

Abstract
FoxP3 is an essential transcription factor (TF) for immunologic homeostasis, but how it utilizes the common forkhead DNA-binding domain (DBD) to perform its unique function remains poorly understood. We here demonstrated that, unlike other known forkhead TFs, FoxP3 formed a head-to-head dimer using a unique linker (Runx1-binding region, RBR) preceding the forkhead domain. Head-to-head dimerization conferred distinct DNA-binding specificity and created a docking site for the cofactor Runx1. RBR was also important for proper folding of the forkhead domain, as truncation of RBR induced domain-swap dimerization of forkhead, which was previously considered the physiological form of FoxP3. Rather, swap-dimerization impaired FoxP3 function, as demonstrated with the disease-causing mutation R337Q, while a swap-suppressive mutation largely rescued R337Q-mediated functional impairment. Altogether, our findings suggest that FoxP3 can fold into two distinct dimerization states: head-to-head dimerization representing functional specialization of an ancient DBD and swap-dimerization associated with impaired functions.
Introduction
The human genome is estimated to encode approximately 1,600 transcription factors (TFs). The vast majority of these TFs, however, utilize one of just 10 types of DNA binding domains (DBDs) (Lambert et al., 2018), many of which display remarkably conserved and narrow sequence specificity (Nitta et al., 2015). The apparent simplicity in DBD composition is in sharp contrast to the complex network of genes they control. How DBDs can divergently evolve to carry out distinct functions remains incompletely understood (Badis et al., 2009; Jolma et al., 2015; Reiter et al., 2017).
One of the largest families of DBDs in eukaryotes is the forkhead DBD, which commonly displays a winged-helix fold and recognizes the consensus sequence known as forkhead motif (FKHM) – TGTTTAC (Dai et al., 2021). There are about 50 forkhead TFs in humans, which play important roles in key biological processes– including development, reproduction, aging and immunity (Benayoun et al., 2011; Hannenhalli and Kaestner, 2009). Some forkhead TFs function as pioneering TFs that can directly recognize DNA sequences within the condensed chromatin and open its structure with the help of chromatin modifiers (Drouin, 2014), while others utilize the pre-existing chromatin landscape without dramatically altering the chromatin structure (Samstein et al., 2012). This suggests that there is great functional diversity even within the forkhead TF family not only in the biological processes they control, but also in the molecular mechanisms they employ.
FoxP3 is a forkhead TF that plays a critical role in the development of regulatory T cells (Tregs), a branch of CD4+ T cells that suppresses immune functions to prevent autoimmunity and excessive inflammation (Bennett et al., 2001; Brunkow et al., 2001; Fontenot et al., 2003; Hori et al., 2003). Certain mutations in FoxP3 lead to the multiorgan autoimmune disease immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome in human and similar autoimmune conditions in mice (Bennett et al., 2001; Brunkow et al., 2001; Chatila et al., 2000; Gambineri et al., 2008; Rubio-Cabezas et al., 2009; Tanaka et al., 2005; Wildin et al., 2001). FoxP3 is key to determining and maintaining Treg cell identity by transcriptionally up- or down-regulating hundreds of genes (Kwon et al., 2017; van der Veeken et al., 2020). Despite its importance in immune homeostasis, molecular functions of FoxP3 remain poorly understood. For example, it is highly debated whether FoxP3 is a transcriptional activator, suppressor, or both depending on the target genes (Arvey et al., 2014; Kwon et al., 2017; Li et al., 2007; Zemmour et al., 2021; Zheng et al., 2007). Moreover, which genes affected by FoxP3 are direct targets versus those regulated indirectly is not understood (Ramirez et al., 2022; van der Veeken et al., 2020; Zemmour et al., 2021). It is also unclear how FoxP3 alters the target gene expression as FoxP3 binds predominantly to genomic loci that have pre-established chromatin accessibility and induces little change in the accessibility of the bound sites (Samstein et al., 2012; van der Veeken et al., 2020; Yoshida et al., 2019).
Equally puzzling are the biochemical and structural properties of FoxP3. FoxP3 contains an N-terminal proline-rich region that recruits a variety of cofactors, zinc finger (ZF) that likely binds DNA, and coiled coil (CC) that forms an antiparallel dimer (Song et al., 2012) (Figure 1A). Following the CC is a long linker that recruits the cofactor Runx1 (Ono et al., 2007) (referred to here as Runx1-binding region; RBR) and the forkhead domain responsible for DNA binding. Among these domains, the forkhead domain is most frequently mutated in IPEX patients and has been most extensively studied (Barzaghi et al., 2012; Huang et al., 2020). Previous studies report that isolated FoxP3 forkhead folds into an unusual domain-swap dimer that drastically differs from the winged-helix monomeric structure typical of forkhead DBD (Bandukwala et al., 2011; Chen et al., 2015). FoxP1–also expressed in Treg cells (Ghosh et al., 2018; Konopacki et al., 2019)– and FoxP2 are closely related to FoxP3 and can also form domain-swap dimers (Chu et al., 2011; Stroud et al., 2006). These observations have led to the widely accepted notion that FoxP TFs may have evolved to adopt the swap-dimeric fold. However, a peptide that binds FoxP3 ZF-CC can disrupt FoxP3 dimerization, inconsistent with the domain-swap model of forkhead (Lozano et al., 2017). FoxP2 was also crystallized as a non-swap monomer as well as the domain-swap dimer (Stroud et al., 2006; Wu et al., 2006), further raising the question about the physiologically relevant structure of the FoxP forkhead domains. More globally, the overall architecture of FoxP3 beyond its DBD and how FoxP3 differs from other closely related TFs to carry out its unique function also remains unclear.
Figure 1. Overall architecture of FoxP3.
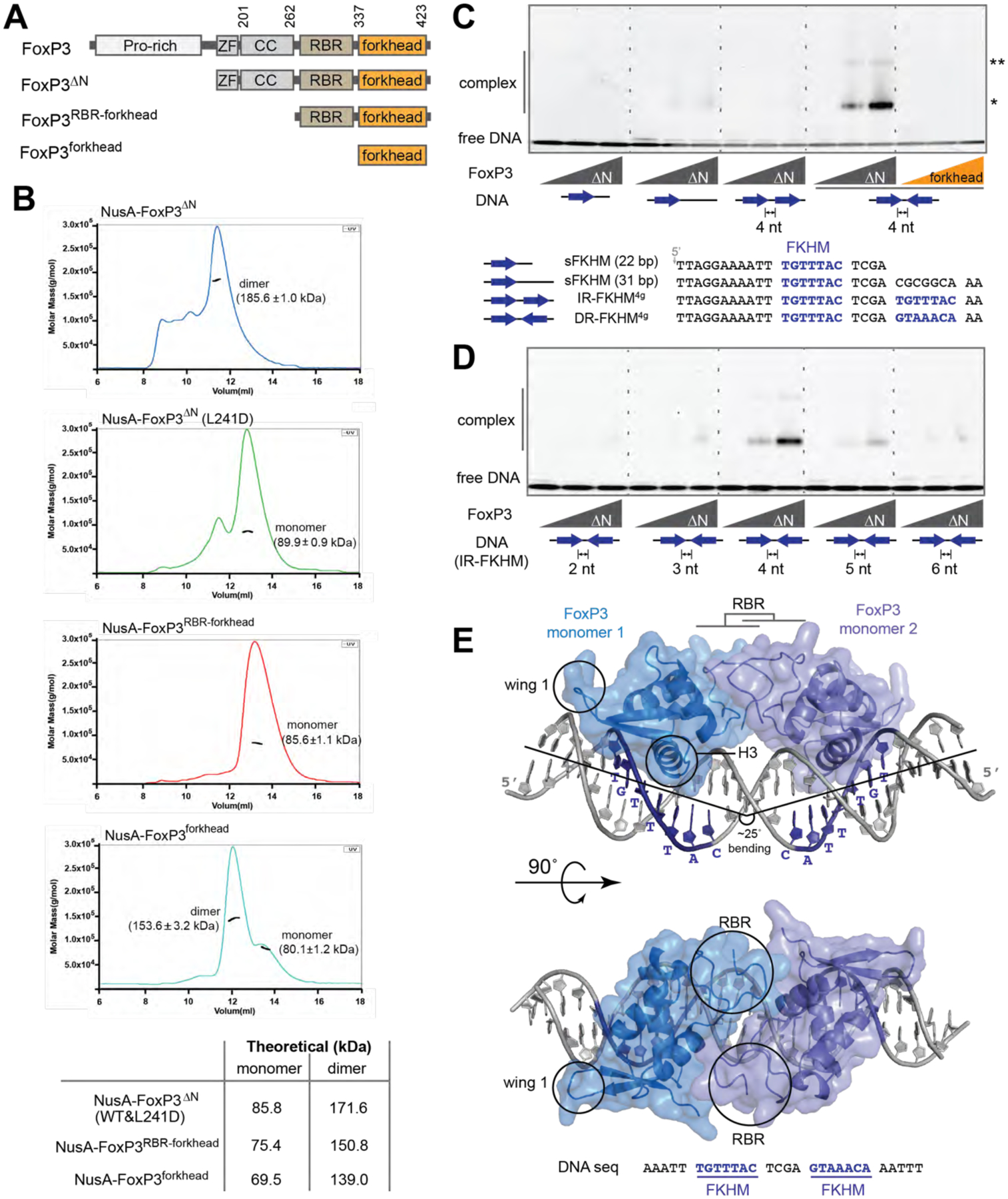
See also Figures S1 & S2.
A. Domain architecture of FoxP3ΔN, FoxP3RBR-forkhead and FoxP3forkhead. (ZF: zinc finger; CC: coiled coil; RBR: Runx1-binding region).
B. SEC-MALS of various truncation variants of FoxP3. Experimentally determined mass values are shown in parenthesis on the graphs. Theoretical values are shown in the table below. The NusA tag was fused for all constructs to increase the accuracy of mass estimation. L241D disrupts CC dimerization.
C. EMSA of FoxP3ΔN or FoxP3forkhead (0, 0.4 and 0.8 μM) using four DNA oligos (0.2 μM) with different FKHM arrangements. Sybr Gold stain was used to visualize DNA. While FoxP3ΔN binds DNA predominantly as a dimer (*), a small population of higher-order oligomer (**) was also seen.
D. EMSA of FoxP3ΔN (0, 0.4 and 0.8 μM) with DNA containing IR-FKHM (0.2 μM) with varying gap sizes.
E. Crystal structure of Foxp3ΔN in complex with IR-FKHM4g DNA. Two non-swap FoxP3 monomers form a head-to-head dimer through the RBR loop. ZF and CC were present in the crystal (See Figure S2E), but were not resolved. The helix 3 (H3) and wing 1 that are characteristics of the canonical forkhead structure are indicated with circles.
Data in (B-D) are representative of at least three independent experiments.
We here showed that FoxP3 forkhead, as well as those of other FoxP TFs, folded into the winged-helix, non-swap conformation, and that this required the presence of the RBR linker. Individually folded forkhead domains then formed a head-to-head dimer upon DNA binding. While other FoxP TFs also utilized their RBR-like linkers for monomeric folding, head-to-head dimerization was unique to FoxP3 and imparted distinct functions to FoxP3. Our functional data provide new insights into the pathogenic mechanism for IPEX mutations and a new framework of understanding for FoxP3 functions.
Results
FoxP3 preferentially binds two forkhead consensus motifs in inverted repeat
We first attempted purifying full-length mouse FoxP3 protein from E. coli, but found it heavily degraded in the N-terminal Pro-rich region. This was consistent with multiple structural predictions (e.g. AlphaFold2, Jpred4) suggesting that the N-terminal region is intrinsically disordered (Drozdetskiy et al., 2015; Jumper et al., 2021). We therefore purified an N-terminal truncation variant (FoxP3ΔN) harboring ZF, CC, RBR and forkhead domains (Figures 1A and S1A). We also purified FoxP3RBR-forkhead and FoxP3forkhead for comparison. Consistent with the previous report that FoxP3forkhead constitutively forms a swap dimer (Bandukwala et al., 2011), FoxP3forkhead was dimeric as measured by size exclusion chromatography-coupled multiangle light scattering (SEC-MALS) (Figure 1B). We used FoxP3 fused with the protein tag NusA (60 kDa) to improve accuracy of molecular weight estimation by SEC-MALS. Unlike FoxP3forkhead, FoxP3RBR-forkhead was monomeric (Figure 1B), which was incompatible with the model that the forkhead domain forms a swap-dimeric structure. FoxP3ΔN was a dimer but disruption of the CC dimerization by a mutation in the CC dimeric interface (L241D) (Song et al., 2012) converted it to a monomer (Figure 1B), suggesting that FoxP3 dimerization was exclusively mediated by CC, and that forkhead within the FoxP3ΔN construct was a monomer.
Given that the observed differences in the oligomeric state of FoxP3 forkhead depended on other domains, we postulated that there may be different conformations of forkhead. We next asked how these different conformations affected DNA binding. We first examined DNA containing only a single FKHM (sFKHM, 22 or 31 bp) by electrophoretic mobility shift assay (EMSA), but found that binding was nearly undetectable for all three proteins of FoxP3ΔN, FoxP3RBR-forkhead and FoxP3forkhead up to 0.8 μM (Figures 1C, S1E and S1F). A tandem repeat of FKHMs is suggested to increase FoxP3 affinity (Koh et al., 2009), prompting us to examine the potential effect of tandem repeats of FKHM, both direct repeats (DR) and inverted repeats (IR), with various gap sizes. FoxP3ΔN had a strong preference for IR-FKHM with a 4 nt gap (IR-FKHM4g) (Figures 1C and 1D), but not for any DR-FKHMs, regardless of the gap size (Figures S1B and S1C). The same DNA specificity was observed for full-length FoxP3 expressed in 293T cells, suggesting that FoxP3’s preference for IR-FKHM4g is independent of the source of the protein (Figure S1D).
FoxP3RBR-forkhead also displayed the same preference for IR-FKHM4g (Figure S1E), while FoxP3forkhead did not (Figure S1F). FoxP3forkhead was less efficient in DNA binding than FoxP3ΔN and FoxP3RBR-forkhead (Figure 1C, last 6 lanes, and Figure S1E–F). Considering that the preference for IR-FKHM4g was observed only with FoxP3ΔN and FoxP3RBR-forkhead, which harbored monomeric forkhead, but not with swap-dimeric FoxP3forkhead, we suspected that the different DNA selectivity was a direct consequence of different forkhead conformations. In fact, modeling suggested that the two swap dimers could not simultaneously occupy two FKHMs in IR-FKHM4g (Figure S1G), explaining why FoxP3forkhead did not display the same preference for IR-FKHM4g as with FoxP3ΔN and FoxP3RBR-forkhead. These results indicated that FoxP3 forkhead in more native domain architecture (FoxP3ΔN and FoxP3RBR-forkhead) is not in the swap dimeric conformation, and that the true conformation of FoxP3 is the key to understanding its DNA specificity.
Overall architecture of FoxP3
We co-crystallized FoxP3ΔN in complex with IR-FKHM4g. We tested multiple truncation variants for improved solubility and crystallizability, and found deletion of residues 277–304 and 277–314 within the RBR linker necessary (FoxP3ΔN’ and FoxP3ΔN” in Figure S2A). Both constructs displayed preferential binding for IR-FKHM4g as with FoxP3ΔN (Figure S2B). Crystals of FoxP3ΔN’ and FoxP3ΔN” diffracted to 4.0 Å and 3.1 Å, respectively (Table S1). Our attempt to solve the structure using the swap-dimeric FoxP3forkhead as the molecular replacement template failed. However, using non-swap monomeric structures of forkhead from other TFs (such as FoxN1 (Newman et al., 2020)), we identified unambiguous solutions and obtained final models (See Methods and Table S1).
The structures of FoxP3ΔN’ and FoxP3ΔN” were similar and revealed several notable features. First, both FoxP3ΔN’ and FoxP3ΔN” formed the winged-helix, non-swap conformation, rather than the swap dimer (Figure 1E, Figure S2C). As with other forkhead TFs with the winged-helix fold (Dai et al., 2021), FoxP3 inserted the signature helix 3 (H3) into the major groove forming a sequence-specific interaction with DNA, while wing 1 formed additional contact with the DNA phosphate backbone. The potential source of the discrepancy between our structure versus the previous swap dimer structure and the functional implications of the new structure are discussed in Figure 2. Second, the structure also revealed that FoxP3ΔN bound IR-FKHM4g as a head-to-head (H-H) dimer, where each monomer occupied a single FKHM (Figure 1E). In this configuration, the forkhead dimer bound one side of the DNA, occupying two consecutive major grooves. DNA was bent ~25° (Figure 1E), although the degree of bending differed slightly between the FoxP3ΔN’ and FoxP3ΔN” structures (Figure S2D). H-H dimerization of FoxP3 are discussed in Figures 3 and 4. Third, whereas structures of forkhead and part of RBR (residues 322–336) were resolved, the rest of RBR (residues 262–321), CC and ZF were not seen in either crystal structure. SDS-PAGE analysis, however, showed that the protein was intact in the crystal (Figure S2E), suggesting that RBR (262–321), ZF and CC were flexible or adopt heterogeneous conformations in the crystal. While ZF within the FoxP3ΔN construct made a significant contribution to DNA binding (Figure S2F), isolated ZF-CC on its own displayed little DNA affinity (Figure S2G). Thus, we propose an overall architecture of FoxP3–DNA complex, where the H-H dimer of FoxP3RBR-forkhead forms the primary contact with DNA, while two ZFs flexibly tethered through CC and RBR form secondary contacts with nearby sites on DNA without a fixed location relative to FoxP3RBR-forkhead (Figure S2H).
Figure 2. FoxP3 folds into the non-swap monomer in the native domain architecture.
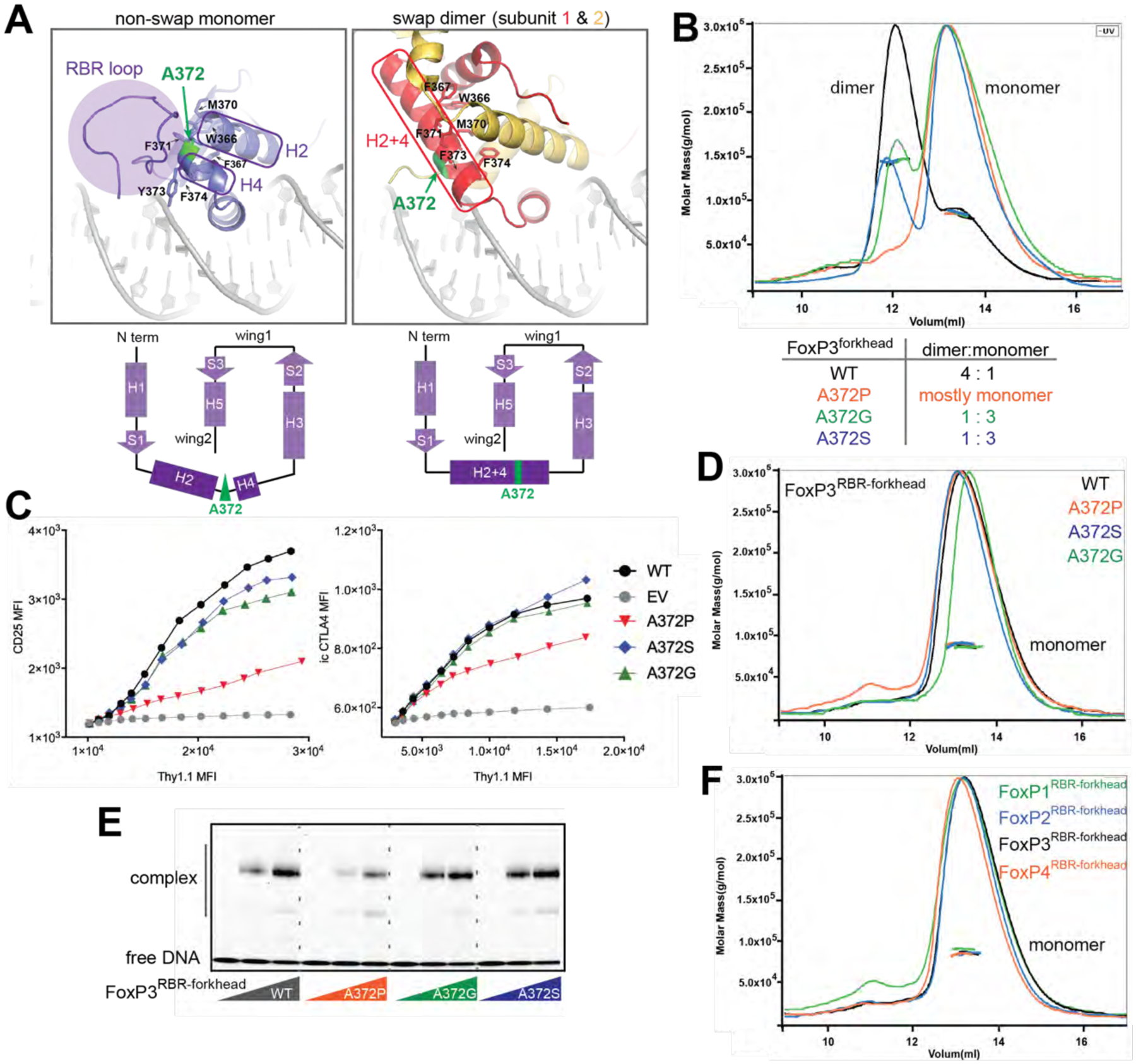
See also Figure S3.
A. Structural comparison between non-swap monomer (our structure) of FoxP3 and swap dimer (previous structure, PDB: 3QRF). Hydrophobic residues (W366, F367, M370, F371, F373 and F374) lining the swap dimerization interface (right) are shown in sticks. Note that these residues in the non-swap monomer are folded to form a hydrophobic core protected by the RBR loop (left). Bottom: the two structures have identical secondary structure topology, except for helix 2 (H2) and helix 4 (H4), which are merged into one helix (H2+4) in the swap dimer. The residue A372 (green) is located in the junction between H2 and H4.
B. SEC-MALS of NusA-tagged FoxP3forkhead with and without mutations in A372. Below: dimer-to-monomer ratio was compared using peak intensities.
C. FoxP3 cellular activity of swap-suppressive mutants, as measured by FACS. CD4+ T cells were retrovirally transduced to express FoxP3 with and without mutations in A372. Transcriptional activity of FoxP3 was analyzed by intracellular (i.c.) staining of CTLA4 or cell surface staining of CD25. FoxP3 expression was measured by Thy 1.1, which is under the control of IRES from the bicistronic mRNA expressing FoxP3.
D. SEC-MALS of NusA-tagged FoxP3RBR-forkhead with and without mutations in A372, all showing only one population corresponding to monomers.
E. EMSA of NusA-tagged FoxP3RBR-forkhead (0, 0.4 and 0.8 μM) with and without mutations in A372. DNA with IR-FKHM4g was used.
F. SEC-MALS of NusA-tagged FoxP1RBR-forkhead, FoxP2RBR-forkhead, FoxP3RBR-forkhead and FoxP4RBR-forkhead, all showing only one population corresponding to monomers.
Data in (B-F) are representative of at least three independent experiments.
Figure 3. RBR loop-mediated H-H dimerization is important for and unique to FoxP3.
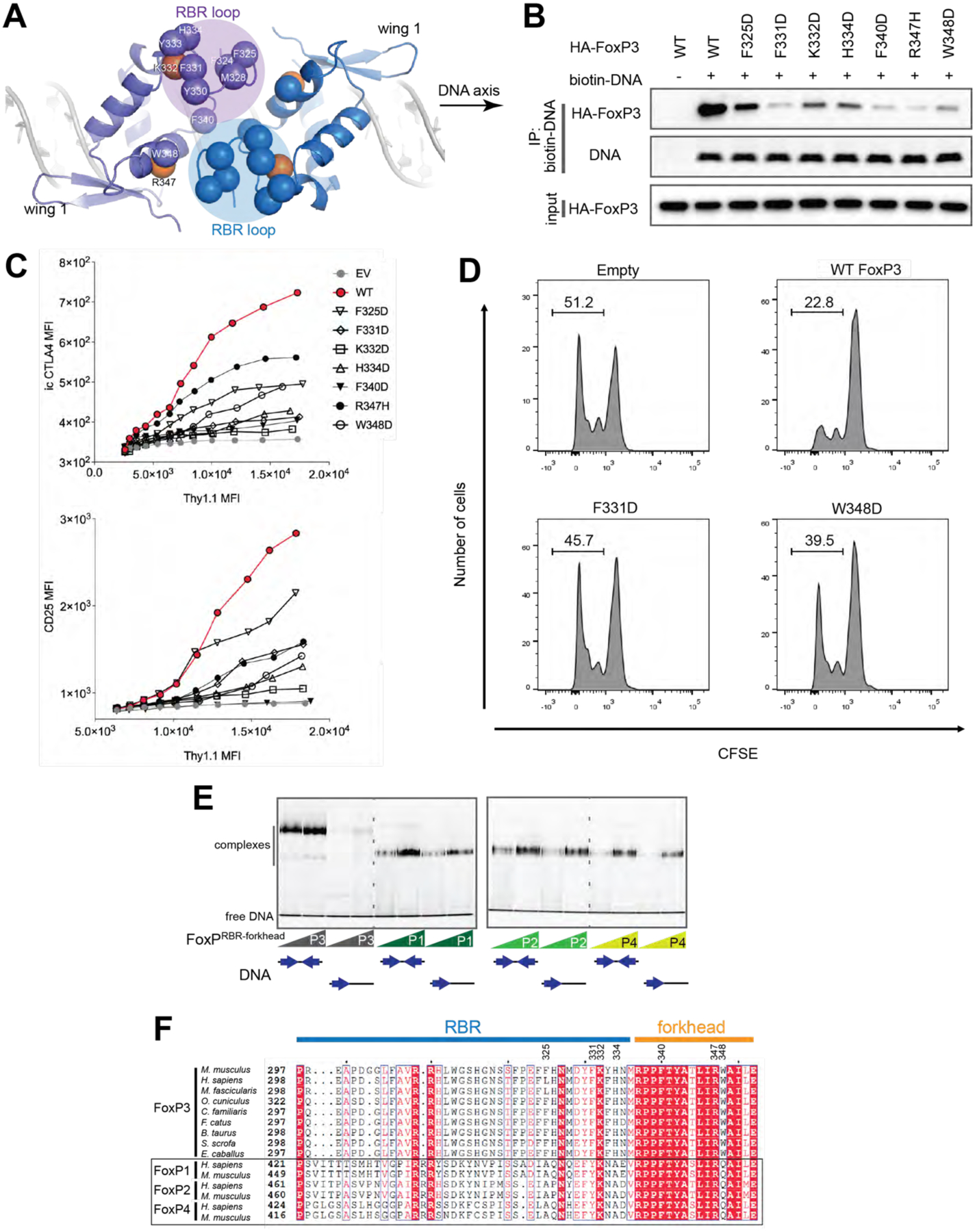
See also Figure S4.
A. Top view of the FoxP3 H-H dimer. The RBR loop forms the interface through the RBR–RBR and RBR–forkhead interactions. Hydrophobic residues (Cα) at or near the interface are shown in blue or purple spheres. K332 and R347 are shown in orange spheres.
B. DNA binding activity of H-H interface mutants. Biotinylated DNA with IR-FKHM4g was used to pull-down FoxP3 (WT or mutants) ectopically expressed in 293T cells.
C. Transcriptional activity of the H-H interface mutants. Experiments were performed as in Figure 2C.
D. T cell suppression assay of FoxP3. CD4+ T cells were retrovirally transduced to express FoxP3 with and without mutations (F331D and W348D) and their suppressive effect on proliferation of the responder T cells was examined. Shown is the representative histogram depicting the CFSE dilution profile of responder T cells cultured with FoxP3-expressing suppressor cells at a ratio of 1:2 (suppressors:responders).
E. EMSA of NusA-tagged FoxP1–4 (0.4 and 0.8 μM) using DNA oligos (0.2 μM) with IR-FKHM4g or single FKHM. All proteins were RBR-forkhead domains fused with NusA.
F. Sequence alignment of FoxP3 orthologs and paralogs in the FoxP family.
Data in (B-E) are representative of at least three independent experiments.
Figure 4. FoxP3 binds DNA as a H-H dimer, independent of DNA sequence.
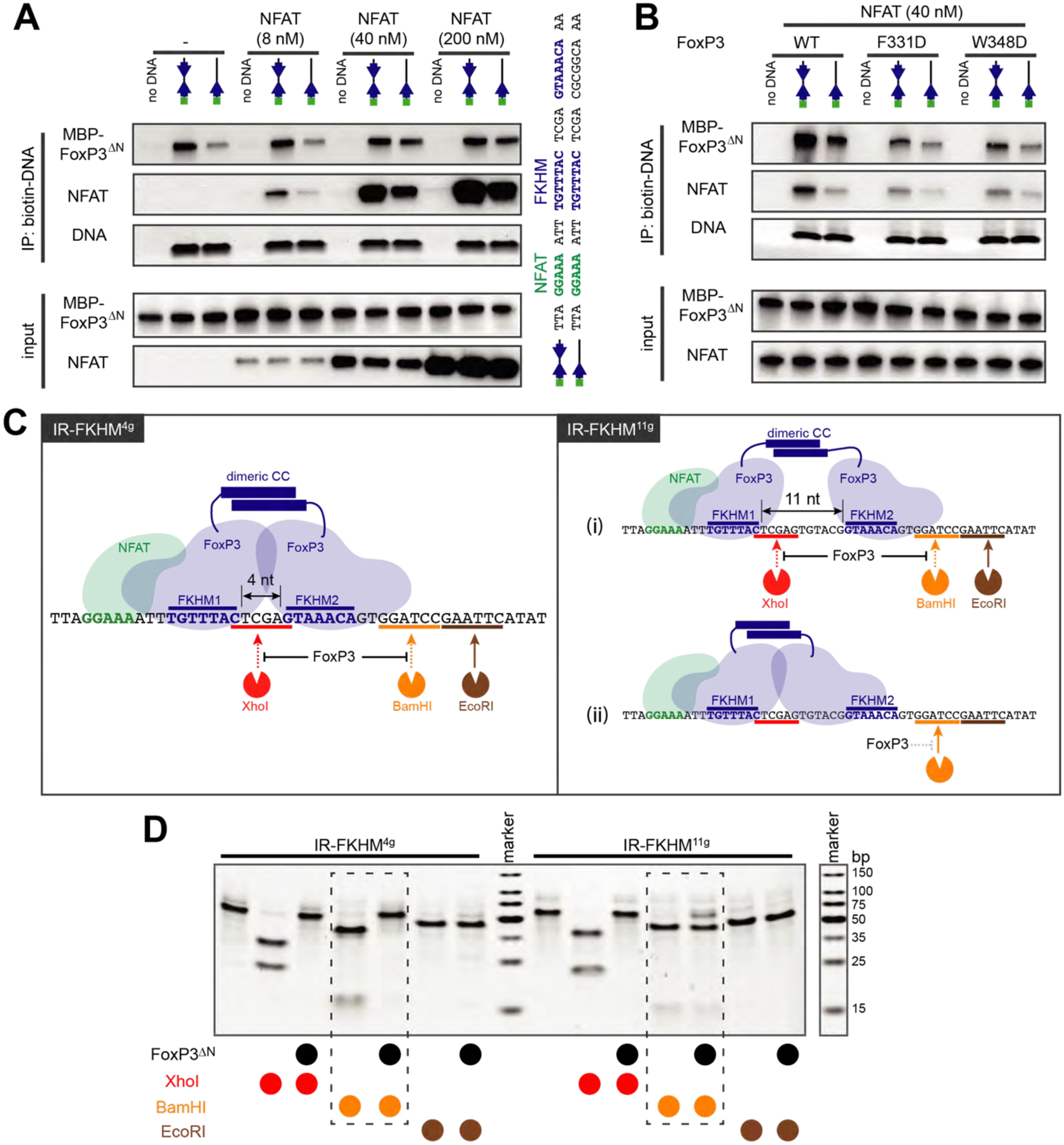
See also Figure S5.
A. Effect of NFAT on FoxP3–DNA interaction. FoxP3ΔN (0.2 μM) and biotinylated DNA (0.2 μM) were incubated with an increasing concentration of NFATRHR and were subjected to streptavidin pull-down. Both FoxP3ΔN and NFATRHR were purified from recombinant expression in E. coli. DNA oligos with IR-FKHM4g and single FKHM were compared (see the sequence on the right).
B. Biotin-DNA pull-down to monitor FoxP3-DNA interaction in the presence and absence of NFAT. FoxP3 WT and two H-H interface mutants (F331D and W348D, see Figure 3) were compared. Experiments were performed as in (A).
C. Schematic of the restriction enzyme protection assay. Left: H-H dimerization of FoxP3 on IR-FKHM4g (47 bp) should protect the DNA from cleavage by XhoI and BamHI, but not by EcoRI. Right: two potential binding modes of FoxP3 for IR-FKHM11g (54 bp), where the difference is in the accessibility of second FKHM (FKHM2), which can be examined by the BamHI site protection.
D. Restriction enzyme protection assay with Foxp3ΔN in the presence of NFAT. Note that higher FoxP3ΔN concentration was used for IR-FKHM11g (3.2 μM) than for IR-FKHM4g (1.6 μM) so that all DNA is fully occupied by FoxP3 during footprint analysis. NFAT (0.4 μM) and DNA (0.2 μM) were used.
FoxP3 forkhead exists in the non-swap monomeric conformation in the presence of RBR
Our finding that the forkhead domain within FoxP3ΔN formed the non-swap monomeric conformation contradicted the previous report that isolated FoxP3forkhead exists as a domain-swap dimer (Bandukwala et al., 2011). We thus investigated the potential source of the discrepancy and which of the two is functionally relevant. The primary difference between the swap dimer and non-swap monomer was in the helix 2 (H2) and helix 4 (H4), which in the non-swap monomer were separated by a turn at residue A372 (Figure 2A, left). In contrast, in the swap dimer, H2 and H4 are merged into a single extended helix (H2+4) (Bandukwala et al., 2011) (Figure 2A, right). The swap dimer interface on H2+4 contains a patch of hydrophobic residues (W366, F367, M370, F371, Y373 and F374), which in the non-swap conformation were buried within the folded forkhead core or protected by RBR (Figure 2A). This suggested that RBR may be responsible for stabilizing the non-swap conformation by protecting otherwise solvent exposed hydrophobic residues. In other words, deletion of RBR and subsequent exposure of hydrophobic residues may drive the swap dimerization. Consistent with this view, FoxP3 forkhead was monomeric in the presence of RBR (as in FoxP3ΔN and FoxP3RBR-forkhead), while isolated FoxP3forkhead without RBR formed a swap dimer (Figure 1B).
To further examine which of the two conformations is physiologically relevant, we employed a protein engineering approach with the focus on residue 372 at the junction of H2 and H4 (Figure 2A). The residue 372 is unique in its potential to switch the forkhead conformation between swap and non-swap structures (Bandukwala et al., 2011). We hypothesized that replacement of Ala at 372 by an amino acid with low helix propensity, such as Pro, Gly or Ser, would force separation of H2 and H4 and convert FoxP3forkhead to the monomeric conformation. As predicted, A372P, A372G and A372S all biased isolated FoxP3forkhead from dimeric to largely monomeric state, albeit to varying degrees (Figure 2B). Protein crosslinking analysis also suggested that all three mutations suppressed swap dimerization (Figure S3A). We next examined the impact of the A372 mutations on the transcriptional activity of FoxP3, as measured by the levels of CTLA4 and CD25 (two Treg cell markers) upon expression of FoxP3 in CD4+ T cells. If FoxP3 functions require the swap-dimeric structure, all three mutations of A372 should result in loss of function. The result showed that, while A372P partially impaired the transcriptional activity of FoxP3, as previously reported (Bandukwala et al., 2011), A372G and A372S had little impact (Figure 2C), arguing against the requirement for domain-swap dimerization in FoxP3 function. The negative impact of A372P appeared independent of its ability to suppress swap dimerization; introduction of A372P in FoxP3RBR-forkhead, which already existed as a non-swap monomer, did not alter its monomeric state (Figure 2D), but negatively affected its DNA affinity (Figure 2E) and the protein melting curve (Figure S3B). Such negative impact was not observed with A372G and A372S (Figures 2E and S3B). Collectively, the results with A372G and A372S suggested that swap dimerization is not required for FoxP3 functions.
We next asked whether the impact of RBR on forkhead folding is conserved in other FoxP TFs, which also contain Ala at position equivalent to 372 (Figure S3C) and form domain-swap dimers when expressing forkhead domains in isolation (Chu et al., 2011; Stroud et al., 2006). FoxP1, FoxP2 and FoxP4 all contain RBR-like linkers between CC and FKH. We expressed FoxP1/2/4 proteins equivalent to the FoxP3RBR-forkhead construct and found that FoxP1/2/4 proteins were monomeric when purified with RBR-like linkers (Figure 2F). Thus, the role of RBR or RBR-like linkers in stabilizing the non-swap conformation is conserved in all FoxP TFs. These results further supported the notion that the non-swap conformation is the physiologically relevant form for all four members of FoxP TFs.
Head-to-head (H-H) dimerization of forkhead is unique to FoxP3 and is important for DNA binding
Our structure showed that individual FoxP3 forkhead folded into a monomer, but this monomer formed a H-H dimer when bound to DNA containing IR-FKHM4g (Figure 1E). H-H dimerization was mediated by part of RBR (residues 321–336, referred to as the RBR loop), which interacted with the RBR loop and forkhead from the other subunit (Figure 3A). Electron density for the RBR loop was not as well defined as other parts of the protein (Figure S4A), suggesting conformational flexibility in the H-H interface. Residues located within the RBR loop and near the interface were highly hydrophobic (Figure 3A), suggesting that H-H dimerization was driven by a collection of hydrophobic interactions, reminiscent of “fuzzy” interactions shown for other TFs (Pricer et al., 2017; Tuttle et al., 2021). The RBR loop was also involved in crystallographic packing (Figures S4A, S4B), likely involving similarly fuzzy, hydrophobic interactions. Thus, the hydrophobic nature of the RBR loop may enable multiple types of protein-protein interactions beyond H-H dimerization (to be discussed in Figure 6).
Figure 6. H-H dimerization is also important for Runx1 binding.
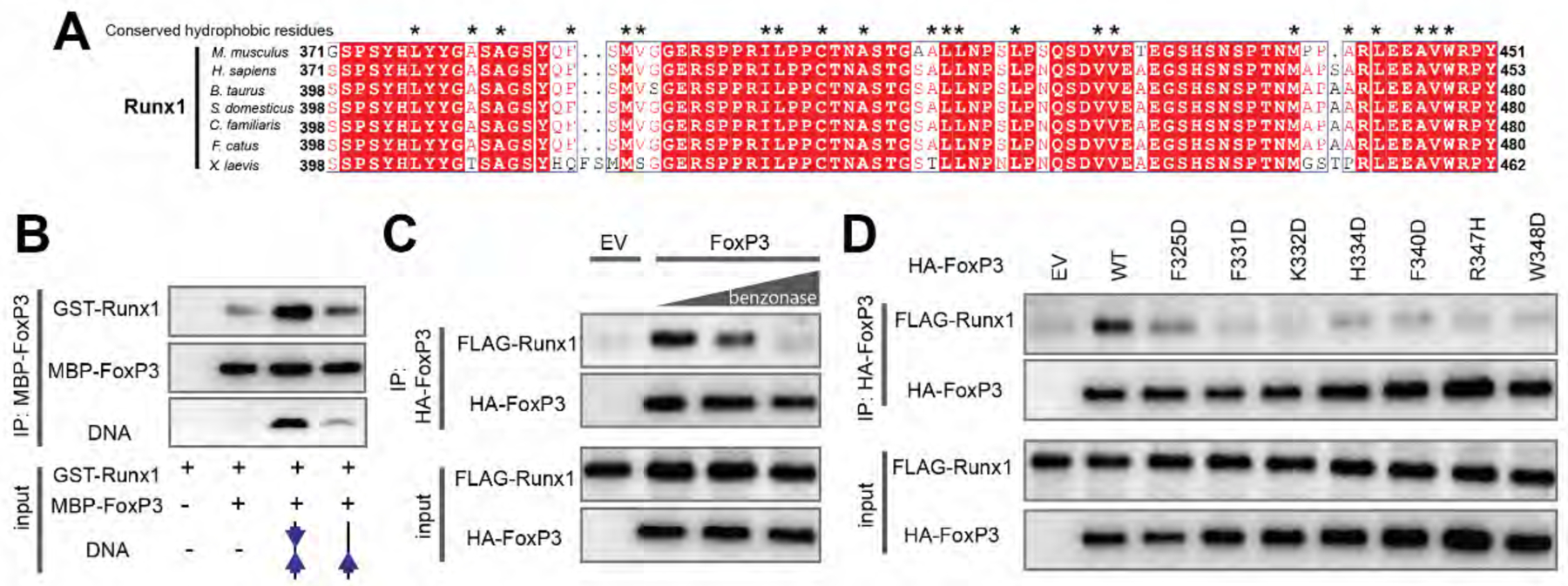
A. Sequence alignment of Runx1 orthologs showing conserved hydrophobic residues (*) in its C-terminal tail that binds FoxP3.
B. FoxP3 interaction with Runx1 using purified proteins. MBP-tagged FoxP3RBR-forkhead and GST-tagged Runx1 (residue 371–451) was purified from E. coli and was subjected to MBP pull-down in the presence and absence of DNA harboring IR-FKHM4g or single FKHM.
C. FoxP3 interaction with Runx1 from 293T cells. HA-tagged full-length FoxP3 and FLAG-tagged full-length Runx1 were separately expressed in 293T cells. Cell lysates were mixed and were subjected to anti-HA immunoprecipitation (IP). The increasing concentrations of benzonase was used to examine the effect of cellular DNA on FoxP3–Runx1 interaction.
D. FoxP3 interaction with Runx1 from 293T cells. H-H interface mutations were compared to WT FoxP3. Experiments were performed as in (C), except no benzonase was used in all conditions.
Data in (B-D) are representative of at least three independent experiments.
To examine whether the H-H dimerization is simply a result of binding IR-FKHM4g or accounts for FoxP3’s preference for IR-FKHM4g, we introduced single-point mutations in or near the H-H dimeric interface in both RBR and forkhead. These included aforementioned hydrophobic residues and basic residues, including an IPEX mutation R347H (Gambineri et al., 2008) (Figure 3A). Consistent with the view that H-H dimerization is the cause, rather than the consequence, of IR-FKHM4g binding, the H-H interface mutations impaired FoxP3’s affinity for IR-FKHM4g DNA, as measured by biotinylated DNA pull-down (Figure 3B). Some residues in the RBR loop, such as F331 and H334, were not located at the interface but still played important roles, suggesting that these residues may shape the RBR loop and thereby indirectly involved in H-H dimerization. The interface mutations also impaired the cellular functions of FoxP3, as measured by induction of CD25 and CTLA4 (Figure 3C) or by T cell suppression assay (Figure 3D).
Note that the loss of H-H dimerization did not necessarily lead to domain-swap dimerization; F331D, R347H and W348D FoxP3RBR-forkhead were impaired in H-H dimerization and yet were stable as a monomer (Figure S4C). Thus, while the non-swap conformation was prerequisite for H-H dimerization, H-H dimerization was not required for non-swap conformation. Additionally, these data showed that H-H dimerization was important for both DNA binding and FoxP3’s transcriptional activity.
Given the importance of H-H dimerization for FoxP3, we next asked whether H-H dimerization also occurred with other FoxP TFs. We compared FoxPRBR-forkhead binding to IR-FKHM versus sFKHM, since H-H dimerization would manifest in preferential binding of IR-FKHM. In contrast to FoxP3, FoxP1/2/4 equally bound IR-FKHM4g and sFKHM (Figure 3E), and this was independent of the IR-FKHM gap size (Figure S4D). Additionally, the native gel migration rate of the FoxP1/2/4 complexes were similar, regardless of whether on IR-FKHM4g or sFKHM – another indication that they bound both DNAs as a monomer (Figure 3E). These results suggested that H-H dimerization did not occur to other FoxP TFs. Perhaps reflecting this difference in the ability to form a H-H dimer, the RBR sequence of FoxP3 differs from the equivalent linker in other FoxP TFs (Figure 3F). Altogether, our results suggested that H-H dimerization was an important and a unique feature of FoxP3 and imparted distinct DNA specificity to FoxP3.
H-H dimerization is important for FoxP3 binding to suboptimal DNA sequence
We next asked whether H-H dimerization of FoxP3 was limited to IR-FKHM4g or whether this also occurred when FoxP3 bound suboptimal, low-affinity sequences. This was an important question because many TFs were known to bind suboptimal sequences in cells (Pfeifer et al., 1987; Segal et al., 2008), likely driven by cooperative binding with cofactors (Reiter et al., 2017). In fact, FoxP3 functions together with other cofactors (Kwon et al., 2017; Rudra et al., 2012; Wu et al., 2006), many of which have their own DBDs and may assist FoxP3–DNA interaction in cells (Samstein et al., 2012; Wu et al., 2006). One of the best-characterized cofactors for FoxP3 is NFAT, whose DBD (Rel Homology Region, RHR) interacts directly with FoxP3 forkhead (Bandukwala et al., 2011). Although the previous structure of FoxP3 in complex with NFAT was determined with the swap-dimeric FoxP3forkhead, the NFATRHR interface is nearly identical in both the swap and non-swap conformations, making both conformations compatible with NFAT binding (Figure S5A). As such, the non-swap FoxP3ΔN also showed cooperative DNA binding with NFAT when the NFAT-binding site was 3 nt upstream of FKHM, the requirement for NFAT–FoxP3 interaction (Figure 4A). Both FoxP3 and NFAT bound IR-FKHM4g better than sFKHM. Furthermore, both FoxP3 and NFAT bound better in the presence of the other protein than in its absence (Figures 4A and S5B–C), suggesting that they mutually assist each other. The effect of NFAT on FoxP3 was more evident with sFKHM than with IR-FKHM4g, consistent with the notion that FoxP3 alone can bind IR-FKHM4g efficiently, alleviating requirement of cofactor assistance. When H-H interface mutations (F331D and W348D) were introduced to FoxP3, DNA affinity was reduced for both IR-FKHM4g and single FKHM, with or without NFAT (Figure 4B). This suggested that H-H dimerization was important even when FoxP3 bound suboptimal DNA assisted by the cofactor NFAT.
To more directly examine whether FoxP3 formed the H-H dimer when it bound suboptimal DNA sequence, we examined FoxP3 footprint using a restriction enzyme protection assay. We chose IR-FKHM11g as a suboptimal DNA because it allowed for predicting two distinct binding modes. In one mode, two FoxP3 molecules bridged by CC could occupy two FKHMs separated by the 11 nt gap without forming a H-H contact (Figure 4C, right panel (i)). In the alternative mode, FoxP3 could bind IR-FKHM11g as a H-H dimer, occupying only one of the FKHMs – most likely FKHM near the NFAT site (FKHM1) – and the suboptimal sequence 4 nt away from FKHM1, leaving the second FKHM (FKHM2) unoccupied (Figure 4C, right panel (ii)). To distinguish between the two modes, we placed the BamHI restriction site near FKHM2 to examine the occupancy of FKHM2 (Figure 4C). The XhoI and EcoRI sites were also introduced to report FKHM1 occupancy and DNA binding specificity, respectively.
We first tested an equivalent construct with IR-FKHM4g. Under the condition where FoxP3ΔN occupied DNA in a sequence-specific manner (i.e. the XhoI site was fully protected while the EcoRI site was fully accessible), the BamHI site was protected by FoxP3 (Figure 4D, left half), consistent with the crystal structure where both FKHM1 and FKHM2 were occupied by FoxP3 H-H dimer. In contrast, the BamHI site of IR-FKHM11g was largely accessible even though DNA was fully occupied by FoxP3 (as evidenced by the XhoI site protection) (Figure 4D, right half). Unlike FoxP3ΔN, FoxP3forkhead (A372S), which could not form a H-H dimer, showed no difference between IR-FKHM4g and IR-FKHM11g in the BamHI site protection (Figure S5D). These results suggested that FoxP3ΔN maintained the H-H dimeric structure when binding IR-FKHM11g as well as IR-FKHM4g. Together with the data in Figure 4B, they further supported the notion that FoxP3 binds DNA as a H-H dimer, regardless of the DNA sequence.
H-H dimerization enables FoxP3 to recognize diverse DNA sequences
Our data above suggested that H-H dimerization was an important mode of DNA binding for FoxP3 and this greatly influenced its DNA sequence specificity in vitro. To assess the impact of H-H dimerization on FoxP3–DNA interaction in Treg cells, we investigated previously reported FoxP3 ChIP-seq data for potential enrichment of the IR-FKHM4g sequence (Kitagawa et al., 2017; Samstein et al., 2012). De novo motif analysis of 5000 consensus FoxP3 ChIP-seq peaks did not identify IR-FKHM4g (Figure 5A), although single FKHM was identified in these peaks (P-value<10−9) (Figure 5A). As an alternative strategy, we separately analyzed 548 FoxP3-bound peaks containing an FKHM and counted the number of instances of an inverted FKHM repeats with a gap size ranging from 1 to 21 nt; no enrichment of 4 nt spacing was observed (Figure 5B). One explanation for this lack of IR-FKHM4g enrichment could be that FoxP3, like other TFs (Nakagawa et al., 2013), utilizes many distinct suboptimal sequences in the presence of cofactors. H-H dimerization of FoxP3 may further increase the diversity of such suboptimal sequences it can bind, as shown with other dimeric TFs (Arnett et al., 2010; Jiang et al., 2019).
Figure 5. H-H dimerization enables FoxP3 to recognize diverse sequences.
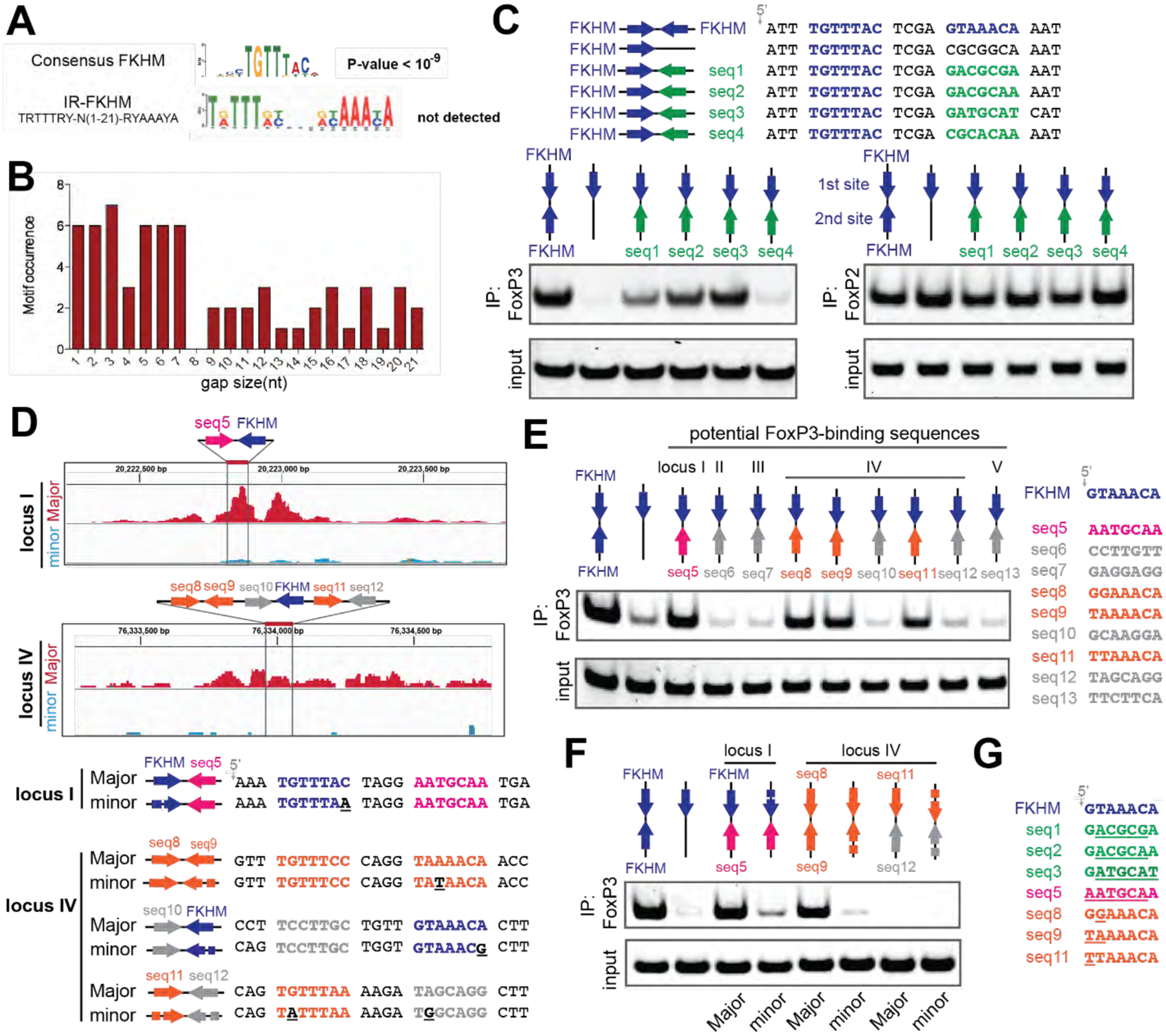
See also Figure S6.
A. De novo motif analysis of FoxP3 ChIP-seq (n=5000) sequences. The canonical FKHD motif was enriched (P-value<10−9), but IR-FKHM motif was not detected for any of the gap sizes tested (1–21 nt). A more relaxed FKHM sequence (TRTTRY; R and Y indicate purine and pyrimidine, respectively) was used to be inclusive.
B. Occurrence of IR-FKHM, iteratively testing gap sizes of 1 to 21 nt, of FoxP3 ChIP-seq sequences that contain an FKHM (n=548). Motif occurrences were counted if p-value<10−5 and score greater than 12.
C. FoxP3 interaction with DNA harboring IR-FKHM4g and FKHM paired with non-canonical motifs (seq1–4). Purified MBP-FoxP3ΔN was mixed with DNA oligos and was subjected to MBP pull-down, followed by native PAGE analysis of co-purified DNA. DNA was visualized by Sybr Gold stain. Right: MBP-FoxP2ΔN was used for comparison.
D. FoxP3 Cut&Run intensity showing allelic imbalance of FoxP3 occupancy in loci I and IV. Major and minor alleles indicate alleles with greater and lesser FoxP3 occupancy, respectively. Below: Major and minor allele sequences with their differences highlighted with underscores in the minor sequences.
E. FoxP3 interaction with DNA harboring potential FoxP3-binding sequences paired with FKHM. These sequences were chosen from five loci in the Major allele sequence where mutations were associated with reduced FoxP3 occupancy.
F. FoxP3 interaction with DNA harboring natural sequences from loci I and IV (see D).
G. Alignment of FoxP3-compatible sequences examined in this figure. Sequence deviation from FKHM was highlighted with underscore.
Data in (C, E and F) are representative of at least three independent experiments.
To examine the possibility of diverse sequence recognition by the FoxP3 H-H dimer, we examined FoxP3 binding to four non-consensus motifs (seq1–4) that were previously shown to bind other forkhead TFs (Nakagawa et al., 2013; Rogers et al., 2019). These non-consensus sequences were paired with FKHM in the inverted orientation with a 4 nt gap (Figure 5C, top). FoxP3 bound FKHM paired with seq1/2/3 significantly more efficiently than sFKHM (Figure 5C, bottom left). In comparison, FoxP2 showed similar binding to all DNA regardless of the sequence paired with FKHM (Figure 5C, bottom right). When seq1–3 were paired with themselves instead of FKHM, FoxP3 binding was undetectable (Figure S6C), indicating that at least one FKHM was necessary. Similar results were obtained using FoxP3 by EMSA (Figures S6A and S6B). These results suggested that strong anchoring of one FoxP3 monomer at FKHM relaxed the sequence requirement for the second site, allowing FoxP3 to bind other sequences besides IR-FKHM4g.
We next examined physiological target sites of FoxP3. A previous Cut&Run analysis (van der Veeken et al., 2020) identified sequence-specific FoxP3-binding sites based on allelic difference in FoxP3 occupancy associated with allelic variations in the FKHM sequence between two evolutionary distant mouse genomes (see Figure 5D for examples; “major” and “minor” alleles indicate those with greater and lesser FoxP3 occupancy, respectively). We asked whether these sites contained FoxP3-compatible sequences as the second binding site. Sequences paired with FKHM in five loci with allelic imbalance in FoxP3 occupancy were tested. Locus IV had several FKHM-like sequences (seq8, seq9 and seq11; Figure 5D), which were also included in our analysis. Among the 9 sequences tested, four sequences (seq5/8/9/11) efficiently bound FoxP3 when paired with FKHM (Figure 5E), but not when paired with themselves (Figure S6D). Thus, loci I and IV had suboptimal, but nevertheless FoxP3-compatible sequences (seq5/8/9/11) paired with each other or with FKHM. However, loci II, III and V did not contain FoxP3-compatible sequences paired with FKHM, suggesting that FoxP3 may bind elsewhere within these loci without FKHM.
We further examined loci I and IV. As expected, FoxP3 binding to FKHM-seq5 of locus I was reduced upon mutating FKHM (as in the minor allele) (Figure 5F), consistent with the view that FKHM-seq5 was responsible for FoxP3 binding at locus I. In contrast to locus I, locus IV contained multiple potential FoxP3 binding sites: seq8 paired with seq9, seq10 with FKHM, and seq11 with seq12 (Figure 5D). Of these, seq8-seq9 was the only site where FoxP3-compatible sequences were paired. Accordingly, only seq8-seq9 showed strong FoxP3 binding, while seq10-FKHM and seq11-seq12 did not (Figure 5E and 5F). Furthermore, sequence variation in seq9 (as in the minor allele) reduced FoxP3 binding (Figure 5F), supporting the view that seq8-seq9 significantly contributed to locus IV binding. Both seq8 and seq9 were FKHM-like sequences, but neither was strong enough to recruit FoxP3 alone or when paired with itself (Figure S6D). This suggested that certain combinations of motifs had a non-additive effect and pairing of suboptimal sequences with the optimal FKHM was not obligatory.
The non-consensus sequences that we identified here did not display any obvious patterns (Figure 5G), which may explain why the second FoxP3 site could not be detected from the global motif analysis. These non-consensus sequences are also likely to be a small subset of a possibly much larger pool of sequences FoxP3 binds in the presence of cofactors in cells, as evidenced by the lack of enrichment of these compatible sequences, relative to non-compatible sequences, within the ChIP-seq peaks (Figure S6E). Nevertheless, our findings suggested that H-H dimerization significantly altered FoxP3 DNA specificity and enabled FoxP3 to recognize diverse sequences beyond IR-FKHM4g.
H-H dimerization is important for Runx1 binding
Given that H-H dimerization was largely mediated by RBR and that RBR recruits Runx1 (Ono et al., 2007), we next asked what effect H-H dimerization had on Runx1 binding. Our structure showed that H-H dimerization created a large hydrophobic surface that could mediate protein-protein interaction, as judged by their involvement in crystallographic packing (Figure S4B). Given that FoxP3-interacting region of Runx1 (residue 371–451) also is highly hydrophobic (Figure 6A), we asked whether the hydrophobic surface of FoxP3 RBR serves as a cofactor docking site for Runx1.
Using purified recombinant Runx1 (Runx1ΔN, residue 371–451) and FoxP3RBR-forkhead, we confirmed that their interaction was direct (Figure 6B). We also found that this interaction was enhanced in the presence of DNA with IR-FKHM4g (Figure 6B), the condition that promoted H-H dimerization. The Runx1–FoxP3 interaction was not enhanced by DNA harboring sFKHM, further supporting the notion that H-H dimerization was important for Runx1 binding. Since Runx1ΔN did not contain a DBD, the observed dependence of Runx1–FoxP3 interaction on IR-FKHM4g was unlikely due to DNA-mediated bridging. Using full-length Runx1 and FoxP3 overexpressed in 293T cells, we also confirmed that their association depended on nucleic acids, as the benzonase treatment impaired their co-immunoprecipitation (co-IP) (Figure 6C). Consistent with the idea that Runx1 binding was facilitated by H-H dimerization, H-H interface mutations reduced Runx1–FoxP3 co-IP (Figure 6D). Together, these data suggested that FoxP3 harnessed its H-H dimerization capability not only to alter DNA sequence specificity, but also to recruit Runx1.
IPEX mutation R337Q induces swap dimerization and impairs FoxP3 functions
Given our findings supporting the importance of H-H dimerization and non-swap conformation, we asked whether some IPEX mutations are caused by errors in these features. Note that the non-swap conformation is a precondition for H-H dimerization, and thus, any mutation that alters non-swap folding would impair all aspects of FoxP3 function including H-H dimerization, DNA binding and Runx1 binding. Among the IPEX-associated mutations of FOXP3, R337Q attracted our attention as it is located near the end of RBR (Rubio-Cabezas et al., 2009) (Figure 1A), which was involved in both non-swap conformation and H-H dimerization. Additionally, R337 directly contacted DNA in our non-swap structure, but is far away from DNA in the swap-dimeric structure (Figure 7A), a feature that could further help distinguish between the two forkhead conformations.
Figure 7. IPEX mutation R337Q induces swap dimerization and impairs FoxP3 functions.
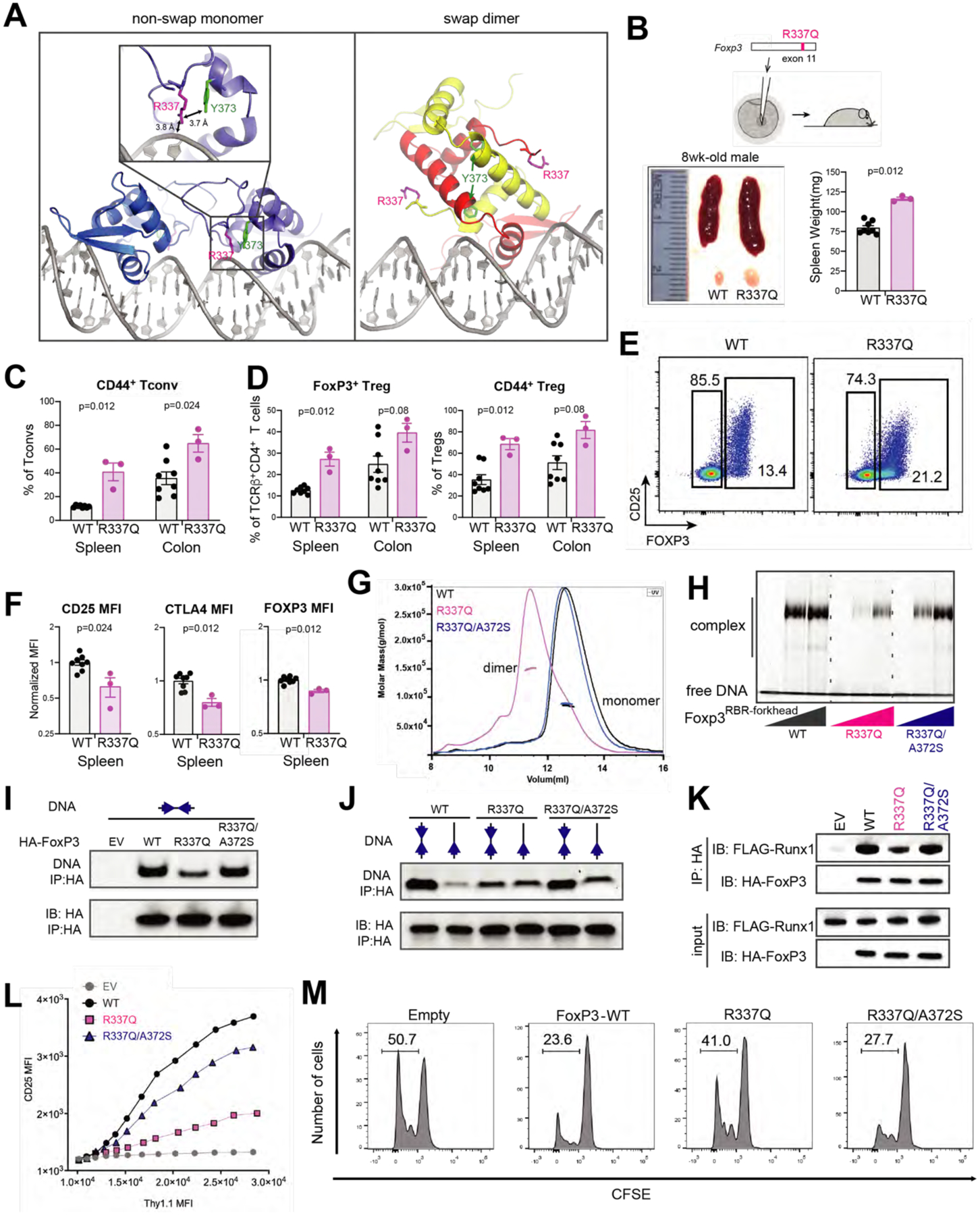
See also Figure S7.
A. Location of R337 in the crystal structures of swap dimeric and non-swap monomeric FoxP3. R337 interacts with DNA and Y373 only in the non-swap conformation.
B. Abnormal immune homeostasis in 8-week-old R337Q male mice, generated by CRISPR mutagenesis of Foxp3. Bottom left: a representative picture comparing the spleens and inguinal lymph nodes of R337Q mutant mice and their WT littermates. Bottom right: spleen weights.
C. Fraction of activated (CD44+) conventional T (Tconv) cells among CD4+ T cells in the spleen and the colonic lamina propria of WT and R337Q mutant mice.
D. Foxp3+ Treg in the spleen and the colonic lamina propria of R337Q mutant mice in comparison to their WT littermates. Left: their fraction among TCRβ+CD4+ T cells. Right: their activation status assessed by the marker CD44+.
E. CD25/FoxP3 cytometry plots of spleen TCRβ+CD4+ T cells from WT and R337Q mutant mice.
F. Quantification of CD25, CTLA4 and FoxP3 by flow cytometry (MFI) in TCRβ+CD4+Foxp3+ spleen Tregs of R337Q mutant mice and WT littermates.
G. SEC-MALS of NusA-tagged FoxP3RBR-forkhead (WT, R337Q and R337Q/A372S).
H. DNA binding activity of recombinant, purified FoxP3RBR-forkhead (WT, R337Q and R337Q/A372S) as measured by EMSA. FoxP3 was tagged with NusA. DNA harbors IR-FKHM4g.
I. DNA binding activity of full-length FoxP3 (WT, R337Q and R337Q/A372S) expressed in 293T cells. HA-tagged, full-length FoxP3 was ectopically expressed in 293T cells and purified by anti-HA immunoprecipitation (IP). Equal amount of IR-FKHM4g DNA was added to FoxP3-bound beads and further purified by anti-HA IP. Bound DNA and proteins were isolated and analyzed by native-PAGE and SDS-PAGE, respectively. Sybr Gold stain was used for DNA measurement.
J. FoxP3 sequence preference. HA-tagged, full-length FoxP3 (WT, R337Q and R337Q/A372S) was expressed in 293T and were purified to compare FoxP3 sequence preference between IR-FKHM4g and sFKHM. Experiments were performed as in Figure 7I.
K. Runx1 binding for full-length FoxP3 (WT, R337Q and R337Q/A372S) from 293T cells.
L. Transcriptional activity of FoxP3 as measured by CD25 level. See Figure S7B for CTLA4 measurement. The effects of R337Q, either alone or in combination with the swap-suppressive mutation A372S, were examined.
M. Treg suppression assay of FoxP3 WT, R337Q and R337Q/A372S. Shown is a representative histogram depicting the CFSE dilution profile of responder T cells cultured with transduced FoxP3-expressing suppressor cells. Experiments were performed as in Figure 3D.
Data in (B-D, F) are presented as mean ± SEM. P values were obtained by Mann-Whitney test comparing WT and R337Q. Data in (G-M) are representative of at least three independent experiments.
To better analyze the effect of R337Q in Treg cells in vivo, we generated by CRISPR/Cas9 germline mutagenesis a mutant mouse line carrying the R337Q mutation in the endogenous Foxp3 locus (Figure 7B). Young male mice hemizygous for the mutation were viable, with no sign of wasting disease, or overt dermatitis or intestinal pathology, differing from full FoxP3 deficiencies (Fontenot et al., 2003; Godfrey et al., 1991), although reduced weight gain and dermatitis did appear in older R337Q animals. In addition, they presented significant splenomegaly and lymphadenopathy (Figure 7B), and an increase in the level of activated (CD44+) T conventional (Tconv) cells (Figure 7C), suggesting a partial loss of Treg cell functionality. The proportion of FoxP3+ Treg cells and their activated (CD44+) fraction also increased in R337Q mice (Figure 7D), a compensatory phenotype previously observed in partial FoxP3 deficiencies (Kwon et al., 2018; Van Gool et al., 2019). R337Q Treg cells also showed decreased levels of CD25 and FoxP3, as well as CTLA4, characteristic of FoxP3 hypomorphs (Figures 7E and 7F), all indicative of partial FoxP3 dysfunction. Thus, the R337Q mutation impaired Treg fitness, with a repercussion in the control of CD4+ T cell homeostasis, although not as radical as the typical scurfy phenotype usually seen with the complete Foxp3 deficiencies.
We next introduced R337Q in recombinant protein and analyzed its impact on the biochemical properties of FoxP3. FoxP3RBR-forkhead with R337Q was purified as a dimer, which differed from monomeric WT FoxP3RBR-forkhead (Figure 7G). The R337Q dimer appeared to be a swap dimer, since addition of the swap-suppressive mutation A372S reverted R337Q FoxP3RBR-forkhead to a monomer (Figure 7G). Thermal stability of R337Q was also lower than that of WT, but was rescued by A372S (Figure S7A). In line with the importance of the non-swap conformation in H-H dimerization and DNA binding, R337Q reduced DNA affinity, while A372S partially restored it (Figure 7H). To examine whether R337Q in fact existed in the domain-swap dimer in cells and whether this conformation accounted for the functional impairment of R337Q, we measured five activities of FoxP3 expressed in 293T or CD4+ T cells. These were (1) DNA binding (Figure 7I), (2) sequence preference for IR-FKHM over sFKHM (Figure 7J), (3) Runx1 binding (Figure 7K), (4) transcriptional activity (Figures 7L and S7B) and (5) T cell suppression activity (Figure 7M). All five activities, which depended on the non-swap conformation, were impaired upon R337Q mutation and were largely rescued by A372S. The incomplete rescue likely reflects the role of R337 not only in stabilizing the non-swap conformation, but also in direct DNA binding, as our FoxP3 structure and DNA binding assay suggested (Figures 7A and 7H). Collectively, these data supported the notion that functional impairment by the R337Q mutation is largely mediated by its induction of the domain-swap conformation, although its role in direct DNA binding also contributes.
Other IPEX mutations near the H2/H4 junctions can also cause folding errors
The impact of R337Q on FoxP3 folding was at first unexpected as R337 is on the protein surface and folding errors are often associated with mutations in protein cores. R337 in our structure was in close proximity to Y373, another surface residue mutated in IPEX patients (Y373V) (Tanaka et al., 2005). We first confirmed that Y373V impaired FoxP3 transcriptional function (Figure S7C). Analysis of other forkhead protein structures (in the non-swap conformation) showed that residues equivalent to R337 and Y373 were often in close proximity with the potential of forming pi–cation or pi–pi interactions (Figure S7D). This observation prompted us to ask whether Y373 also played an important role in stabilizing the non-swap conformation. Recombinant Y373V FoxP3RBR-forkhead protein displayed a broad elution profile in size-exclusion chromatogram with the peak molecular weight corresponding to a tetramer (Figure S7E), which was indicative of folding defects, albeit somewhat differently than R337Q.
To further examine whether similar folding errors can account for IPEX pathogenesis, we investigated seven additional IPEX mutations in the forkehad domain: I346T, R347H, I363V, M370I, F371C, F367L, F374C (Figures S7F–G). Of these mutants, we found that I346T, M370I and F371C shifted the size-exclusion elution profile of FoxP3RBR-forkhead from a monomer to aggregate, likely indicating folding errors (Figures S7F). Mapping of the folding-disruptive mutations, including R337Q and Y373V, onto our crystal structure (Figure S7H) revealed that all but I346T were located near the junction of H2 and H4, the key site for the domain-swap to non-swap conversion (Figure 2A). These observations suggested that the H2/H4 junction may be the “Achilles’ heel” for proper folding of FoxP3 and such folding errors involving the H2/H4 junction may be a common etiology for IPEX.
DISCUSSION
To better understand how FoxP3 utilizes the common DBD forkhead to function as a unique TF for Treg determinism, we characterized FoxP3 using a combination of structural biology, biochemistry and functional assays. Our study revealed that, unlike previous reports with isolated forkhead DBD (Bandukwala et al., 2011), FoxP3 forkhead folded into a non-swap monomer and utilized the appendage domain RBR to form a head-to-head dimer for DNA binding. This requirement for H-H dimerization distinguishes FoxP3 from other forkhead TFs characterized to date, including FoxP1/2/4, which binds DNA as individual DBDs (Dai et al., 2021). As a result, FoxP3 read DNA sequence spanning ~18 bp, rather than 7 bp as monomeric forkhead would. While FoxP3 strongly favored inverted repeat of FKHM (IR-FKHM) over a single FKHM, it also recognized diverse and distinct non-consensus sequences beyond IR-FKHM. FoxP3 H-H dimerization also facilitated its interaction with the essential cofactor Runx1, likely through the large hydrophobic patch formed upon dimerization. Since H-H dimerization was promoted by DNA binding, this Runx1 binding mechanism should enable FoxP3 to tightly coordinate DNA binding and Runx1 recruitment. Thus, a highly conserved DBD can diversify its function by gaining a multimerization capability, which may have implications beyond forkhead DBD.
Our results showed that the RBR linker was important not only for H-H dimerization but also for the forkhead DBD folding, providing an unusual example where an appendage domain altered folding of a highly conserved DBD. Given that it is a common practice to study isolated DBD for TF–DNA interactions, FoxP3 may serve as a cautionary tale for such approaches. In the absence of RBR, FoxP3 forkhead folded into the swap dimer as previously described (Bandukwala et al., 2011), whereas in the presence of RBR, it formed the canonical forkhead structure. Although the RBR-like linkers in FoxP1, 2, and 4 did not support H-H dimerization as for FoxP3, their role in stabilizing the canonical forkhead structure was conserved across the FoxP family. Our data suggest that the canonical forkhead structure is the functional form as the swap dimer did not efficiently bind DNA and Runx1. Furthermore, the IPEX mutation R337Q favored swap dimerization, compromising DNA and Runx1 binding and impairing the transcriptional activity, while a swap-suppressive mutation largely restored these functional impairments of R337Q. These results altogether support that canonical non-swap conformation is the physiological form of FoxP3.
The unique dependence of FoxP3 folding on RBR and its sensitivity to surface residue mutations, such as R337Q, raised the question whether FoxP3 folding landscape is complex and whether folding errors are more common than just with R337Q. In other words, swap dimerization may not just be an in vitro artifact of expressing forkhead in isolation, but may readily occur in the presence of certain mutations. In keeping with this view, we found that three additional IPEX mutations near the key site that controls the domain-swap to non-swap conversion also altered the protein multimeric state, indicative of errors in protein folding. Based on these observations, we speculate that FoxP3 folding could be a novel therapeutic target; small molecules that either promote or destabilize the canonical monomeric forkhead structure or its H-H dimerization could be used to modulate Tregs in immunotherapy of cancer or autoimmunity.
Limitations of the study
An important limitation of this study is in identifying direct target sites in the genome using our model that FoxP3 binds DNA as a H-H dimer. Our results showed that H-H dimerization leads to an altered DNA specificity of FoxP3 compared to a monomeric forkhead domain, but exactly which genomic sites are directly bound by FoxP3 as a result of H-H dimerization remain unclear. Future research is necessary to understand the full repertoire of DNA sequence FoxP3 recognizes and to link it to previously reported ChIP-seq or Cut&Run-seq data. Additionally, more detailed structural model of how H-H dimerization enables recruitment of Runx1 and how this recruitment affects target gene transcription need to be investigated.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sun Hur (sun.hur@crystal.harvard.edu)
Materials Availability
All plasmids generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The atomic coordinates have been deposited in the Protein Data Bank with accession codes: 7TDW and 7TDX.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK293T cells
Cells were maintained in DMEM (High glucose, L-glutamine, Pyruvate) with 10% fetal bovine serum, 1% penicillin/streptomycin.
Naive CD4+ T Cells
Cells were isolated by using Naive CD4+ T Cell Isolation Kit (Miltenyi Biotec, Cat#130-104-453) according to the manufacturer’s instructions and maintained in complete RPMI medium (10% FBS heat-inactivated, 2mM L-Glutamine, 1mM Sodium Pyruvate, 100μM NEAA, 5mM HEPES, 0.05mM 2-ME).
Mice
B6.Foxp3R337Q/Doi mice were generated by CRISPR mutagenesis as described (Zemmour et al., 2017).Except that, instead of Cas9-encoding mRNA, a nucleoprotein complex (0.13μM Alt-R® S.p. HiFi Caspase 9 (IDT), 0.6μM sgRNA, and 0.3μM of a single-stranded recombination template carrying the desired mutation with ~60 bp of flanking homology arms, mixed in 0.2μm filtered 0.1X TE Buffer) was microinjected into the male pronucleus of fertilized mouse oocytes, then implanted into pseudo-pregnant females. The resulting mutation (GA> AG at position 1286–1287 in exon 11, NM_054039.2) encodes the R337Q missense mutation, at the very beginning of the FKHR domain. The presence of the mutation and absence of other changes in the FoxP3 coding sequence were verified by Sanger sequencing. The germline mutation was maintained by crossing onto the B6 background, and routinely carried with heterozygous female breeders. For the experiments reported here, 8-week-old R337Q males and their WT B6 littermates were analyzed. Data from two independent litters were pooled. All experiments were performed following animal protocols approved by the HMS Institutional Animal Use and Care Committee (protocols IS00000054).
METHOD DETAILS
Material Preparation
Plasmids
Mouse FoxP3 was used for all analyses. For bacterial expression of FoxP3 variants, the genes encoding mouse FoxP3ΔN (residues 188–423), FoxP3RBR-forkhead (residues 284–423), FoxP3forkhead (residues 336–423), FoxP3ZF-CC (residues 188–284) were inserted into pET50b between Xmal and HindIII sites, and into a modified pMAL-c2 vector between BamHI and Xbal sites respectively. All mutations within FoxP3 were generated by site-directed mutagenesis using Phusion High Fidelity (New England Biolabs) DNA polymerases. For crystallization, the gene encoding mouse FoxP3ΔN’ (residues 204–276+305–417) and FoxP3ΔN” (residues 204–276+315–417) were cloned into pET50b using overlap extension PCR. Mouse FoxP1RBR-forkhead (residues 433–579), mouse FoxP2ΔN (residues 353–588), mouse FoxP2RBR-forkhead (residues 447–588) and mouse FoxP4RBR-forkhead (residues 418–577) were cloned into pET50b between Xmal and HindIII sites, and modified pMAL-c2 vector between BamHI and Xbal sites respectively. Mouse Runx1 (residues 371–451) was cloned into pGEX-6P-1 between EcoRI and XhoI. Mouse NFAT1 (residues 394–680) was cloned into pET50b plasmid between Xmal and HindIII sites.
For Mammalian expression plasmids, HA-tagged mouse FoxP3 CDS was inserted into pcDNA3.1+ vector between KpnI and BamHI sites. All FoxP3 mutations including F325D, F331D, K332D, H334D, R337Q, R347H, W348D and R337Q/A372S were generated by site-directed mutagenesis using Phusion High Fidelity (New England Biolabs) DNA polymerases. The genes encoding mouse Runx1 and NFAT1 were inserted into pFlag-CMV4 vector between EcoRI and XbaI, and between NotI/XbaI sites respectively. For retroviral packaging plasmids, HA-tagged mouse FOXP3 CDS was inserted into MSCV-IRES-Thy1.1 vector. All FoxP3 mutations including F325D, F331D, K332D, H334D, R337Q, R347H, W348D, A372S, A372G, R337Q/A372S and Y373V were generated by site-directed mutagenesis using Phusion High Fidelity (New England Biolabs) DNA polymerases.
DNAs
Single-stranded DNA oligos were synthesized by IDTDNA. Double-stranded DNAs for EMSA assay, pulldown assay and restriction enzyme protection assay were annealed from single-stranded, complementary oligos. After briefly spinning down each oligonucleotide pellet, ssDNAs were dissolved in annealing buffer (10 mM Tris pH 7.5, 50 mM NaCl). Complementary ssDNAs were then mixed together in equal molar amounts, heated to 94°C for 2 minutes and gradually cool down to room temperature. For dsDNAs used in crystallization, HPLC purified single-stranded, complementary oligos were purchased from IDTDNA. After annealing, dsDNA was further purified by size-exclusion chromatography on Superdex 75 Increase 10/300 (GE Healthcare) columns in 20 mM Tris pH 7.5, 150 mM NaCl. Biotin labeled ssDNA were synthesized by IDTDNA and then dissolved in annealing buffer (10 mM Tris pH 7.5, 50 mM NaCl). Complementary biotin labeled ssDNAs were then mixed together in equal molar amounts, heated to 94°C for 2 minutes and gradually cool down to room temperature.
Protein expression and purification
All recombinant proteins in this paper were expressed in BL21(DE3) at 18°C for 16–20 hr following induction with 0.2 mM IPTG. Cells were lysed by high-pressure homogenization using an Emulsiflex C3 (Avestin). All protiens are from the Mus. musculus sequence. FoxP3ΔN, FoxP3ΔN’ and FoxP3ΔN” were expressed as a fusion protein with N-terminal His6-NusA tag. After purification using Ni-NTA agarose, the proteins were treated with HRV3C protease to cleave the His6-NusA-tag and were further purified by a combination of chromatography using HiTrap Heparin (GE Healthcare) and Hitrip SP (GE Healthcare), followed by Superdex 200 Increase 10/300 (GE Healthcare) columns in 20 mM Tris-Hcl pH 7.5, 500 mM Nacl, 2 mM DTT. FoxP3RBR-forkhead, FoxP3forkhead and NFAT1 (residues 394–680) were expressed as fusion protein with N-terminal His6-NusA tag. After purification using Ni-NTA agarose, the proteins were treated with HRV3C protease to cleave the His6-NusA-tag and were further purified by size-exclusion chromatography on Superdex 75 Increase 10/300 (GE Healthcare) column in 20 mM Tris-Hcl pH 7.5, 500 mM NaCl, 5% Glycerol, 2 mM DTT. His6-MBP fused FoxP3ΔN, FoxP3RBR-forkhead, FoxP2ΔN and His6-GST fused Runx1 (residues 371–451) were purified by a combination of chromatography on Ni-NTA agarose and Superdex 200 Increase 10/300 (GE Healthcare) columns in 20 mM Tris-Hcl pH 7.5, 500 mM NaCl, 2 mM DTT. For SEC-MALS analysis, proteins were purified without HRV3C cleavage to increase the molecular weight of the protein for accurate size determination. His6-NusA fused FoxP3 variants, FoxP1RBR-forkhead, FoxP2RBR-forkhead and FoxP4RBR-forkhead were purified by Ni-NTA agarose affinity chromatography, followed by size-exclusion chromatography on Superdex 200 Increase 10/300 (GE Healthcare) columns in 20 mM Tris-Hcl pH 7.5, 500 mM NaCl, 2mM DTT.
Electrophoretic Mobility Shift Assay (EMSA)
0.2 μM of DNA was mixed with the indicated amount of FoxP3 variants or other FoxP proteins in the buffer (20 mM Tris-Hcl pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, and 2 mM DTT). Proteins and DNAs were incubated for 30 min at 4 °C and analyzed on 3–12% gradient Bis-Tris native gels (Life Technologies) at 4 °C. After staining with Sybr Gold stain (Life Technologies), Sybr Gold fluorescence was recorded using iBright FL1000 (Invitrogen) and analyzed with iBright Analysis Software.
Multi-Angle Light Scattering (MALS)
NusA-fused FoxP3 variants, FoxP1RBR-forkhead, FoxP2RBR-forkhead and FoxP4RBR-forkhead were analyzed with Superdex 200 Increase 10/300 column (GE Healthcare), which was connected to a miniDAWN MALS detector (Wyatt Technology) and an Optilab differential refractive index (dRI) detector (Wyatt Technology). The buffer (20 mM Tris-Hcl pH 7.5, 150 mM Nacl, 2 mM DTT) was used for size-exclusion chromatography. The AKTA pure (GE Healthcare)’s UV 280 nm absorbance signal was used for concentration detection. The Optilab differential refractive index (dRI) detector measured dRI data for additional concentration analysis. Data analysis and MW calculations were performed using the ASTRA7.3.1 software (Wyatt Technology).
Crystallization of FoxP3ΔN’ and FoxP3ΔN”
Crystallization of FoxP3 was tried with various constructs and DNA, but crystals were only obtained with FoxP3ΔN’ and FoxP3ΔN”, which had C217S and C231S mutations and internal deletions in the RBR loop. FoxP3ΔN’ contained residues 204–276 and 305–417, while FoxP3ΔN” contained residues 204–276 and 315–417. Purified proteins (post Superdex 200 Increase) were mixed with purified IR-FKHM4g dsDNA (5’- AAATTTGTTTACTCGAGTAAACAAATTT, post Superdex 75 Increase 10/300) at 1:1.2 molar ratio in 20 mM Tris-Hcl pH 7.5, 150 mM Nacl, 3 mM 2-mercaptoethanol and the mixture was concentrated to 10 mg/ml (Protein concentration) using an Amicon Ultra-4 filter (3 kDa molecular-weight cutoff, Millipore). The FoxP3ΔN’–DNA complex was then mixed with the reservoir solution (0.1 M Tris-Hcl pH 8.5, 12% PEG4000) at a 1:1 volume ratio and was crystallized at 18°C by vapor diffusion using the hanging drop method. Crystals for the FoxP3ΔN”–DNA complex grew in the reservoir solution (0.5 M Lithium sulfate, 2% PEG8000) and were obtained using a similar method as with FoxP3ΔN’. To test whether the FoxP3ΔN” protein was intact in the crystal, a single crystal was picked, washed three times in 10 μl reservoir solution and then directly transferred into 10 μl of 1x SDS sample buffer prior to SDS-PAGE analysis. The protein was visualized by Krypton stain (Thermo Scientific).
Data Collection, Structure Determination, and Analysis
Crystals were cryoprotected in the reservoir solution supplemented with 25% glycerol and were flash-cooled in liquid nitrogen. The X-ray diffraction data were collected at the Advance Photon Source, beamline NECAT-24-ID-E. Diffraction data were processed using XDS (Kabsch, 2010) and HKL2000 (Otwinowski and Minor, 1997) in P6322 symmetry. The FoxP3ΔN”–DNA complex structure was solved by molecular replacement with Phaser (McCoy et al., 2007) using canonical forkhead structures of FoxN1 (PDB 6EL8) and ~14 bp DNA molecule (from PDB: 3QRF). The known structure of FoxP3 forkhead domain (PDB: 3QRF) and FoxP3 coiled-coil domain (PDB: 4I1L) were also tried as molecular replacement templates, but no solution was obtained. The MR solution for DNA showed that two 14 bp DNAs face each other through 2-fold symmetry in a way that can make one continuous 28 bp DNA, which was the biological sample used for crystallization. The data processed with P63 symmetry showed a single FoxP3 dimer bound to 28 bp DNA in the asymmetric unit, of which the structure was nearly identical to that obtained with P6322. However, data merging statistics and quality of the electron density map were inferior to those obtained with P6322. Therefore, P6322 was used for subsequent model building and refinment. The model of FoxP3ΔN”monomer with 14bp DNA was automatically built using Autobuild (Terwilliger et al., 2008), followed by manual model building using COOT (Emsley et al., 2010). Structural refinement was performed with PHENIX (Liebschner et al., 2019) and REFMAC5 in ccp4 (Murshudov et al., 2011). The FoxP3ΔN’–DNA complex structure was solved by molecular replacement using the model of the FoxP3ΔN”–DNA complex, followed by structural refinement using PHENIX and REFMAC5. The atomic coordinates of FoxP3ΔN’–DNA and FoxP3ΔN”–DNA have been deposited in the Protein Data Bank with accession codes 7TDW and 7TDX, respectively. Data collection and refinement statistics are summarized in Table S1. Figures of structure illustration were prepared using Pymol (Schrödinger, LLC).
Co-IP of HA-FoxP3 and FLAG-Runx1
HEK293T cells (in 6-well plate) were transfected with empty vector or pcDNA encoding HA-tagged FoxP3 (wild-type or mutants), or pFlag-Runx1-CMV4 respectively using Lipofectamine 3000 (Life Technologies) following manufacturer’s instructions. 48 hours later, cells were washed twice in 1XPBS (1 ml of PBS for each well) and spun down at 500g for 5 mins. Cells were resuspended in 500 μl lysis buffer (20 mM HEPES pH 7.5, 0.05% IGEPAL, 1.5 mM MgCl2, 10 mM KCl, 5 mM EDTA and 1x Mammalian protease cocktail) for 15 minutes at 4 °C and were spun down at 500g for 5 minutes at 4 °C. The supernatant was collected and centrifuged at 14000 rpm for another 5 minutes. Soluble fractions containing HA-FoxP3 and Flag-Runx1 (400 μl each) were mixed together and incubated with anti-HA magnetic beads (4 μl) (Thermo Scientific) for 1 hour at 4°C with slow rotation. To examine if FoxP3–Runx1 interaction was dependent on nucleic acids, 4 μl or 8 μl of Benzonase (Millipore) was added to the FoxP3 and Runx1 mixture for 10 minutes at 25 °C. Beads were washed three times with RIPA buffer (50 mM Tris-HCl pH 7.5, 1 mM EDTA, 1% Triton x-100, 0.5% Sodium Deoxycholate, 0.1% SDS, 150 mM NaCl, 1x Mammalian protease cocktail), followed by protein elution using SDS loading buffer and analysis by SDS-PAGE.
Co-IP of FoxP3 and DNAs
HEK293T cells were transfected with pcDNA encoding HA-tagged FoxP3 (wild-type or mutants). After 48 hours, cells were lysed using RIPA buffer (10mM Tris-HCl, pH 8.0, 1mM EDTA, 1% Triton X-100, 0.1% Sodium Deoxycholate, 0.1% SDS, 140 mM NaCl and 1x proteinase inhibitor) and treated with Benzonase (Millipore) for 30 mins. The lysate was then incubated with Anti-HA Magnetic Beads (Thermo Fisher) for 1 hour. Beads were washed three times using RIPA buffer and incubated with DNA oligos for 20 mins at room temperature. Bound DNA was recovered using proteinase K (New England Biolabs), purified using QIAquick Nucleotide Removal kit (QIAGEN) and analyzed on 10% Novex TBE Gels (Invitrogen).
MBP-FoxP3ΔN pulldown with DNAs
Purified MBP-mFoxP3ΔN or MBP-FoxP2ΔN protein (0.4 μM) was incubated with DNA (0.1 μM) in the buffer (20 mM Tirs, pH 7.5, 100 mM NaCl, 1.5 mM MgCl2) for 20 mins at RT, and was further incubated with Amylose Resin (25 μL) (New England Biolabs) with rotation for 30 mins at RT. Bound DNA was recovered using proteinase K (New England Biolabs), purified using QIAquick Nucleotide Removal kit (QIAGEN) and analyzed on 10% Novex TBE Gels (Invitrogen).
Biotin-DNAs pulldown with HA-FoxP3 variants
HEK293T cells were transfected with pcDNA-HA-FoxP3 (wild-type or mutations) and lysed using RIPA buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1% Triton X-100, 0.1% Sodium Deoxycholate, 0.1% SDS, 140 mM NaCl and 1x proteinase inhibitor). Biotin-dsDNA (1 μM) was incubated with the lysate for 1 hour. Streptavidin Agarose (25 μL)(Thermo Fisher) was added and further incubated for 30 mins with rotation. Beads were washed three times using RIPA buffer and eluted using SDS loading buffer prior to SDS-PAGE analysis.
Biotin-DNAs pulldown with MBP-mFoxP3ΔN and NFAT1 (394–680)
Purified MBP-FoxP3ΔN protein (0.2 μM) was incubated with purified NFAT1 (394–680) protein (0.008, 0.04, 0.2 μM) and Biotin-DNAs (0.2 μM) for 10 mins at room temperature in 20 mM Tris-HCl, pH 7.5, 100M Nacl, 2mM Mgcl2, 0.1% NP40, 2mM DTT. Streptavidin Agarose beads (25 μL) (Thermo Fisher) were added and incubated with rotation for 10 mins at 4°C. Beads were washed three times with incubation buffer, prior to elution and analysis by SDS-PAGE or TBE gels.
MBP-FoxP3RBR-forkhead pulldown with GST-Runx1(371–451)
Purified MBP-FoxP3RBR-forkhead protein (1 μM) was incubated with purified GST-Runx1(371–451) protein (0.5 μM) with or without 2 μM DNA for 30 mins at room temperature in RIPA buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1% Triton X-100, 0.1% Sodium Deoxycholate, 0.1% SDS, 140 mM NaCl and 1x proteinase inhibitor). Streptavidin Agarose beads (25 μL) (Thermo Fisher) were added and incubated with rotation for 30 mins at RT. Beads were washed three times with RIPA buffer, prior to elution and analysis by SDS-PAGE or TBE gels.
Crosslinking Analysis
Protein-protein crosslinking using BMOE (Thermo Scientific) was carried out according to the product manual. Briefly, BMOE was added to 2 μM of FoxP3forkhead protein to a final concentration of 100 μM in 1XPBS. After 1-hour incubation at 25°C, DTT (10 mM) was added to quench the crosslinking reaction. Samples were then analyzed by SDS-PAGE and Krypton staining (Thermo Scientific).
Thermal shift assay
WT FoxP3RBR-forkhead and mutations (19 μl, 0.25 mg/ml) were mixed with 20x Protein thermal shift dye (1 μl) (Thermo Scientific) in SEC buffer (50 mM Tris-HCl pH 7.5, 150 mM Nacl, 2 mM DTT) and aliquoted into MicroAmp Optical 96-Well Reaction Plate (Applied Biosystems). Thermal shift assay was performed using StepOnePlus Real-Time PCR System (Applied Biosystems). Briefly, temperature was increased in a step-and-hold manner from 25°C to 98°C in a 0.3°C/cycle increment and with an equilibration time of 15s at each temperature. Normalized reporter (Rn) view visualizing the rise in fluorescence throughout the temperature ramp were generated by onestep plus software. The normalized reporter (Rn), displayed on the y-axis, is calculated as the fluorescence signal from the reporter dye normalized to the fluorescence signal of the passive reference.
Restriction enzyme protection assay
IR-FKHM4g and IR-FKHM11g (0.2 μM) were pre-incubated with NFAT (0.4 μM) for 10 mins at 25°C in the buffer (50 mM Potassium Acetate, 20 mM Tris-acetate, 10 mM Magnesium Acetate, 100 ug/ml BSA, pH 7.9, 50 mM NaCl). Then FoxP3ΔN (1.6 μM for IR-FKHM4g and 3.2 μM for IR-FKHM11g) was added to the mixture. Different concentration of FoxP3 was used for the two DNA because IR-FKHM11g requires higher amount of FoxP3 for binding. After 10 mins of incubation at 25°C, restriction enzymes (XhoI, BamHI-HF or EcoRI-HF, New England Biolabs) were added and further incubated for 5 mins at 37°C. Restriction digestion was quenched by adding 1 μl proteinase K (New England Biolabs) and incubation for 2 mins at 37°C, followed by adding 0.25% SDS and 20 mM EDTA. DNA was analyzed on 20% Novex TBE Gels (Invitrogen). For restriction enzyme protection assay with FoxP3forkhead-A372S, 6.4 μM of FoxP3forkhead-A372S was used for both IR-FKHM4g and for IR-FKHM11g.
CD4+ T Cell Cultures and Retroviral Transductions
Naïve CD4+ T cells were isolated by negative selection from mouse spleens by using the isolation kit (Miltenyi Biotec) according to the manufacturer’s instructions. Cell purity was validated with >90% by FACS analysis using PE anti-CD4 (Biolegend). Cells were then activated with anti-CD3 (Biolegend), anti-CD28 (Biolegend) and 50 U/mL of IL2 (Peprotech) in complete RPMI medium (10% FBS heat-inactivated, 2 mM L-Glutamine, 1 mM Sodium Pyruvate, 100 μM NEAA, 5 mM HEPES, 0.05 mM 2-ME). Stimulation of naïve CD4+ T cells was confirmed by their increased cell sizes and expression of the activation marker CD44 (BioLegend) by FACS analysis. After 48 hours, cells were spin-infected with retrovirus containing supernatant from HEK293T cells transfected with retroviral expression plasmids (Empty MSCV-IRES-Thy1.1 vector, wildtype-FoxP3 and mutations encoding vectors) and cultured for 2~3 days in complete RPMI medium with 100 U/mL of IL2.
Flow Cytometry
For detecting of CD25 and CTLA4 expression in transduced CD4+ T cells, activated CD4+ T cells were stained with cell surface anti-CD25 (Biolegend) and Thy1.1 (Biolegend) on day 2 post retroviral infection. For intracellular staining, anti-CTLA4 (Biolegend) was applied on day 3 post retroviral infection using the Transcription Factor Staining Buffer Set (eBioscience) according to the manufacturer’s instructions. Flow cytometry data were analyzed with FlowJo software and presented as plots of mean fluorescence intensity (MFI) of CD25 and CTLA4 in cells grouped into bins of Thy1.1 intensity. Each result is representative of 3 independent experiments.
For analysis of mutant mice, single cell suspensions were obtained from murine spleens after physical dissociation with a 40 μm mesh and red blood cell lysis, and from the colonic lamina propria per (Sefik et al., 2015). After Fc blocking, extracellular staining was done in ice-cold buffer (phenol red–free DMEM, 2% FBS) for 30 min using antibodies against CD45 (30-F11; BioLegend, dilution 1:200), CD4 (RM4–5; BioLegend, dilution 1:200), CD25 (PC61; BioLegend, 1:50), TCRβ (H57–597; BioLegend, 1:150), CD44 (IM7; BioLegend, dilution 1:100). Cells were then fixed overnight at 4°C using 100 μL of Fix/Perm buffer (eBioscience), followed by permeabilization using 1X permeabilization buffer (eBioscience) for 45 minutes at room temperature in the presence of the following intracellular antibodies: FoxP3 (clone FJK-16s, Invitrogen, 1:75); CTLA4/CD152 (clone UC10–4B9, BioLegend, dilution 1:200). Data was recorded on a FACSymphonyTM flow cytometer (BD Biosciences) and analyzed using FlowJo 10 software.
FoxP3 ChIP-seq analysis
FoxP3 ChIP-seq (Kitagawa et al., 2017; Samstein et al., 2012) data was mapped to mm10 using bowtie2 (Langmead and Salzberg, 2012) and peaks were called using HOMER with an input ChIP-seq control. Overlapping FoxP3 ChIP-seq peaks were retained and ranked by signal intensity. The top 5000 ranked FoxP3 peaks were selected for a de novo motif analysis using MEME (Bailey et al., 2015). FIMO (Grant et al., 2011) was performed to identify variations in IR-FKHM motifs based on gap size and enrichment determined for non-consensus sequences from the 548 FoxP3 ChIP-seq sequences with an observed FKHM.
T cell suppression assay
Isolated naïve CD4+ T cells were activated with anti-CD3 (Biolegend), anti-CD28 (Biolegend) and 50 U/mL of IL2 (Peprotech) in complete RPMI medium (10% FBS heat-inactivated, 2 mM L-Glutamine, 1 mM Sodium Pyruvate, 100 uM NEAA, 5 mM HEPES, 0.05 mM 2-ME). After 48 hours, activated CD4+ T cells were transduced with the retroviral constructs co-expressing Thy1.1 and FoxP3 wildtype or mutants, and these cell populations were used as suppressors. Freshly isolated naïve CD4+ T cells were labelled with CellTrace™ CFSE (Invitrogen) and used as responders. Also, CD3- T cells representing APC cells were isolated using the isolation kit (Miltenyi Biotec) according to the manufacturer’s instructions. For suppression assay, the CFSE-labeled responders (5 X 104 cells) were co-cultured with WT or mutants of FoxP3 transduced suppressors and stimulated with APC cells (104 cells) and anti-CD3 (1 μg/mL) in 96-well round-bottom plates for 4 days. Proliferation ratio of the responders were calculated as a function of CFSE dye dilution by FACS analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Results were statistically analyzed using Student’s t test or a Mann-Whitney test with multiple comparisons where appropriate using Prism 6.0 (GraphPad Software, Inc). p value of < 0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgements
We thank members of the Hur lab for discussion and critical reading of the manuscript. This study was supported by NIH grants (R01AI154653 and R01AI111784 to S.H; AI150686 to C.B.; P30 CA008748 and R01 AI034206 to A.R.). J.L. was supported by an INSERM Poste d’Accueil and an Arthur Sachs scholarship, R.R. by NIH supplement AI116834-03S1. S.H. and A.R. are investigators at the Howard Hughes Medical Institute. X-ray diffraction data were collected at Advance Photon Source, beamline NECAT-24-ID-E.
Footnotes
Declaration of interests
L.H. is co-founder of and holds equity in Edelweiss Immune Inc. A.R. is an SAB member and holds equity in Sonoma Biotherapeutics, RAPT Therapeutics, Surface Oncology, Vedanta Biosciences and an SAB member of BioInvent. S.H. is a SAB member of IFM therapeutics and CJ CheilJedang Corporation.
References
- Arnett KL, Hass M, McArthur DG, Ilagan MX, Aster JC, Kopan R, and Blacklow SC (2010). Structural and mechanistic insights into cooperative assembly of dimeric Notch transcription complexes. Nat Struct Mol Biol 17, 1312–1317. 10.1038/nsmb.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvey A, van der Veeken J, Samstein RM, Feng Y, Stamatoyannopoulos JA, and Rudensky AY (2014). Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat Immunol 15, 580–587. 10.1038/ni.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. (2009). Diversity and complexity in DNA recognition by transcription factors. Science 324, 1720–1723. 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Johnson J, Grant CE, and Noble WS (2015). The MEME Suite. Nucleic Acids Res 43, W39–49. 10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandukwala Hozefa S., Wu Y, Feuerer M, Chen Y, Barboza B, Ghosh S, Stroud James C., Benoist C, Mathis D, Rao A, and Chen L (2011). Structure of a Domain-Swapped FOXP3 Dimer on DNA and Its Function in Regulatory T Cells. Immunity 34, 479–491. 10.1016/j.immuni.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzaghi F, Passerini L, and Bacchetta R (2012). Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: a paradigm of immunodeficiency with autoimmunity. Front Immunol 3, 211. 10.3389/fimmu.2012.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benayoun BA, Caburet S, and Veitia RA (2011). Forkhead transcription factors: key players in health and disease. Trends Genet 27, 224–232. 10.1016/j.tig.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, and Ochs HD (2001). The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 27, 20–21. 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, and Ramsdell F (2001). Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 27, 68–73. 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, and Bowcock AM (2000). JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest 106, R75–81. 10.1172/JCI11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Chen C, Zhang Z, Liu C-C, Johnson ME, Espinoza CA, Edsall LE, Ren B, Zhou XJ, Grant SFA, et al. (2015). DNA binding by FOXP3 domain-swapped dimer suggests mechanisms of long-range chromosomal interactions. Nucleic Acids Research 43, 1268–1282. 10.1093/nar/gku1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu YP, Chang CH, Shiu JH, Chang YT, Chen CY, and Chuang WJ (2011). Solution structure and backbone dynamics of the DNA-binding domain of FOXP1: insight into its domain swapping and DNA binding. Protein Sci 20, 908–924. 10.1002/pro.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai S, Qu L, Li J, and Chen Y (2021). Toward a mechanistic understanding of DNA binding by forkhead transcription factors and its perturbation by pathogenic mutations. Nucleic Acids Res 49, 10235–10249. 10.1093/nar/gkab807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin J (2014). Minireview: pioneer transcription factors in cell fate specification. Mol Endocrinol 28, 989–998. 10.1210/me.2014-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozdetskiy A, Cole C, Procter J, and Barton GJ (2015). JPred4: a protein secondary structure prediction server. Nucleic Acids Res 43, W389–394. 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, and Rudensky AY (2003). Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 4, 330–336. 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Gambineri E, Perroni L, Passerini L, Bianchi L, Doglioni C, Meschi F, Bonfanti R, Sznajer Y, Tommasini A, Lawitschka A, et al. (2008). Clinical and molecular profile of a new series of patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: inconsistent correlation between forkhead box protein 3 expression and disease severity. J Allergy Clin Immunol 122, 1105–1112 e1101. 10.1016/j.jaci.2008.09.027. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Roy-Chowdhuri S, Kang K, Im SH, and Rudra D (2018). The transcription factor Foxp1 preserves integrity of an active Foxp3 locus in extrathymic Treg cells. Nat Commun 9, 4473. 10.1038/s41467-018-07018-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey VL, Wilkinson JE, and Russell LB (1991). X-linked lymphoreticular disease in the scurfy (sf) mutant mouse. Am J Pathol 138, 1379–1387. [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannenhalli S, and Kaestner KH (2009). The evolution of Fox genes and their role in development and disease. Nat Rev Genet 10, 233–240. 10.1038/nrg2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori S, Nomura T, and Sakaguchi S (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061. 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- Huang Q, Liu X, Zhang Y, Huang J, Li D, and Li B (2020). Molecular feature and therapeutic perspectives of immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. J Genet Genomics 47, 17–26. 10.1016/j.jgg.2019.11.011. [DOI] [PubMed] [Google Scholar]
- Jiang L, Dai S, Li J, Liang X, Qu L, Chen X, Guo M, Chen Z, Chen L, Wei H, and Chen Y (2019). Structural basis of binding of homodimers of the nuclear receptor NR4A2 to selective Nur-responsive DNA elements. J Biol Chem 294, 19795–19803. 10.1074/jbc.RA119.010730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolma A, Yin Y, Nitta KR, Dave K, Popov A, Taipale M, Enge M, Kivioja T, Morgunova E, and Taipale J (2015). DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature 527, 384–388. 10.1038/nature15518. [DOI] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010). Xds. Acta Crystallogr D Biol Crystallogr 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa Y, Ohkura N, Kidani Y, Vandenbon A, Hirota K, Kawakami R, Yasuda K, Motooka D, Nakamura S, Kondo M, et al. (2017). Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat Immunol 18, 173–183. 10.1038/ni.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh KP, Sundrud MS, and Rao A (2009). Domain requirements and sequence specificity of DNA binding for the forkhead transcription factor FOXP3. PLoS One 4, e8109. 10.1371/journal.pone.0008109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopacki C, Pritykin Y, Rubtsov Y, Leslie CS, and Rudensky AY (2019). Transcription factor Foxp1 regulates Foxp3 chromatin binding and coordinates regulatory T cell function. Nat Immunol 20, 232–242. 10.1038/s41590-018-0291-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H-K, Chen H-M, Mathis D, and Benoist C (2017). Different molecular complexes that mediate transcriptional induction and repression by FoxP3. Nature Immunology 18, 1238–1248. 10.1038/ni.3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HK, Chen HM, Mathis D, and Benoist C (2018). FoxP3 scanning mutagenesis reveals functional variegation and mild mutations with atypical autoimmune phenotypes. Proc Natl Acad Sci U S A 115, E253–E262. 10.1073/pnas.1718599115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, and Weirauch MT (2018). The Human Transcription Factors. Cell 172, 650–665. 10.1016/j.cell.2018.01.029. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Samanta A, Song X, Iacono KT, Bembas K, Tao R, Basu S, Riley JL, Hancock WW, Shen Y, et al. (2007). FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc Natl Acad Sci U S A 104, 4571–4576. 10.1073/pnas.0700298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75, 861–877. 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano T, Gorraiz M, Lasarte-Cia A, Ruiz M, Rabal O, Oyarzabal J, Hervas-Stubbs S, Llopiz D, Sarobe P, Prieto J, et al. (2017). Blockage of FOXP3 transcription factor dimerization and FOXP3/AML1 interaction inhibits T regulatory cell activity: sequence optimization of a peptide inhibitor. Oncotarget 8, 71709–71724. 10.18632/oncotarget.17845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, and Vagin AA (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Gisselbrecht SS, Rogers JM, Hartl DL, and Bulyk ML (2013). DNA-binding specificity changes in the evolution of forkhead transcription factors. Proc Natl Acad Sci U S A 110, 12349–12354. 10.1073/pnas.1310430110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JA, Aitkenhead H, Gavard AE, Rota IA, Handel AE, Hollander GA, and Gileadi O (2020). The crystal structure of human forkhead box N1 in complex with DNA reveals the structural basis for forkhead box family specificity. J Biol Chem 295, 2948–2958. 10.1074/jbc.RA119.010365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta KR, Jolma A, Yin Y, Morgunova E, Kivioja T, Akhtar J, Hens K, Toivonen J, Deplancke B, Furlong EE, and Taipale J (2015). Conservation of transcription factor binding specificities across 600 million years of bilateria evolution. Elife 4. 10.7554/eLife.04837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, Miyachi Y, Tsukada T, and Sakaguchi S (2007). Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature 446, 685–689. 10.1038/nature05673. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pfeifer K, Prezant T, and Guarente L (1987). Yeast HAP1 activator binds to two upstream activation sites of different sequence. Cell 49, 19–27. 10.1016/0092-8674(87)90751-3. [DOI] [PubMed] [Google Scholar]
- Pricer R, Gestwicki JE, and Mapp AK (2017). From Fuzzy to Function: The New Frontier of Protein-Protein Interactions. Acc Chem Res 50, 584–589. 10.1021/acs.accounts.6b00565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez RN, Chowdhary K, Leon J, Mathis D, and Benoist C (2022). FoxP3 associates with enhancer-promoter loops to regulate Treg-specific gene expression. Sci Immunol 7, eabj9836. 10.1126/sciimmunol.abj9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter F, Wienerroither S, and Stark A (2017). Combinatorial function of transcription factors and cofactors. Curr Opin Genet Dev 43, 73–81. 10.1016/j.gde.2016.12.007. [DOI] [PubMed] [Google Scholar]
- Ramirez Ricardo N., C. K, Leon Juliette, Mathis Diane* and Benoist Christophe* (In Press). FoxP3 associates with enhancer-promoter loops to regulate Treg-specific gene expression. [DOI] [PMC free article] [PubMed]
- Rogers JM, Waters CT, Seegar TCM, Jarrett SM, Hallworth AN, Blacklow SC, and Bulyk ML (2019). Bispecific Forkhead Transcription Factor FoxN3 Recognizes Two Distinct Motifs with Different DNA Shapes. Mol Cell 74, 245–253 e246. 10.1016/j.molcel.2019.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Minton JA, Caswell R, Shield JP, Deiss D, Sumnik Z, Cayssials A, Herr M, Loew A, Lewis V, et al. (2009). Clinical heterogeneity in patients with FOXP3 mutations presenting with permanent neonatal diabetes. Diabetes Care 32, 111–116. 10.2337/dc08-1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, Leslie C, Shaffer SA, Goodlett DR, and Rudensky AY (2012). Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol 13, 1010–1019. 10.1038/ni.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, Neph S, Sabo P, Kim JM, Liao W, et al. (2012). Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 151, 153–166. 10.1016/j.cell.2012.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, Burzyn D, Ortiz-Lopez A, Lobera M, Yang J, et al. (2015). MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science 349, 993–997. 10.1126/science.aaa9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Raveh-Sadka T, Schroeder M, Unnerstall U, and Gaul U (2008). Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature 451, 535–540. 10.1038/nature06496. [DOI] [PubMed] [Google Scholar]
- Song X, Li B, Xiao Y, Chen C, Wang Q, Liu Y, Berezov A, Xu C, Gao Y, Li Z, et al. (2012). Structural and biological features of FOXP3 dimerization relevant to regulatory T cell function. Cell Rep 1, 665–675. 10.1016/j.celrep.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud JC, Wu Y, Bates DL, Han A, Nowick K, Paabo S, Tong H, and Chen L (2006). Structure of the forkhead domain of FOXP2 bound to DNA. Structure 14, 159–166. 10.1016/j.str.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Tsugawa K, Kudo M, Sugimoto K, Kobayashi I, and Ito E (2005). Low-dose cyclosporine A in a patient with X-linked immune dysregulation, polyendocrinopathy and enteropathy. Eur J Pediatr 164, 779–780. 10.1007/s00431-005-1746-4. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, and Adams PD (2008). Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr 64, 61–69. 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle LM, Pacheco D, Warfield L, Wilburn DB, Hahn S, and Klevit RE (2021). Mediator subunit Med15 dictates the conserved “fuzzy” binding mechanism of yeast transcription activators Gal4 and Gcn4. Nat Commun 12, 2220. 10.1038/s41467-021-22441-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veeken J, Glasner A, Zhong Y, Hu W, Wang ZM, Bou-Puerto R, Charbonnier LM, Chatila TA, Leslie CS, and Rudensky AY (2020). The Transcription Factor Foxp3 Shapes Regulatory T Cell Identity by Tuning the Activity of trans-Acting Intermediaries. Immunity 53, 971–984 e975. 10.1016/j.immuni.2020.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gool F, Nguyen MLT, Mumbach MR, Satpathy AT, Rosenthal WL, Giacometti S, Le DT, Liu W, Brusko TM, Anderson MS, et al. (2019). A Mutation in the Transcription Factor Foxp3 Drives T Helper 2 Effector Function in Regulatory T Cells. Immunity 50, 362–377 e366. 10.1016/j.immuni.2018.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, et al. (2001). X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet 27, 18–20. 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, et al. (2006). FOXP3 Controls Regulatory T Cell Function through Cooperation with NFAT. Cell 126, 375–387. 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Lareau CA, Ramirez RN, Rose SA, Maier B, Wroblewska A, Desland F, Chudnovskiy A, Mortha A, Dominguez C, et al. (2019). The cis-Regulatory Atlas of the Mouse Immune System. Cell 176, 897–912 e820. 10.1016/j.cell.2018.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemmour D, Charbonnier LM, Leon J, Six E, Keles S, Delville M, Benamar M, Baris S, Zuber J, Chen K, et al. (2021). Single-cell analysis of FOXP3 deficiencies in humans and mice unmasks intrinsic and extrinsic CD4(+) T cell perturbations. Nat Immunol 22, 607–619. 10.1038/s41590-021-00910-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemmour D, Pratama A, Loughhead SM, Mathis D, and Benoist C (2017). Flicr, a long noncoding RNA, modulates Foxp3 expression and autoimmunity. Proc Natl Acad Sci U S A 114, E3472–E3480. 10.1073/pnas.1700946114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, and Rudensky AY (2007). Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature 445, 936–940. 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates have been deposited in the Protein Data Bank with accession codes: 7TDW and 7TDX.