

Abstract
Background
Aberrant activation of the WNT/β-catenin and STAT3 signaling pathways plays a critical role in cancer progression. However, direct targeting of these pathways as an anti-cancer therapeutic approach needs to be reconsidered due to its serious side effects. Here, we demonstrate that overexpression of WNT induces STAT3 activation in a galectin-3-dependent manner.
Methods
We investigated how galectin-3 mediates the crosstalk between WNT/β-catenin and STAT3 signaling and whether inhibition of galectin-3 can reduce gastric cancer. The molecular mechanisms were analyzed by biochemical assays using cultured gastric cancer cells, patient tissues, and genetically engineered mice. Moreover, we confirm of therapeutic effects of GB1107, a cell-penetrating galectin-3 specific inhibitor, using orthotopic gastric cancer-bearing mice
Results
Increased levels of galectin-3 and STAT3 phosphorylation were detected in the stomach tissues of WNT1-overexpressing mouse models. Also, high expression levels and co-localization of β-catenin, pSTAT3, and galectin-3 in patients with advanced gastric cancer were correlated with a poorer prognosis. Galectin-3 depletion significantly decreased STAT3 Tyr705 phosphorylation, which regulates its nuclear localization and transcriptional activation. A peptide of galectin-3 (Y45-Q48) directly bound to the STAT3 SH2 domain and enhanced its phosphorylation. GB1107, a specific membrane-penetrating inhibitor of galectin-3, significantly reduced the activation of both STAT3 and β-catenin and inhibited tumor growth in orthotopic gastric cancer-bearing mice.
Conclusions
We propose that galectin-3 mediates the crosstalk between the WNT and STAT3 signaling pathways. Therefore GB1107, a galectin-3-specific inhibitor, maybe a potent agent with anti-gastric cancer activity. Further studies are needed for its clinical application in gastric cancer therapy.
Keywords: Galectin-3, Gastric cancer, WNT, STAT3, GB1107
Introduction
Gastric cancer (GC) has the second highest death rate of all cancers, and it was recently estimated that nearly 1,000,000 new cases are diagnosed each year worldwide [1]. The most important factor in the development of GC is thought to be chronic infection with Helicobacter pylori [2]. However, most H. pylori-infected individuals do not develop cancer, suggesting the importance of additional factors [3]. Several signaling pathways, such as the Wnt/β-catenin, STAT3, JNK, and nuclear factor-κB pathways, are associated with GC development [4]. These pathways are deregulated in > 70% of patients diagnosed with GC, resulting in increased inflammatory cytokine production, abnormal apoptosis, undesirable epithelial cell proliferation/differentiation, and epithelial cell transformation.
Aberrant activation of the Wnt/β-catenin signaling pathway occurs in > 30% of tissues in patients with GC [5]. The WNT signaling pathway is named for its ligands, such as WNT1, WNT2, and WNT3a, which are members of the WNT family. WNT family proteins participate in signal-transduction pathways that play fundamental roles in cell fate, such as embryonic development and tissue homeostasis in adults [6]. Upregulation of WNT1 plays important roles in the proliferation of GC stem cells and in advanced GCs [7]. In fact, K19-Wnt1/C2mE transgenic mice have been established as a mouse model of GC [8]. The model was constructed using the K19 promoter to simultaneously overexpress WNT1 and cyclooxygenase-2/prostaglandin E2 (PGE2), which results in invasive gastric adenocarcinoma and is referred to as the GAN mouse model. These data indicate that the WNT signaling pathway is critical in the development and progression of GC.
Previously, galectin-3, a β-galactoside-binding protein, was implicated as a regulator of β-catenin [9, 10] and accumulation of nuclear β-catenin is a hallmark of activated WNT signaling [11]. Galectin-3 may directly bind β-catenin and/or increase its protein levels by inhibiting GSK-3β, to promote its nuclear accumulation [9, 10, 12]. We also reported that galectin-3 drives gastric tumor progression, malignant transformation, and tumor metastasis [13, 14]. Furthermore, we investigated gastric tumorigenesis in vivo using galectin-3 knockout (lgals3−/−) mouse embryonic fibroblasts (MEFs). Unexpectedly, we detected STAT3 activation in malignant stomach tissues of K19-Wnt transgenic mice, and this activation was diminished in stomach tissues from the littermates of lgals3−/− mice and K19-WNT1 transgenic mice. Moreover, increased expression of both STAT3 and β-catenin have been detected in several cancer types and the GAN mouse model [15–23], although the associated mechanisms have not yet been determined.
Here, we demonstrated how WNT signaling increases STAT3 activation during gastric tumorigenesis. We analyzed malignant tissues from patients with GC and confirmed our findings using a genetically engineered mouse model. Our results suggest that WNT signaling induces galectin-3 expression, which mediates WNT signaling pathway-induced STAT3 activation. In addition, we confirmed that a galectin-3 inhibitor can regulate the crosstalk between the WNT and STAT3 signaling pathways in gastric tumorigenesis.
Materials and methods
Cell culture
Human GC cell lines (AGS, MKN28, YCC-2, KATOIII, SNU-1, SNU-5, SNU-16, SNU-216, SNU-601, SNU-638, SNU-668, and SNU-719) were obtained from the Korea Cell Line Bank (KLCB, South Korea). The phenotypes of these cell lines were authenticated by the KCLB. To further examine the relationship between β-catenin, STAT3, phosphorylated STAT3, and galectin-3, we determined their expression levels in these 12 GC cell lines (Supplementary Fig. S3).
RNA interference (RNAi)
Cells were transfected with small interfering RNAs (siRNAs) against galectin-3, STAT3, and β-catenin using Lipofectamine RNAiMAX reagent (Invitrogen, USA), as previously described [9, 24]. The sequences of the siRNAs were: galectin-3 (#1: 5′-UCCAGACCCAGAUAACGCAUCAUGG-3′, #2: 5′-UAAAGUGGAAGGCAACAUCAUUCCC-3′, #3: 5′-AUAUGAAGCACUGGUGAGGUCUAUG-3′, STAT3 (5′-UGAAAGUGGUAGAGAAUCU-3′), and β-catenin (5′-GGGUUCAGAUGAUAUAAAUTT-3′).
Immunohistochemical (IHC) analysis
This study was approved by the Institutional Review Board of the Asan Medical Center (IRB #2010-0388, Seoul, South Korea), and all procedures were conducted in accordance with the guidelines of the Declaration of Helsinki. The tissue microarray contained tissues from 209 GC patients who underwent gastrectomy at the Asan Medical Center in 2003. One core, with a 2 mm diameter, was obtained from each formalin-fixed paraffin-embedded tumor tissue for tissue microarray production. The mean age of patients at the time of surgery was 56.9 years, and the patients were followed up for a mean of 45.3 months postoperatively. The detailed clinicopathological features are shown in Supplementary Table S2. Standard biotinavidin-complex immunohistochemistry was performed on tissue microarrays. The tissue microarray section was deparaffinized, rehydrated, and subjected to heat-induced antigen retrieval using an antigen retrieval buffer of pH 6.0 (for both Galectin3 and β-catenin) or pH 9.0 (for pSTAT3[Y705]) (Dako, Carpinteria, CA). Endogenous peroxidase activity was blocked with 3% H2O2 for 10 min. The sections were incubated with primary antibodies against galectin-3 (Cell Signaling; cat. # 87985; 1:1,000 dilution), β-catenin (Cell Signaling; cat. # 8814; 1:500 dilution), and pSTAT3 (Abcam, Cambridge, MA; cat. ab76315; 1:50 dilution) for 2 h at room temperature. The antigen–antibody reaction was visualized with the Dako EnVision + Dual Link System-HRP (Dako) and DAB+ (3, 3′-diaminobenzidine; Dako). The slides were lightly counterstained with hematoxylin and reviewed by a pathologist (KEK). The control included immunoglobulin G (IgG) and omission of the primary antibody.
To quantify protein expression, the intensity of staining in each case was recorded as a score from 0 to 3, and the percentage of cells showing each intensity was also recorded. The total score in each case was calculated by multiplying each intensity and percentage (range: 0–300). The mean values were used as cut-offs for discriminating between the high and low expression of immunohistochemical staining. Cut-off values for galectin-3, β-catenin, and pSTAT3 were 117.30, 70.96, and 96.84 respectively.
Chromatin immunoprecipitation (ChIP) assays
Chromatin immunoprecipitation (ChIP) assays were performed using a ChIP assay kit (Millipore, USA) and were performed as described previously [14]. Primers were prepared to amplify the STAT3 binding sites at −1231: 5′-CATTGAGCTGAGATCATGCC-3′ and −1009: 5′-TATTAGCCCTCCAGCCCCAC-3′ in the survivin promoter and the STAT3 binding sites at −627: 5′-AACTTGCACAGGGGTTGTGT-3′ and −405: 5′-GAGACCACGAGAAGGGGTGACTG-3′ in the cyclin D1 promoter. PCR was performed with Ex Taq (Takara, Japan).
Preparation of orthotopic gastric xenograft mouse models
As described previously [25], 6-week-old female BALB/c-nude mice (Orient, Korea) were subcutaneously inoculated with luciferase-induced AGS cells (1 × 106). Subcutaneous tumors were excised and implanted into the gastric wall of nude mice [26, 27]. The mice were then randomized into groups (n = 5 per group), and treatment was started 2 weeks after tumor implantation. For the in vivo treatment, we prepared a suspension of GB1107 in 10/90 propylene glycol/0.5% HPMC (Oral-B) for oral injection. Mice received 100 μL of either the Oral-B solution (control) or GB1107 (5 or 10 mg/kg) in Oral-B solution, three times a week for 4 weeks by oral gavage. At 18 and 28 days, the mice were injected intraperitoneally (i.p.) with luciferin (Xenogen, Alameda, CA, USA), and luciferase activity was measured using IVIS imaging. The experiment was terminated at 4 weeks, and the length (b) and width (a) of the orthotopically placed tumors were measured using calipers, and tumor volume was calculated using the formula a2 × b × 0.5.
ITC analysis
To prepare the STAT3 protein, the DNA sequence corresponding to residues 127–688 of human STAT3 was cloned into pET22b(+) (Novagen). This peptide was purified as previously published [28]. ITC experiments were performed at 25 °C in a VP-ITC calorimeter (Microcal Software) [29]. The WT (GAYPGQAPPGAYPGQAPPGA) and Y45P (GApYPGQAPPGAYPGQAPPGA) galectin-3 peptides (140 μM each) were dissolved in ITC buffer (20 mM Tris, pH 7.5 and 300 mM sodium chloride) and injected in 10 μL, 220 s increments, into a calorimetric cell containing 1.8 mL of STAT3 (7 μM). In separate experiments, galectin-3 peptides were titrated into the STAT3 protein, as described above. Titration data were analyzed using Origin 7.0 data analysis software (Microcal Software). Injections were integrated following manual adjustment of the baseline. Heats of dilution were determined from control experiments with ITC buffer and were subtracted before curve fitting using a single set of binding site models.
Care and breeding of mice
All animal experiments were approved by the Institutional Review Board of Yonsei University College of Medicine and were performed in specific pathogen-free (SPF) facilities in accordance with the Guidelines for the Care and Use of Laboratory Animals of the university (IACUC number: 2013-0018). The K19-WNT1, K19-WNT1/C2mE, and lgals3+/− mice were bred in SPF facilities and maintained on a C57BL/6 genetic background.
Statistical analysis
Unpaired (two sample) t tests were used to determine the p values. All statistical analyses were performed using GraphPad Prism (GraphPad Software, La Jolla, CA, USA) and PASW Statistics 18 for Windows (IBM SPSS, Inc., Chicago, IL, USA). Crosstabs, Pearson’s chi square test, and Fisher’s exact test were used as needed. Kaplan–Meier and Cox regression tests were used to analyze survival data. Statistical significance was defined as a p value <0.05.
Results
STAT3 phosphorylation is enhanced in stomach tissues from K19-WNT1 transgenic mice
We established stomach tumors in K19-WNT1/C2mE and K19-WNT1 transgenic mice as previously described [8]. Significant gastric mucosa hyperplasia was observed in the stomach tissues of both K19-WNT1/C2mE and K19-WNT1 transgenic mice after 45 weeks (Fig. 1a). Unexpectedly, we observed elevated phosphorylation at Tyr 705 of STAT3 and its downstream target proteins (survivin and cyclin D1) in the stomach tissues of both mice, compared with the levels of age-matched WT mice (Fig. 1b, c). As expected, we observed abundant expression of inflammatory cytokines, such as IL-6, IL-10, INF-γ, and TNF-α, in the stomach tissues of K19-WNT1/C2mE mice (Fig. 1d) [8]. However, STAT3 phosphorylation was detected in K19-WNT1 transgenic mice, without increased expression levels of inflammatory cytokines (Fig. 1d). We hypothesized that overexpression of WNT1 can induce phosphorylation of STAT3, independent of host inflammatory responses.
Fig. 1.
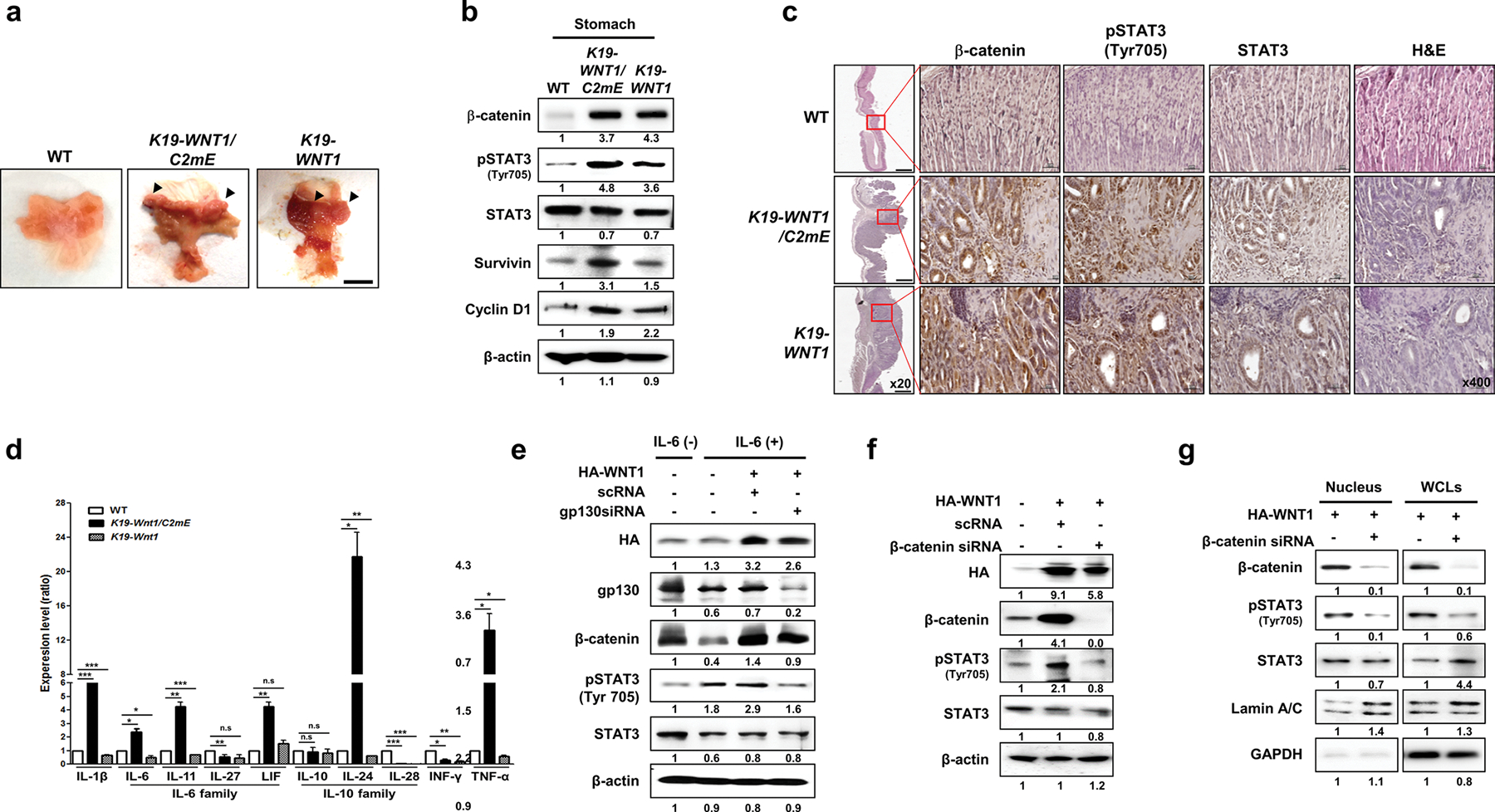
Overexpression of WNT1 induces the phosphorylation and nuclear accumulation of STAT3 in a cytokine receptor-independent but β-catenin-dependent manner. a Representative whole stomachs from wild-type (WT) mice and K19-WNT1/C2mE and K19-WNT1 transgenic mice at 45 weeks. The arrows indicate regions of hyperplasia. Scale bar, 5 mm. b Levels of the indicated proteins in whole stomach tissues from the three mouse lines (indicated at top) as detected by western blotting of extracts. c Immunohistochemical analysis of β-catenin, STAT3, and pSTAT3(Tyr705) in stomach tissues from WT mice and K19-WNT1/C2mE and K19-WNT1 transgenic mice. Scale bars, 500 μm under low magnification (20×) and 20 μm under high magnification (400×). d Gene expression levels of various cytokines (indicated at bottom) as detected by quantitative RT-PCR of whole stomach tissues from the three mouse types. Levels are given as the mean ± SEM ratio to that of WT mice (n = 3). Significant differences are indicated by asterisks (n.s., *p < 0.01, **p < 0.001, and ***p = 0.0001). p values were calculated using Student’s t test. e Western blotting of the indicated proteins in IL-6 untreated non-transfected AGS cells (lane 1) and in IL-6-treated cells with or without HA-WNT transfection and gp130 silencing by siRNA (indicated at top). The relative pSTAT3 level is shown below the blots. f Detection of the indicated proteins in AGS cells with or without transfection with HA-WNT1 and β-catenin siRNA. g Detection of the indicated proteins in nuclear and whole cell lysates (WCLs) of AGS cells with or without transfection with HA-WNT1 and β-catenin siRNA. Lamin A/C and GAPDH were used as nuclear and cytosolic markers, respectively
Overexpression of WNT induces STAT3 phosphorylation in a cytokine receptor-independent and β-catenin signaling-dependent manner
We examined the effect of WNT1 overexpression on STAT3 phosphorylation in the presence and absence of signaling through IL-6 and the gp130 cytokine receptor (Fig. 1e). IL-6 treatment or WNT1 overexpression alone induced STAT3 phosphorylation. In the absence of gp130, WNT1 overexpression still induced STAT3 phosphorylation (Fig. 1e). However, in the absence of β-catenin, WNT1 overexpression could not induce STAT3 phosphorylation (Fig. 1f). Moreover, nuclear accumulation of pSTAT3 was significantly reduced by depletion of β-catenin (Fig. 1g).
Overexpression of WNT induces phosphorylation of STAT3 in an intracellular galectin-3-dependent manner
Crosstalk between WNT signaling and galetin-3 was reported previously [10]. We observed elevated expression of galectin-3 in both K19-WNT1/C2mE and K19-WNT1 transgenic mice (Fig. 2a). The level of β-catenin was significantly decreased in galectin-3 knockout (lgals3−/−) MEFs (Fig. 2b). Interestingly, the levels of pSTAT3 and its target genes survivin and cyclin D were also lower in lgals3−/− MEFs (Fig. 2b). Overexpression of WNT1 or WNT3 in AGS cells elevated the expression of galectin-3, the phosphorylation of STAT3, and the levels of β-catenin and GSK-3β, and these effects were reduced by depletion of galectin-3 (Fig. 2c). Moreover, depletion of galectin-3 also inhibited the nuclear accumulation of phosphorylated STAT3 and total STAT3 induced by WNT overexpression (Fig. 2d, e). Depletion of galectin-3 markedly lowered tSTAT3 transcriptional activity in a luciferase assay (Fig. 2f), whereas overexpression of galectin-3 enhanced it (Fig. 2g) to levels similar to those in untreated, WNT1-transfected, or IL-6-treated cells.
Fig. 2.
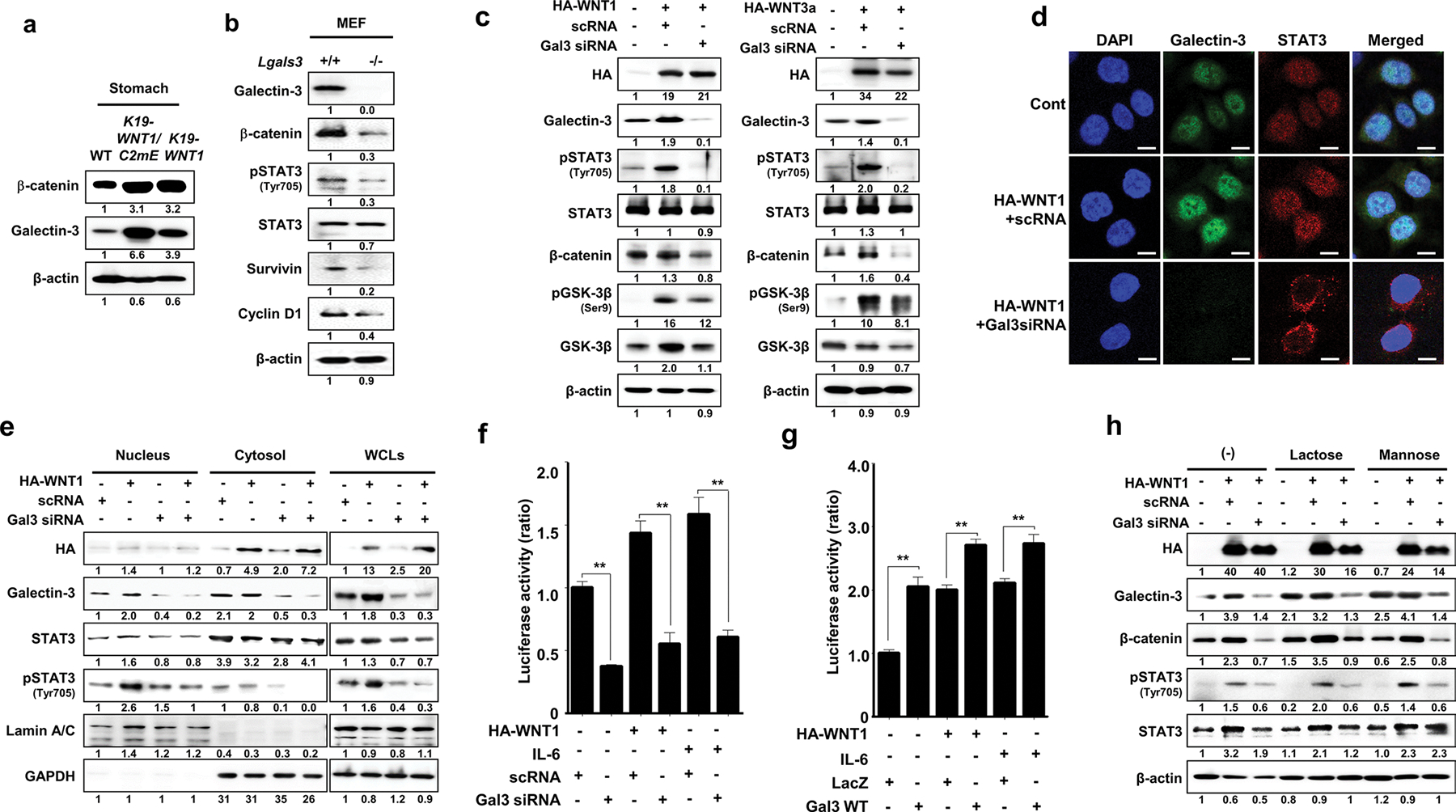
Overexpression of WNT1 induces phosphorylation and nuclear accumulation of STAT3 in a galectin-3-dependent manner. a Levels of β-catenin and galectin-3 in whole stomach tissues of WT mice and K19-WNT1/C2mE and K19-WNT1 transgenic mice. b Levels of the proteins indicated to the left in MEFs from WT and lgals3−/− mice. c Detection of the indicated proteins in AGS cells with or without transfection with HA-WNT1 (left) or HA-WNT3a (right) combined with galectin-3 silencing by siRNA. d Immunocytochemical analysis for the cellular localization of galectin-3 and STAT3 in WNT1-overexpressing and galectin-3-depleted AGS cells. Anti-galectin-3 FITC (green) and anti-STAT3 cy5 (red) antibodies were used, and cells were evaluated under a confocal microscope. DAPI was used to stain the nuclei (blue). Scale bars, 10 μm. e Detection of the indicated proteins in the nucleus, cytosol, or whole cell lysates of AGS cells with or without transfection with HA-WNT1 and galectin-3 siRNA. f, g STAT3 reporter luciferase activity in AGS cells transfected with HA-WNT1. At 24 h after plasmid transfection, the cells were treated with IL-6 with or without silencing of galectin-3 by siRNA (f) or overexpression of galectin-3 (g). The data are presented as the ratios of luciferase activities in untreated, non-HA-WNT1, and mock-transfected cells. Significant differences are indicated by asterisks (**p < 0.001). h Assay with the same experimental design as in the left of panel D, without or with a 4 h preincubation with 50 mM lactose or mannose before cell extraction
To determine whether the binding of extracellular galectin-3 to cell surface glycans induces phosphorylation of STAT3, we overexpressed WNT1 and co-treated these cells with lactose (50 mM), which blocks the binding of galectins to glycans on the cell surface but is too polar to be taken up by the cells. Non-inhibitory mannose was used as a control (Fig. 2h). However, as previously observed in the glial system [30], and in contrast to galectin-3 siRNA transfection, treatment with either lactose or mannose did not inhibit STAT3 phosphorylation or β-catenin stability, suggesting that WNT1-induced STAT3 phosphorylation does not require interaction between galectin-3 and cell surface glycans. However, non-carbohydrate-based binding to cell surface receptors cannot be ruled out.
Clinicopathological analyses of the correlations among galectin-3, β-catenin, and STAT3 expression in patients with GC
To analyze the relationships among galectin-3, β-catenin, and STAT3 in GC patients, we analyzed their expression levels in the GSE27342 dataset from the NCBI database (Fig. 3a). GCs with high galectin-3 expression levels also showed high expression levels of STAT3 and β-catenin. To determine the clinical relevance of galectin-3, pSTAT3, and β-catenin expression in GC, we assessed their expression in tissues from patients with GC by IHC (Fig. 3b). Although galectin-3 showed a positive correlation with pSTAT3 and β-catenin, it was not statistically significant (Supplementary Fig. S1). The combinations of galectin-3high/pSTAT3high (χ2 test; p < 0.001), galectin-3high/β-cateninhigh (χ2 test; p < 0.001), and galectin-3high/pSTAT3high/β-cateninhigh (χ2 test; p = 0.005) were significantly higher in AGC tissues than in EGC tissues (Fig. 3c). We next examined the relationship of each protein with survival. Patients with galectin-3high/pSTAT3high (mean survival, 38.6 months; p = 0.075) and galectin-3high/β-cateninhigh (mean survival, 41.5 months; p = 0.051) had worse disease-specific survival than patients with galectin-3low/pSTAT3low (mean survival, 45.0 months) and galectin-3low/β-cateninlow (mean survival, 46.8 months) (Supplementary Fig. S2). We next analyzed the survival rate of patients with double positives vs. all others. Patients with galectin3high/pSTAT3high expression showed significantly shorter disease-free survival (mean survival, 38.6 vs. 46.9 months; p = 0.038) than other patients, and patients with galectin3high/β-cateninhigh showed a tendency toward worse disease-free survival (mean survival, 41.5 vs. 46.7 months; p = 0.052) than other patients (Fig. 3d). However, there was no significant survival difference between the pSTAT3 high/β-cateninhigh group and all others. Notably, patients with the combination of high expression for all three proteins showed worse disease-free survival (mean survival, 28.8 months; Log rank, p = 0.008 and p = 0.004, respectively) than patients with triple-negative (mean survival, 46.9 months), single-positive (44.3 months), or double positive (48.3 months) tissue samples (Fig. 3e). The Cox proportional hazard model showed that the triple positive combination (galectin3high/pSTAT3high/β-cateninhigh) is an independent poor prognostic factor (HR = 3.570 [95% CI, 1.652–7.713]; p = 0.001) in patients with GC (Fig. 3f). Taken together, these data indicate that galectin3-pSTAT3-β-catenin expression is closely linked to the tumorigenesis of GC and is a potential prognostic factor and therapeutic target for GC.
Fig. 3.
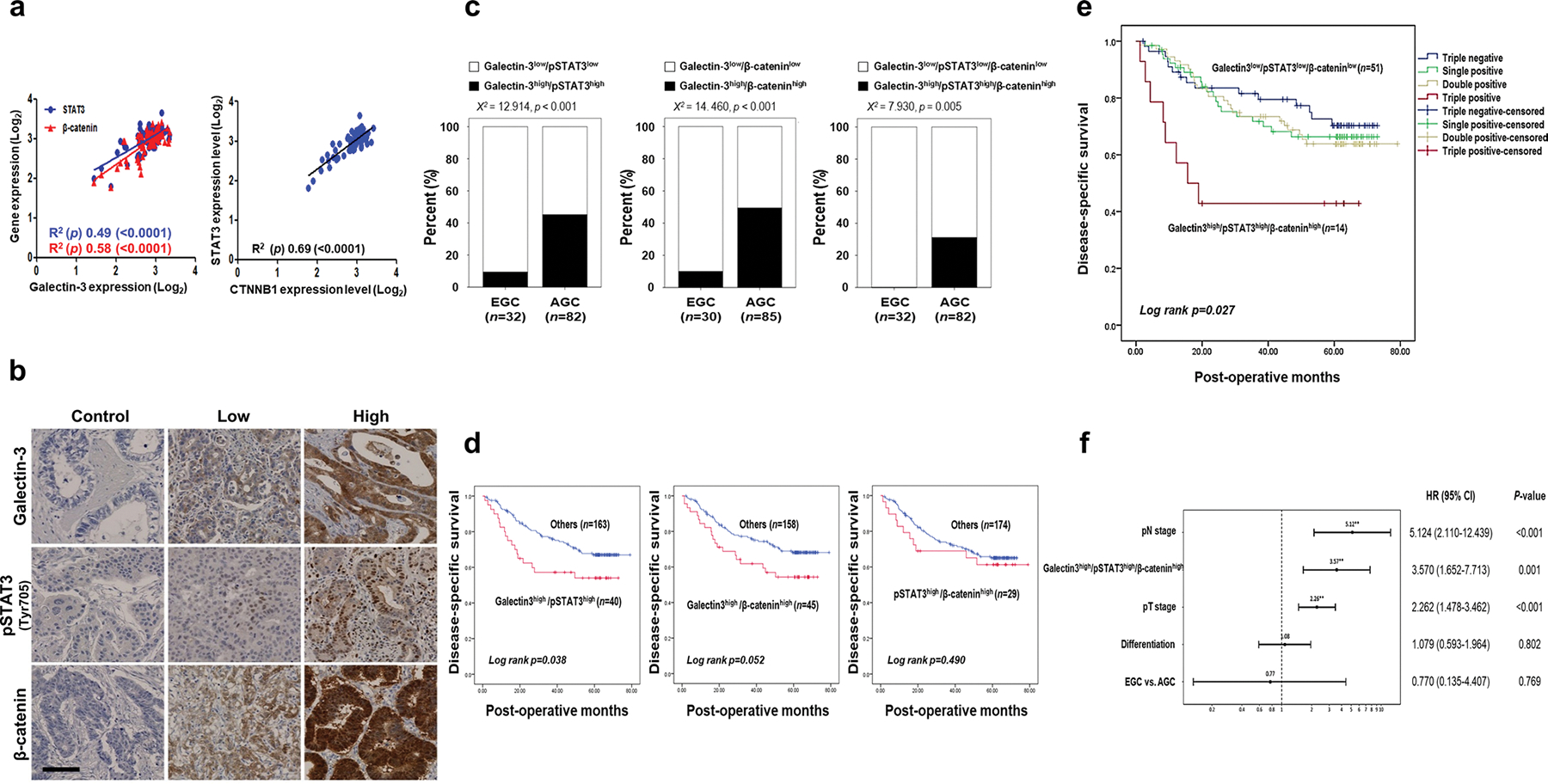
Clinicopathological correlations among galectin-3, β-catenin, and STAT3 in malignant tissues of patients with GC. a Confirmation of the correlations among STAT3, β-catenin, and galectin-3 expression (left) and the correlation between STAT3 and CTNNB1 (β-catenin) expression (right) in the tissues of 80 patients with GC in the GSE27342 public dataset. b Representative IHC analysis of galectin-3, pSTAT3 (Tyr705), and β-catenin expression in 209 GC specimens. Scale bar, 100 μm. c Combinations of galectin-3, pSTAT3(Tyr705), and β-catenin expression were compared between early gastric cancer (EGC) and advanced gastric cancer (AGC). All combined expressions of these three protein were significantly higher in AGC specimens than in EGC specimens (χ2 test; p < 0.001, p < 0.001, and p = 0.005, respectively). d Patients with galectin-3high/pSTAT3high expression had significantly shorter disease-free survival than other patients (mean survival, 38.6 vs. 46.9 months, p = 0.038). Patients with galectin3high/β-cateninhigh expression exhibited a tendency toward worse disease-free survival than patients with others (mean survival, 41.5 vs. 46.7 months, p = 0.052). There was no significant difference in survival between pSTAT3high/β-cateninhigh expression group and others. (e) Patients with galectin-3high/pSTAT3high/β-cateninhigh expression exhibited significantly shorter disease-specific survival (mean survival, 28.8 months) than patients with galectin-3low/pSTAT3low/β-cateninlow expression (mean survival, 47.5 months; Log rank p = 0.008) and others (mean survival, 46.6 months; Log rank p = 0.004). f Multivariate analysis of hazard ratios using the Cox regression method for all patients with gastric cancer. EGC early gastric cancer; AGC advanced gastric cancer
Galectin-3 depletion directly reduces STAT3 phosphorylation and its nuclear localization
We transfected AGS cells with three different galectin-3-specific siRNAs, which resulted in reduced STAT3 nuclear localization (Fig. 4b), reduced STAT3 phosphorylation, reduced expression levels of survivin and cyclin D1 (Fig. 4a), and reduced cell proliferation (Supplementary Fig. S4). Galectin-3 depletion also reduced STAT3 phosphorylation and nuclear localization in other tested GC cell lines, including KATOIII, SNU601, and SNU668 cells (Fig. 4c). We also examined STAT3 phosphorylation and nuclear localization after IL-6 stimulation in galectin-3-depleted MKN-28 cells, which exhibit a low basal level of STAT3 phosphorylation (Fig. S3). Under IL-6 stimulation, STAT3 phosphorylation (Fig. 4d) and nuclear localization (Supplementary Fig. S5) were decreased in galectin-3-depleted MKN-28 cells. Remarkably, co-localization of STAT3 and galectin-3 was detected in all GC cells.
Fig. 4.
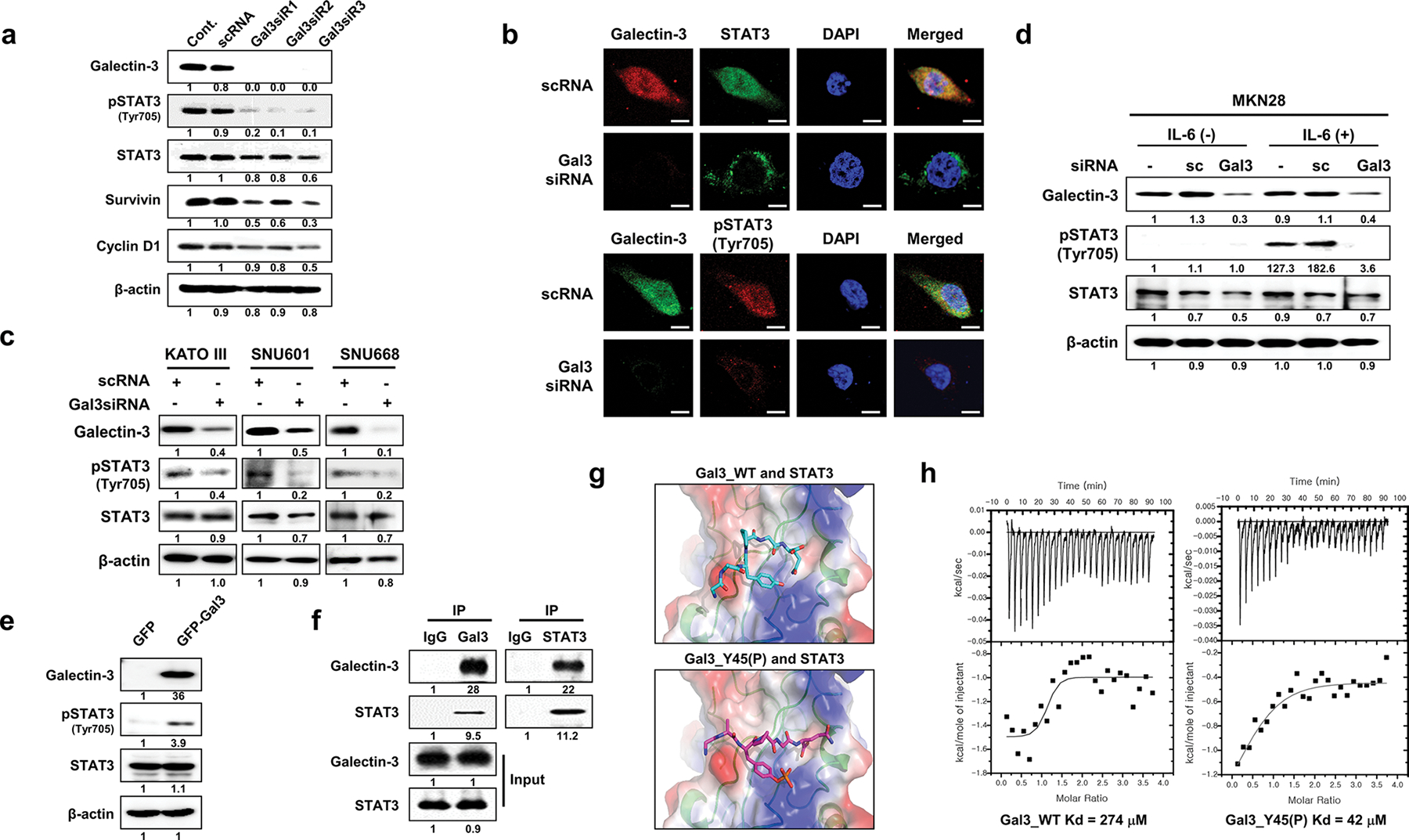
Galectin-3 depletion reduces Tyr705 phosphorylation of STAT3 and its nuclear localization through direct interaction with STAT3. a Levels of the indicated proteins in nuclear and cytosolic fractions of AGS cells after galectin-3 silencing with galectin-3 siRNA as measured by western blotting. b Immunocytochemical analysis of the subcellular localization of FITC-STAT3 and Cy5-pSTAT3(Tyr705) in FITC (up) or Cy5 (down)-galectin-3-silenced AGS cells. DAPI was used to visualize the nuclei (blue). Scale bars, 10 μm. c After transfection of three gastric cancer cell lines, KATOIII, SNU 601, and SNU668, with galectin-3 siRNA, the expression levels of galectin-3, phosphorylated STAT3 (Tyr705), and STAT3 were assessed by western blotting. β-Actin was used as a loading control. d After galectin-3-specific siRNA transfection, IL-6 cytokine was subjected for 6 h in MKN28 cells, and the expression levels of galectin-3, phosphorylated STAT3(Tyr705), and STAT3 were detected by western blotting. e Evaluation of galectin-3, STAT3, and pSTAT3(Tyr705) levels in SNU-638 cells transfected with pcDNA3.1/NT-GFP or pcDNA3.1/NT-GFP-gal3. f Immunoprecipitation (IP) with an anti-galectin-3 or STAT3 antibody to detect galectin-3 interaction with STAT3 in AGS cells. g Model structures of a STAT3/WT galectin-3 peptide (upper panel) with a STAT3/Y45(P) galectin-3 peptide (lower panel). h Binding affinities of STAT3 to a WT galectin-3 peptide (left) and Y45(P) galectin-3 peptide as measured by ITC analysis
Overexpression of galectin-3 induces STAT3 phosphorylation through direct binding to STAT3
To investigate the direct links between galectin-3 and STAT3 phosphorylation, we examined the effects of transient galectin-3 overexpression in galectin-3-null SNU638 cells. Overexpression of galectin-3 significantly increased STAT3 phosphorylation without affecting total STAT3 protein levels (Fig. 4e). Since co-immunoprecipitation of galectin-3 and STAT3 was detected (Fig. 4f), we searched for potential STAT3 binding sites in the galectin-3 amino acid sequence (Supplementary Fig. S6). Two instances of the amino acid sequence YPGQ in the N-terminal domain of galectin-3, Tyr45–Gln48 and Tyr54–Gln57, were suggested as possible binding sites, as they resemble ligands of the STAT3 SH2 domain (Fig. 4g). The results of isothermal titration calorimetry (ITC) experiment, in which a fixed concentration (7 μM) of STAT3 was titrated with either a wild-type galectin-3 peptide (GAYPGQ) or a Y45-phosphorylated variant peptide (GApYPGQ), yielded calculated affinities of ~ 270 and 40 μM for the WT and Y45(P) peptides, respectively (Fig. 4h). This indicated that the phosphorylated peptide binds to STAT3 with higher affinity than the WT peptide, as was expected for the SH2 domain. To model this interaction, a STAT3 C-terminal peptide containing 705Y- bound to the SH2 domain of another STAT3 peptide in a dimer (PDB: 4E68), was replaced by either of the abovementioned galectin-3 peptides as a starting point for calculations. The results suggested direct binding of galectin-3 to STAT3 through its SH2 domain.
Galectin-3 regulates DNA binding and transcriptional activation of STAT3
We next determined whether galectin-3 could increase the phosphorylation and nuclear localization of STAT3. Although overexpression of WT galectin-3 increased the phosphorylation and nuclear localization of STAT3, overexpression of a galectin-3 mutant (Gal3Δ44–58) did not affect the phosphorylation (Fig. 5a) or nuclear localization of STAT3 (Fig. 5b). Thus, galectin-3 may induce the phosphorylation and nuclear localization of STAT3. We explored the effects of galectin-3 on the interaction between STAT3 and the promoter regions of survivin and cyclin D1 (Supplementary Fig. S7) using ChIP assays (Fig. 5c). In the presence of the high native galectin-3 expression levels in AGS cells, the promoter regions of both survivin and cyclin D1 were immunoprecipitated with either anti-STAT3 or anti-galectin-3 antibodies. However, in the absence of galectin-3, anti-STAT3 antibodies did not precipitate either promoter region. Moreover, overexpression of a putative phosphorylation site mutant of galectin-3 (Gal3Y45A or Gal3Y54A) abolished or decreased STAT3 transcriptional activity (Fig. 5d).
Fig. 5.
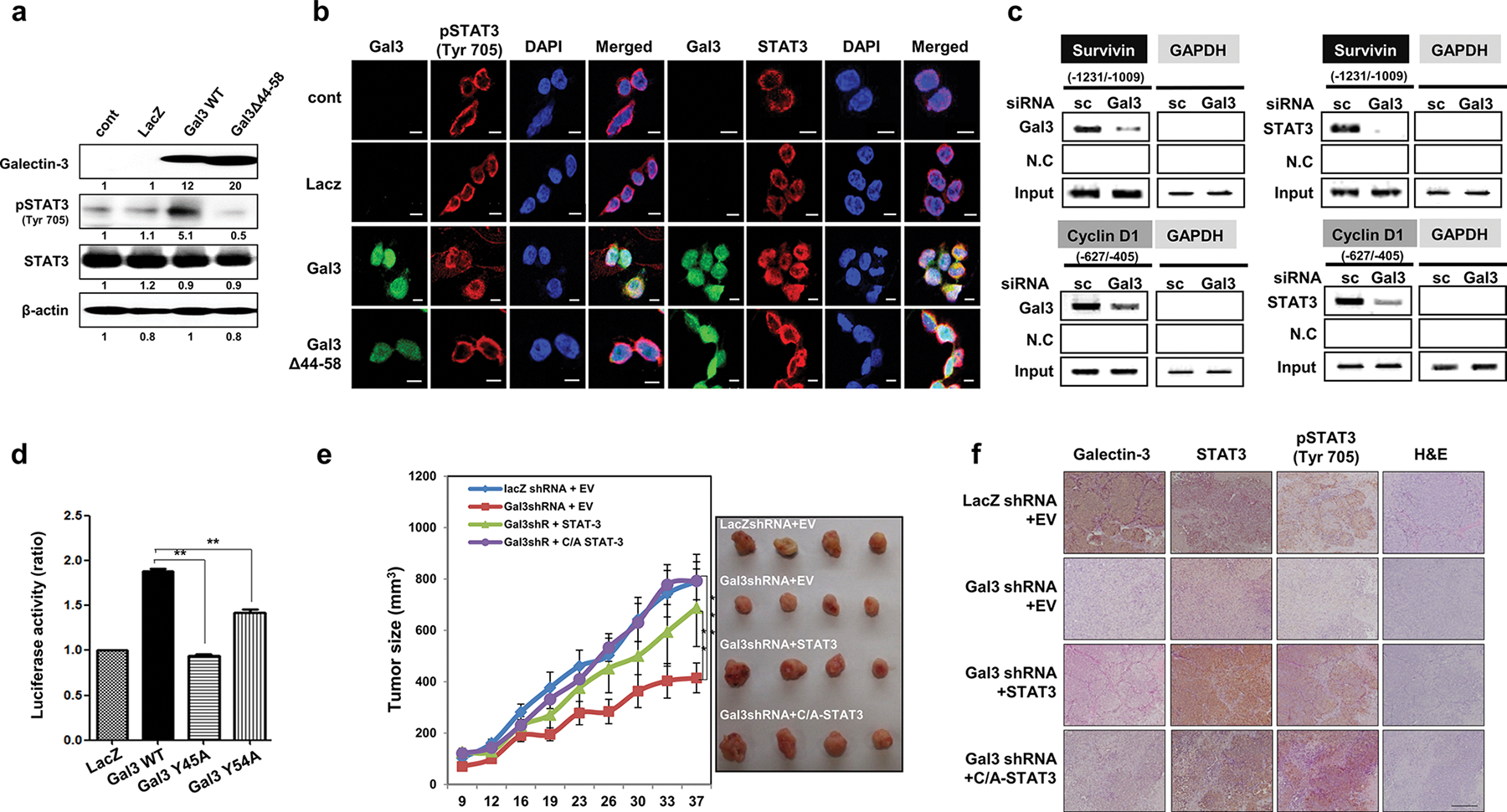
Galectin-3 increases the nuclear localization of STAT3 and regulates the DNA binding and transcriptional activation of STAT3. a,b SNU638 cells, which lack endogenous galectin-3, were infected with lentivirus vectors that overexpress LacZ, galectin-3 WT, and a STAT3 binding motif-deleted mutant of galectin-3 (Gal3Δ44–58). a The expression levels of galectin-3, STAT3, and pSTAT3 (Tyr705) as detected by western blotting. b Immunocytochemical analysis of the subcellular localization of FITC-galectin-3, Cy-5-STAT3, and Cy5-pSTAT3(Tyr705) in cells overexpressing LacZ, WT galectin-3, and Gal3Δ44–58. DAPI was used to visualize the nuclei (blue). Scale bars, 10 μm. Cells were fixed, and immunocytochemical analysis was performed using a confocal microscope. DAPI was used to visualize the nuclei (blue). Scale bars, 10 μm. c ChIP assays using antibodies against galectin-3 and STAT3 in AGS cells transfected with scRNA or gal3 siRNA. PCR fragments of the survivin and cyclin D1 promoters were detected. d SNU638 cells overexpressing lacZ, wild-type galectin-3 (Gal3 WT), and SH2 domain-binding motif mutant galectin-3 (Y45A or Y54A). This luciferase experiment was repeated three times, with similar results, and the data are shown as mean ± SD (n = 3). e AGS cells (106 cells) expressing LacZ, gal-3shRNA, gal-3shRNA, and wild-type STAT3, or gal-3shRNA and constitutively activated STAT3 (C/A STAT3) were subcutaneously inoculated into both flanks of a mouse to generate xenograft tumors (n = 5 per group). Tumor volume was measured for 37 days with calipers (as shown on the right). The error bars indicate the 95% confidence intervals (CI); *p = 0.0012, **p = 0.0023, and ***p = 0.0034, two-sided t test for values on the final day. f Galectin-3, pSTAT3(Tyr705), and STAT3 expression in mouse tumor tissues, as subcutaneously injected into the xenograft mouse model, was detected by IHC staining (brown), along with H&E staining. Magnification: 200×
STAT3 restored reduced tumor growth induced by galectin-3 depletion
We prepared GC cells with stably depleted galectin-3 expression and stably transfected them with an expression vector encoding either STAT3 or constitutively activated (C/A)-STAT3 (Supplementary Fig. S8). Compared to tumors formed by LacZ shRNA-transfected AGS cells, tumors formed by galectin-3-depleted cells were smaller and showed lower growth rates. However, galectin-3-depleted tumors expressing STAT3 or (C/A)-STAT3 were as large as the controls (Fig. 5e). IHC analysis revealed decreased STAT3 phosphorylation levels in galectin-3-depleted tumor sections (Fig. 5f). However, decreased STAT3 phosphorylation was reversed by overexpression of STAT3 or (C/A)-STAT3. These results showed that galectin-3 is required for high tumor growth rate, possibly by enhancing STAT3 activation; however, a lack of galectin-3 can be compensated for by increased levels of STAT3.
A cell-penetrating galectin-3 inhibitor, GB1107, blocks activation of both the WNT and STAT3 signaling pathways
To further assess the effects of galectin-3 in GC, we employed a novel high affinity galectin-3 specific inhibitor, GB1107 [31], with improved uptake across cell membranes [32] (Fig. 6a). We confirmed the inhibitory effect of GB1107 on the proliferation of five out of six tested GC cell lines (Fig. 6b). Treatment with 10 μM GB1107 induced G1 cell-cycle arrest (Fig. 6c) and significantly reduced the phosphorylation of STAT3 and the expression levels of β-catenin, cyclin D1, and survivin in AGS cells (Fig. 6d). The interaction among galectin-3, STAT3, and β-catenin was blocked by GB1107 (Fig. 6e). The inhibitor also blocked the nuclear accumulation of galectin-3, phosphorylated STAT3, and β-catenin in a dose-dependent manner (Fig. 6f, g) as well as the transcriptional activity of TCF4, a transcription factor that modulates WNT signaling (Fig. 6h) and STAT3 (Fig. 6i).
Fig. 6.
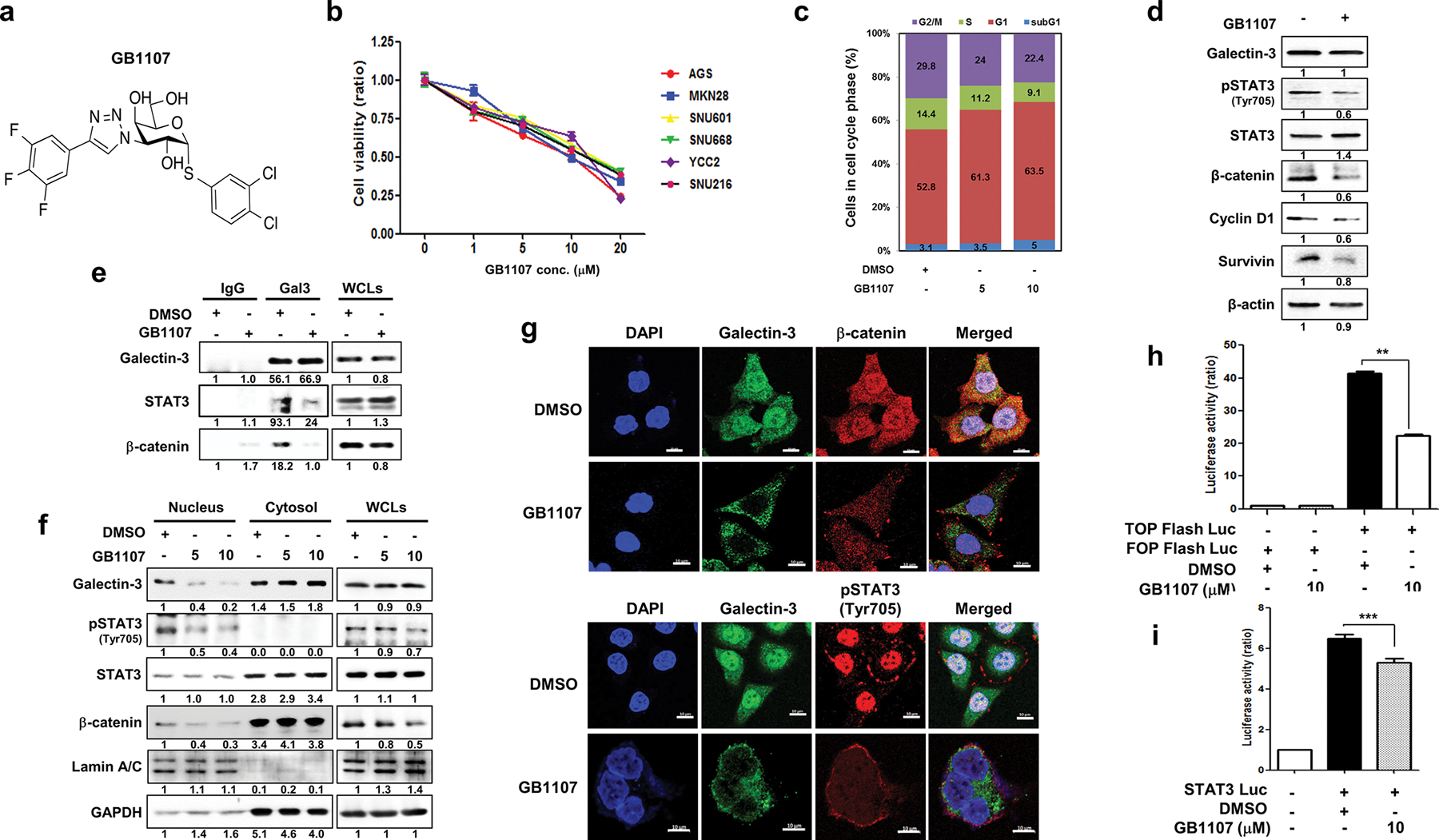
The galectin-3 specific inhibitor GB1107 significantly blocks the activation of the WNT and STAT3 signaling pathways. a Chemical structure of the galectin-3 inhibitor GB1107 [31]. b Cell viability of six GC cell lines treated with different concentrations of GB1107 for 48 h as measured by WST assays. Data are shown as mean ± SD (n = 5). c Effect of GB1107 treatment for 48 h on the cell cycle populations of AGS cells, as examined by PI staining. d Effect of GB1107 treatment (10 μM) for 48 h on the levels of galectin-3, pSTAT3(Tyr705), STAT3, β-catenin, survivin, and cyclin D1 in AGS cells. e Effect of GB1107 on the interaction of galectin-3 with STAT3 or β-catenin as detected by immunoprecipitation. Cells were incubated with 10 μM GB1107 for 48 h. f Levels of nuclear galectin-3, pSTAT3(Tyr705), and β-catenin in AGS cells after treatment with GB1107 (5 or 10 μM, for 48 h). g Immunocytochemical analysis of the subcellular localization of FITC-galectin-3 and Alexa-fluor 647-pSTAT3(Tyr705) or FITC-galectin-3 and Alexa fluor 647-β-catenin in AGS cells after treatment with DMSO or GB1107. DAPI was used to visualize the nuclei (blue). Scale bars, 10 μm. h, i TOP flash, FOP flash h, and STAT3 i luciferase activity was measured in AGS cells after 24 h of transfection with each respective luciferase plasmid and treatment with GB1107 (5 or 10 μM, for 24 h). This luciferase experiment was repeated three times, with similar results, and the data are shown as mean ± SD (n = 3)
GB1107 reduces tumor burden in an orthotopic GC model in mice
To investigate the effects of GB1107 in vivo, we established an orthotopic xenografted GC model using nude mice implanted with pre-grown tumors consisting of AGS cells and expressing luciferase to facilitate volume measurement. In untreated mice, the tumors grew to about 500 mm3 in 4 weeks, whereas in mice treated with 5 or 10 mg/kg of GB1107 three times a week by oral gavage, the tumor size either remained unchanged or decreased to an average volume of about 100 mm3 (Fig. 7a–d). Administration of GB1107 also reduced the levels of phosphorylated STAT3 and β-catenin expression as well as galectin-3 expression in the tumor tissues of the mice (Fig. 7e). These data suggested that the galectin-3 specific inhibitor GB1107 could be used as a therapeutic agent for GC.
Fig. 7.
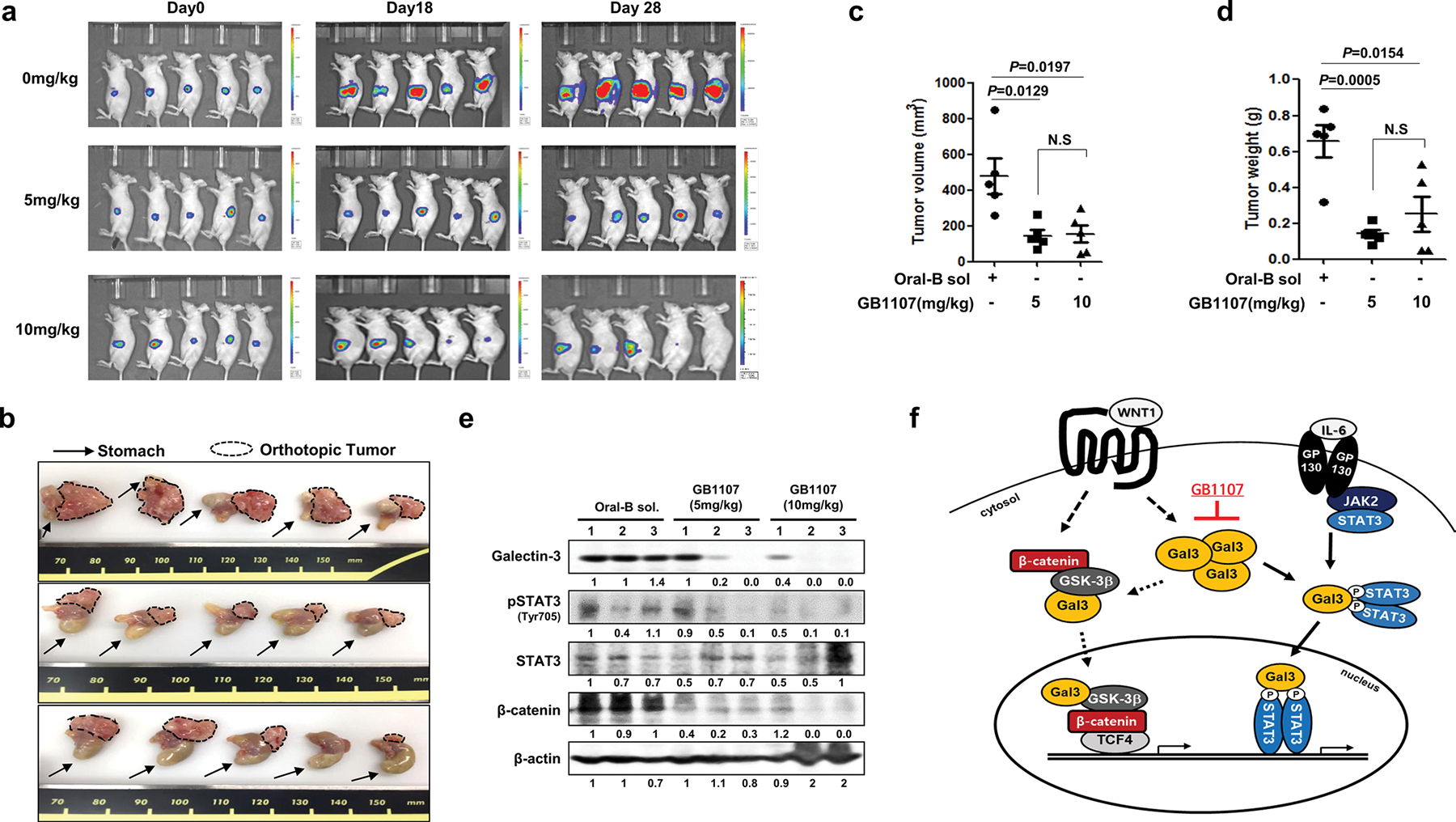
The galectin-3 specific inhibitor GB1107 significantly reduces tumor burden in orthotopic gastric cancer tumor-bearing mice. a–d Development of the gastric cancer orthotopic mouse model. Mice were given 100 μL of either Oral-B solution (control) or Oral-B solution containing 5 or 10 mg/kg GB1107 by oral gavage three times per week. a Tumor size as estimated by luciferase bioluminescent imaging. b Mice were sacrificed, and the orthotopic gastric tumor was removed for analysis. The arrow indicates the mouse stomach, and the dotted line encircles the orthotopic gastric cancer tumor. c, d Tumor volume c and weight d at 28 days after starting GB1107 treatment. e Levels of galectin-3, pSTAT3(Tyr705), STAT3, and β-catenin in the stomach of mice treated with Oral-B solution or GB1107. f Experimental scheme of this study. Galectin-3 interacts with STAT3 and alters STAT3 phosphorylation. STAT3 phosphorylation at Tyr705 was induced by WNT signaling-induced galectin-3 expression. These complexes then regulate survivin and cyclin D1 transcriptional activity downstream of STAT3. Gal3 galectin-3
Discussion
In this study, we found that intracellular galectin-3 interacts directly with STAT3 and enhances its Tyr705 phosphorylation, leading to its nuclear translocation and transcriptional activation. Constitutively activated STAT3 is frequently detected in the tissues of patients with GC, and STAT3 phosphorylation at Tyr705 is correlated with TNM stage and poor survival rates, suggesting that STAT3 Tyr705 phosphorylation is a potent diagnostic marker of gastric malignancy and a potential therapeutic target for GC [33]. Here, we demonstrated that phosphorylation of STAT3 at Tyr705 may involve a galectin-3-mediated interaction with STAT3. We prepared galectin-3 mutants that could not interact with STAT3 observed that overexpression of these galectin-3 mutants impaired STAT3 phosphorylation, suggesting that galectin-3 is induced STAT3 phosphorylation. We also found that an NLS-like sequence of galectin-3 is critical for nuclear localization of STAT3.
Here, we propose another STAT3-activation pathway, a WNT signaling-induced galectin-3-mediated pathway. First, we found that overexpression of WNT increased galectin-3 expression. The promoter region of the galectin-3 gene (Lgals3) contains predicted binding sites for TCF4 binding (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3), which may explain how WNT increases galectin-3 expression. WNT signaling-induced galectin-3 interacts with β-catenin and enhances its nuclear accumulation as well as transcriptional activation of TCF4 [9]. In addition, galectin-3 interacts with STAT3 and enhances their nuclear accumulation and transcriptional activation of STAT3 (Fig. 7f).
The galectin-3 specific inhibitor GB1107 [31] was used to assess the galectin-3 mediated effects, including the nuclear localization of phosphorylated STAT3 and β-catenin, and their transcriptional activities, in GC. Since GB1107 can penetrate into cells [32], it may interact with cytosolic galectin-3, whereas most galectin-3 inhibitors interact with only extracellular galectin-3. GB1107 binds specifically to the carbohydrate-binding site within the galectin-3 carbohydrate-recognition-binding domain (CRD) as shown by X-ray crystallography [34]. Therefore, it is not expected to directly inhibit the interaction between the N-terminal galectin-3 peptide and STAT3. However, it might inhibit the interaction of STAT3 with β-catenin, which may involve peptides within the CRD [35]. It may also inhibit galectin-3 self-interaction, which involves both the CRD and N-terminus [36]. The interaction of GB1107 with the CRD of galectin-3 may lead to a conformational change, but this is unlikely since the X-ray structure of GB1107 in complex with the galectin-3 CRD revealed no change in the structure of galectin-3 when compared to that of the corresponding lactose complex [31].
Excitingly, GB1107 clearly reduced the tumor burden in orthotopic GC tumor-bearing mice, without intestinal inflammation or harmful side effects, such as leanness. The mechanism of action may be similar to that described above for cultured cells since the levels of pSTAT3 and β-catenin were lower in the GB1107-treated tumor tissues. GB1107 could also inhibit extracellular galectin-3, which plays a major role in tumor microenvironments [37]. For example, galectin-3 released from cancer cells may induce apoptosis or anergy of tumor-infiltrating T-cells [38]. Galectin-3 at the cell surface may also regulate a tumor-promoting lattice, with increased exposure of growth-promoting receptors [39]. Moreover, extracellular galectin-3 also mediates homotypic and heterotypic aggregation and promotes interactions between tumor cells and endothelial cells during angiogenesis and tumor metastasis [40, 41]. Therefore, a cell-penetrating galectin-3 inhibitor that could target both intracellular and extracellular galectin-3 is expected to have better therapeutic effects.
Taken together, our data suggest a link between WNT signaling and STAT3 signaling that is mediated by intracellular galectin-3. Targeting galectin-3 with the cell-penetrating galectin-3 inhibitor GB1107 inhibited both pathways and could be a promising anti-cancer approach. Therefore, further analysis of the related molecular mechanisms and therapeutic effects is desirable.
Supplementary Material
Acknowledgments
We thank Dr. M. Oshima (Kanazawa University, Japan) for providing K19-Wnt1/C2mE transgenic mice and Dr. F-T Liu (UC Davis, USA) for providing lgals3−/− mice. This work was supported by grants from National Research Foundation (NRF) of Korea funded by the Korean government (MSIP, No. NRF-2014R1A2A1A11050600, NRF-2016R1A5A1010764, NRF-2017R1C1B2005265, NRF-2017R1A2B2006238, NRF-2019R1A2C2089237, NRF-2020R1A4A1019063 and NRF-2020R1A2C110007811), the Bio & Medical Technology Development Program of the NRF funded by the Korean government (MSIP, No. NRF-2015M3A9B6073835, NRF-2015M3A9B6073833), the Basic Science Research Program through the NRF of Korea funded by the Ministry of Education (No. NRF-2014R1A1A2055009), and the International Research & Development Program of the NRF funded by the Ministry of Education, Science and Technology (MEST) of Korea (No. NRF-2016K1A3A1A47921595), KBRI basic research program through Korea Brain Research Institute funded by Ministry of Science and ICT (21-BR-03-05) and the matching fund provided by Swedish Research Council (No. 2016-07109).
Footnotes
Conflict of interest F.R.Z. is an employee of and H.L. and U.J.N. are shareholders in Galecto Biotech AB, a company that is developing galectin inhibitors. The other authors have no conflicts to declare.
Ethical standard All institutional and national guidelines for the care and use of laboratory animals were followed.
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s10120-021-01186-5.
References
- 1.Giraud AS, Menheniott TR, Judd LM. Targeting STAT3 in gastric cancer. Expert Opin Ther Targets. 2012;16(9):889–901. [DOI] [PubMed] [Google Scholar]
- 2.Ding SZ, Goldberg JB, Hatakeyama M. Helicobacter pylori infection, oncogenic pathways and epigenetic mechanisms in gastric carcinogenesis. Future Oncol. 2010;6(5):851–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Denson LA. Adding fuel to the fire: STAT3 priming of gastric tumorigenesis. Gastroenterology. 2006;131(4):1342–4. [DOI] [PubMed] [Google Scholar]
- 4.Ooi CH, Ivanova T, Wu J, Lee M, Tan IB, Tao J, et al. Oncogenic pathway combinations predict clinical prognosis in gastric cancer. PLoS Genet. 2009;5(10):e1000676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiurillo MA. Role of the Wnt/beta-catenin pathway in gastric cancer: an in-depth literature review. World J Exp Med. 2015;5(2):84–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Croce JC, McClay DR. Evolution of the Wnt pathways. Methods Mol Biol. 2008;469:3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katoh M, Kirikoshi H, Terasaki H, Shiokawa K. WNT2B2 mRNA, up-regulated in primary gastric cancer, is a positive regulator of the WNT- beta-catenin-TCF signaling pathway. Biochem Biophys Res Commun. 2001;289(5):1093–8. [DOI] [PubMed] [Google Scholar]
- 8.Oshima H, Matsunaga A, Fujimura T, Tsukamoto T, Taketo MM, Oshima M. Carcinogenesis in mouse stomach by simultaneous activation of the Wnt signaling and prostaglandin E2 pathway. Gastroenterology. 2006;131(4):1086–95. [DOI] [PubMed] [Google Scholar]
- 9.Kim SJ, Choi IJ, Cheong TC, Lee SJ, Lotan R, Park SH, et al. Galectin-3 increases gastric cancer cell motility by up-regulating fascin-1 expression. Gastroenterology. 2010;138(3):1035–1045e1-2. [DOI] [PubMed] [Google Scholar]
- 10.Shimura T, Takenaka Y, Fukumori T, Tsutsumi S, Okada K, Hogan V, et al. Implication of galectin-3 in Wnt signaling. Cancer Res. 2005;65(9):3535–7. [DOI] [PubMed] [Google Scholar]
- 11.Kim SJ, Shin JY, Cheong TC, Choi IJ, Lee YS, Park SH, et al. Galectin-3 germline variant at position 191 enhances nuclear accumulation and activation of beta-catenin in gastric cancer. Clin Exp Metastasis. 2011;28(8):743–50. [DOI] [PubMed] [Google Scholar]
- 12.Song S, Mazurek N, Liu C, Sun Y, Ding QQ, Liu K, et al. Galectin-3 mediates nuclear beta-catenin accumulation and Wnt signaling in human colon cancer cells by regulation of glycogen synthase kinase-3beta activity. Cancer Res. 2009;69(4):1343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim SJ, Shin JY, Lee KD, Bae YK, Choi IJ, Park SH, et al. Galectin-3 facilitates cell motility in gastric cancer by up-regulating protease-activated receptor-1 (PAR-1) and matrix metalloproteinase-1 (MMP-1). PLoS One. 2011;6(9):e25103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim SJ, Lee HW, Gu Kang H, La SH, Choi IJ, Ro JY, et al. Ablation of galectin-3 induces p27(KIP1)-dependent premature senescence without oncogenic stress. Cell Death Differ. 2014;21(11):1769–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frank DA. Transcription factor STAT3 as a prognostic marker and therapeutic target in cancer. J Clin Oncol. 2013;31(36):4560–1. [DOI] [PubMed] [Google Scholar]
- 16.Banerjee K, Resat H. Constitutive activation of STAT3 in breast cancer cells: A review. Int J Cancer. 2016;138(11):2570–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ji K, Zhang M, Chu Q, Gan Y, Ren H, Zhang L, et al. The role of p-STAT3 as a prognostic and clinicopathological marker in colorectal cancer: a systematic review and meta-analysis. PLoS One. 2016;11(8):e0160125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arend RC, Londono-Joshi AI, Straughn JM Jr, Buchsbaum DJ. The Wnt/beta-catenin pathway in ovarian cancer: a review. Gynecol Oncol. 2013;131(3):772–9. [DOI] [PubMed] [Google Scholar]
- 19.Hadjihannas MV, Bruckner M, Jerchow B, Birchmeier W, Dietmaier W, Behrens J. Aberrant Wnt/beta-catenin signaling can induce chromosomal instability in colon cancer. Proc Natl Acad Sci USA. 2006;103(28):10747–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li LF, Wei ZJ, Sun H, Jiang B. Abnormal beta-catenin immunohistochemical expression as a prognostic factor in gastric cancer: a meta-analysis. World J Gastroenterol. 2014;20(34):12313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uehara Y, Inoue M, Fukuda K, Yamakoshi H, Hosoi Y, Kanda H, et al. Inhibition of beta-catenin and STAT3 with a curcumin analog suppresses gastric carcinogenesis in vivo. Gastric Cancer. 2015;18(4):774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashrafizadeh M, Zarrabi A, Orouei S, Zarrin V, Rahmani Moghadam E, Zabolian A, et al. STAT3 pathway in gastric cancer: signaling, therapeutic targeting and future prospects. Biology (Basel). 2020;9(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tye H, Kennedy CL, Najdovska M, McLeod L, McCormack W, Hughes N, et al. STAT3-driven upregulation of TLR2 promotes gastric tumorigenesis independent of tumor inflammation. Cancer Cell. 2012;22(4):466–78. [DOI] [PubMed] [Google Scholar]
- 24.Ahn YH, Yi H, Shin JY, Lee KD, Shin SP, Lee SJ, et al. STAT3 silencing enhances the efficacy of the HSV.tk suicide gene in gastrointestinal cancer therapy. Clin Exp Metastasis. 2012;29(4):359–69. [DOI] [PubMed] [Google Scholar]
- 25.Lee HW, Kim SJ, Choi IJ, Song J, Chun KH. Targeting Notch signaling by gamma-secretase inhibitor I enhances the cytotoxic effect of 5-FU in gastric cancer. Clin Exp Metastasis. 2015;32(6):593–603. [DOI] [PubMed] [Google Scholar]
- 26.Cho Y, Kang HG, Kim SJ, Lee S, Jee S, Ahn SG, et al. Post-translational modification of OCT4 in breast cancer tumorigenesis. Cell Death Differ. 2018;25(10):1781–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim SJ, Wang YG, Lee HW, Kang HG, La SH, Choi IJ, et al. Up-regulation of neogenin-1 increases cell proliferation and motility in gastric cancer. Oncotarget. 2014;5(10):3386–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ren Z, Mao X, Mertens C, Krishnaraj R, Qin J, Mandal PK, et al. Crystal structure of unphosphorylated STAT3 core fragment. Biochem Biophys Res Commun. 2008;374(1):1–5. [DOI] [PubMed] [Google Scholar]
- 29.Pierce MM, Raman CS, Nall BT. Isothermal titration calorimetry of protein-protein interactions. Methods. 1999;19(2):213–21. [DOI] [PubMed] [Google Scholar]
- 30.Jeon SB, Yoon HJ, Chang CY, Koh HS, Jeon SH, Park EJ. Galectin-3 exerts cytokine-like regulatory actions through the JAK-STAT pathway. J Immunol. 2010;185(11):7037–46. [DOI] [PubMed] [Google Scholar]
- 31.Zetterberg FR, Peterson K, Johnsson RE, Brimert T, Hakansson M, Logan DT, et al. Monosaccharide derivatives with low-nanomolar lectin affinity and high selectivity based on combined fluorine-amide, phenyl-arginine, sulfur-pi, and halogen bond Interactions. ChemMedChem. 2018;13(2):133–7. [DOI] [PubMed] [Google Scholar]
- 32.Stegmayr J, Zetterberg F, Carlsson M, Huang X, Sharma G, Kahl-Knutson B, Schambye H, Nilsson U, Oredsson S, Leffler H. Extracellular and intracellular small-molecule galectin-3 inhibitors. Sci Rep. 2018;9:2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji K, Zhang L, Zhang M, Chu Q, Li X, Wang W. Prognostic value and clinicopathological significance of p-stat3 among gastric carcinoma patients: a systematic review and meta-analysis. Medicine (Baltimore). 2016;95(5):e2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haudek KC, Spronk KJ, Voss PG, Patterson RJ, Wang JL, Arnoys EJ. Dynamics of galectin-3 in the nucleus and cytoplasm. Biochim Biophys Acta. 2010;1800(2):181–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimura T, Takenaka Y, Tsutsumi S, Hogan V, Kikuchi A, Raz A. Galectin-3, a novel binding partner of beta-catenin. Cancer Res. 2004;64(18):6363–7. [DOI] [PubMed] [Google Scholar]
- 36.Lepur A, Salomonsson E, Nilsson UJ, Leffler H. Ligand induced galectin-3 protein self-association. J Biol Chem. 2012;287(26):21751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gordon-Alonso M, Hirsch T, Wildmann C, van der Bruggen P. Galectin-3 captures interferon-gamma in the tumor matrix reducing chemokine gradient production and T-cell tumor infiltration. Nat Commun. 2017;8(1):793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahmed H, Guha P, Kaptan E, Bandyopadhyaya G. Galectin-3: a potential target for cancer prevention. Trends Carbohydr Res. 2011;3(2):13–22. [PMC free article] [PubMed] [Google Scholar]
- 39.Nabi IR, Shankar J, Dennis JW. The galectin lattice at a glance. J Cell Sci. 2015;128(13):2213–9. [DOI] [PubMed] [Google Scholar]
- 40.Ahmed H, AlSadek DM. Galectin-3 as a Potential target to prevent cancer metastasis. Clin Med Insights Oncol. 2015;9:113–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fortuna-Costa A, Gomes AM, Kozlowski EO, Stelling MP, Pavao MS. Extracellular galectin-3 in tumor progression and metastasis. Front Oncol. 2014;4:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.