

Abstract
Uveal melanoma (UM) is the most common primary intraocular malignant tumor type in adults. Even after the treatment of the ocular tumor, the prognosis of patients with metastasis remains poor. Hence, an urgent unmet need exists to identify novel approaches to treat advanced UM. Previous studies have revealed G subunit alpha Q and alpha 11 (GNAQ/11) mutations in more than 85% of patients with UM, thus indicating the importance of GNAQ and downstream signaling pathways in UM occurrence. Here, we demonstrate that microRNA (miR)-181a-5p, a small non-coding RNA, effectively inhibited the viability, proliferation, and colony formation but induced apoptosis of UM cells. Furthermore, silencing GNAQ or AKT3 mimicked the anti-UM effects of miR-181a-5p, whereas overexpression of GNAQ or AKT3 rescued the anti-UM effects induced by miR-181a-5p. In addition, miR-181a-5p had a stronger effect in decreasing the viability of GNAQ mutant than GNAQ wild-type cells. Moreover, miR-181a-5p suppressed the total expression and phosphorylation of members of the ERK and PI3K/AKT/mTOR signaling pathways. Importantly, miR-181a-5p potently inhibited the growth of UM xenografts in nude mice. MiR-181a-5p also decreased the expression of Ki67, GNAQ, and AKT3, and induced the expression of cleaved-caspase3 in UM tumors. These results suggest that miR-181a-5p inhibits UM development by targeting GNAQ and AKT3.
Keywords: Uveal melanoma, miR-181a-5p, GNAQ, AKT3
Introduction
Uveal melanoma (UM), the most common primary intraocular malignant tumor in adults, arises from the transformation of melanocytes in the uveal tract [1,2]. Even if the primary tumor is successfully treated with radiation or surgery, as many as 50% of patients succumb to metastatic disease [3,4]. Metastatic UM responds poorly to clinically available therapies, and no curative therapies exist [5-8]. Hence, an urgent unmet need exists to identify novel approaches to treat advanced UM.
New insights into the molecular biology of UM have highlighted frequent somatic mutations in genes encoding the guanine nucleotide-binding proteins G subunit alpha Q and alpha 11 (GNAQ/11), which are present in more than 85% of UM cases. GNAQ/11 mutations are early initiating events found in all stages of UM [9,10]; most of these mutations occur within the GTPase catalytic domain and lead to constitutive activation of downstream pathways of G-protein coupled receptor signaling, including the RAF/MEK/ERK, phosphatidylinositol 3-kinase (PI3K)/AKT/mechanistic target of rapamycin (mTOR), and Rho/Rac/YAP pathways [3]. However, GNAQ/11 inhibitors have not been found to significantly affect survival, possibly because of the redundancy of downstream signaling pathways [11,12]. Therefore, combinational therapies that co-target multiple pathways have been proposed as an attractive therapeutic approach in UM.
Recent findings have suggested that microRNAs (miRNAs) may serve as promising new therapeutic agents in oncology [13]. MiRNAs regulate post-transcriptional gene expression via canonical base pairing of the 5’ end region miRNA seed sequence, which is complementary to the 3’ untranslated region (UTR) of the target messenger RNA [14]. Therapeutic modulation of a single miRNA can simultaneously affect many functional pathways within a cell, with no or limited toxicity [15]. Human miR-181a-5p is involved in important cell functions and has been found to be downregulated in many tumors [14,16]. In particular, the expression of miR-181a-5p is downregulated in both primary UM and high-risk UM but is absent in metastatic UM [17,18]. Although the contribution of miR-181a-5p to UM has not yet been clarified [19], mounting evidence indicates that miR-181a-5p exhibits anti-neoplastic effects by promoting cancer cell apoptosis, preventing tumor invasion, and enhancing drug sensitivity [20-22]. In addition, previous studies have shown that GNAQ and AKT3 are directly regulated by miR-181a-5p [23,24], thus suggesting that miR-181a-5p may contribute to the suppression of GNAQ expression and aberrant signaling pathways activated by GNAQ mutations in UM.
In this study, we sought to investigate the anti-UM effects of miR-181a-5p and its roles in modulating the aberrant GNAQ protein and downstream signaling pathways.
Materials and methods
Cell culture
OMM2.5 (GNAQ mutant) cells, a kind gift from Dr. Bruce R. Ksander, Harvard University, were cultured in RPMI 1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Wisent, Canada), 1% penicillin and streptomycin (Wisent, Canada), 1% non-essential amino acids (Wisent, Canada), and 0.05 mM 2-mercaptoethanol (Gibco, Canada) [25]. UM001 (GNAQ mutant) cells, obtained from Dr. Solange Landreville, Université Laval, were cultured in RPMI 1640 (Wisent, Canada), supplemented with 10% FBS, 1% non-essential amino acids, 1% HEPES (Gibco, Canada), and 0.5% penicillin and streptomycin [26]. The GNAQ wild-type (WT) cells Mel285 and Mel290, also provided by Dr. Solange Landreville, were cultured in RPMI 1640 (Wisent, Canada) supplemented with 10% FBS, 2 mM L-glutamine (Wisent, Canada), and 1% penicillin and streptomycin [27]. All cells were cultured at 37°C in a humidified incubator with 5% CO2.
Transient transfection
hsa-miR-181a-5p mimic and miRNA negative control (miR-NC) were obtained from Thermo Fisher Scientific, and siR-GNAQ and siR-AKT3 were obtained from Thermo Fisher Scientific. They were dissolved in RNase-free water at a stock concentration of 10 mM and stored in aliquots at -20°C. pcDNA3.1 empty vector was a gift from Oskar Laur (Addgene_128034). pcDNA3.1 constructs encoding full-length human AKT3 was a gift from Morris Birnbaum (Addgene_27293). pcDNA3.1 constructs encoding WT GNAQ (GNAQWT) and Q209L mutant GNAQ (GNAQQ209L) were purchased from cDNA.org (Bloomsburg University). They were dissolved in RNase-free water at a stock concentration of 1 μg/μL and stored at -80°C. For transfection of miRNA, small interfering RNA, and overexpression plasmids, nucleotides were mixed with TransIT X2 Mirus reagent (Mirus Bio LLC, USA) in Opti-MEM medium from Thermo Fisher Scientific, then added to target cells according to the manufacturer’s instructions.
Viability assays
For cell viability, 6,000 OMM2.5, Mel285, or Mel290 cells or 10,000 UM001 cells were plated per well in 96-well black plates with flat clear bottoms from VWR (Mississauga, Canada) and cultured with increasing concentrations of miR-181a-5p for 48 h (OMM2.5 and UM001) or 72 h (Mel285 and Mel290). The viability was assessed with resazurin-based cell viability assays (PrestoBlueTM, Thermo Fisher Scientific). The absorbance was detected after 2 h at a wavelength of 615 nm with a CLARIOstar plate reader (BMG Labtech, France).
Cell proliferation assays
OMM2.5 cells (50,000 cells/well) were plated in 24-well plates from Sarstedt (Montreal, Canada), and 50 μL of [3H]-thymidine from PerkinElmer (Waltham, MA, USA) was added immediately after transfection. After a 48-h incubation, the supernatant was removed, cells were rinsed with cold 5% trichloroacetic acid (TCA) from Sigma-Aldrich (Oakville, Canada) three times, rinsed with cold phosphate-buffered saline (PBS; Wisent, Canada) once, and lysed with 200 μL lysis solution (Triton X-100 0.1% in 0.1 M NaOH). The lysates were transferred to 10 ml of scintillation cocktail (Research Product International, USA), and radioactive signals were detected with a Hidex 300SL scintillation counter as described previously [28].
Apoptosis assays
For apoptosis measurements, OMM2.5 cells were treated with miR-181a-5p for 48 h at the indicated concentrations. Cells were then washed with PBS and collected by trypsinization, then resuspended in staining solution diluted in 1× Annexin V binding buffer (Invitrogen, Burlington, Canada). The staining solution was composed of 0.1 µg of Annexin V-fluorescein-5-isothiocyanate from CedarLane (Burlington, Canada) and 0.1 µg of propidium iodide from Thermo Fisher Scientific. Cells were stained for 15 min in the dark and then analyzed with a BD FACS Canto II flow cytometer with the manufacturer’s software [29]. Staurosporine was used as a positive control.
Colonyformation assays
The colony-forming ability of UM cells was determined as described previously [30]. Briefly, OMM2.5 cells were pre-treated with miR-NC or miR-181a-5p for 48 h, then collected and washed with 1× PBS from Wisent and seeded in miRNA-free RPMI 1640 medium. Three weeks later, the cells were fixed with fixation solution (acetic acid/methanol 1:7) for 20 min and stained with crystal violet solution (0.5% crystal violet and 20% methanol dissolved in distilled H2O) from Sigma-Aldrich (Oakville, Canada) for 2 h. The colonies (> 50 cells) were then counted.
Western blotting
Whole cell lysates were prepared with mammalian protein extraction reagent supplemented with protease inhibitor cocktail and phosphatase inhibitor from Thermo Fisher Scientific. Western blotting was performed as previously described [31]. Briefly, polyacrylamide gel electrophoresis was performed with 12 µg protein, and proteins were transferred to polyvinylidene difluoride membranes from Bio-Rad (Mississauga, Canada). Antibodies to mTOR, phosphorylated-ERK (p-ERK), AKT, p-AKT, and PI3K were obtained from New England BioLabs (Whitby, Canada). Antibodies to GNAQ, p-mTOR, and ERK1/2 were from Abcam (Toronto, Canada). Anti-AKT3, β-actin and GAPDH antibodies were from Proteintech (Rosemont, USA), and the antibody to p-PI3K was from New England Biolabs. HRP labeled anti-rabbit and anti-mouse secondary antibodies were from Santa Cruz Biotechnologies (Dallas, USA). Primary and secondary antibodies were used at the recommended dilutions. Detection was performed with ClarityMax ECL substrate from Bio-Rad and an ImageQuant LAS 500 chemiluminescent imaging system from GE Healthcare (Chicago, USA). Densitometry values were measured in terms of pixel intensity by ImageJ software.
Quantitative real-time PCR
The quantitative real-time PCR procedure was conducted with a LightCycler96 instrument from Roche Life Science (Bâle, Switzerland). Synthesis of cDNAs and quantitative analysis of gene expression were performed as described previously [32]. To test the level of miR-181a-5p in UM cells and tissues, we extracted total miRNAs with a Qiagen miRNeasy Mini kit, and generated cDNAs with a miRCURY RT Kit from Qiagen according to the manufacturer’s instructions. miRCURY LNA SYBR Green Master Mix (Qiagen) was used to perform the PCR. Expression levels of miR-103a-3p were used as an endogenous reference [33]. Primers for miRNAs were from Qiagen (ON, Canada). Primer sequences are as followed: miR-181a-5p, MIMAT0000256, 5’AACAUUCAAC GCUGUCGGUGAGU; miR-103a-3p, MIMAT0000101, 5’AGCAGCAUUGUACAGGGCUAUGA.
Tumor xenograft experiments
All experiments were performed according to protocols approved by the CHU Sainte-Justine Animal Care Committee. Male nude mice (6-8 weeks of age) were purchased from Charles River Laboratories (Senneville, Canada). OMM2.5 cells (4 × 106 cells in 100 μl PBS) were subcutaneously inoculated into the flanks of nude mice. Tumor volumes were measured with calipers every week and calculated with the following formula: a2 × b × 0.5, where a represents the smallest diameter, and b is the diameter perpendicular to a [34]. After 6 weeks of tumor growth, when the tumors reached 50 mm3, mice were randomly divided into three groups receiving miR-NC (2 μg), miR-181a-5p (2 μg), or the same volume of PBS twice intratumorally per week. After the mice were euthanized, tumor tissues were fixed with 4% paraformaldehyde overnight and were transferred into 30% sucrose at 4°C for 12 h, then embedded in Optimal Cutting Temperature (OCT) Compound (Sakura, USA) and cut into 10-μm sections using CryoStar NX50 Cryostat (ThermoFisher Scientific, USA). Sections were colored with hematoxylin-eosin (H&E Staining Kit; Abcam, CA) following the manufacturer’s instructions or labeled with Ki67, cleaved-caspase3, GNAQ or AKT3.
Immunofluorescence staining
Tumor tissues were fixed and cut as described in section Tumor xenograft experiments. After blocking with 1% bovine serum albumin, tumor sections were incubated with primary antibodies overnight at 4°C and then with secondary antibodies for 2 h at room temperature. The antibody to Ki67 was from Abcam (Toronto, Canada). The antibody to cleaved-caspase3 was obtained from New England BioLabs (Toronto, Canada). The antibodies to GNAQ and AKT3 are described in section Western blotting. Goat anti-rabbit AlexaFluor 594 fluorescent antibody was from Thermo Scientific (Thermo Fisher, USA). Primary and secondary antibodies were used at the recommended dilutions. The cell nuclei were stained with DAPI (Sigma, Canada).
Statistical analysis
All in vitro experiments were repeated three times, and data are expressed as mean ± standard deviation. Comparisons between two groups were analyzed with two-tailed Student’s t-test, and differences among multiple groups were analyzed with one-way ANOVA with post hoc comparison through Tukey’s test unless otherwise stated. Data were plotted in GraphPad Prism9 (Software 9.4.1, La Jolla, USA), and P-values < 0.05 were considered statistically significant.
Results
MiR-181a-5p restricts the viability and survival of UM cells
To test the effects of miR-181a-5p on UM cells, we first evaluated the viability of OMM2.5 and UM001 cells after transfection with different concentrations of miR-181a-5p. PrestoBlueTM assays indicated that miR-181a-5p dose-dependently inhibited the cell viability of OMM2.5 and UM001 cells. Because 25 nM was the lowest concentration of miR-181a-5p that effectively decreased the viability of UM cells (37.96% and 44.59%, respectively; Figure 1A), we used 25 nM of miR-181a-5p in the following experiments. Since both OMM2.5 and UM001 cells are GNAQ mutants, OMM2.5 cells were used in most experiments in this study. After transfection, the miR-181a-5p levels in UM cells increased dramatically (6833.3- and 5504.5-fold, respectively; Figure S1). In agreement with the viability assay results, miR-181a-5p significantly decreased the proliferation of OMM2.5 cells (38.86%; Figure 1B). We next evaluated the capability of miR-181a-5p to induce apoptosis. MiR-181a-5p displayed remarkable apoptosis-inducing ability in OMM2.5 cells, as determined with Annexin V/propidium iodide dual staining assays (2.64-fold; Figure 1C). Meanwhile, OMM2.5 cells transfected with miR-181a-5p exhibited marked retardation in clonogenicity (91.49%; Figure 1D). Collectively, these data revealed that miR-181a-5p effectively inhibits the viability and survival of UM cells in vitro.
Figure 1.

MiR-181a-5p decreases cell viability and survival of UM cells. A. OMM2.5 and UM001 cells were transfected with 25 nM of miR-181a-5p or scrambled miRNA negative control (miR-NC) for 48 h. Cell viability was measured with PrestoBlueTM assays, and values are presented as a percentage of control (CTL). *P < 0.05, ***P < 0.001 vs. miR-NC. B. Cell proliferation of miR-181a-5p transfected OMM2.5 cells, assessed according to [3H]-thymidine DNA incorporation. Relative proliferation rates are presented as a percentage of CTL. ***P < 0.001 vs. miR-NC. C. Representative figures of flow cytometry results (left) of OMM2.5 cells stained with PI and Annexin V after treatment with miR-181a or miR-NC. The percentage of apoptotic cells is quantified (right). ***P < 0.001 vs. miR-NC. D. After treatment with miR-181a-5p for 48 h, OMM2.5 cells were seeded into 6-well plates and incubated for 21 days. Representative images of colony formation of OMM2.5 cells treated with miR-181a-5p or miR-NC for 48h (left). Colonies were counted after 21 days (shown as percentage of CTL) from 3 independent experiments in triplicate (right). Values are means ± standard error of the mean. ***P < 0.001 vs. miR-NC.
MiR-181a-5p exhibits stronger anti-UM effects in cell lines with GNAQ mutation
We also tested the influence of miR-181a-5p on the viability of the GNAQWT cell lines Mel285 and Mel290. Unlike the effects of miR-181a-5p in OMM2.5 and UM001 cells, in which 25 nM of miR-181a-5p dramatically decreased cell viability by 50% (Figure 2A), 50 nM miR-181a-5p decreased the viability of Mel285 and Mel290 cells by only 26% and 17% respectively (Figure 2B). Together, these findings revealed that miR-181a-5p has greater viability suppression activity in GNAQ mutant than WT cells.
Figure 2.
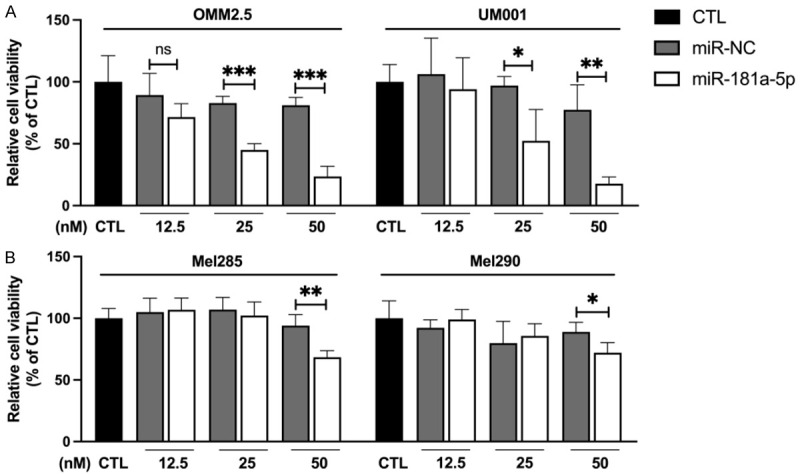
MiR-181a-5p shows stronger anti-viability effects on GNAQ mutant UM cells than GNAQ wild-type UM cells. A. GNAQ mutant UM cells (OMM2.5 and UM001) were transfected with the indicated doses (12.5, 25 or 50 nM) of miR-181a-5p or miR-NC for 48 h. B. GNAQ wild-type UM cells (Mel285 and Mel290) were transfected with the indicated doses (12.5, 25 or 50 nM) of miR-181a-5p or miR-NC for 72 h. Cell viability was measured with PrestoBlueTM assay, and values are presented as a percentage of CTL. *P < 0.05, **P < 0.01, ***P < 0.001 vs. miR-NC.
MiR-181a-5p inhibits the growth of UM cells by targeting GNAQ
After determining that GNAQ mutant cell lines were more sensitive than WT cells to miR-181a-5p, we sought to elucidate the role of GNAQ in the anti-UM effects of miR-181a-5p in UM cells. We first detected the expression of GNAQ after miR-181a-5p transfection because bioinformatics methods have predicted that miR-181a-5p targets GNAQ. Western blotting indicated that miR-181a-5p decreased the protein level of GNAQ in OMM2.5 cells (58.08%; Figures 3A and S2A). The downregulation of GNAQ expression in OMM2.5 cells transfected with siR-GNAQ was confirmed by Western blotting (59.88%; Figure S2B). Downregulation of GNAQ expression in OMM2.5 cells by siR-GNAQ significantly decreased cell viability (22.17%; Figure 3B) and colony formation (73.14%; Figure 3C). In addition, GNAQ silencing inhibited cell proliferation (62.41%; Figure S2C) and promoted apoptosis (1.85-fold; Figure S2D) of OMM2.5 cells. Together, these data indicated an oncogenic role of GNAQ in UM cells. We also observed that the anti-UM effects of miR-181a-5p were markedly blunted by overexpression of GNAQQ209L in both viability (1.53-fold; Figure 3D) and colony formation (1.43-fold; Figure 3E) assays. The overexpression of GNAQ in OMM2.5 cells transfected with GNAQ expression plasmids was confirmed by Western blotting (4.66-fold; Figure S3A). Moreover, GNAQQ209L reversed the effects of miR-181a-5p in proliferation (1.19-fold; Figure S3B). GNAQWT plasmids also increased the viability and proliferation of UM cells beyond those in the control group, because of the overexpression of GNAQ protein; however, GNAQQ209L plasmids had stronger effects. These results supported our hypothesis that the anti-UM effects of miR-181a-5p may function through downregulation of GNAQ expression.
Figure 3.
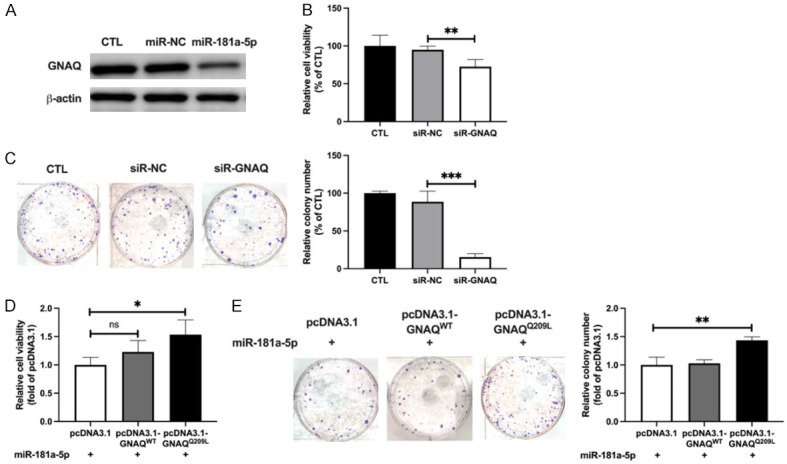
MiR-181a-5p exhibits anti-UM effects through targeting GNAQ. A. Western blotting analysis of GNAQ protein in miRNA transfected OMM2.5 cells after treatment with 25 nM miR-181a-5p for 48 h. B. Cell viability of siR-GNAQ transfected OMM2.5 cells. **P < 0.01 vs. siR-NC. C. Representative images of siR-GNAQ transfected OMM2.5 cells (left) and relative colony numbers (right). ***P < 0.001 vs. siR-NC. D. After co-transfection with miR-181a-5p or GNAQWT or GNAQQ209L expression plasmids for 48 h, OMM2.5 cell viability was measured, and is presented as relative cell viability with respect to that in the miR-181a-5p+pcDNA3.1 group. *P < 0.05 vs. miR-181a-5p+pcDNA3.1. E. Colony formation of co-transfected OMM2.5 cells (left) and relative colony numbers (right). **P < 0.01 vs. miR-181a-5p+pcDNA3.1.
MiR-181a-5p suppresses the growth of UM cells by targeting AKT3
Because AKT3 is highly expressed in UM cells, and miR-181a-5p targets AKT3 directly, we subsequently assessed the role of AKT3 in the anti-UM effects of miR-181a-5p. Western blotting demonstrated that miR-181a-5p decreased the protein level of AKT3 (60.39%; Figures 4A and S4A). The downregulation and overexpression of AKT3 in OMM2.5 cells transfected with siR-AKT3 or AKT3 expression plasmids were confirmed by Western blotting (55.25% and 272%, respectively; Figure S4B). SiR-AKT3 diminished not only the viability (17.9%; Figure 4B) but also the colony formation (57.14%; Figure 4C) of OMM2.5 cells. Additionally, AKT3 silencing inhibited the proliferation (19.81%; Figure S4C) and induced cell apoptosis (86%; Figure S4D) of OMM2.5 cells; therefore, AKT3 silencing mimicked the effects of miR-181a-5p on OMM2.5 cells. Nonetheless, AKT3 overexpression abrogated the anti-UM effect of miR-181a-5p, as evidenced by cell viability assays (2.47-fold; Figure 4D) and colony formation assays (1.89-fold; Figure 4E). The proliferation inhibiting and apoptosis promoting effects of miR-181a-5p on OMM2.5 cells were also diminished by AKT3 overexpression (22% and 36.85%, respectively; Figure S4E, S4F). Together, these data indicated that suppressing the expression of AKT3 may contribute to the anti-UM effect of miR-181a-5p in UM cells.
Figure 4.

Downregulation of AKT3 expression contributes to the anti-UM effect of miR-181a-5p. A. Western blotting analysis of AKT3 protein levels in OMM2.5 cells with or without transfection of miR-181a-5p. B. Cell viability of siR-AKT3 transfected OMM2.5, measured and presented as a percentage of CTL. *P < 0.05 vs. siR-NC. C. Colony formation of siR-AKT3 transfected OMM2.5 cells (left) and relative colony numbers (right). **P < 0.01 vs. siR-NC. D. OMM2.5 cells were co-transfected with miR-181a-5p and AKT3 expression plasmid for 48 h, cell viability was measured, and relative values with respect to those in the miR-181a-5p+pcDNA3.1 group are presented. **P < 0.01 vs. miR-181a-5p+pcDNA3.1. E. Representative images of colony formation of co-transfected OMM2.5 cells (left), and relative colony numbers (right). **P < 0.01 vs. miR-181a-5p+pcDNA3.1.
MiR-181a-5p suppresses the activity of GNAQ downstream signaling pathways
Although miR-181a-5p was found to exert anti-UM effects by targeting GNAQ and AKT3, how miR-181a-5p affected the GNAQ downstream signaling pathway was unknown. Previous studies have shown that miR-181a-5p targets the 3’-UTR of extracellular signal-regulated kinase 2 (Erk2) messenger RNA [35], and miR-181a-5p inhibition promotes phosphorylation in multiple cell lines [36,37]. In addition, a potential miR-181a-5p binding site exists in the 3’-UTR of the NRAS proto-oncogene, encoding a protein that activates the PI3K/AKT/mTOR pathway [38]. In the present study, we found that total ERK and phosphorylated ERK levels were significantly decreased by miR-181a-5p (21.81% and 22.42%, respectively; Figure 5A). Western blotting also indicated that miR-181a-5p decreased the protein expression of total AKT and mTOR (36.21% and 18.56%, respectively; Figure 5C, 5D), and the phosphorylation of these two proteins (21.82% and 31.34%, respectively; Figure 5C, 5D), without influencing the levels of total and phosphorylated PI3K (Figure 5B). These data suggested that miR-181a-5p suppresses the activity of GNAQ downstream signaling pathways.
Figure 5.
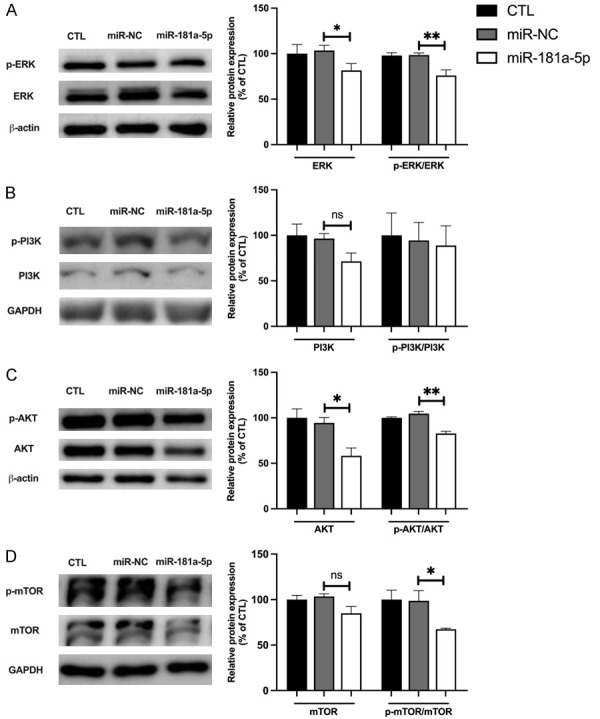
MiR-181a-5p abrogates GNAQ downstream signaling pathways. Representative Western blot of protein levels of phosphorylated ERK (p-ERK) and ERK (A), p-PI3K and PI3K (B), p-AKT and AKT (C), and p-mTOR and mTOR (D) in OMM2.5 cells with or without transfection of miR-181a-5p (left). The quantified phosphorylated protein or total protein levels of each gene are presented as a percentage of CTL. *P < 0.05, **P < 0.01 vs. miR-NC.
MiR-181a-5p suppresses outgrowth of xenografted UM cells in nude mice
To determine the in vivo antineoplastic activity of miR-181a-5p, we tested the effect of this miRNA in a nude mouse xenograft model. As shown in Figure 6A, after 3 weeks of treatment, miR-181a-5p significantly inhibited the growth of xenografts (1.03-fold; Figure 6B) and decreased the tumor size (48.68%; Figure 6C). In addition, H&E staining revealed small nests of necrotic UM cells (Figure 7A). Immunofluorescence staining analysis demonstrated diminished expression of Ki67 and elevated cleaved-caspase3 levels in the tumor tissues of the miR-181a-5p treated mice (95.75% and 8.96-fold, respectively; Figure 7B, 7C, 7F), thus suggesting that miR-181a-5p suppresses cell proliferation and promotes cell apoptosis in UM tumor tissues. We further detected the expression of GNAQ and AKT3 from xenografted tumor issues by immunostaining. miR-181a-5p decreased the levels of GNAQ and AKT3 (68.03% and 67.38%, respectively; Figure 7D-F). These results together indicate that miR-181a-5p has potent anti-UM activity in vivo.
Figure 6.
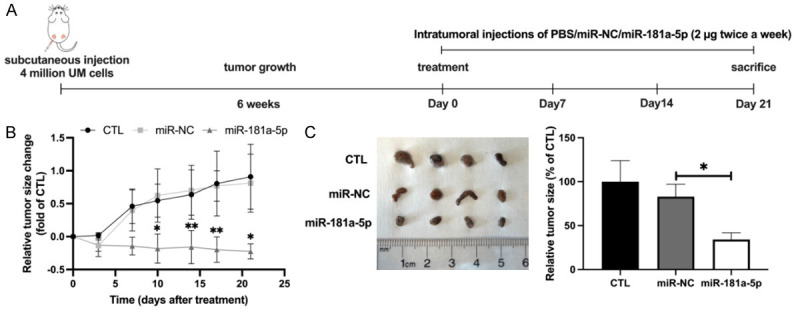
MiR-181a-5p suppresses the growth of subcutaneous UM tumors. A. Schematic of an in vivo nude mouse model of UM and miR-181a-5p administration schedule. B. Tumor growth curves are plotted for the nude mice intratumorally treated with PBS, miR-NC, or miR-181a-5p. C. UM tumors were collected on day 21 after treatment injections. Representative images of UM tumors (left) and relative tumor size (right) for each group. *P < 0.05, **P < 0.01 vs. miR-NC.
Figure 7.

H&E staining of UM tumors and regulation of protein expression in UM xenograft tumors. UM xenografted tumors were injected with miR-NC or miR-181a-5p twice per week for 3 weeks. H&E staining is shown (A), and Ki67 (B), cleaved-caspase3 (C), GNAQ (D) and AKT3 (E) expression was detected. The quantified protein levels are presented as a percentage of CTL (F). *P < 0.05, **P < 0.01, ***P < 0.001 vs. miR-NC.
Discussion
Aberrant GNAQ/11 expression has a key role in UM neoplastic progression. The potential anti-tumor short noncoding RNA miR-181a-5p has shown potent activity against retinoblastoma, breast cancer, and oral squamous cell carcinoma [20,21,39]. In the present study, we demonstrated that miR-181a-5p effectively inhibited UM development targeting GNAQ and AKT3 (Figure 8).
Figure 8.
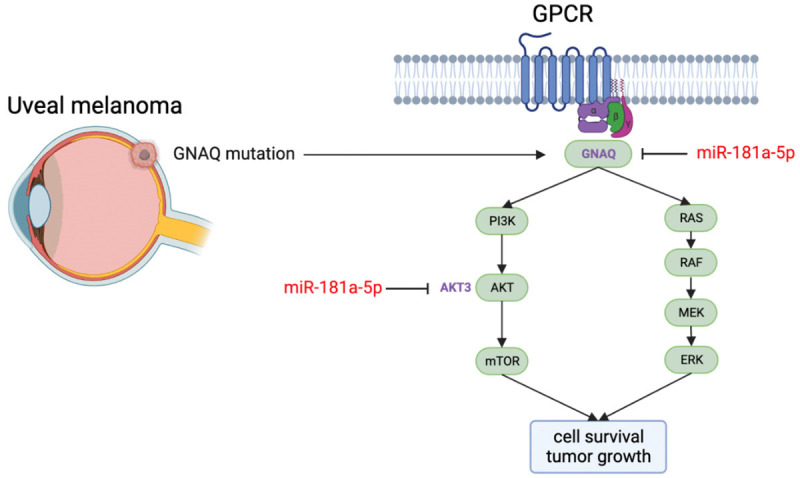
Schematic diagram of the anti-UM effects of miR-181a-5p. GNAQ is an α subunit of G-protein coupling receptor. GNAQ mutation drives uveal melanoma oncogenesis. MiR-181a-5p inhibits UM cell survival and tumor growth by targeting GNAQ and AKT3.
Compared with cutaneous melanoma, UM has a much lower mutational burden [3]. However, most UM carries a GNAQ/11 mutation regardless of tumor stage, thus suggesting that GNAQ/11 mutations are necessary to initiate tumorigenesis [3,23]. Because most UM contain mutations deregulating the GNAQ/11 pathway, drug targeting of this pathway might be effective in most UM. In the present study, an important molecular association between miR-181a-5p and GNAQ was demonstrated. First, GNAQ protein expression in UM was downregulated by overexpression of miR-181a-5p. Second, the downregulation of GNAQ expression by miR-181a-5p strongly suppressed UM cell growth. Third, overexpression of GNAQ reversed the anti-UM effect of miR-181a-5p. Fourth, a previous study has predicted that GNAQ is a direct target of miR-181a-5p [23]. Together, the present findings demonstrated that miR-181a-5p regulates GNAQ expression and functions as a tumor suppressor in UM development.
However, previous studies have reported that GNAQ inhibition does not improve overall survival in patients with UM, although it inhibits UM viability and growth [40]. Several studies have therefore focused on interfering with critical downstream effectors, such as mitogen-activated protein kinase, protein kinase C, PI3K, and AKT signaling [41-43]. Unfortunately, no improvement in survival rate has been observed in patients with UM. One reason for these disappointing results may be that inhibitors of these effectors act far downstream of oncogenic GNAQ and GNA11 and inhibiting only one arm of oncogenic networks is likely to be inefficient because of the redundancy of the downstream signaling pathways. Therefore, combinatory inhibition of GNAQ and downstream signaling pathways has been tested; however, no studies have presented satisfactory results [4]. On the basis of the miRNA database, miR-181a-5p has multiple target genes that play crucial roles in tumor growth apart from GNAQ. AKT3, one target of miR-181a-5p, has critical roles in the (a) transformation of normal melanocytes into melanomas; (b) metastatic spread of melanoma cells; and (c) development of drug resistance. Analysis of the expression and activity of AKT isoforms in melanoma tumors showed significantly greater activity of AKT3 than AKT1 or AKT2. In addition, small interfering RNA mediated knockdown of AKT3 but not AKT1 or AKT2 sensitizes melanoma cells to apoptosis [44]. Importantly, high AKT3 expression has been detected in UM cells and tissues and has been found to be associated with the proliferation and invasion of UM cells [45]. As the critical role of AKT3 in UM development, this explains why targeting GNAQ alone has not achieved a significant anti-UM effect. In the present study, we discovered that AKT3 is also critical for miR-181a-5p enabled viability and colony formation inhibition in UM cells, thus further supporting the anti-UM effects of miR-181a-5p.
MiR-181a-5p has been found to affect GNAQ downstream signaling pathways, such as those involving ERK and PI3K/AKT/mTOR, in addition to AKT3. The ERK and PI3K/AKT/mTOR pathways regulate cell growth and proliferation and are abnormally activated in more than 50% of patients with UM; consequently, they are currently considered actionable targets [3]. In T-cells, gain of miR-181a-5p inhibits multiple tyrosine phosphatases, thereby significantly increasing ERK1/2 phosphorylation [46]. We wondered whether the gain of miR-181a-5p might enhance levels of ERK1/2 phosphorylation through a similar mechanism. Unexpectedly, we observed a decrease in phospho-ERK1/2 after miR-181a-5p mimic transfection. Therefore, miR-181a-5p was considered unlikely to use the same mechanism in UM cells as in T-cells by targeting multiple phosphatases. However, researchers have recently demonstrated that miR-181a-5p is a key regulator of cell growth through negative modulation of mitogen-activated protein kinase/ERK signaling [35]. In this study, miR-181a-5p decreased the expression of total ERK and phosphorylated ERK, in agreement with findings from previous studies. Moreover, our research indicated that miR-181a-5p inhibited the PI3K/AKT/mTOR signaling pathway. Although miR-181a-5p did not decrease PI3K expression and phosphorylation of PI3K, total AKT and mTOR expression and phosphorylation were significantly suppressed by miR-181a-5p, a finding that may be explained by miR-181a-5p targeting AKT3.
Previous studies have identified GNAQ/11 mutation in more than 85% of patients with UM and reported that these mutations arise early in the development of the disease, thus indicating the importance of GNAQ in UM occurrence. In this study, we observed a more profound function of miR-181a-5p in decreasing the viability in GNAQ mutant cells than in GNAQWT cells, thus indicating that miR-181a-5p may be more efficient in treating UM patients with GNAQ mutation. Nonetheless, miR-181a-5p also decreased the viability of GNAQWT cell lines, thus indicating that miR-181a-5p also has the potential to treat UM without GNAQ mutation. This phenomenon may be explained by the muti-targeting feature of miR-181a-5p, given that miR-181-5p was found to target not only GNAQ but also molecules in GNAQ downstream signaling pathways, such as AKT signaling.
A previous study has indicated that UM cells that grow slowly are high-risk UM cells [47]. We observed that the OMM2.5 UM cells took 6 weeks to grow to 50 mm3, indicating quite a slow growth rate with respect to that of other cells such as OMM1, which took 3 weeks to grow to 50 mm3 [34,48]. Our in vivo study showed that miR-181a-5p dramatically delayed the xenografted UM tumor growth; thus, miR-181a-5p is efficient in high-risk UM treatment. In addition, we found small nests of neoplastic melanocytes from the H&E staining, in agreement with findings from a previous study indicating that nodules of tumor cells are distributed as septa by laminin [49]. After treatment with miR-181a-5p, we observed some balloon cell formations, owing to the degeneration of melanosomes and the resultant progressive vacuolization [50], thus indicating the in vivo anti-UM effect of miR-181a-5p. Moreover, miR-181a-5p decreased proliferation, increased apoptosis in tumor tissues, and decreased the expression of GNAQ and AKT3, in agreement with the in vitro results.
Conclusion
This study provides the first demonstration that miR-181a-5p is a potential tumor-suppressing miRNA in UM. MiR-181a-5p exerts anti-UM effects by targeting GNAQ and AKT3. Therefore, miR-181a-5p may serve as a useful therapeutic application for miRNA-based UM therapy.
Acknowledgements
This work was supported by operating grants from the Fonds Dre Christine Corriveau de recherche en oncologie oculaire/Université de Montréal (P.H.) and the Canadian Institutes of Health Research (#426236; P.H.). The procurement of UM cells was possible thanks to the Uveal Melanoma Biobank (S.L.), which is financially supported by the Vision Health Research Network (VHRN, a thematic network supported by the Fonds de recherche du Québec-Santé (FRQS)). This work was also supported by the Merit Scholarship from Faculty of Medicine/Université de Montréal, Doctoral Excellence Scholarship from CHU Sainte-Justine and Recruitment Scholarship from VHRN (R.W.). We kindly thank Mrs. Khadija Cherif, Mrs. Claudia Gilbert, Mr. Carl Fortin, and Mrs. Carmen Gagnon for technical support.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Li Y, Shi J, Yang J, Ge S, Zhang J, Jia R, Fan X. Uveal melanoma: progress in molecular biology and therapeutics. Ther Adv Med Oncol. 2020;12:1758835920965852. doi: 10.1177/1758835920965852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chattopadhyay C, Kim DW, Gombos DS, Oba J, Qin Y, Williams MD, Esmaeli B, Grimm EA, Wargo JA, Woodman SE, Patel SP. Uveal melanoma: from diagnosis to treatment and the science in between. Cancer. 2016;122:2299–2312. doi: 10.1002/cncr.29727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smit KN, Jager MJ, de Klein A, Kiliҫ E. Uveal melanoma: towards a molecular understanding. Prog Retin Eye Res. 2020;75:100800. doi: 10.1016/j.preteyeres.2019.100800. [DOI] [PubMed] [Google Scholar]
- 4.Mallone F, Sacchetti M, Lambiase A, Moramarco A. Molecular insights and emerging strategies for treatment of metastatic uveal melanoma. Cancers (Basel) 2020;12:2761. doi: 10.3390/cancers12102761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang J, Manson DK, Marr BP, Carvajal RD. Treatment of uveal melanoma: where are we now? Ther Adv Med Oncol. 2018;10:1758834018757175. doi: 10.1177/1758834018757175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rantala ES, Hernberg M, Kivelä TT. Overall survival after treatment for metastatic uveal melanoma: a systematic review and meta-analysis. Melanoma Res. 2019;29:561–568. doi: 10.1097/CMR.0000000000000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai KK, Bollin KB, Patel SP. Obstacles to improving outcomes in the treatment of uveal melanoma. Cancer. 2018;124:2693–2703. doi: 10.1002/cncr.31284. [DOI] [PubMed] [Google Scholar]
- 8.Park JJ, Diefenbach RJ, Joshua AM, Kefford RF, Carlino MS, Rizos H. Oncogenic signaling in uveal melanoma. Pigment Cell Melanoma Res. 2018;31:661–672. doi: 10.1111/pcmr.12708. [DOI] [PubMed] [Google Scholar]
- 9.Pandiani C, Béranger GE, Leclerc J, Ballotti R, Bertolotto C. Focus on cutaneous and uveal melanoma specificities. Genes Dev. 2017;31:724–743. doi: 10.1101/gad.296962.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Onken MD, Worley LA, Long MD, Duan S, Council ML, Bowcock AM, Harbour JW. Oncogenic mutations in GNAQ occur early in uveal melanoma. Invest Ophthalmol Vis Sci. 2008;49:5230–5234. doi: 10.1167/iovs.08-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Croce M, Ferrini S, Pfeffer U, Gangemi R. Targeted therapy of uveal melanoma: recent failures and new perspectives. Cancers (Basel) 2019;11:846. doi: 10.3390/cancers11060846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chua V, Lapadula D, Randolph C, Benovic JL, Wedegaertner PB, Aplin AE. Dysregulated GPCR signaling and therapeutic options in uveal melanoma. Mol Cancer Res. 2017;15:501–506. doi: 10.1158/1541-7786.MCR-17-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tutar L, Tutar E, Özgür A, Tutar Y. Therapeutic targeting of microRNAs in cancer: future perspectives. Drug Dev Res. 2015;76:382–388. doi: 10.1002/ddr.21273. [DOI] [PubMed] [Google Scholar]
- 14.Braicu C, Gulei D, Raduly L, Harangus A, Rusu A, Berindan-Neagoe I. Altered expression of miR-181 affects cell fate and targets drug resistance-related mechanisms. Mol Aspects Med. 2019;70:90–105. doi: 10.1016/j.mam.2019.10.007. [DOI] [PubMed] [Google Scholar]
- 15.Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, Chang TC, Vivekanandan P, Torbenson M, Clark KR, Mendell JR, Mendell JT. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rezaei T, Amini M, Hashemi ZS, Mansoori B, Rezaei S, Karami H, Mosafer J, Mokhtarzadeh A, Baradaran B. microRNA-181 serves as a dual-role regulator in the development of human cancers. Free Radic Biol Med. 2020;152:432–454. doi: 10.1016/j.freeradbiomed.2019.12.043. [DOI] [PubMed] [Google Scholar]
- 17.Smit KN, Chang J, Derks K, Vaarwater J, Brands T, Verdijk RM, Wiemer EAC, Mensink HW, Pothof J, de Klein A, Kilic E. Aberrant MicroRNA expression and its implications for uveal melanoma metastasis. Cancers (Basel) 2019;11:815. doi: 10.3390/cancers11060815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Radhakrishnan A, Badhrinarayanan N, Biswas J, Krishnakumar S. Analysis of chromosomal aberration (1, 3, and 8) and association of microRNAs in uveal melanoma. Mol Vis. 2009;15:2146–2154. [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang L, He X, Li F, Pan H, Huang X, Wen X, Zhang H, Li B, Ge S, Xu X, Jia R, Fan X. The miR-181 family promotes cell cycle by targeting CTDSPL, a phosphatase-like tumor suppressor in uveal melanoma. J Exp Clin Cancer Res. 2018;37:15. doi: 10.1186/s13046-018-0679-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tabatabaei SN, Derbali RM, Yang C, Superstein R, Hamel P, Chain JL, Hardy P. Co-delivery of miR-181a and melphalan by lipid nanoparticles for treatment of seeded retinoblastoma. J Control Release. 2019;298:177–185. doi: 10.1016/j.jconrel.2019.02.014. [DOI] [PubMed] [Google Scholar]
- 21.Gu M, Wang L, Yang C, Li X, Jia C, Croteau S, Ruan X, Hardy P. Micro-RNA-181a suppresses progestin-promoted breast cancer cell growth. Maturitas. 2018;114:60–66. doi: 10.1016/j.maturitas.2018.06.004. [DOI] [PubMed] [Google Scholar]
- 22.Su SF, Chang YW, Andreu-Vieyra C, Fang JY, Yang Z, Han B, Lee AS, Liang G. miR-30d, miR-181a and miR-199a-5p cooperatively suppress the endoplasmic reticulum chaperone and signaling regulator GRP78 in cancer. Oncogene. 2013;32:4694–4701. doi: 10.1038/onc.2012.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yin P, Xue Y, Wang T, Zhong D, Li G. The therapeutic targets of fingolimod (FTY720) are involved in pathological processes in the frontal cortex of Alzheimer’s disease patients: a network pharmacology study. Front Aging Neurosci. 2021;13:609679. doi: 10.3389/fnagi.2021.609679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu Z, Luo T, Pang T, Du Z, Yin X, Cui H, Fang G, Xue X. MALAT1 promotes gastric adenocarcinoma through the MALAT1/miR-181a-5p/AKT3 axis. Open Biol. 2019;9:190095. doi: 10.1098/rsob.190095. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Chen PW, Murray TG, Uno T, Salgaller ML, Reddy R, Ksander BR. Expression of MAGE genes in ocular melanoma during progression from primary to metastatic disease. Clin Exp Metastasis. 1997;15:509–518. doi: 10.1023/a:1018479011340. [DOI] [PubMed] [Google Scholar]
- 26.Yoshida M, Selvan S, McCue PA, DeAngelis T, Baserga R, Fujii A, Rui H, Mastrangelo MJ, Sato T. Expression of insulin-like growth factor-1 receptor in metastatic uveal melanoma and implications for potential autocrine and paracrine tumor cell growth. Pigment Cell Melanoma Res. 2014;27:297–308. doi: 10.1111/pcmr.12206. [DOI] [PubMed] [Google Scholar]
- 27.White JS, Becker RL, McLean IW, Director-Myska AE, Nath J. Molecular cytogenetic evaluation of 10 uveal melanoma cell lines. Cancer Genet Cytogenet. 2006;168:11–21. doi: 10.1016/j.cancergencyto.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 28.Yang C, Mwaikambo BR, Zhu T, Gagnon C, Lafleur J, Seshadri S, Lachapelle P, Lavoie JC, Chemtob S, Hardy P. Lymphocytic microparticles inhibit angiogenesis by stimulating oxidative stress and negatively regulating VEGF-induced pathways. Am J Physiol Regul Integr Comp Physiol. 2008;294:R467–476. doi: 10.1152/ajpregu.00432.2007. [DOI] [PubMed] [Google Scholar]
- 29.Cai G, Wang Y, Houda T, Yang C, Wang L, Gu M, Mueck A, Croteau S, Ruan X, Hardy P. MicroRNA-181a suppresses norethisterone-promoted tumorigenesis of breast epithelial MCF10A cells through the PGRMC1/EGFR-PI3K/Akt/mTOR signaling pathway. Transl Oncol. 2021;14:101068. doi: 10.1016/j.tranon.2021.101068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 31.Yang C, Xiong W, Qiu Q, Shao Z, Hamel D, Tahiri H, Leclair G, Lachapelle P, Chemtob S, Hardy P. Role of receptor-mediated endocytosis in the antiangiogenic effects of human T lymphoblastic cell-derived microparticles. Am J Physiol Regul Integr Comp Physiol. 2012;302:R941–949. doi: 10.1152/ajpregu.00527.2011. [DOI] [PubMed] [Google Scholar]
- 32.Yang C, Xiong W, Qiu Q, Tahiri H, Superstein R, Carret AS, Sapieha P, Hardy P. Anti-proliferative and anti-tumour effects of lymphocyte-derived microparticles are neither species- nor tumour-type specific. J Extracell Vesicles. 2014:3. doi: 10.3402/jev.v3.23034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ragni E, Colombini A, De Luca P, Libonati F, Viganò M, Perucca Orfei C, Zagra L, de Girolamo L. miR-103a-3p and miR-22-5p are reliable reference genes in extracellular vesicles from cartilage, adipose tissue, and bone marrow cells. Front Bioeng Biotechnol. 2021;9:632440. doi: 10.3389/fbioe.2021.632440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou J, Liu S, Wang Y, Dai W, Zou H, Wang S, Zhang J, Pan J. Salinomycin effectively eliminates cancer stem-like cells and obviates hepatic metastasis in uveal melanoma. Mol Cancer. 2019;18:159. doi: 10.1186/s12943-019-1068-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carrella S, Barbato S, D’Agostino Y, Salierno FG, Manfredi A, Banfi S, Conte I. TGF-β controls miR-181/ERK regulatory network during retinal axon specification and growth. PLoS One. 2015;10:e0144129. doi: 10.1371/journal.pone.0144129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hegarty SV, Sullivan AM, O’Keeffe GW. Inhibition of miR-181a promotes midbrain neuronal growth through a Smad1/5-dependent mechanism: implications for Parkinson’s disease. Neuronal Signal. 2018;2:NS20170181. doi: 10.1042/NS20170181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Q, Sun H, Jiang Y, Ding L, Wu S, Fang T, Yan G, Hu Y. MicroRNA-181a suppresses mouse granulosa cell proliferation by targeting activin receptor IIA. PLoS One. 2013;8:e59667. doi: 10.1371/journal.pone.0059667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ouyang M, Liu G, Xiong C, Rao J. microRNA-181a-5p impedes the proliferation, migration, and invasion of retinoblastoma cells by targeting the NRAS proto-oncogene. Clinics (Sao Paulo) 2022;77:100026. doi: 10.1016/j.clinsp.2022.100026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi L, Cheng Z, Zhang J, Li R, Zhao P, Fu Z, You Y. hsa-mir-181a and hsa-mir-181b function as tumor suppressors in human glioma cells. Brain Res. 2008;1236:185–193. doi: 10.1016/j.brainres.2008.07.085. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, He J, Qiu C, Shang Q, Qian G, Fan X, Ge S, Jia R. The oncolytic virus H101 combined with GNAQ siRNA-mediated knockdown reduces uveal melanoma cell viability. J Cell Biochem. 2019;120:5766–5776. doi: 10.1002/jcb.27863. [DOI] [PubMed] [Google Scholar]
- 41.Carvajal RD, Sosman JA, Quevedo JF, Milhem MM, Joshua AM, Kudchadkar RR, Linette GP, Gajewski TF, Lutzky J, Lawson DH, Lao CD, Flynn PJ, Albertini MR, Sato T, Lewis K, Doyle A, Ancell K, Panageas KS, Bluth M, Hedvat C, Erinjeri J, Ambrosini G, Marr B, Abramson DH, Dickson MA, Wolchok JD, Chapman PB, Schwartz GK. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA. 2014;311:2397–2405. doi: 10.1001/jama.2014.6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carvajal RD, Piperno-Neumann S, Kapiteijn E, Chapman PB, Frank S, Joshua AM, Piulats JM, Wolter P, Cocquyt V, Chmielowski B, Evans TRJ, Gastaud L, Linette G, Berking C, Schachter J, Rodrigues MJ, Shoushtari AN, Clemett D, Ghiorghiu D, Mariani G, Spratt S, Lovick S, Barker P, Kilgour E, Lai Z, Schwartz GK, Nathan P. Selumetinib in combination with dacarbazine in patients with metastatic uveal melanoma: a phase III, multicenter, randomized trial (SUMIT) J. Clin. Oncol. 2018;36:1232–1239. doi: 10.1200/JCO.2017.74.1090. [DOI] [PubMed] [Google Scholar]
- 43.Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, Dutertre M, Stern MH. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun. 2016;7:10615. doi: 10.1038/ncomms10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madhunapantula SV, Robertson GP. Targeting protein kinase-b3 (akt3) signaling in melanoma. Expert Opin Ther Targets. 2017;21:273–290. doi: 10.1080/14728222.2017.1279147. [DOI] [PubMed] [Google Scholar]
- 45.Li J, Liu X, Li C, Wang W. miR-224-5p inhibits proliferation, migration, and invasion by targeting PIK3R3/AKT3 in uveal melanoma. J Cell Biochem. 2019;120:12412–12421. doi: 10.1002/jcb.28507. [DOI] [PubMed] [Google Scholar]
- 46.Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, Skare P, Klein LO, Davis MM, Chen CZ. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–161. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 47.Bakhoum MF, Francis JH, Agustinus A, Earlie EM, Di Bona M, Abramson DH, Duran M, Masilionis I, Molina E, Shoushtari AN, Goldbaum MH, Mischel PS, Bakhoum SF, Laughney AM. Loss of polycomb repressive complex 1 activity and chromosomal instability drive uveal melanoma progression. Nat Commun. 2021;12:5402. doi: 10.1038/s41467-021-25529-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y, Liu M, Jin Y, Jiang S, Pan J. In vitro and in vivo anti-uveal melanoma activity of JSL-1, a novel HDAC inhibitor. Cancer Lett. 2017;400:47–60. doi: 10.1016/j.canlet.2017.04.028. [DOI] [PubMed] [Google Scholar]
- 49.Clarijs R, Otte-Höller I, Ruiter DJ, de Waal RM. Presence of a fluid-conducting meshwork in xenografted cutaneous and primary human uveal melanoma. Invest Ophthalmol Vis Sci. 2002;43:912–918. [PubMed] [Google Scholar]
- 50.Hashimoto K, Bale GF. An electron microscopic study of balloon cell nevus. Cancer. 1972;30:530–540. doi: 10.1002/1097-0142(197208)30:2<530::aid-cncr2820300233>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.