

Significance
Drug-resistant strains of Acinetobacter baumannii are increasingly prevalent leading to increased morbidity and mortality, thereby dictating the need for new classes of antibiotics. Herein, we provide genetic and chemical validation of the A. baumannii pyrimidine biosynthetic enzyme dihydroorotate dehydrogenase (AbDHODH) as a potential drug target to treat this infection. We showed that AbDHODH is essential for A. baumannii to survive within the host, and we identified potent, species-specific inhibitors of AbDHODH active against highly drug-resistant A. baumannii strains. One compound conferred significant protection to mice. Structural studies of inhibitor-bound AbDHODH defined the inhibitor-binding pocket. These data support the development of antimicrobial agents directed against AbDHODH for the treatment of infections due to drug-resistant A. baumannii.
Keywords: Acinetobacter baumannii, antimicrobial resistance, dihydroorotate dehydrogenase, pyrimidine metabolism, drug discovery
Abstract
New antimicrobials are needed for the treatment of extensively drug-resistant Acinetobacter baumannii. The de novo pyrimidine biosynthetic enzyme dihydroorotate dehydrogenase (DHODH) is a validated drug target for malaria and human autoimmune diseases. We provide genetic evidence that A. baumannii DHODH (AbDHODH) is essential for bacterial survival in rodent infection models. We chemically validate the target by repurposing a unique library of ~450 triazolopyrimidine/imidazopyrimidine analogs developed for our malaria DHODH program to identify 21 compounds with submicromolar activity on AbDHODH. The most potent (DSM186, DHODH IC50 28 nM) had a minimal inhibitory concentration of ≤1 µg/ml against geographically diverse A. baumannii strains, including meropenem-resistant isolates. A structurally related analog (DSM161) with a long in vivo half-life conferred significant protection in the neutropenic mouse thigh infection model. Encouragingly, the development of resistance to these compounds was not identified in vitro or in vivo. Lastly, the X-ray structure of AbDHODH bound to DSM186 was solved to 1.4 Å resolution. These data support the potential of AbDHODH as a drug target for the development of antimicrobials for the treatment of A. baumannii and potentially other high-risk bacterial infections.
The development of new antimicrobials active against carbapenem-resistant, extensively drug-resistant (CR-XDR) Acinetobacter baumannii has been identified by the NIH and WHO as a high priority. This imperative was reinforced by data just released by the CDC which reported a 78% increase in CR-A. baumannii infections from 2019 to 2020, more than any other pathogen of concern (1). Disturbingly, 50% of A. baumannii isolates from US intensive care units (ICUs) are CR-XDR, far higher than for other pathogens (2) and pan drug-resistant isolates are increasingly described (3). Globally, an estimated 132,000 deaths were attributable to and 450,000 deaths associated with drug-resistant A. baumannii infection in 2019 (4). Furthermore, in contrast to other resistant bacteria, virtually no antibiotics are in the pipeline to provide treatment options for CR-XDR A. baumannii (2, 5). The threat of a postantibiotic era is on the cusp of being fulfilled by A. baumannii (2). Unfortunately, antimicrobials such as ceftolozane-tazobactam, ceftazidime-avibactam, meropenem-vaborbactam, imipenem-cilastatin-relebactam, fosfomycin, and plazomicin are poorly active against CR-XDR and PDR A. baumannii (6), and increasing resistance has been reported for the last-resort antimicrobials such as tigecycline, polymyxins, and cefiderocol (3, 7).
A. baumannii is capable of de novo biosynthesis of pyrimidines needed for the downstream biosynthesis of RNA and DNA (8), but it lacks both a complete pyrimidine salvage pathway and an alternative biosynthetic pathway (EcoCyc/BioCyc analysis, SI Appendix, Table S1). We previously demonstrated based on genetic studies that carbamoyl phosphate synthase and dihydroorotase are required for survival in a rat infection model (9), while dihydroorotate dehydrogenase (DHODH) (quinone)(EC1.3.5.2), the fourth enzyme in the pathway, was found to be essential in a murine pneumonia model based on a whole-genome transposon mutagenesis study (10). These studies demonstrate that enzymes in the de novo pyrimidine pathway are essential for A. baumannii survival in vivo and show that uracil salvage via uracil phosphoribosyl transferase (SI Appendix, Table S1) is insufficient to sustain an infection in vivo.
DHODH is a clinically validated target for the treatment of rheumatoid arthritis and multiple sclerosis, with the FDA-approved drug leflunomide used for the treatment of these diseases (11). Recently, DHODH has also been clinically advanced as a target for the treatment of malaria (12) and fungal pathogens (13). The triazolopyrimidine-based Plasmodium falciparum DHODH (PfDHODH) inhibitor (DSM265) (14) showed single dose efficacy against P. falciparum malaria in human clinical studies (15, 16). The orotomide olorofim recently received breakthrough therapy designation from the FDA for the treatment of invasive mold infections with additional results awaited in the current clinical study (17). Thus, DHODH has been verified as a druggable target in eukaryotes, but it has received less attention as a potential antibacterial target despite some promising reports for H. pylori and E. coli (18, 19). These data motivated us to consider A.baumannii DHODH (AbDHODH) as a potential antibacterial target as it represents an unexplored mechanism of action for the treatment of extensively and pan-drug-resistant A. baumannii.
DHODH catalyzes the flavin mononucleotide (FMN)-dependent conversion of dihydroorotateto orotic acid followed by the reoxidation of the FMN cofactor in a second step that requires one of the three possible cofactors: Coenzyme Q (CoQ) (type II enzymes; mitochondrial in eukaryotes and cytoplasmic membrane bound in bacteria), fumarate (type 1A), or NAD+ (type 1b) (20). Both human and Plasmodium species use the membrane-associated type II enzyme that has proven to be the druggable target. Gram-negative bacilli (GNB) such as A. baumannii, Pseudomonas aeruginosa, Escherichia coli, and Klebsiella pneumoniae also encode type II enzymes, suggesting the potential druggability of the target in these species as well.
The inhibitor-binding site of the type II DHODHs overlaps the CoQ binding site, and X-ray structure analysis of human, E. coli, and Plasmodium DHODHs identified amino acid differences between the species that change the nature and shape of this binding pocket, thereby providing opportunity for the development of species-selective inhibitors (21–24). Our ability to identify PfDHODH inhibitors that do not inhibit human DHODH was a key strength of the malaria DHODH program contributing to the good safety profile of DSM265 (14). Based on sequence alignment of these enzymes with AbDHODH, we hypothesized that good selectivity between A. baumannii and human DHODH could also be achieved.
During the lead optimization program to identify antimalarial DHODH inhibitors, we synthesized ~450 molecules, primarily in the triazolopyrimidine series (14, 23, 25–29), but additionally some related scaffolds (e.g., imidazopyrimidines (28)). Extensive pharmacology data are already available for many, providing an ideal starting point for potential rapid progress against a new disease indication. Herein, we have used this resource to identify inhibitors of AbDHODH that are active in vitro against live A. baumannii, including CR-XDR strains. In a proof-of-concept study, we also showed in vivo activity in the mouse thigh infection model. We further confirmed the essentiality of AbDHODH in vivo using genetic knockout strains and used these strains to benchmark the activity of our identified DHODH inhibitors. We solved the X-ray structure of the most potent analog bound to AbDHODH, providing insight into the species selectivity of the series. The identified lead compounds are well positioned to form the basis of lead optimization studies to identify a development candidate for the treatment of A. baumannii and potentially other high-risk bacterial infections.
Results
AbDHODH is Essential In Vivo.
As the reported essentiality of DHODH was based on a whole-genome study, these findings were not fully validated (10). Thus, to formally establish that DHODH (encoded by pyrD) is essential for the infectivity of A. baumannii in vivo, a site-directed DHODH mutant was constructed in the A. baumannii clinical isolate AB307-0294 (AB307). AB307ΔpyrD (ΔpyrD) was unable to grow on defined M9 minimal medium (MM, formulation in supplemental methods), which more closely mimics the in vivo environment than rich laboratory medium (30) (Fig. 1A). By contrast, the wild-type parent AB307, the complemented mutant AB307ΔpyrD/pNLAC[pyrD] (ΔpyrD_C), and ΔpyrD chemically complemented with uracil grew at similar rates. ΔpyrD also failed to grow in human ascites (Fig. 1B) or survive in the rat subcutaneous abscess model (Fig. 1C), whereas the complemented mutant ΔpyrD_C achieved the same plateau density as its wild-type parent AB307 in both growth environments (Fig. 1 B–C). Additionally, survival of mice that underwent intraperitoneal (IP) or pulmonary challenge was significantly greater for those infected with ΔpyrD compared to the parent AB307 (Fig. 1 D–G). After IP and pulmonary challenge, the odds of death were 13.5-fold and 29-fold greater, respectively, when infected with AB307 compared to ΔpyrD (P = 0.0014 and 0.0003), after adjusting for challenge inoculum. These findings demonstrate that AbDHODH is essential for growth and survival ex vivo in human ascites and in vivo in rat and murine infection models, thereby supporting AbDHODH as a potential drug target.
Fig. 1.

AbDHODH is essential for in vitro growth and for survival and infectivity in rat and mouse disease models. (A) Growth comparison in MM between wild-type AB307(wt), the DHODH deletion strain (ΔpyrD), the complemented strain AB307 ΔpyrD/pNLAC[pyrD] (ΔpyrD_C), and ΔpyrD grown with uracil (0.2 mM) rescue. Data represent the mean ± SEM for three independent studies, where each was derived from the average of n = 2 to 3 technical replicates. (B) Growth comparison of AB307 (wt) versus ΔpyrD and ΔpyrD_C cultured in human ascites ex vivo. Data represent the mean ± SEM of the mean for 3 to 5 independent studies, where each was derived from n = 2 technical replicates. (C) Survival in the rat subcutaneous (SQ) abscess model for AB307 (wt) versus ΔpyrD and ΔpyrD_C strains. Growth/survival in minimal medium, human ascites, and the rat model was assessed by measurement of CFU (data are mean ± SEM for n = 4 rats per condition). *P < 0.05 (two-tailed unpaired t tests for the area under the curve), AB307 and ΔpyrD_C compared to ΔpyrD. (D and E) Lethality of AB307 (wt) in the BALB/c murine pneumonia model (D) and in the BALB/c murine IP infection model (E). (F and G) Lethality of ΔpyrD in the BALB/c murine pneumonia model (F) and ΔpyrD in the BALB/c murine IP infection model (G). Dose levels of CFU administered to mice on the day of challenge (day 1) are shown in the plots. Percent survival of mice (as measured by an in extremis state) is plotted versus the days from the challenge. (Panels D–G) After adjusting for challenge inoculum, after IP and pulmonary challenge, the odds of death was 13.5-fold and 29-fold greater, respectively, when infected with AB307 compared to ΔpyrD (P = 0.0014 and 0.0003) (N = 5 for each strain and challenge inoculum).
Identification of Species-Selective AbDHODH Inhibitors.
To identify initial lead compounds targeting AbDHODH for the treatment of A. baumannii, we used our established colorimetric end point 384 well plate HTS assay (31) to screen our triazolopyrimidine malaria DHODH compound collection, which as noted above contains ~450 triazolopyrimidine or related molecules, for inhibitors of purified recombinant AbDHODH. The collection was screened at an initial concentration of 3 μM, and compounds that showed >90% inhibition (Supplemental Excel HTS file) were then retested in our standard steady-state rate assay (26) to determine the IC50 against AbDHODH, identifying 18 compounds with an IC50 < 1 μM (SI Appendix, Table S2). We identified potent inhibitors from three of the core structure variants represented in the library, including the triazolopyrimidines, imidazopyrimidines, and 2-methylquinolines. Representative examples, including the most potent from each structural class, were analyzed in more detail (Table 1). The most potent compound (DSM186) has an imidazopyrimidine core and where direct comparisons between matched compounds were possible, the imidazopyrimidines were more potent than the triazolopyrimidines (e.g., DSM186 was 10-fold more potent than DSM161) (Table 1). A second hit from the imidazopyridine core (DSM187; SI Appendix, Table S2) also showed good activity (IC50 = 0.17 μM) as did the 2-methylquinoline DSM250 (Table 1). DSM186 with an IC50 of 0.028 μM showed similar potency toward AbDHODH as that observed for DSM265 toward PfDHODH (IC50 = 0.020 μM) (Table 1 and SI Appendix, Table S2). Notably, the species selectivity profile between AbDHODH and PfDHODH is markedly different, and while DSM265 is the most potent analog versus PfDHODH, DSM186 shows the best potency toward AbDHODH. In general, while C2 substitution increased the affinity of compounds toward PfDHODH, it generally reduced affinity to AbDHODH.
Table 1.
Enzyme and live whole cell activity of select AbDHODH hit compounds
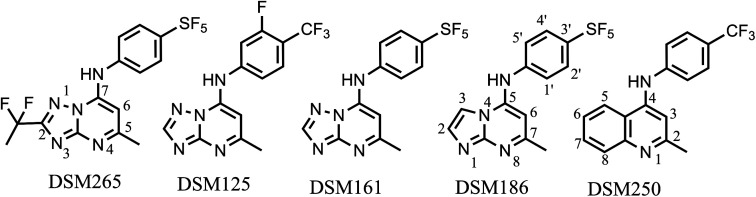
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Enzyme activity data | A. baumannii in vitro assays | |||||||
| Cmpd ID | AbDHODH IC50 μM | HsDHODH IC50 μM | AB307 EC50 μM | AB307 MIC μM | HUMC EC50 μM | HUMC MIC μM | LAC-4 EC50 μM | LAC-4 MIC μM |
| DSM265 | 0.59 ± 0.16 (3) | >100* | 2.7 ± 0.21 (3) | 8.3 ± 2.1 (3) [3.4] | ND | ND | ND | ND |
| DSM125 | 0.91 ± 0.070 (6) | >100* | 9.6 | 14[4.4] | 34 | 51[16] | ND | ND |
| DSM161 | 0.30 ± 0.11 (3) | >100* | 1.4 ± 0.30 (3) | 2.6 ± 0.50 (3) [0.91] | 8.4 ± 0.85 (5) | 25 ± 0. (5) [8.8] | 4.4 ± 0.47 (3) | 25 ± 0.0 (3)[8.8] |
| DSM186 | 0.028 ± 0.0038 (7) | >100 | 0.053 ± 0.0082 (5) | 0.60 ± 0.11 (5) [0.21] | 0.17 ± 0.0033 (3) | 0.78 ± 0.0 (3) [0.27] | 0.17 ± 0.033 (3) | 1.3 ± 0.0 (3)[0.45] |
| DSM250 | 0.085 ± 0.015 (2) | >100 | 0.38 ± 0.14 (4) | 2.8 ± 1.2 (3) [0.84] | ND | ND | 1.6 | 12.5 [3.8] |
AbDHODH, A. baumannii DHODH; HsDHODH, human DHODH; AB307, AB307-0294; HUMC1 CR-XDR strain; MDR-LAC-4. ND, not determined. Data were collected with n=3 (enzyme) or n=2-4 (Ab in vitro) technical replicates for each concentration in the dose response and data represent the mean ± standard error of the mean for multiple independent studies (number of replicates in parenthesis).
*Data were previously reported (25, 27). See also SI Appendix, Table S2 for data on the full set of identified hit compounds. The reported MIC and EC50 values for the A. baumannii in vitro assays are based on the Abs600 data collected after 24 h of growth. Data in brackets [] represent values in μg/mL.
All identified hit compounds were selective inhibitors of AbDHODH and none inhibited human DHODH, providing the basis for selective toxicity toward A. baumannii (Table 1). This finding matches the results we observed for PfDHODH throughout our work on this series, where strong species selectivity versus the human enzyme was typically achieved for most analogs. The two top compounds (DSM186 and DSM161) were also tested for cytotoxicity versus a human HepG2 line. Neither compound inhibited growth of this cell line up to the highest tested concentration (CC50 > 50 μM).
AbDHODH Inhibitors are Active against Drug-Sensitive and Drug-Resistant A. baumannii.
Compounds from each structural class were selected for testing against A. baumannii using established in vitro assays to determine the minimal inhibitory concentration (MIC) (via Å600 and colony-forming units (CFU)), EC50, and time-kill activity against both drug-sensitive (AB307) and MDR and CR-XDR (LAC4 and HUMC1, respectively) isolates (Table 1 and Fig. 2). We observed a strong correlation between the IC50 for AbDHODH versus the IC50 for bacterial cells grown in pyrimidine-deficient medium, a growth environment that is similar to human body fluids (30). Paralleling the enzyme results, DSM186 was the most active compound and showed minimal (3-fold) difference between the drug-sensitive (AB307) and drug-resistant strains (Table 1 and Fig. 2 A and B). The EC50 as measured by Å600 for DSM186 ranged from 0.053 (AB307) to 0.17 (HUMC1 and LAC-4) μM, values which were within 2 to 6-fold of the potency measured on the enzyme. DSM161 showed reasonable potency versus drug-sensitive AB307 (EC50 = 1.4 μM) but activity dropped off 3–6-fold for HUMC1 and LAC-4 (Table 1 and Fig. 2 A and B). Similar results were observed for DSM125 and DSM250. MIC as measured by CFU for DSM186 (1.6 µM (0.55 µg/mL)) for both AB307 and HUMC1) was in reasonable agreement with the MIC measured by Å600 (0.60 and 0.78 µM, respectively, Table 1 and Fig. 2 C and D). For DSM161, the MIC as measured by CFU was 10-fold higher than that observed by the absorbance assay for both strains (CFU MIC 25 µM (8.8 µg/mL) for AB307 and 50 µM (17.5 µg/mL) for HUMC1).
Fig. 2.

DSM161 and DSM186 activity against A. baumannii in vitro. Assays were performed in MM in the presence or absence of compounds as indicated. (A and B) Dose–response curves showing normalized Å600 (relative to DMSO control) at 24 h for DSM161 and DSM186 against AB307 (A) and HUMC1 (B) plus and minus uracil rescue (0.2 mM). Representative data derived from 3 to 5 independent studies (averaged from 2 to 4 technical replicates). EC50 and MIC data derived from the multiple biological replicates are presented in Table 1. (C and D) MIC as measured by CFU for DSM161 and DSM186 against AB307 (C) and HUMC1 (D). Data represent the mean ± SEM for three independent studies, where each was derived from n = 3 to 4 technical replicates, with the exception that for the HUMC1 DSM186 + uracil, only two independent studies were performed. (E–G) Time-kill assay for DSM161 (50 µM; 17.5 µg/mL), DSM186 (2.9 µM; 1 µg/mL), and a meropenem control (2.6 µM; 1 µg/mL) against AB307 (E), HUMC1 (F), and AB307ΔpyrD (G). Data represent the mean ± SEM for 2 to 4 independent studies, where each was derived from n = 2 to 5 technical replicates with exception that for (G) plus uracil and meropenem controls represent only a single independent study. (H) Impact of efflux inhibitor PAβN and the permeability-increasing agent PMBN. Assays were performed in MM in the presence or absence of DSM186 (0.043 µM), PAβN, PMBN, and/or DMSO. Data represent the mean ± SEM for two independent studies, where each was derived from n = 2 technical replicates.
DSM186 at concentrations >1 µg/mL reduced CFUs below the initial inoculum level, suggesting that it has cidal activity, whereas at the highest concentrations assessed, DSM161 was bacteriostatic (T = 0 titer 5 × 105 CFU/m) (Fig. 2 C and D). Thus, time-kill assays were performed at 1 µg/mL and 17.5 µg/mL for DSM186 and DSM161, respectively, and compared to 1 µg/mL of meropenem, an established, clinically utilized antibacterial agent (Fig. 2 E and F). The CFU of the drug-sensitive strain AB307 decreased below the starting inoculum over time after exposure to DSM186, DSM161, and meropenem (Fig. 2E). Importantly, the CFU of the CR-XDR strain HUMC1 decreased below the starting inoculum over time after exposure to DSM186, but not meropenem as expected (Fig. 2F), thereby demonstrating the potential utility of this class of compounds in the treatment of problematic CR-XDR carbapenem-resistant A. baumannii.
AbDHODH Inhibitors Show on Target Toxicity.
Growth of AB307 and HUMC1 was rescued by the addition of uracil in vitro in the presence of DSM186 (Fig. 2 A, B, E, and F, and uracil rescue was also demonstrated for the growth of AB307 after treatment with DSM265, DSM161, and DSM250 (SI Appendix, Fig. S1), although DSM250 did show some off-target activity at concentrations above 10 µM. These data provide strong evidence that these compounds are acting on target in the pyrimidine biosynthetic pathway. Consistently, neither DSM186 nor DSM161 caused increased killing when time-kill curves were performed with the DHODH deletion mutant ΔpyrD (Fig. 2G) in pyrimidine-deficient medium, further supporting specificity.
Impact of Efflux and Permeability on DSM186 Antibacterial Activity.
The EC50 for antibacterial activity on AB307 is ~2-fold higher than that required for enzyme inhibition. To determine whether this difference was due to efflux and/or decreased permeability, AB307 was treated with a sub-MIC concentration of DSM186 (0.015 µg/mL) in the presence or absence of the efflux inhibitor phenylalanine arginine β-naphthylamide (PAβN, 70 µg/mL) (32) and the permeability-increasing agent polymyxinB nonapeptide hydrochloride (PMBN, 64 µg/mL). Both PAβN and PMBN somewhat increased the activity of DSM186 against AB307 (Fig. 2H), suggesting that both efflux and permeability reduced the efficacy of DSM186 against A. baumannii. Interestingly, the combination of PAβN and PMBN was less active than PMBN alone. These data are consistent with known liabilities of antimicrobials directed against enteric GNB and have the potential to inform downstream medicinal chemical optimization studies.
Testing of DSM186 on a Panel of A. baumannii Strains.
DSM186 was tested for activity against an additional 27 A. baumannii strains (SI Appendix, Table S3), including 12 isolates resistant to the carbapenem-meropenem (MIC > 8 µg/mL) and three intermediate isolates (MIC 4 to 8 µg/mL). When assessed by Å600, the MIC for DSM186 ranged from 0.11 to 1.09 µg/mL for meropenem-sensitive strains and from 0.27 to 3.0 µg/mL for meropenem-intermediate and -resistant strains (SI Appendix, Table S3). MIC was also assessed by CFU for 23 strains; DSM186 inhibited the growth of 17/23 strains at 1 µg/mL (2.9 µM) relative to the starting inoculum (± ½ log) at 24 h and at 2 µg/mL (5.8 µM) for the remaining six strains (SI Appendix, Fig. S2). These data demonstrate that DSM186 inhibits growth of all geographically diverse A. baumannii strains tested, including carbapenem-resistant isolates.
X-Ray Structure of AbDHODH Bound to DSM186.
The X-ray structure of AbDHODH bound to DSM186 was solved by molecular replacement to 1.4 Å resolution (Rwork/Rfree of 15.7%/17.5%, respectively) using the previously reported structure of E.coli DHODH as the search model (PDB ID 1F76) (33) (SI Appendix, Table S4 and Fig. S3). The two bacterial structures are overall very similar (Fig. 3 A and B), though the inhibitor-binding site does contain a couple of significant changes expected to lead to differential inhibitor binding (e.g., AbL318 to EcF321 and AbL74 to EcM77) (Fig. 3B and SI Appendix, Fig. S3). The structure shows that DSM186 binds into the inhibitor pocket in a similar orientation to that previously observed for binding of DSM265 to PfDHODH (14) (Fig. 3C). Like DSM265, H-bonds are formed between the inhibitor and two conserved binding pocket residues (H19 to NH and R99 to N8) that likely contribute to binding affinity. The SF5 group also binds in a largely hydrophobic pocket (e.g., L71, L74, and L318), and a narrow open channel is present between the FMN cofactor and the C2 and C3 positions of DSM186 that could be exploited to build additional functionality into the molecule (see Table 1 for atom numbering). However, in contrast to PfDHODH, the AbDHODH pocket in the region of C6 and C7 of DSM186 is more hydrophilic owing to the substitutions of T22-AbDHODH for F188-PfDHODH, and of Y3-AbDHODH for L172-PfDHODH (Fig. 3C and SI Appendix, Fig. S3). These differences in amino acid composition provide the structural basis for differences in compound affinity between the two enzymes. Additionally, R7 is turned toward the binding pocket where it stacks against the pi cloud of F11 in AbDHODH, which places this charged residue closer to the binding pocket than the equivalent (K173) residue in PfDHODH. In AbDHODH, the Y3 hydroxyl forms a close H-bond interaction with the backbone carbonyl of I319, while the hydroxyl of T22 makes an H-bond with the backbone carbonyl of A18.
Fig. 3.

AbDHODH structure bound to DSM186 determined by X-ray crystallography. Structural comparison of the AbDHODH (green) bound to DSM186 with DHODH from other species. (A) Ribbon diagram overlay with E. coli DHODH (PDB ID 1F76) (pink), which was used as the search model for phase determination. Structures were superimposed to an RMSD = 1.04 Å (over Cα of 323 residues). DSM186, FMN, and L-orotate are shown as spheres. (B) Inhibitor-binding site alignment with E. coli DHODH. (C) Inhibitor-binding site alignment with P. falciparum DHODH (blue) bound to DSM265 (PDB ID 4rxo) (RMSD = 1.57 Å over 306 residues) and (D) Inhibitor-binding site alignment with human DHODH (tan) (PDB ID 1D3h) (RMSD = 1.40 Å over 301 residues) bound to A77 1726 (A77). (B–D) Bound inhibitors are shown as ball and stick. Structures were superimposed using Phenix version 1.20.1-4487 (phenix.superpose_and_morph) and structures were displayed in PyMOL (The PyMOL Molecular Graphics System, Version 2.5.2 Schrödinger, LLC). The AbDHODH-DSM186 structure has been submitted to the Protein Data Bank (PDB ID 7UT5).
As noted above, we observe strong species selectivity of DSM186 toward AbDHODH with no measurable inhibitor activity on the human enzyme. The structural basis for this difference is apparent from the comparison of the X-ray structures of the two enzymes (Fig. 3D and SI Appendix, Fig. S3). Several amino acid differences are observed in the inhibitor-binding pocket that contribute to this selectivity.
An AbDHODH multisequence comparison using sequences obtained from an ortholog database shows that the overall amino acid identity was >98% and was 100% for 7 of 8 (34, 35). The exception to 100% identity was sequenced strain SDF, which was isolated from human body lice. The AbDHODH inhibitor-binding site, as defined in SI Appendix, Fig. S4, was 100% identical across all the eight strains, confirming that this binding pocket is a conserved feature across A. baumannii strains. Collectively, these data suggest that inhibitors of DHODH would be broadly active against strains of A. baumannii.
ADME Assessment of the Identified AbDHODH Inhibitors.
Based on our historical data, DSM125 and DSM161 have good metabolic stability and PK properties in mice and rats (SI Appendix, Table S5), including prolonged plasma exposure after either oral or IV dosing (25, 27). DSM186 is metabolically stable in human liver microsomes but is less stable in mouse microsomes. Aqueous solubility at physiological pH ranged from 10 to 70 μM for these compounds. To assess the potential for DSM186 and DSM161 to be dosed in a proof-of-concept in vivo model of A. baumannii, we performed a preliminary PK study in mice to establish the relationship between dose and plasma exposure after a single oral dose of 100 mg/kg (SI Appendix, Table S6 and Fig. 4A). The oral dosing route was chosen because the compound collection was optimized for good oral exposure based on the goal of developing an antimalarial drug. Good plasma exposure was observed for both compounds, although as predicted by in vitro metabolism data, exposure for DSM186 was lower than that for DSM161 due to a significantly shorter half-life. Based on these data and considering plasma protein binding (SI Appendix, Table S5), we estimated that a dose in the range of ~200 mg/kg/day for both DSM161 and DSM186 would be required to achieve plasma concentrations above the plasma equivalent AB307 MIC (i.e., binding corrected) for 24 h if dosed as a split dose twice daily (Fig. 4A). While DSM161 is less potent than DSM186, this is counterbalanced by higher and more sustained plasma exposure, leading to similar dose estimations for both compounds, though the data suggested that DSM161 was more likely to achieve levels above MIC for longer in the 24-h period than DSM186. However, uncertainty remains regarding the concentrations that are achieved in various tissue compartments that would be important for the treatment of A. baumannii infection (e.g., lung, thigh muscle).
Fig. 4.

In vivo activity of DSM161 in a neutropenic thigh infection model. (A) Plasma exposure of DSM161 (green) and DSM186 (pink) in uninfected mice after oral dosing at 100 mg/kg. Data represent the mean ± SD for n = 3 mice. Plasma concentrations were measured over time and calculated PK parameters are reported in SI Appendix, Table S6. Dashed lines represent plasma-bound MIC for DSM161 (green) and DSM186 (pink) based on absorbance-based MIC data for AB307 from Table 1 corrected for protein binding (SI Appendix, Table S5 values). (B–D) Growth/survival of AB307 (wt) and AB307ΔpyrD (DHODH negative) was assessed by measurement of CFU 24 h post infection. Cohorts of AB307-infected mice were treated with either vehicle control (n = 5) (20% PEG400, 10% Cremophor EL and 70% water OG at T = 0 and 12 h), meropenem (n = 8) (100 mg/kg IP at T = 0, 6, 12, and 18 h), or DSM161 (n = 12) (100 mg/kg OG at T = 0 and 12 h). Animals challenged with AB307ΔpyrD (n = 8) were treated with vehicle alone. Animals were killed at 24 h, and blood, thigh, and spleen were harvested for CFU determination. B. Blood CFU. C. Spleen CFU. D. Thigh CFU. Data are mean ± SEM, with individual data for each mouse shown on the graph. *P < 0.05 (two-tailed unpaired t tests), AB307 challenged control animals compared to the other groups.
Efficacy of DSM161 in the Neutropenic Mouse Thigh Infection Model.
Based on its good oral exposure profile that suggests the binding-corrected MIC could be achieved with BID dosing of 100 mg/kg, we chose to perform an in vivo efficacy study with DSM161 using the neutropenic mouse thigh infection model (Fig. 4 B–D). Given that hit compounds from our screen have not yet been optimized for use in treating A. baumannii infection, our goal was to demonstrate at least partial efficacy to provide an in vivo proof-of-concept of the DHODH target. Three cohorts of BALB/c mice, rendered neutropenic with vinblastine, were challenged with 1 to 2 × 106 CFU via injection of AB307 into the left thigh muscle. The cohorts were treated with vehicle control, DSM161 (100 mg/kg dosed orally at 0 and 12 h), or meropenem (100 mg/kg dosed by IP at 0, 6, 12, and 18 h). A fourth cohort of mice were infected with ΔpyrD (DHODH negative) as a control. Animals were killed at 24 h, and blood, thigh, and spleen were harvested for CFU determination. Both meropenem and DSM161 significantly decreased CFUs compared to untreated controls at all the three sites. CFU was decreased even further for the ΔpyrD strain compared to that of the wild-type Ab307 control. Quantitatively, the effects were greatest in blood and spleen. Despite DSM161 not being the most potent compound identified, it was able to demonstrate partial efficacy, thus these data provide proof-of-concept that DHODH inhibitors can protect against bacterial infection. This study was conducted over only a short 24 h dosing period. Future studies on more optimized compounds would likely need to include dosing time as a variable. Indeed, the cell-killing activity of DHODH inhibitors versus the malaria parasite Plasmodium falciparum was shown to have a 24 to 48 h lag phase before strong killing commenced (14, 16). Presumably, because it takes some initial growth before nucleotide depletion is evident, similar lag phases may also be at play for A. baumannii, suggesting that longer treatment times will significantly improve efficacy. Importantly, the degree of protection is predicted to be significantly greater with compounds optimized for more potent activity against A. baumannii while retaining the strong PK properties of DSM161.
In Vitro and In Vivo Resistance Studies.
In silico analysis does not support resistance via the development of a pyrimidine by-pass pathway (SI Appendix, Table S1). However, this consideration was experimentally assessed. ΔpyrD was assessed for growth in MM medium after overnight growth in either rich LB medium or M9 medium plus uracil. These overnight cultures were washed, concentrated, resuspended in MM, and approximately 2.0 × 109 CFU was intermittently plated on MM over 7 d. No breakthrough growth was observed. To assess for preexisting drug resistance, 1 × 109 CFU of AB307 and HUMC1 was plated on MM + 1 µg/mL of DSM186. No breakthrough growth occurred. To evaluate for the potential of resistance development during treatment with DHODH inhibitors in vivo, colonies of AB307 recovered from DSM161-treated animals were tested. Eight independent colonies from three different sites were assessed from three animals, and no change in MIC, as measured by CFU, was observed (SI Appendix, Table S7).
Discussion
The identification of new classes of antimicrobials active against CR-XDR GNB such as A. baumannii has been a difficult process (36, 37). Herein, we describe the identification of AbDHODH as a potential antimicrobial target for A. baumannii. By repurposing a customized, noncommercial library (~450 compounds) created for drug development efforts targeting PfDHODH, compounds that are active against purified AbDHODH at nM concentrations were identified. Critically, the compounds from these series are also active against A. baumannii, demonstrating a good degree of permeability and retention, and thus overcoming what is often a rate-limiting step in GNB drug development (36, 37). Importantly, DSM186, the most active compound in vitro, was active against all the 30 strains tested, including 12 CR-XDR A. baumannii isolates. Both genetic studies using an isogenic DHODH-deficient derivative and uracil rescue studies established that A. baumannii killing was target specific, while structural studies defined the inhibitor-binding pocket on AbDHODH. DSM161 demonstrated significant but partial efficacy in the murine thigh infection model consistent with its relatively modest potency against Ab307 in vitro. Encouragingly, the development of resistance to these compounds was not identified in vitro or in vivo. Taken together, the identification of drug-like triazolopyrimidine/imidazopyrimidine compounds with potent in vitro activity against A. baumannii forms the basis for the development of derivatives with improved properties that holds promise for the development of a new class of antimicrobial agents active against CR-XDR A. baumannii.
In early work, target-based approaches did not always yield good lead molecules because insufficient consideration was given to the druggability of the target versus its essentiality (38). More recently, target-based approaches have been recognized to have significant potential (37). Our work leading to the identification of the antimalarial DSM265 is an example that supports the promise of the target-based approach (14). While DHODH has been verified as a druggable target in eukaryotes and has been the focus of significant chemistry to identify inhibitors of eukaryotic enzymes (11), it has received less attention as a potential target in bacteria. Early studies on E. coli and H. pylori reported promising results (18, 19), but DHODH as a target has neither been progressed for these species nor considered for A. baumannii. The identification of drug-like triazolopyrimidine/imidazopyrimidine derivatives that are active against purified recombinant AbDHODH, have antibacterial activity against A. baumannii, including CR-XDR-isolates, and that show efficacy in a murine thigh infection model, provides chemical validation of the target.
Despite the excellent in vitro potency of DSM186 and to a lesser degree for DSM161, additional lead optimization is needed to achieve the desired properties for the treatment of A. baumannii infections. While the potency of DSM186 against the AB307 strain of A. baumannii is good for an initial hit compound (EC50 = 53 nM), it would need to be improved ~10-fold to bring it into the range expected of an optimized drug candidate (e.g., DSM265), particularly given the high plasma protein binding. While we were able to show partial in vivo efficacy for DSM161, the lack of potency likely led to unbound plasma concentrations below the MIC during the mouse efficacy study. Furthermore, the extent that concentrations in infected tissues remained above the unbound MIC remains unknown. Compounds in the PfDHODH inhibitor library were developed based on the product profile required for the treatment of malaria, including oral bioavailability and prolonged exposure in blood (14). By contrast, A. baumannii primarily infects extravascular sites (e.g., lung) in hospitalized patients, and an ideal product profile for severe infection is a parenteral formulation with a greater tolerance for more than once-a-day dosing. Therefore, different compound properties may need to be built-in during lead optimization to suit the clinical needs for treatment of A. baumannii. Additionally, DSM186 exhibits 2- to 3-fold lower potency on drug-resistant A. baumannii strains, and antibacterial activity was improved in the presence of efflux inhibitors and/or permeability enhancers. Thus, while promising, the imidazopyrimidine series (to which the DSM186 belongs) will require optimization to improve antibacterial activity and the desired ADME properties (e.g., solubility, protein binding) to improve in vivo activity at the typical sites of CR-XDR A. baumannii infection. Future hit expansion and lead optimization studies will be aided by our determination of the X-ray structure of AbDHODH bound to DSM186 to 1.4 Å resolution, which has identified several novel features of the AbDHODH binding site.
A perceived limitation of this class of compounds is that in vitro activity could be bacteriostatic. However, the CFU of both the drug-sensitive strain AB307 and the drug-resistant strain HUMC1 decreased below the starting inoculum over time after exposure to DSM186 at concentrations >1 µg/mL in vitro (Fig. 2 C–F), suggesting that it has cidal activity. Further, in vivo host factors contribute to bacterial clearance. This phenomenon can be gleaned by comparing growth/survival of ΔpyrD, an isogenic DHODH-minus derivative of AB307 in minimal medium to that observed in human ascites ex vivo, and in the rat subcutaneous abscess model (Fig. 1 A–C). When host factors are present, clearance is enhanced, supporting the concept that in vitro activity incompletely translates to the treatment of infection in vivo (39).
In summary, the data presented lay the foundation for logical lead progression and lead optimization of AbDHODH inhibitors with the long-term goal of identifying a late lead compound with the properties needed to advance to preclinical development. Such a compound that possesses improved solubility and PK/PD properties would represent a sorely needed new class of antimicrobials with activity against CR-XDR A. baumannii and perhaps other CR-XDR GNB.
Materials and Methods
Bacterial Strains and Media.
See SI Appendix, Supplemental Material and Methods and Table S3 for details on strains and media. The ΔpyrD (pyrD encodes DHODH) strain, which is an isogenic DHODH-minus derivative of AB307, was created by site-directed mutagenesis using pBlueScript[pyrD::kanamycin] as a suicide plasmid in which 818bp of pyrD was replaced with a kanamycin-resistant cassette. The successful disruption of pyrD in ΔpyrD was confirmed by sequence analysis of a PCR-generated amplicon using primers outside of pyrD (5′-ACCAACCAAACTTGTGG-3′, plus, 5′-TTTTCGGCTATTTCTATTCT-3′, minus). Polar effects were excluded as described (9) and the complemented derivative ΔpyrD_C) was generated to confirm that the observed phenotypic differences between AB307 and ΔpyrD were due to DHODH.
Expression and Purification of AbDHODH.
AbDHODH cloning and heterologous expression in E. coli.
Primers (sense, 5′-GCGCGACATATGTTATATTCACTTGCTCGC-3′; antisense, 5′-GCATGACTCGAGTTAAGTCATGGCTTCAACAC-3′) were synthesized (Integrated DNA Technologies, Iowa, USA) and used to PCR amplify the fragment of the AB307 pyrD (ABBFA_001187) encoding DHODH (corresponding to amino acid residues 1-334 of the transcribed gene product) (40). The PCR product was ligated into pGEM-T Easy and subsequently cloned into both pET-duet-SUMO and pET28b as described in SI Appendix, Supplemental Material and Methods. The pET-SUMO-AbpyrD construct was used for the expression of protein for library screening to identify hit compounds as described in supplemental methods. All other studies used the N-terminal His6-tagged pET28b-AbDHODH vector for protein expression in BL21 phage-resistant cells (Novagen). Protein was expressed and purified as previously described (23) with modifications (supplemental methods).
Crystallization and Structure Determination of AbDHODH Bound to DSM186.
AbDHODH was premixed with DSM186 (1 mM) before crystallization and incubated for 1 h, followed by centrifugation to remove precipitated compound. After centrifugation, protein/DSM186 solution was mixed with reservoir solution at a one-to-one (v/v) ratio. Random crystallization screen kit Cryo (Hampton Research) and detergent screen kits (Hampton Research) were utilized to determine preliminary crystallization conditions. Detergent screens showed that addition of Anapoe C10E9 improved AbDHODH crystallization, so it was added (0.1%) to the final purification step (above). AbDHODH crystals for data collection were obtained at a reservoir solution of 0.085 M Hepes pH 7.5, 8.5% 2-propanol (v/v), 17% PEG4000 (w/v), and 15% glycerol (v/v) at 20 °C. The crystals were frozen in liquid nitrogen for data collection.
Crystal diffraction data were collected at 100 K on beam line 19 BM at the Advanced Photon Source. Crystallographic phases for AbDHODH structure were solved by molecular replacement with Phaser (41) using the previously reported structure of E.coli DHODH (PDB ID 1F76 (22)) as a search model. The resulting model from Phaser was used as a starting model for the AutoBuild routine of Phenix (42). Structures were rebuilt with Coot (43) and refined in Phenix to Rwork and Rfree of 0.166 and 0.174, respectively (SI Appendix, Table S4).
Structural modeling was performed using The PyMOL Molecular Graphics System, Version 2.5.2, Schrödinger, LLC. The AbDHODH-DSM186 data set has been submitted to the Protein Data Bank (PDB ID 7UT5).
Compound Collections and Chemical Synthesis.
Compound collection for screening.
The compound collection of ~450 triazolopyrimidines and related scaffolds (e.g., imidazopyrimidines) was synthesized over the course of our PfDHODH drug discovery project and was stored in HTS-compatible plate format at −20 °C in DMSO. Synthesis and characterization of these compounds has been described in a series of publications (14, 23, 25–29). The complete compound list can be found in the Supplemental Excel HTS file.
Synthesis of hit compounds.
For specific compounds that were followed up in this study, DSM161 (27) and DSM125 (25) were synthesized as previously described. The synthetic methods, schemes, and experimental data for the synthesis of DSM186, DSM187, and DSM250 are described in SI Appendix, Supplemental Materials and Methods.
Screening to Identify AbDHODH Inhibitors from the Preexisting PfDHODH Library.
To identify inhibitors of purified recombinant AbDHODH, we used our established high-throughput, colorimetric end point assay in 384 well plate format (31) that follows the reduction of 2,6-dichloroindophenol (DCIP) at 595 nM. Reactions were performed in assay buffer (0.1 M HEPES, pH 8.0, 0.15 M NaCl, 10% glycerol, 0.10% Triton X-100) mixed with substrates to yield final concentrations as follows: L-DHO (200 μM), CoQ (20 μM), and DCIP (120 μM). The mixture (0.045 mL) was dispensed into assay plates, and compound stocks prepared at 100× in DMSO were dispensed to plates containing the buffer mixture using a Biomek FX robotic liquid handling device (Beckman, Inc.) to a final concentration of 3 μM. Reactions were initiated by addition of 0.005 mL AbDHODH enzyme stock (10 μM) in assay buffer (final concentration of 200 nM) and the reactions were allowed to proceed for 20 min before quenching with a final concentration of 1% SDS. Plates were spun to remove bubbles (3000 rpm for 1 min), and absorbance at 595 nm was read using an Envision plate reader. The assay gave robust Z values (average Z’ = 0.66 over 6 plates in the AbDHODH 500 molecule screen). DSM125, identified as an AbDHODH inhibitor during some preliminary spot screen of the library, was plated at 3 µM as the positive control and DMSO was used as the negative control.
Steady-State Kinetic Analysis of AbDHODH Inhibitors.
Compounds that showed >90% inhibition at 3 μM in the primary screen were retested at multiple concentrations (between 0.001 and 100 μM) in our standard steady-state rate assay (26) to determine the 50% inhibitory concentration (IC50) on AbDHODH. Substrate and buffer conditions are described above, with the modification that the final enzyme concentration in these assays was 40 nM. Multiple technical replicates (n = 3) were collected per inhibitor concentration for each independent dose–response curve. Data were fitted to inhibitor vs response – variable slope (four parameters) equation in GraphPad Prism to determine the IC50.
In Vitro Assays against Live A. baumannii.
Antimicrobial susceptibility testing.
Bacterial strains were grown for 18 h in MM plus casamino acids. Bacteria were diluted 1:1,000 in the same medium and titers were confirmed by performing serial 10-fold dilutions and enumeration on LB plates plus/minus appropriate antibiotics. MIC were determined by the measurement of optical density in 96-well microtiter plates containing various concentrations of DSM compounds (resuspended in DMSO) in 100 µL of the appropriate medium to which 100 µL of the same medium containing bacteria is added, with a final bacterial concentration of approximately 5 × 105 CFU/mL of the strain being assessed. Control wells contained medium only or DMSO. Plates were incubated with double-orbital shaking for 24 h at 37 °C in an Epoch 2, Biotek, spectrophotometer with Å600 measured in each well every 15 min. The MIC was determined by the lowest concentration of drug that inhibited more than 85% of bacterial growth relative to the no drug condition calculated by the following formula: (Å600 of treatment group / Å600 of the nontreatment group) × 100. Visible growth is first discerned at approximately 1 to 2 × 107 CFU/ml. MIC was also determined by measuring CFUs in wells at 24 h; the MIC as assessed by CFU was designated as the drug concentration that inhibited growth ± ½ log relative to the starting inoculum when measured at 24 h. Doses ranged from 0.050 µM to 50 µM for DSM161 and from 0.010 µM to 13 µM for DSM186. Time-kill assays were performed in the presence or absence of a fixed DSM drug concentration (DSM161 50 µM (17.5 µg/mL), DSM186 2.9 µM (1 µg/mL)) over time as described (44) and compared to meropenem 2.6 µM (1 µg/mL).
Cytotoxicity assays.
Cytotoxicity was assessed over a range of inhibitor concentrations versus the human HepG2 line as described (45).
Efflux and permeability assays.
Antimicrobial susceptibility testing methodology was used in the presence or absence of the efflux inhibitor PaβN (final concentration 69.8 µg/mL) (32), the permeability-inducing compound PMBN (final concentration 64 µg/mL) (46), and/or DSM186 (0.043 µM (0.015 µg/mL)). Pilot titration experiments were performed that identified the concentration of DSM186 which enabled some growth so that the effect of PAβN and PMBN could be assessed.
ADME and PK Studies.
See SI Appendix, Supplemental Material and Methods for details.
Animal Infection Models.
The rat subcutaneous abscess model and the murine IP and pulmonary challenge infection models have been described (47–49). See SI Appendix, Supplemental Material and Methods for details on animal welfare and these models with minor modifications.
Mouse thigh infection.
BALB/c mice (18 to 22 g) were treated IP with vinblastine (5.0 mg/kg, administered in 125 µL of water) 72 h prior to bacterial challenge to render them neutropenic. Three animal cohorts were challenged with approximately 1 × 106 CFU of AB307 and one cohort with ΔpyrD via injection into the animal’s thigh; two cohorts infected with AB307 were treated with meropenem (100 mg/kg, dosed IP in 200 µL of 1 × PBS at 0, 6, 12, and 18 h) or DSM161 (100 mg/kg, dosed orally at 0 and 12 h delivered in a suspension of 20% PEG400, 10% Cremophor EL, and 70% water). One cohort infected with AB307 and a cohort infected with ΔpyrD served as no treatment, vehicle controls. At study end (24 h post infection), mice were anesthetized and blood was obtained from the retroorbital space for bacterial enumeration and an assessment of the neutrophil count. Next, the right atrial appendage was incised and the injection of 5 mL of normal saline through the left ventricle flushed the vasculature. This ensured that bacterial counts reflected tissue-associated bacteria. Lastly, the spleen and thigh were harvested and homogenized and bacteria were enumerated via serial dilutions. Animals that were not neutropenic were excluded from the analysis.
Resistance Studies.
See SI Appendix, Supplemental Material and Methods for details.
Ethics.
All animal procedures were conducted according to the code of conduct of the respective countries, either United States or Australia, and were approved by the Ethics Committees of the respective institutions as detailed under the specific studies discussed above.
Statistical Analyses.
Continuous data were assessed for normality and presented as mean ± SEM. P values of 0.05/n (n = the number of comparisons) were considered statistically significant based on the Bonferroni correction for multiple comparisons. For Fig. 1 A–C, to normalize ex vivo growth/survival data, log10 transformed values were utilized, the area under each curve was calculated, and these areas were compared using two-tailed unpaired t tests (Prism 4 for MacIntosh, GraphPad Software Inc.). For Fig. 1 D–G, day 14 mortality was modeled by infection type (pulmonary, IP) as a function of log10(CI) level and strain using exact logistic regression. Nonsignificant strain by log10(CI)-level interactions was removed from the model. Exact test about the odds ratio = 1 in comparing strains was performed at alpha = 0.05 (two-sided) adjusted for log10(CI) level. Exact estimates and the corresponding 95% CIs for the odds were also calculated for strain. For Fig. 4 B–D, two-tailed unpaired t tests were utilized.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (PDF)
Acknowledgments
We thank the Structural Biology Laboratory at UT Southwestern Medical Center for support with X-ray crystallographic studies and the Preclinical Pharmacology Core for assistance with protein-binding assessment of DSM161 and DSM186. MAP holds the Sam G. Winstead and F. Andrew Bell Distinguished Chair in Biochemistry. Results shown in this report are derived from work performed at the Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. SBC-CAT is operated by UChicago Argonne, LLC, for the US Department of Energy, Office of Biological and Environmental Research, under contract DE-AC02-06CH11357. This work was supported by the National Institutes of Health grant R01AI103947 (M.A.P., P.K.R.), the National Institutes of Health grant R56AI129986 (T.A.R., M.A.P., P.K.R.), the Department of Veterans Affairs VA Merit Review 1I01BX000984 and 1I01BX004677-01A1 (T.A.R.), and The Welch Foundation I-1257 (M.A.P.).
Author contributions
T.A.R., T.C.U., S.K., B.P., P.K.R., S.A.C., and M.A.P. designed research; T.C.U., X.D., F.E., S.K., R.O., U.C.-M., J.B., C.L.A., A.H., and H.C. performed research; T.A.R., T.C.U., X.D., F.E. S.K., D.R.T., B.P., S.A.C., and M.A.P. analyzed data; and T.A.R. and M.A.P. wrote the paper.
Competing interest
Dr. Thomas Russo is involved in a consortium NIH grant (2R01AI130060-06) with a reviewer of the paper Dr. Robert Bonomo. Russo is a subcontractor on the grant with a limited role and has not had direct collaborative interactions with Bonomo. The authors have research support to disclose.
Footnotes
Reviewers: R.A.B., Case Western Reserve University and Louis Stokes Cleveland VA; and M.K.R., Portland VA Medical Center.
Contributor Information
Thomas A. Russo, Email: trusso@buffalo.edu.
Margaret A. Phillips, Email: margaret.phillips@utsouthwestern.edu.
Data, Materials, and Software Availability
The AbDHODH-DSM186 structure has been submitted to the PDB data bank (PDB ID 7UT5). All other data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.CDC, "COVID-19: U.S. Impact on Antimicrobial Resistance" (Special Report, CDC, Atlanta, GA, 2022). [Google Scholar]
- 2.Wong D., et al. , Clinical and pathophysiological overview of Acinetobacter infections: A century of challenges. Clin. Microbiol. Rev. 30, 409–447 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meng X., et al. , Ten-year changes in bloodstream infection with acinetobacter baumannii complex in intensive care units in eastern china: A retrospective cohort study. Front. Med. (Lausanne) 8, 715213 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antimicrobial Resistance Collaborators, Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 399, 629–655 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prasad N. K., Seiple I. B., Cirz R. T., Rosenberg O. S., Leaks in the pipeline: A failure analysis of gram-negative antibiotic development from 2010 to 2020. Antimicrob. Agents Chemother. 66, e0005422 (2022), 10.1128/aac.00054-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doi Y., Treatment options for carbapenem-resistant gram-negative bacterial infections. Clin. Infect. Dis. 69, s565–s575 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iovleva A., et al. , Carbapenem-resistant Acinetobacter baumannii in U.S. hospitals: Diversification of circulating lineages and antimicrobial resistance. mBio 13, e0275921 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palfey B. A., Bjornberg O., Jensen K. F., Insight into the chemistry of flavin reduction and oxidation in Escherichia coli dihydroorotate dehydrogenase obtained by rapid reaction studies. Biochemistry 40, 4381–4390 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Umland T. C., et al. , In vivo-validated essential genes identified in Acinetobacter baumannii by using human ascites overlap poorly with essential genes detected on laboratory media. MBio 3, e00113-12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang N., Ozer E. A., Mandel M. J., Hauser A. R., Genome-wide identification of Acinetobacter baumannii genes necessary for persistence in the lung. mBio 5, e01163-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munier-Lehmann H., Vidalain P. O., Tangy F., Janin Y. L., On dihydroorotate dehydrogenases and their inhibitors and uses. J. Med. Chem. 56, 3148–3167 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Phillips M. A., et al. , Malaria. Nat. Rev. Dis. Primers 3, 17050 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Oliver J. D., et al. , F901318 represents a novel class of antifungal drug that inhibits dihydroorotate dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 113, 12809–12814 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phillips M. A., et al. , A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci. Transl. Med. 7, 296ra111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCarthy J. S., et al. , Safety, tolerability, pharmacokinetics, and activity of the novel long-acting antimalarial DSM265: A two-part first-in-human phase 1a/1b randomised study. Lancet. Infect. Dis. 17, 626–635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Llanos-Cuentas A., et al. , Antimalarial activity of single-dose DSM265, a novel plasmodium dihydroorotate dehydrogenase inhibitor, in patients with uncomplicated Plasmodium falciparum or Plasmodium vivax malaria infection: A proof-of-concept, open-label, phase 2a study. Lancet. Infect. Dis. 18, 874–883 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.C. Trials.gov, Evaluate F901318 treatment of invasive Fungal Infections in Patients Lacking Treatment Options (FORMULA-OLS). https://clinicaltrials.gov/ct2/show/NCT03583164 (2018), p. NCT03583164.
- 18.Copeland R. A., et al. , Helicobacter pylori-selective antibacterials based on inhibition of pyrimidine biosynthesis. J. Biol. Chem. 275, 33373–33378 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Marcinkeviciene J., et al. , Selective inhibition of bacterial dihydroorotate dehydrogenases by thiadiazolidinediones. Biochem. Pharmacol. 60, 339–342 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Phillips M. A., Rathod P. K., Plasmodium dihydroorotate dehydrogenase: A promising target for novel anti-malarial chemotherapy. Infect. Disord. Drug. Targets. 10, 226–239 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu S., Neidhardt E. A., Grossman T. H., Ocain T., Clardy J., Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure 8, 25–33 (2000). [DOI] [PubMed] [Google Scholar]
- 22.Norager S., Jensen K. F., Bjornberg O., Larsen S., E. coli dihydroorotate dehydrogenase reveals structural and functional distinctions between different classes of dihydroorotate dehydrogenases. Structure 10, 1211–1223 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Deng X., et al. , Structural plasticity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds. J. Biol. Chem. 284, 26999–27009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deng X., et al. , Fluorine modulates species selectivity in the triazolopyrimidine class of Plasmodium falciparum dihydroorotate dehydrogenase inhibitors. J. Med. Chem. 57, 5381–5394 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gujjar R., et al. , Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice. J. Med. Chem. 52, 1864–1872 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coteron J. M., et al. , Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J. Med. Chem. 54, 5540–5561 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gujjar R., et al. , Lead optimization of aryl and aralkyl amine-based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with antimalarial activity in mice. J. Med. Chem. 54, 3935–3949 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marwaha A., et al. , Bioisosteric transformations and permutations in the triazolopyrimidine scaffold to identify the minimum pharmacophore required for inhibitory activity against plasmodium falciparum dihydroorotate dehydrogenase. J. Med. Chem. 55, 7425–7436 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kokkonda S., et al. , Tetrahydro-2-naphthyl and 2-indanyl triazolopyrimidines targeting Plasmodium falciparum dihydroorotate dehydrogenase display potent and selective antimalarial activity. J. Med. Chem. 59, 5416–5431 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simmonds H., Duley J., Davies P. "Analysis of purine samples and pyrimidines in blood, urine, and other physiologic fluids" in Techniques in Diagnostic Human Biochemical Genetics. A Laboratory Manual, Hommes F., Ed. (Wiley-Liss, New York, 1991), pp. 397–424. [Google Scholar]
- 31.Baldwin J., et al. , High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J. Biol. Chem. 280, 21847–21853 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Jacobs A. C., et al. , Characterization of the Acinetobacter baumannii growth phase-dependent and serum responsive transcriptomes. FEMS Immunol. Med. Microbiol. 64, 403–412 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Nørager S., Jensen K. F., Björnberg O., Larsen S., E. coli dihydroorotate dehydrogenase reveals structural and functional distinctions between different classes of dihydroorotate dehydrogenases. Structure 10, 1211–1223 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Sievers F., et al. , Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altenhoff A. M., et al. , OMA orthology in 2021: Website overhaul, conserved isoforms, ancestral gene order and more. Nucleic Acids Res. 49, D373–d379 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silver L. L., A Gestalt approach to Gram-negative entry. Bioorg. Med. Chem. 24, 6379–6389 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Tommasi R., Brown D. G., Walkup G. K., Manchester J. I., Miller A. A., ESKAPEing the labyrinth of antibacterial discovery. Nat. Rev. Drug. Discov. 14, 529–542 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Hopkins A. L., Groom C. R., The druggable genome. Nat. Rev. Drug. Discov. 1, 727–730 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Pankey G. A., Sabath L. D., Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections. Clin. Infect. Dis. 38, 864–870 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Adams M. D., Comparative genome sequence analysis of multidrug-resistant Acinetobacter baumannii. J. Bacteriol. 190, 8053–8064 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCoy A. J., Solving structures of protein complexes by molecular replacement with Phaser. Acta. Crystallogr. D. Biol. Crystallogr. 63, 32–41 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams P. D., et al. , PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta. Crystallogr. D. Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta. Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Marr C. M., MacDonald U., Trivedi G., Chakravorty S., Russo T. A., An evaluation of BfmR-regulated antimicrobial resistance in the extensively drug resistant (XDR) Acinetobacter baumannii strain HUMC1. Front. Microbiol. 11, 595798 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lawong A., et al. , Novel antimalarial tetrazoles and amides active against the hemoglobin degradation pathway in Plasmodium falciparum. J. Med. Chem. 64, 2739–2761 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muller C., Plesiat P., Jeannot K., A two-component regulatory system interconnects resistance to polymyxins, aminoglycosides, fluoroquinolones, and beta-lactams in Pseudomonas aeruginosa. Antimicrob. Agents. Chemother. 55, 1211–1221 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Russo T. A., Rat pneumonia and soft-tissue infection models for the study of Acinetobacter baumannii biology. Infect. Immun. 76, 3577–3586 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Russo T. A., Olson R., MacDonald U., Beanan J., Davidson B. A., Aerobactin, but not yersiniabactin, salmochelin, or enterobactin, enables the growth/survival of hypervirulent (hypermucoviscous) Klebsiella pneumoniae ex vivo and in vivo. Infect. Immun. 83, 3325–3333 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Russo T., Sharma G., Brown C., Campagnari A., The loss of the O4 antigen moiety from the lipopolysaccharide of an extraintestinal isolate of Escherichia coli has only minor effects on serum sensitivity and virulence in vivo. Infect. Immun. 63, 1263–1269 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (PDF)
Data Availability Statement
The AbDHODH-DSM186 structure has been submitted to the PDB data bank (PDB ID 7UT5). All other data are included in the article and/or SI Appendix.