (17 of 108) of patients without typical SCID. The great majority of cases of Omenn syndrome (88%) were due to pathogenic variants in RAG1 and RAG2; 21% of all patients with pathogenic RAG1 and RAG2 variants developed Omenn syndrome.
Patient characteristics by PIDTC 2022 Definitions assignment
To determine how well the revised PIDTC 2022 Definitions separated patients into distinct subtypes, we analyzed various diagnostic features (Table II). Because the clinical and laboratory characteristics of patients with typical SCID may differ depending on whether maternal T cells are present, we further separated typical SCID into 3 subgroups on the basis of whether TME was detected, tested and not detected, or unknown (not tested).
TABLE II.
SCID subtype and diagnostic features according to PIDTC 2022 Definitions
| Diagnostic features | Typical SCID | Leaky/atypical SCID | Omenn syndrome | |||
|---|---|---|---|---|---|---|
| All typical SCID | TME tested and detected | TME unknown (not tested) | TME tested and not detected | |||
| N (total = 346) | 238 | 71 | 85 | 82 | 91 | 17 |
| Pathogenic gene variant identified | 226 (95%) | 66 (93%) | 84 (99%) | 76 (93%) | 79 (87%) | 17 (100%) |
| Age at first CD3 count (mo), median (range) | 0.61 (0–37.3) | 1.3 (0–37.3) | 0.46 (0–9.9) | 0.39 (0–19.7) | 1.08 (0–161.7) | 0.99 (0.03–6.44) |
| Age <1 y | 232 (97%) | 67 (94%) | 85 (100%) | 80 (98%) | 73 (80%) | 17 (100%) |
| Age 1–2 y | 5 | 3 | 0 | 2 | 8 | 0 |
| Age 2+ y | 1 | 1 | 0 | 0 | 10 | 0 |
| Initial CD3 count (×109/L), median (range) | 0.008 (0–8.898) | 0.046 (0–8.898) | 0.004 (0–0.460) | 0.005 (0–0.135) | 0.174 (0.021–5.67) | 2.159 (0.036–44.366) |
| Initial CD3 count <0.05 ×109/L, n (%) | 199 (84) | 37 (52) | 83 (98) | 79 (96) | 6 (7) | 1 (5) |
| Second CD3 count concordant* | 111 of 124 (90%) | 30 of 39 (77%) | 33 of 35 (94%) | 48 of 50 (96%) | 44 of 49 (90%) | — |
| CD4/CD45RO >80% | 52 of 94 (55%) | 29 of 47 (62%) | 11 of 21 (52%) | 12 of 26 (46%) | 31 of 75 (41%) | 17 of 17 (100%) |
| Abnormal TRECs | 200 of 206 (97%) | 61 of 64 (95%) | 67 of 69 (97%) | 72 of 73 (99%) | 84 of 88 (95%) | 14 of 14 (100%) |
| Oligoclonal T cells | 7 of 14 (50%) | 2 of 6 (33%) | 0 of 1 | 5 of 7 (71%) | 6 of 15 (40%) | 6 of 6 (100%) |
| Generalized rash | 30 (13%) | 15 (21%) | 5 (6%) | 10 (12%) | 17 (19%) | 17 (100%) |
| Elevated eosinophils | 30 (13%) | 16 (23%) | 9 (11%) | 5 (6%) | 14 (15%) | 15 (88%) |
| Elevated IgE level | 13 of 145 (9%) | 6 of 49 (12%) | 2 of 52 (4%) | 5 of 44 (11%) | 17 of 65 (26%) | 12 (71%) |
| Lymphadenopathy | 8 (3%) | 6 (8%) | 1 (1%) | 1 (1%) | 3 (3%) | 9 (53%) |
| Organomegaly | 15 (5%) | 9 (13%) | 2 (2%) | 4 (5%) | 5 (6%) | 4 (24%) |
| OS score of ≥2 in patients with >80% memory T cells & generalized rash | 7 of 14 (50%) | 7 of 10 (70%) | 0 of 2 | 0 of 2 | 1 of 8 (13%) | 17 (100%) |
| PHA, median (range) | 0% (0%−100%) | 1% (0%−77%) | 0% (0%−27%) | 0% (0%−100%) | 16% (0%−100%) | 16% (1%−100%) |
| <10% | 179 (88%)† | 52 (78%)† | 60 (95%)† | 67 (92%)† | 35 (41%)† | 6 (38%)† |
| 10%−29% | 20 | 12 | 3 | 5 | 19 | 6 |
| 30%−49% | 2 | 2 | 0 | 0 | 10 | 0 |
| ≥50% | 2 | 1 | 0 | 1 | 21 | 4 |
| Not done | 35 | 4 | 22 | 9 | 6 | 1 |
Values are n (%) unless otherwise indicated.
PHA, Phytohemagglutinin (of bulk lymphocytes); OS, Omenn syndrome.
Concordant CD3 counts: both <0.005 × 109/L or both ≥0.005 × 109/L.
Percentage of those with test performed.
Pathogenic variants in 1 of 18 SCID-causing genes were identified in 322 of 346 (93%) patients overall, more commonly in patients with typical (95%) versus leaky/atypical (87%) SCID (P = .02). Patients with typical SCID came to clinical attention (had their first T-cell count performed) at a younger median age than those with leaky/atypical SCID: 0.61 months (range, 0–37.3 months) versus 1.08 months (range, 0–161.7 months) (P < .001) (Fig 2). Remarkably, 18 of 91 (20%) of those with leaky/atypical SCID were diagnosed at age older than 1 year, compared with 4 of 71 (6%) of those with typical SCID and detected TME and 2 of 165 (1%) of those with typical SCID without known evidence of TME (P < .001). This effect was primarily confined to those patients without an NBS test consistent with SCID (median age at diagnosis for typical was 4.5 months vs leaky/atypical median age of 11.2 months; P = .006); patients reported to have an NBS test consistent with SCID had only a trend toward lower median age at diagnosis for typical SCID (0.36 months) versus leaky/atypical (0.82 months; P = .055). The median T-cell count in typical SCID without TME was 0.005 × 109/L (range, 0–0.135), compared with 0.174 × 109/L (range, 0.021–5.67) in those with leaky/atypical SCID.
FIG 2.

Age at first CD3 count in months (log-scale) by PIDTC 2022 Criteria subtype classification.
Those with typical SCID without known TME were more likely (97%; 162 of 167) to have initial CD3 counts less than 0.05 × 109/L than those with positive TME (54%; 37 of 69; P < .001). Only 6 of 90 (7%) patients with leaky/atypical SCID had initial CD3 counts less than 0.05 × 109/L (P < .001, compared with typical SCID without known TME). A confirmatory T-cell count was performed in 180 of 345 (52%) patients; 161 (89%) were broadly concordant (both <0.005 × 109/L or both ≥0.005 × 109/L). Patients with TME had the highest likelihood of discordance: 9 of 39 (23%). Four patients (2%) had initial T-cell counts greater than or equal to 0.05 × 109/L but dropped to less than 0.05 × 109/L on a second determination, including patients with pathogenic variant(s) in AK2 (n = 1), IL2RG (n = 1), RAG1 (n = 1), and none (n = 1), classifying them as typical SCID. Conversely, 6 (3%) patients with leaky/atypical SCID had initial T-cell counts less than 0.05 × 109/L, but their T cells subsequently increased to greater than or equal to 0.05 × 109/L, including patients with pathogenic variant(s) in IL2RG (n = 1), RAG1 (n = 1), RAG2 (n = 1), LIG4 (n = 1), and none (n = 2).
Overall, 239 of 345 (69%) patients had NBS performed, with another 69 having research-level TREC testing. When performed, TREC testing result was abnormal in 97% (298 of 308) of patients; patients with normal TRECs are reported in Table E4 in this article’s Online Repository at www.jacionline.org. Both typical and leaky/atypical SCID had more than 80% of T cells having a memory CD4+CD45RO+ phenotype where recorded (55% of typical and 41% of leaky/atypical SCID). Testing for clonality of T-cell receptors was rarely performed, but when tested, oligoclonal T cells were noted in 50% of typical SCID and 40% of leaky/atypical SCID cases. TME was ascertained in 64% of patients with typical SCID and was positive in 46% (71 of 153) of those in whom it was evaluated (Table III); there was significantly less TME in ADA SCID compared with all other genotypes (P = .01), but no difference in incidence of TME between the 6 other most common genotypes (P = .51). The distinctive clinical and laboratory features of Omenn syndrome were uncommon in typical or atypical/leaky SCID. There were 32 patients who met the 2 key criteria for Omenn syndrome: more than 80% memory T cells and generalized rash. Of these, 25 had an Omenn syndrome score of 2 or more, of whom 17 met the other criteria for Omenn syndrome (pathologic gene variant(s) and negative TME testing result); 7 were patients with typical SCID and positive TME testing result (the score may have reflected graft-versus-host disease mediated by maternal T cells); and 1 was classified as atypical SCID due to a lack of identified pathologic gene variant(s).
TABLE III.
Maternal engraftment according to genotype in typical SCID by PIDTC 2022 Definitions
| Genotype | All typical | TME tested and detected | TME unknown (not tested) | TME tested and not detected | % of typical SCID with known TME* |
|---|---|---|---|---|---|
| IL2RG | 99 | 32 | 38 | 29 | 52% |
| ADA | 26 | 1 | 15 | 10 | 9% |
| IL7R | 23 | 6 | 12 | 5 | 55% |
| DCLRE1C | 20 | 6 | 3 | 11 | 35% |
| RAG1 | 20 | 4 | 8 | 8 | 25% |
| JAK3 | 14 | 7 | 3 | 4 | 64% |
| RAG2 | 11 | 6 | 1 | 4 | 60% |
| CD3D | 6 | - | 3 | 3 | 0% |
| AK2 | 4 | 3 | 1 | — | 75% |
| RMRP | 1 | 1 | — | — | 100% |
| PNP | 1 | — | — | 1 | 0% |
| MSN | 1 | — | — | 1 | 0% |
| Unknown | 12 | 5 | 1 | 6 | 45% |
| Total | 238 | 71 | 85 | 82 | 46% |
TME testing performed and result reported as positive or negative.
The proliferative response to PHA was less than 10% of the lower end of the reference range in 88% of patients with typical SCID, compared with 41% of those with leaky/atypical SCID (P <.001). When PHA was performed by flow cytometry (n = 91), permitting comparison of gating on CD45+ total lymphocyte versus CD3+ T-cell populations, 38.1% of patients had a higher PHA proliferation category (0%, 1%−9%, 10%−29%, 30%−49%, or ≥50%) in their CD3+ population than in their CD45+ population and 26.2% (22 of 84) had PHA proliferation in the CD3+ gate more than 30% of the lower boundary of the reference range (see Table E5 in this article’s Online Repository at www.jacionline.org). Patients without known TME or Omenn syndrome with a CD3 count of less than 0.05 × 109/L were more likely to have profoundly decreased (<10%) proliferation of total PBMCs to PHA: 92% (82 of 89) versus 34% (21 of 62) for patients with CD3 count greater than or equal to 0.05 × 109/L (P <.001). In Fig 3, the highlighted boxes indicate 2 groups of patients for whom the 2014 Criteria gave different classifications than do the 2022 Definitions: patients formerly called atypical SCID solely on the basis of PHA more than 10%, and formerly called typical SCID on the basis of having 0.05 × 109 to 0.3 × 109 T cells/L and a PHA proliferation of more than 10%.
FIG 3.

Relationship of absolute CD3 count to total PBMCs proliferative response to PHA for typical vs leaky/atypical SCID. Quadratic trend (black line) of higher PBMCs proliferative response to PHA in patients with higher final (using second value, when available) CD3 counts, including only patients with negative testing result for TME and omitting patients with Omenn syndrome. The PIDTC 2022 Criteria consider patients with typical SCID to be those with final (using second value, when available) CD3 counts less than 0.05 × 109/L, irrespective of proliferation; most (91.8%) cases also have poor proliferation (<10%) of PBMCs to PHA (P < .001); some (red-dotted box) have modest proliferation and would have been considered leaky/atypical SCID per PIDTC 2014 Criteria. Most patients (66.1%) with CD3 counts more than 0.05 × 109/L have low/normal proliferation (>10%) of PBMCs to PHA, though some (blue-dotted box) have poor proliferation (<10%) and would have been considered typical SCID per PIDTC 2014 Criteria.
DISCUSSION
The diagnosis of SCID in the United States and Canada has transformed in the past decade compared with the period 2000 to 2009, largely due to the introduction of population-based NBS with the TREC assay throughout the United States and much of Canada.8 Advances in genetic sequencing and interpretation of pathogenic gene variants have also been critical, with sequence-based diagnosis now standard of care. As a result of these changes, diagnosis has been made earlier in life and with higher accuracy, with fewer patients proposed for inclusion in the prospective study being found to not have SCID between 2010 and 2021 compared with the previous decade (7% for PIDTC 6901 vs 14% for PIDTC 6902).5 In addition, a higher proportion of cases were identified to have SCID-causing pathogenic gene variants (93% vs 69% in 2000–2009), and more cases of atypical (leaky/atypical SCID and Omenn syndrome) were diagnosed: 108 of 346 (31%) all SCID in the period 2010 to 2021 versus 40 of 285 (14%) all SCID in the period 2000 to 2009.9
A major change in the revised PIDTC 2022 Definitions is a 6-fold decrease in the threshold value of the absolute T-cell count from 0.3 × 109/L to 0.05 × 109/L for patients without TME. This criterion effectively distinguishes patients considered to have typical SCID without TME, who also tended to have pathogenic gene variants, very few or absent TRECs on NBS, and/or few or undetectable naive T cells. Rare patients (3%) developed more T cells over time in the absence of known TME; these are more concerning diagnostically than those who started with more than 0.05 × 109/L T cells that subsequently decreased. Some patients with rising T-cell numbers may have had expansion of transplacentally acquired maternal T cells (although not all patients were tested for this); others could have had expansion of oligoclonal host T cells (but only few patients were tested for this); still others might have had idiopathic T-cell lymphopenia (if no identified pathogenic gene variants) that could resolve over time without treatment. This uncertainty highlights the importance of repeat testing of T-cell counts over at least 8 weeks, as well as undertaking vital diagnostic measures, including genetic sequencing and assessment of maternal T-cell engraftment. The measurement of naive and memory T-cell populations is especially important in the diagnosis of leaky/atypical SCID. TCR clonality testing was rarely performed from 2010 to 2021, but newer techniques may help characterize restricted repertoires in patients with SCID.10
Although age at first T-cell count was higher in patients with leaky/atypical SCID than in typical SCID, this was likely a reflection of the fact that some patients in this cohort were diagnosed before the introduction of NBS in their region, because false-negative SCID NBS results are extremely rare (2 cases in 3.2 million births),11 and mainly due to late-onset ADA deficiency.12 We therefore anticipate that—in future cohorts recruited from regions with fully implemented SCID NBS—there will not be a difference in age at diagnosis between SCID subtypes.
The PIDTC 2022 SCID Definitions have retained defective mitogen proliferation as a supporting feature, though low proliferation of bulk or CD45+ populations appears to be primarily a reflection of extremely low T-cell numbers. Testing proliferative responses remains challenging in severely T-lymphopenic patients because of the substantial blood volumes required. Moreover, there were rare patients (<2% of total SCID cases) who had more than 30% PHA proliferation, who were formerly considered not to have SCID per PIDTC 2014 Criteria, but who met PIDTC 2022 Definitions for leaky/atypical SCID.
Another important distinction between the PIDTC 2014 Criteria and 2022 Definitions is the specific exclusion of patients with known idiopathic T-cell lymphopenia and all thymic defects, including complete DiGeorge syndrome or pathogenic variants in FOXN1. These patients are not effectively treated with HCT. Furthermore, the PIDTC 2022 Definitions now exclude most cases of combined immunodeficiency disorders in which T cells may develop but are nonfunctional, such as those due to pathogenic variants in ZAP70.13,14 Rare patients (7%) without an identified pathogenic gene variant met the PIDTC 2022 Definitions, especially those classified as atypical SCID. Additional SCID-causing gene defects likely remain to be discovered, consistent with the discovery of new SCID genotypes in the last decade, including MSN, MAN2B2, and BCL11B.15–17 Alternatively, some patients may have had incomplete genetic evaluations without whole-exome/genome testing18; others may have had unrecognized thymic disorders19 or idiopathic T-cell lymphopenia.11 Because patients lacking identified pathogenic variants have had worse outcomes following HCT,20 this is an important area for further investigation.
Omenn syndrome remains rare, found in only 5% of all cases of SCID, and 16% of non-typical SCID. We previously reported that 5% of all SCID cases from 1982 to 2012 were considered to have Omenn syndrome, but Omenn syndrome represented 33% of patients without typical SCID.20 It is possible that earlier diagnosis of SCID due to NBS is facilitating pre-emptive HCT in some patients whose clinical picture would otherwise have evolved from leaky SCID to Omenn syndrome. The fact that 14% to 25% of patients with leaky/atypical SCID had some combination of generalized rash, elevated eosinophils, or elevated IgE supports this hypothesis, and suggests that clinicians should have a high index of suspicion that such infants may be at risk of progression to Omenn syndrome.
The implications of the revisions to SCID subtype classifications will require additional analyses to be fully understood. Previous studies by the PIDTC did not show a difference in post-HCT overall survival based on whether a patient was assigned to stratum A (typical) or B (atypical) SCID.20,21 Now that some patients have been reassigned, determined to not have SCID, or newly included as SCID, this conclusion may change. Analyses of the precise role of conditioning may be enhanced by a more rigorous distinction between typical and leaky/atypical SCID. Furthermore, because patients with SCID can develop infections before HCTeven when identified by NBS,21 it may be informative to analyze whether patients with leaky/atypical SCID have sufficient residual immunity to provide some degree of protection from these infections.
This analysis regarding the PIDTC 2022 SCID Definitions has several limitations. Importantly, there is no “criterion standard” for the diagnosis of SCID, such that any evaluation of the performance of new criteria/definitions can only be assessed in a semi-critical fashion compared with previous criteria. Second, although the PIDTC 6901 prospective study requested reporting of uniform evaluations at time of diagnosis, investigations were not universally performed; some data are missing, which could potentially shift patients from the typical to leaky/atypical category, or vice versa. Furthermore, the study captured only the percentage of memory T cells; these were not necessarily the inverse of naive T cells, absence of which is a better marker of leaky/atypical SCID and will be used moving forward. In the future, consideration of the effects of specific gene variants (eg, null vs hypomorphic) may further facilitate categorization, and our current PIDTC study is assessing all enrollees for these variants. In addition, it is possible that some of the patients without identified pathogenic gene variants did not actually have SCID, but rather had thymic defects or other T-lymphopenic disorders. The PIDTC 2022 Definitions may not be suitable for retrospective use in patients diagnosed before 2010, because elements of current diagnostic testing were not widely available. Finally, some centers may not have submitted cases that they knew would not fit the PIDTC 2014 Criteria, causing omission of patients who would now fit the PIDTC 2022 Definitions.
In conclusion, based on recent diagnostic advances, the revised PIDTC 2022 Definitions provide more stringent definitions of SCID than the PIDTC 2014 Criteria. The new definitions will facilitate rigorous analyses of patient outcomes following various approaches to definitive therapy (HCT or GT). This in turn may provide clinicians with increased insights into risks of progression to Omenn syndrome, resistance to engraftment, and other factors. We anticipate that, by the time of the next revision, advances in immunologic profiling,10 genetic testing and variant interpretation,22 and evaluations of thymic function23,24 will allow these and other methodologies to be incorporated into the future classification of patients with SCID, further honing the SCID categories.
Supplementary Material
Clinical implications:
The revised PIDTC 2022 Definitions better distinguish typical SCID, leaky/atypical SCID, and Omenn syndrome and should be used to classify patients with SCID for analyses of treatment outcomes.
Acknowledgments
The work was supported by the Division of Allergy, Immunology and Transplantation, National Institute of Allergy and Infectious Diseases (NIAID); and the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH), Bethesda, Md: grant number U54AI082973 (MPI: J.M.P., C.C.D., and E.H.); and grant numbers U54NS064808 and U01TR001263 (PI: J.K.). The Primary Immunodeficiency Treatment Consortium (PIDTC) is a part of the Rare Diseases Clinical Research Network of ORDR, NCATS. The collaborative work of the PIDTC with the Pediatric Transplantation and Cellular Therapy Consortium (PTCTC) is supported by the U54 grants listed, along with support of the PTCTC Operations Center by the St. Baldrick’s Foundation and grant number U10HL069254 (PI: M.M.H.). Collaborative work of the PIDTC with the Center for International Blood and Marrow Transplant Research is supported by grant number U24CA076518 (PI: B.E.S.); grant number U01HL069294 (PI: M.M.H.); contract numbers HHSH250201200016C and HHSH234200637015C with the Health Resources and Services Administration, Department of Health and Human Services; and grant numbers N00014-13-1-0039 and N00014-14-1-0028 from the Office of Naval Research. M.J.C. and J.M.P. are supported by the California Institute of Regenerative Medicine (grant no. CLIN2-10830). M.A.P. is supported by grant numbers 1U01AI126612-01A1, P30CA040214, and 2UG1HL069254. S.Y.P. is supported by funding from the Intramural Research Program, NIH, National Cancer Institute, Center for Cancer Research. L.D.N. is supported by the Division of Intramural Research, NIAID, and NIH (grant no. ZIA AI001222-07 to PI L.D.N.).
Disclosure of potential conflict of interest:
C. C. Dvorak is an author for UpToDate, serves on the Data Safety Monitoring Board for Chiesi, and serves as consultant for Orchard Therapeutics. E. Haddad is a consultant for Jasper, Takeda, & CSL Behring. J. Heimall is an author for UpToDate, received investigator-initiated grant from CSL Behring, and serves as a consultant for ADMA, CIRM, and Horizon. M. J. Cowan is an author for UpToDate and SAB for Homology Medicine. L. F. Satter is a consultant for ADMA, Grifols, Takeda, Horizon, Enzyvant, and Orchard. J. J. Bednarski is an advisor for Sobi and Horizon. B. J. D. Saldana is a consultant for Orchard and Sobi. A. Petrovic is an advisor for Orchard, Horizon, and Enzyvant. G. D. E. Cuvelier is a consultant for Miltenyi. E. H. Caywood is an advisor for Pfizer. S. Chandrakasan is an advisor for Sobi. E. Shereck is a speaker for Sobi. A. J. Shah is a consultant for Orchard. K. E. Sullivan is a consultant for Enzyvant and IDF, advisor for ADMA and Neovii, and was funded for the conduct of sponsored trials by Atara, AlloVir, and Jasper. S. E. Prockop is an inventor of intellectual property licensed to Atara with all rights assigned to MSKCC. R. S. Abraham is an advisor for Enzyvant. M. S. Thakar is a consultant for Infectious Disease Research Institute (nonprofit). J. W. Leiding is a speaker/consultant/advisor for Sobi, Horizon, Pharming, CSL Behring, and ADMA and employee/shareholder at bluebird bio. D. B. Kohn is an author for UpToDate, serves on the Data Safety Monitoring Board for Chiesi, and is a consultant/SAB member for ImmunoVec. M. A. Pulsipher reports study support from Adaptive & Miltenyi, works as an advisor for Vertex, Medexus, Equillium, Novartis, and Mesoblast, and receives educational honoraria from Novartis and Miltenyi. J. M. Puck is an author for UpToDate and has a family member employed by Invitae. The rest of the authors declare that they have no relevant conflicts of interest.
This article is dedicated to the memory of William T. Shearer, MD, PhD (1937-2018). Brent Logan, PhD, provided valuable statistical advice.
Abbreviations used
- ERT
Enzyme-replacement therapy
- GT
Gene therapy
- HCT
Hematopoietic cell transplantation
- NBS
Newborn screening
- PIDTC
Primary Immune Deficiency Treatment Consortium
- SCID
Severe combined immunodeficiency
- TME
Transplacental maternal/maternally engraftment
- TREC
T-cell receptor excision circle
REFERENCES
- 1.Dvorak CC, Haddad E, Buckley RH, Cowan MJ, Logan B, Griffith LM, et al. The genetic landscape of severe combined immunodeficiency in the United States and Canada in the current era (2010–2018). J Allergy Clin Immunol 2019;143: 405–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dvorak CC, Cowan MJ, Logan BR, Notarangelo LD, Griffith LM, Puck JM, et al. The natural history of children with severe combined immunodeficiency: baseline features of the first fifty patients of the Primary Immune Deficiency Treatment Consortium prospective study 6901. J Clin Immunol 2013;33:1156–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffith LM, Cowan MJ, Kohn DB, Notarangelo LD, Puck JM, Schultz KR, et al. Allogeneic hematopoietic cell transplantation for primary immune deficiency diseases: current status and critical needs. J Allergy Clin Immunol 2008;122:1087–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffith LM, Cowan MJ, Notarangelo LD, Puck JM, Buckley RH, Candotti F, et al. Improving cellular therapy for primary immune deficiency diseases: recognition, diagnosis, and management. J Allergy Clin Immunol 2009;124:1152–60.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, Logan BR, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. J Allergy Clin Immunol 2014;133:1092–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamann D, Baars PA, Hooibrink B, van Lier RW. Heterogeneity of the human CD4+ T-cell population: two distinct CD4+ T-cell subsets characterized by coexpression of CD45RA and CD45RO isoforms. Blood 1996;88:3513–21. [PubMed] [Google Scholar]
- 7.Puck JM. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: the winner is T-cell receptor excision circles. J Allergy Clin Immunol 2012;129:607–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Currier R, Puck JM. SCID newborn screening: what we’ve learned. J Allergy Clin Immunol 2021;147:417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pai S-Y, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med 2014;371:434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delmonte OM, Castagnoli R, Yu J, Dvorak CC, Cowan MJ, Dávila Saldaña BJ, et al. Poor T-cell receptor β repertoire diversity early posttransplant for severe combined immunodeficiency predicts failure of immune reconstitution. J Allergy Clin Immunol 2022;149:1113–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amatuni GS, Currier RJ, Church JA, Bishop T, Grimbacher E, Nguyen AA, et al. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California, 2010–2017. Pediatrics 2019;143:e20182300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.la Marca G, Canessa C, Giocaliere E, Romano F, Duse M, Malvagia S, et al. Tandem mass spectrometry, but not T-cell receptor excision circle analysis, identifies newborns with late-onset adenosine deaminase deficiency. J Allergy Clin Immunol 2013;131:1604–10. [DOI] [PubMed] [Google Scholar]
- 13.Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol 2020;40:24–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cuvelier GD, Rubin TS, Wall DA, Schroeder ML. Long-term outcomes of hematopoietic stem cell transplantation for ZAP70 deficiency. J Clin Immunol 2016;36: 713–24. [DOI] [PubMed] [Google Scholar]
- 15.Verheijen J, Wong SY, Rowe JH, Raymond K, Stoddard J, Delmonte OM, et al. Defining a new immune deficiency syndrome: MAN2B2-CDG. J Allergy Clin Immunol 2020;145:1008–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Punwani D, Zhang Y, Yu J, Cowan MJ, Rana S, Kwan A, et al. Multisystem anomalies in severe combined immunodeficiency with mutant BCL11B. N Engl J Med 2016;375:2165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lagresle-Peyrou C, Luce S, Ouchani F, Soheili TS, Sadek H, Chouteau M, et al. X-linked primary immunodeficiency associated with hemizygous mutations in the moesin (MSN) gene. J Allergy Clin Immunol 2016;138:1681–9.e8. [DOI] [PubMed] [Google Scholar]
- 18.Heimall JR, Hagin D, Hajjar J, Henrickson SE, Hernandez-Trujillo HS, Tan Y, et al. Use of genetic testing for primary immunodeficiency patients. J Clin Immunol 2018;38:320–9. [DOI] [PubMed] [Google Scholar]
- 19.Kreins AY, Maio S, Dhalla F. Inborn errors of thymic stromal cell development and function. Semin Immunopathol 2021;43:85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haddad E, Logan BR, Griffith LM, Buckley RH, Parrott RE, Prockop SE, et al. SCID genotype and 6-month posttransplant CD4 count predict survival and immune recovery. Blood 2018;132:1737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heimall J, Logan BR, Cowan MJ, Notarangelo LD, Griffith LM, Puck JM, et al. Immune reconstitution and survival of 100 SCID patients post–hematopoietic cell transplant: a PIDTC natural history study. Blood 2017;130:2718–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adhikari AN, Gallagher RC, Wang Y, Currier RJ, Amatuni G, Bassaganyas L, et al. The role of exome sequencing in newborn screening for inborn errors of metabolism. Nat Med 2020;26:1392–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bifsha P, Leiding JW, Pai S-Y, Colamartino ABL, Hartog N, Church JA, et al. Diagnostic assay to assist clinical decisions for unclassified severe combined immune deficiency. Blood Adv 2020;4:2606–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bosticardo M, Pala F, Calzoni E, Delmonte OM, Dobbs K, Gardner CL, et al. Artificial thymic organoids represent a reliable tool to study T-cell differentiation in patients with severe T-cell lymphopenia. Blood Adv 2020;4:2611–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.