

Abstract
Background/objectives:
Rhabdomyosarcoma (RMS) is characterized by the expression of the myogenic regulatory protein MYOD1. Histologic types include alveolar, embryonal (ERMS), and spindle cell sclerosing RMS (SRMS). SRMS harbors MYOD1 mutations in a subset of adult cases in association with poor prognosis.
Design/methods:
To study the level of MYOD1 protein expression and its clinical significance, we have analyzed variable numbers of pediatric (<18 years of age) and adult (age range ≥18 to 35 years) ERMS and SRMS cases for presence or absence of MYOD1 immunoreactivity in correlation with clinical outcome and MYOD1 L122R mutations.
Results:
Lack of MYOD1 immunoreactivity, identified in 23.8% of nonalveolar RMS (non-ARMS) cases, was more prevalent in SRMS (44%) than ERMS (17.2%) and was significantly associated with low overall survival and unfavorable tumor sites (p < .05). Lack of MYOD1 immunoreactivity was not associated with MYOD1 L122R mutations, which were identified in 3/37 (8%) cases including only two of 31 (6.5%) pediatric cases, one of 11 or 9% pediatric SRMS, and one case of infant ERMS.
Conclusion:
These studies highlight the prognostic role of MYOD1 in non-ARMS. Lack of MYOD1 immunoreactivity is associated with poor prognosis in ERMS and SRMS. MYOD1 gene mutations are generally infrequent in pediatric RMS. Although mutations are predominant in SRMS, they may exceptionally occur in infantile ERMS.
Keywords: immunohistochemistry, MYOD1, rhabdomyosarcoma, spindle cell sclerosing
1 |. INTRODUCTION
Rhabdomyosarcoma (RMS), the most common pediatric soft tissue sarcoma, is clinically, genetically, and histologically heterogeneous and has been classified into three major histologic types, embryonal, alveolar, and spindle cell sclerosing. Alveolar RMS (ARMS) is biologically more aggressive than embryonal RMS (ERMS) and is characterized by a gene fusion of PAX3 or PAX7 with FOXO1 in >80% of cases.1 ERMS shows more heterogenous molecular features and encompasses several histologic patterns including the botryoid variant, which is associated with good clinical outcome, the anaplastic variant, which is linked to the presence of TP53 mutations, and the conventional variant which has a more diverse clinical and biologic behavior.2,3 The spindle cell and sclerosing RMS type (SRMS) is the least frequent type and is histologically and biologically a heterogenous disease that may be more aggressive in adults than children, in whom it may not be significantly different from classic ERMS.4–9 RMS prognosis is also affected by clinical staging and grouping systems that evaluate the site of disease, local extension, tumor size, regional nodal involvement, the presence or absence of metastasis, and extent of surgical resection at diagnosis.10
Located in chromosome 11p15.1, the MYOD1 gene encodes a protein that is a member of the basic helix–loop–helix muscle regulatory factor family. MYOD1 is necessary for muscle fiber differentiation, and transfection of MYOD1 into mesenchymal progenitor cells induces myogenic differentiation and inhibits cell proliferation.11 Myogenic differentiation by MYOD1 is regulated by p38 mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K-AKT) pathways.12 Recent evidence suggests MYOD1 expression may also be regulated by specific microRNAs, which are also linked to genes involved in cell proliferation and by point mutations in the MYOD1 gene that were identified in about 10% of cases leading to p.Leu122Arg substitution (MYOD1 L122R).13,14
MYOD1 protein is expressed in RMS cell lines and patient tissues, and MYOD1 immunoreactivity is utilized to confirm RMS diagnosis.15 However, clinical observations have suggested that MYOD1 staining is not uniform with different patterns and levels of staining, suggesting a possible role in RMS prognosis. In this study, we analyzed a high number of ERMS and SRMS cases for the rate of MYOD1 immunoreactivity and its correlation with MYOD1 L122R mutations and patient’s prognosis.
2 |. METHODS
A retrospective analysis of archived paraffin-embedded tissue materials was approved by the Institutional Research Integrity Board of Children’s Mercy Hospital and collaborating institutions.
2.1 |. Patients/subjects/samples
Tumor cases were collected from the Pathology files of three separate institutions in the USA and one pediatric hospital in Mexico, collected over a period of 20 years (1999–2019). All patients up to 35 years of age at the time of the surgery with the pathologic diagnosis of ERMS and SRMS, who had available archived blocks or freshly cut unstained slides suitable for MYOD1 immunohistochemistry, were included. RMS diagnosis was confirmed by re-review of the histologic slides for the characteristic morphology and immunophenotype. Pertinent clinical data including patient age, sex, metastasis information, and tumor locations was collected. Follow-up information including whether the patient was alive or dead at the end of follow-up period, and the overall survival rate was also collected when available.
2.2 |. Immunohistochemistry
A monoclonal antibody against MYOD1 (Leica Biosystems, Buffalo Grove, IL) was used in automated immunohistochemistry experiments employing antigen retrieval methods in a Bond Max Leica instrument (Leica Biosystems, Buffalo Grove, IL) that included appropriate positive and negative controls. MYOD1 immunohistochemistry was performed on initial diagnostic specimens regardless of the final clinical stage determination. The immunostained slides were simultaneously reviewed by three pathologists (AA, SH, MS) who evaluated the presence and extent of nuclear staining, irrespective of the cytoplasmic staining. Statistical analysis was performed with SPSS version 23 (IBM, Armonk, NY). Tests of significant differences (p < .05) were calculated with Fisher Exact and chi-square tests. Kaplan–Meier analysis was performed to study the association of MYOD1 staining levels with overall survival.
2.3 |. Mutational analysis
Genomic DNA was isolated from formalin-fixed and paraffin-embedded tissue using the QIAmp DNA FFPE Tissue kit (Qiagen Germantown, MD), and subjected to targeted polymerase chain reaction (PCR) amplification of the known MYOD1 hot spot mutation (p.Leu122Arg) followed by Sanger sequencing. PCR primers included MYOD1 Exon 1 forward: 5′-CCTACTGTGGGCCTGCAAG-3′; and MYOD1 Exon 1 reverse: 5′-GGATCTCCACCTTGGGCAAC. Amplification was performed with the KAPA HiFi HotStart Ready Mix PCR Kit (Cat. Num. KK2601). Amplicons were purified from the PCR reactions using a PureLink PCR Purification Kit (Cat. Num. K310001). Sequencing of purified PCR products was performed by Sanger sequencing using a Big Dye protocol.
3 |. RESULTS
3.1 |. Patients and tumor specimens
Total of 105 nonalveolar RMS (non-ARMS) were enrolled for MYOD1 immunohistochemistry. Patients age ranged from 0 to 35 years, with an average age of 8 years and median of 5 years. Eighteen tumors were classified as SRMS and 87 as ERMS including 14 cases with anaplasia, four botryoid, and 69 conventional cases. Male/female ratio was about 2:1. Metastasis information was available in 54 patients, and 15 tumors were metastatic at presentation.
3.2 |. MYOD1 immunohistochemistry
Of the 105 ERMS and SRMS samples, MYOD1 staining was detected in 80 (76.2%) and was absent in 25 (23.8%). Positive staining ranged from rare to diffuse and was categorized as low (in <50% of cells, n = 37) and high (in ≥50% of cells, n = 43). MYOD1-negative cases were more predominant in SRMS (eight of 18, 44%) than ERMS (15/87, 17.2%), with the difference being statistically significant (p = .0014) (Table 1). Of the ERMS, MYOD1 staining was absent in one of four botryoid, five of 14 anaplastic, and nine of 69 conventional variants. Undetectable staining was significantly more prevalent in unfavorable sites (19/58 cases, 32.8%) than favorable (six of 41 cases, 14.6%) (p = .0212). Of the unfavorable sites, the extremities/soft tissues and pelvic/abdominal locations had the highest prevalence of negative cases (55.6% and 58.3%, respectively). On the other hand, all head/neck, orbit, and parameningeal locations had high staining prevalence (90.6% positive rate). No staining differences were detected based on age or gender (Table 1). Fifteen patients had metastasis (27.8%); six of their tumors were negative for MYOD1 staining (six of 15 or 40%) in contrast to six of 39 (15.4%) tumors in patients with localized disease (p = .0715).
TABLE 1.
MYOD1 expression at different clinical parameters and statistical comparison between positive (high and low) and negative nonalveolar RMS cases from all institutions
| Clinical parameters | Positive number | Negative number | Totals | p-Value |
|---|---|---|---|---|
| Age (years) | ||||
| ≥18 (adult) | 4 | 4 | 8 | .0896 |
| <18 (pediatric) | 76 | 21 | 97 | |
| Gender | ||||
| Female | 32 | 7 | 39 | .3467 |
| Male | 48 | 18 | 66 | |
| Tumor histology | ||||
| SRMS | 8 | 10 | 18 | .0014 |
| ERMS | 72 | 15 | 87 | |
| Tumor locations: Favorable versus unfavorable sites | ||||
| Favorable | 41 | 6 | 47 | .0212 |
| Unfavorable | 39 | 19 | 58 | |
| Totals | 66 | 20 | 86 | |
| Tumor stage: Localized versus metastatic | ||||
| Localized | 33 | 6 | 39 | .0715 |
| Metastatic | 9 | 6 | 15 | |
| Totals | 42 | 12 | 54 | |
3.3 |. Statistical analysis
Of the US cases, patient survival information was available for 60 patients and the follow-up ranged from 5 to 208.8 months (median 46.05 months). Five patient tumors were negative for MYOD1 expression. A Kaplan–Meier survival analysis revealed patients with MYOD1-negative tumors (5/60) had significantly lower overall survival (p = .003) compared to patients with positively staining tumors (55/60; confidence interval: 96.671–156.559) (Figure 1). The five patients with MYOD1-negative tumors survived for a period of ≤52 months and were not alive at the end of follow-up. In contrast, 12 out of 55 MYOD1-positive patients were deceased at the end of follow-up. There was no significant difference in survival between low and high staining and between high and combined low/negative staining.
FIGURE 1.
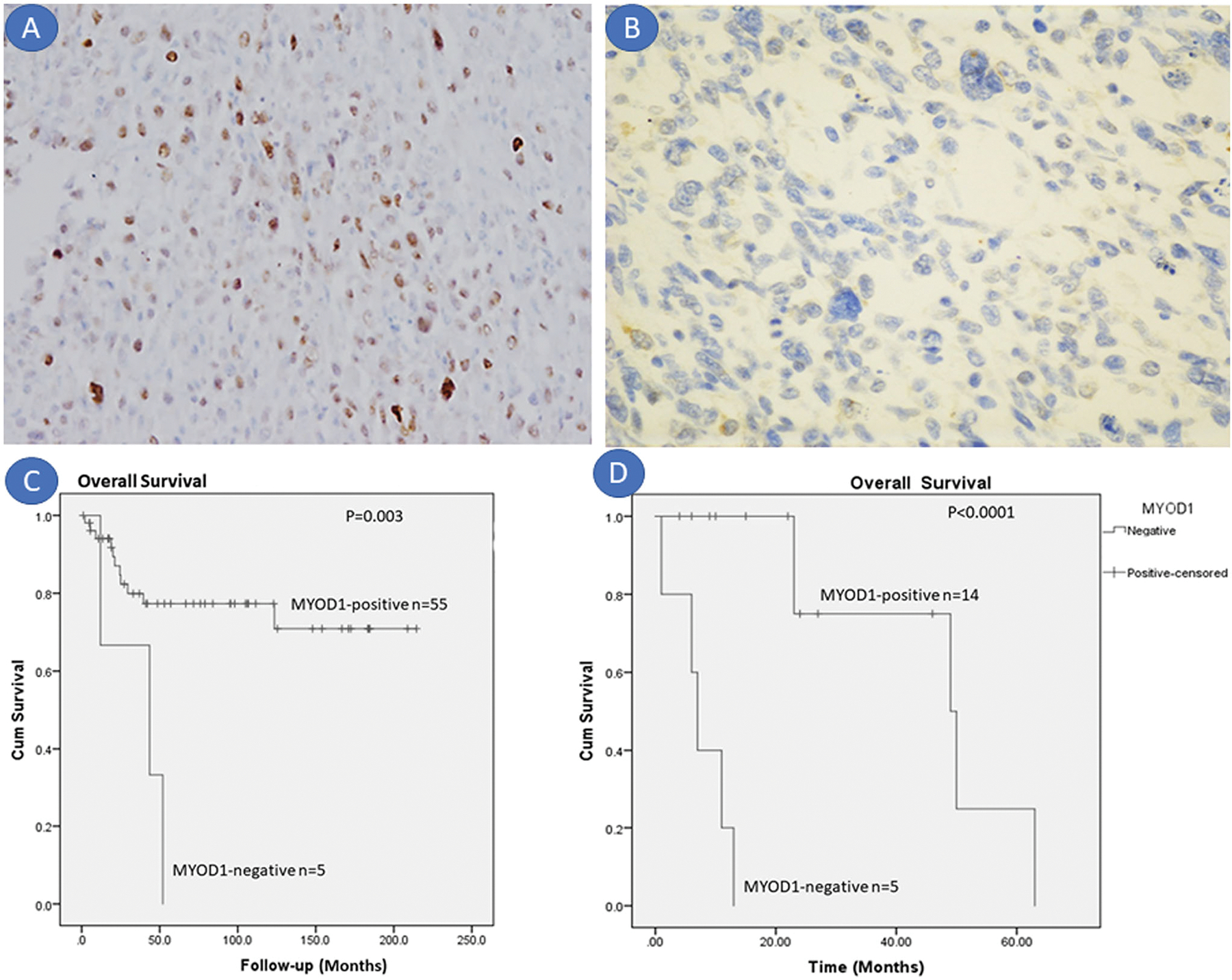
(A) Low level nuclear MYOD1 staining in a nasopharyngeal embryonal rhabdomyosarcoma (ERMS) from a 9-year-old male lacking a MYOD1 mutation (×40). (B) Lack of staining in ERMS with anaplasia. (C) Kaplan–Meier survival plot of US samples revealing significant differences in the overall survival between negative and positive MYOD1 staining. No significant difference was noted between low and high staining. (D) Kaplan–Meier survival plot of Mexican samples revealing significant differences in the overall survival between negative and positive MYOD1 staining
Because of socioeconomic and possible genetic differences, clinical information was analyzed separately for the Mexican patients who had a shorter survival than US patients. Follow-up of the 19 Mexican patients ranged from 1 to 63 months (median 15 months), and five patients were deceased at the end of the follow-up. Metastatic disease at presentation was present in 10/19 patients. Five patients had negative staining, suffered from metastasis (5/10), and had lower overall survival ranging from 1 to 13 months (median 7 months). Only one patient of this group died at the end of follow-up, suggesting the presence of different socioeconomic factors that may have played a role in the shorter follow-up and shorter survival. Kaplan–Meier survival analysis (Figure 1) confirmed significant association of lack of MYOD1 staining with adverse overall survival (p < .0001).
3.4 |. MYOD1 mutation analysis
DNA was successfully retrieved from formalin-fixed paraffin-embedded tissue in 37 cases. MYOD1 L122R mutations were found in three cases (8%), with no distinct relationship to protein immunoreactivity (Table 2). Two of the positive cases were SRMS and one botryoid ERMS. Of the pediatric cases, the mutation frequency was two of 31 (6.5%) in general, including one of 11 (9%) in pediatric SRMS. Patients had variable prognosis, and tumors exhibited infrequent to absent MYOD1 immunoreactivity. The pediatric SRMS tumor was located in the parotid gland (i.e., head/neck region) of a 12-year old female who died of recurrent disease (Figure 2D–F). The other SRMS tumor was in the paraspinal soft tissue of a 35-year old man who did not have follow-up information. Both SRMS cases were negative for MYOD1 staining. The botryoid ERMS tumor had low level of MYOD1 staining and was located in the endocervix of an 11-month-old female child who presented with a protruding vaginal mass (Figure 2A–C). This patient was alive and healthy after a 72-month follow-up.
TABLE 2.
Clinical information of the three MYOD1 L122R mutation-positive cases
| Characteristics | Case 1 | Case 2 | Case 3 |
|---|---|---|---|
| Age and gender | 11-month female | 12-year female | 35-year male |
| Tumor location | Endocervix | Parotid | Paraspinal soft tissue |
| Diagnostic category | Botryoid ERMS | Sclerosing SRMS | Spindle SRMS |
| MYOD1 stain | 10% | 0 | 0 |
| Clinical stage | I | I | n/a |
| Follow-up period (months) | 72 | 52 | n/a |
| Follow-up status | Alive and healthy | Died ofdisease | n/a |
| Overall survival (months) | 72 | 52 | n/a |
| Disease-free survival (months) | 67 | 0 | n/a |
FIGURE 2.
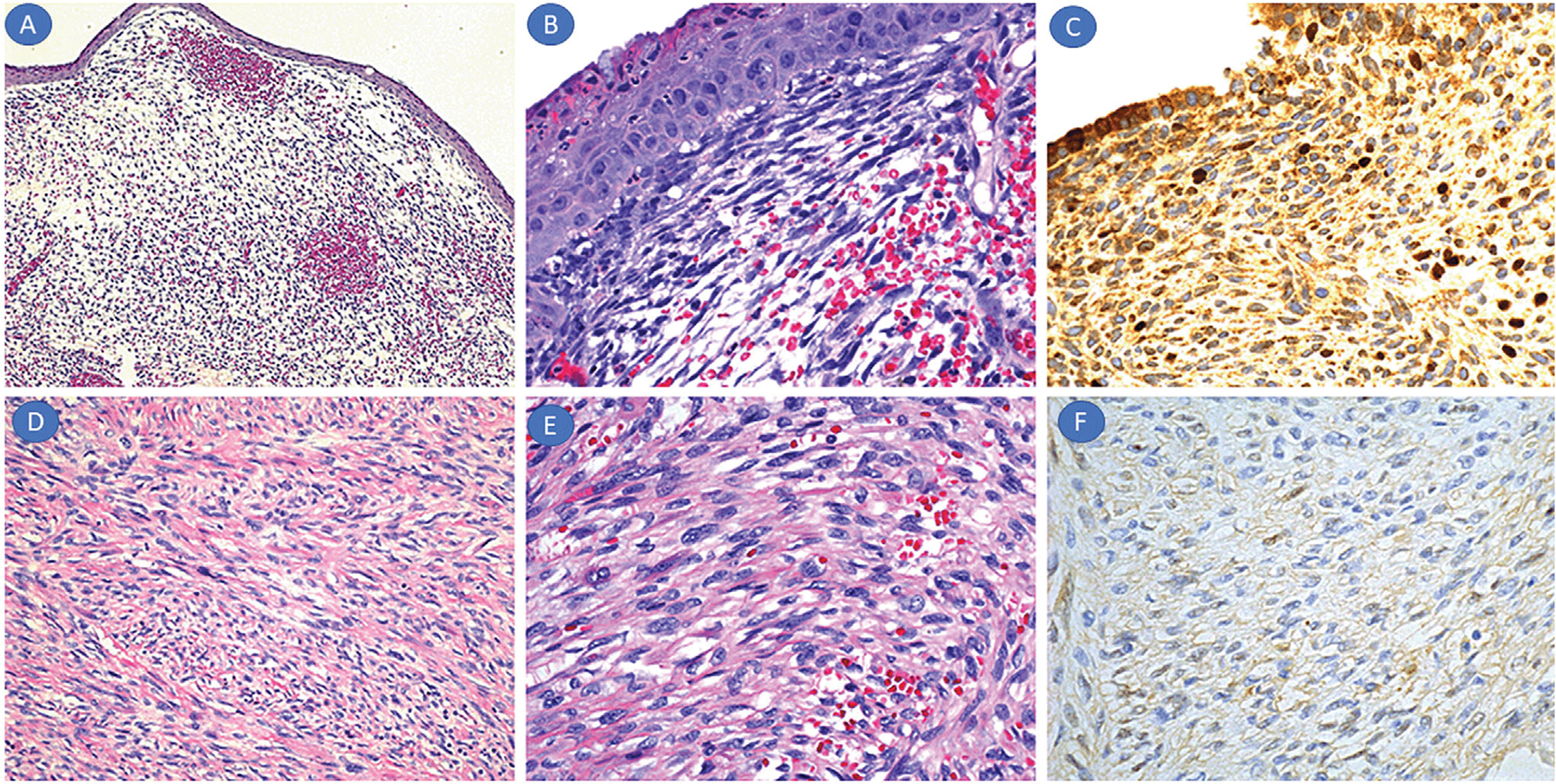
Mutation-positive rhabdomyosarcoma (RMS). (A–C) Morphologic features of the endocervical ERMS of the 11-month-old girl. The submucosal tumor showed polypoid appearance with loose myxoid areas (A: H&E, ×10), subepithelial cambium layer (B: H&E, ×40), and low MYOD1 nuclear staining (×20) with marked nonspecific cytoplasmic staining. (D–F) Morphologic features of the parotid gland SRMS in the 12-year-old girl. The tumor showed typical spindle cell and fascicular morphology in a sclerosed background (D: H&E, ×20), with mild cytologic atypia and low mitotic rate (E: H&E, ×40). MYOD1 immunohistochemistry revealed absence of nuclear staining (F: ×20)
4 |. DISCUSSION
The expression or lack of nuclear MYOD1 in our case collection did not correlate with the limited prevalence of MYOD1 gene mutations and suggests that MYOD1 protein expression, if present, results from transactivation by other signaling pathways and not from inactivating gene mutations. These premises are supported by earlier studies that revealed MYOD1 activation by YAP/TAZ and Wnt/β-catenin pathways.16,17 MYOD1 translocates to the nucleus and activates myogenin and other myogenic transcription factors. Thus, the absence of nuclear protein suggests a lack of or altered myogenic differentiation and a more primitive proliferative phenotype. MYOD1 has recently been shown to play a universal role in cancer development where it regulates proliferation and apoptosis.17 In this study, we have documented the absence of MYOD1 nuclear immunoreactivity in about 23.8% of non-ARMS and showed its significant association with shorter overall survival and its prevalence in unfavorable sites and SRMS tumors. Although most US patients had localized disease, lack of MYOD1 immunoreactivity may explain the mortality and shorter overall survival in this group.
In contrast to immunohistochemical staining, MYOD1 L122R mutations were only identified in 8% of our cases. This prevalence is similar to what was recently reported (10%) in a larger cohort of 104 cases where mutations were associated with PI3K-AKT pathway mutations.14 These mutations were found in a subset of clinically aggressive adult and pediatric ERMS cases with a predominant spindle cell histology and predilection for older individuals and head and neck sites. The L122R substitution enhances binding of MYOD1 to MYC sites, leading to activation of a MYC-like transcriptional program and inhibits the function of wild type MYOD1 in a dominant-negative fashion.18 Subsequent studies have revealed that these mutations occur mostly in the SRMS type with reported percentages varying from 33% to 56%. Most of the mutation-positive cases have a sclerosing pattern (33–100%), while tumors with a predominant spindle cell pattern harbor the mutations in lower frequencies (25–41%).19–23 In comparison, pediatric RMS in this study had lower mutation frequency (two of 13 or 6.5%) than what is reported in adult series. The mutation rate in our pediatric SRMS (<18 years) is also low (one of 11 or 9%).
The high prevalence of MYOD1 mutations in the sclerosing variant SRMS was confirmed in a recent study where 10 out of 15 mutation-positive pediatric SRMS were of the sclerosing variant.24 The lower rate of MYOD1 mutations frequency in our series may seem discordant but correspond to the lower rate of the sclerosing variant SRMS, which included four out of 18 cases, one of which had the mutation (i.e., one of four or 25%). In addition, our SRMS cases were less frequent in the head/neck locations (6/18), which were reported to have higher mutation rate. These differences could be due to regional geographic variations as most of our pediatric cases are from the midwestern United States rather than a collection of referral cases in large centers. Our findings conform to other studies that suggest that SRMS in children and adults have different characteristics. A recent review of a large cohort of Children’s Oncology Group clinical trial patients confirmed that pediatric SRMS has a lower frequency of sclerosing variants and only rare MYOD1 mutations.9 Although the prognosis of these tumors is reportedly better than in adults, the presence of MYOD1mutations imparts an unfavorable outcome with higher incidence of recurrence/metastasis and a higher clinical stage.19,20
In addition to lower mutation frequency, our illustrated cases reveal absent or low staining in mutation-positive cases and low rate of MYOD1 staining in SRMS in general, in contrast to more diffuse staining reported by others.19 This difference could be attributed to similar regional factors as described above as well as technical factors. MYOD1 immunohistochemistry frequently results in diffuse cytoplasmic staining that may be over-interpreted as genuine nuclear labeling (Figure 2A).
Our case cohort includes the first case of a non-SRMS tumor harboring the MYOD1 L122R mutation. The tumor in the endocervix of an 11-month-old girl exhibited a botryoid pattern. Infant RMS cases tend to be of the conventional ERMS subtype and exhibit fusions involving VGLL2 and NCOA2 rather than MYOD1 mutations.23,25 This tumor was associated with favorable botryoid histology, favorable site, and stage I disease. The patient was alive and healthy at the end of a 6-year follow-up. Botryoid ERMS is known to have a favorable clinical course with low rate of distant metastasis.26 This case highlights the genetic heterogeneity of infantile RMS, including the first report of MYOD1 mutations. Thus, regardless of the mutation status, the prognosis can be affected or modified by the tumor histology and locations. This statement is supported by a recent study in which a multivariate analysis of 99 head and neck RMS containing MYOD1-mutant cases emphasized the importance of the histologic type in association with the prognosis, thus adding to the algorithmic complexity of RMS prognosis.21
In conclusion, the studied cases highlight the prognostic role of MYOD1 immunoreactivity in the evaluation of non-ARMS. Lack of MYOD1 staining is associated with poor prognosis and SRMS histology. MYOD1 gene mutations are infrequent in our collection of pediatric RMS. Although mutations are predominant in SRMS, they can exceptionally occur in infantile ERMS.
ACKNOWLEDGMENTS
The authors would like to thank the DNA core facility of the University of Missouri-Columbia, Missouri for processing the Sanger sequencing. We would also like to acknowledge Dr. Lisa Teot for initial review of the histologic and immunostained slides. We also wish to thank the departments of Pathology and Laboratory Medicine and Pediatric Hematology-Oncology, Children’s Mercy Hospital, Kansas City for supporting the studies.
Abbreviations:
- ARMS
alveolar rhabdomyosarcoma
- ERMS
embryonal rhabdomyosarcoma
- RMS
rhabdomyosarcoma
- SRMS
spindle cell sclerosing rhabdomyosarcoma
Footnotes
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1.Parham DM. Pathologic classification of rhabdomyosarcomas and correlations with molecular studies. Mod Pathol. 2001;14:506–514. [DOI] [PubMed] [Google Scholar]
- 2.Newton WA, Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification–an Intergroup Rhabdomyosarcoma Study. Cancer. 1995;76:1073–1085. [DOI] [PubMed] [Google Scholar]
- 3.El Demellawy D, Mcgowan-Jordan J, De Nanassy J, Chernetsova E, Nasr A. Update on molecular findings in rhabdomyosarcoma. Pathology. 2017;49:238–246. [DOI] [PubMed] [Google Scholar]
- 4.Mentzel T, Kuhnen C. Spindle cell rhabdomyosarcoma in adults: clinicopathological and immunohistochemical analysis of seven new cases. Virchows Arch. 2006;449:554–560. [DOI] [PubMed] [Google Scholar]
- 5.Nascimento AF, Fletcher CD. Spindle cell rhabdomyosarcoma in adults. Am J Surg Pathol. 2005;29:1106–1113. [PubMed] [Google Scholar]
- 6.Mentzel T, Katenkamp D. Sclerosing, pseudovascular rhabdomyosarcoma in adults. Clinicopathological and immunohistochemical analysis of three cases. Virchows Arch. 2000;436:305–311. [DOI] [PubMed] [Google Scholar]
- 7.Chiles MC, Parham DM, Qualman SJ, et al. Sclerosing rhabdomyosarcomas in children and adolescents: a clinicopathologic review of 13 cases from the Intergroup Rhabdomyosarcoma Study Group and Children’s Oncology Group. Pediatr Dev Pathol. 2004;7:583–594. [DOI] [PubMed] [Google Scholar]
- 8.Owosho AA, Chen S, Kashikar S, et al. Clinical and molecular heterogeneity of head and neck spindle cell and sclerosing rhabdomyosarcoma. Oral Oncol. 2016;58:e6–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rudzinski ER, Anderson JR, Hawkins DS, Skapek SX, Parham DM, Teot LA. The World Health Organization classification of skeletal muscle tumors in pediatric rhabdomyosarcoma: a report from the Children’s Oncology Group. Arch Pathol Lab Med. 2015;139:1281–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dagher R, Helman L. Rhabdomyosarcoma: an overview. Oncologist. 1999;4:34–44. [PubMed] [Google Scholar]
- 11.Sorrentino V, Pepperkok R, Davis RL, Ansorge W, Philipson L. Cell proliferation inhibited by MyoD1 independently of myogenic differentiation. Nature. 1990;345:813–815. [DOI] [PubMed] [Google Scholar]
- 12.Zhu J, Blenis J, Yuan J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc Natl Acad Sci U S A. 2008;105:6584–6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elsaeid Elnour I, Dong D, Wang X, et al. Bta-miR-885 promotes proliferation and inhibits differentiation of myoblasts by targeting MyoD1. J Cell Physiol. 2020;235:6625–6636. [DOI] [PubMed] [Google Scholar]
- 14.Kohsaka S, Shukla N, Ameur N, et al. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat Genet. 2014;46:595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morotti RA, Nicol KK, Parham DM, et al. An immunohistochemical algorithm to facilitate diagnosis and subtyping of rhabdomyosarcoma: the Children’s Oncology Group experience. Am J Surg Pathol. 2006;30:962–968. [DOI] [PubMed] [Google Scholar]
- 16.Sun C, De Mello V, Mohamed A, et al. Common and distinctive functions of the Hippo effectors Taz and Yap in skeletal muscle stem cell function. Stem Cells. 2017;35:1958–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao L, Liu Y, Tong D, et al. MeCP2 promotes gastric cancer progression through regulating FOXF1/Wnt5a/β-catenin and MYOD1/Caspase-3 signaling pathways. EBioMedicine. 2017;16:87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Antwerp ME, Chen DG, Chang C, Prochownik EV. A point mutation in the MyoD basic domain imparts c-Myc-like properties. Proc Natl Acad Sci U S A. 1992;89:9010–9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rekhi B, Upadhyay P, Ramteke MP, Dutt A. MYOD1 (L122R) mutations are associated with spindle cell and sclerosing rhabdomyosarcomas with aggressive clinical outcomes. Mod Pathol. 2016;29:1532–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agaram NP, Chen C-L, Zhang L, Laquaglia MP, Wexler L, Antonescu CR. Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: evidence for common pathogenesis. Genes Chromosomes Cancer. 2014;53:779–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Owosho AA, Huang S-C, Chen S, et al. A clinicopathologic study of head and neck rhabdomyosarcomas showing FOXO1 fusion-positive alveolar and MYOD1-mutant sclerosing are associated with unfavorable outcome. Oral Oncol. 2016;61:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szuhai K, De Jong D, Leung WY, Fletcher CDM, Hogendoorn PCW. Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. J Pathol. 2014;212:300–307. [DOI] [PubMed] [Google Scholar]
- 23.Alaggio R, Zhang L, Sung Y-S, et al. A molecular study of pediatric spindle and sclerosing rhabdomyosarcoma: identification of novel and recurrent VGLL2-related fusions in infantile cases. Am J Surg Pathol. 2016;40:224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agaram NP, Laquaglia MP, Alaggio R, et al. MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: an aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod Pathol. 2019;32:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sparber-Sauer M, Stegmaier S, Vokuhl C, et al. Rhabdomyosarcoma diagnosed in the first year of life: localized, metastatic, and relapsed disease. Outcome data from five trials and one registry of the Cooperative Weichteilsarkom Studiengruppe (CWS). Pediatr Blood Cancer. 2019;66:e27652. [DOI] [PubMed] [Google Scholar]
- 26.Kriseman ML, Wang W-L, Sullinger J, et al. Rhabdomyosarcoma of the cervix in adult women and younger patients. Gynecol Oncol. 2012;126:351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.