

Abstract
Background:
The appropriate dose of aspirin to lower the risk of death, myocardial infarction, and stroke and to minimize major bleeding in patients with established atherosclerotic cardiovascular disease is a subject of controversy.
Methods:
Using an open-label, pragmatic design, we randomly assigned patients with established atherosclerotic cardiovascular disease to a strategy of 81 mg or 325 mg of aspirin per day. The primary effectiveness outcome was a composite of death from any cause, hospitalization for myocardial infarction, or hospitalization for stroke, assessed in a time-to-event analysis. The primary safety outcome was hospitalization for major bleeding, also assessed in a time-to-event analysis.
Results:
A total of 15,076 patients were followed for a median of 26.2 months (interquartile range [IQR], 19.0 to 34.9). Before randomization, 13,537 (96.0% of those with available information on previous aspirin use) were already taking aspirin, and 85.3% of these patients were previously taking 81 mg of daily aspirin. Death, hospitalization for myocardial infarction, or hospitalization for stroke occurred in 590 patients (estimated percentage, 7.28%) in the 81-mg group and 569 patients (estimated percentage, 7.51%) in the 325-mg group (hazard ratio, 1.02; 95% confidence interval [CI], 0.91 to 1.14). Hospitalization for major bleeding occurred in 53 patients (estimated percentage, 0.63%) in the 81-mg group and 44 patients (estimated percentage, 0.60%) in the 325-mg group (hazard ratio, 1.18; 95% CI, 0.79 to 1.77). Patients assigned to 325 mg had a higher incidence of dose switching than those assigned to 81 mg (41.6% vs. 7.1%) and fewer median days of exposure to the assigned dose (434 days [IQR, 139 to 737] vs. 650 days [IQR, 415 to 922]).
Conclusions:
In this pragmatic trial involving patients with established cardiovascular disease, there was substantial dose switching to 81 mg of daily aspirin and no significant differences in cardiovascular events or major bleeding between patients assigned to 81 mg and those assigned to 325 mg of aspirin daily. (Funded by the Patient-Centered Outcomes Research Institute; ADAPTABLE ClinicalTrials.gov number, NCT02697916.).
Atherosclerotic cardiovascular disease remains the leading cause of illness and death in the United States, and aspirin is recommended in patients with established atherosclerotic cardiovascular disease to lower the risk of adverse health outcomes that are important to patients and clinicians.1–5 Yet there are discordant findings from observational studies and post hoc analyses of randomized, controlled trials regarding the preferred dosage of aspirin in patients with atherosclerotic cardiovascular disease, with some studies suggesting different risks and benefits depending on which dose is used.6–9 Furthermore, questions exist regarding the side-effect profile, including potential differences in major bleeding or discontinuation due to minor bleeding or dyspepsia.
Although the European Society of Cardiology clinical guidelines provide a definitive recommendation for low-dose aspirin in patients with stable disease, the American College of Cardiology–American Heart Association clinical guidelines do not provide definitive recommendations on aspirin dosage for patients with established atherosclerotic cardiovascular disease.10–12 In 2014, results from the National Cardiovascular Data Registry showed that more than 60% of patients who were discharged after myocardial infarction were treated with 325 mg of aspirin daily, and there was variation across participating centers by a factor of 25 in the proportional use of high-dose aspirin; these findings suggest that uncertainty remained about which aspirin dose clinicians should recommend.13 Given the prevalence of atherosclerotic cardiovascular disease, evidence to support a preferred dosage of aspirin will have a major public health effect on outcomes that are important to people with atherosclerotic cardiovascular disease, such as death, myocardial infarction, stroke, and major bleeding.
We designed and conducted ADAPTABLE (Aspirin Dosing: A Patient-Centric Trial Assessing Benefits and Long-Term Effectiveness), an open-label, pragmatic, randomized, controlled trial, to assess whether a strategy of aspirin at a dose of 325 mg per day would result in a lower risk of death from any cause, hospitalization for myocardial infarction, or hospitalization for stroke among patients with established atherosclerotic cardiovascular disease14 than a strategy of 81 mg per day. In doing so, we incorporated pragmatic methods and quality-by-design guiding principles to provide a real-world assessment of the comparative effectiveness of aspirin in routine cardiovascular care.15,16 ADAPTABLE was the first clinical trial to use PCORnet, the National Patient-Centered Clinical Research Network, a “network of networks” funded in 2014 by the Patient-Centered Outcomes Research Institute (PCORI) to conduct comparative-effectiveness research, with a focus on pragmatic clinical trials.17
METHODS
TRIAL DESIGN
The trial design and processes have been described previously.14 In brief, the trial was conducted in 40 centers and in one health plan participating in PCORnet (Table S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org). Patients with established atherosclerotic cardiovascular disease were identified with the use of electronic health record data at each institution through a cohort identification query (termed “computable phenotype”).18,19 The trial protocol and statistical analysis plan are available at NEJM.org. The protocol and all patient-facing materials were designed with a group of nine patient-partners (who called themselves “Adaptors”), and patient-engagement activities were coordinated by the Health eHeart Alliance (San Francisco). An independent data and safety monitoring committee also approved the trial protocol and monitored patient safety throughout the trial. All the patients provided electronic informed consent before enrollment.
TRIAL POPULATION AND RECRUITMENT STRATEGIES
Eligible patients had established atherosclerotic cardiovascular disease as defined by the inclusion and exclusion criteria in the Supplementary Appendix. Details of the recruitment strategies have been reported previously and are described in the Supplementary Appendix.14
Baseline demographic characteristics were reported by the patients, including age, sex, race, ethnic group, current tobacco use, and medication use before randomization. Baseline clinical characteristics and medical history were retrieved by means of a trial-specific query of the electronic health record (with the use of the PCORnet Common Data Model format) at enrolling health centers. All baseline medical history was recorded with a look-back period of 5 years from the date of enrollment.
RANDOMIZATION AND TRIAL TREATMENT
After providing informed consent, patients were randomly assigned through the patient portal in a 1:1 ratio to take 81 mg or 325 mg of daily aspirin, and they purchased the assigned dose over the counter. Patients received $25 remuneration for trial participation. Patients were also randomly assigned in a 1:1 ratio to follow-up visits every 3 months or every 6 months to better understand the effect of the frequency of clinical trial assessments on patient engagement and follow-up. There were no in-person visits at the trial centers during follow-up. Patients were asked to return to the patient portal for an Early Study Encounter (conducted through telephone contact by the call center for non-Internet participants) between 1 and 3 weeks after randomization to confirm adherence to the appropriate dosage and answer questions about secondary contact information.
Routine follow-up encounters then occurred every 3 or 6 months, depending on the randomized group. Internet participants were sent email reminders to complete trial visits, and non-Internet participants received telephone calls from the call center. At each encounter, patients were asked about adherence to the trial medication, the use of concomitant medications, recent hospitalizations (and primary diagnoses of hospitalizations), and patient-reported outcomes. Internet participants who had not completed a trial follow-up encounter for 6 months were converted to non-Internet participation and contacted by the call center in order to complete follow-up. When patients missed an encounter and returned for a subsequent encounter, they were asked to complete information on hospitalizations (trial outcomes) that had occurred since the last complete encounter.
CLINICAL OUTCOMES
The primary effectiveness outcome was the time to a first occurrence of any event in the composite of death from any cause, hospitalization for myocardial infarction, or hospitalization for stroke. Prespecified secondary outcomes included coronary revascularization (percutaneous coronary intervention or coronary-artery bypass grafting), the individual components of the primary outcome, and hospitalization for transient ischemic attack. The primary safety outcome was hospitalization for major bleeding with an associated blood-product transfusion. The PCORI Patient-Reported Outcomes Common Measures short form was administered through the patient portal (or call center) at baseline and every 6 months.
DATA SOURCES
Outcomes were ascertained from multiple data sources, including patient report at scheduled trial encounters, queries of electronic health record data organized according to the PCORnet Common Data Model format, linkage with data sources from PCORnet private health plan partners (Aetna, Anthem, and Humana), and linkage with fee-for-service Centers for Medicare and Medicaid Services claims data (Fig. S1). Details on the programming algorithms for outcomes, ascertainment of death, outcome validation plan, outcome reconciliation and confirmation, and censoring rules are described in the Supplementary Appendix.
STATISTICAL ANALYSIS
The planned sample size was 20,000 patients. In 2017, the sample size was reevaluated by the coordinating center (with PCORI oversight) with the use of data on trial recruitment rates and blinded, aggregate event rates, and the trial size was reduced to 15,000 patients. These changes are described in the Supplementary Appendix. Comparisons based on randomized treatment assignments were performed according to the intention-to-treat principle with the use of time-to-first-event analyses. Descriptive summaries of baseline demographic and clinical variables were generated for each randomized treatment group. Continuous baseline variables were presented as medians with interquartile ranges, and discrete variables were summarized with the use of frequencies and percentages.
Cumulative event rates (estimated percentages) were estimated at median follow-up in each treatment group with the use of Kalbfleisch and Prentice’s nonparametric estimator of the cumulative incidence function. Event-free survival rates for the primary effectiveness outcome and death from any cause were compared with the use of Cox proportional-hazards models; censoring rules are described in the Supplementary Appendix. The secondary effectiveness outcomes were compared with the use of the Fine and Gray method to take into account the competing risk of death from any cause. The proportional-hazards assumption was checked for the randomized treatment assignment with the use of weighted Schoenfeld residuals. Hazard ratios with 95% confidence intervals are presented comparing the 81-mg group with the 325-mg group, such that hazard ratios greater than 1 indicate a higher risk of events in the 81-mg group. There was no prespecified plan to adjust for multiple comparisons. Results of analyses of secondary outcomes and subgroup analyses are reported with 95% confidence intervals, without P values. The confidence intervals have not been adjusted for multiple comparisons and should not be used to infer definitive treatment effects.
Event-free survival rates for the primary safety outcome were compared with the use of the Fine and Gray method to account for the competing risk of death from any cause; the same censoring rules that were used for the analyses of the primary effectiveness outcome and death from any cause were applied. The test was two-tailed and was performed at an overall alpha level of 0.05.
Sensitivity analyses were performed to assess robustness of primary results to potential misclassification of events based on electronic phenotypes and underreporting of events that occurred outside PCORnet health systems. A landmark analysis was conducted to allow for washout of previous aspirin use. A modified per-protocol analysis was also performed with reported aspirin dose modeled as a time-dependent covariate. (See the Supplementary Appendix for details.) All analyses were performed with the use of SAS software, version 9.4 (SAS Institute).
RESULTS
PATIENTS, MEDICATION USE, AND FOLLOW-UP
Recruitment began in April 2016 and ended in June 2019. During this time, approximately 450,000 eligible patients were approached for enrollment, 50,245 visited the patient portal, 32,164 entered a personalized access code, and 15,076 were enrolled and underwent randomization (7540 to the 81-mg group and 7536 to the 325-mg group) (Fig. S3). At the time of randomization, 13,172 patients (87.4%) chose to complete trial encounters through the patient portal, and 1904 patients (12.6%) chose to complete trial encounters through the call center. Follow-up encounters occurred every 3 or 6 months, and patients were followed through June 2020. The median duration of follow-up was 26.2 months (interquartile range [IQR], 19.0 to 34.9) and was similar in the two groups. A total of 79.2% of the patients completed at least 51% of follow-up encounters through the patient portal or call center (Table S2), and data availability from the multiple data sources is shown in Table S3 and Figure S2. The data sources that were used to capture the primary effectiveness outcome are shown in Table S4.
The demographic and clinical characteristics of the patients at baseline were similar in the two groups (Table 1 and Table S5). The median age was 67.6 years, 68.7% were men, 8.7% were Black, 3.2% were Hispanic, 1.0% were Asian, and 6.5% and 6.9% had undetermined race and ethnic group, respectively. At baseline, 35.3% of the patients had previous myocardial infarction and 53.0% had previous coronary revascularization procedures within 5 years before enrollment. A total of 96.0% of the patients with available information reported that they had been taking daily aspirin before enrolling in the trial; of these patients, 85.3% reported taking 81 mg, 2.3% reported taking 162 mg, and 12.2% reported taking 325 mg (Table 1). A total of 3081 of 13,818 patients (22.3%) were taking a P2Y12 inhibitor at the time of enrollment, with 2849 of those patients (92.5%) taking clopidogrel (Table S5).
Table 1.
Characteristics of the Patients at Baseline.*
| Characteristic | 81-mg Group (N = 7540) |
325-mg Group (N = 7536) |
|---|---|---|
| Median age (IQR) — yr | 67.7 (60.7–73.6) | 67.5 (60.7–73.5) |
| Female sex — no. (%) | 2307 (30.6) | 2417 (32.1) |
| Median weight (IQR) — kg | 90.0 (78.6–103.6) | 90.0 (78.2–104.1) |
| Race — no. (%) | ||
| White | 6014 (79.8) | 5976 (79.3) |
| Black | 664 (8.8) | 647 (8.6) |
| Hispanic ethnic group — no. (%) | 249 (3.3) | 232 (3.1) |
| Medical history — no. (%)† | ||
| Previous myocardial infarction | 2674 (35.6) | 2631 (35.0) |
| Previous coronary revascularization | 4034 (53.6) | 3943 (52.4) |
| Previous percutaneous coronary intervention | 3005 (40.0) | 2941 (39.1) |
| Previous coronary-artery bypass grafting | 1786 (23.8) | 1741 (23.2) |
| Hypertension | 6264 (83.3) | 6248 (83.1) |
| Dyslipidemia | 6472 (86.1) | 6474 (86.1) |
| Diabetes mellitus | 2820 (37.5) | 2856 (38.0) |
| Atrial fibrillation | 605 (8.0) | 628 (8.4) |
| Congestive heart failure | 1718 (22.8) | 1786 (23.8) |
| Chronic kidney disease | 1315 (17.5) | 1333 (17.7) |
| Peripheral artery disease | 1706 (22.7) | 1787(23.8) |
| Previous clinically significant gastrointestinal bleeding | 455 (6.1) | 495 (6.6) |
| Previous intracranial hemorrhage | 98 (1.3) | 110(1.5) |
| Current smoker — no. (%) | 696 (9.2) | 686 (9.1) |
| Aspirin use before trial | ||
| Missing data — no. (%) | 404 (5.4) | 569 (7.6) |
| No — no. (%) | 286 (3.8) | 280 (3.7) |
| Yes — no. (%) | 6850 (90.8) | 6687 (88.7) |
| 81 mg — no./total no. (%) | 5823/6850 (85.0) | 5724/6687 (85.6) |
| 162 mg — no./total no. (%) | 168/6850 (2.5) | 142/6687 (2.1) |
| 325 mg — no./total no. (%) | 845/6850 (12.3) | 812/6687 (12.1) |
| Other dose — no./total no. (%) | 14/6850 (0.2) | 9/6687 (0.1) |
Age, sex, race, ethnic group, tobacco use, and medication use before randomization were reported by the patients. A total of 980 patients chose “Prefer not to say” for race, and 1042 patients chose “Prefer not to say” for ethnic group. Percentages may not total 100 because of rounding. IQR denotes interquartile range.
A total of 7520 of 7540 patients (99.7%) in the 81-mg group and 7519 of 7536 patients (99.8%) in the 325-mg group had available medical history through electronic health record data. Percentages for medical history are based on the number of patients in each group with available medical history.
A total of 614 of 15,076 patients (4.1%; 221 of 7540 patients [2.9%] in the 81-mg group and 393 of 7536 patients [5.2%] in the 325-mg group) requested to withdraw consent from the trial. An additional 391 patients (2.6%; 133 patients [1.8%] in the 81-mg group and 258 patients [3.4%] in the 325-mg group) asked for limited participation in trial activities.
EFFECTIVENESS OUTCOMES
Death from any cause, hospitalization for myocardial infarction, or hospitalization for stroke (primary effectiveness outcome) occurred in 590 patients (estimate at median follow-up, 7.28%) in the 81-mg group and 569 patients (estimate at median follow-up, 7.51%) in the 325-mg group (hazard ratio, 1.02; 95% confidence interval [CI], 0.91 to 1.14) (Figure 1A and Table 2). Death from any cause occurred in 315 patients (estimate at median follow-up, 3.80%) in the 81-mg group and 357 patients (estimate at median follow-up, 4.43%) in the 325-mg group (hazard ratio, 0.87; 95% CI, 0.75 to 1.01) (Fig. S4). Hospitalizations for myocardial infarction and stroke (individual components of the primary effectiveness outcome) and key secondary outcomes were similar in the two groups. Mean scores for patient-reported outcome measures were similar in the two groups at baseline and follow-up (Table S6). The treatment effect on the primary effectiveness outcome appeared similar across the prespecified subgroups (Fig. S5). There was no difference in treatment effect according to the secondary randomization to 3 months or 6 months of follow-up (Table S7).
Figure 1. Time-to-Event Curves for the Primary Effectiveness Outcome and Primary Safety Outcome, According to Randomized Treatment Group.
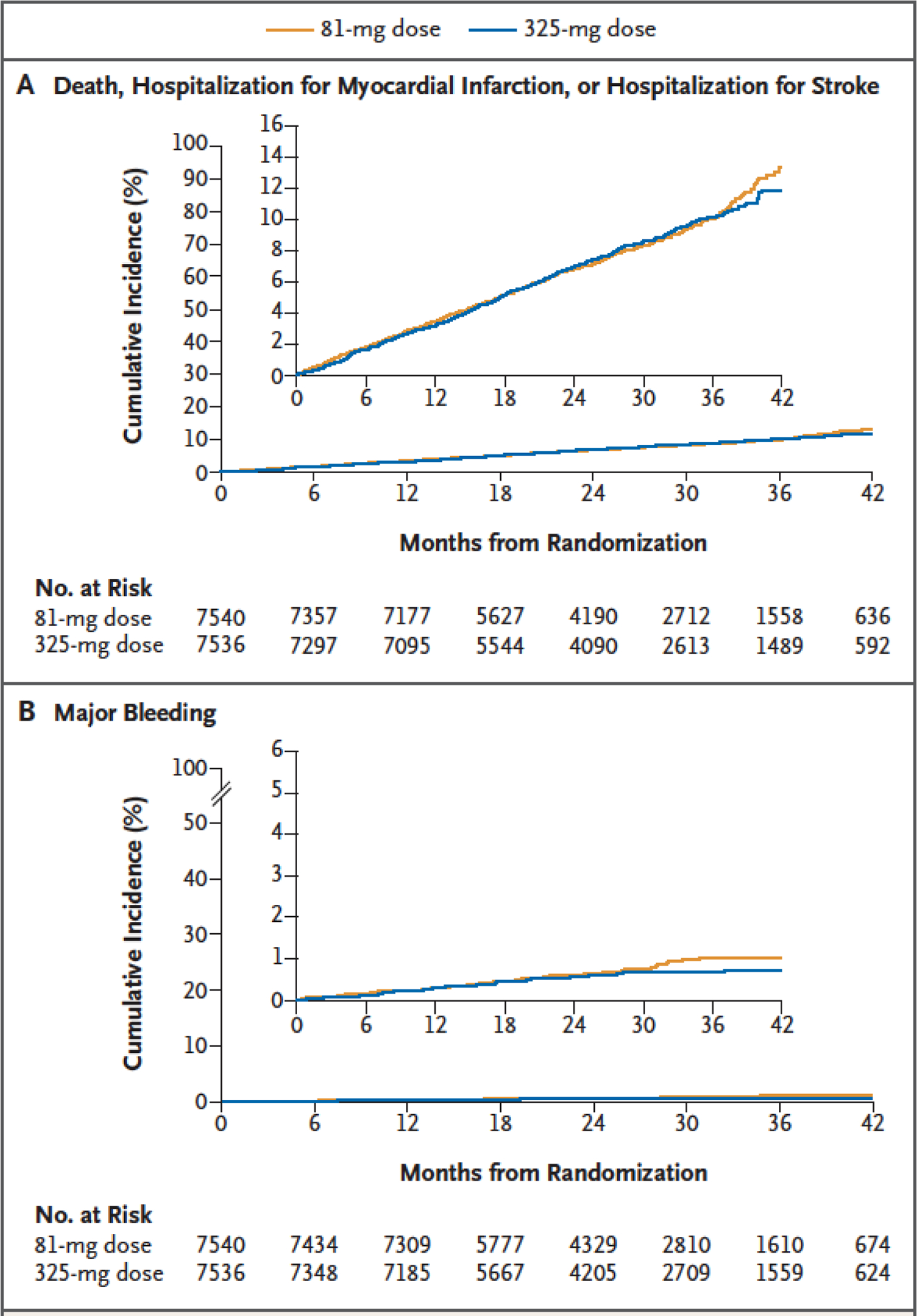
Panel A shows the cumulative incidence of death from any cause, hospitalization for myocardial infarction, or hospitalization for stroke (primary effectiveness outcome). Panel B shows the cumulative incidence of hospitalization for major bleeding that was associated with a blood-product transfusion (primary safety outcome). In each panel, the inset shows the same data on an enlarged y axis.
Table 2.
Primary Effectiveness Outcome, Key Secondary Effectiveness Outcomes, and Primary Safety Outcome.*
| Outcome | 81-mg Group | 325-mg Group | Hazard Ratio (95% CI) | P Value |
|---|---|---|---|---|
| events (estimated percentage) | ||||
| Primary effectiveness outcome: death from any cause, hospitalization for MI, or hospitalization for stroke | 590 (7.28) | 569 (7.51) | 1.02 (0.91–1.14) | 0.75 |
| Death from any cause | 315 (3.80) | 357 (4.43) | 0.87 (0.75–1.01) | |
| Hospitalization for Ml | 228 (2.99) | 213 (2.87) | 1.06 (0.88–1.27) | |
| Hospitalization for stroke | 102 (1.23) | 92 (1.27) | 1.09 (0.82–1.45) | |
| Occurrence of PCI or CABG | 471 (6.05) | 446 (5.96) | 1.04 (0.92–1.19) | |
| Hospitalization for transient ischemic attack | 20 (0.23) | 25 (0.35) | 0.79 (0.44–1.42) | |
| Primary safety outcome: hospitalization for major bleeding with associated blood-product transfusion | 53 (0.63) | 44 (0.60) | 1.18 (0.79–1.77) | 0.41 |
Events include data from electronic health record data, Centers for Medicare and Medicaid Services claims, private insurance claims, and confirmed patient-reported outcomes. Estimated percentages are calculated at median follow-up (26.2 months) with the use of the Kalbfleisch and Prentice cumulative incidence function estimator. CABG denotes coronary-artery bypass grafting, CI confidence interval, MI myocardial infarction, and PCI percutaneous coronary intervention.
SAFETY OUTCOMES
Hospitalization for major bleeding with an associated blood-product transfusion (primary safety outcome) occurred in 53 patients (estimate at median follow-up, 0.63%) in the 81-mg group and 44 patients (estimate at median follow-up, 0.60%) in the 325-mg group (hazard ratio, 1.18; 95% CI, 0.79 to 1.77) (Figure 1B and Table 2).
ADHERENCE TO TRIAL MEDICATION
Aspirin discontinuation was reported by 7.0% of the patients assigned to the 81-mg dosing strategy and 11.1% of those assigned to the 325-mg dosing strategy, and dose switching was reported by 7.1% in the 81-mg group and 41.6% in the 325-mg group (Table 3). The reasons for discontinuation are listed according to treatment group in the Supplementary Appendix. The median number of days of exposure to the assigned aspirin dose was lower in the 325-mg group than in the 81-mg group (434 days [IQR, 139 to 737] vs. 650 days [IQR, 415 to 922]), as was the median number of days of exposure to any aspirin dose (646 days [IQR, 412 to 922] vs. 670 days [IQR, 439 to 944]) (Table 3). As reported at trial encounters, the incidence of dose switching diverged early after randomization in the 325-mg group and continued to separate from the 81-mg group over the course of the trial (Fig. S6).
Table 3.
Adherence to Trial Medication, According to Treatment Group.*
| Outcome | Overall | 81-mg Group | 325-mg Group |
|---|---|---|---|
| Patients who switched aspirin doses — no./total no. (%)† | 3479/14,391 (24.2) | 516/7261 (7.1) | 2963/7130 (41.6) |
| Patients who discontinued aspirin — no./total no. (%)‡ | 1299/14,391 (9.0) | 506/7261 (7.0) | 793/7130 (11.1) |
| Median days of exposure to assigned aspirin dose (IQR) | 551 (304–834) | 650 (415–922) | 434 (139–737) |
| Median days of exposure to any aspirin dose (IQR) | 658 (426–932) | 670 (439–944) | 646 (412–922) |
Trial medication adherence is based on patient report at visits every 3 or 6 months. All reported information from one completed visit to the next was used. Dose switching or discontinuation that occurred after the last reported aspirin dose would have been missed (8.2% of the patients in the 81-mg group and 9.6% of those in the 325-mg group were alive at the end of the trial but did not complete an end-of-trial visit).
Dose switching is defined as reporting a dose of aspirin different from the randomized dose at one or more postrandomization trial encounters.
If Discontinuation is defined as reporting “No” to the trial question “Are you regularly taking aspirin?” at one or more postrandomization trial encounters.
SENSITIVITY ANALYSES
To assess robustness of results to potential underreporting and misclassification of outcomes, sensitivity analyses were performed and showed that there were no changes in results in analyses that accounted for potential missing data when patients moved or left the enrolling health system (Table S8 and Fig. S7) or that accounted for potential misclassification of outcomes (Fig. S8). In addition, a prespecified landmark analysis that omitted outcomes during the first 10 days of follow-up after randomization to account for events that were probably related to previous aspirin use showed no change in the results for the primary effectiveness outcome (Table S8). However, when dose was used as a time-dependent covariate (regardless of randomized dose), patients who took 81 mg had a higher risk of death from any cause, hospitalization for myocardial infarction, or hospitalization for stroke than those who took 325 mg (hazard ratio, 1.25; 95% CI, 1.10 to 1.43).
DISCUSSION
In this large, open-label, pragmatic, multicenter trial, which was performed remotely and integrated into the routine care of patients with established atherosclerotic cardiovascular disease, there were no significant differences in effectiveness or safety outcomes between a strategy of 81 mg of aspirin daily and a strategy of 325 mg daily. Over the course of the trial, patients who were assigned to 325 mg of aspirin were more likely to switch to 81 mg than vice versa, and those assigned to 325 mg also discontinued aspirin more frequently. Dose switching or discontinuation may have biased the results toward the null. The sensitivity analysis with dose as a time-varying covariate indicates that patients who continued to take a 325-mg dose had a lower event rate over time, but as with any postrandomization analysis, this analysis has many biases that include patient behavior, clinician perspectives, and adverse events that may affect outcomes.
Data published in 2014 from the National Cardiovascular Data Registry that showed that approximately 60% of patients discharged from the hospital after myocardial infarction were treated with 325 mg of daily aspirin suggested that equipoise existed for the use of the two trial doses at the time of trial design.12 Of our enrolled patients previously taking aspirin, the majority (85.3%) were taking 81 mg of daily aspirin before randomization. Trial patients frequently switched their assigned dose, and this was more prominent in the 325-mg group. Multiple reasons for switching dose were possible, such as patient preference (including preference for the aspirin dose taken before the trial), clinician practices (including incorporation of European Society of Cardiology guidelines), the development of nuisance bruising and bleeding, and the development of concurrent illnesses (e.g., atrial fibrillation, cancer, or liver disease). The publication of the 2016 American College of Cardiology–American Heart Association Guideline Focused Update on Duration of Dual Antiplatelet Therapy in Patients with Coronary Artery Disease20 and a Class IB recommendation for low-dose aspirin in patients treated with a P2Y12 inhibitor may have also influenced dose switching in patients who were treated with long-term dual antiplatelet therapy and in others who underwent percutaneous coronary intervention during the course of the trial.
Patients also discontinued the 325-mg dosing strategy more often than the 81-mg dosing strategy. Published reports about a lack of effectiveness in the primary prevention of cardiovascular events did contribute to discontinuation of aspirin during the trial (in both groups), as reported in the Supplementary Appendix.2–4 Although the trial teams produced patient-facing materials and clinician-facing materials to limit nonadherence to the trial drug,21 it is often difficult to combat misinformation or public confusion regarding the role of aspirin to prevent secondary outcomes in people with atherosclerotic cardiovascular disease.
This trial was the first demonstration project for pragmatic clinical trials within PCORnet and has notable features that have important implications for large, pragmatic clinical trials. Leveraging new electronic methods to identify a large pool of potential participants at a health-system level, the trial recruited more than 15,000 patients from only 40 centers in the United States with the use of multimodal, low-touch recruitment strategies, electronic informed consent, and a patient-specific access code that linked the patients to health-system data. The trial was created with a group of nine patient-partners who guided the trial from start to finish; reviewed the protocol, protocol amendments, and all patient-facing materials; and provided in-depth, patient-centered decision making on all important matters. The trial was truly embedded in real-world practice, because electronic health record data from the PCORnet Common Data Model, patient-reported information, and other electronic health-related data were incorporated to reduce the burden of research on patients and sites and reduce the total costs of the trial. Lessons that were learned from this pivotal demonstration trial include the feasibility of identifying a large cohort of eligible patients; engaging, recruiting, and randomly assigning these patients to take daily aspirin doses; and performing follow-up with new methods alongside strong engagement of patient-partners. With the use of previous guidance15,16 and lessons from this trial for future remote or decentralized trials, it will be important to expand clinician and patient engagement by providing value over time to participants through return of results or other high-value methods to ensure long-term retention and adherence to trial protocols.
Several important limitations must be acknowledged, including the open-label design and the inclusion of patients who primarily took 81 mg of daily aspirin before the trial. Patients commonly switched their randomized dose, and it is plausible that patient or clinician biases or perceptions about the relative risks and benefits of aspirin dosing may have led to dose changes over time. Despite attempts to identify and enroll a diverse population of patients with atherosclerotic cardiovascular disease, the enrollment of women and traditionally underrepresented groups lagged behind our goals and mirrored enrollment in previous traditional cardiovascular studies. In addition, there was a relatively modest duration of follow-up for a comparative-effectiveness trial for cardiovascular outcomes. The safety outcome of hospitalization for major bleeding with an associated blood-product transfusion had very stringent criteria, the overall incidence of events was low, and we did not assess nonserious or minor bleeding adverse events. Finally, although clinical event reporting was systematic, the classification of outcomes differed from traditional clinical outcome adjudication and required a formal validation plan that resulted in one third of events being clinically reviewed.
In this trial, there were no significant differences in the primary effectiveness or safety outcomes between the two aspirin dosing strategies among patients with established atherosclerotic cardiovascular disease. As interest grows for real-world evidence, the trial provides a demonstration that randomized clinical trials can leverage electronic health record data, direct-to-patient methods, and patient-reported outcomes to address important, patient-centered questions. Future trials may take lessons learned from this one as fit for purpose, balancing trade-offs on precision versus generalizability as well as costs. In conclusion, a strategy of 81 mg of daily aspirin had similar effectiveness as a strategy of 325 mg in patients with established atherosclerotic cardiovascular disease, and long-term adherence was better with the 81-mg dosing strategy.
Supplementary Material
Acknowledgments
We thank Bill Larsen — a beloved patient-partner from Gainesville, Florida, who died during the course of the trial — for his participation, voice, and thoughtful demeanor. In Bill’s honor, we encourage future study teams to engage patient-partners for their valuable insight and meaningful contributions. We thank Robert M. Califf, M.D. — who was instrumentally involved in the initial conception and design of the trial before he transitioned to the Food and Drug Administration to be Principal Deputy Commissioner and later Commissioner — for his mentorship, guidance, and vision throughout this trial. We also thank Elizabeth E.S. Cook, B.A., for her editorial support with an earlier version of the manuscript.
Supported by a PCORI Award (ASP-1502-27029).
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
All statements in this report, including its findings and conclusions, are solely those of the authors and do not necessarily represent the views of the Patient-Centered Outcomes Research Institute (PCORI), its Board of Governors, or its Methodology Committee.
A data sharing statement provided by the authors is available with the full text of this article at NEJM.org.
Collaborators
ADAPTABLE Team:
Robert A Harrington, Russell Rothman, Adrian F Hernandez, W Schuyler Jones, Lesley H Curtis, Greg Marcus, Henry Cruz, Patient Partner, Jacqueline Alikhaani, Matthew Roe, Lisa Berdan, Holly Robertson, Amber Sharlow, Brad Hammill, Debra Harris, Laura Qualls, Hillary Mulder, Lisa Wruck, Michael Pencina, Guillaume Marquis-Gravel, Clyde Yancy, Dave Demets, Judith Hochman, Bernard Gersh, Alice Jacobs, Debbe McCall, Hugo Campos, Desiree Davidson, Kevin Edgley, Greg Merritt, Linda Brown, Henry Cruz, Nadine Zemon, Tom McCormick, Ken Gregoire, Abel Kho, Russ Waitman, Veronique Roger, Rainu Kaushal, Elizabeth Shenkman, Kathleen McTigue, Douglas Bell, Thomas Carton, A Kho, M Nodal, D Fintel, F Ahmad, A Williams, R C Shah, A Flores, M Jansen, T Polonsky, D Hood, J French, P Dexter, D Riley, N Smith, S Girotra, R Waitman, S Chandaka, T McMahon, K Greening, Y Murr, K Munshi, H Sandoval, K Gupta, L Verhagen, T Foss, J VanWormer, K Osinski, A Schwarz, S Cornell, M Berendt, J Whittle, A Mosa, V Mandhadi, L Lawrence, A Kumar, D Mehr, J McClay, P Guda, A Wolfe, C Geary, K Ostlund, L Larson, R Harper, J Campbell, L Manuel, O Suarez, J Polanco, A Tirado-Ramos, S Tsai, J Cai, M Bell, P Reeder, T Bosler, K Wilkinson, S Antony, S Das, K Hanson, C Rodgers, E Ebere, S Bradley, M Miedema, R Thiel, B Lemke, I Haller, C Benziger, S Rea, D Ward, H Maestas, N Garbe, L Evans, P Haug, K Knowlton, V Roger, E Koepsell, S Brue, J Bauman, B Bucknell, H Jouni, M Jindra, J Harper, M Bright, B Lampert, S Fernandez, J Ryan, S Brougher, M Harris, A Fishstom, L Ferguson, D Braswell, K Shah, B Nallamothu, C Friedman, D Williams, P Farrehi, W S Jones, J Curtis, J Hamill, B O’Brien, M B Summers, X Idriss, W Pohlman, K Thompson, D Dewalt, R Bradford, T Barber, D Crenshaw, K Goggins, W Hucke, S Kripalani, S McCrate, J Moore, D Muñoz, C O’Donnell, C Pritchett, C Roumie, R Servis, K Worley, J Borgman, L Doomy, K Blinson, G Rosenthal, L Zhou, O A Sandu, Y Goldberg, E Fass, D McKee, J Lin, C Pulgarin, S Blecker, R Kaushal, A Cohen, N Goldberg, S Chaudhary, A LaMar, T Haque, K LaScalea, R Kim, T Campion, B Shenkman, B Roth, D Bright, J Hensley, T Chou, J-A Norton, C Garrett, C Pepine, D Anderson, E Handberg, B Johnson, T Robinson, D Allen, M Warrington, H Seifein, H Noel, V K Kasi, D Gumas, H Lehmann, M Gauvey-Kern, D Ford, T Metkus, J McCullough, K Confer, C Chuang, J Kraschnewski, M Weiner, J Turella, A Paranjape, K McTigue, N Cappella, P Kant, A Arita, E Klawson, S Jain, A Huffman, C Moynier, T Greimes, B Steinberg, R Hess, M Zachariah, D Bell, G Fonarow, T Carton, B Nauman, L Rudov, L Hall, L Clariday, R Baker, P McCullough, C Parke, D Rachal, R Jenkins, S Cohen, G Liu, M Effron, R Re, A Irimpen, M Pletcher, M Faulkner, M Cziraky, Q Shi, A Marshall, E R Kennedy, K Haynes, V Nair
A complete list of the ADAPTABLE investigators is provided in the Supplementary Appendix, available at NEJM.org.
Contributor Information
W Schuyler Jones, Duke Clinical Research Institute, Duke University, Durham, North Carolina.
Hillary Mulder, Duke Clinical Research Institute, Duke University, Durham, North Carolina.
Lisa M Wruck, Duke Clinical Research Institute, Duke University, Durham, North Carolina.
Michael J Pencina, Duke Clinical Research Institute, Duke University, Durham, North Carolina.
Sunil Kripalani, Vanderbilt University Medical Center, Nashville, New Orleans.
Daniel Muñoz, Vanderbilt University Medical Center, Nashville, New Orleans.
David L Crenshaw, Vanderbilt University Medical Center, Nashville, New Orleans.
Mark B Effron, Ochsner Health, New Orleans.
Richard N Re, Ochsner Health, New Orleans.
Kamal Gupta, University of Kansas Medical Center, Kansas City, Pennsylvania.
R David Anderson, University of Florida, Gainesville, Pennsylvania.
Carl J Pepine, University of Florida, Gainesville, Pennsylvania.
Eileen M Handberg, University of Florida, Gainesville, Pennsylvania.
Brittney R Manning, University of Florida, Gainesville, Pennsylvania.
Sandeep K Jain, University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania.
Saket Girotra, University of Iowa, Iowa City, Wisconsin.
Danielle Riley, University of Iowa, Iowa City, Wisconsin.
Darren A DeWalt, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina.
Jeff Whittle, Medical College of Wisconsin, Milwaukee, Wisconsin.
Ythan H Goldberg, Albert Einstein College of Medicine, Bronx, New York.
Veronique L Roger, Mayo Clinic, Rochester, Minnesota.
Rachel Hess, University of Utah School of Medicine, Salt Lake City.
Catherine P Benziger, Essentia Health Heart and Vascular Center, Duluth, Minnesota.
Peter Farrehi, University of Michigan, Ann Arbor.
Li Zhou, Wake Forest University School of Medicine, Winston-Salem, North Carolina.
Daniel E Ford, Johns Hopkins University School of Medicine, Baltimore.
Kevin Haynes, HealthCore, Wilmington, DE.
Jeffrey J VanWormer, Marshfield Clinic Research Institute, Marshfield, Wisconsin.
Kirk U Knowlton, Intermountain Medical Center Heart Institute, Salt Lake City.
Jennifer L Kraschnewski, Penn State College of Medicine, Hershey, Pennsylvania.
Tamar S Polonsky, University of Chicago Medicine, Chicago.
Dan J Fintel, Northwestern University Feinberg School of Medicine, Chicago.
Faraz S Ahmad, Northwestern University Feinberg School of Medicine, Chicago.
James C McClay, University of Nebraska Medical Center, Omaha, California.
James R Campbell, University of Nebraska Medical Center, Omaha, California.
Douglas S Bell, University of California, Los Angeles, Los Angeles, California.
Gregg C Fonarow, University of California, Los Angeles, Los Angeles, California.
Steven M Bradley, Allina Health and Minneapolis Heart Institute, Minneapolis, Minnesota.
Anuradha Paranjape, Temple University, Philadelphia, Pennsylvania.
Matthew T Roe, Duke Clinical Research Institute, Duke University, Durham, North Carolina.
Holly R Robertson, Duke Clinical Research Institute, Duke University, Durham, North Carolina.
Lesley H Curtis, Duke Clinical Research Institute, Duke University, Durham, North Carolina.
Amber G Sharlow, Duke Clinical Research Institute, Duke University, Durham, North Carolina.
Lisa G Berdan, Duke Clinical Research Institute, Duke University, Durham, North Carolina.
Bradley G Hammill, Duke Clinical Research Institute, Duke University, Durham, North Carolina.
Debra F Harris, Duke Clinical Research Institute, Duke University, Durham, North Carolina.
Laura G Qualls, Duke Clinical Research Institute, Duke University, Durham, North Carolina.
Guillaume Marquis-Gravel, Duke Clinical Research Institute, Duke University, Durham, North Carolina.
Madelaine F Modrow, University of California, San Francisco, San Francisco, California.
Gregory M Marcus, University of California, San Francisco, San Francisco, California.
Thomas W Carton, Louisiana Public Health Institute, New Orleans.
Elizabeth Nauman, Louisiana Public Health Institute, New Orleans.
Lemuel R Waitman, University of Missouri School of Medicine, Columbia.
Abel N Kho, Northwestern University Feinberg School of Medicine, Chicago.
Elizabeth A Shenkman, University of Florida, Gainesville, Pennsylvania.
Kathleen M McTigue, University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania.
Rainu Kaushal, Weill Cornell Medicine and New York-Presbyterian Hospital, New York.
Frederick A Masoudi, University of Colorado School of Medicine, Anschutz Medical Campus, Aurora.
Elliott M Antman, Brigham and Women’s Hospital, Harvard Medical School, Boston.
Desiree R Davidson, Chicago.
Kevin Edgley, St. Joseph, MO.
James G Merritt, Brighton, MI.
Linda S Brown, Columbia, TN.
Doris N Zemon, Alachua, FL.
Thomas E McCormick, 3rd, Columbia, MD.
Jacqueline D Alikhaani, North Hills, CA.
Kenneth C Gregoire, Metairie, LA.
Russell L Rothman, Vanderbilt University Medical Center, Nashville, New Orleans.
Robert A Harrington, Stanford University School of Medicine, Stanford, California.
Adrian F Hernandez, Duke Clinical Research Institute, Duke University, Durham, North Carolina.
REFERENCES
- 1.Virani SS, Alonso A, Benjamin EJ, et al. Heart disease and stroke statistics — 2020 update: a report from the American Heart Association. Circulation 2020;141(9): e139–e596. [DOI] [PubMed] [Google Scholar]
- 2.The ASCEND Study Collaborative Group. Effects of aspirin for primary prevention in persons with diabetes mellitus. N Engl J Med 2018;379:1529–39. [DOI] [PubMed] [Google Scholar]
- 3.Gaziano JM, Brotons C, Coppolecchia R, et al. Use of aspirin to reduce risk of initial vascular events in patients at moderate risk of cardiovascular disease (ARRIVE): a randomised, double-blind, placebo-controlled trial. Lancet 2018;392: 1036–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McNeil JJ, Wolfe R, Woods RL, et al. Effect of aspirin on cardiovascular events and bleeding in the healthy elderly. N Engl J Med 2018;379:1509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antithrombotic Trialists’ (ATT) Collaboration. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009;373:1849–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xian Y, Wang TY, McCoy LA, et al. Association of discharge aspirin dose with outcomes after acute myocardial infarction: insights from the Treatment with ADP Receptor Inhibitors: Longitudinal Assessment of Treatment Patterns and Events after Acute Coronary Syndrome (TRANSLATE-ACS) Study. Circulation 2015; 132:174–81. [DOI] [PubMed] [Google Scholar]
- 7.The CURRENT–OASIS 7 Investigators. Dose comparisons of clopidogrel and aspirin in acute coronary syndromes. N Engl J Med 2010;363:930–42. [DOI] [PubMed] [Google Scholar]
- 8.Kohli P, Udell JA, Murphy SA, et al. Discharge aspirin dose and clinical outcomes in patients with acute coronary syndromes treated with prasugrel versus clopidogrel: an analysis from the TRITON-TIMI 38 study (trial to assess improvement in therapeutic outcomes by optimizing platelet inhibition with prasugrel-thrombolysis in myocardial infarction 38). J Am Coll Cardiol 2014;63:225–32. [DOI] [PubMed] [Google Scholar]
- 9.Mahaffey KW, Huang Z, Wallentin L, et al. Association of aspirin dose and vorapaxar safety and efficacy in patients with non-ST-segment elevation acute coronary syndrome (from the TRACER Trial). Am J Cardiol 2014;113:936–44. [DOI] [PubMed] [Google Scholar]
- 10.Amsterdam EA, Wenger NK, Brindis RG, et al. 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014;130:2354–94. [DOI] [PubMed] [Google Scholar]
- 11.Smith SC Jr, Benjamin EJ, Bonow RO, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation. Circulation 2011;124:2458–73. [DOI] [PubMed] [Google Scholar]
- 12.Montalescot G, Sechtem U, Achenbach S, et al. 2013 ESC guidelines on the management of stable coronary artery disease: the Task Force on the Management of Stable Coronary Artery Disease of the European Society of Cardiology. Eur Heart J 2013;34:2949–3003. [DOI] [PubMed] [Google Scholar]
- 13.Hall HM, de Lemos JA, Enriquez JR, et al. Contemporary patterns of discharge aspirin dosing after acute myocardial infarction in the United States: results from the National Cardiovascular Data Registry (NCDR). Circ Cardiovasc Qual Outcomes 2014;7:701–7. [DOI] [PubMed] [Google Scholar]
- 14.Marquis-Gravel G, Roe MT, Robertson HR, et al. Rationale and design of the Aspirin Dosing: A Patient-centric Trial Assessing Benefits and Long-term Effectiveness (ADAPTABLE) Trial. JAMA Cardiol 2020;5:598–607. [DOI] [PubMed] [Google Scholar]
- 15.Ford I, Norrie J. Pragmatic trials. N Engl J Med 2016;375:454–63. [DOI] [PubMed] [Google Scholar]
- 16.Loudon K, Treweek S, Sullivan F, Donnan P, Thorpe KE, Zwarenstein M. The PRECIS-2 tool: designing trials that are fit for purpose. BMJ 2015;350:h2147. [DOI] [PubMed] [Google Scholar]
- 17.Hernandez AF, Fleurence RL, Rothman RL. The ADAPTABLE Trial and PCORnet: shining light on a new research paradigm. Ann Intern Med 2015;163: 635–6. [DOI] [PubMed] [Google Scholar]
- 18.Ahmad FS, Ricket IM, Hammill BG, et al. Computable phenotype implementation for a national, multicenter pragmatic clinical trial: lessons learned from ADAPTABLE. Circ Cardiovasc Qual Outcomes 2020;13(6):e006292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fishman E, Barron J, Dinh J, et al. Validation of a claims-based algorithm identifying eligible study subjects in the ADAPTABLE pragmatic clinical trial. Contemp Clin Trials Commun 2018;12:154–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levine GN, Bates ER, Bittl JA, et al. 2016 ACC/AHA guideline focused update on duration of dual antiplatelet therapy in patients with coronary artery disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2016;68:1082–115. [DOI] [PubMed] [Google Scholar]
- 21.Marquis-Gravel G, Roe MT, Harrington RA, Muñoz D, Hernandez AF, Jones WS. Revisiting the role of aspirin for the primary prevention of cardiovascular disease. Circulation 2019;140:1115–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.