

Abstract
Background
Clinical trials are needed to study topics relevant to older adults with serious illness. Investigators conducting clinical trials with this population are challenged by how to appropriately define, classify, report, and monitor serious and non-serious adverse events (SAEs/AEs), given that some traditionally reported AEs (pressure ulcers, delirium) and SAEs (death, hospitalization) are common in persons with serious illness, and may be consistent with their goals of care.
Objectives
A multi-stakeholder group convened to establish greater clarity on and new approaches to address this critical issue.
Participants
Thirty-two study investigators, members of regulatory and sponsor agencies, and patient stakeholders took part.
Approach
The group met virtually four times and, using a collaborative approach, conducted a survey, select interviews, and reviewed regulatory guidance to collectively define the problem and identify a new approach.
Results
SAE/AE challenges fell into two areas: (1) definitions and classifications, including (a) implausible relationships, (b) misalignment with patient-centered care goals, and (c) well-known associations, and (2) reporting and monitoring, including (a) limited guidance, (b) inconsistent standards across regulators, and (c) Data Safety Monitoring Board (DSMB) member knowledge gaps. Problems largely reflected practice norms rather than regulatory requirements that already support context-specific and aggregate reporting. Approaches can be improved by adopting principles that better align strategies for addressing adverse events with the type of intervention being tested, favoring routine and aggregate over expedited reporting, and prioritizing how SAE/AEs relate to patient-centered care goals. Reporting plans and decisions should follow an algorithm underpinned by these principles.
Conclusions
Adoption of the proposed approach—and supporting it with education and better alignment with regulatory guidance and procedures—could improve the quality and efficiency of clinical trials’ safety involving older adults with serious illness and other vulnerable populations.
KEY WORDS: serious adverse events, adverse events, serious illness, clinical trials, adverse event reporting
INTRODUCTION
Serious illness is a condition that carries a high risk of mortality and either negatively impacts a person’s daily function or quality of life or excessively strains their caregivers.1,2 Reports and convenings call for clinical trials to address care gaps for the seriously ill.3–8 Safety monitoring of these trials is essential and, since 1998, has been required by the National Institutes of Health (NIH), including serious and non-serious adverse event (SAE/AE) reporting.9 While critical, documented issues associated with SAE/AE reporting10 include confusion regarding reporting standards; onerous or inappropriate reporting11–13; use of expedited rather than aggregate summary reporting14; coordination challenges among investigators, data and safety monitoring boards (DSMBs), and regulatory bodies (Office for Human Research Protections [OHRP], Institutional Review Boards [IRBs], Food and Drug Administration [FDA])15,16; and the downstream threat of premature study termination.17 Investigators conducting trials affecting seriously ill older populations have faced additional challenges. Traditionally reported AEs (pressure ulcers, delirium) and SAEs (death, hospitalization) frequently occur in these populations irrespective of study interventions. Some events may not be viewed as adverse by trial subjects. Further, the variety of interventions, subjects, and settings can complicate appropriate SAE/AE selection.
To address these challenges, we convened a multi-stakeholder group of investigators, regulator and sponsor agency members, and patient stakeholders with experience in clinical trials research across diverse areas. We summarize our efforts to (1) systematically identify and describe current problems with defining and reporting SAEs/AEs in clinical trials involving seriously ill older adults; (2) propose an alternative approach; and (3) delineate next steps.
CONVENING PROCESS, PARTICIPANT SURVEY, AND INTERVIEWS
The convening process, funded by the National Institute on Aging (NIA), was led by a planning group at the Icahn School of Medicine at Mount Sinai’s Claude D. Pepper Older Americans Independence Center (OAIC), the OAIC Coordinating Center at Wake Forest School of Medicine, and the National Palliative Care Research Center. Four hour-long virtual meetings took place in 2020–2021. Thirty-two individuals participated in at least one meeting (mean attendance = 23) (Table 1). The planning group developed and chaired meetings, facilitated discussions, circulated notes, and conducted information collection and synthesis.
Table 1.
Convening Meetings’ Description
| Meeting | Date | # Attended | Topics | Organizations represented |
|---|---|---|---|---|
| Meeting 1: convening process and participant review | 10/23/2020 | 22 |
• Introduction and background on issue • Aims of project • Draft outline of process and planned meetings • Feedback on proposed process and participants • Discussion on topics including the importance of defining the nature of the problem |
• American Geriatrics Society (AGS) • Ariadne Labs* • Brigham and Women’s Hospital* • Duke University School of Medicine • Harvard Medical School • Icahn School of Medicine at Mount Sinai • Indiana University • Marcus Institute for Aging Research* • Massachusetts General Hospital • National Institute on Aging (NIA) • National Palliative Care Research Center (NPCRC) • National Patient Advocate Foundation (NPAF)* • Office for Human Research Protections (OHRP) • Palliative Care Research Cooperative Group (PCRC) • Patient-Centered Outcomes Research Institute (PCORI)* • U.S. Department of Veteran Affairs - Veterans Health Administration - Geriatric Research Education and Clinical Centers (GRECCs) • U.S. Food and Drug Administration • University of Connecticut Center on Aging • University of Pennsylvania Perelman School of Medicine* • Wake Forest School of Medicine *Organization included after meeting 1. |
| Meeting 2: problem identification | 1/20/2021 | 24 |
• Current SAE reporting process • Challenges of current SAE definitions and process for studies on older adults with serious illness: investigator perspectives • Federal agency perspectives and reactions (National Institute on Aging and Office for Human Research Protections) |
|
| Meeting 3: Potential alternative approaches identification | 2/22/2021 | 28 |
• Summary/debrief of last meeting and process changes • U.S. Food and Drug Administration perspectives • Group discussion and brainstorm regarding possible alternatives to current approaches |
|
| Meeting 4: Strategies to advance alternative approaches | 8/25/2021 | 19 |
• Review of feedback on problem definitions • Discussion of proposed new approaches • Building roadmap for advancing new approach • Active dissemination strategies |
Our collaborative included diverse perspectives, expertise, and experiences; was deliberative and iterative; generated open and honest communication, mutual trust, and respect; identified common goals; and shared power and decision-making18. Emerging out of meeting one was a suggestion to conduct a survey (N = 12) and in-depth interviews with a cross section of convening participants (N = 4; 2 investigators, 2 study sponsors) to identify SAE/AE reporting challenges. The planning group conducted interviews and thematically organized information to generate problem areas and illustrative vignettes (Tables 2 and 3), which the entire convening reviewed and refined. They formed the basis for alternative approaches formulated and discussed during subsequent meetings. The present manuscript, led by a subset of convening participants, was circulated to the convening at several points to ensure content agreement and revise as needed.
Table 2.
Problems and Vignettes Regarding Defining and Classifying SAEs/AEs in Clinical Trials Involving Seriously Ill Older Adults
|
Problem 1: Implausible relationships SAE/AE unlikely to be causally related to the study interventions | |
|
a. Reporting falls in nursing home residents as an AE in a study that introduces education modules to improve vaccination information and uptake among nursing home staff. b. Expedited reporting of hospitalizations and deaths as SAEs in a study of an evidence-based team huddle to coordinate care among clinical and non-clinical providers in an outpatient oncology clinic. c. Reporting delirium as an AE in a psychosocial-behavioral study to support patients’ emotional coping strategies transitioning to nursing home care. d. Expedited reporting of death as an SAE of end-of-life patients in a video behavioral intervention study to improve coping skills in caregivers of patients with a terminal illness. e. Expedited reporting of acute lung injury as an SAE even though it is secondary to initial resuscitation and hospital-acquired pneumonia during prolonged mechanical ventilation in a trial of intensive insulin therapy in patients with trauma. | |
|
Problem 2: Misaligned values SAE/AE that could plausibly be causally related to the study intervention, but events are concordant with a study participant’s goals of care | |
|
a. Expedited reporting of death as an SAE after a patient elects to terminate life-sustaining therapy in a study of a video intervention to help patients with serious illness and poor prognosis make an advanced care planning decision. b. Expedited reporting of a breathlessness crisis as an SAE in palliative sedation when consistent with established goals of care. | |
|
Problem 3: Well-known relationships SAEs/AEs that are already known to be associated or caused by the study’s invention mechanisms | |
|
a. Reporting common and normal feelings of distress and grief as AEs in a video behavioral intervention to improve coping skills in caregivers of patients with a terminal illness prognosis. b. Expedited reporting of treatable infection as an AE in a hip replacement intervention. c. Expedited reporting of severe neutropenia as an SAE in a study involving chemotherapy in a population with advanced cancer. |
Table 3.
Problems and Vignettes Regarding Reporting and Monitoring SAEs/AEs in Clinical Trials Involving Seriously Ill Older Adults
|
Problem 1: Limited guidance Limited support to develop appropriate data safety monitoring plans (DSMP) consisting of appropriate defined, classified, and selected SAEs/AEs for a study context | |
|
a. An investigator seeks but cannot find guidance to define appropriate SAEs/AEs in their DSMP. As a result, the investigator includes SAEs/AEs that are commonly reported and suggests reporting of them in expedited timeframes, but these SAEs/AEs are either implausibly related, misaligned, or well known with the study population, setting, or intervention. Later, when a selected SAE/AE occurs in the context of the study, the event is not logically related but the research team nonetheless spends significant time filling out reporting forms and submitting single expedited reports in cases where aggregate reporting would be sufficient. b. A DSMB member receives a notification each time an AE (e.g., fall) occurs in a large multi-sited study where falls are both expected for an older seriously ill population, unrelated to the study intervention, and if reported should be reported in aggregate. The DSMB member feels overwhelmed by the amount of data coming in and would like study investigators to be better informed on reporting criteria but does not know how to better prepare them for DSMB communications. | |
|
Problem 2: Inconsistent standards Different regulatory bodies that oversee research safety use different reporting standards, constructs, and thresholds | |
|
a. OHRP and FDA have a higher reporting threshold compared to DSMB, and each reporting body uses its own reporting timeline. An investigator, fearful of incorrectly reporting, over-reports or confuses reporting requirements, creating large volumes of irrelevant or incorrectly reported data and potentially drowns out safety concerns of interest. b. Some regulatory bodies indicate that “expected events” must be listed in the study brochure or investigational plan at the outset of a study. A death (SAE) occurs in a study that was not previously documented as expected, but the investigator knows that it occurred as part of the study participant’s underlying disease condition. Since it was not previously documented in study materials as unrelated to study mechanisms, the investigator does not know whether it must be reported, to whom, or at what time-point. | |
|
Problem 3: DSMB knowledge gaps DSMB committees are designed to be comprised of topic and biostatistics experts, but there are gaps in their collective knowledge to evaluate a DSMP and set up appropriate monitoring structures and reporting timelines | |
|
a. DSMB requires a study team to conduct expedited individual reporting of all deaths that occur during the study, even though the investigator explains that some deaths will occur given the participants are in a late stage of disease. Repeated expedited reporting requires significant effort on the part of the research team and monitoring bodies, and will not provide the data to determine if there is enhanced safety risks to a treatment group. However, the DSMB composition does not seem to include experts in end-of-life care to support pragmatic reporting frequencies and timelines. b. A health services researcher submits a DSMP for a trial to compare two electronic chart documentation strategies for ease of follow-up usage by front-line administrators. The investigator excludes from reporting patient-level AEs. A DSMB challenges this omission, and ultimately, the investigator adjusts the DSMP to report patient-level clinical AEs, even though they do not believe they can adversely affect patient outcomes through the study mechanisms. |
CURRENT PROBLEMS WITH SAE/AE DEFINING, CLASSIFYING, REPORTING, AND MONITORING
Difficulties Defining and Classifying SAEs/AEs
The first problem area (Table 2) relates to the definition and classification of SAEs/AEs. Three categories are illustrated using vignettes depicting different intervention types, settings, and populations. One category, “implausible relationships,” encompasses reporting of SAEs/AEs when a causal pathway connecting intervention to the adverse event seems exceedingly improbable. 1a and 1b describe organization-level interventions to improve “behind the scenes” adoption of staff-associated interventions or processes like clinical workflows and coordination, which may be unlikely to cause patient-level SAEs/AEs like hospitalizations. In interviews, investigators leading such studies indicated that they have nonetheless been expected to report such events to DSMBs (in an expedited timeframe in the case of SAEs). 1c is a psychosocial-behavioral intervention to support coping in nursing home residents; while some AEs (e.g., increased anxiety19) may be plausibly related to the intervention, this study reported delirium as an AE despite no likely causal relationship. Expedited, single-case reporting is required for SAEs (deaths, life-threatening events, hospitalizations) in novel drug and device studies regulated by the FDA20,21; however, reporting standards are often applied to non-FDA studies, including non-treatment research in which relationships between intervention and event are often implausible. 1d describes a coping intervention for caregivers wherein an SAE of patient death was reported in an expedited fashion even though the patient was not a trial subject. Also in this category are treatment-oriented studies like 1e, in which an SAE (acute lung injury) was likely due to the patient’s resuscitation and hospital-acquired pneumonia rather than the trial intervention (intensive insulin therapy).
The second problem category, labeled “misaligned values,” describes adverse events that may be causally related to the intervention but are driven by and aligned with patients’ goals of care. 2a and 2b highlight studies where SAEs occur, but they align with patients’ choices to cease curative treatment or focus on comfort. Given evidence suggesting that some patients prefer these events over continued disease-directed treatments,22,23 they might be reported on an aggregate (non-expedited) basis. Consistent with this suggestion, 7 of 12 convening survey respondents reported “misalignment between SAE/AE reporting and patient goals” as the most significant problem. One respondent said, “Some people have goals of comfort and are not worried about dying…[or] being hospitalized. Their goals conflict with the [traditional] concept of SAE.”
A third category of problems, “well-known relationships,” refers to reporting SAEs/AEs that are already known to be associated with the intervention or study procedures. 3a demonstrates a psychosocial intervention producing predictable and potentially healthy coping reactions (e.g., expressions of sadness/tearfulness when discussing impending death/loss). However, DSMB reporting was required. While responding to such events as part of a study protocol could be valuable, reporting may only be useful if the event goes beyond a predefined range (e.g., suicidal ideation) or frequency. 3b and 3c describe the problem of requiring expedited reporting of well-known events in clinical treatment studies. Given these are known reactions, reporting in aggregate (rather than expedited, single case) is often sufficient and appropriate while not always practiced.14
Difficulties in Reporting and Monitoring of SAEs/AEs
A second area of concern derived from interviews and convening discussions involves three problem categories around reporting and monitoring of SAEs/AEs (Table 3). The first category is “Limited Guidance” to develop and implement appropriate reporting procedures. Inappropriate SAE/AE reporting could be averted by developing a robust data and safety monitoring plan (DSMP). Unfortunately, many investigators do not have adequate training or guidance to establish such plans, a problem for investigators (1a) and DSMB members (1b).
Noted in interviews and the literature24, the second category of problems, “Inconsistent Standards,” refers to differing vocabulary, constructs, and thresholds of reporting across regulators and monitoring bodies. 2a reveals confusion by an investigator trying to follow guidance offered by the study sponsor, DSMB, OHRP, IRBs, and FDA. The issue of reporting “unexpected” events (2b) also causes confusion.
The third category of problems is “DSMB Knowledge Gaps.” DSMBs are typically appointed by a sponsor to oversee clinical trial safety. Some DSMBs do not have adequate expertise to appropriately define and classify SAEs/AEs for seriously ill older adults. In 3a, a DSMB unfamiliar with the population required expedited reporting of every death irrespective of intervention relatedness or what is expected in the population. 3b illustrates this problem in service delivery studies. While study sponsors ultimately approve DSMPs, regulate reporting, and make study termination decisions, DSMB recommendations inform them. In interviews, investigators described feeling reluctant to advocate for pragmatic and contextually relevant reporting.
The expansion of DSMBs has not kept pace with training needs25. Czaja et al. describe how, in a social behavioral trial, a critical activity was “…educating the DSMB about the nature of the intervention and the characteristics of the target population...”12 Education may be more critical (and lacking) as interventions diversify beyond biomedical treatment studies.
Over-reporting is a downstream consequence of the described problems, and a concern across clinical trials.26 Over-reporting is burdensome and competes for time with other study activities. Regulatory bodies are overwhelmed by the deluge of reports and cannot separate safety signals from noise.10 Once appropriate SAEs/AEs are established, routine reporting on a routine basis—rather than expedited reporting of single events—may be appropriate for most SAEs/AEs, allow for analytic assessment of event rates, and lead to actionable DSMB recommendations.
NEW APPROACH TO IMPROVE SAE/AE DEFINITIONS, REPORTING, AND MONITORING
Review of Existing Regulations to Inform a New Approach
Reviewing select NIH,27 OHRP,28 and FDA21 regulations and policies reveals that they have resulted not from statutory or regulatory requirements, but rather from convention. To the challenge of defining an SAE/AE relevant to study intervention type, only new drug and device trials are regulated using specific definitions, standards, and procedures.21 The large numbers of comparative treatment, behavioral and organizational trials funded by the NIH state that investigators “must propose their own definitions”27 to encourage that SAEs/AEs are appropriate to study context. To the challenge of overuse of expedited over aggregate reporting, guidance for non-serious adverse events is to report them in routine, aggregated fashion, and not report to OHRP or FDA.13 Finally, to the challenges associated with defining expected events (Table 3, vignettes 2b and 3a) investigators are typically instructed to omit from reports events that are “expected,” defined as those known to stem from “research procedures” or the “characteristics of the study population/underlying disease states.” Convening members stressed the value of the expectedness construct. However, “expectedness” is difficult to operationalize because current guidance relies upon the convention that expected events are listed in protocols, study brochures, and materials prior to study commencement. 20,21,29 Further, the complex and varied trajectories of illness in seriously ill older adults make it difficult to pre-specify all possible events.30 Instead, we propose in the next section to subsume expectedness as a criterion of assessing relatedness rather than as a stand-alone determinant.
Proposed Novel SAE/AE Defining and Reporting Approach: Key Principles
Within existing regulations and guidance, there are opportunities to improve the selection, monitoring, and reporting of SAEs/AEs in research involving seriously ill older adults by protecting them without undermining research integrity, effectiveness, meaning, and timeliness.14 This proposed approach is underpinned by key principles, applied in a decision-making tree, to guide defining and selecting SAEs/AEs and reporting events that occur (Fig. 1).
Fig. 1.
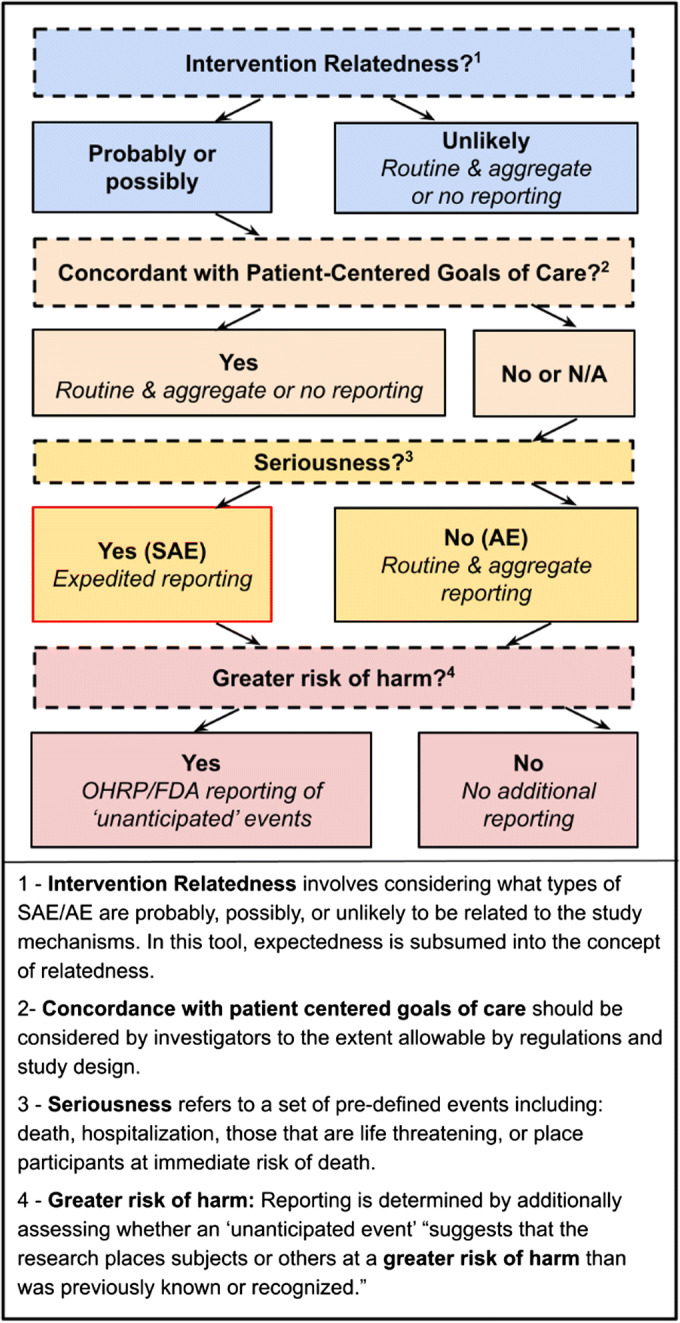
Decision-making tree to determine SAE/AE reporting based on key principles
1.Consider Interventions, Populations, Settings
Interventions, populations, and settings vary in research involving seriously ill older adults. Definitions, frequency, and reporting of SAEs/AEs may be more prescribed in FDA–regulated device and biological studies, but investigators studying behavioral, social, clinical effectiveness, and organizational interventions should be empowered to select appropriate SAEs/AEs and reporting procedures.
2.Use Aggregate and Routine Over Singular and Expedited Reporting for Most SAEs/AEs
Expedited singular reporting should be reserved for SAEs that are plausibly related to study interventions and discordant with patient-centered goals of care. Most reporting should be routine and aggregate, a recommendation consistent with clinical trial advocacy groups.31 Expedited reporting should be made consistent between the DSMB/study sponsor (currently 24–48 h), OHRP (requesting “prompt” reporting), and FDA (currently 7–10 days).
3.Prioritize Intervention Relatedness
SAE/AE relatedness to intervention mechanisms, determined as “unlikely,” “possibly,” and “probably,” should be the first consideration in deciding what to report and if expedited reporting is needed. Relatedness to study mechanisms is typically assessed by considering underlying disease states; temporal relationships between the intervention and the SAE/AE; known associations between SAEs/AEs and intervention mechanisms; alternative causes; and SAE/AE disappearance with cessation of the intervention. Several tools exist to support the unbiased identification of causality.12,20,32 SAEs that are unlikely intervention related should be reported in aggregate to discern previously unknown higher frequencies or patterns in treatment versus control groups.
4.Subsume Expectedness into Relatedness Determination
Current guidance indicates that SAEs/AEs should be reported as unexpected and along expedited timelines for SAEs. Given difficulties in forecasting the full range of SAEs/AEs in seriously ill older adults at a trial’s outset, expectedness should be ascertained when events occur by using the relatedness criterion, “ruling out underlying disease states.”
5.Consider Goal Concordance
The principle of autonomy, defined as self-determined decision-making, should be integrated into safety reporting considerations.33 Patients should be empowered to express decisions drawn from their values and views, informing patient-centered care.34 Safety reporting should strive for consistency between how patients and caregivers define adversity and safety and how it is monitored in trials in which they take part. This entails defining and reporting SAEs/AEs appropriate to patients’ goals, accepting that some events typically counted as SAE/AE may be goal concordant, and establishing reporting expectations accordingly. Development of methods to define, measure, document, and review goal-concordant SAEs/AEs, including regularly engaging patients and caregivers, will foster greater patient goal concordance in clinical trials.
6.Reconsider Seriousness
Regulatory bodies are most concerned with serious events resulting in “death, hospitalization, [or that] are life threatening, or place participants at immediate risk of death from the event as it occurs.” We recommend retaining these definitions but assessing seriousness after relatedness (principle 3); in this order, unrelated SAEs and related non-serious AEs would be reported on a routine basis, while related SAEs of greatest concern would receive expedited attention.
Steps Forward
To advance the proposed approach, we must build consensus beyond the convening members. Presentations and publications will raise to view the importance of the described issues. Factoring patient goals of care into reporting in particular requires forging collective agreement that reporting can prioritize both safety and patient-centered goal concordance. Additionally, we must develop procedures and documentation based on the key principles and the proposed decision tree including (1) methods for documenting investigator decisions (especially where non-expedited reporting is deemed appropriate); (2) DSMP templates that acknowledge intervention, setting, and population; and (3) processes for tabulating, comparing, and reviewing individualized SAE/AE definitions that center patient goal-concordant care. We propose that convening members take part in working groups tasked with developing methodologies and materials. Review and refinement should harmonize the proposed approach with existing regulations, guidelines, and extant tools. Participants from government agencies could lead on integration into existing guidance (e.g., updating NIA Toolkit). Finally, developing and implementing training for DSMB members, sponsors, regulators, and investigators are essential, and will necessitate including additional expertise (e.g., from education specialists). In sum, the proposed plans aim to meaningfully improve the conduct of urgently needed research in the care of seriously ill older adults without sacrificing safety.
Acknowledgements
We would like to acknowledge the full convening group for their significant collective contribution to the development of the ideas presented in this manuscript, and in particular Abby Archer who provided administrative support for the convening. Individuals participating in the convening contributed personal opinions and were not expressing the views of their respective agencies and institutions. The convening and resulting manuscript was supported by an administrative supplement from the National Institutes on Aging U24AG059624 (Coordinating Center of the Claude D. Pepper Older Americans Independence Centers; PI: Kitzman). Additional support was provided by National Institutes on Aging P30AG028741 (Enhancing Independence in Elders with Serious Illness; PIs: Siu, Morrison) and the National Palliative Care Research Center (NPCRC). Some of the ideas included in this manuscript were presented at the American Academy of Hospice and Palliative Medicine 2022 State of the Science on February 12, 2022.
Declarations
Conflict of Interest
The authors declare that they do not have a conflict of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kelley AS. Defining ‘serious illness’. J Palliat Med. 2014;17(9). doi: 10.1089/jpm.2014.0164 [DOI] [PubMed]
- 2.Kelley AS, Covinsky KE, Gorges RJ, et al. Identifying older adults with serious illness: a critical step toward improving the value of health care. Health Serv Res. 2017;52(1):113–131. doi: 10.1111/1475-6773.12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Committee on Approaching Death: Addressing Key End of Life Issues, Institute of Medicine. Dying in America: Improving Quality and Honoring Individual Preferences Near the End of Life. Washington DC: National Academies Press; 2015. [PubMed]
- 4.National Academies of Sciences E. Families Caring for an Aging America; 2016. doi:10.17226/23606. [PubMed]
- 5.National Academies of Sciences E. Policy and Research Needs to Maximize Independence and Support Community Living: Workshop Summary; 2016. doi:10.17226/21893 [PubMed]
- 6.Institute of Medicine. Integrating Health Care and Social Services for People with Serious Illness: Proceedings of a Workshop. www.nationalacademies.org/hmd/Reports/2019/integrating-healthcare-and-social-services-for-people-with-serious-illness-proceedings.aspx, accessed 11/16/2021. [PubMed]
- 7.Institute of Medicine. Cognitive Aging: Progress in Understanding and Opportunities for Action; 2015. doi:10.17226/21693. [DOI] [PubMed]
- 8.NIH State-of-the-Science Conference Statement on improving end-of-life care. NIH Consens State Sci Statements. 2004;21(3):1-26. [PubMed]
- 9.National Institutes of Health. NIH Policy for Data and Safety Monitoring. 1998. https://grants.nih.gov/grants/guide/notice-files/not98-084, accessed 11/2/2021, accessed 11/2/2021.
- 10.Califf R. Clinical trials bureaucracy: unintended consequences of well-intentioned policy. Clin Trials. 2006;3:496–502. doi: 10.1177/1740774506073173. [DOI] [PubMed] [Google Scholar]
- 11.Levit L, et al. Streamlining adverse events reporting in oncology: An American Society of Clinical Oncology Research Statement. J Clin Oncol. 2018;36(6). doi: 10.1200/JCO.2017.75.8193 [DOI] [PubMed]
- 12.George G, et al. Improving attribution of adverse events in oncology clinical trials. Cancer Treat Rev. 2019;76. doi: 10.1016/j.ctrv.2019.04.004 [DOI] [PubMed]
- 13.Cook D, et al. Serious adverse events in academic critical care research. Can Med Assoc J. 2008;178(9). doi: 10.1503/cmaj.071366 [DOI] [PMC free article] [PubMed]
- 14.Reith C, et al. Randomized clinical trials — removing unnecessary obstacles. NEJM. 369;11: 1061-1065. doi: 10.1056/NEJMsb1300760 [DOI] [PubMed]
- 15.Czaja S, et al. Data and safety monitoring in social behavioral intervention trials: the REACH II experience. Clin Trials J. 2006;3(2). doi: 10.1191/1740774506cn136oa [DOI] [PMC free article] [PubMed]
- 16.Vose, et al. Addressing Administrative and Regulatory Burden in Cancer Clinical Trials: Summary of a Stakeholder Survey and Workshop Hosted by the American Society of Clinical Oncology and the Association of American Cancer Institutes. J Clin Onc. 2016; 34(31). doi: 10.1200/JCO.2016.69.6781 [DOI] [PubMed]
- 17.Halpern S, Temel J, Courtright K. Dealing with death as an outcome in supportive care clinical trials. JAMA Intern Med. 2021;181(7). doi: 10.1001/jamainternmed.2021.1816 [DOI] [PubMed]
- 18.D’Amour D, et al. The conceptual basis for interprofessional collaboration: core concepts and theoretical frameworks. J Interprof Care. 2005;19(Suppl 1):116–31. doi: 10.1080/13561820500082529. [DOI] [PubMed] [Google Scholar]
- 19.Peterson A, et al. The need for expanded monitoring of adverse events in behavioral health clinical trials. Cont Clin Trials. 2012;34(1):152–4. doi: 10.1016/j.cct.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 20.Nebeker J, Barach P, Samore M. Clarifying adverse drug events: a clinician's guide to terminology, documentation, and reporting. Ann Intern Med. 2004;140(10):795–801. doi: 10.7326/0003-4819-140-10-200405180-00009. [DOI] [PubMed] [Google Scholar]
- 21.Federal Drug Administration. 21 CFR 312.32(a). https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr = 312.32, accessed 11/4/2021.
- 22.Higginson IJ, et al. Project PRISMA. Priorities for treatment, care and information if faced with serious illness: a comparative population-based survey in seven European countries. Palliat Med. 2014;28(2):101–10. doi: 10.1177/0269216313488989. [DOI] [PubMed] [Google Scholar]
- 23.Teno JM, Fisher ES, Hamel MB, et al. Medical Care Inconsistent with Patients' Treatment Goals: Association with 1-Year Medicare Resource Use and Survival. J Am Ger Soc. 2002;50:496–500. doi: 10.1046/j.1532-5415.2002.50116.x. [DOI] [PubMed] [Google Scholar]
- 24.Infectious Diseases Society of America. Grinding to a Halt: The effects of the increasing regulatory burden on research and quality improvement efforts. CID;2009:49. [DOI] [PubMed]
- 25.DeMets D, Ellenberg S. Data Monitoring Committees — Expect the Unexpected. NEJM. 2016. doi: 10.1056/NEJMra1510066 [DOI] [PubMed]
- 26.Eapen Z, et al. Rescuing clinical trials in the United States and beyond: A call for action. Am Heart J. 2013;165:837–47. doi: 10.1016/j.ahj.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 27.NIA Guidance on Clinical Trials. http://www.nia.nih.gov/research/grants-funding/nia-guidance-clinical-trial, accessed 11/4/2021.
- 28.Reviewing and Reporting Unanticipated Problems Involving Risks to Subjects or Others and Adverse Events: OHRP Guidance 2007. http://www.hhs.gov/ohrp/regulations-and-policy/guidance/reviewing-unanticipated-problems/index.html, accessed 9/23/2021.
- 29.Serious Adverse Event/Unanticipated Problem Decision Tree for Principal Investigators. https://www.nia.nih.gov/sites/default/files/2020-12/AE-SAE-Process-Chart_Dec2020.pdf, Accessed 9/23/2021.
- 30.Morrison RS, et al. The Hard Task of Improving the Quality of Care at the End of Life. 2000. JAMA;160. doi: 10.1001/archinte.160.6.743 [DOI] [PubMed]
- 31.CTTI Recommendations: Data Monitoring Committees. https://ctti-clinicaltrials.org/wp-content/uploads/2021/06/CTTI_DMC_Recs.pdf, accessed 9/23/2021.
- 32.Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239–45. doi: 10.1038/clpt.1981.154. [DOI] [PubMed] [Google Scholar]
- 33.Childress J. The Place of Autonomy in Bioethics. 1990. The Hastings Center Report. 20(1). [PubMed]
- 34.Stange K, et al. Defining and measuring the patient-centered medical home. J Gen Intern Med. 2010;25(6). doi: 10.1007/s11606-010-1291-3. [DOI] [PMC free article] [PubMed]