

Abstract
Aquatic ectotherms are predicted to harbour genomic signals of local adaptation resulting from selective pressures driven by the strong influence of climate conditions on body temperature. We investigated local adaptation in redband trout (Oncorhynchus mykiss gairdneri) using genome scans for 547 samples from 11 populations across a wide range of habitats and thermal gradients in the interior Columbia River. We estimated allele frequencies for millions of single nucleotide polymorphism loci (SNPs) across populations using low‐coverage whole genome resequencing, and used population structure outlier analyses to identify genomic regions under divergent selection between populations. Twelve genomic regions showed signatures of local adaptation, including two regions associated with genes known to influence migration and developmental timing in salmonids (GREB1L, ROCK1, SIX6). Genotype–environment association analyses indicated that diurnal temperature variation was a strong driver of local adaptation, with signatures of selection driven primarily by divergence of two populations in the northern extreme of the subspecies range. We also found evidence for adaptive differences between high‐elevation desert vs. montane habitats at a smaller geographical scale. Finally, we estimated vulnerability of redband trout to future climate change using ecological niche modelling and genetic offset analyses under two climate change scenarios. These analyses predicted substantial habitat loss and strong genetic shifts necessary for adaptation to future habitats, with the greatest vulnerability predicted for high‐elevation desert populations. Our results provide new insight into the complexity of local adaptation in salmonids, and important predictions regarding future responses of redband trout to climate change.
Keywords: developmental timing, ecological niche modelling, genome scan, genotype–environment association analysis, Oncorhynchus mykiss gairdneri, thermal gradient
1. INTRODUCTION
Understanding the genomic architecture underlying local adaptation is important for predicting future responses of populations to climate change and other anthropogenic stressors (Aguirre‐Liguori et al., 2021; Fitzpatrick & Edelsparre, 2018; Waldvogel et al., 2020). For aquatic ectotherms, environmental temperature is predicted to be an important driver of local adaptation due to their strong dependence of body temperature on water temperature (Schluter, 2000). Because of this dependence, environmental temperature has a major influence on many evolutionarily important life history traits, including developmental rate, body size, disease resistance, migratory patterns, longevity and survival (Martins et al., 2012; Portner & Knust, 2007; Whitney et al., 2014). Numerous studies have uncovered genomic evidence for local adaptation driven by environmental temperature for these taxa, finding that the genomic architecture underlying thermal adaptation is polygenic and complex (Everett & Seeb, 2014; Grummer et al., 2019; Jackson et al., 1998; Muñoz et al., 2015). Because of the prevalence of local adaptation, future climate change could lead to substantial changes in the adaptive potential and geographical distributions of aquatic ectotherms (e.g., Isaak et al., 2012; Muñoz et al., 2015).
Redband trout (Oncorhynchus mykiss gairdneri) are ectotherms for which multiple studies have identified local adaptation driven by temperature in freshwater ecosystems (Chen & Narum, 2021). This subspecies of rainbow trout occurs in the interior Pacific Northwest of North America and occupies a wide range of thermal regimes, from cold montane forests to high‐elevation deserts (Meyer et al., 2010; Muhlfeld et al., 2015). Studies investigating thermal adaptation to local habitats in redband trout have used a wide variety of analytical approaches. Some studies have identified candidate adaptive single nucleotide polymorphisms (SNPs) based on allele frequency differences between populations in desert vs. montane habitats (Chen, Farrell, Matala, & Narum, 2018; Chen & Narum, 2021; Narum et al., 2010, 2013). Other studies have used landscape genomic approaches to identify genetic variation associated with environmental variation across a wide range of habitats (Collins et al., 2020; Micheletti, Matala, et al., 2018). In addition, common garden experiments have been used to identify differences in gene expression in response to thermal stress for redband trout from desert vs. montane populations (Chen, Farrell, Matala, & Narum, 2018; Garvin et al., 2015; Narum & Campbell, 2015). Common garden experiments have also been used to identify SNPs associated with different phenotypic responses to thermal stress (Chen, Farrell, Matala, Hoffman, et al., 2018; Chen, Farrell, Matala, & Narum, 2018; Chen & Narum, 2021). Together, these studies have identified many candidate genomic regions probably involved in thermal adaptation in redband trout.
Investigations on local adaptation of redband trout conducted thus far have used a variety of approaches for generating genomic and transcriptomic data, including amplicon sequencing or qPCR (quantitative polymerase chain reaction) of relatively small numbers of candidate SNPs, Illumina sequencing of thousands or millions of genome‐wide SNPs, and Illumina sequencing of total RNA. One of the most recent studies (Chen & Narum, 2021) used low‐coverage whole genome resequencing (lcWGR), a tool which has proven to be particularly powerful for exploring the genomic architecture of adaptation. This method enables cost–effect surveys of a much larger portion of the genome than many other techniques (e.g., up to 75% of the genome in Chen & Narum, 2021) by sequencing shotgun libraries at low depth per sample, but high depth per population. Unlike standard PoolSeq (Schlötterer et al., 2014), which generates uniquely barcoded libraries for pools of genomic DNA from all individuals in a population, lcWGR generates a uniquely barcoded library for each individual sample (Fuentes‐Pardo & Ruzzante, 2017; Lou et al., 2021). This barcoding strategy allows allele frequency estimates to be generated that account for variation in read depth across individuals and allows flexibility in conducting analyses that group different combinations of data subsets, such as grouping by population, sex or phenotype.
Thus far, lcWGR has only been used to investigate thermal adaptation across three redband trout populations representing different ecotypes (Chen & Narum, 2021). Here we build upon previous studies by performing lcWGR genome scans comparing 11 populations across a gradient of habitat types with substantial differences in thermal regimes, including cold montane, cool montane and high‐elevation desert habitats. Whereas several of these populations have been included in prior studies of thermal adaptation in redband trout (e.g., Chen, Farrell, Matala, Hoffman, et al., 2018), earlier studies did not account for a wide range of potential environmental drivers of local adaptation. To identify genomic regions potentially harbouring standing variation that enables local adaptation, we used population structure outlier tests to identify regions with strong allele frequency differences between three general habitat types. We also used genotype–environment association (GEA) analyses to identify genomic regions significantly associated with environmental variation across habitats. We further investigated environmental variables potentially involved in local adaptation by conducting ecological niche modelling (ENM) to identify variables with a strong impact on the presence or absence of redband trout across the region. To assess the susceptibility of our study populations to climate change, we used ENM to predict future changes in habitat suitability under two climate change scenarios. Finally, we used information gained from GEA analyses regarding genomic adaptation, along with expected future environmental conditions, to predict the level of genomic change that would be required for redband trout to adapt to future climate scenarios. Our hypotheses predicted that outlier analyses would reveal local adaptation across climate regimes, driven by environmental variables related to temperature. We also predicted that the level of genomic change required for adaptation to future climates would be high for all ecotypes, due to strong local adaptation across ecotypes.
2. METHODS
2.1. Laboratory work
Tissue samples for this study included 559 redband trout fin clips collected from 11 sites across three habitat types representing a gradient of climate regimes in Idaho (high‐elevation desert, cool montane forest and cold montane forest; hereafter referred to as desert, cool and cold) (Table 1; Figure 1). Of these 11 sites, nine are located among tributaries of the Snake River basin, whereas the remaining two sites are located in the Kootenai River drainage that flows into the upper Columbia River. Levels of connectivity among collections from the Snake River vs. Kootenai River drainages were expected to be very low given their extreme separation in the river network in this region. Previous studies indicate that hybridization of redband trout with cutthroat trout (Oncorhynchus clarkii) or non‐native rainbow trout (O. mykiss irideus) is low for our study sites (Kozfkay et al., 2011). Samples were collected between 1998 and 2018 (Table 1). For five of the sites, approximately half the samples were collected from two different years, with the length of time between collections varying from 2 to 12 years (Table 1). For one site (Mann Creek), samples were collected from two geographically proximate tributaries within the same year and were expected to demonstrate high gene flow at fine scale.
TABLE 1.
Sample collection locations, sample sizes after removing samples with low numbers of raw sequence reads, and collection years
| Site | Ecotype | River basin | Year collected | n (per replicate) | n (total) | Latitude | Longitude |
|---|---|---|---|---|---|---|---|
| Little Jacks Creek | Desert | Snake | 2010 | 33 | 61 | 42.72889 | −116.105 |
| 1998 | 28 | 42.72889 | −116.105 | ||||
| Big Jacks Creek | Desert | Snake | 2002 | 37 | 62 | 42.56278 | −116.043 |
| 1998 | 25 | 42.56278 | −116.043 | ||||
| Duncan Creek | Desert | Snake | 2001 | 16 | 51 | 42.54793 | −116.028 |
| 2003 | 35 | 42.54793 | −116.028 | ||||
| Williams Creek | Desert | Snake | 2003 | 31 | 31 | 42.87577 | −116.929 |
| Keithley Creek | Cool montane | Snake | 2004 | 20 | 55 | 44.55295 | −116.885 |
| 2013 | 35 | 44.55282 | −116.885 | ||||
| Little Weiser Creek | Cool montane | Snake | 2004 | 35 | 35 | 44.51247 | −116.339 |
| Dry Creek | Cool montane | Snake | 2012 | 33 | 72 | 43.68899 | −116.174 |
| 2016 | 39 | 43.71776 | −116.135 | ||||
| Trail Creek | Cool montane | Kootenai | 2018 | 35 | 35 | 48.56912 | −116.388 |
| Fawn Creek | Cold montane | Snake | 2011 | 33 | 33 | 44.38234 | −116.059 |
| Mann Creek | Cold montane | Snake | 2004 | 29 | 59 | 44.52606 | −116.934 |
| 2004 | 30 | 44.54770 | −116.987 | ||||
| S.F. Callahan Creek | Cold montane | Kootenai | 2006 | 15 | 53 | 48.42030 | −116.031 |
| 2018 | 38 | 48.42030 | −116.031 | ||||
| Total | 547 |
FIGURE 1.
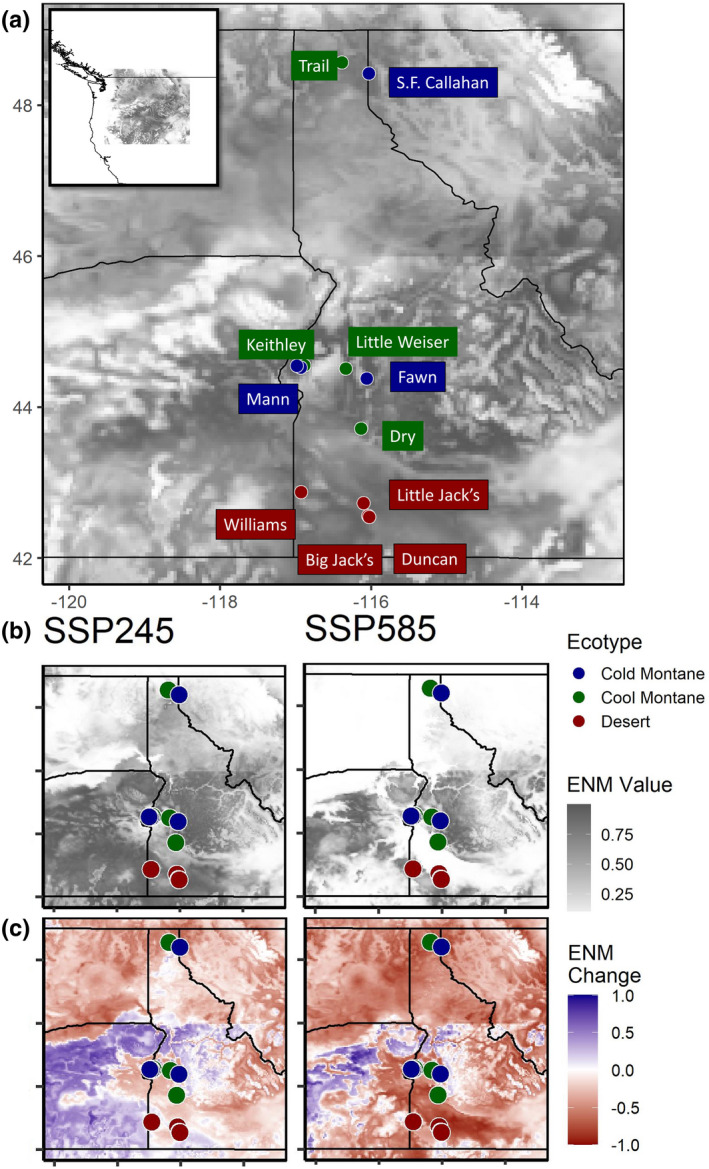
Map of study sites and ecological niche model (ENM) results for the present day, and for the period 2081–2100 across two shared‐socioeconomic pathways (SSP245 and SSP585). a) ENM values for the present day, with higher ENM values representing higher relative probability of presence (a proxy for habitat quality). b) Raw ENM value for future time period. c) Change from present day ENM values for future time period. “Cold” = cold montane forest, “Cool” = cool montane forest, “Desert” = high‐elevation desert
Genomic DNA was extracted from fin clips using Qiagen DNeasy kits following the manufacturer's protocol. Prior to performing lcWGR, we identified duplicate samples and hybrid individuals for removal from the data set using a “Genotyping‐in‐Thousands by Sequencing” (GTseq) panel of 376 SNPs developed for O. mykiss (Campbell et al., 2015; Willis et al., 2020). This panel includes neutral genetic markers that can be used to distinguish individuals and therefore identify duplicate samples in the data set, as well as markers that can be used to detect individuals with hybridization between rainbow trout and cutthroat trout. Sequencing of the GTseq panel was performed using an Illumina NextSeq at the Hagerman Genetics Laboratory, and genotyping of sequence data was performed as previously described (Campbell et al., 2015; Willis et al., 2020). Samples identified as duplicates or hybrids were removed from the data set prior to lcWGR. Data from the GTseq panel also enhanced several aspects of the lcWGR analyses described below. In particular, the GTseq panel allowed identification of the sex of each individual in the data set, since the panel includes a marker within the sdY region that is highly predictive of sex (Brunelli et al., 2008). In addition, the GTseq panel includes markers in two genomic regions that were identified as outliers in our lcWGR analyses (described further below). These two regions had been included in the GTseq panel due to association with developmental timing in redband trout and other salmonids (Micheletti, Hess, et al., 2018; Waters et al., 2021; Willis et al., 2020). Therefore, the GTseq data allowed comparison of allele frequencies generated using lcWGR vs. amplicon sequencing for these two genomic regions (described further below).
Library preparation for lcWGR was performed by preparing whole genome shotgun sequencing libraries using Illumina Nextera DNA kits at the University of Idaho Genomics and Bioinformatics Resources Core, with each sample uniquely barcoded using a combination of two indexes. Libraries were sequenced on two Illumina NovaSeq lanes with 2 × 150‐bp reads at the University of California Berkeley QB3 Vincent J. Coates Genomics Sequencing Laboratory.
2.2. Bioinformatic analysis
Sequence reads were demultiplexed by barcode, and read quality for each sample was evaluated using fastqc version 0.11.8 (Andrews, 2010) and multiqc version 1.7 (Ewels et al., 2016). Samples with low read counts (<1 million raw read pairs) were removed from subsequent analyses. The “ppalign” module in poolparty version 0.8 (Micheletti & Narum, 2018) was used for sequence quality filtering, genome alignment and SNP identification, following the parameter settings used in Chen and Narum (2021). bbmap version 38.90 (Bushnell, 2016) was used to remove sequencing adapters and primer dimers, trim reads using a mean quality threshold of 20, and remove reads that were <25 bp long after trimming. Quality‐filtered reads were then aligned to a rainbow trout reference genome (NCBI Accession GCF_013265735.2; Gao et al., 2021) using bwa mem version 0.7.17 (Li, 2013). PCR duplicates were removed using samblaster version 0.1.26 (Faust & Hall, 2014) and reads were sorted using picard tools version 2.25.0 (Broad Institute). samtools version 1.12 (Li et al., 2009) was used to remove ambiguously mapped reads, unaligned reads and reads with mapping quality <20. bcftools version 1.12 (Li, 2011) was used to identify SNPs and remove SNPs with quality <20, depth < 15 for all samples combined, and minor allele frequency (MAF) <0.05 across all samples (note that more stringent filtering of SNPs by depth and MAF was conducted in subsequent analyses, as described below). Next, SNP identification and allele frequency estimation were performed using popoolation2 version 1.201 (Kofler et al., 2011) as implemented in the poolparty “ppalign” module, using a normalization step to account for variance in sequence depth across individuals (Micheletti & Narum, 2018). This normalization step restricts the contribution of each individual to only two alleles for each SNP for the calculation of population allele frequencies. popoolation2 was also used to remove SNPs within 15 bp of an indel to account for potential mapping error. The resulting SNP allele frequency data set served as a starting point for subsequent analyses using the poolparty “ppanalyze” module for a variety of population comparisons with the full sample set and various sample subsets (described further below). Each of these analyses used additional filters for depth and MAF.
2.3. Genome‐wide population structure
Genome‐wide population structure across space and time was evaluated using the poolparty “ppanalyze” module. These analyses were conducted in two different ways: (i) treating each location independently and (ii) treating each temporal replicate independently. For analyses treating each location independently, we used highly stringent filtering criteria to remove SNPs with depth <15 for any population, depth >250 for any population and MAF <0.05 across all populations. For analyses treating each temporal replicate independently, we used the same filtering criteria except for a lower stringency for minimum depth (<12) to retain a sufficient number of SNPs. After filtering, principal components analyses (PCAs) were conducted using normalized allele frequencies with the “prcomp” function in R version 3.6.0 (R Core Team, 2021). To remove the potential influence of adaptive genomic variation on genome‐wide population structure, we also conducted PCA with a set of putatively neutral SNPs generated using two different methods: (i) excluding all SNPs above the 90th percentile of the distribution of F ST values, and (ii) excluding all SNPs identified as population structure outliers and/or GEA outliers by the methods described below. For the first of these two methods, F ST values were calculated using the sliding window Karlsson‐type F ST (Karlsson et al., 2007) with popoolation2 in the “ppanalyze” module of poolparty, with a window size of 5000 and a step size of 500.
2.4. Genome scans: population structure outliers
The poolparty “ppanalyze” module was also used to identify genomic regions under divergent selective pressures by identifying loci with strong allele frequency differences between groups. For these analyses, we removed SNPs with depth < 15 for any population, depth > 250 for any population and MAF <0.05 across all populations. We then evaluated the significance of pairwise allele frequency differences between groups using Fisher's exact test (FET). We first used this approach to compare different habitats (hereafter referred to as “between‐habitat comparisons”) by combining temporal replicates within each geographical site and then performing FETs for each pair of geographical sites that were from different habitat types. The resulting p‐values were then averaged to produce the final p‐values for each habitat comparison, and p‐values between populations from the same habitat type were not calculated or averaged. We used this approach for the following habitat comparisons, for a total of five different analyses: (i) one analysis comparing all three habitat types; (ii) three analyses comparing each pairwise combination of the three habitat types; and (iii) one analysis comparing all desert populations vs. all montane populations (i.e., all desert populations vs. all cool or cold populations). We performed these analyses using the full data set, and also using a data subset including only the Snake River populations, to investigate potential signals of adaptive divergence within the Snake River that may have been obscured by inclusion of the two Kootenai River populations. We also compared temporal replicates within populations (hereafter referred to as “temporal replicate comparisons”) to identify potential signals of adaptive differences over time within populations.
After performing FETs for each of the analyses described above, outlier genomic regions were identified with a Local Score analysis (Fariello et al., 2017) implemented in poolparty. This method uses a score function related to the FET p‐value and a window size based on the proximity of statistically significant p‐values. Genomic regions were considered outliers if p < .001. After identifying outlier regions, we evaluated which geographical sites were driving the allele frequency differences for each between‐habitat outlier region by conducting PCAs using region‐specific SNPs, using the “prcomp” function in R. To evaluate whether temporal variation in adaptive pressures influenced the results of the between‐habitat comparisons, we also performed PCAs of the between‐habitat outlier regions treating each temporal replicate separately.
To identify the genes that were potentially responsible for the observed signatures of divergent selection, we first identified all genes occurring within the outlier regions using the R packages genomicranges version 1.44.0 and genomicfeatures version 1.44.1 (Lawrence et al., 2013) and the reference genome annotations in the gff file. To narrow this list to the genes most probably under divergent selection, we identified the regions with the highest peak in Local Score values within each outlier region (regions containing SNPs above the 90th percentile of the distribution of Local Score values), and then identified the genes within those peak regions, as well as the closest gene on either side of the peak regions.
We used genome coordinates to determine if any of the SNPs from the GTseq panel occurred within the outlier regions, or within 100 kb of these regions. We identified a total of 23 GTseq SNPs for two outlier regions (described further in the Section 3), and then used the GTseq data to calculate allele frequencies across geographical sites for each of these SNPs. We then plotted these allele frequencies for visual comparison with the population structure observed in the lcWGR PCAs for each outlier region.
2.5. Genome scans: GEA Outliers
Environmental data for GEA analyses were obtained from the WorldClim database (Fick & Hijmans, 2017) and the NorWeST stream temperature database from 1993 to 2011 (Isaak et al., 2011). After removing highly correlated environmental variables, we retained nine variables for our final GEA analyses (Table 2; Figure S1, described further in the Supporting Information). Environmental variation across geographical sites for these nine variables was visualized using PCA with the R package factominer version 2.4 (Husson et al., 2020).
TABLE 2.
Descriptions of environmental variables used in GEAs, significance levels (p‐values) for each environmental variable in the RDA and pRDA models for the full data set analysis, and the number of outlier SNPs identified by LFMM for each environmental variable for the full data set and Snake River data subset analyses
| Environment variable | Source | Description | RDA: p | pRDA: p | LFMM: no. of SNPs | |
|---|---|---|---|---|---|---|
| Full data set | Snake River | |||||
| Canopy | NorWeST | Canopy cover (%): represents stream shade | .059 | .503 | 1094 | 2925 |
| Drainage area | NorWeST | Cumulative drainage area (km2): represents stream size and amount of insolation | .248 | .594 | 4161 | 5519 |
| Slope | NorWeST | Stream slope (%): affects flow velocity and equilibration time to local heating conditions | .01 | .101 | 1402 | 3426 |
| Temp: Stream | NorWeST | Mean August stream temp: 19‐year average from 1993 to 2011 | .797 | .755 | 667 | 1856 |
| Isothermality | WorldClim | [(Mean diurnal air temp range)/(Air temp annual range)](×100) | .671 | .672 | 13,564 | 1841 |
| Precipitation | WorldClim | Coefficient of variation of monthly precipitation | .312 | .426 | 1123 | 2233 |
| Temp: Mean Diurnal Range | WorldClim | Mean of monthly [(Max air temp) − (Min air temp)] | .043 | .205 | 21,367 | 1567 |
| Temp: Min Coldest Month | WorldClim | Min air temp of coldest month | .438 | .497 | 1657 | 4003 |
| Temp: Annual Range | WorldClim | [(Max air temp of warmest month) − (Min air temp of coldest month)] | .086 | .706 | 1251 | 3579 |
Three different GEA tests were performed to identify outlier SNPs associated with environmental variation: redundancy analysis (RDA), partial‐redundancy analysis (pRDA), and latent factor mixed‐modelling (LFMM) (Supporting Information). RDA and pRDA use a multivariate approach to identify outlier SNPs, whereas LFMM uses univariate regression. Both LFMM and pRDA control for population structure when identifying outliers; LFMM accomplishes this by modelling neutral genetic variation in latent factors, whereas pRDA includes neutral genetic variation as a covariable.
RDA and pRDA were conducted using the “rda” function in the R package vegan version 2.5‐6 (Oksanen et al., 2019). For pRDA, we accounted for neutral population structure using the loadings from a PCA (generated using “rda”) of putatively neutral SNPs as a covariable. The putatively neutral SNPs for the PCA were selected by removing SNPs above the 90th percentile of the distribution of F ST values, as determined from the poolparty “ppanalyze” results. We determined the number of PCA axes to include in the pRDA using the broken‐stick method (Legendre & Legendre, 1998) implemented with the PCAsignificance function in biodiversityr (Kindt & Coe, 2005). For both RDA and pRDA, we used the outlier function from the nescent RDA tutorial to reduce false positives (Forester et al., 2018), with 3.5 standard deviations set as the threshold of significance (equivalent to a two‐tailed p = .0005).
For LFMM, we used the “lfmm2” function in the R package lea version 3.7.0 (Gain & Francois, 2021), accounting for population structure using K = 9 based on previous analyses of genetic structure for these populations (Kozfkay et al., 2011; Supporting Information). To identify outliers, we used a significance threshold of alpha = .05 after Benjamini–Hochberg (BH) correction with the R package ihw version 1.15.0 (Ignatiadis et al., 2016). To avoid loss of power with LFMM due to pooled sequencing, we simulated 20 individuals per population using the R function “rbeta,” following recommendations for pooled sequence data on the LFMM website and personal communication with the authors (http://membres‐timc.imag.fr/Olivier.Francois/lfmm/faq.htm; O. Francois, personal communication July 21, 2020).
We performed all GEA analyses (RDA, pRDA, LFMM) using the full data set, and we also performed LFMM analyses using only the Snake River data subset (excluding the Kootenai River populations). We did not perform RDA and pRDA for the Snake River data subset due to the relatively low explanatory power observed for analysis of the full data set using these methods (described further below).
2.6. Comparing outlier SNPs across methods
To investigate the level of consistency in the identities of outlier SNPs detected across analytical methods, we identified outlier SNPs that were shared across GEA tests (pRDA, RDA and LFMM). We also identified GEA outlier SNPs that occurred within the outlier regions identified by the Local Score test. Finally, we investigated whether any of the SNPs identified as outliers in a previous landscape genetics study of redband trout (Micheletti, Matala, et al., 2018) occurred within the Local Score outlier regions identified in our study.
2.7. Climate change assessment
To assess the vulnerability of each population to climate change, we conducted ENM and genetic offset analysis for current and future climates, using the WorldClim variables from the GEA analysis described above. We predicted future environmental conditions using two shared‐socioeconomic pathways (SSPs) for the years 2081–2100, including a “low‐emission” scenario in which social, economic and technological trends continue to follow historical patterns (SSP245), and a “high‐emission” scenario in which fossil fuels are heavily exploited (SSP585) (Riahi et al., 2017).
For ENM, we used the machine learning software maxent version 3.4.3 in the R package dismo (Hijmans et al., 2021) to predict the relative probability of future occurrence of redband trout across Idaho. maxent is a presence‐only ecological niche model software (Phillips et al., 2006; Phillips & Dudik, 2008). We obtained 451 resident redband trout presence locations from Idaho Fish and Game (Meyer et al., 2010). To remove points with potential errors (e.g., probable missing negative signs, locations in the ocean), we filtered to retain only points with a longitude less than −110 and a latitude greater than 39. A total of 429 presence points remained after filtering, although if multiple points are present within a single raster cell, all but one are automatically removed by maxent. We then clipped the WorldClim variables to the minimum and maximum longitude points, plus or minus 0.5 decimal degrees using the R package terra version 1.4.2.2 (Hijmans, 2022). This was done to reduce the pseudoabsence points from occurring well outside probable niche space, which can lead to overfitting. To match the resolution of the present‐day layers (30 s) to that of the future layers (2.5 min), we conducted bilinear resampling. We then used the R package enmeval version 2.0.2 (Muscarella et al., 2014) to select the model features (e.g., linear, threshold) and to tune the regularization multiplier to avoid over‐fitting. Based on Akaike Information Criterion (AIC) weight, the best supported model used linear, quadratic and hinge features. We selected the second feature set due to higher area under the curve (AUC). For our maxent settings, we used 10,000 psuedo‐absence points, a regularization multiplier of 0.5, jackknife analysis and 10 replicates with cross‐validation to assess fit. After predicting relative probability of presence across the landscape, we then extracted the change in ENM value for the geographical sites of each of our study populations.
Genetic offset was calculated using the “genetic.offset” function in lea with the full data set (including all Kootenai and Snake River populations), and represents the level of mismatch in allele frequencies between the current and predicted future genetic composition of populations at putatively adaptive loci. For this analysis, adaptive loci are identified through GEA analyses under an assumption that selection is potentially weak but highly polygenic (Gain & Francois, 2021).
3. RESULTS
Analysis of GTseq data identified one hybrid individual and one duplicate pair of individuals, which were removed from the data set prior to performing lcWGR. For the lcWGR data, a total of nine samples had low read counts (<1 million read pairs) and were removed from subsequent analyses. After sample filtering, the final data set included 547 samples from 11 sites (four desert sites, four cool sites and three cold sites; Table 1). The total sample size per site ranged from 31 to 72, the number of raw read pairs per sample ranged from 1,338,833 to 28,410,031 (mean = 10,438,707), and the number of reads after quality filtering ranged from 1,267,065 to 26,677,778 (mean = 9,831,827) (Tables S1 and S2). Using the GTseq data for the sex‐predictive marker, we determined that 257 individuals were female, 320 were male and seven could not be determined due to poor sequencing results for the sex‐determining marker (Table S1).
3.1. Genome‐wide population structure
For the genome‐wide population structure analysis treating each geographical site independently, a total of 4,193,763 SNPs were retained after filtering. The first PCA axis separated the Kootenai River populations (Trail Creek and S.F. Callahan) from each other and from all other locations, and the second PCA axis separated Little Jacks, Dry Creek and Fawn Creek from each other and all other locations (Figure 2). For the analysis treating each temporal replicate independently, a total of 2,141,860 SNPs were retained after filtering. PCA results for this analysis were similar to those for the analysis treating each geographical site independently, and temporal replicates from the same population generally clustered together (Figure S2). PCA results were also similar for analyses conducted using only putatively neutral SNPs (Figure S3).
FIGURE 2.
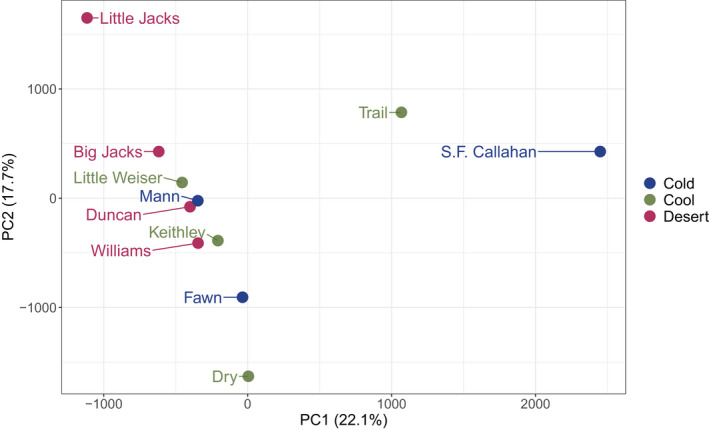
Principal components analysis of all genome‐wide SNPs across geographical sites, colour‐coded by ecotype: “Cold” = cold montane forest, “Cool” = cool montane forest, “Desert” = high‐elevation desert
3.2. Genome scans: population structure outliers
For the five different between‐habitat outlier analyses with the full data set (i.e., all Kootenai and Snake River populations), the number of genome‐wide SNPs ranged from 4,021,582 to 5,634,494. Local Score analysis identified 10 outlier regions across the different pairwise comparisons, with each region occurring on a separate chromosome (Figure 3, Table 3). Of the 10 regions, six were outliers for the comparison of all three ecotypes, two for the desert vs. cool comparison, five for the desert vs. cold comparison, four for the desert vs. montane comparison, and four for the cool vs. cold comparison (Table 3). Only one region was an outlier for all five of these comparisons; this was a region on chromosome Omy28. Population structure varied across the 10 regions (Figure 4a,b; Figure S4). The desert populations clustered together in the Omy01 and Omy23 outlier region PCAs, but populations did not cluster by habitat type for the remaining outlier regions. The two Kootenai River populations (Trail Creek, a cool site; and S.F. Callahan Creek, a cold site) plotted separately from Snake River populations for most of the outlier regions, following the pattern observed in the PCA of genome‐wide SNPs and neutral SNPs (Figure 4; Figure S4). Dry Creek (a cool site) also plotted separately from other populations for the Omy01, Omy06 and Omy16 outlier regions.
FIGURE 3.
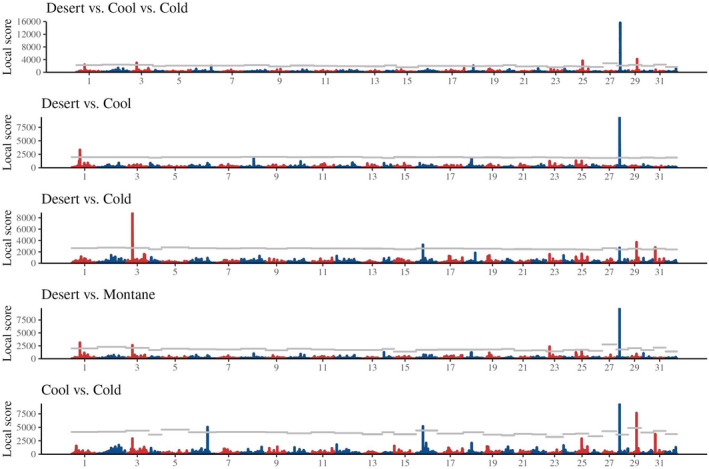
Manhattan plots of Local Scores across genome‐wide SNPs for pairwise comparisons between habitat types for the full data set analyses (including all Snake River and Kootenai River populations). Chromosome numbers are given along the x‐axis. Horizontal grey lines indicate average chromosome significance = .001 after correction for multiple tests
TABLE 3.
Results of the Local Score population structure outlier analysis for the full data set, including chromosome positions and numbers of SNPs for outlier regions identified by Local Score analysis, chromosome positions for the subregions with the highest 10% of Local Scores (LS) values, and pairwise comparisons for which the region was an outlier (“All 3” = Desert vs. Cool vs. Cold, “Des” = Desert, “Mont” = Montane, “X” indicates the region was an outlier for the comparison)
| Chromosome | Start position (bp) | End position (bp) | Length (bp) | Number of SNPs in region | High LS region: start | High LS region: end | All 3 | Des vs. Cool | Des vs. Cold | Des vs. Mont | Cool vs. Cold |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Omy01 | 30,535,904 | 30,581,128 | 45,224 | 103 | 30,572,309 | 30,581,128 | X | X | X | ||
| Omy03 | 24,355,857 | 24,604,913 | 249,056 | 788 | 24,502,268 | 24,604,913 | X | X | X | ||
| Omy06 | 68,318,638 | 68,922,834 | 604,196 | 1186 | 68,875,569 | 68,922,834 | X | ||||
| Omy16 | 25,172,402 | 25,351,988 | 179,586 | 537 | 25,315,776 | 25,351,988 | X | X | |||
| Omy18 | 31,140,760 | 31,175,693 | 34,933 | 65 | 31,166,658 | 31,175,693 | X | ||||
| Omy23 | 12,975,657 | 13,002,716 | 27,059 | 86 | 12,998,956 | 13,002,716 | X | ||||
| Omy25 | 22,360,805 | 22,422,253 | 61,448 | 128 | 22,420,077 | 22,422,253 | X | ||||
| Omy28 | 12,014,130 | 12,168,212 | 154,082 | 454 | 12,119,895 | 12,168,212 | X | X | X | X | X |
| Omy29 | 30,797,457 | 31,029,281 | 231,824 | 680 | 30,959,520 | 31,029,281 | X | X | X | ||
| Omy31 | 5,817,178 | 5,979,323 | 162,145 | 541 | 5,964,083 | 5,979,323 | X |
FIGURE 4.
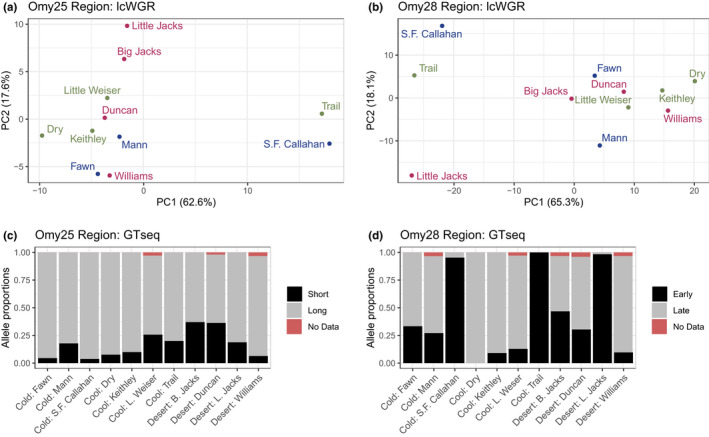
Population structure for SNPs occurring within outlier genomic regions identified using Local Score analysis on chromosomes Omy25 and Omy28. (a, b) PCA for all SNPs identified by poolparty analysis and occurring within the outlier regions (n = 128 SNPs for Omy 25 region, n = 454 SNPs for Omy28 region); red = high‐elevation desert populations; green = cool montane forest populations; blue = cold montane forest populations. (c, d) Allele proportions for one GTseq SNP occurring within 100 kb of each outlier region (Locus L01 for Omy25, Locus L02 for Omy28); allele proportions for the remaining GTseq SNPs within these regions show similar results due to strong linkage, and are reported in Figures S11 and S13. Previous studies have identified Oncorhynchus mykiss phenotypes associated with each GTseq SNP allele: an earlier (“Short”) or later (“Long”) age at maturity for the Omy25 region, and an “Early” or “Late” migration timing for the Omy28 region (Willis et al., 2020). “No Data” indicates the proportion of samples that failed to produce genotype data
For the between‐habitat comparisons of the Snake River data subset, the number of genome‐wide SNPs for the five analyses ranged from 4,225,340 to 6,117,241. Local Score analysis identified a total of seven outlier regions for these comparisons, including five that overlapped with the outlier regions from the full data set analysis (regions on Omy03, Omy06, Omy18, Omy23 and Omy28), as well as two new outlier regions (on Omy15 and Omy19) (Figure S5, Table S3). Thus, in total we identified 12 unique outlier regions for between‐habitat comparisons of the full data set and the Snake River data subset, including five regions unique to the full data set analyses, two unique to the Snake River data subset analyses and five shared for analyses with both data sets.
Of the seven regions identified by the Snake River data subset analyses, one was an outlier for the comparison of all three ecotypes, three for the desert vs. cool comparison, two for the desert vs. cold comparison, four for the desert vs. montane comparison and two for the cool vs. cold comparison (Table S3). For four of these outlier regions, PCA separated most desert populations from montane populations (regions on Omy03, Omy15, Omy18 and Omy23; Figure S6). For the Omy28 region, PCA separated Little Jacks Creek (a desert site) from the rest of the populations. For the two remaining regions (Omy06 and Omy19), PCA separated Dry Creek (a cool site) from all other populations (Figure S6).
For the temporal replicate population structure outlier analyses, the number of genome‐wide SNPs ranged from 785,051 to 4,569,224. Local Score analyses identified a total of eight outlier regions, with all except one population (S.F. Callahan) having at least one outlier region (Figure S7, Table S4). None of the outlier regions were shared across populations, and only one region overlapped with an outlier region identified in the between‐habitat comparisons (i.e., a region on chromosome Omy03). Local Score values for the between‐replicate outlier regions were generally much less significant than for the between‐habitat outlier regions (compare Figure 3 and Figures S5 and S7). In addition, PCAs of the 12 between‐habitat outlier regions treating each temporal replicate separately showed similar clustering of habitat types as the PCAs that combined temporal replicates (compare Figures S4, S6 and S8). These results indicate that adaptive differences for temporal comparisons were probably much weaker than those for between‐habitat comparisons, and that temporal variation in adaptive pressures probably had minimal influence on results of the between‐habitat comparisons.
A number of genes were associated with the between‐habitat outlier regions and are candidates for local adaptation. For the full data set analysis, 56 genes occurred within the outlier regions (Table S5) and 38 genes occurred within or on either side of the peak Local Score regions (Figure S9, Table S6). For the Snake River data subset, 172 genes occurred within the outlier regions (Table S7) and 47 genes occurred within or on either side of the peak regions (Figure S10, Table S8). Two of the candidate gene regions, occurring on Omy25 and Omy28, have been previously studied in redband trout and other salmonids. The closest gene downstream of the Omy25 outlier region, SIX6, is known to be associated with age at maturity in rainbow trout and other salmonids (Waters et al., 2021). In addition, two of the genes within the Omy28 outlier region, GREB1L and ROCK1, are known to be associated with migration timing in rainbow trout and Chinook Salmon (Alshwairikh et al., 2021; Hess et al., 2016; Koch & Narum, 2020; Micheletti, Hess, et al., 2018; Thompson et al., 2019; Willis et al., 2020).
By comparing the genome coordinates of the Local Score outlier regions and the SNPs present in the GTseq panel, we determined that the GTseq panel includes 10 SNPs within 100 kb of the Omy25 Local Score outlier region (three within the SIX6 gene, six in the upstream promoter region for SIX6 and one in the downstream intergenic region), and 13 SNPs within the Omy28 outlier region (six within the GREB1L gene, six within the intergenic region and one within the ROCK1 gene) (Willis et al., 2020). These 23 SNPs had originally been included in the GTseq panel due to known associations of SIX6 and GREB1L/ROCK1 with developmental timing in salmonids (Hess et al., 2016; Micheletti, Hess, et al., 2018; Prince et al., 2017; Waters et al., 2021). Furthermore, later studies identified rainbow trout phenotypes associated with each allele at these SNPs: alleles in the SIX6 region have been associated with an earlier (“short”) or later (“long”) age at maturity, and alleles in the GREB1L/ROCK1 region have been associated with an “early” or “late” arrival time to spawning grounds (Micheletti, Hess, et al., 2018; Willis et al., 2020). Most populations in our study were dominated by the “long” alleles in the SIX6 region, but some populations had fairly high proportions of “short” alleles (Little Weser, Big Jacks, Duncan) (Figure 4c; Figure S11a). This pattern was consistent across both sexes (Figure S11b,c). However, this pattern differed from the population structure observed in our lcWGR PCA results for the Omy25 outlier region, for which Trail Creek and S.F. Callahan Creek segregated separately from other populations (Figure 4a). This discrepancy is probably explained by the fact that the GTseq SNPs occur further downstream than the outlier region identified in our study, and therefore probably occur in a different part of the SIX6 promoter region (Figure S12). This result potentially indicates that selection is acting on a different portion of the SIX6 promoter region in our study than has been observed in previous studies; alternatively, this inconsistency may reflect a lack of precision in the Local Score analysis for identifying the region under divergent selection. For the GTseq SNPs in the Omy28 outlier region, allele frequencies from our data indicated a greater proportion of “early” alleles in S.F. Callahan, Trail Creek and Little Jacks Creek, when compared to the other populations (Figure 4d; Figure S13). This result is consistent with the population structure observed in our lcWGR PCA for the Omy28 outlier region (Figure 4b).
3.3. GEAs
PCA of environmental variables demonstrated that collections in this study represented a gradient of climate regimes, with the first axis separating the desert sites from the montane sites (Figure 5a; Table S9). In contrast, the cool and cold montane sites overlapped substantially, forming two clusters that separated on the second axis: one cluster included S.F. Callahan (cold) and Trail Creek (cool), and the other cluster included the remaining cool and cold sites (Figure 5a). The first PCA axis was driven by stream temperature, drainage, canopy and precipitation, and the second PCA axis was driven by the WorldClim temperature variables (isothermality, mean diurnal temperature range, minimum temperature of coldest month, annual temperature range) (Figure 5b).
FIGURE 5.
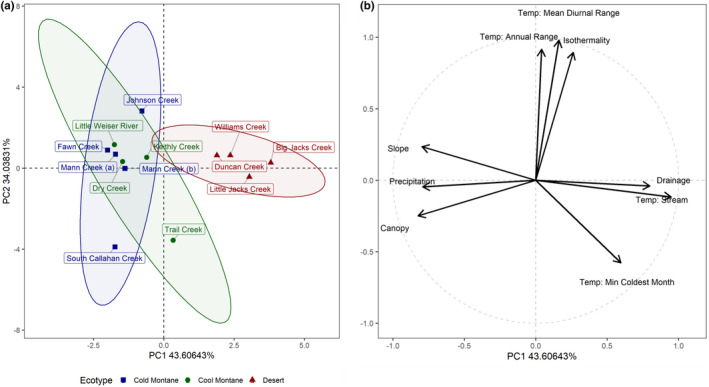
Principal components analysis of environmental data. (a) Populations colour coded by ecotype, and with distribution ellipses around 95% confidence intervals. “Blue” = cold montane forest, “green” = cool montane forest, “red” = high‐elevation desert. “Mann Creek (a)” and “Mann Creek (b)” represent two geographically proximate tributaries. (b) Contributions of environmental variables to the principal components
The total number of SNPs used for GEAs with the full data set was 2,971,532. For RDA, the overall model had p = .15, with an adjusted R 2 value of .316. Two environmental variables were significant (slope, p = .01; mean diurnal temperature range, p = .04) and two had p < .10 (canopy, p = .06; temperature annual range, p = .09) (Table 2). The first axis of the model had p = .14, and the second axis had p = .42. Due to the high p‐value of the second axis, we retained only the first axis, which explained 30.7% of the variance and identified 3930 outlier SNPs. pRDA revealed results similar to those of the RDA, but with lower explanatory power and lower statistical significance. We used only the first axis of the neutral SNP PCA as a covariable in pRDA, because the broken‐stick method indicated the percentage of variance explained for this axis was lower than the broken‐stick percentage, and cumulative percentage was never higher than the broken‐stick cumulative percentage. The overall pRDA model was nonsignificant (p = .451), with an adjusted R 2 value of .045. None of the environmental variables or axes were significant. We retained the first axis (p = .50), but outliers from the pRDA should be interpreted with caution due to the lack of significance for the model and axes. The first axis explained 25.7% of the variance and identified 3284 outlier SNPs.
LFMM analyses for the full data set indicated that a total of 35,198 SNPs were significantly associated with at least one environmental variable. Of these, 10,317 SNPs were significantly associated with two or more environmental variables. The number of significant associations was highest for mean diurnal temperature (21,367 SNPs) and isothermality (13,564 SNPs), and lowest for stream temperature (667 SNPs) and canopy cover (1094 SNPs) (Table 2, Figure 6a). A similar pattern was observed for the 847 LFMM outlier SNPs that occurred within the Local Score outlier regions, for which the number of significant associations was consistently highest for mean diurnal temperature and isothermality (Figure 6b–d; Figure S14).
FIGURE 6.
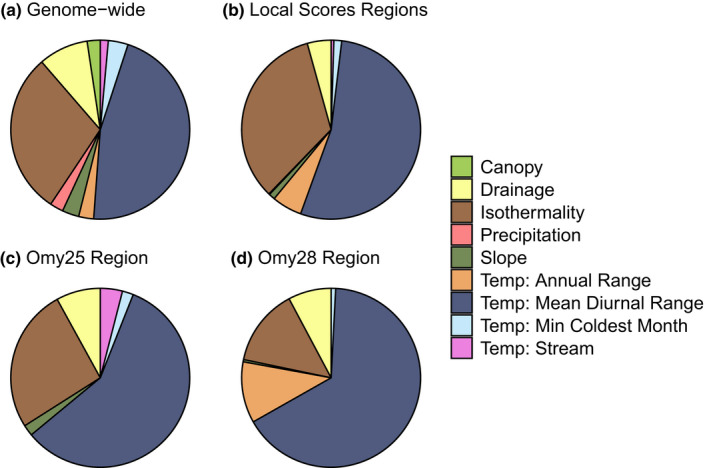
Proportions of environmental associations for LFMM outlier SNPs for full data set analyses (including both Snake River and Kootenai River populations). Each environmental association is treated independently, and therefore SNPs associated with more than one environmental variable are represented more than once. (a) All LFMM outlier SNPs (35,198 SNPs), (b) only LFMM outlier SNPs occurring in the 10 Local Scores population structure outlier regions (847 SNPs), (c) only LFMM outlier SNPs within the Omy25 outlier region (33 SNPs), and (d) only LFMM outlier SNPs within the Omy28 outlier region (165 SNPs)
LFMM analysis of the Snake River data subset used a total of 3,057,141 SNPs, of which 20,279 SNPs were significantly associated with at least one environmental variable, including 4543 SNPs associated with two or more environmental variables. The number of associations was highest for drainage area (5519 SNPs) and minimum temperature of the coldest month (4003 SNPs), and lowest for mean diurnal temperature range (1567 SNPs) and isothermality (1841 SNPs) (Table 2). Similarly, the LFMM outlier SNPs that occurred within the Local Score outlier regions had the greatest number of associations for drainage area and minimum temperature of the coldest month, although other environmental variables also had relatively large numbers of associations for some outlier regions (Figure S15).
3.4. Overlap of outlier SNPs across methods
For the full data set analyses, the greatest overlap in outliers across GEA tests occurred between LFMM and RDA (Figure 7a). Few outliers were shared between RDA and pRDA, although higher percentages of shared outliers occurred within the outlier regions identified by the Local Score test (Figure 7b; Figure S16). Within these outlier regions, 309 SNPs were RDA outliers (9.31% of total SNPs within the regions), 287 were pRDA outliers (8.65%), and 847 were outliers for one or more LFMM tests (25.5%). However, these percentages varied across outlier regions; for example, the Omy23 region had only one SNP that was an outlier for a GEA test, whereas ≥50% of SNPs were outliers for one or more GEA tests in the Omy18, Omy25, Omy28 and Omy29 regions (Figure 7c,d; Figure S10). For the Snake River data subset analyses, the percentages of SNPs within the Local Score outlier regions that were LFMM outliers ranged from 1.3% to 10.8% across the seven regions (Table S3). Of the outlier SNPs identified in the study by Micheletti, Matala, et al. (2018), a total of 52 occurred within the 12 Local Score population structure outlier regions: six in the Omy03 region, 12 in Omy06, seven in Omy15, one in Omy16, one in Omy18, eight in Omy19 and 17 in Omy28 (including the 13 SNPs in the GTseq panel). An additional 22 outlier SNPs from that study occurred within 100 kb of the outlier regions: four in Omy01, two in Omy15, one in Omy18, 11 in Omy25 (including the 10 SNPs in the GTseq panel), one in Omy28 and three in Omy29.
FIGURE 7.
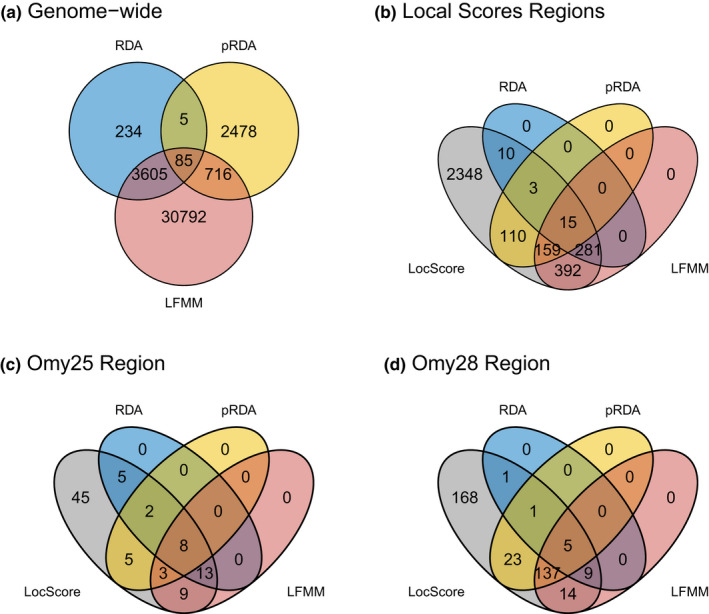
Overlap of outlier SNPs across detection methods for the full data set analyses (including all Snake River and Kootenai River populations). (a) Genome‐wide outlier SNPs from all GEA methods (RDA, pRDA, LFMM), (b) SNPs found within all 10 outlier regions identified by the Local Score (LocScore) population structure outlier analysis, (c) SNPs found within the Omy25 outlier region, and (d) SNPs found within the Omy28 outlier region. SNPs that were outliers for more than one environmental variable were counted only once
3.5. Climate change assessment
For the ENM analyses, overall ecological model fit was moderate (AUC = 0.783, Figure 1; Figure S17). The environmental variables with the highest permutation importance were isothermality, annual temperature range and mean diurnal temperature range (Table S10). Permutation importance represents the contribution to the relative probability of presence score across the landscape during permutations of the variables, and is different from the variable contribution value, which may be biased toward the order the variables are fitted. Redband trout habitat decreased substantially across Idaho for both SSP scenarios, and especially for the high‐emission scenario (Figure 1). The greatest probability of presence for both SSP scenarios occurred in high‐elevation areas, with the exception of the desert sites. This result indicates the potential for elevation, which was correlated with climate in our study, to influence the distribution of climate refugia in future climate scenarios. For our study sites, model predictions with the low‐emission scenario indicated about half the populations had positive or near zero ENM‐change values, potentially indicating that populations could be sustained or even improve as temperatures move closer to the optimal growth temperatures (Bear et al., 2007; Hahlbeck et al., 2021) (Table 4). In contrast, ENM‐change values were negative for all study sites for the high‐emission scenario (Table 4).
TABLE 4.
Genetic offset values, and ecological niche model (ENM) change in relative probability of presence, for the period 2081–2100 under two shared‐socioeconomic pathways (SSPs). “Mann Creek (a)” and “Mann Creek (b)” represent two geographically proximate tributaries
| Population | Ecotype | SSP245 | SSP585 | ||
|---|---|---|---|---|---|
| Genetic offset | ENM value change | Genetic offset | ENM value change | ||
| Little Jacks Creek | Desert | 1 | −0.08 | 1 | −0.28 |
| Big Jacks Creek | Desert | 0.78 | −0.06 | 0.86 | −0.87 |
| Duncan Creek | Desert | 0.78 | 0.18 | 0.86 | −0.71 |
| Williams Creek | Desert | 0.85 | −0.49 | 0.9 | −0.82 |
| Keithley Creek | Cool montane | 0.83 | 0.09 | 0.9 | −0.31 |
| Little Weiser Creek | Cool montane | 0.87 | −0.31 | 0.93 | −0.6 |
| Dry Creek | Cool montane | 0.85 | 0.17 | 0.91 | −0.73 |
| Trail Creek | Cool montane | 1 | −0.44 | 1 | −0.72 |
| Fawn Creek | Cold montane | 0.85 | 0.07 | 0.91 | −0.57 |
| Mann Creek (a) | Cold montane | 1 | −0.11 | 1 | −0.45 |
| Mann Creek (b) | Cold montane | 1 | −0.32 | 1 | −0.51 |
| S.F. Callahan Creek | Cold montane | 1 | −0.04 | 1 | −0.39 |
Genetic offset was very high for all populations, indicating all populations would experience strong selective pressures under both SSPs (Table 4; Figure S18). Thus, even if environmental conditions move toward a species optimum for some populations, those populations may still perform poorly in future conditions due to local adaptation to previous conditions.
4. DISCUSSION
Genome scans of 11 redband trout populations across a wide range of thermal regimes identified 12 genomic regions that provided evidence for local adaptation in this species across highly heterogeneous landscapes. Results suggest that a combination of evolutionary forces including drift in relatively isolated populations, along with selection (e.g., Buffalo & Coop, 2020) have contributed to signals of genetic variation that influence adaptive potential (Seaborn et al., 2021). The strongest adaptive differences observed in this study were found in the two most northern populations, which are geographically distant from all others and occur in a separate river drainage (i.e., S.F. Callahan and Trail Creek, which are found in the Kootenai River drainage, whereas all other populations occur in the Snake River drainage). Historical gene flow between the Kootenai River populations and those in the Snake River is expected to have been very rare given the large migration distance that would need to be travelled for successful immigration (~1000+ of river km). Furthermore, contemporary gene flow is expected to be essentially nonexistent because all populations in this study occur above different hydropower dams, without upstream fish passage. Within the Snake River drainage, we also identified adaptive differences between desert and montane populations. This result is consistent with previous studies of redband trout that have found evidence for local adaption to these distinct habitats (e.g., Chen, Farrell, Matala, Hoffman, et al., 2018; Narum et al., 2010). Under scenarios of climate change, desert populations were at the greatest risk of extirpation as conditions may become unsuitable for these isolated populations with limited migration opportunities to thermal refugia.
The two Kootenai River populations with strong adaptive differences in our study do not occur in the same habitat type (S.F. Callahan is a cold site, Trail Creek a cool site), but are highly divergent from other populations for environmental variables including mean diurnal temperature range, isothermality, annual temperature range and minimum temperature of the coldest month (Figure 5b). Two of these variables, mean diurnal temperature range and isothermality, also had the largest number of significant genetic associations for LFMM analyses. Furthermore, mean diurnal temperature range was one of only two significant variables in the RDA model, and ENM across the region indicated that mean diurnal temperature range and isothermality are strong predictors of redband trout presence. Together these results provide evidence that these two environmental variables are strong drivers of divergent selection in redband trout. Both variables are related to the magnitude of fluctuation in environmental temperatures, with mean diurnal temperature describing the daily fluctuation, and isothermality describing the relationship between daily and annual fluctuation. Given that redband trout are aquatic ectotherms and therefore have body temperatures that are strongly influenced by environmental temperature, it is reasonable that habitats with large temporal variation in temperature could exert unique selective forces and promote local adaptation. Large temperature variation could affect physiology and growth rates at higher temperatures (e.g., Colinet et al., 2015; Oligny‐Hebert et al., 2015) and could promote the evolution of enhanced adfluvial migratory capabilities to access thermal refugia in nearby lakes and reservoirs that would be expected to improve growth and survival (Hahlbeck et al., 2021). For example, one of the Kootenai River populations (Trail Creek) appears to be adfluvial, migrating as yearlings and returning as adults. However, the two Kootenai River populations in our study had lower mean diurnal temperature and isothermality compared to the other populations, indicating that thermal stability at these two sites may drive divergent selective pressures compared to many other populations across the subspecies range (diurnal temperature range was 12.0–12.4°C for the Kootenai River populations, compared to 15.1–16.3°C for other populations; isothermality was 35.2–36.0, compared to 39.6–41.8 for other populations). Alternatively, the genomic associations identified here could be driven by unmeasured environmental variables that are correlated with variables included in our study, or by environmental variables that were removed from our analyses due to correlation.
Two of the 12 outlier regions identified in this study were associated with genes known to be involved in developmental timing in salmonids. One of these regions is on chromosome Omy25 and was an outlier for the full data set comparison, but not the Snake River data subset comparison. This region is associated with SIX6, a gene involved in age‐at‐maturity in salmonids (Barson et al., 2015; Sinclair‐Waters et al., 2020; Waters et al., 2021; Willis et al., 2020). Age‐at‐maturity is a life history trait that has evolutionary trade‐offs, because delaying maturity results in a greater body size at reproduction and a corresponding increase in fecundity, but also results in a greater chance of mortality prior to reproduction. Although the geographical distribution of genetic variation at this outlier region has not been reported for redband trout, a study of anadromous Oncorhynchus mykiss found that variation in this genomic region was significantly associated with age‐at‐maturity (Willis et al., 2020). In our study, genetic variation in this outlier region was driven by divergence of the two Kootenai River populations from all other populations, indicating these two populations may experience distinct selective pressures for age‐at‐maturity, potentially related to adfluvial life histories or compressed growing seasons.
The second outlier region associated with developmental timing occurs on chromosome Omy28 and is known to have a large effect on adult migration timing in O. mykiss (Hess et al., 2016; Micheletti, Hess, et al., 2018; Prince et al., 2017; Willis et al., 2020). This region was an outlier for both the full data set analysis and the Snake River data subset analysis, and includes the genes GREB1L and ROCK1, which are associated with maturation state at the time of entry into spawning grounds for anadromous redband trout. Fish that have “early” alleles in this region enter freshwater and arrive at spawning grounds early, whereas fish that have “late” alleles enter freshwater later or hold in tributaries for several months before arriving at spawning grounds (Hess et al., 2016; Micheletti, Hess, et al., 2018; Waples et al., 2022). A previous study found that most populations of O. mykiss have higher frequencies of late alleles, whereas early alleles are much more rare and only occur in specific drainages (Collins et al., 2020). We found similar results, with most of our study populations being predominated by late alleles, with the exception of three populations that had predominantly early alleles, including one population from each habitat type (cold: S.F. Callahan; cool: Trail Creek; desert: Little Jacks). Our results are also concordant with those of a previous lcWGR study comparing redband trout from Little Jacks, Keithley and Fawn, which found significant allele frequency divergence for Little Jacks at this outlier region, along with a genetic association with thermal tolerance phenotypes for this region (Chen & Narum, 2021).
For both of the outlier regions associated with migration and developmental timing, about 50% of SNPs within the regions were also significantly associated with environmental variation for the full data set analyses, with the largest number of associations for mean diurnal temperature range and isothermality. These results are concordant with previous landscape genetics studies of O. mykiss in the Columbia River Basin, which identified significant associations with temperature for SNPs in these two outlier regions (Collins et al., 2020; Micheletti, Matala, et al., 2018). These results suggest that temperature is an important driver of evolution for developmental timing traits for our study populations. Developmental timing is well known to be influenced by water temperature in salmonids. For example, warmer water usually causes faster developmental rates and fry emergence (e.g., Bromage et al., 1992), and temperature along with flow can trigger spawning (e.g., Muhlfeld et al., 2009). However, less is known about the influence of daily and annual temperature variation on migration timing and age at maturity.
Although the association of Omy25 and Omy28 outlier regions and developmental timing have been well established in multiple salmonid species, our study populations have different life history traits from populations in previous studies, and therefore could have different selective mechanisms. Whereas previous studies of SIX6 on Omy25 and GREB1L/ROCK1 on Omy28 have focused on anadromous fish, the populations in our study were all resident redband trout, although some or all populations may have had anadromous migrations before the construction of anthropogenic barriers (multiple dams) which blocked access to the sea in the mid‐1900s (e.g., Mann Creek; Holecek et al., 2012). Due to the focus on anadromous fish in previous studies, the life history traits associated with these genes have been defined in relation to migratory timing and migratory life stages. For example, the age‐at‐maturity trait associated with SIX6 has been defined in relation to the length of time that anadromous fish spend at sea before returning to spawning grounds, resulting in a strong relationship between age‐at‐maturity and body size due to a high growth rate at sea (Willis et al., 2020). In addition, the migration timing trait associated with GREB1L and ROCK1 has been defined as the maturation state at which fish enter freshwater and arrive at spawning grounds after migrating from the sea (Micheletti, Hess, et al., 2018; Willis et al., 2020). Since our study populations are no longer anadromous, variation at these two genomic regions may represent present‐day selective pressures related to processes of developmental timing that occur entirely in freshwater. For example, spawn timing in trout appears to be under strong selection to avoid fry emergence during winter and spring high‐flow events, when eggs can wash away (e.g., Wenger et al., 2011), or may be related to differences among locations in productivity and growth rate. In addition, resident redband trout in freshwater often exhibit fluvial or adfluvial migration throughout a drainage but return to spawning areas with variable timing and age‐at‐maturity (e.g., Meyer & Schill, 2021; Schill et al., 2010). Alternatively, the signature of divergent selection we observed for these outlier regions could result from selective pressures that operated in the past, when anadromous fish may have been present in these populations prior to the construction of dams. Because these dams were constructed relatively recently in the last century, genetic drift and background selection may not have had sufficient time to erode signatures of selection that formed in the past.
The remaining 10 outlier regions identified in our study were associated with a number of genes with roles that have not been well characterized in salmonids and represent intriguing candidate regions for local adaptation. For example, genes associated with these outlier regions have been implicated in regulating early development, spawn timing, immune function, muscle mass, lipid metabolism and atrophy resistance in fishes (Belghit et al., 2014; Goldsmith et al., 2003; Leder et al., 2006; Panserat et al., 2009; Wang et al., 2011). While the potential evolutionary mechanisms underlying local adaptation driven by these genes remain unknown for redband trout, five of these outlier regions contained SNPs that were also significantly associated with environmental variation in a previous landscape genomics study (Micheletti, Matala, et al., 2018). These candidate regions were significantly associated with environmental drivers such as precipitation, temperature and migratory distance. However, many of the genes implicated in local adaptation in the current study were not shared with those identified in previous studies of local adaptation in redband trout (e.g., Chen & Narum, 2021). Lack of overlap in candidate adaptive loci between studies could result from a number of factors. In some cases, lack of overlap probably results from differences in the numbers and types of genetic markers used, along with the inclusion of different populations across studies. Most previous studies used a much smaller number of genetic loci compared to our study, and therefore surveyed a much smaller proportion of the genome. For example, the landscape genetics study by Micheletti, Matala, et al. (2018) used 24,526 SNPs generated by RADseq, whereas our analyses used millions of SNPs from lcWGR. Furthermore, outlier detection is strongly influenced by the composition of populations included in the study, because even if some populations are shared between studies, the strongest signal of selection could be driven by populations that are not shared. Additionally, different experimental designs and analytical approaches are expected to identify different loci (e.g., Whitlock & Lotterhos, 2015). For example, common garden studies identifying genetic associations with specific thermal tolerance phenotypes are more likely to identify SNPs that are variable within populations than outlier analyses, which are more likely to identify SNPs that vary between, but not within, populations (Chen & Narum, 2021). Overall, the relatively large numbers of candidate genes identified in our study and other studies of local adaptation in redband trout indicate that the genomic architecture of local adaptation across heterogeneous landscapes in this species is polygenic and complex.
4.1. Susceptibility to climate change
Evidence for temperature‐driven local adaptation for redband trout raises concerns regarding the potential for this subspecies to adapt to a warming climate without human‐assisted gene flow (e.g., Chen et al., 2022). Although ENM indicates that habitats in about half our study populations would remain viable in a low‐emission future climate change scenario, genetic offset analyses indicate that all our study populations would require substantial evolution to adapt to the new habitats in that scenario. Furthermore, ENM predicts the loss of a large proportion of redband trout habitat across the region under this low‐emission scenario. Under a high‐emission scenario, ENM shows an even greater reduction in viable redband trout habitat, including all our study populations. As in the low‐emission scenario, genetic offset analyses indicate that all our study populations would require substantial evolution to adapt to new habitats in the high‐emission scenario. Elevation and climate were strongly correlated across our study region, and ENM predicted that high‐elevation areas would act as climate refugia or a climate shield (Isaak et al., 2018, 2015), with the exception of the high‐elevation desert habitats found in southern Idaho. These results raise questions as to whether redband trout currently living in desert habitats would have the physiological capabilities or evolutionary capacity to adapt as the climate changes, a finding consistent with a previous study (Chen, Farrell, Matala, Hoffman, et al., 2018).
5. CONCLUSIONS
Here we present the most comprehensive study thus far on local adaptation in redband trout, utilizing 11 populations across a wide habitat range and millions of SNPs generated using lcWGR. We found evidence for polygenic local adaptation that is primarily associated with diurnal temperature variation and isothermality, and driven by two populations in the northern extreme of the subspecies range that have not been included in previous studies. We also found evidence for polygenic local adaptation driven by differences between desert and montane habitat types on a smaller geographical scale within a river drainage. Genetic offset analyses predicted substantial genetic changes would be required for all populations to adapt to future climates, indicating that local adaptation is prevalent across the habitat range for redband trout. For some populations, such as high‐elevation desert populations, adaptation will probably be impossible, as ENM indicates these populations are probably living at the extreme edge of the habitat range in relatively isolated systems.
AUTHOR CONTRIBUTIONS
This study was designed by SRN, KRA, TS and ZC, with contributions from CCC, PAH and LPW. The preparation of DNA libraries for sequencing was led and performed by MWF and DDN, with coordination assistance from SRN, CCC, PAH and LPW. Bioinformatic analyses were performed by KRA, TS and JPE, with consultation from SRN. The writing of the paper was led by KRA, with substantial contributions from TS and SRN. All authors reviewed the manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
BENEFIT‐SHARING STATEMENT
Benefits from this study include the sharing of these data on NCBI.
Supporting information
Table S1
Figure S1
ACKNOWLEDGEMENTS
Tissue samples were collected and made available for this study by biologists with Idaho Department of Fish & Game and the Columbia River Inter‐Tribal Fish Commission. Laboratory assistance was provided by Lauren Jurczak, Anne Veillet, Megan Moore and Stephanie Harmon. Bioinformatic assistance was provided by Stuart Willis. This study was made possible by the NSF Idaho EPSCoR Program and by the National Science Foundation under award no. OIA‐1757324, and was supported in part by NIH COBRE Phase III grant P30GM103324.
Andrews, K. R. , Seaborn, T. , Egan, J. P. , Fagnan, M. W. , New, D. D. , Chen, Z. , Hohenlohe, P. A. , Waits, L. P. , Caudill, C. C. , & Narum, S. R. (2023). Whole genome resequencing identifies local adaptation associated with environmental variation for redband trout. Molecular Ecology, 32, 800–818. 10.1111/mec.16810
Handling Editor: Shotaro Hirase
DATA AVAILABILITY STATEMENT
Raw low‐coverage whole genome resequencing data are available on the NCBI Short Read Archive (Bioproject accession no. PRJNA900579). Sample metadata (sampling locations, dates, sex, environmental data) are available in Tables S1 and S9. Individual genotypes for candidate markers from the GTseq panel are available in Table S1.
REFERENCES
- Aguirre‐Liguori, J. A. , Ramirez‐Barahona, S. , & Gaut, B. S. (2021). The evolutionary genomics of species' responses to climate change. Nature Ecology & Evolution, 5, 1350–1360. 10.1038/s41559-021-01526-9 [DOI] [PubMed] [Google Scholar]
- Alshwairikh, Y. A. , Kroeze, S. L. , Olsson, J. , Stephens‐Cardenas, S. A. , Swain, W. L. , Waits, L. P. , Horn, R. L. , Narum, S. R. , & Seaborn, T. (2021). Influence of environmental conditions at spawning sites and migration routes on adaptive variation and population connectivity in Chinook salmon. Ecology and Evolution, 11, 16890–16908. 10.1002/ece3.8324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data [Online] . http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Barson, N. J. , Aykanat, T. , Hindar, K. , Baranski, M. , Bolstad, G. H. , Fiske, P. , Jacq, C. , Jensen, A. J. , Johnston, S. E. , Karlsson, S. , Kent, M. , Moen, T. , Niemelä, E. , Nome, T. , Næsje, T. F. , Orell, P. , Romakkaniemi, A. , Sægrov, H. , Urdal, K. , … Primmer, C. R. (2015). Sex‐dependent dominance at a single locus maintains variation in age at maturity in salmon. Nature, 528, 405. 10.1038/nature16062 [DOI] [PubMed] [Google Scholar]
- Bear, E. A. , McMahon, T. E. , & Zale, A. V. (2007). Comparative thermal requirements of westslope cutthroat trout and rainbow trout: Implications for species interactions and development of thermal protection standards. Transactions of the American Fisheries Society, 136, 1113–1121. 10.1577/t06-072.1 [DOI] [Google Scholar]
- Belghit, I. , Skiba‐Cassy, S. , Geurden, I. , Dias, K. , Surget, A. , Kaushik, S. , Panserat, S. , & Seiliez, I. (2014). Dietary methionine availability affects the main factors involved in muscle protein turnover in rainbow trout (Oncorhynchus mykiss). British Journal of Nutrition, 112, 493–503. 10.1017/s0007114514001226 [DOI] [PubMed] [Google Scholar]
- Bromage, N. , Jones, J. , Randall, C. , Thrush, M. , Davies, B. , Springate, J. , Duston, J. , & Barker, G. (1992). Broodstock management, fecundity, egg quality and the timing of egg‐production in the rainbow trout (Oncorhynchus mykiss). Aquaculture, 100, 141–166. 10.1016/0044-8486(92)90355-o [DOI] [Google Scholar]
- Brunelli, J. P. , Wertzler, K. J. , Sundin, K. , & Thorgaard, G. H. (2008). Y‐specific sequences and polymorphisms in rainbow trout and Chinook salmon. Genome, 51, 739–748. 10.1139/g08-060 [DOI] [PubMed] [Google Scholar]
- Buffalo, V. , & Coop, G. (2020). Estimating the genome‐wide contribution of selection to temporal allele frequency change. Proceedings of the National Academy of Sciences of the United States of America, 117, 20672–20680. 10.1073/pnas.1919039117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell, B. (2016). BBMap short read aligner. University of California. http://sourceforge.net/projects/bbmap [Google Scholar]
- Campbell, N. R. , Harmon, S. A. , & Narum, S. R. (2015). Genotyping‐in‐Thousands by sequencing (GT‐seq): A cost effective SNP genotyping method based on custom amplicon sequencing. Molecular Ecology Resources, 15, 855–867. 10.1111/1755-0998.12357 [DOI] [PubMed] [Google Scholar]
- Chen, Z. Q. , Farrell, A. P. , Matala, A. , Hoffman, N. , & Narum, S. R. (2018). Physiological and genomic signatures of evolutionary thermal adaptation in redband trout from extreme climates. Evolutionary Applications, 11, 1686–1699. 10.1111/eva.12672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. Q. , Farrell, A. P. , Matala, A. , & Narum, S. R. (2018). Mechanisms of thermal adaptation and evolutionary potential of conspecific populations to changing environments. Molecular Ecology, 27, 659–674. 10.1111/mec.14475 [DOI] [PubMed] [Google Scholar]
- Chen, Z. Q. , Grossfurthner, L. , Loxterman, J. L. , Masingale, J. , Richardson, B. A. , Seaborn, T. , Smith, B. , Waits, L. P. , & Narum, S. R. (2022). Applying genomics in assisted migration under climate change: Framework, empirical applications, and case studies. Evolutionary Applications, 15, 3–21. 10.1111/eva.13335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. Q. , & Narum, S. R. (2021). Whole genome resequencing reveals genomic regions associated with thermal adaptation in redband trout. Molecular Ecology, 30, 162–174. 10.1111/mec.15717 [DOI] [PubMed] [Google Scholar]
- Colinet, H. , Sinclair, B. J. , Vernon, P. , & Renault, D. (2015). Insects in fluctuating thermal environments. Annual Review of Entomology, 60, 123–140. 10.1146/annurev-ento-010814-021017 [DOI] [PubMed] [Google Scholar]
- Collins, E. E. , Hargrove, J. S. , Delomas, T. A. , & Narum, S. R. (2020). Distribution of genetic variation underlying adult migration timing in steelhead of the Columbia River basin. Ecology and Evolution, 10, 9486–9502. 10.1002/ece3.6641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett, M. V. , & Seeb, J. E. (2014). Detection and mapping of QTL for temperature tolerance and body size in Chinook salmon (Oncorhynchus tshawytscha) using genotyping by sequencing. Evolutionary Applications, 7, 480–492. 10.1111/eva.12147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels, P. , Magnusson, M. , Lundin, S. , & Kaller, M. (2016). MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 32, 3047–3048. 10.1093/bioinformatics/btw354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fariello, M. I. , Boitard, S. , Mercier, S. , Robelin, D. , Faraut, T. , Arnould, C. , Recoquillay, J. , Bouchez, O. , Salin, G. , Dehais, P. , Gourichon, D. , Leroux, S. , Pitel, F. , Leterrier, C. , & SanCristobal, M. (2017). Accounting for linkage disequilibrium in genome scans for selection without individual genotypes: The local score approach. Molecular Ecology, 26, 3700–3714. 10.1111/mec.14141 [DOI] [PubMed] [Google Scholar]
- Faust, G. G. , & Hall, I. M. (2014). SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics, 30, 2503–2505. 10.1093/bioinformatics/btu314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fick, S. E. , & Hijmans, R. J. (2017). WorldClim 2: New 1‐km spatial resolution climate surfaces for global land areas. International Journal of Climatology, 37, 4302–4315. 10.1002/joc.5086 [DOI] [Google Scholar]
- Fitzpatrick, M. J. , & Edelsparre, A. H. (2018). The genomics of climate change. Science, 359, 29–30. 10.1126/science.aar3920 [DOI] [PubMed] [Google Scholar]
- Forester, B. R. , Lasky, J. R. , Wagner, H. H. , & Urban, D. L. (2018). Comparing methods for detecting multilocus adaptation with multivariate genotype‐environment associations. Molecular Ecology, 27, 2215–2233. 10.1111/mec.14584 [DOI] [PubMed] [Google Scholar]
- Fuentes‐Pardo, A. P. , & Ruzzante, D. E. (2017). Whole‐genome sequencing approaches for conservation biology: Advantages, limitations, and practical recommendations. Molecular Ecology, 26, 5369–5406. 10.1111/mec.14264 [DOI] [PubMed] [Google Scholar]
- Gain, C. , & Francois, O. (2021). LEA 3: Factor models in population genetics and ecological genomics with R. Molecular Ecology Resources, 21, 2738–2748. 10.1111/1755-0998.13366 [DOI] [PubMed] [Google Scholar]
- Gao, G. T. , Magadan, S. , Waldbieser, G. C. , Youngblood, R. C. , Wheeler, P. A. , Scheffler, B. E. , Thorgaard, G. H. , & Palti, Y. (2021). A long reads‐based de‐novo assembly of the genome of the Arlee homozygous line reveals chromosomal rearrangements in rainbow trout. G3: Genes, Genomes, Genetics, 11, jkab052. 10.1093/g3journal/jkab052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvin, M. R. , Thorgaard, G. H. , & Narum, S. R. (2015). Differential expression of genes that control respiration contribute to thermal adaptation in Redband trout (Oncorhynchus mykiss gairdneri). Genome Biology and Evolution, 7, 1404–1414. 10.1093/gbe/evv078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith, M. I. , Fisher, S. , Waterman, R. , & Johnson, S. L. (2003). Saltatory control of isometric growth in the zebrafish caudal fin is disrupted in long fin and rapunzel mutants. Developmental Biology, 259, 303–317. 10.1016/s0012-1606(03)00186-6 [DOI] [PubMed] [Google Scholar]
- Grummer, J. A. , Beheregaray, L. B. , Bernatchez, L. , Hand, B. K. , Luikart, G. , Narum, S. R. , & Taylor, E. B. (2019). Aquatic lanscape genomics and environmental effects on genetic variation. Trends in Ecology & Evolution, 34, 641–654. 10.1016/j.tree.2019.02.013 [DOI] [PubMed] [Google Scholar]
- Hahlbeck, N. , Tinniswood, W. R. , Sloat, M. R. , Ortega, J. D. , Wyatt, M. A. , Hereford, M. E. , Ramirez, B. S. , Crook, D. A. , Anlauf‐Dunn, K. J. , & Armstrong, J. B. (2021). Contribution of warm habitat to cold‐water fisheries. Conservation Biology, 36, e13857. 10.1111/cobi.13857 [DOI] [PubMed] [Google Scholar]
- Hess, J. E. , Zendt, J. S. , Matala, A. R. , & Narum, S. R. (2016). Genetic basis of adult migration timing in anadromous steelhead discovered through multivariate association testing. Proceedings of the Royal Society B: Biological Sciences, 283, 20153064. 10.1098/rspb.2015.3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijmans, R. (2022). terra: Spatial Data analysis . R package version 1.5‐9. https://rspatial.org/terra/
- Hijmans, R. J. , Phillips, S. , Leathwick, J. , & Elith, J. (2021). dismo: Species distribution modeling . R package version 1.3‐5. https://CRAN.R‐project.org/package=dismo
- Holecek, D. E. , Scarnecchia, D. L. , & Miller, S. E. (2012). Smoltification in an impounded, adfluvial redband trout population upstream from an impassable dam: Does it persist? Transactions of the American Fisheries Society, 141, 68–75. 10.1080/00028487.2011.651550 [DOI] [Google Scholar]
- Husson, F. , Josse, J. , Le, S. , & Mazet, J. (2020). FactoMineR: Multivariate exploratory data analysis and data mining . R package version 2.4. https://CRAN.R‐project.org/package=FactoMineR
- Ignatiadis, N. , Klaus, B. , Zaugg, J. B. , & Huber, W. (2016). Data‐driven hypothesis weighting increases detection power in genome‐scale multiple testing. Nature Methods, 13, 577–580. 10.1038/nmeth.3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaak, D. J. , Luce, C. H. , Horan, D. L. , Chandler, G. L. , Wollrab, S. P. , & Nagel, D. E. (2018). Global warming of salmon and trout rivers in the Northwestern US: Road to ruin or path through purgatory? Transactions of the American Fisheries Society, 147, 566–587. 10.1002/tafs.10059 [DOI] [Google Scholar]
- Isaak, D. J. , Wenger, S. J. , Peterson, E. E. , Ver Hoef, J. M. , Hostetler, S. , Luce, C. H. , Dunam, J. B. , Kershner, J. , Roper, B. B. , Nagel, D. , Horan, D. , Chan‐dler, G. , Parkes, S. , & Wollrab, S. (2011). NorWeST: An interagency stream temperature database and model for the Northwest United States. US Fish and Wildlife Service, Great Northern and North Pacific Landscape Conservation Cooperative grants. https://www.fs.usda.gov/rm/boise/AWAE/projects/NorWeST.html [Google Scholar]
- Isaak, D. J. , Wollrab, S. , Horan, D. , & Chandler, G. (2012). Climate change effects on stream and river temperatures across the northwest US from 1980‐2009 and implications for salmonid fishes. Climatic Change, 113, 499–524. 10.1007/s10584-011-0326-z [DOI] [Google Scholar]
- Isaak, D. J. , Young, M. K. , Nagel, D. E. , Horan, D. L. , & Groce, M. C. (2015). The cold‐water climate shield: Delineating refugia for preserving salmonid fishes through the 21st century. Global Change Biology, 21, 2540–2553. 10.1111/gcb.12879 [DOI] [PubMed] [Google Scholar]
- Jackson, T. R. , Ferguson, M. M. , Danzmann, R. G. , Fishback, A. G. , Ihssen, P. E. , O'Connell, M. , & Crease, T. J. (1998). Identification of two QTL influencing upper temperature tolerance in three rainbow trout (Oncorhynchus mykiss) half‐sib families. Heredity, 80, 143–151. 10.1038/sj.hdy.6882890 [DOI] [Google Scholar]
- Karlsson, E. K. , Baranowska, I. , Wade, C. M. , Salmon Hillbertz, N. H. C. , Zody, M. C. , Anderson, N. , Biagi, T. M. , Patterson, N. , Pielberg, G. R. , Kulbokas, E. J., III , Comstock, K. E. , Keller, E. T. , Mesirov, J. P. , von Euler, H. , Kämpe, O. , Hedhammar, Å. , Lander, E. S. , Andersson, G. , Andersson, L. , & Lindblad‐Toh, K. (2007). Efficient mapping of mendelian traits in dogs through genome‐wide association. Nature Genetics, 39, 1321–1328. 10.1038/ng.2007.10 [DOI] [PubMed] [Google Scholar]
- Kindt, R. , & Coe, R. (2005). Tree diversity analysis: A manual and software for common statistical methods for ecological and biodiversity studies .
- Koch, I. J. , & Narum, S. R. (2020). Validation and association of candidate markers for adult migration timing and fitness in Chinook Salmon. Evolutionary Applications, 13, 2316–2332. 10.1111/eva.13026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler, R. , Pandey, R. V. , & Schlotterer, C. (2011). PoPoolation2: Identifying differentiation between populations using sequencing of pooled DNA samples (Pool‐Seq). Bioinformatics, 27, 3435–3436. 10.1093/bioinformatics/btr589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozfkay, C. C. , Campbell, M. R. , Meyer, K. A. , & Schill, D. J. (2011). Influences of habitat and hybridization on the genetic structure of Redband trout in the Upper Snake River bBasin, Idaho. Transactions of the American Fisheries Society, 140, 282–295. 10.1080/00028487.2011.567837 [DOI] [Google Scholar]
- Lawrence, M. , Huber, W. , Pages, H. , Aboyoun, P. , Carlson, M. , Gentleman, R. , Morgan, M. T. , & Carey, V. J. (2013). Software for computing and annotating genomic ranges. PLoS Computational Biology, 9, e1003118. 10.1371/journal.pcbi.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leder, E. H. , Danzmann, R. G. , & Ferguson, M. M. (2006). The candidate gene, clock, localizes to a strong spawning time quantitative trait locus region in rainbow trout. Journal of Heredity, 97, 74–80. 10.1093/jhered/esj004 [DOI] [PubMed] [Google Scholar]
- Legendre, P. , & Legendre, L. (1998). Numerical ecology. Elsevier. [Google Scholar]
- Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics, 27, 2987–2993. 10.1093/bioinformatics/btr509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM . arXiv:1303.3997v1 [q‐bio.GN].
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , Durbin, R. , & 1000 Genome Project Data Processing Subgroup . (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou, R. N. , Jacobs, A. , Wilder, A. P. , & Therkildsen, N. O. (2021). A beginner's guide to low‐coverage whole genome sequencing for population genomics. Molecular Ecology, 30, 5966–5993. 10.1111/mec.16077 [DOI] [PubMed] [Google Scholar]
- Martins, E. G. , Hinch, S. G. , Cooke, S. J. , & Patterson, D. A. (2012). Climate effects on growth, phenology, and survival of sockeye salmon (Oncorhynchus nerka): A synthesis of the current state of knowledge and future research directions. Reviews in Fish Biology and Fisheries, 22, 887–914. 10.1007/s11160-012-9271-9 [DOI] [Google Scholar]
- Meyer, K. A. , Lamansky, J. A. , & Schill, D. J. (2010). Biotic and abiotic factors related to redband trout occurrence and abundance in desert and montane streams. Western North American Naturalist, 70, 77–91. 10.3398/064.070.0109 [DOI] [Google Scholar]
- Meyer, K. A. , & Schill, D. J. (2021). The gill‐oxygen limitation theory and size at maturity/maximum size relationships for salmonid populations occupying flowing waters. Journal of Fish Biology, 98, 44–49. 10.1111/jfb.14555 [DOI] [PubMed] [Google Scholar]
- Micheletti, S. J. , Hess, J. E. , Zendt, J. S. , & Narum, S. R. (2018). Selection at a genomic region of major effect is responsible for evolution of complex life histories in anadromous steelhead. BMC Evolutionary Biology, 18, 140. 10.1186/s12862-018-1255-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheletti, S. J. , Matala, A. R. , Matala, A. P. , & Narum, S. R. (2018). Landscape features along migratory routes influence adaptive genomic variation in anadromous steelhead (Oncorhynchus mykiss). Molecular Ecology, 27, 128–145. 10.1111/mec.14407 [DOI] [PubMed] [Google Scholar]
- Micheletti, S. J. , & Narum, S. R. (2018). Utility of pooled sequencing for association mapping in nonmodel organisms. Molecular Ecology Resources, 18, 825–837. 10.1111/1755-0998.12784 [DOI] [PubMed] [Google Scholar]
- Muhlfeld, C. C. , Albeke, S. E. , Gunckel, S. L. , Writer, B. J. , Shepard, B. B. , & May, B. E. (2015). Status and conservation of interior Redband trout in the Western United States. North American Journal of Fisheries Management, 35, 31–53. 10.1080/02755947.2014.951807 [DOI] [Google Scholar]
- Muhlfeld, C. C. , McMahon, T. E. , Belcer, D. , & Kershner, J. L. (2009). Spatial and temporal spawning dynamics of native westslope cutthroat trout, Oncorhynchus clarkii lewisi, introduced rainbow trout, Oncorhynchus mykiss, and their hybrids. Canadian Journal of Fisheries and Aquatic Sciences, 66, 1153–1168. 10.1139/f09-073 [DOI] [Google Scholar]
- Muñoz, N. J. , Farrell, A. P. , Heath, J. W. , & Neff, B. D. (2015). Adaptive potential of a Pacific salmon challenged by climate change. Nature Climate Change, 5, 163–166. 10.1038/nclimate2473 [DOI] [Google Scholar]
- Muscarella, R. , Galante, P. J. , Soley‐Guardia, M. , Boria, R. A. , Kass, J. M. , Uriarte, M. , & Anderson, R. P. (2014). ENMeval: An R package for conducting spatially independent evaluations and estimating optimal model complexity for MAXENT ecological niche models. Methods in Ecology and Evolution, 5, 1198–1205. 10.1111/2041-210x.12261 [DOI] [Google Scholar]
- Narum, S. R. , & Campbell, N. R. (2015). Transcriptomic response to heat stress among ecologically divergent populations of redband trout. BMC Genomics, 16, 103. 10.1186/s12864-015-1246-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narum, S. R. , Campbell, N. R. , Kozfkay, C. C. , & Meyer, K. A. (2010). Adaptation of redband trout in desert and montane environments. Molecular Ecology, 19, 4622–4637. 10.1111/j.1365-294X.2010.04839.x [DOI] [PubMed] [Google Scholar]
- Narum, S. R. , Campbell, N. R. , Meyer, K. A. , Miller, M. R. , & Hardy, R. W. (2013). Thermal adaptation and acclimation of ectotherms from differing aquatic climates. Molecular Ecology, 22, 3090–3097. 10.1111/mec.12240 [DOI] [PubMed] [Google Scholar]
- Oksanen, J. , Blanchet, F. G. , Friendly, M. , Kindt, R. , Legendre, P. , McGlinn, D. , Minchin, P. R. , O'Hara, R. B. , Simpson, G. L. , Solymos, P. , Stevens, M. H. H. , Szoecs, E. , & Wagner, H. (2019). vegan: Community ecology package . R package version 2.5‐6. https://CRAN.R‐project.org/package=vegan
- Oligny‐Hebert, H. , Senay, C. , Enders, E. C. , & Boisclair, D. (2015). Effects of diel temperature fluctuation on the standard metabolic rate of juvenile Atlantic salmon (Salmo salar): Influence of acclimation temperature and provenience. Canadian Journal of Fisheries and Aquatic Sciences, 72, 1306–1315. 10.1139/cjfas-2014-0345 [DOI] [Google Scholar]
- Panserat, S. , Hortopan, G. A. , Plagnes‐Juan, E. , Kolditz, C. , Lansard, M. , Skiba‐Cassy, S. , Esquerré, D. , Geurden, I. , Médale, F. , Kaushik, S. , & Corraze, G. (2009). Differential gene expression after total replacement of dietary fish meal and fish oil by plant products in rainbow trout (Oncorhynchus mykiss) liver. Aquaculture, 294, 123–131. 10.1016/j.aquaculture.2009.05.013 [DOI] [Google Scholar]
- Phillips, S. J. , Anderson, R. P. , & Schapire, R. E. (2006). Maximum entropy modeling of species geographic distributions. Ecological Modelling, 190, 231–259. 10.1016/j.ecolmodel.2005.03.026 [DOI] [Google Scholar]
- Phillips, S. J. , & Dudik, M. (2008). Modeling of species distributions with Maxent: New extensions and a comprehensive evaluation. Ecography, 31, 161–175. 10.1111/j.0906-7590.2008.5203.x [DOI] [Google Scholar]
- Portner, H. O. , & Knust, R. (2007). Climate change affects marine fishes through the oxygen limitation of thermal tolerance. Science, 315, 95–97. 10.1126/science.1135471 [DOI] [PubMed] [Google Scholar]
- Prince, D. J. , O'Rourke, S. M. , Thompson, T. Q. , Ali, O. A. , Lyman, H. S. , Saglam, I. K. , Hotaling, T. J. , Spidle, A. P. , & Miller, M. R. (2017). The evolutionary basis of premature migration in Pacific salmon highlights the utility of genomics for informing conservation. Science Advances, 3, e1603198. 10.1126/sciadv.1603198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2021). R: A language and environment for statistical computing. R Foundation for Statistical Computing. https://www.R‐project.org/ [Google Scholar]
- Riahi, K. , van Vuuren, D. P. , Kriegler, E. , Edmonds, J. , O’Neill, B. C. , Fujimori, S. , Bauer, N. , Calvin, K. , Dellink, R. , Fricko, O. , Lutz, W. , Popp, A. , Cuaresma, J. C. , Leimbach, M. , Jiang, L. , Kram, T. , Rao, S. , Emmerling, J. , Ebi, K. , … Tavoni, M. (2017). The shared socioeconomic pathways and their energy, land use, and greenhouse gas emissions implications: An overview. Global Environmental Change‐Human and Policy Dimensions, 42, 153–168. 10.1016/j.gloenvcha.2016.05.009 [DOI] [Google Scholar]
- Schill, D. J. , LaBar, G. W. , Mamer, E. , & Meyer, K. A. (2010). Sex ratio, fecundity, and models predicting length at sexual maturity of Redband trout in Idaho Desert streams. North American Journal of Fisheries Management, 30, 1352–1363. 10.1577/m10-021.1 [DOI] [Google Scholar]
- Schlötterer, C. , Tobler, R. , Kofler, R. , & Nolte, V. (2014). Sequencing pools of individuals‐mining genome‐wide polymorphism data without big funding. Nature Reviews Genetics, 15, 749–763. 10.1038/nrg3803 [DOI] [PubMed] [Google Scholar]
- Schluter, D. (2000). The ecology of adaptive radiation. Oxford University Press. [Google Scholar]
- Seaborn, T. , Griffith, D. , Kliskey, A. , & Caudill, C. C. (2021). Building a bridge between adaptive capacity and adaptive potential to understand responses to environmental change. Global Change Biology, 27, 2656–2668. 10.1111/gcb.15579 [DOI] [PubMed] [Google Scholar]
- Sinclair‐Waters, M. , Odegard, J. , Korsvoll, S. A. , Moen, T. , Lien, S. , Primmer, C. R. , & Barson, N. J. (2020). Beyond large‐effect loci: Large‐scale GWAS reveals a mixed large‐effect and polygenic architecture for age at maturity of Atlantic salmon. Genetics Selection Evolution, 52, 9. 10.1186/s12711-020-0529-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, T. Q. , Bellinger, M. R. , O'Rourke, S. M. , Prince, D. J. , Stevenson, A. E. , Rodrigues, A. T. , Sloat, M. R. , Speller, C. F. , Yang, D. Y. , Butler, V. L. , Banks, M. A. , & Miller, M. R. (2019). Anthropogenic habitat alteration leads to rapid loss of adaptive variation and restoration potential in wild salmon populations. Proceedings of the National Academy of Sciences of the United States of America, 116, 177–186. 10.1073/pnas.1811559115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldvogel, A. M. , Feldmeyer, B. , Rolshausen, G. , Exposito‐Alonso, M. , Rellstab, C. , Kofler, R. , Mock, T. , Schmid, K. , Schmitt, I. , Bataillon, T. , Savolainen, O. , Bergland, A. , Flatt, T. , Guillaume, F. , & Pfenninger, M. (2020). Evolutionary genomics can improve prediction of species' responses to climate change. Evolution Letters, 4, 4–18. 10.1002/evl3.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. N. , Salem, M. , Qi, N. , Kenney, P. B. , Rexroad, C. E., III , & Yao, J. (2011). Molecular characterization of the MuRF genes in rainbow trout: Potential role in muscle degradation. Comparative Biochemistry and Physiology B‐Biochemistry & Molecular Biology, 158, 208–215. 10.1016/j.cbpb.2010.11.010 [DOI] [PubMed] [Google Scholar]
- Waples, R. S. , Ford, M. J. , Nichols, K. , Kardos, M. , Myers, J. , Thompson, T. Q. , Anderson, E. C. , Koch, I. J. , McKinney, G. , Miller, M. R. , Naish, K. , Narum, S. R. , O'Malley, K. G. , Pearse, D. E. , Pess, G. R. , Quinn, T. P. , Seamons, T. R. , Spidle, A. , Warheit, K. I. , & Willis, S. C. (2022). Implications of large‐effect loci for conservation: A review and case study with Pacific Salmon. Journal of Heredity, 113, 121–144. [DOI] [PubMed] [Google Scholar]
- Waters, C. D. , Clemento, A. , Aykanat, T. , Garza, J. C. , Naish, K. A. , Narum, S. , & Primmer, C. R. (2021). Heterogeneous genetic basis of age at maturity in salmonid fishes. Molecular Ecology, 30, 1435–1456. 10.1111/mec.15822 [DOI] [PubMed] [Google Scholar]
- Wenger, S. J. , Isaak, D. J. , Luce, C. H. , Neville, H. M. , Fausch, K. D. , Dunham, J. B. , Dauwalter, D. C. , Young, M. K. , Elsner, M. M. , Rieman, B. E. , Hamlet, A. F. , & Williams, J. E. (2011). Flow regime, temperature, and biotic interactions drive differential declines of trout species under climate change. Proceedings of the National Academy of Sciences of the United States of America, 108, 14175–14180. 10.1073/pnas.1103097108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock, M. C. , & Lotterhos, K. E. (2015). Reliable detection of loci responsible for local adaptation: Inference of a null model through trimming the distribution of FST . American Naturalist, 186, S24–S36. 10.1086/682949 [DOI] [PubMed] [Google Scholar]
- Whitney, C. K. , Hinch, S. G. , & Patterson, D. A. (2014). Population origin and water temperature affect development timing in embryonic sockeye salmon. Transactions of the American Fisheries Society, 143, 1316–1329. 10.1080/00028487.2014.935481 [DOI] [Google Scholar]
- Willis, S. C. , Hess, J. E. , Fryer, J. K. , Whiteaker, J. M. , Brun, C. , Gerstenberger, R. , & Narum, S. R. (2020). Steelhead (Oncorhynchus mykiss) lineages and sexes show variable patterns of association of adult migration timing and age‐at‐maturity traits with two genomic regions. Evolutionary Applications, 13, 2836–2856. 10.1111/eva.13088 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Figure S1
Data Availability Statement
Raw low‐coverage whole genome resequencing data are available on the NCBI Short Read Archive (Bioproject accession no. PRJNA900579). Sample metadata (sampling locations, dates, sex, environmental data) are available in Tables S1 and S9. Individual genotypes for candidate markers from the GTseq panel are available in Table S1.