

SUMMARY
Raf-activating mutations are frequent in cancer. In the basal state, B-Raf is autoinhibited by its upstream Ras-binding domain (RBD) and cysteine-rich domain (RBD-CRD) interacting with its kinase domain (KD) and the 14-3-3 dimer. Our comprehensive molecular dynamics simulations explore two autoinhibition scenarios in the presence and absence of the 14-3-3 dimer. When present, the 14-3-3 interaction with B-Raf stabilizes the RBD-CRD-KD interaction, interfering with the KD dimerization. Raf’s pSer365 removal fails to induce large disruption. RBD-CRD release promotes KD fluctuations and reorientation for dimerization, consistent with experimental data. In the absence of 14-3-3, our sampled B-Raf conformations suggest that RBD-CRD can block the KD dimerization surface. Our results suggest a B-Raf activation mechanism, whereby one KD monomer is donated by 14-3-3-free B-Raf KD and the other by 14-3-3-bound KD. This mechanism can lead to homo- and heterodimers. These autoinhibition scenarios can transform autoinhibited B-Raf monomers into active B-Raf dimers.
INTRODUCTION
Raf is a family of serine/threonine protein kinases in the Raf/MEK/ERK (MAPK) signaling pathway that promotes cell proliferation (Lavoie and Therrien, 2015; Tsai and Nussinov, 2018). Its activation is essential for cell proliferation in cancer (Durrant and Morrison, 2018; Roskoski, 2018). Oncogenic mutations in B-Raf are frequent and have been identified in many cancers (Davies et al., 2002; Wan et al., 2004). The hotspot oncogenic mutation, V600E, has been found in ~50% of melanomas (Davies et al., 2002; Holderfield et al., 2014). ATP-competitive B-Raf inhibitors have been extensively explored for Raf cancers, and vemurafenib and dabrafenib for B-RafV600E mutant have been approved by the US Food and Drug Administration for the treatment of melanomas (Agianian and Gavathiotis, 2018; Flaherty et al., 2010).
Raf has three main isoforms, A-Raf, B-Raf, and C-Raf (Raf-1). They share similar sequences, structures, and functions, with three conserved regions, CR1, CR2, and CR3 (Shaw et al., 2014; Yaeger and Corcoran, 2019). CR1 contains the regulatory Ras-binding domain (RBD) and the cysteine-rich domain (CRD). In active membrane-anchored Raf, RBD interacts with Ras, and CRD binds to the membrane (Garcia-Gomez et al., 2018; Ghosh et al., 1994; Li et al., 2018; Terrell and Morrison, 2019; Travers et al., 2018). Ras-RBD interactions recruit Raf to the plasma membrane, resulting in an increased population of kinase domain (KD) dimers for activation (Pantsar, 2020). CR2 is the loop with multiple phosphorylation sites. CR3 is the catalytic KD at the C-terminal end.
Full Raf activation requires KD dimerization (Cotto-Rios et al., 2020; Durrant and Morrison, 2018; Hu et al., 2013; Lavoie et al., 2013; Rajakulendran et al., 2009). Like all kinases, wild-type Raf is expected to spend most of its lifetime in the inactive state. The monomeric “closed” autoinhibited state guards against spurious activation and signaling, as well as degradation, by shielding the KD dimerization surface (Nussinov et al., 2020). Extensive data indicate the involvement of 14-3-3 proteins in Raf autoinhibition (Kondo et al., 2019; Liau et al., 2020; Park et al., 2019). 14-3-3 proteins are universally expressed regulatory scaffolds for signaling proteins. They form dimers to accommodate targets containing phosphorylated serine or threonine motifs (Gardino et al., 2006). Raf proteins contain two 14-3-3 recognition motifs (Fu et al., 1994; Kohler and Brummer, 2016; Michaud et al., 1995; Zhang and Guan, 2000). In the C-terminal tail, phosphorylation of serine 729 (pSer729) in B-Raf (pSer621 in C-Raf and pSer582 in A-Raf) promotes Raf activation (Morrison et al., 1993). Removal of the phosphorylation at this site reduces Raf activity (Chong and Guan, 2003; Morrison et al., 1993). In contrast, the phosphorylated serine 365 (pSer365) in B-Raf’s CR2 loop (pSer259 in C-Raf and pSer214 in A-Raf) inhibits Raf activation (Chong and Guan, 2003; Dhillon et al., 2002; Morrison et al., 1993). The recent cryoelectron microscopy (cryo-EM) structures show that the interactions of the 14-3-3 dimer with the two phosphorylated sites maintain the autoinhibition state of B-Raf, supporting the autoinhibition scenario in the presence of the 14-3-3 dimer (Park et al., 2019). In the absence of the 14-3-3 dimer, intramolecular interactions between the N-terminal CR1 and the C-terminal KD can still take place, albeit with lower stability, as we also show here, in line with experimental data (Lavoie and Therrien, 2015; Tran et al., 2005).
Two events are expected to regulate the release of B-Raf autoinhibition: dephosphorylation of pSer365 and release of RBD-CRD from KD. Dephosphorylation of pSer365 is required, but insufficient for full B-Raf activation (Chong and Guan, 2003). Mutations of Ser365 cannot fully activate B-Raf (Chong and Guan, 2003; Morrison et al., 1993). Ras is upstream of Raf in the MAPK pathway (Lu et al., 2016a; Nussinov et al., 2017). It recruits Raf to the membrane, gathering Raf proteins through dimers or nanoclusters. The high affinity of Ras-Raf-RBD interaction, coupled with the loosening of the 14-3-3 interaction through pSer365 dephosphorylation, shifts the equilibrium, disrupting the autoinhibited B-Raf states, thereby promoting full activation of Raf (Fetics et al., 2015; Hatzivassiliou et al., 2013; Lu et al., 2016b; Nussinov et al., 2015, 2019b). The dimerized Raf KD—which reflects the active Raf state—is stabilized by the 14-3-3 dimers, as verified by the two recent cryo-EM structures (Kondo et al., 2019; Park et al., 2019). The cryo-EM structure also reveals the asymmetry in B-Raf’s active sites, illustrating the paradoxical activation of Raf by kinase inhibitors (Kondo et al., 2019).
Within our long-term goal of uncovering the structural mechanisms of Ras regulators and the activation and signaling of Ras effectors in the physiological native states and in cancer, we have taken up the autoinhibited states of B-Raf, explored two B-Raf autoinhibition and full activation scenarios, in the presence and absence of the 14-3-3 dimer, including the effects of pSer365 dephosphorylation and the release of the inhibitory interactions between RBD-CRD and KD, in atomistic detail, and have compared our results with the available experimental data. The structure of KD interacting with RBD-CRD in the absence of the 14-3-3 dimer is unavailable, likely due to the challenge of experimentally determining the structure of an unstable state. We observe that during simulations that initiate from the 14-3-3-bound structure, but with the RBD-CRD and pSer365 removed, KD fluctuates and rotates on the 14-3-3. We also sample the structural ensemble of the B-Raf autoinhibited state in the absence of the 14-3-3 dimer and identify the potential residues at the interface between CRD and KD’s N lobe, in line with the experimental data (Daub et al., 1998). The consistency, including that related to pSer365 dephosphorylation and mutational data, lead us to propose a mechanism for Raf activation, whereby the two autoinhibition scenarios—with and without 14-3-3—can collaborate. We call the KD that is rotated but is still attached to the 14-3-3 14-3-3-bound KD, and the KD from the 14-3-3-free autoinhibition mechanism 14-3-3-free KD. We suggest that the 14-3-3-free KD can interact with the 14-3-3-bound KD, leading to active Raf homo- or heterodimers organized and stabilized on the 14-3-3.
RESULTS
B-Raf autoinhibition in the presence of the 14-3-3 dimer
In the absence of stimulation, B-Raf autoinhibition prevents signaling through the MAPK proliferation pathway, keeping it at a low, basal level. The 14-3-3 dimer regulates Raf autoinhibition. Partial loss of the 14-3-3 dimer interaction via dephosphorylation increases B-Raf activity (Thorson et al., 1998). The cryo-EM structure features two phosphorylated sites (pSer365 and pSer729) in B-Raf for the 14-3-3 interactions. CRD is in the center interacting with the KD and 14-3-3 dimer (Figure 1). The RBD and CR2 loops (residues 283–361 and residues 372–449) in the structure lack full density (Park et al., 2019). To explore the dynamics of B-Raf autoinhibition in the presence of the 14-3-3 dimer, we modeled the missing RBD and loops into the cryo-EM structure and performed molecular dynamics (MD) simulations (for details see STAR methods). The initial and final structures are summarized in Figure S1.
Figure 1. B-Raf autoinhibition in the presence of the 14-3-3 dimer.

(A) B-Raf consists of the N-terminal RBD-CRD and C-terminal kinase domain (KD). The phosphorylated sites in the middle loop (pSer365) and C-terminal tail (pSer729) interact with the 14-3-3 dimer. The release of pSer365 and RBD-CRD activates B-Raf.
(B–D) (B) The structures of the fully autoinhibited B-Raf in complex with the 14-3-3 dimer. The (C) pSer365 and (D) pSer729 form strong interactions with the basic residues (K49, R56, R127) in 14-3-3 dimer.
The overall structure of the autoinhibited B-Raf in complex with the 14-3-3 dimer was stable in the simulations. No significant disruption within or between domains was observed. The pSer365 (362RSSpSAPNVHI371) and pSer729 (726RSApSEPSLNR735) in B-Raf are the 14-3-3 recognition motifs, which stably interacted with the basic residues, K49, R56, and R127 in 14-3-3 with small root-mean-square fluctuation (RMSF) values and residue distances (Figure 2). The interaction energies of pSer365 and pSer729 motifs with the 14-3-3 dimer are ~−577.1 kcal/mol and ~−593.8 kcal/mol, respectively, which suggest significant contributions of the two phosphorylated sites to the B-Raf autoinhibition scenario in the presence of the 14-3-3 dimer (Figure S2). The interaction of the pSer729 motif with the CRD domain was not strong, with only one hydrogen bond forming between S732 in pSer729 motif and N236 in CRD. The pSer365 motif has strong interactions with CRD. There are hydrophobic interactions between P367 in the pSer365 motif and L245 and L254 in CRD, and a salt bridge between D361 in the pSer365 motif and R271 in CRD. The interactions of KD (~−626.9 kcal/mol) and CRD (~−375.5 kcal/mol) with the 14-3-3 dimer are strong, whereas the that of RBD is weak (Figure S2). The CRD in the center of the complex acts as a core to gather the KD and 14-3-3 dimer, indicating its importance in stabilizing the autoinhibited state. It was stable in the simulations of the complex, as evidenced by the small RMSF values (Figure 2A). RBD has no initial contact with the 14-3-3 dimer and KD. It was unstable with high RMSF values in the simulations (Figure 2A), in line with their high flexibility in the cryo-EM structures. KD is constrained and stable with low RMSF values (Figure 2A). Although the dimer interface is not fully blocked, its orientation is not amenable to dimerization for activation (Figure S1).
Figure 2. Release of B-Raf autoinhibition in the presence of the 14-3-3 dimer.
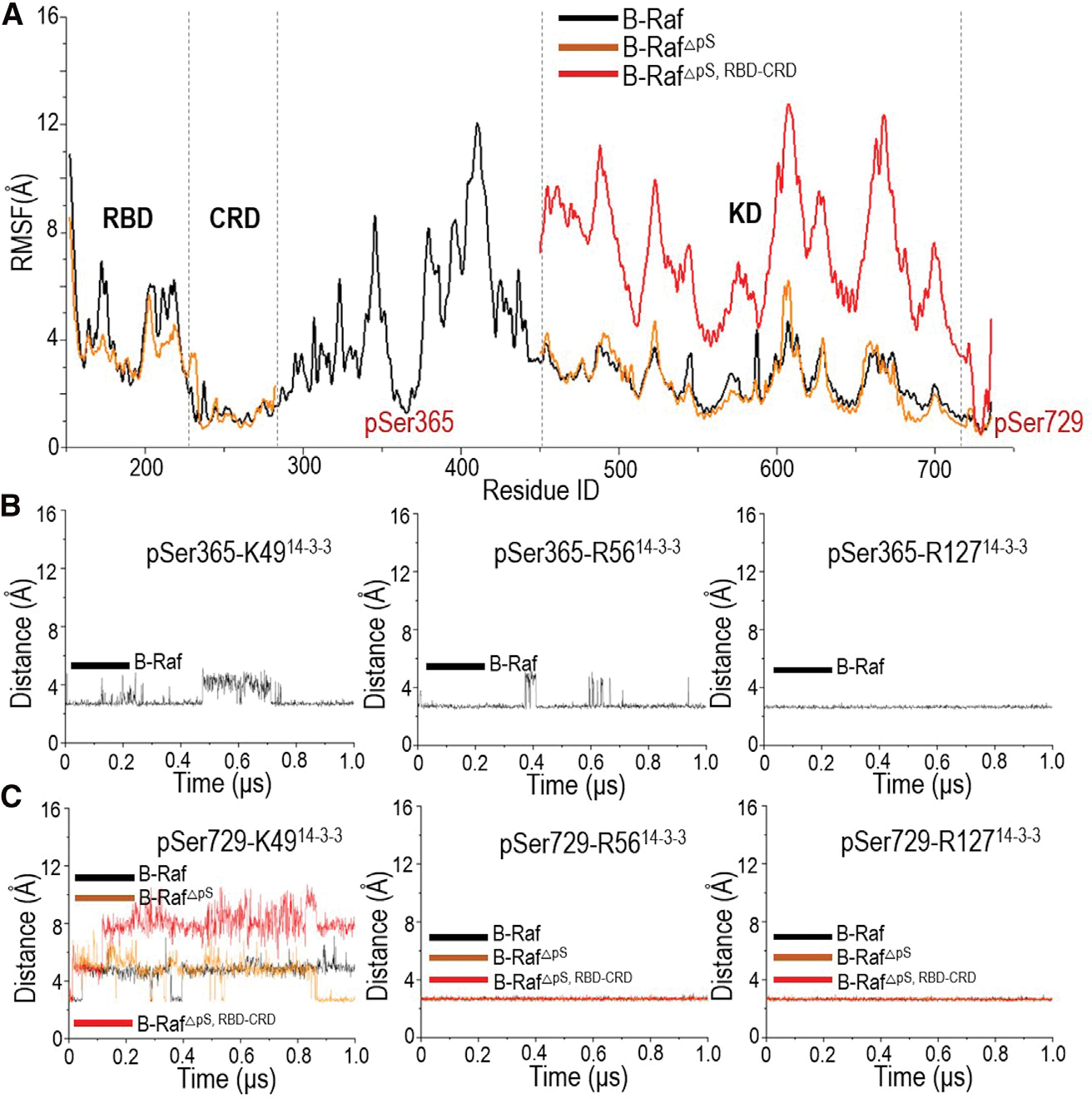
The (A) root-mean-square fluctuation (RMSF) values, (B) distances between pSer365 and the basic residues in 14-3-3 dimer, and (C) distances between pSer729 and the basic residues in 14-3-3 dimer indicate the conformational changes in B-Raf in complex with 14-3-3 dimer upon pSer365 release (denoted as B-RafΔpS) and RBD-CRD release (denoted as B-RafΔpS,RBD–CRD).
Release of B-Raf autoinhibition in the presence of the 14-3-3 dimer
pSer365 is inhibitory for B-Raf. Its removal releases B-Raf autoinhibition and increases the basal activity (Morrison et al., 1993). In the fully autoinhibited B-Raf with the 14-3-3 dimer, the pSer365 motif interacts with the CRD and the 14-3-3 dimer (Figure 3A). The basic residues (K49, R56, and R127) in the 14-3-3 protein formed electrostatic forces with pSer365 (Figure 1C). These interactions were stable throughout the simulations (Figure 2B). Residue P367 in the pSer365 motif interacted with L216 and L220 in the 14-3-3 dimer, and L245 and L254 in CRD (Figure S2). At the N terminus, D361 in the pSer365 motif formed a salt bridge with R271 in CRD. A salt bridge was also formed between K253 in CRD and D223 in the 14-3-3 dimer (Figure 3A).
Figure 3. Structural insights into B-Raf activation upon the release of pSer365 motif and RBD-CRD.
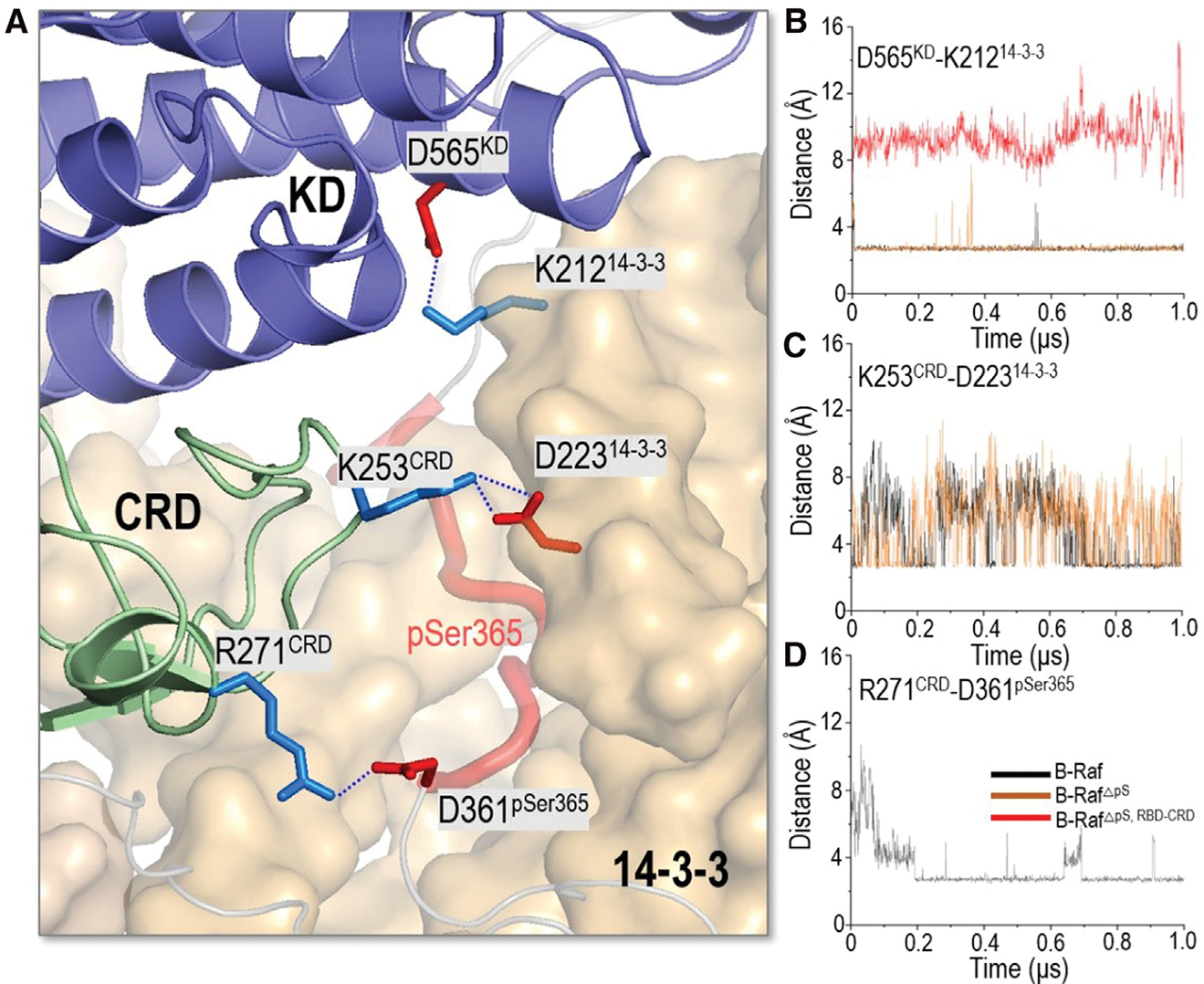
(A) In the fully autoinhibited B-Raf in complex with 14-3-3 dimer, KD and CRD interact with 14-3-3 dimer.
(B–D) The salt bridges of (B) D565KD-K21214-3-3, (C) K253CRD-D22314-3-3, and (D) R271CRD-D361pSer365 show the changes upon the release of pSer365 motif and RBD-CRD.
To explore the partial activation of B-Raf by pSer365 removal, we removed the pSer365 motif from the complex in the simulation to mimic the dephosphorylation of pSer365 in Raf activation. The simulations show that the overall structure does not undergo significant conformational changes upon pSer365 removal. The RMSF curve is comparable with that of the fully autoinhibited B-Raf with the 14-3-3 dimer (Figure 2A). This is in line with the experimental findings that removal of pSer365 is required but insufficient for full activation of B-Raf (Chong and Guan, 2003; Morrison et al., 1993). Instead, it induced local structural changes. CRD is the B-Raf membrane-anchoring domain. It contains hydrophobic loops (residues 242–256) and basic residues (R239, K240, R252, K253, R271) for the membrane interactions (Improta-Brears et al., 1999; Mott et al., 1996). These residues in CRD are largely buried by the interactions with the pSer365 motif and nearby KD and the 14-3-3 dimer (Figure S2). Removal of pSer365 eliminated these interactions. While the salt bridges of D565KD-K21214-3-3 and K253CRD-D22314-3-3 remained stable, the salt bridge of R271CRD-D361pSer365 was disrupted, making CRD more exposed (Figures 3B–3D and S2C).
The cryo-EM structure indicates that CRD interacts with and stabilizes the KD in the complex of the autoinhibited B-Raf with the 14-3-3 dimer (Park et al., 2019). Release of RBD-CRD from the complex is crucial for full activation of B-Raf. We removed both the pSer365 motif and the N-terminal containing the RBD-CRD from the complex to mimic Raf’s activation by Ras, where the N-terminal RBD-CRD is at the membrane, away and without direct contact with KD. The results indicate that KD experienced a notable structural change upon RBD-CRD removal (Figure S1). KD reoriented and became more flexible with high RMSF values (Figure 2A). The salt bridge of D565KD-K21214-3-3 was disrupted (Figure 3B). This is consistent with the cryo-EM structures (PDB: 6NYB for inactive B-Raf, 6UAN for activated B-Raf. and 6Q0J for activated B-Raf with MEK), which show that KD reorientation is needed for the Raf dimerization (Figure S1). Despite the conformational change, the interactions of the pSer729 motif with the 14-3-3 dimer remained stable throughout. The salt bridges between the basic residues (K49, R56, and R127) in 14-3-3 protein and pSer729 were largely maintained (Figure 2C).
B-Raf autoinhibition in the absence of the 14-3-3 dimer
In the absence of the 14-3-3 dimer, B-Raf can also be autoinhibited through the association of RBD-CRD and KD, which indicates another autoinhibition scenario. The interactions between RBD-CRD and KD are expected to be metastable, in line with the principles of autoinhibition (Nussinov et al., 2018, 2020). To date no structure is available. We modeled and simulated RBD-CRD interacting with KD to sample the conformational ensemble of B-Raf autoinhibition in the absence of the 14-3-3 dimer. In the cryo-EM structure of the autoinhibited B-Raf in complex with the 14-3-3 dimer, the CRD interacts with the KD, suggesting a plausible interface between CRD and KD for the B-Raf autoinhibition scenario in the absence of the 14-3-3 dimer (Park et al., 2019). We modeled a system containing RBD-CRD and KD, with the same CRD/KD interface as in the cryo-EM structure, and performed the simulations. The RBD has no initial contact with KD. In the simulations, the interface was largely maintained (Figure 4A). The RBD experienced initial fluctuations and achieved interactions with the KD during the simulations. Salt bridges were identified at the interface between RBD and KD (D179RBD-K700KD, E193RBD-K687:K690:R691KD) (Figure 4B), and a salt bridge (R252RBD-E695KD) and hydrophobic interactions (F243:L245:A246:F247CRD-A712:L716KD) at the CRD/KD interface (Figure 4C). Notably, RBD-CRD interacts with the KD’s C lobe without blocking the KD dimer interface (Figure 4A), indicating that the interactions that we observed here more likely facilitate a 14-3-3-involved B-Raf autoinhibition scenario, but cannot fully represent the B-Raf autoinhibition in the absence of the 14-3-3 dimer.
Figure 4. Structure of the pre-assembled system for the RBD-CRD interacting with KD, containing the same interface between CRD and KD in the cryo-EM structure.
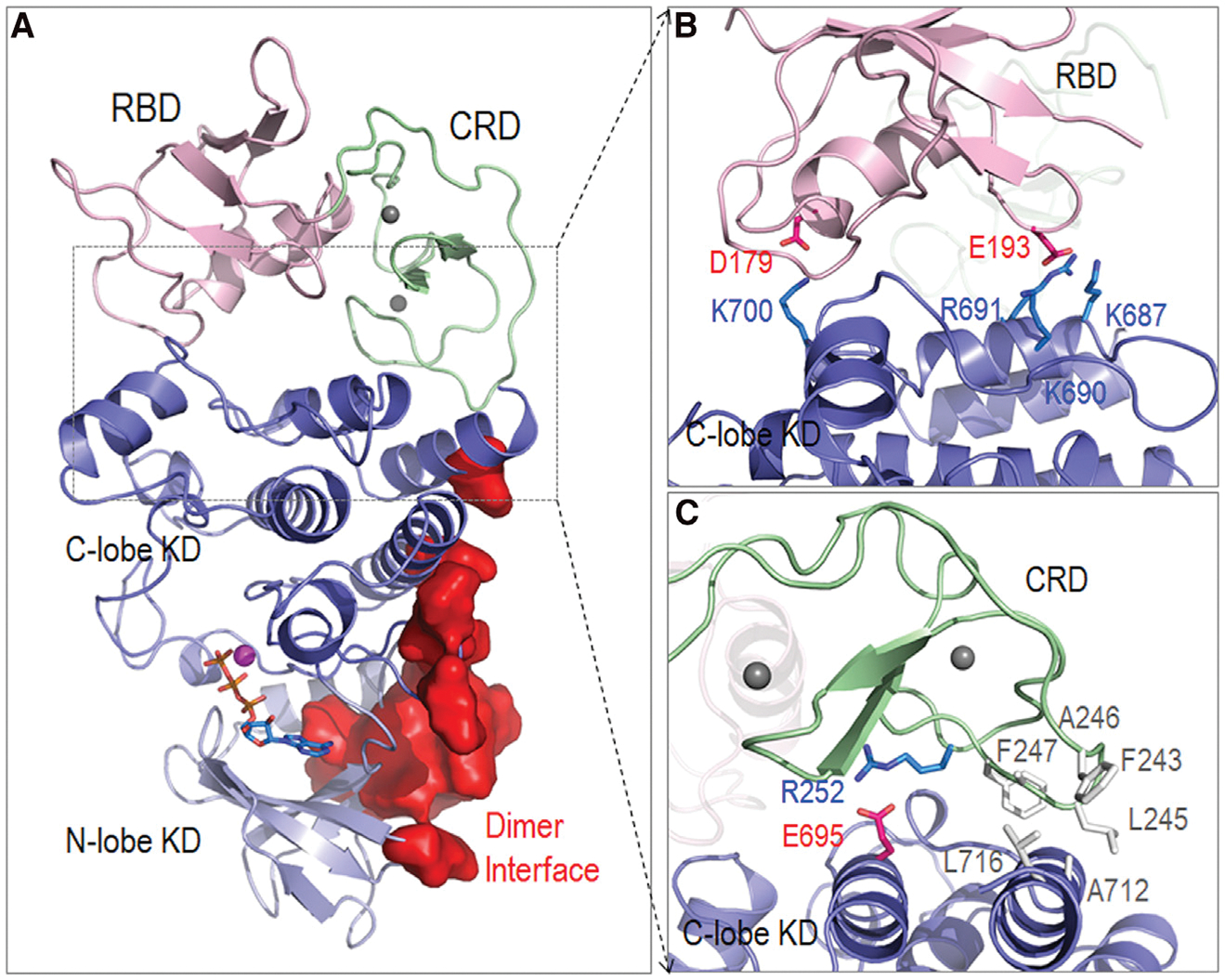
(A) The structure of the system indicates that RBD-CRD interacts with KD’s C lobe. The dimer interface is not blocked.
(B and C) (B) RBD and (C) CRD establish interfacial contacts with KD.
The interfaces between RBD-CRD and KD in the autoinhibited B-Raf in the absence of the 14-3-3 dimer
To further explore the structural ensemble of the autoinhibited B-Raf in the absence of the 14-3-3 dimer, we modeled and simulated 20 systems in which the RBD-CRD and KD were randomly placed in the simulation box with no initial contact (Figure S3). The protocol has been used to sample the autoinhibition of JAK2 tyrosine kinase (Shan et al., 2014). This protocol is expected to avoid artificial effects and achieve unbiased samplings (for details see STAR methods). In most trajectories, the RBD-CRD successfully established the interactions with the KD (Figure S4).
The superimposed final structures delineate the highly populated interface of KD with RBD-CRD (Figure 5A). We calculated the residue-based contact probabilities of RBD-CRD and KD in the sampled systems (Figures 5B and 5C). CRD showed slightly higher contact probabilities than RBD, suggesting its important contribution to the B-Raf autoinhibition scenario in the absence of the 14-3-3 dimer. An alanine scanning mutagenesis study identified activation mutations in the CRD of C-Raf that impact the interaction of RBD-CRD with KD (Daub et al., 1998). We mapped these activation mutations to B-Raf (N236, R239, T241, F247, D249, R252, K253, Q262, T263, K267, R271) and highlighted them by red points in Figure 5B. In general, during the simulations these residues exhibited high probabilities of contacting the KD, consistent with the experimental data (Daub et al., 1998). The interactions of the N lobe of KD with the RBD-CRD were more extensive than those of the C lobe. Given that the dimer interfaces are mainly from the N lobe, this points to the role of the RBD-CRD interaction in blocking the dimer interface and interfering with activation.
Figure 5. Structural ensembles of the RBD-CRD interactions with KD for B-Raf autoinhibition in the absence of the14-3-3 dimer.
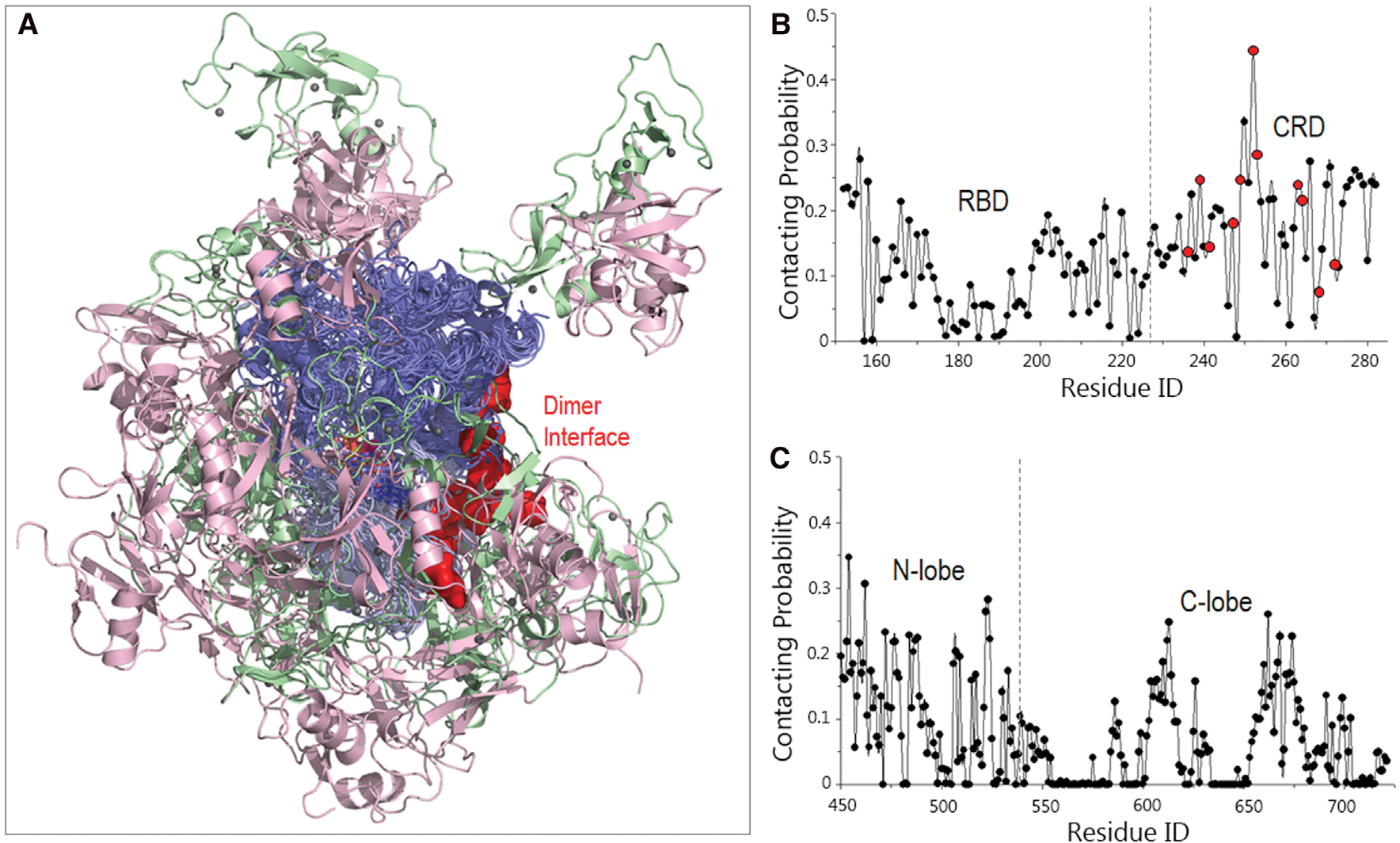
(A) The superimposed structures indicate that the RBD-CRD exhibits a potential to interact with the KD.
(B and C) The residue-based contacting probabilities of (B) RBD-CRD and (C) KD suggest a preference of CRD and KD’s N lobe for the RBD-CRD interactions with KD. The implicated activating residues in CRD for the RBD-CRD interactions with KD are highlighted by red points.
To obtain the autoinhibited conformation in the absence of 14-3-3, we focused on two models (models 2 and 15) among the sampled systems, since the dimer surfaces of the KD in these models are largely blocked by the RBD-CRD interactions (Figure 6), suggesting their roles in B-Raf autoinhibition. In model 2, the RBD interacts with the dimerization interface, whereas in model 15 the CRD contributes more (Figures 6A and 6B). Visualization of the simulations and the RMSF curves for the RBD-CRD indicate that the interactions with the KD dimer interface in model 2 are relatively less stable than in model 15 (Figure 6C). The RBD interactions with the dimer surface were unstable in model 2, with high fluctuations. In contrast, model 15 showed greater stability. The interactions of the CRD with the dimer interface were maintained throughout. The interaction energies of RBD-CRD with KD also indicate a preference of model 15 over model 2 (Figure 6D). In model 15, the RBD forms salt bridges and hydrophobic interactions with the KD, and more of these interactions are observed in the interface between CRD and KD (Figures 6E and 6F). Of particular interest are two salt bridges, R271-E533 and K253-E586, between CRD and KD, since the importance of R271 and K253 of CRD in KD interaction was implied in the alanine screening study (Daub et al., 1998). E586 is at the dimer interface of the KD in B-Raf. Model 15 suggests that RBD-CRD may block the dimerization interface of KD and interfere with B-Raf activation.
Figure 6. Representative structures of the RBD-CRD interactions with KD in the structural ensemble of B-Raf autoinhibition in the absence of 14-3-3 dimer.
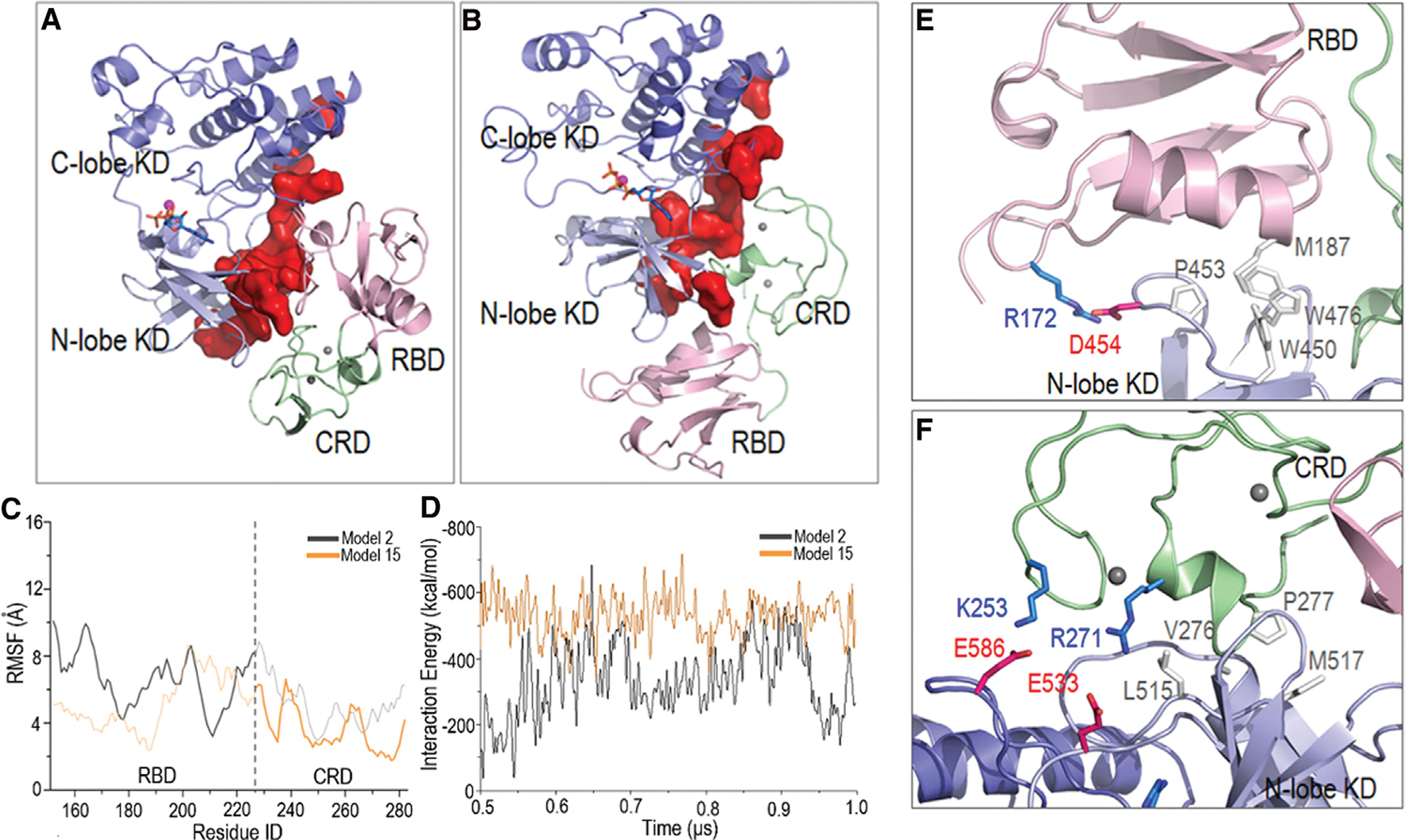
In (A) model 2 and (B) model 15, the KD dimer interfaces are largely blocked. The (C) root-mean-square fluctuations (RMSF) and (D) interaction energies indicate that model 15 is more stable. In RMSF curves, the RBD domain in model 2 and the CRD in model 15 that interact with the KD dimer interface are highlighted. In model 15, (E) the RBD and (F) CRD form salt bridges and hydrophobic interactions with the KD.
DISCUSSION
Raf is essential for MAPK signaling (Haling et al., 2014; Lavoie and Therrien, 2015) and is constitutively activated in many types of cancer (Karoulia et al., 2017). Raf is autoinhibited in the basal state. Upon release its KD dimerizes, fully activating it. 14-3-3 is a co-factor for Raf activation (Clark et al., 1997; Molzan et al., 2010). It also assists in Raf autoinhibition by stabilizing the otherwise low population of the autoinhibited conformation, as can be strikingly seen in the recent cryo-EM structures of autoinhibited B-Raf in complex with the 14-3-3 dimer (Kondo et al., 2019; Park et al., 2019). The dual action of the 14-3-3 dimer involves maintaining B-Raf’s autoinhibited state via interactions with pSer365 and promoting B-Raf dimerization via interactions with pSer729. Notably, pSer365 is required only for autoinhibition. In contrast, pSer729 has dual roles in both autoinhibition and KD dimerization. In the simulations, the autoinhibited B-Raf in complex with the 14-3-3 dimer was compact and stable. CRD contributed significantly; however, RBD was largely exposed and unstable. The removal (dephosphorylation) of pSer365 failed to induce the large conformational change. However, the release (or removal in the simulations) of RBD-CRD triggered the structural reorientation of KD for dimerization (Figure 7).
Figure 7. Schematic illustration of B-Raf activation from two B-Raf autoinhibition scenarios.
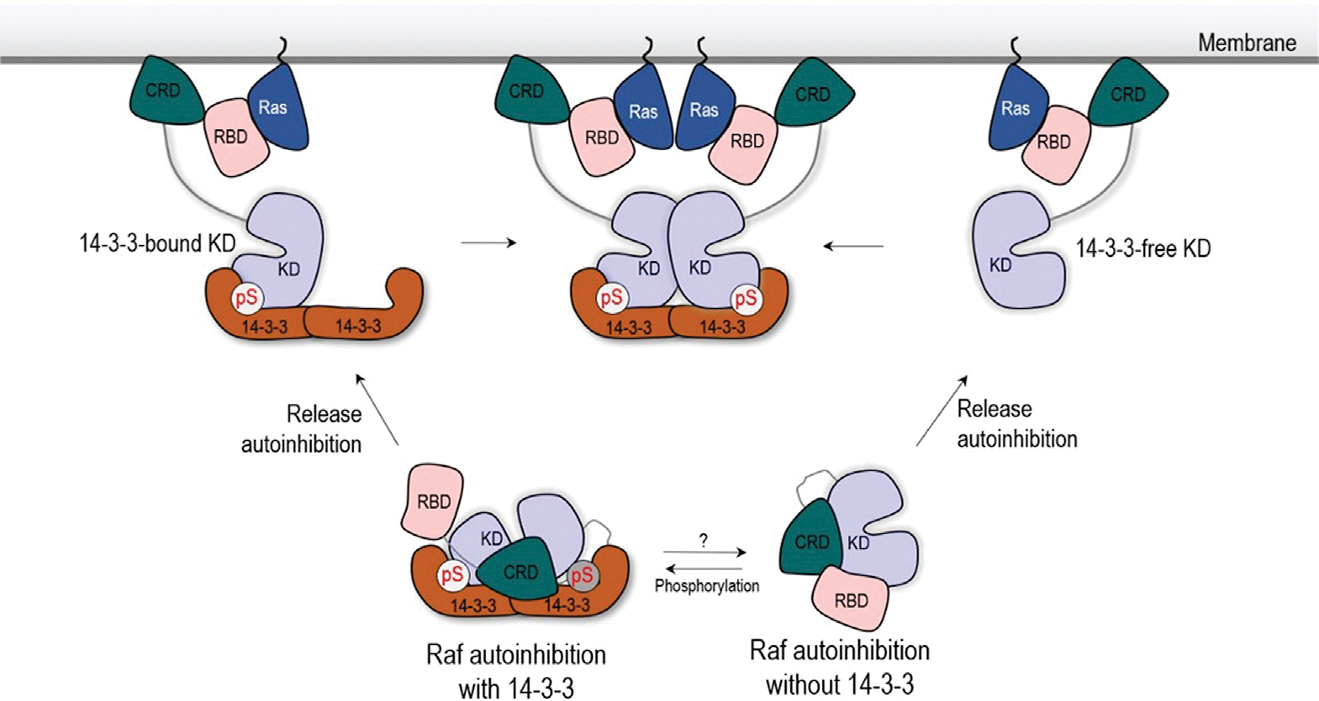
Autoinhibited B-Raf in complex with the 14-3-3 dimer requires the two phosphorylated sites at the CR2 loop and C-terminal tail (Park et al., 2019). The phosphorylation of the CR2 loop (pSer259 in Raf-1 and pSer365 in B-Raf) is a dynamic cellular process, which can be achieved by protein kinase A or Akt and reversed by PP2A (Abraham et al., 2000; Romano et al., 2014; Zimmermann and Moelling, 1999). It has also been proposed that the phosphorylation of B-Raf Ser729 is a controlled cellular event (Lavoie and Therrien, 2015). AMP-activated protein kinase, an important energy sensor, has been reported to phosphorylate Ser729 in B-Raf and control its interactions with the 14-3-3 dimer (Shen et al., 2013). In the absence of the two phosphorylated sites and 14-3-3 interactions, B-Raf’s autoinhibition can also control basal activity, albeit less effectively. The roles of the N-terminal RBD-CRD domain in inhibiting Raf activity have been extensively reported (Cutler et al., 1998; Stanton and Cooper, 1987). We sampled the structural ensemble of B-Raf autoinhibition in the absence of the 14-3-3 dimer. In these sampled B-Raf structures of the RBD-CRD binding to KD, RBD-CRD can block the dimer interface of KD and, thus, B-Raf activation. In both B-Raf autoinhibition scenarios, we observe that CRD contributes considerably to stabilizing the autoinhibited state, whereas RBD’s contribution is lower. This suggests that RBD can be more exposed and flexible in the complex, susceptible to interaction with Ras at the membrane. The high-affinity RBD-Ras interaction favors shifting the equilibrium toward the open B-Raf state (Nussinov et al., 2019a), thus assisting the release of the CRD from the complex, which then attaches the membrane.
The release of the autoinhibition of B-Raf in complex with the 14-3-3 dimer likely results in a monomeric B-Raf KD fluctuating and rotating but still loaded onto the 14-3-3 dimer (14-3-3-bound KD), as we observe in the simulations. It needs another, 14-3-3-free KD, for B-Raf dimerization. The 14-3-3-free KD can be obtained from the disassociation of the loaded B-Raf KD from 14-3-3 dimer. However, given the high affinity between pSer729 and the 14-3-3 dimer, the disassociation may not be highly populated. The disassociation can be promoted by the dephosphorylation of Ser729 at the C-terminal tail. However, no phosphate has been reported to dephosphorylate pSer729 in B-Raf. Alternatively, the 14-3-3-free KD can come from the B-Raf autoinhibition scenario in the absence of 14-3-3. The release of the autoinhibition in this scenario can lead to the 14-3-3-free KD dimerization with the 14-3-3-bound KD. The 14-3-3-free KD is activated through the dimerization with 14-3-3-bound KD. The activated 14-3-3-free KD can autophosphorylate its C-terminal tails (pSer729 in B-Raf) to further stabilize the complex (Hu et al., 2013). The two B-Raf autoinhibition scenarios described here can provide a structural reservoir for several activation combinations. Their selection can depend on the binding affinity and availability of the phosphorylation site in B-Raf and the 14-3-3 dimer.
Raf isoforms are highly similar in sequence and structure, with conserved RBD-CRD in CR2 and KD in CR3, which structurally and functionally support heterodimer formation (Rushworth et al., 2006; Weber et al., 2001). They also share the two 14-3-3 recognition motifs. We expect that other Raf isoforms share the autoinhibition and activation scenarios proposed in this work.
Insight into the activation mechanisms of B-Raf is vastly important. Exactly how Raf releases the autoinhibition and becomes activated is not understood. This mechanistic work provides structural details of B-Raf autoinhibition scenarios and their release for B-Raf drug discovery. We note, however, that at this point we are not clear how a drug can stabilize the autoinhibited state. Possibly covalent allosteric drugs, such as those recently proposed for Ras effector Ral guanosine triphosphatase (GTPase) (Bum-Erdene et al., 2020), which follow the landmark covalent inhibitors of Ras (Ostrem et al., 2013), could provide a venue. However, the different mechanisms of activation of Raf kinase as compared with GTPases question such drugging protocols, putting a dent in such schemes.
In summary, in the presence of the 14-3-3 dimer, autoinhibited B-Raf with two phosphorylated sites, pSer365 and pSer729, was stable in the simulations. Consistent with the cryo-EM structures, whereas the RBD and loops were exposed and unstable, the CRD showed high stability and significant contribution to B-Raf autoinhibition. The exposed RBD can be readily targeted by Ras, and the high-affinity Ras-RBD interactions facilitate CRD release from the autoinhibited complexes and attachment to the membrane. Removal of pSer365 exposed the CRD without inducing a large disruption. In contrast, the release of RBD-CRD induced a huge conformational change of KD relative to the 14-3-3 dimer, predisposing it to dimerization-favored states. We further sampled the conformational ensemble of autoinhibited B-Raf in the absence of the 14-3-3 dimer. The representative structure in the ensemble indicated that the RBD-CRD interactions can block the KD dimer interface and prevent KD dimerization and, thus, full activation of B-Raf. The residues that we identified as key at the interface between CRD and KD’s N lobe are also in line with experimental data (Daub et al., 1998). We propose that the two B-Raf autoinhibition scenarios can co-exist and collaborate. Our model suggests that the 14-3-3-interacting KD is the 14-3-3-bound KD while the monomeric KD without 14-3-3 is the 14-3-3-free KD in their 14-3-3-supported dimerization. We further propose that Raf isoforms can share common autoinhibition scenarios and can heterodimerize through such a route. Collectively, here we proposed the autoinhibition and dimerization mechanistic scenarios for B-Raf activation.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Ruth Nussinov (NussinoR@mail.nih.gov).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Requests for data should be directed to and will be fulfilled by the Lead Contact, Ruth Nussinov (NussinoR@mail.nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
No experimental models were used in this work.
METHOD DETAILS
Modeling of B-Raf autoinhibition in the presence of 14-3-3 dimer
The initial coordinates for the autoinhibited B-Raf in complex with the 14-3-3 dimer were obtained from the Protein DataBank (PDB ID: 6NYB). The MEK in the cryo-EM structure was removed. KD was modified for the inactive conformation (PDB ID: 4WO5). The missing RBD and linkers in B-Raf were modeled into the complex. In the initial models, RBD was modeled in between the KD and 14-3-3 dimer with no initial contact. In the cryo-EM structure of B-Raf in complex with the 14-3-3 dimer, the linkers, residues 283–361 between CRD and pSer365 motif and residues 372–449 between pSer365 motif and KD, lacked full density. They were modeled into the cryo-EM structure. The initial conformations for the linkers were selected as the random coils with the sequence converted. The rounds of minimization and short MD simulations with constrains on the non-linker regions (that present the density in the cryo-EM structures) were performed to optimize and relax the linkers in the structures (Figure S1). When the loops became relatively stable without unreasonable atom contact, the systems were subjected to the production runs. To explore the activation of the autoinhibited B-Raf in complex with the 14-3-3 dimer, the pSer365 motif with the adjacent loop (residues 283–449) was removed (B-RafΔpS). The RBD-CRD (residues 152–282) was further removed from the complex to understand the release of B-Raf autoinhibition in presence of 14-3-3 dimer (B-RafΔpS,RBD–CRD). For each system, 1 μs simulations were conducted. The residues at the dimer interface were identified based on PDB ID: 4MNF.
Modeling of B-Raf autoinhibition in the absence of the 14-3-3 dimer
In the cryo-EM structure of the autoinhibited B-Raf in complex with 14-3-3 dimer, CRD interacts with the KD, suggesting a possible interface between CRD and KD for the B-Raf autoinhibition in the absence of the 14-3-3 dimer. We modeled the pre-assembled system of the CRD in contact with KD, adopting the same interface in the cryo-EM structure (PDB ID: 6NYB). RBD has no initial contact with the KD in the system. To enhance the sampling, twenty additional systems of RBD-CRD interacting with KD were modeled and simulated. The RBD-CRD and KD were placed at random positions and orientations in a water box of ~120 × 120 × 120 Å3. The minimal distance between RBD-CRD and KD in the modeled systems was 6.5 Å to avoid modeling bias and unreasonable atom contacts. The initial and final structures of the autoinhibited B-Raf in absence of 14-3-3 dimer were summarized in Figures S3 and S4. 1 ms simulations were individually conducted for 21 systems.
MD simulation protocol
The all-atom MD simulations were conducted with the NAMD package(Phillips et al., 2005) and the CHARMM all-atom additive force field (version C36) (Brooks et al., 2009). The explicit TIP3 water model was used to solvate the systems. Water molecules within 2.4 Å of proteins were removed to eliminate the atom collapse. Na+ and Cl− ions were used to neutralize the system and generate a ~0.15 mol/L ionic strength. Covalent bonds involving hydrogen atoms were constrained. Constraint was applied to coordinate the zinc ions in the systems by employing the collective variable-based calculations (Colvars) in the NAMD package(Phillips et al., 2005). Additional simulations were performed for some systems for reproducibility. Short-range van der Waals (vdW) and long-rang electrostatic interactions were individually calculated by switch function and the Particle mesh Ewald (PME) algorithm. 2 fs time-step was used in all MD simulations. The VMD(Humphrey et al., 1996) and NAMD(Phillips et al., 2005) programs were used for model the systems.
QUANTIFICATION AND STATISTICAL ANALYSIS
The trajectories were visualized and analyzed by Pymol(DeLano, 2002) and VMD(Humphrey et al., 1996) software. The RMSF values in Figure 2A were calculated based on last 500-ns trajectories, where the structures were superimposed by the 14-3-3 protein interacting with the pSer729 motif. The RMSF values in Figure 6C were calculated based on last 500-ns trajectories, where the RBD-CRD/KD structures were superimposed by the kinase domain. The interaction energies in Figures 6D and S2 were calculated based on the last 500-ns trajectories. The residue distances in Figures 2B, 2C and 3B–3D were defined and calculated by the distances between two closest non-hydrogen atoms in two residues. The residue-based contacting probabilities in Figure 5 were calculated based on the last 500-ns trajectories, where the residues within 3.5 Å of the binding partners were considered as the contacting residues. The solvent accessible surface area (SASA) values in Figure S2C were calculated with the srad of 1.4 Å.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Deposited data | ||
|
| ||
| B-Raf with 14-3-3 | (Park et al., 2019) | PDB: 6NYB |
| B-Raf with 14-3-3 | (Kondo et al., 2019) | PDB: 6UAN |
| B-Raf KD | (Thevakumaran et al., 2015) | PDB: 4WO5 |
| B-Raf KD | (Haling et al., 2014) | PDB: 4MNF |
| B-Raf with 14-3-3 and MEK | (Park et al., 2019) | PDB: 6Q0J |
|
| ||
| Software and algorithms | ||
|
| ||
| NAMD2.12 | (Phillips et al., 2005) | http://www.ks.uiuc.edu/Research/namd/ |
| VMD | (Humphrey et al., 1996) | http://www.ks.uiuc.edu/Research/vmd/ |
| Pymol | (DeLano, 2002) | http://www.pymol.org |
ACKNOWLEDGMENTS
All simulations were performed using the high-performance computational facilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (https://hpc.nih.gov/). This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and the Intramural Research Program of the NIH Clinical Center.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.str.2021.02.005.
REFERENCES
- Abraham D, Podar K, Pacher M, Kubicek M, Welzel N, Hemmings BA, Dilworth SM, Mischak H, Kolch W, and Baccarini M (2000). Raf-1-associated protein phosphatase 2A as a positive regulator of kinase activation. J. Biol. Chem. 275, 22300–22304. [DOI] [PubMed] [Google Scholar]
- Agianian B, and Gavathiotis E (2018). Current insights of BRAF inhibitors in cancer. J. Med. Chem. 61, 5775–5793. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Brooks CL 3rd, Mackerell AD Jr., Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. (2009). CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bum-Erdene K, Liu D, Gonzalez-Gutierrez G, Ghozayel MK, Xu D, and Meroueh SO (2020). Small-molecule covalent bond formation at tyrosine creates a binding site and inhibits activation of Ral GTPases. Proc. Natl. Acad. Sci. U S A 117, 7131–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong H, and Guan KL (2003). Regulation of Raf through phosphorylation and N terminus-C terminus interaction. J. Biol. Chem. 278, 36269–36276. [DOI] [PubMed] [Google Scholar]
- Clark GJ, Drugan JK, Rossman KL, Carpenter JW, Rogers-Graham K, Fu H, Der CJ, and Campbell SL (1997). 14-3-3 zeta negatively regulates raf-1 activity by interactions with the Raf-1 cysteine-rich domain. J. Biol. Chem. 272, 20990–20993. [DOI] [PubMed] [Google Scholar]
- Cotto-Rios XM, Agianian B, Gitego N, Zacharioudakis E, Giricz O, Wu Y, Zou Y, Verma A, Poulikakos PI, and Gavathiotis E (2020). Inhibitors of BRAF dimers using an allosteric site. Nat. Commun. 11, 4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler RE Jr., Stephens RM, Saracino MR, and Morrison DK (1998). Autoregulation of the Raf-1 serine/threonine kinase. Proc. Natl. Acad. Sci. U S A 95, 9214–9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub M, Jockel J, Quack T, Weber CK, Schmitz F, Rapp UR, Wittinghofer A, and Block C (1998). The RafC1 cysteine-rich domain contains multiple distinct regulatory epitopes which control Ras-dependent Raf activation. Mol. Cell. Biol. 18, 6698–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. (2002). Mutations of the BRAF gene in human cancer. Nature 417, 949–954. [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002). The PyMol Molecular Graphics System (DeLano Scientific; ). [Google Scholar]
- Dhillon AS, Pollock C, Steen H, Shaw PE, Mischak H, and Kolch W (2002). Cyclic AMP-dependent kinase regulates Raf-1 kinase mainly by phosphorylation of serine 259. Mol. Cell. Biol. 22, 3237–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant DE, and Morrison DK (2018). Targeting the Raf kinases in human cancer: the Raf dimer dilemma. Br. J. Cancer 118, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetics SK, Guterres H, Kearney BM, Buhrman G, Ma B, Nussinov R, and Mattos C (2015). Allosteric effects of the oncogenic RasQ61L mutant on Raf-RBD. Structure 23, 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, et al. (2010). Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 363, 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Xia K, Pallas DC, Cui C, Conroy K, Narsimhan RP, Mamon H, Collier RJ, and Roberts TM (1994). Interaction of the protein kinase Raf-1 with 14-3-3 proteins. Science 266, 126–129. [DOI] [PubMed] [Google Scholar]
- Garcia-Gomez R, Bustelo XR, and Crespo P (2018). Protein-protein interactions: emerging oncotargets in the RAS-ERK pathway. Trends Cancer 4, 616–633. [DOI] [PubMed] [Google Scholar]
- Gardino AK, Smerdon SJ, and Yaffe MB (2006). Structural determinants of 14-3-3 binding specificities and regulation of subcellular localization of 14-3-3-ligand complexes: a comparison of the X-ray crystal structures of all hu-man 14-3-3 isoforms. Semin. Cancer Biol. 16, 173–182. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Xie WQ, Quest AF, Mabrouk GM, Strum JC, and Bell RM (1994). The cysteine-rich region of raf-1 kinase contains zinc, translocates to liposomes, and is adjacent to a segment that binds GTP-ras. J. Biol. Chem. 269, 10000–10007. [PubMed] [Google Scholar]
- Haling JR, Sudhamsu J, Yen I, Sideris S, Sandoval W, Phung W, Bravo BJ, Giannetti AM, Peck A, Masselot A, et al. (2014). Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell 26, 402–413. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, Hewitt JF, Zak M, Peck A, Orr C, et al. (2013). Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature 501, 232–236. [DOI] [PubMed] [Google Scholar]
- Holderfield M, Deuker MM, McCormick F, and McMahon M (2014). Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 14, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJS, Kornev AP, Taylor SS, and Shaw AS (2013). Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 154, 1036–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD: visual molecular dynamics. J. Mol. Graph. 14 (33–38), 27–38. [DOI] [PubMed] [Google Scholar]
- Improta-Brears T, Ghosh S, and Bell RM (1999). Mutational analysis of Raf-1 cysteine rich domain: requirement for a cluster of basic aminoacids for interaction with phosphatidylserine. Mol. Cell. Biochem. 198, 171–178. [DOI] [PubMed] [Google Scholar]
- Karoulia Z, Gavathiotis E, and Poulikakos PI (2017). New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 17, 676–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler M, and Brummer T (2016). B-Raf activation loop phosphorylation revisited. Cell Cycle 15, 1171–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Ognjenovic J, Banerjee S, Karandur D, Merk A, Kulhanek K, Wong K, Roose JP, Subramaniam S, and Kuriyan J (2019). Cryo-EM structure of a dimeric B-Raf:14-3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science 366, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie H, and Therrien M (2015). Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell. Biol. 16, 281–298. [DOI] [PubMed] [Google Scholar]
- Lavoie H, Thevakumaran N, Gavory G, Li JJ, Padeganeh A, Guiral S, Duchaine J, Mao DY, Bouvier M, Sicheri F, et al. (2013). Inhibitors that stabilize a closed RAF kinase domain conformation induce dimerization. Nat. Chem. Biol. 9, 428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jang H, Zhang J, and Nussinov R (2018). Raf-1 cysteine-rich domain increases the affinity of K-Ras/Raf at the membrane, promoting MAPK signaling. Structure 26, 513–525 e512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liau NPD, Wendorff TJ, Quinn JG, Steffek M, Phung W, Liu P, Tang J, Irudayanathan FJ, Izadi S, Shaw AS, et al. (2020). Negative regulation of RAF kinase activity by ATP is overcome by 14-3-3-induced dimerization. Nat. Struct. Mol. Biol. 27, 134. [DOI] [PubMed] [Google Scholar]
- Lu S, Jang H, Gu S, Zhang J, and Nussinov R (2016a). Drugging Ras GTPase: a comprehensive mechanistic and signaling structural view. Chem. Soc. Rev. 45, 4929–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Jang H, Muratcioglu S, Gursoy A, Keskin O, Nussinov R, and Zhang J (2016b). Ras conformational ensembles, allostery, and signaling. Chem. Rev. 116, 6607–6665. [DOI] [PubMed] [Google Scholar]
- Michaud NR, Fabian JR, Mathes KD, and Morrison DK (1995). 14-3-3 is not essential for Raf-1 function: identification of Raf-1 proteins that are biologically activated in a 14-3-3- and Ras-independent manner. Mol. Cell. Biol. 15, 3390–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molzan M, Schumacher B, Ottmann C, Baljuls A, Polzien L, Weyand M, Thiel P, Rose R, Rose M, Kuhenne P, et al. (2010). Impaired binding of 14-3-3 to C-RAF in Noonan syndrome suggests new approaches in diseases with increased Ras signaling. Mol. Cell. Biol. 30, 4698–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison DK, Heidecker G, Rapp UR, and Copeland TD (1993). Identification of the major phosphorylation sites of the Raf-1 kinase. J. Biol. Chem. 268, 17309–17316. [PubMed] [Google Scholar]
- Mott HR, Carpenter JW, Zhong S, Ghosh S, Bell RM, and Campbell SL (1996). The solution structure of the Raf-1 cysteine-rich domain: a novel ras and phospholipid binding site. Proc. Natl. Acad. Sci. U S A 93, 8312–8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Jang H, and Tsai CJ (2015). Oligomerization and nanocluster organization render specificity. Biol. Rev. Camb. Philos. Soc. 90, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Jang H, Tsai CJ, Liao TJ, Li S, Fushman D, and Zhang J (2017). Intrinsic protein disorder in oncogenic KRAS signaling. Cell. Mol. Life Sci. 74, 3245–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, and Jang H (2019a). Does ras activate raf and PI3K allosterically? Front. Oncol. 9, 1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, and Jang H (2019b). Is nanoclustering essential for all oncogenic KRas pathways? Can it explain why wild-type KRas can inhibit its oncogenic variant? Semin. Cancer Biol. 54, 114–120. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, and Jang H (2020). Autoinhibition can identify rare driver mutations and advise pharmacology. FASEB J. 34, 16–29. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Zhang M, Tsai CJ, Liao TJ, Fushman D, and Jang H (2018). Autoinhibition in Ras effectors Raf, PI3Kalpha, and RASSF5: a comprehensive review underscoring the challenges in pharmacological intervention. Biophys. Rev. 10, 1263–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, and Shokat KM (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantsar T (2020). The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J. 18, 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Rawson S, Li K, Kim BW, Ficarro SB, Pino GG, Sharif H, Marto JA, Jeon H, and Eck MJ (2019). Architecture of autoinhibited and active BRAF-MEK1–14-3–3 complexes. Nature 575, 545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005). Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakulendran T, Sahmi M, Lefrancois M, Sicheri F, and Therrien M (2009). A dimerization-dependent mechanism drives RAF catalytic activation. Nature 461, 542–545. [DOI] [PubMed] [Google Scholar]
- Romano D, Nguyen LK, Matallanas D, Halasz M, Doherty C, Kholodenko BN, and Kolch W (2014). Protein interaction switches coordinate Raf-1 and MST2/Hippo signalling. Nat. Cell Biol. 16, 673–684. [DOI] [PubMed] [Google Scholar]
- Roskoski R Jr. (2018). Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacol. Res. 135, 239–258. [DOI] [PubMed] [Google Scholar]
- Rushworth LK, Hindley AD, O’Neill E, and Kolch W (2006). Regulation and role of Raf-1/B-Raf heterodimerization. Mol. Cell. Biol. 26, 2262–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan YB, Gnanasambandan K, Ungureanu D, Kim ET, Hammaren H, Yamashita K, Silvennoinen O, Shaw DE, and Hubbard SR (2014). Molecular basis for pseudokinase-dependent autoinhibition of JAK2 tyrosine kinase. Nat. Struct. Mol. Biol. 21, 579–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AS, Kornev AP, Hu J, Ahuja LG, and Taylor SS (2014). Kinases and pseudokinases: lessons from RAF. Mol. Cell. Biol. 34, 1538–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen CH, Yuan P, Perez-Lorenzo R, Zhang Y, Lee SX, Ou Y, Asara JM, Cantley LC, and Zheng B (2013). Phosphorylation of BRAF by AMPK impairs BRAF-KSR1 association and cell proliferation. Mol. Cell 52, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton VP Jr., and Cooper GM (1987). Activation of human raf transforming genes by deletion of normal amino-terminal coding sequences. Mol. Cell. Biol. 7, 1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrell EM, and Morrison DK (2019). Ras-mediated activation of the raf family kinases. Cold Spring Harb. Perspect. Med. 9, a033746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevakumaran N, Lavoie H, Critton DA, Tebben A, Marinier A, Sicheri F, and Therrien M (2015). Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat. Struct. Mol. Biol. 22, 37–43. [DOI] [PubMed] [Google Scholar]
- Thorson JA, Yu LW, Hsu AL, Shih NY, Graves PR, Tanner JW, Allen PM, Piwnica-Worms H, and Shaw AS (1998). 14-3-3 proteins are required for maintenance of Raf-1 phosphorylation and kinase activity. Mol. Cell. Biol. 18, 5229–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran NH, Wu X, and Frost JA (2005). B-Raf and Raf-1 are regulated by distinct autoregulatory mechanisms. J. Biol. Chem. 280, 16244–16253. [DOI] [PubMed] [Google Scholar]
- Travers T, Lopez CA, Van QN, Neale C, Tonelli M, Stephen AG, and Gnanakaran S (2018). Molecular recognition of RAS/RAF complex at the membrane: role of RAF cysteine-rich domain. Sci. Rep. 8, 8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CJ, and Nussinov R (2018). Allosteric activation of RAF in the MAPK signaling pathway. Curr. Opin. Struct. Biol. 53, 100–106. [DOI] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, et al. (2004). Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116, 855–867. [DOI] [PubMed] [Google Scholar]
- Weber CK, Slupsky JR, Kalmes HA, and Rapp UR (2001). Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res. 61, 3595–3598. [PubMed] [Google Scholar]
- Yaeger R, and Corcoran RB (2019). Targeting alterations in the RAF-MEK pathway. Cancer Discov. 9, 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang BH, and Guan KL (2000). Activation of B-Raf kinase requires phosphorylation of the conserved residues Thr598 and Ser601. EMBO J. 19, 5429–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann S, and Moelling K (1999). Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 286, 1741–1744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Requests for data should be directed to and will be fulfilled by the Lead Contact, Ruth Nussinov (NussinoR@mail.nih.gov).