

Abstract
Atomically precise nanomaterials with tunable solid-state luminescence attract global interest. In this work, we present a new class of thermally stable isostructural tetranuclear copper nanoclusters (NCs), shortly Cu4@oCBT, Cu4@mCBT and Cu4@ICBT, protected by nearly isomeric carborane thiols: ortho-carborane-9-thiol, meta-carborane-9-thiol and ortho-carborane 12-iodo 9-thiol, respectively. They have a square planar Cu4 core and a butterfly-shaped Cu4S4 staple, which is appended with four respective carboranes. For Cu4@ICBT, strain generated by the bulky iodine substituents on the carboranes makes the Cu4S4 staple flatter in comparison to other clusters. High-resolution electrospray ionization mass spectrometry (HR ESI-MS) and collision energy-dependent fragmentation, along with other spectroscopic and microscopic studies, confirm their molecular structure. Although none of these clusters show any visible luminescence in solution, bright μs-long phosphorescence is observed in their crystalline forms. The Cu4@oCBT and Cu4@mCBT NCs are green emitting with quantum yields (Φ) of 81 and 59%, respectively, whereas Cu4@ICBT is orange emitting with a Φ of 18%. Density functional theory (DFT) calculations reveal the nature of their respective electronic transitions. The green luminescence of Cu4@oCBT and Cu4@mCBT clusters gets shifted to yellow after mechanical grinding, but it is regenerated after exposure to solvent vapour, whereas the orange emission of Cu4@ICBT is not affected by mechanical grinding. Structurally flattened Cu4@ICBT didn't show mechanoresponsive luminescence in contrast to other clusters, having bent Cu4S4 structures. Cu4@oCBT and Cu4@mCBT are thermally stable up to 400 °C. Cu4@oCBT retained green emission even upon heating to 200 °C under ambient conditions, while Cu4@mCBT changed from green to yellow in the same window. This is the first report on structurally flexible carborane thiol appended Cu4 NCs having stimuli-responsive tunable solid-state phosphorescence.
A new class of flexible tetranuclear copper nanoclusters protected by carborane-thiols having stimuli-responsive tunable solid state phosphorescence.
Introduction
Atomically precise metal nanoclusters (NCs) are emerging nanomaterials with atom-specific tunable photoluminescence properties.1–3 Closely spaced molecule-like electronic energy levels associated with interfacial charge transfer from the metallic core to the ligand shell or vice versa are responsible for their photoluminescence properties.4,5 The atomic arrangements of the metallic core and the surrounding ligands define the emission characteristics of NCs from the visible to the NIR region.6–8 Noble metal nanoclusters (NMNCs) of specific nuclearity composed of gold,9 silver10 and their alloys11 exhibit luminescence in the red region with relatively low quantum yields with emission lifetimes ranging from the picosecond to the nanosecond time scale. Expensive precursors, complex synthetic methods, poor stability of NMNCs and their weak luminescence limiting their practical applications have prompted the exploration of more stable clusters.12 Thus, compared to gold and silver, the synthesis of copper NCs from cheaper precursors with greater stability presents a promising alternative.13,14 Copper NCs with nuclearity <15 could find use in different applications including light-emitting diodes (LEDs),15–17 electroluminescence,18 circularly polarized luminescence19 and X-ray radioluminescence,20 principally due to their stronger emission.
Phosphorescence, originating from the charge relaxation of the triplet excited state is of great interest in fundamental research as well as applications.21,22 Generally, different organic luminophores such as carbonyl and thioether functionalized molecules,23–25 fluorene,26 borates,27etc., were reported with bright phosphorescence characteristics. Facile functionalization of organic luminophores using functional groups and counter ions promotes their tunable emission due to efficient intersystem crossing.28,29 On the other hand, metal centred phosphorescence especially from copper NCs is weak due to the absence of d–d transitions of Cu atoms with d10 configuration.30,31 Ligand to metal charge transfer (LMCT) or metal to ligand charge transfer (MLCT) is the primary reason behind their phosphorescence. Copper clusters with bi,32 tri,33 tetra,34 and hexanuclearity35 and higher orders36,37 were reported with LMCT and MLCT characteristics due to facile charge transport between metal atoms and ligands. The surrounding chemical environment also controls the emission characteristics of luminescent materials.38,39 Intermolecular aggregation through favourable interactions such as CH⋯π, π⋯π and hydrogen bonding contributing to enhanced emission is referred to as aggregation-induced emission (AIE) or aggregation-induced emission enhancement (AIEE).40 The restriction of molecular motion in the aggregated state and the opening of the radiative relaxation pathways result in emission enhancement. Different strategies of supramolecular assembly, such as host–guest complexation,41 embedding within a rigid polymer matrix,42 co-crystallization,43etc., have also been involved in improving the emission quantum yield of different luminescent materials. Other external stimuli, especially temperature and pressure, can also tune emission properties.44,45
For copper NCs, surface ligands protect the fragile metallic core and co-determine their photophysical properties. Thiols,46 phosphines,47 carbenes48 and amino acids49 are functional groups that have been reported as capping ligands of copper clusters with luminescence characteristics. Variation due to ligand isomerism was observed in copper clusters too.50 Carboranes (dicarba-closo-dodecaboranes of the general formula C2B10H12), twelve-vertex nearly icosahedral molecules composed of ten boron and two carbon atoms, are a new family of materials with interesting properties.51 Due to their rigid three-dimensional architectures with delocalized electrons over the molecular framework of CH and BH vertices, they strongly shield the unstable metal cores.52 Carboranes have the potential for synthesizing coordination complexes,53 luminescent supramolecular materials,54 biologically active materials55 and hypergolic materials56 due to their rigid and robust three-dimensional architectures, chemical and thermal stability and unique electronic structure. From this perspective, carborane-thiol protected isostructural tetranuclear copper clusters are a new class of functional materials with tunable emission properties.
In this work, we synthesized three carborane-thiol (ortho-carborane-9-thiol, meta-carborane-9-thiol and ortho-carborane 12-iodo 9-thiol labelled Cu4@oCBT, Cu4@mCBT and Cu4@ICBT, respectively) protected Cu4 clusters with high yields and phase purity at room temperature using a ligand exchange induced structural transformation (LEIST) reaction. Various structural, spectroscopic, and thermal analysis studies were conducted to reveal novel phenomena. Cumulatively, this work presents an unexplored class of stimuli-responsive tunable phosphorescent materials.
Results and discussion
Synthesis and crystallization
The copper clusters were synthesized by a LEIST reaction, starting from [Cu18(DPPE)6H16]2+ (DPPE = 1,2-bis(diphenylphosphino) ethane).57 The details of the syntheses are presented in the Experimental section. A similar type of synthetic approach was also used for gold, silver and alloy nanoclusters.58–60 Cu4@oCBT and Cu4@ICBT were obtained as microcrystalline solids with green and orange emissions, respectively. Cu4@mCBT did not precipitate, but formed green-emitting crystals. Fig. S1† shows the photographic images of the synthetic steps. Compared to high-temperature synthetic methods,61 this method gives clusters with high yields under ambient conditions. Single crystals suitable for X-ray diffraction were grown in mixed solvents after 6–8 days of crystallization at room temperature. Cu4@oCBT formed colourless hexagonal crystals in a mixture of acetone and dichloromethane (DCM) (3 : 1, volume ratio). The Cu4@mCBT and Cu4@ICBT NCs formed brick-like and rhombus-shaped crystals, respectively, in DCM : acetone : methanol (3 : 1 : 1) mixtures. Fig. 1a–c show the images of the individual crystals inspected with an optical microscope. The morphology of each crystal and basic elemental composition were further confirmed using SEM imaging and EDS elemental analysis (shown in Fig. S2–S4† for Cu4@oCBT, Cu4@mCBT and Cu4@ICBT, respectively). The EDS elemental maps of Cu4@oCBT and Cu4@mCBT showed the presence of Cu, S, C and B in the crystals, whereas Cu, S, I, C and B were detected in the Cu4@ICBT crystal.
Fig. 1. Optical microscopy images of the crystal of (a) Cu4@oCBT, (b) Cu4@mCBT and (c) Cu4@ICBT (insets show the schematic representations of the morphology). Single crystal structure of (d) Cu4@oCBT, (e) Cu4@mCBT and (f) Cu4@ICBT having a nearly square planar Cu4 core and a butterfly type Cu4S4 staple, protected by four carboranes in a twisted fashion. (g–i) The comparative structural analogy of the Cu4S4 staple reveals the flattening of the Cu4S4 staple for the Cu4@ICBT NC in comparison to other clusters due to three iodine insertions in the structure. Inset shows the wing flapping of the respective butterfly. Color code: orange = copper, yellow = sulfur, pink = boron, violet = iodine, gray = carbon and white = hydrogen.

Single crystal structural details
The molecular structures of the clusters were resolved using single-crystal X-ray diffraction (SC-XRD) analysis. Cu4@oCBT crystallized in a trigonal crystal system with the space group of P3121. The cell volume of the cluster was 3333 Å3 (Table 1). Fig. 1d shows the atomic structure of the cluster consisting of a nearly square planar Cu4 core protected by four ortho-carborane thiol ligands. The structural anatomy (shown in Fig. S5†) reveals that four ‘V-shaped’ Cu–S–Cu motifs surround the Cu4 core. The Cu–Cu bond distance ranges from 2.684 to 2.728 Å, which is shorter than the Cu–Cu van der Waals distance of 2.8 Å, indicating metallic bonding. The opposite Cu–S–Cu bond angles are 76.58° and 78.13°, respectively. The orientation of Cu4S4 motifs resembles a butterfly with an opposite S–S distance of 5.54 Å (Fig. 1g). Four ortho-carborane ligands are connected to the Cu4 core through V-shaped Cu–S–Cu staples with an average Cu–S bond length of 2.16 Å. The anti-orientation of the carborane cages in the opposite corners of the Cu4S4 unit makes the cluster flexible (shown in Fig. S6†). Structurally similar Cu4@mCBT crystallized in a monoclinic crystal system with the space group of P21/c and a cell volume of 4400 Å3 (Table 1). The molecular structure of this cluster is nearly identical to that of Cu4@oCBT (shown in Fig. 1e), having a square planar Cu4 core protected by four meta-carborane thiol ligands in a twisted orientation.
Comparative unit cell parameters of the respective NCs.
| Clusters | Cu4@oCBT | Cu4@mCBT | Cu4@ICBT |
| Crystal system | Trigonal | Monoclinic | Tetragonal |
| Volume (Å3) | 3333.4 | 4400.1 | 6438.0 |
| Space group | P3121 | P21/c | P43212 |
| Density (mg m−3) | 1.428 | 1.442 | 1.375 |
| Z | 3 | 4 | 4 |
Cu4@ICBT crystallized in a tetragonal crystal system with the space group of P43212 and a cell volume of 6438 Å3 (Table 1). In this structure, four carborane ligands are connected to the Cu4 core through not only S atoms but also three iodine atoms (Fig. 1f). The longer Cu–I distances of 3.11–3.31 Å, compared to B–I distances of 2.15–2.25 Å, indicate a weak coordination type of Cu–I interaction. The removal of one iodine atom from the carborane cage is probably due to steric restrictions within the structural framework. As a natural consequence of these competitive interactions, the Cu–Cu bonds in Cu4@ICBT become weak, exhibiting distances ranging from 2.80 to 2.85 Å. Similarly, the opposite S⋯S distance increases to 5.94 Å in comparison to 5.54 Å for Cu4@oCBT and 5.49 Å for Cu4@mCBT (Fig. 1g–i). Iodine substitution on the carborane cage in the position adjacent to the SH group makes the Cu4S4 “butterfly” flatter than in other clusters. The higher cell volume of Cu4@ICBT, along with other structural parameters, suggests structural expansion due to iodine-induced steric congestion. The ORTEP structures of these clusters are presented in Fig. S7–S9,† respectively. The effects of these structural changes on the photophysical properties of this cluster system are discussed later.
The unit cell and molecular packing of Cu4@oCBT (shown in Fig. S10†) reveal six clusters packed on the opposite faces of the cuboid. The longest distance between the two centroids of the Cu4S4 unit is 29.440 Å along the c axis, whereas the distance between the other two pairs of the oppositely packed centroids is 11.434 Å. The extended molecular packing (shown in Fig. 2a and b) reveals an ABC⋯ABC kind of layered packing. Three types of short-contact van der Waals interactions, CH⋯S, BH⋯B(H) and CH⋯B(B), are responsible for the packing of Cu4@oCBT. Careful analysis further reveals four BH⋯B(H), eight S⋯HC and four CH⋯B(B) interactions per cluster, promoting their crystallization. Cu4@mCBT is packed in horizontally stacked layers, where four clusters are packed inside the unit cell in an AB⋯AB packing mode (Fig. S11† and 2c and d). Cu4@ICBT is packed in a type of AB⋯BA packing mode, where five clusters are packed inside the unit cell in an undulated fashion (Fig. S12,† and 2e and f). The extended packing represents horizontally stacked wave-like layers of the clusters. In the case of Cu4@mCBT, one CH⋯HB, two B⋯S, four BH⋯S, two CH⋯S and nine BH⋯B(H) short contact interactions were observed. For Cu4@ICBT, there are eight S⋯C(H), four BH⋯HB and three CH⋯HB interactions leading to intermolecular packing.
Fig. 2. Supramolecular organization of Cu4@oCBT (a and b), Cu4@mCBT (c and d) and Cu4@ICBT (e and f) clusters. (a), (c) and (e) The packing view along the b crystallographic axis and (b), (d) and (f) along the c crystallographic axis. The rectangles correspond to the unit cell. Color code: orange = copper, yellow = sulfur, pink = boron, violet = iodine, gray = carbon and white = hydrogen.

Additional characterization
All three copper clusters were further characterized using high-resolution mass spectrometry (HRMS). The details of instrumentation and preparation of the samples are provided in the ESI.† We didn't observe any characteristic peaks from the as-prepared samples in either positive or negative ion modes, which suggests neutral charge for the clusters. In the presence of formic acid, Cu4@oCBT and Cu4@ICBT ionized with characteristic peaks at m/z = 956.7 (Fig. 3a) and 1334.06 (Fig. 3c), respectively. Proton addition promoted the ionization of these clusters. The molecular composition of Cu4@ICBT with only three iodine atoms as determined by X-ray diffraction analysis was also confirmed by HRMS. In the presence of caesium acetate, Cu4@mCBT shows a peak at m/z = 1088.26 (Fig. 3b), which is the Cs+ attached peak of the cluster. The experimental isotopic distribution of all the clusters matched well with the calculated ones (shown in the insets of Fig. 3a–c for Cu4@oCBT, Cu4@mCBT and Cu4@ICBT, respectively).
Fig. 3. Positive ion-mode ESI-mass spectra of (a) Cu4@oCBT, (b) Cu4@mCBT and (c) Cu4@ICBT. Formic acid and cesium acetate were used for the measurements as ionization enhancers. Insets show the matching of the experimental and theoretical spectra, respectively. Peaks labeled * are due to Cs+ ion impurity. (d) Collision energy dependent fragmentation of the molecular ion peak at m/z 956.35 of Cu4@oCBT shows the losses of different carborane thiol fragments without opening the Cu4 core. (e) Schematic representation showing the respective fragmentations. (f) Collision energy dependent fragmentation of the molecular ion peak at m/z 1334.06 of Cu4@ICBT shows the losses of carborane thiol fragments along with the opening of the Cu4 core. (g) Schematic representation showing the losses of different fragments of Cu4@ICBT. Color code: orange = copper, yellow = sulfur, pink = boron, violet = iodine, gray = carbon and green = hydrogen.

Collision-energy (CE) dependent fragmentation revealed additional insight into their structural details. The molecular ion peak at m/z 956.7 for Cu4@oCBT was selected for fragmentation (Fig. 3d). At CE 50 (in instrumental units), two new peaks observed at m/z 781.20 and 674.26 were assigned to [Cu4S3(C2B10H11)3]H+ and [Cu4S3(C2B10H11)2(B2CH2)]H+, respectively. Further increasing the collision energy up to CE 70 yielded fragments at m/z 565.32 and 390.13, which were assigned to [Cu4S3(C2B10H11)(B2CH2)2]H+ and [Cu4(SB2CH2)2]H+, respectively. Fig. S13† shows both experimental and calculated isotopic distributions of the respective fragments. Fragmentation involves the initial loss of a carborane thiol and subsequent fragmentation of the cage structure of the carborane of selected Cu4@oCBT ions, which illustrates high stability of the tetranuclear copper core of the system, schematically illustrated in Fig. 3e. The fragmentation of the carborane cage structure was also observed for the O9 ligand (shown in Fig. S57b†). Collision energy-dependent fragmentation of Cu4@ICBT (shown in Fig. 3f) shows four fragments at m/z 1099.39, 925.36, 864.73 and 690.70. These peaks are assigned to [Cu3S3I3(C2B10H10)3]5H+, [Cu3S2I3(C2B10H10)2]5H+, [Cu2S2I3(C2B10H10)2]8H+ and [Cu2SI3(C2B10H10)]8H+, respectively. The theoretical isotopic distribution of these fragments matched well with the experimental spectra (shown in Fig. S14†). The scission of Cu–Cu bonds in Cu4@ICBT (shown in Fig. 3g) compared to Cu4@oCBT indicates weaker bonding manifested by longer Cu–Cu bonds (2.80–2.85 Å) in Cu4@ICBT, compared to the shorter Cu–Cu bonds (2.68–2.73 Å) in Cu4@oCBT.
The size of the particles was analysed using TEM studies, shown in Fig. S15–S17† for Cu4@oCBT, Cu4@mCBT and Cu4@ICBT, respectively. We did not observe any cluster particles after 2 min beam exposure, apparently due to the presence of a smaller number of metal atoms, whereas 15 min beam exposure created larger particles (with a particle dimension of 4–5 nm) due to beam-induced aggregation. The lattice spacing of 2.6 nm indicates the (1 0 2) plane of copper sulfide. The EDS elemental analysis of Cu4@oCBT and Cu4@mCBT (in SEM) shows a nearly 1 : 1 atomic ratio of Cu : S. The atomic ratio of Cu : S : I of 6.75 : 4.34 : 4.15 for Cu4@ICBT also indicates the presence of iodine in the cluster. We observed 10–15 μm rhombohedral crystalline particles for Cu4@ICBT due to their fast crystallization (shown in Fig. S18†). The binding of the carborane ligands was confirmed using 11B{1H} and 13C{1H} NMR studies. The comparative 11B{1H} NMR data of each cluster and respective carborane ligand are shown in Fig. S19, S21 and S23† and peak positions and other details are summarized in Table S9–S11,† respectively.
The 11B NMR spectrum of Cu4@oCBT at room temperature shows six peaks at δ 6.54, −1.36, −7.56, −12.63, −14.56 and −15.35 ppm in comparison to the peaks of the O9 ligand at δ 5.01, −1.34, −7.58, −13.26, −14.52, and −15.35 ppm. The appearance of 11B NMR peaks in nearly the same position indicates that the boron spectral region of the O9 ligand is less influenced by binding with the metal core. The chemical shift of the boron connected to sulphur is shifted to δ 6.54 ppm compared to 5.01 ppm for the O9 ligand. The 13C NMR peaks of O9 at δ 53.41 and 47.01 ppm are shifted to 54.95 and 46.44 ppm in Cu4@oCBT (Fig. S20†). The peak splitting increases from 6.4 ppm to 8.51 ppm for Cu4@oCBT due to enhanced C–C coupling. Cu4@mCBT and Cu4@ICBT have characteristic 11B spectra similar to M9 and I9 ligands, respectively (Fig. S21 and S23†).
The 13C NMR spectra showed a single peak at δ 54.51 ppm due to C1 and C7 carbons of the M9 ligand, which got split into δ 56.37 and 49.99 ppm for Cu4@mCBT and the peaks at δ 45.97 and 44.41 ppm of I9 ligands were shifted to δ 51.50 and 46.91 ppm, respectively, for Cu4@ICBT (Fig. S22 and S24†). Literature reports on the magnitude of dipole moment and alteration in the orientation of carboranes upon binding with the metal surface indicate changes in their electronic properties upon binding with metal atoms.62–64
Infrared (IR) and Raman spectroscopy were performed to understand the vibrational spectral signatures of these clusters. The IR spectra of Cu4@oCBT, Cu4@mCBT and Cu4@ICBT are shown in Fig. S25–S27,† along with those of the respective ligands. The appearance of BH and CH stretching peaks at 2600 and 3050 cm−1 and other bending vibrational modes independently confirm the presence of carborane thiols in the cluster sample. The stretching vibrational spectral regions (especially BH and CH) become broad upon binding to the copper core. The cage breathing vibrational modes at 860–878 cm−1 confirm ligand binding. Theoretical calculations shown in Fig. S28 and S29† for Cu4@oCBT and Cu4@ICBT, respectively, also support the experimental spectra. Raman spectra shown in Fig. S30–S32† also confirm the broadening of CH and BH spectral regions of the respective clusters. XPS spectra shown in Fig. S33–S35† of Cu4@oCBT, Cu4@mCBT and Cu4@ICBT manifest the characteristic Cu, S, C and B signatures. Peak fitting of the Cu 2p region with 2p3/2 and 2p1/2 peaks at 930 and 950 eV of Cu4@oCBT and the absence of satellite peaks indicate a metal-like zero oxidation state for Cu. Similar spectra of Cu for Cu4@mCBT and Cu4@ICBT also suggest a zero oxidation state. The 3p3/2, 3d5/2, 4p and 4d peaks at 874, 619, 122, and 50 eV of iodine, respectively, were observed for Cu4@ICBT.
Photophysical properties
We have studied the absorption and emission characteristics of these clusters in detail. The UV-vis absorption spectra of the clusters recorded in their respective solutions (Cu4@oCBT and Cu4@mCBT in acetonitrile and Cu4@ICBT in a 5 : 1 DCM and acetone mixture) as well as in the crystalline state are presented in Fig. 4a–c. In solution, these clusters show a sharp absorption band in the window of 260–282 nm, whereas this absorption appears broadened in the crystalline state, with the onset at ∼400 nm. Generally, these clusters are non-emitting in solution, whereas bright emission (green for Cu4@oCBT and Cu4@mCBT and orange for Cu4@ICBT) were observed in the crystalline state under UV light (shown in the inset of Fig. S36†). The photographs of the respective crystals are shown in Fig. 4d. PL measurements showed emission peaks at 525, 533 and 595 nm for Cu4@oCBT, Cu4@mCBT and Cu4@ICBT in the crystalline state (Fig. 4e). Such types of strong emission peaks are absent in their respective solutions (Fig. S36†). These emission bands are associated with a broad excitation band in the window of 250–400 nm, which resembles their absorption band (Fig. 4e). These crystallization induced emission (CIE) features are reminiscent of cubane type [Cu4X4L4] complexes, which also showed poor luminescence properties in solution when compared to the solid and aggregated states.65,66 We have further checked the aggregation-induced emission (AIE) behaviour of Cu4@oCBT and Cu4@mCBT clusters (shown in Fig. S37†). Both of these clusters are non-emissive in acetonitrile. However, bright green luminescence was observed upon increasing the water content. PL spectral measurements show that aggregates started forming at a water fraction (fw) of 30 and 20% for Cu4@oCBT and Cu4@mCBT, respectively, and the emission intensifies gradually up to fw 80%.
Fig. 4. UV-vis absorption spectra of (a) Cu4@oCBT, (b) Cu4@mCBT and (c) Cu4@ICBT in the solid state and respective solutions. The calculated absorption spectrum of each cluster, also shown here, correlates well with the experimental spectrum. Yellow lines indicate relative oscillator strengths. The absorbance values refer only to the experimental spectra. (d) Photographs of the clusters under daylight and UV-light. Green luminescence was observed from Cu4@oCBT and Cu4@mCBT, whereas Cu4@ICBT is orange emitting. (e) Photoluminescence excitation and emission spectra of the clusters in their solid state. (f) Emission decay profiles of the clusters at room temperature (25 °C). (g) Electron density maps of natural transition orbitals (NTOs) responsible for the absorption feature of Cu4@oCBT. (h) Schematic diagram of the calculated excited states of Cu4@oCBT. (i) NTOs responsible for triplet emission.

To get additional insights into the CIE/AIE properties, we have studied the structural variation from the crystalline state to the isolated solution state using density functional theory (DFT) calculations, which show structural relaxation of Cu4@oCBT and Cu4@mCBT in solution compared to their crystalline state (shown in Fig. S38†). The Cu–S bond length decreases from 2.65 to 2.27 Å for Cu4@oCBT and 2.60 to 2.28 Å for Cu4@mCBT, upon changing from crystals to clusters in solution. Increase in the Cu–S–Cu bond angle from 74.65° to 78.45° for Cu4@oCBT and 73.65° to 78.08° for Cu4@mCBT also suggests such relaxation. Intercluster interaction in the solid state is also responsible for the CIE/AIE behaviour. Non-covalent intermolecular interactions have a significant effect on their electronic structure. Cu4@oCBT is packed in a trigonal crystal lattice through C–H⋯B(B) (3.04–3.06 Å), B–H⋯B–H (2.37–3.06 Å) and C–H⋯S (2.74–2.80 Å) van der Waals interactions (Fig. S39†). Similar intermolecular interactions were also present for Cu4@mCBT and Cu4@ICBT (shown in Fig. S40 and S41,† respectively). The disappearance of emission can be attributed to the structural relaxation of the clusters in solution as well as quenching of the excited state by solvent molecules.
The emission lifetimes of 43, 42 and 35 μS for Cu4@oCBT, Cu4@mCBT and Cu4@ICBT indicate phosphorescence nature originating from a triplet excited state (Fig. 4f). The respective fittings of each decay trace are shown in Fig. S42.† To verify the nature of the excited state, we have measured PL intensity upon interaction with oxygen. Quenching the emission intensity of all these clusters upon oxygen exposure and subsequent PL recovery upon nitrogen exposure verified prominent interaction of oxygen with the triplet excited state (Fig. S43†). Similar experiments were performed to detect the triplet state for metal nanoclusters.67,68 The quantum yields of 81%, 59% and 18% correlate with the emission intensity of the clusters. Detailed photophysical parameters are summarised in Table 2.
Photophysical parameters of Cu4 clusters in an air atmosphere.
| Clusters | λ max (nm) | τ air (μs) | Φ air |
|---|---|---|---|
| Cu4@oCBT | 525 (295 K) | 43 (295 K)a | 81% (295 K) |
| 535 (5 K) | 103 (5 K)a | 100% (5 K) | |
| Cu4@mCBT | 533 (295 K) | 42 (295 K)a | 59% (295 K) |
| 543 (5 K) | 125 (5 K)a | 98% (5 K) | |
| Cu4@ICBT | 595 (295 K) | 35 (295 K)b | 18% (295 K) |
| 606 (5 K) | 99 (5 K)b | 50% (5 K) |
Monoexponential decay profile.
Average lifetime obtained from biexponential fit.
The excited states of these clusters were studied using Density Functional Theory (DFT) and Time-dependent DFT (TD-DFT) calculations. The calculated absorption features and their respective oscillator strength in solution match well with the experimental spectrum (shown in Fig. 4a–c). The natural transition orbitals (NTOs) of these singlet excitations (Fig. 4g, S44, and S45† for Cu4@oCBT, Cu4@mCBT, and Cu4@ICBT, respectively) show that the hole orbitals (i.e. S0) are located in the Cu4S4 core region, whereas excited electron orbitals (i.e., S1, S2 and S3) with comparable energy are delocalized all over the copper carborane cluster framework. So, the absorption bands are due to the charge transfer from the metal core to the ligand shell (MCLS). The low energy absorption transition extends to the ‘surface’ of the clusters and it might depend on the intermolecular packing in the crystalline state. In the experimental spectra, such low energy transitions within ca. 300–400 nm are observed as a weak absorption tail. Their relative intensities are apparently overestimated by the calculations.
Next, we have calculated excited triplet states (Tn). The calculated energies (shown in Table S12 and S13†) of the first triplet state (T1) are too high to correspond to the wavelength of the phosphorescence observed in the solid state. However, phosphorescence proceeds from relaxed triplet 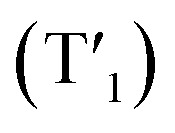 , and geometry optimization of T1 changes its energy drastically. The copper cores of the clusters undergo extensive rearrangement upon excited state relaxation (Fig. S46†). While the
, and geometry optimization of T1 changes its energy drastically. The copper cores of the clusters undergo extensive rearrangement upon excited state relaxation (Fig. S46†). While the 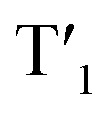 energy of Cu4@oCBT gets close to that of the observed solid state phosphorescence, the value calculated for Cu4@ICBT decreases even more, perhaps due to inefficient calculation of the spin orbit coupling for the iodinated compound. The low energies of the relaxed excited states may explain why phosphorescence is not observed in solution.
energy of Cu4@oCBT gets close to that of the observed solid state phosphorescence, the value calculated for Cu4@ICBT decreases even more, perhaps due to inefficient calculation of the spin orbit coupling for the iodinated compound. The low energies of the relaxed excited states may explain why phosphorescence is not observed in solution.
The NTOs of T1 and 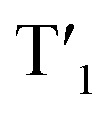 provide another insight into their properties (Fig. 4i and S47†). In all these clusters, the hole orbitals are located along the Cu4S4 rim, while the excited electron orbitals are mainly concentrated within the Cu4 core. In Cu4@oCBT (Fig. 4i) and Cu4@mCBT (Fig. S47a†), the hole and electron orbitals are almost perfectly symmetric, unaffected by the different orientations of the carborane ligands and the T1 excitations proceed concentrically from the rim of the cluster to its core. On the other hand, even though the iodine atoms of Cu4@ICBT (Fig. S47b†) participate in the NTOs to a limited extent, the hole orbital is shifted towards the pair of axially oriented carborane cages, where one copper atom is in close contact with two iodine atoms, while the electron orbital leans towards the equatorially attached ligand pair and the lone copper atom with no contact with iodine atoms. This shift creates a dipole between the excited electron and the hole that in solution interacts with the polarisable continuum of the solvent. This is why the T1 energies of Cu4@oCBT and Cu4@mCBT do not change in solution but that of Cu4@ICBT increases (Table S12 and S13†).
provide another insight into their properties (Fig. 4i and S47†). In all these clusters, the hole orbitals are located along the Cu4S4 rim, while the excited electron orbitals are mainly concentrated within the Cu4 core. In Cu4@oCBT (Fig. 4i) and Cu4@mCBT (Fig. S47a†), the hole and electron orbitals are almost perfectly symmetric, unaffected by the different orientations of the carborane ligands and the T1 excitations proceed concentrically from the rim of the cluster to its core. On the other hand, even though the iodine atoms of Cu4@ICBT (Fig. S47b†) participate in the NTOs to a limited extent, the hole orbital is shifted towards the pair of axially oriented carborane cages, where one copper atom is in close contact with two iodine atoms, while the electron orbital leans towards the equatorially attached ligand pair and the lone copper atom with no contact with iodine atoms. This shift creates a dipole between the excited electron and the hole that in solution interacts with the polarisable continuum of the solvent. This is why the T1 energies of Cu4@oCBT and Cu4@mCBT do not change in solution but that of Cu4@ICBT increases (Table S12 and S13†).
In addition, we have performed low-temperature photophysical measurements. As shown in Fig. 5a, the emission maximum at 525 nm of Cu4@oCBT was gradually shifted to 535 nm upon reducing the temperature from 295 to 5 K. Similar types of emission features were also observed for Cu4@mCBT, where emission centered at 533 nm was shifted to 543 nm with 1.5-fold emission enhancement (Fig. 5b). The 595 nm peak got shifted to 606 nm for Cu4@ICBT upon cooling the sample from 295 to 5 K (Fig. 5c). From the intensity variation, we have estimated that the quantum yields at 5 K approach ∼100% for Cu4@oCBT and Cu4@mCBT but ∼50% for Cu4@ICBT. The phosphorescence lifetimes of all these clusters moderately increase upon cooling down the sample to 5 K. For Cu4@oCBT, the lifetime becomes 103 μs at 5 K compared to 43 μs at room temperature (Fig. 5d). For Cu4@mCBT and Cu4@ICBT, 42 and 35 μs lifetimes at room temperature increase to 125 and 99 μs, respectively (Fig. 5e and f). The respective fittings of the lifetime profiles are shown in Fig. S48.† The change in the emissive lifetime for all three clusters at low temperature showed their emission to be phosphorescence rather than thermally activated delayed fluorescence (TADF). Efficient spin–orbit coupling between copper and carborane-thiol promotes phosphorescence.
Fig. 5. Low temperature (from 298 K to 5 K) photoluminescence excitation and emission spectra of (a) Cu4@oCBT, (b) Cu4@mCBT and (c) Cu4@ICBT, respectively, in the solid (polycrystalline) phase. Comparative emission decay traces of (d) Cu4@oCBT, (e) Cu4@mCBT and (f) Cu4@ICBT at 5 and 295 K, excited with a ns-pulsed laser at 337 nm. The PL decays of Cu4@oCBT and Cu4@mCBT follow exponential kinetics with the indicated lifetimes; those of Cu4@ICBT deviate from monoexponential curves; the indicated lifetimes are average values derived from biexponential fits.

Mechanoresponsive luminescence
Mechanoresponsive luminescence has been observed in these materials similar to previously reported phosphine protected copper complexes.69–71 We observed this by mechanical grinding. Cu4@oCBT and Cu4@mCBT clusters exhibit significant changes in their luminescence behavior upon mechanical grinding. The iodinated analogue, Cu4@ICBT, is resistant to mechanical activity. Grinding the crystalline samples of Cu4@oCBT and Cu4@mCBT using a mortar and pestle leads to changes in emission from green to yellow, without change in their appearance to the naked eye (Fig. 6a and b). We have observed that 20–25 min grinding of Cu4@oCBT is required to complete the transition from green to yellow, whereas only 8–10 min grinding is required for Cu4@mCBT. Cu4@ICBT didn't show any visible luminescence change after 20 min of grinding (Fig. 6c). PL spectral measurements showed that the emission maxima at 525 and 533 nm of the as-prepared Cu4@oCBT and Cu4@mCBT clusters shifted to 566 and 594 nm, respectively, upon grinding (Fig. 6d and e). We have also observed an increase in the full width at half maximum (fwhm) of the ground sample from 100 to 131 nm for Cu4@oCBT and from 97 to 120 nm for Cu4@mCBT. The broader emission is likely due to the higher structural heterogeneity by grinding. The emission maximum at 595 nm of Cu4@ICBT remains the same, i.e., unaffected by grinding (Fig. 6f). We have also observed the regeneration of luminescence from ground samples back to the initial state by exposure to DCM vapours. The yellow emission from the ground samples changed to green after 30 min of vapour exposure.
Fig. 6. Photographic images of (a) Cu4@oCBT and (b) Cu4@mCBT clusters show visible luminescence changes from green to yellow upon mechanical grinding. Luminescence was regenerated upon exposure of vapours of DCM. (c) Cu4@ICBT shows unaltered mechanoresponsive behaviour. Photoluminescence spectra of the grinding-solvent exposure cycle of (d) Cu4@oCBT, (e) Cu4@mCBT and (f) Cu4@ICBT. PXRD spectra of (g) Cu4@oCBT, (h) Cu4@mCBT and (i) Cu4@ICBT reveal a reduction of crystallinity upon grinding and regenerating the crystallinity upon solvent exposure.

Powder X-ray diffraction studies were performed to understand the phase transitions during the grinding and vapour exposure process. For all of these samples, the characteristic diffraction peaks matched well with the simulated PXRD pattern, which indicated crystalline purity (Fig. 6g–i and Tables S14–S16†). The broad diffraction features of the ground samples suggest crystalline to amorphous transition. The appearance of sharp diffraction peaks from the vapour exposed samples indicates an amorphous to crystalline phase transition, which is accompanied by regained green emission. The reversible mechanoluminescence of Cu4@oCBT and Cu4@mCBT makes them suitable for pressure-sensing applications. Raman studies (Fig. S49 and S50† for Cu4@oCBT and Cu4@mCBT, respectively) were performed to understand structural details at the molecular level. The ground Cu4@oCBT and Cu4@mCBT samples show broad spectral signatures with a slight shift of BB, BC, BBB and BBC vibrational features. We have observed a reversible shift of the icosahedral cage breathing vibrational mode from 764 to 757 cm−1 upon grinding, which returned to 763 cm−1 upon solvent exposure. Spectral shifts of 26 and 18 cm−1 for BH stretching modes were also observed for Cu4@oCBT and Cu4@mCBT, respectively. These studies clearly confirm the changes of structural microenvironments during respective operations, which was also observed for copper complexes, studied through solid state NMR studies.72,73 The nearly planar structure of Cu4@ICBT with longer Cu–Cu bond distances than the other clusters makes it so relaxed that it is unresponsive to mechanical grinding. Therefore, the variation of the changes in the emission of Cu4@oCBT and Cu4@mCBT can be attributed to their structural relaxation upon grinding and subsequent rigidification upon solvent exposure.
Thermoresponsive luminescence
In addition to mechanoresponsive behaviour, we have investigated the thermal dependence of luminescence as well. These experiments were carried out using ∼150 mg microcrystals under ambient conditions. Fig. 7a shows a series of photographs of Cu4@oCBT, exhibiting green emission from crystals after heating to 200 °C. No significant changes in the physical texture and color of the crystals were observed during thermal treatment. PL measurements (shown in Fig. 7b) show 1.1–1.3 fold emission enhancement up to 150 °C. Further heating, above 150 °C, slightly reduced the emission intensity. 1.5, 2.0 and 4.5-fold reductions in emission were observed at 175, 200, and 225 °C heating, respectively. Further heating of the sample above 250 °C quenches the emission completely due to the decomposition of the structure.
Fig. 7. Thermostable luminescence of Cu4@oCBT crystals. (a) Photographs show retained green emission up to 200 °C, without changes in the visible color. (b) Emission spectra measured after heating at different temperatures show that the emission peak position remains the same up to 200 °C. (c) TG and DTG measured in nitrogen, showing thermal stability of the crystals up to 380 °C. (d) PXRD patterns show identical diffraction peaks up to 200 °C, which indicate superior structural stability. Simulated spectrum is built with the single crystal data. All the data correspond to samples at room temperature, exposed to the temperature mentioned for 4 h.

TG and DTG shown in Fig. 7c further confirm the stability of the Cu4@oCBT cluster. There is no mass loss up to 350 °C. Heating above 380 °C leads to a loss of 40% of its mass due to the degradation of two carborane-thiol ligands along with a sulfur atom. Differential scanning calorimetry (DSC) (Fig. S51†) also confirms its superior thermal stability. The full range PXRD spectra of samples heated to different temperatures (Fig. 7d) show the preservation of the diffraction pattern up to 200 °C. Heating the sample to 250 °C leads to quenching of luminescence, in addition to irreversible structural changes. Spectral analysis reveals no significant enhancement of the spectral sharpness at low angle (below 25°) diffraction peaks, which indicates that there is no major phase transformation upon heating. The higher angle diffraction peaks, such as at 2θ of 36.51°, 40.82°, 43.97° and 45.36°, become sharper upon heating the sample to 150 °C (Fig. S52†). This can be rationalized by better crystallinity of the sample along with slight emission enhancement upon initial heating. However, the PXRD diffraction pattern of the 250 °C heated sample shows the complete loss of crystallinity, which is also manifested by the complete quenching of the luminescence. All together, these studies confirm that the rigid Cu4@oCBT crystals show high luminescence stability over a relatively broad temperature range.
On the other hand, nearly isostructural Cu4@mCBT shows interesting thermal dependence of luminescence in its crystalline state. Fig. 8a shows the photographic images of the sample during heating. Crystalline samples heated up to 100 °C show bright green luminescence followed by a change to yellow emission (at about 150 °C), which was observed up to 200 °C. Consistent with Cu4@oCBT, further heating to 250 °C leads to structural degradation (formation of black powder) and the disappearance of the luminescence. PL measurements showed a 1.1–1.2 fold emission enhancement up to 100 °C (Fig. 8b). At 125 °C, there is 1.1-fold reduction in emission. The 533 nm peak position remains the same up to 125 °C, matching with the visible green emission. We have observed double emission peaks at 510 and 612 nm from the sample heated to 150 °C. Similar peaks were also observed with samples heated to 200 °C. The appearance of a double emission band at 150 °C is probably due to the thermal expansion of the crystalline lattice and associated changes in the electronic excited state.
Fig. 8. Temperature-dependent tunable luminescence of Cu4@mCBT crystals; (a) photographs show green emitting crystals become yellow emitting after heating at 150 °C. Similar yellowish emission was observed up to 200 °C. (b) Photoluminescence spectra show a nearly similar emission maximum (533 nm) up to 125 °C heating, whereas heating at 150 °C leads to new peaks at 511 and 612 nm. (c) TG and DTG analyses show the thermal stability of the crystals up to 371 °C. (d) PXRD patterns show the appearance of new diffraction peaks after 150 °C heating, indicating structural transformation upon heating. Simulated spectrum was built with the single crystal data. All the data in (a, b and d) were measured at room temperature after heating the sample to the required temperature for 4 h.

TG and DTG showed thermal stability up to 371 °C (Fig. 8c). The 65.59% mass loss indicates the desorption of [CuS(C2B10H11S)3] beyond 371 °C. We have not observed any phase transition in TG/DTG analysis. In contrast, the DSC result shows an exothermic peak in the window of 250–300 °C (Fig. S53†). The PXRD measurements of the heated samples showed the appearance of new diffraction peaks after heating the sample at 150 °C (Fig. 8d). Magnified spectra shown in Fig. S54† reveal the appearance of new diffraction peaks at 2θ of 9.197°, 9.395°, 16.986°, 18.059°, and 18.962°, due to (012), (10–2), (121), (10–6), and (21–3) lattice planes. A reduction of the peak intensity was also observed at higher angles. So the appearance of new lattice planes, along with the reduction of some others, is due to structural change inside the crystal lattice. The flexible nature makes the emission properties of Cu4@mCBT tunable.
Thermogravimetric analysis shows lower thermal stability of Cu4@ICBT compared to the other two clusters. At 130.5 °C, there is a mass loss of 13.68% (Fig. S55†) corresponding to one carboranethiolate ligand. DSC data show an endothermic peak at 129.3 °C (Fig. S56†). We haven't observed any temperature dependence of luminescence for Cu4@ICBT.
Conclusions
This work presents a class of isostructural tetranuclear copper NCs protected by carborane thiol ligands. Single crystal XRD shows that all of these clusters have a square planar Cu4 core and a butterfly-shaped Cu4S4 staple, surrounded by respective carboranes. Three iodine insertions in Cu4@ICBT make the Cu4S4 staple more flattened than other nanoclusters. High-resolution mass spectrometric studies and other spectroscopic and microscopic studies further confirm the molecular composition of these clusters and associated structural details. All of these clusters emit negligibly in solution, whereas bright visible emission was observed in the solid state due to intermolecular aggregation manifested by their CIE and AIE characteristics. Tuning the emission from green to yellow was observed due to structural flattening of Cu4@ICBT in the crystalline state. The emission lifetimes of the microsecond range indicate phosphorescence, originating from triplet states, which was verified by oxygen quenching experiments. DFT calculation verified radiative relaxation from the relaxed triplet state. We observed mechanoresponsive luminescence switching of Cu4@oCBT and Cu4@mCBT upon mechanical grinding and subsequent solvent exposure. Cu4@ICBT didn't show any luminescence tuning by mechanical treatment. Among these clusters, Cu4@mCBT shows thermoresponsive luminescence. Systematic investigations using different spectroscopic and microscopic studies proved that structural modification due to aggregation and external stimuli manifests in the change of their phosphorescence properties. This work thus presents a new class of coinage metal NCs with bright and thermally stable phosphorescence, which in addition can be efficiently tuned via manipulating the excited states.
Experimental section
Chemicals
Ortho-carborane-9-thiol (O9), meta-carborane-9-thiol (M9) and ortho-carborane 12-iodo 9-thiol (I9) were synthesized according to the literature.74,75 The purity of each ligand was confirmed by their characteristic mass spectrometric analyses, presented in Fig. S57–S59,† respectively. While the O9 and M9 X-ray single-crystal structures were reported previously, the molecular structure of the I9 ligand is provided together with the respective discussion of its supramolecular structure in the ESI (Fig. S60–S62†). Copper iodide (CuI) and sodium borohydride (NaBH4, 98%) were purchased from Aldrich Chemicals. 1,2-bis-(Diphenylphosphino)ethane (DPPE) was purchased from Rankem Chemicals. Solvent grade dichloromethane (DCM), chloroform (CHCl3), n-hexane, acetone, acetonitrile and methanol (99.5%) were purchased from Rankem and Finar India, respectively. Deuterated solvents such as CDCl3 and d6-acetone were purchased from Sigma Aldrich. All of the chemicals were commercially available and used as received. Crystallization was achieved in HPLC-grade solvents.
Synthesis of [Cu18(DPPE)6H16]2+
The [Cu18(DPPE)6H16]2+ (shortly, Cu18) cluster was prepared according to the literature.57 In brief, 95 mg (0.49 mM) CuI was mixed with 120 mg (0.03 mM) DPPE under argon and dissolved in 15 ml of acetonitrile. After 30 min, the as-formed white complexes were reduced by directly adding 180–185 mg of NaBH4 powder. After another hour of stirring (750 rpm) at room temperature, an orange colored precipitate was formed, which indicated the formation of the cluster. After another 6 hours, the mixture was centrifuged to yield an orange solid, which was washed three times using 5 ml of acetonitrile and methanol to remove any starting reagents. Finally, dark orange colored Cu18 was extracted using DCM for the LEIST reaction. UV-vis and ESI-MS spectra (Fig. S63†) were recorded to confirm the Cu18 cluster. The yield was calculated to be 75% relative to the respective copper precursor.
Synthesis of Cu4@oCBT
The purified Cu18 nanocluster (∼50 mg) in 15 ml DCM was reacted with 40 mg of O9 at room temperature. After 2 h of reaction, the as-formed yellowish solution yields white color precipitates. Bright green emission from these precipitates was observed under a 365 nm UV lamp. After completing the reaction (5 h), microcrystalline white precipitates were washed several times using methanol and DCM to remove any excess of unreacted ligands. The purified microcrystal-line cluster was used for further studies (yield = 85%).
Synthesis of Cu4@mCBT and Cu4@ICBT
Cu4@mCBT and Cu4@ICBT were synthesized following a similar LEIST reaction to that reported above. Instead of using O9, we have used M9 and I9 as protecting ligands for synthesizing these clusters. The as-formed Cu4@mCBT is soluble in DCM from which it was crystallized, whereas Cu4@ICBT formed a yellow colored microcrystalline in-soluble solid (Yield: 75% of Cu4@mCBT and 80% of Cu4@ICBT).
Oxygen sensitivity
The nature of the excited state was verified by oxygen sensitivity experiments. A thin layer of the microcrystalline sample was cast on a glass slide, which was exposed to oxygen (exposure time 12 h). PL measurements were performed after oxygen exposure. Nitrogen was subsequently exposed to confirm oxygen sensitivity. Prior to the gas exposure, each sample was put in a vacuum for 1 h.
AIE properties
The aggregation behaviours of Cu4@oCBT and Cu4@mCBT were studied upon dissolving them in acetonitrile. A varying fraction of water was added to it by keeping the cluster concentration the same (1 mg ml−1). Non-emitting acetonitrile solution of these clusters becomes green emitting upon increasing the fraction of water.
Grinding and solvent exposure experiments
95–100 mg of freshly prepared microcrystalline powder was ground mechanically using a clean mortar and pestle. Solvent exposure was accomplished by placing the mortar inside a closed chamber with DCM vapour.
Thermal heating experiments
A freshly prepared 150–160 mg crystalline powder was placed in a closed glass vial and heated under ambient conditions for 4 h. Just after heating under specific conditions, luminescence and PXRD studies were performed.
Data availability
X-ray crystallographic structures are deposited in CCDC. Additional data can be found in ESI.†
Author contributions
A. J. performed the synthesis, crystallization and most of the experimental studies. M. J. and W. A. D. assisted A. J., especially in the synthesis. A. J., J. R. and P. C. performed mass spectrometric studies. A. J. and S. M. conducted Raman studies. G. P. and J. M. performed computational studies. J. M. and T. B. synthesized and purified the carborane thiol ligand used for this work. K. K. and K. L. performed room temperature lifetime and quantum yield measurements. Low temperature photophysical studies were performed by S. L. in the laboratory of M. M. K. Single crystal XRD structural analyses were performed by S. A. and M. K. S. G. G. was involved in the discussion of experimental data. The first draft of the manuscript was written by A. J. and all the authors have given approval to the final version of the manuscript. T. P. and T. B. supervised the project and finalized the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The authors acknowledge the support of the Department of Science and Technology (DST), Govt. of India and the Ministry of Education, Youth and Sports (MEYS) of Czech Republic for their financial support to the bilateral research projects, DST/INT/Czech/P-16/2020 and LTAIN19152, respectively. The authors would like to thank the Sophisticated Analytical Instrumental Facility, Indian Institute of Technology Madras (SAIF-IITM) for single crystal XRD, solid state UV-vis absorption spectroscopy and thermogravimetric measurements. We acknowledge the CzechNanoLab Research Infrastructure supported by MEYS (CR LM2018110) for crystallographic analysis. For theoretical calculations, computational resources were supplied by the project “e-Infrastruktura CZ” (e-INFRA CZ LM2018140) supported by the Ministry of Education, Youth and Sports of the Czech Republic. A. J. acknowledges financial support from IIT Madras, and M. J. thanks UGC, Govt. of India for their research fellowships. T. P. acknowledges funding from the Centre of Excellence on Molecular Materials and Functions under the Institution of Eminence scheme of IIT Madras. This work is a result of a joint India-Czech project aimed at exploring the potential of atomically precise nanomaterials produced by combining metal and boron clusters, initiated by T. P. and T. B.
Electronic supplementary information (ESI) available. CCDC 2192345, 2192348, 2192353 and 2192354. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06578a
References
- Jin R. Zeng C. Zhou M. Chen Y. Chem. Rev. 2016;116:10346–10413. doi: 10.1021/acs.chemrev.5b00703. [DOI] [PubMed] [Google Scholar]
- Kang X. Zhu M. Chem. Soc. Rev. 2019;48:2422–2457. doi: 10.1039/C8CS00800K. [DOI] [PubMed] [Google Scholar]
- Chakraborty I. Pradeep T. Chem. Rev. 2017;117:8208–8271. doi: 10.1021/acs.chemrev.6b00769. [DOI] [PubMed] [Google Scholar]
- Jana A. Chakraborty P. Dar W. A. Chandra S. Khatun E. Kannan M. P. Ras R. H. A. Pradeep T. Chem. Commun. 2020;56:12550–12553. doi: 10.1039/D0CC03983G. [DOI] [PubMed] [Google Scholar]
- Jena P. Sun Q. Chem. Rev. 2018;118:5755–5870. doi: 10.1021/acs.chemrev.7b00524. [DOI] [PubMed] [Google Scholar]
- Jana A. Jash M. Poonia A. K. Paramasivam G. Islam M. R. Chakraborty P. Antharjanam S. Machacek J. Ghosh S. Adarsh K. N. V. D. Base T. Pradeep T. ACS Nano. 2021;15:15781–15793. doi: 10.1021/acsnano.1c02602. [DOI] [PubMed] [Google Scholar]
- Xie M. Han C. Liang Q. Zhang J. Xie G. Xu H. Sci. Adv. 2019;5:1–9. doi: 10.1126/sciadv.aav9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. L. M. Zhou G. Zhou G. Lee H. K. Zhao N. Prezhdo O. V. Mak T. C. W. Chem. Sci. 2019;10:10122–10128. doi: 10.1039/C9SC03455B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y. Luo Z. Chevrier D. M. Leong D. T. Zhang P. Jiang D. E. Xie J. J. Am. Chem. Soc. 2014;136:1246–1249. doi: 10.1021/ja411643u. [DOI] [PubMed] [Google Scholar]
- AbdulHalim L. G. Bootharaju M. S. Tang Q. Del Gobbo S. AbdulHalim R. G. Eddaoudi M. Jiang D. E. Bakr O. M. J. Am. Chem. Soc. 2015;137:11970–11975. doi: 10.1021/jacs.5b04547. [DOI] [PubMed] [Google Scholar]
- Chen T. Yang S. Chai J. Song Y. Fan J. Rao B. Sheng H. Yu H. Zhu M. Sci. Adv. 2017;3:1–8. doi: 10.1126/sciadv.1700956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Chen L. Black A. Parak W. J. Klinke C. Chakraborty I. Aggregate. 2022;3:1–18. [Google Scholar]; (b) Jana A. Unnikrishnan P. M. Poonia A. K. Roy J. Jash M. Paramasivam G. Machacek J. Adarsh K. N. V. D. Base T. Pradeep T. Inorg. Chem. 2022;61:8593–8603. doi: 10.1021/acs.inorgchem.2c00186. [DOI] [PubMed] [Google Scholar]
- Ford P. C. Cariati E. Bourassa J. Chem. Rev. 1999;99:3625–3647. doi: 10.1021/cr960109i. [DOI] [PubMed] [Google Scholar]
- Liu X. Astruc D. Coord. Chem. Rev. 2018;359:112–126. doi: 10.1016/j.ccr.2018.01.001. [DOI] [Google Scholar]
- Smolentsev G. Milne C. J. Guda A. Haldrup K. Szlachetko J. Azzaroli N. Cirelli C. Knopp G. Bohinc R. Menzi S. Pamfilidis G. Gashi D. Beck M. Mozzanica A. James D. Bacellar C. Mancini G. F. Tereshchenko A. Shapovalov V. Kwiatek W. M. Czapla-Masztafiak J. Cannizzo A. Gazzetto M. Sander M. Levantino M. Kabanova V. Rychagova E. Ketkov S. Olaru M. Beckmann J. Vogt M. Nat. Commun. 2020;11:1–9. doi: 10.1038/s41467-019-13993-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. N. Xu R. D. Guo J. S. Miao J. L. Zhang M. J. Li C. J. Mater. Chem. C. 2021;9:8589–8595. doi: 10.1039/D1TC00964H. [DOI] [Google Scholar]
- Ai L. Jiang W. Liu Z. Liu J. Gao Y. Zou H. Wu Z. Wang Z. Liu Y. Zhang H. Yang B. Nanoscale. 2017;9:12618–12627. doi: 10.1039/C7NR03985A. [DOI] [PubMed] [Google Scholar]
- Olaru M. Rychagova E. Ketkov S. Shynkarenko Y. Yakunin S. Kovalenko M. V. Yablonskiy A. Andreev B. Kleemiss F. Beckmann J. Vogt M. J. Am. Chem. Soc. 2020;142:373–381. doi: 10.1021/jacs.9b10829. [DOI] [PubMed] [Google Scholar]
- Wang J. J. Zhou H. T. Yang J. N. Feng L. Z. Yao J. S. Song K. H. Zhou M. M. Jin S. Zhang G. Bin Yao H. J. Am. Chem. Soc. 2021;143:10860–10864. doi: 10.1021/jacs.1c05476. [DOI] [PubMed] [Google Scholar]
- Kirakci K. Fejfarová K. Martinčík J. Nikl M. Lang K. Inorg. Chem. 2017;56:4609–4614. doi: 10.1021/acs.inorgchem.7b00240. [DOI] [PubMed] [Google Scholar]
- Yam V. W. W. Au V. K. M. Leung S. Y. L. Chem. Rev. 2015;115:7589–7728. doi: 10.1021/acs.chemrev.5b00074. [DOI] [PubMed] [Google Scholar]
- Zhang K. Y. Yu Q. Wei H. Liu S. Zhao Q. Huang W. Chem. Rev. 2018;118:1770–1839. doi: 10.1021/acs.chemrev.7b00425. [DOI] [PubMed] [Google Scholar]
- Kenry Chen C. Liu B. Nat. Commun. 2019;10:1–15. doi: 10.1038/s41467-018-07882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton O. Lee D. Jung J. Kim J. Chem. Mater. 2014;26:6644–6649. doi: 10.1021/cm503678r. [DOI] [Google Scholar]
- Chen C. Chi Z. Chong K. C. Batsanov A. S. Yang Z. Mao Z. Yang Z. Liu B. Nat. Mater. 2021;20:175–180. doi: 10.1038/s41563-020-0797-2. [DOI] [PubMed] [Google Scholar]
- Wang J. Wang C. Gong Y. Liao Q. Han M. Jiang T. Dang Q. Li Y. Li Q. Li Z. Angew. Chem., Int. Ed. 2018;57:16821–16826. doi: 10.1002/anie.201811660. [DOI] [PubMed] [Google Scholar]
- Shoji Y. Ikabata Y. Wang Q. Nemoto D. Sakamoto A. Tanaka N. Seino J. Nakai H. Fukushima T. J. Am. Chem. Soc. 2017;139:2728–2733. doi: 10.1021/jacs.6b11984. [DOI] [PubMed] [Google Scholar]
- Penfold T. J. Gindensperger E. Daniel C. Marian C. M. Chem. Rev. 2018;118:6975–7025. doi: 10.1021/acs.chemrev.7b00617. [DOI] [PubMed] [Google Scholar]
- He Z. Zhao W. Lam J. W. Y. Peng Q. Ma H. Liang G. Shuai Z. Tang B. Z. Nat. Commun. 2017;8:1–7. doi: 10.1038/s41467-016-0009-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuttell D. G. Kuang S. M. Fanwick P. E. McMillin D. R. Walton R. A. J. Am. Chem. Soc. 2002;124:6–7. doi: 10.1021/ja012247h. [DOI] [PubMed] [Google Scholar]
- Lu T. Wang J. Y. Tu D. Chen Z. N. Chen X. T. Xue Z. L. Inorg. Chem. 2018;57:13618–13630. doi: 10.1021/acs.inorgchem.8b02217. [DOI] [PubMed] [Google Scholar]
- Schinabeck A. Leitl M. J. Yersin H. J. Phys. Chem. Lett. 2018;9:2848–2856. doi: 10.1021/acs.jpclett.8b00957. [DOI] [PubMed] [Google Scholar]
- Dias H. V. R. Diyabalanage H. V. K. Rawashdeh-Omary M. A. Franzman M. A. Omary M. A. J. Am. Chem. Soc. 2003;125:12072–12073. doi: 10.1021/ja036736o. [DOI] [PubMed] [Google Scholar]
- Roppolo I. Celasco E. Fargues A. Garcia A. Revaux A. Dantelle G. Maroun F. Gacoin T. Boilot J. P. Sangermano M. Perruchas S. J. Mater. Chem. 2011;21:19106–19113. doi: 10.1039/C1JM13600C. [DOI] [Google Scholar]
- Ai P. Mauro M. De Cola L. Danopoulos A. A. Braunstein P. Angew. Chem., Int. Ed. 2016;55:3338–3341. doi: 10.1002/anie.201510150. [DOI] [PubMed] [Google Scholar]
- Baghdasaryan A. Bürgi T. Nanoscale. 2021;13:6283–6340. doi: 10.1039/D0NR08489A. [DOI] [PubMed] [Google Scholar]
- Lu Y. Chen W. Chem. Soc. Rev. 2012;41:3594–3623. doi: 10.1039/C2CS15325D. [DOI] [PubMed] [Google Scholar]
- Wang J. Gu X. Ma H. Peng Q. Huang X. Zheng X. Sung S. H. P. Shan G. Lam J. W. Y. Shuai Z. Tang B. Z. Nat. Commun. 2018;9:1–9. doi: 10.1038/s41467-017-02088-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai Y. Li Y. Chan M. H. Y. Xiao G. Zou B. Yam V. W. W. J. Am. Chem. Soc. 2021;143:10659–10667. doi: 10.1021/jacs.1c04200. [DOI] [PubMed] [Google Scholar]
- Mei J. Leung N. L. C. Kwok R. T. K. Lam J. W. Y. Tang B. Z. Chem. Rev. 2015;115:11718–11940. doi: 10.1021/acs.chemrev.5b00263. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Y. Liu Y. Chem. Sci. 2019;10:7773–7778. doi: 10.1039/C9SC02633A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai S. Ma H. Shi H. Wang H. Wang X. Xiao L. Ye W. Huang K. Cao X. Gan N. Ma C. Gu M. Song L. Xu H. Tao Y. Zhang C. Yao W. An Z. Huang W. Nat. Commun. 2019;10:1–8. doi: 10.1038/s41467-019-11749-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L. Zhu W. Yang F. Li B. Ren X. Zhang X. Hu W. Phys. Chem. Chem. Phys. 2018;20:6009–6023. doi: 10.1039/C7CP07167A. [DOI] [PubMed] [Google Scholar]
- Wu Z. Liu J. Gao Y. Liu H. Li T. Zou H. Wang Z. Zhang K. Wang Y. Zhang H. Yang B. J. Am. Chem. Soc. 2015;137:12906–12913. doi: 10.1021/jacs.5b06550. [DOI] [PubMed] [Google Scholar]
- Perruchas S. Goff X. F. L. Maron S. Maurin I. Guillen F. Garcia A. Gacoin T. Boilot J. P. J. Am. Chem. Soc. 2010;132:10967–10969. doi: 10.1021/ja103431d. [DOI] [PubMed] [Google Scholar]
- Wei W. Lu Y. Chen W. Chen S. J. Am. Chem. Soc. 2011;133:2060–2063. doi: 10.1021/ja109303z. [DOI] [PubMed] [Google Scholar]
- Perruchas S. Tard C. Le Goff X. F. Fargues A. Garcia A. Kahlal S. Saillard J. Y. Gacoin T. Boilot J. P. Inorg. Chem. 2011;50:10682–10692. doi: 10.1021/ic201128a. [DOI] [PubMed] [Google Scholar]
- Shi S. Jung M. C. Coburn C. Tadle A. Sylvinson D. M. R. Djurovich P. I. Forrest S. R. Thompson M. E. J. Am. Chem. Soc. 2019;141:3576–3588. doi: 10.1021/jacs.8b12397. [DOI] [PubMed] [Google Scholar]
- Basu K. Paul S. Jana R. Datta A. Banerjee A. ACS Sustainable Chem. Eng. 2019;7:1998–2007. doi: 10.1021/acssuschemeng.8b04301. [DOI] [Google Scholar]
- Lin Y. J. Chen P. C. Yuan Z. Ma J. Y. Chang H. T. Chem. Commun. 2015;51:11983–11986. doi: 10.1039/C5CC02342D. [DOI] [PubMed] [Google Scholar]
- Núñez R. Tarrés M. Ferrer-Ugalde A. De Biani F. F. Teixidor F. Chem. Rev. 2016;116:14307–14378. doi: 10.1021/acs.chemrev.6b00198. [DOI] [PubMed] [Google Scholar]
- Poater J. Viñas C. Bennour I. Escayola S. Solà M. Teixidor F. J. Am. Chem. Soc. 2020;142:9396–9407. doi: 10.1021/jacs.0c02228. [DOI] [PubMed] [Google Scholar]
- Spokoyny A. M. MacHan C. W. Clingerman D. J. Rosen M. S. Wiester M. J. Kennedy R. D. Stern C. L. Sarjeant A. A. Mirkin C. A. Nat. Chem. 2011;3:590–596. doi: 10.1038/nchem.1088. [DOI] [PubMed] [Google Scholar]
- Furue R. Nishimoto T. Park I. S. Lee J. Yasuda T. Angew. Chem., Int. Ed. 2016;55:7171–7175. doi: 10.1002/anie.201603232. [DOI] [PubMed] [Google Scholar]
- Kodr D. Yenice C. P. Simonova A. Saftić D. P. Pohl R. Sýkorová V. Ortiz M. Havran L. Fojta M. Lesnikowski Z. J. O'Sullivan C. K. Hocek M. J. Am. Chem. Soc. 2021;143:7124–7134. doi: 10.1021/jacs.1c02222. [DOI] [PubMed] [Google Scholar]
- Wang Q. Wang J. Wang S. Wang Z. Cao M. He C. Yang J. Zang S. Mak T. C. W. J. Am. Chem. Soc. 2020;142:12010–12014. doi: 10.1021/jacs.0c04638. [DOI] [PubMed] [Google Scholar]
- Li J. Ma H. Z. Reid G. E. Edwards A. J. Hong Y. White J. M. Mulder R. J. O'Hair R. A. J. Chem.–Eur. J. 2018;24:2070–2074. doi: 10.1002/chem.201705448. [DOI] [PubMed] [Google Scholar]
- Kang X. Zhu M. Chem. Mater. 2019;31:9939–9969. doi: 10.1021/acs.chemmater.9b03674. [DOI] [Google Scholar]
- Kang X. Wei X. Wang S. Zhu M. Chem. Sci. 2021;12:11080–11088. doi: 10.1039/D1SC00632K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang X. Huang L. Liu W. Xiong L. Pei Y. Sun Z. Wang S. Wei S. Zhu M. Chem. Sci. 2019;10:8685–8693. doi: 10.1039/C9SC02667C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H. Yang F. Pan D. Lin Y. Hohman J. N. Solis-Ibarra D. Li F. H. Dahl J. E. P. Carlson R. M. K. Tkachenko B. A. Fokin A. A. Schreiner P. R. Galli G. Mao W. L. Shen Z. X. Melosh N. A. Nature. 2018;554:505–510. doi: 10.1038/nature25765. [DOI] [PubMed] [Google Scholar]
- Baše T. Bastl Z. Šlouf M. Klementová M. Šubrt J. Vetushka A. Ledinský M. Fejfar A. Macháček J. Carr M. J. Londesborough M. G. S. J. Phys. Chem. C. 2008;112:14446–14455. doi: 10.1021/jp802281s. [DOI] [Google Scholar]
- Schwartz J. J. Mendoza A. M. Wattanatorn N. Zhao Y. Nguyen V. T. Spokoyny A. M. Mirkin C. A. Baše T. Weiss P. S. J. Am. Chem. Soc. 2016;138:5957–5967. doi: 10.1021/jacs.6b02026. [DOI] [PubMed] [Google Scholar]
- Mills H. A. Jones C. G. Anderson K. P. Ready A. D. Djurovich P. I. Khan S. I. Hohman J. N. Nelson H. M. Spokoyny A. M. Chem. Mater. 2022;34(15):6933–6943. doi: 10.1021/acs.chemmater.2c01319. [DOI] [Google Scholar]
- Ford C. P. Coord. Chem. Rev. 1994;132:129–140. doi: 10.1016/0010-8545(94)80032-4. [DOI] [Google Scholar]
- Troyano J. Zamora F. Delgado S. Chem. Soc. Rev. 2021;50:4606–4628. doi: 10.1039/D0CS01470B. [DOI] [PubMed] [Google Scholar]
- Zhu C. Xin J. Li J. Li H. Kang X. Pei Y. Zhu M. Angew. Chem., Int. Ed. 2022;61:1–6. doi: 10.1002/anie.202205947. [DOI] [PubMed] [Google Scholar]
- Khatun E. Ghosh A. Chakraborty P. Singh P. Bodiuzzaman M. Ganesan P. Nataranjan G. Ghosh J. Pal S. K. Pradeep T. Nanoscale. 2018;10:20033–20042. doi: 10.1039/C8NR05989F. [DOI] [PubMed] [Google Scholar]
- Perruchas S. Goff X. F. L. Maron S. Maurin I. Guillen F. Garcia A. Gacoin T. Boilot J. P. J. Am. Chem. Soc. 2010;132:10967–10969. doi: 10.1021/ja103431d. [DOI] [PubMed] [Google Scholar]
- Utrera-Melero R. Huitorel B. Cordier M. Mevellec J. Y. Massuyeau F. Latouche C. Martineau-Corcos C. Perruchas S. Inorg. Chem. 2020;59:13607–13620. doi: 10.1021/acs.inorgchem.0c01967. [DOI] [PubMed] [Google Scholar]
- Huitorel B. El Moll H. Cordier M. Fargues A. Garcia A. Massuyeau F. Martineau-Corcos C. Gacoin T. Perruchas S. Inorg. Chem. 2017;56:12379–12388. doi: 10.1021/acs.inorgchem.7b01870. [DOI] [PubMed] [Google Scholar]
- Benito Q. Le Goff X. F. Maron S. Fargues A. Garcia A. Martineau C. Taulelle F. Kahlal S. Gacoin T. Boilot J. P. Perruchas S. J. Am. Chem. Soc. 2014;136:11311–11320. doi: 10.1021/ja500247b. [DOI] [PubMed] [Google Scholar]
- Huitorel B. El Moll H. Utrera-Melero R. Cordier M. Fargues A. Garcia A. Massuyeau F. Martineau-Corcos C. Fayon F. Rakhmatullin A. Kahlal S. Saillard J. Y. Gacoin T. Perruchas S. Inorg. Chem. 2018;57:4328–4339. doi: 10.1021/acs.inorgchem.7b03160. [DOI] [PubMed] [Google Scholar]
- Plešek J. Hermanek S. Collect. Czech. Chem. Commun. 1981;46:687–692. doi: 10.1135/cccc19810687. [DOI] [Google Scholar]
- Plešek J. Janoušek Z. Hermanek S. Collect. Czech. Chem. Commun. 1978;43:1332–1338. doi: 10.1135/cccc19781332. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
X-ray crystallographic structures are deposited in CCDC. Additional data can be found in ESI.†