

Abstract
Ketamine is a well-characterized NMDA receptor (NMDAR) antagonist, although the relevance of this pharmacology to its rapid (within hours of administration) antidepressant actions, which depend on mechanisms convergent with strengthening of excitatory synapses, is unclear. Activation of synaptic NMDARs is necessary for the induction of canonical long-term potentiation (LTP) leading to a sustained expression of increased synaptic strength. We tested the hypothesis that induction of rapid antidepressant effects requires NMDAR activation, by using behavioral pharmacology, western blot quantification of hippocampal synaptoneurosomal protein levels, and ex vivo hippocampal slice electrophysiology in male mice. We found that ketamine exerts an inverted U-shaped dose-response in antidepressant-sensitive behavioral tests, suggesting that an excessive NMDAR inhibition can prevent ketamine's antidepressant effects. Ketamine's actions to induce antidepressant-like behavioral effects, up-regulation of hippocampal AMPAR subunits GluA1 and GluA2, as well as metaplasticity measured ex vivo using electrically-stimulated LTP, were abolished by pretreatment with other non-antidepressant NMDAR antagonists, including MK-801 and CPP. Similarly, the antidepressant-like actions of other putative rapid-acting antidepressant drugs (2R,6R)-hydroxynorketamine (ketamine metabolite), MRK-016 (GABAAα5 negative allosteric modulator), and LY341495 (mGlu2/3 receptor antagonist) were blocked by NMDAR inhibition. Ketamine acted synergistically with an NMDAR positive allosteric modulator to exert antidepressant-like behavioral effects and activation of the NMDAR subunit GluN2A was necessary and sufficient for such relevant effects. We conclude rapid-acting antidepressant compounds share a common downstream NMDAR-activation dependent effector mechanism, despite variation in initial pharmacological targets. Promoting NMDAR signaling or other approaches that enhance NMDAR-dependent LTP-like synaptic potentiation may be an effective antidepressant strategy.
SIGNIFICANCE STATEMENT The anesthetic and antidepressant drug ketamine is well-characterized as an NMDA receptor (NMDAR) antagonist; though, the relevance and full impact of this pharmacology to its antidepressant actions is unclear. We found that NMDAR activation, which occurs downstream of their initial actions, is necessary for the beneficial effects of ketamine and several other putative antidepressant compounds. As such, promoting NMDAR signaling, or other approaches that enhance NMDAR-dependent long-term potentiation (LTP)-like synaptic potentiation in vivo may be an effective antidepressant strategy directly, or acting synergistically with other drug or interventional treatments.
Keywords: AMPA receptor, antidepressant, hydroxynorketamine, ketamine, LTP, NMDA receptor
Introduction
Depressive symptoms, including low mood, anhedonia, and suicidal ideation, can be improved within hours following initiation of pharmacological treatment with racemic (R,S)-ketamine (ketamine), and similar effects are also observed with ketamine's (S)-ketamine enantiomer (Berman et al., 2000; Zarate et al., 2006b; Murrough et al., 2013; Daly et al., 2018; Fava et al., 2020). Ketamine is also effective in patients who are resistant to the beneficial effects of chronic standard treatments (Zarate et al., 2006b; Murrough et al., 2013; Fava et al., 2020). Despite this potential, the development of novel rapid-acting antidepressants is hampered by an incomplete understanding of the mechanism(s) by which ketamine exerts its therapeutic actions (Gould et al., 2019; Krystal et al., 2019).
Ketamine is a well-characterized NMDA receptor (NMDAR) antagonist, acting noncompetitively as an open channel blocker (Anis et al., 1983). Ketamine has long been used as an anesthetic agent, with evidence supporting NMDAR inhibition as a primary mechanism underlying these anesthetic effects, as well as its dissociative side-effects and abuse potential (Zanos et al., 2018b). Hypotheses regarding ketamine's antidepressant mechanism of action have mainly focused on potential NMDAR inhibition-mediated processes. In particular, it has been hypothesized that ketamine acts rapidly to treat depression by (1) preferential inhibition of NMDARs localized to GABAergic interneurons leading to selective disinhibition of excitatory glutamatergic neurons, increased glutamate release, and the resultant increase in synaptic strength (Duman, 2014; Krystal et al., 2019); or (2) transient ketamine-mediated inhibition of spontaneously activated NMDARs, which results in a homeostatic reset of synaptic strength (Kavalali and Monteggia, 2020, 2023). Both these hypotheses converge on mechanisms that lead to an increase in synaptic strength of excitatory neuronal circuits, which are weakened in those who suffer from affective disorders (Zanos et al., 2018a; Thompson, 2023).
However, clinically, other NMDAR open channel blockers, such as memantine and AZD-6765, do not exert the full antidepressant profile of ketamine (Zarate et al., 2006a, 2013; Smith et al., 2013; Sanacora et al., 2014, 2017), nor do antagonists selective for the GluN2B subunit of the NMDAR (Preskorn et al., 2008; Ibrahim et al., 2012; Paterson et al., 2015; Cerecor, 2016), or the NMDAR glycine co-agonist site (Park et al., 2020). These controlled human observations suggest that NMDAR inhibition cannot fully explain the rapid and robust antidepressant actions of ketamine. Alternative hypotheses for ketamine's antidepressant actions include distinct targets, such as opioid receptors (Zhang et al., 2021; Hess et al., 2022; Wulf et al., 2022), or a principal role of ketamine's biologically active metabolites including (2R,6R)-hydroxynorketamine (HNK), which is a weak NMDAR antagonist, that increases glutamate release probability (Zanos et al., 2016; Pham et al., 2018; Fukumoto et al., 2019; Aleksandrova et al., 2020; Riggs et al., 2020, 2022; Wulf et al., 2022).
While there is considerable evidence of a correlation between rapid antidepressant actions and potentiation of excitatory synapses in affect-regulating neuronal circuits, ketamine actions to inhibit NMDAR function is difficult to reconcile, especially considering the critical role of NMDAR activity in the induction of synaptic potentiation. Activation of NMDARs is necessary for induction of canonical long-term potentiation (LTP) of excitatory synapses, leading to maintained upregulation of synaptic strength. Specifically, increased glutamatergic neurotransmission results in membrane depolarization mediated by postsynaptic AMPA receptors (AMPARs) activation, the opening of the NMDAR following the release of the Mg2+ block, and Ca2+ influx that facilitates postsynaptic plasticity including upregulation of synaptic AMPARs (Huganir and Nicoll, 2013). Such a mechanism converges with evidence that ketamine can indirectly increase glutamatergic neurotransmission (Moghaddam et al., 1997; Duman, 2014), resulting in downstream acute activation and subsequent maintained upregulation of AMPARs (Maeng et al., 2008; Autry et al., 2011 and Li et al., 2010). We thus tested the hypothesis that induction of rapid antidepressant effects requires NMDAR activation by using other NMDAR antagonists (i.e., MK-801 and CPP) before administration of ketamine and other putative rapid-acting antidepressant drugs. We also examined the role of synaptic metaplasticity and GluN2A expression in mediating the NMDAR activation-dependent effects of ketamine.
Materials and Methods
Animals
Male CD-1 mice (Charles River), eight weeks of age, were housed in groups of four to five per cage on arrival. Animals were acclimated to the vivarium (University of Maryland, Baltimore, MD) for at least one week after arrival and were maintained on a 12/12 h light/dark cycle. For the social defeat experiments, eight- to nine-week-old male C57BL/6J mice (University of Maryland, veterinary resources breeding colony) and retired male CD-1 breeders (Charles River Laboratories) were used. Food and water were provided ad libitum. All experiments were approved by the University of Maryland Baltimore Animal Care and Use Committee and were completed in accordance with the latest National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Materials
(R,S)-ketamine HCl and the NMDAR channel blocker MK-801 (Sigma-Aldrich) were dissolved in 0.9% saline and administered intraperitoneally in a volume of 7.5 ml/kg of body mass. The negative allosteric modulator of GABAA receptors containing α5 subunits (GABA-NAM) MRK-016 (Tocris Bioscience, R&D Systems) was prepared in 100% DMSO and injected at the volume of 1.25 ml/kg intraperitoneal (final volume was 40–50 μl). The competitive NMDAR antagonist (±)-CPP (Tocris Bioscience, R&D Systems), the GluN2A-preferring NMDAR antagonist PEAQX hydrochloride (NIMH Chemical Synthesis and Drug Supply Program), the positive NMDAR modulator rapastinel (Allergan Pharmaceuticals), and the mGlu2/3 receptor antagonist LY341495 disodium salt (Tocris Bioscience, R&D Systems) were dissolved in 0.9% saline and administered intraperitoneally in a volume of 7.5 ml/kg of body mass. The GluN2A-selective NMDAR positive modulator GNE-5729 was synthesized by WuXi and characterized by the National Center for Advancing Translational Sciences (NCATS) and was dissolved in a mixture of 10% DMSO, 10% cremaphor EL, 80% saline solution and given at the volume of 4 ml/kg. Ketamine's metabolite (2R,6R)-HNK was synthesized and characterized by NCATS, dissolved in 0.9% saline, and was administered intraperitoneally in a volume of 7.5 ml/kg of body mass. All additional chemicals and reagents used in this study, unless otherwise noted, were of analytical or higher grade obtained from Sigma-Aldrich.
Behavioral assays
All injections were performed by a male experimenter based on the findings of Georgiou et al. (2022).
Forced-swim test (FST)
During the FST, mice were subjected to a 6 min swim session in clear Plexiglas cylinders (30 cm in height × 20 cm in diameter) filled with 15 cm of water (23 ± 1°C). The FST was performed in normal light conditions (800 Lux). Sessions were recorded using a digital video camera. Immobility time, defined as passive floating with no additional activity other than that necessary to keep the animal's head above water, was scored during the last 4 min of the 6-min test by a trained observer blind to the treatment groups. To assess interactions between inhibition of the NMDAR and antidepressant actions of ketamine and other putative rapid-acting antidepressants, an NMDAR antagonist or vehicle was administered 10 min before ketamine, its active metabolite (2R,6R)-HNK, the GABA-NAM MRK-016, the mGlu2/3 receptor antagonist LY341495, or saline, and mice were tested 24 h later to avoid acute effects of the drugs (MK-801 has been eliminated and does not have effects 24 h posttreatment (Wegener et al., 2011).
Inescapable shock-induced escape deficits
The inescapable shock-induced escape deficits (or learned helplessness) paradigm consisted of three different phases: inescapable shock training, escapable shock screening, and the escapable shock test. On day 1, the animals were placed in one side of two-chambered shuttle boxes (34 cm in height × 37 cm in width × 18 cm in depth; Coulbourn Instruments), with the door between the chambers closed. Following a 5-min adaptation period, 120 inescapable foot-shocks (0.45 mA, 15 s in duration, pseudo-randomized average intershock interval of 45 s) were delivered through the floor. During the escapable shock screening session (day 2), the mice were placed in one of the two chambers of the apparatus for 5 min. A shock (0.45 mA) was then delivered, and the door between the two chambers was raised simultaneously. Crossing over into the second chamber terminated the shock. If the animal did not cross over, the shock terminated after 3 s. A total of 30 screening trials of escapable shocks were presented to each mouse with an average of 30-s delays between each trial. Mice that developed escape deficit behavior (more than five escape failures during the last 10 screening shocks) received the assigned drug in a randomized blinded manner 24 h following screening (day 3) and 24 h before testing. For the experiments assessing the effects of NMDAR antagonists on the actions of ketamine, (2R,6R)-HNK, or LY341495, pretreatment with MK-801 and/or CPP preceded treatment by 10 min. For the subeffective dose treatment experiments (synergistic effect experiment), rapastinel and ketamine were administered at the same time. During the escapable shock test phase (day 4), the animals were placed in the shuttle boxes and, after a 5-min adaptation period, a 0.45-mA shock was delivered concomitantly with door opening for the first five trials, followed by a 2-s delay before the door opening for the next 40 trials as we previously established this response was sensitive to ketamine treatment (Zanos et al., 2015). Crossing over to the second chamber terminated the shock. If the animal did not cross over to the other chamber, the shock was terminated after 24 s. A total of 45 trials of escapable shocks were presented to each mouse with 30-s intertrial intervals. The number of escape failures was recorded for each mouse by computer software (Graphic State v3.1; Coulbourn Instruments).
Chronic social defeat stress (CSDS) and sucrose preference
The day before the social defeat phase of the experiment, male C57BL/6J mice were singly housed and presented with two identical bottles containing either tap water or 1% (w/v) sucrose solution for assessing their “baseline” sucrose preference. Then, experimental mice were introduced to the home cage (43 cm in length × 11 cm in width × 20 cm in height) of a resident aggressive retired CD-1 breeder (prescreened for aggressive behaviors) for 10 min. Following this physical attack phase, mice were transferred and housed in the opposite side of the resident's cage divided by a perforated Plexiglas divider, to maintain continuous, 24 h, sensory contact. This process was repeated daily for 10 d, with experimental mice being introduced to a novel aggressive CD-1 mouse each day. Following day 10, for assessing the postdefeat sucrose preference, mice were singly housed and presented with two identical bottles containing either tap water or 1% (w/v) sucrose solution. Twenty-four hours later, sucrose preference was measured and the mice that underwent social defeat stress were assigned to two groups: resilient (sucrose preference >70%) and susceptible (sucrose preference <55%). Only susceptible mice were treated with ketamine and the other compounds described below.
Ketamine experiment: Susceptible mice were treated with saline or ketamine at the doses of 10 or 100 mg/kg and sucrose preference was measured for an additional 24 h.
MK-801 effects on ketamine's reversal of sucrose preference deficits: A different cohort of mice underwent the social defeat paradigm (as described above). Susceptible mice were treated with saline or MK-801 10 prior to treatment with saline or ketamine (10 mg/kg) and sucrose preference was measured for an additional 24 h.
Novel object recognition (NOR) test
The NOR test was conducted under dim light conditions (∼3–5 Lux). The NOR behavioral testing consisted of three different sessions during the same day. During the habituation phase, the animals explored the NOR apparatus (40 × 9 × 23 cm) for 10 min in the absence of objects. Immediately after, during the familiarization session and without taking the test mouse outside the boxes, two identical objects were carefully and silently fixed on the floor of the apparatus symmetrically 8.5 cm from the wall, and the animals were allowed to explore the objects for a further 10 min. The objects were either two 50-ml clear glass conical flasks (4.5 cm in bottom diameter × 7 cm in height) containing blue marbles or two white-painted small glass vials (2.5 cm in bottom diameter × 6 cm in height). After familiarization with the “familiar” objects, mice were immediately returned to their home cages. Following a 50-min delay, mice were placed back into the NOR apparatus, in which one of the “familiar” objects used during the familiarization session was replaced by a “novel” object (retention phase). Mice were permitted to freely explore the objects for 10 min. During both familiarization and retention sessions, the objects were used in a counterbalanced between-groups manner. All the sessions were videotaped by an overhead digital video camera. The retention sessions were manually scored by a trained observer blind to the experimental groups, using the AnoStar scoring software (Cleversys Inc.). Mice were considered to be interacting with the objects when their head was facing the object in a pre-set distance of ≤1 cm. A discrimination index was calculated as the time a mouse was interacting with the novel object divided by the total time of interaction with both the objects during the retention phase (Bevins and Besheer, 2006).
Open-field test
This experiment was performed at 100 Lux. Mice were placed into individual open-field arenas (50 cm in length × 50 cm in width × 38 cm in height; San Diego Instruments) for a 30-min locomotor assessment. Distance traveled was automatically analyzed using TopScan v2.0 (CleverSys, Inc).
Western blottings
To purify synaptoneurosomes, mouse hippocampi were dissected and homogenized in Syn-PER Reagent (ThermoFisher Scientific; catalog #87793) with 1× protease and phosphatase inhibitor cocktail (ThermoFisher Scientific; catalog #78440). The homogenate was centrifuged for 10 min at 1200 × g at 4°C. The supernatant was centrifuged at 15,000 × g for 20 min at 4°C. After centrifugation, the pellet (synaptosomal fraction) was resuspended and sonicated in N-PER Neuronal Protein Extraction Reagent (ThermoFisher Scientific; catalog #87792). Protein concentration was determined via the BCA protein assay kit (ThermoFisher Scientific; catalog #23227). An equal amount of protein (10–40 µg as optimal for each antibody) for each sample was loaded into NuPage 4–12% Bis-Tris gel for electrophoresis. Gel transfer was performed with the TransBlot Turbo Transfer System (Bio-Rad). Nitrocellulose membranes with transferred proteins were blocked with 5% milk in TBST (TBS + 0.1% Tween 20) for 1 h and kept with primary antibodies overnight at 4°C. The following primary antibodies were used: GluA1 (Cell Signaling Technology; catalog #13185) and GluA2 (Cell Signaling Technology; catalog #13607). The next day, blots were washed three times in TBST and incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibody (1:5000-1:10,000) for 1 h. After the final three washes with TBST, bands were detected using enhanced chemiluminescence (ECL) with the Syngene Imaging System (G:Box ChemiXX9). After imaging, the blots were incubated in the stripping buffer (ThermoFisher Scientific; catalog #46430) for 10–15 min at room temperature followed by three washes with TBST. The stripped blots were incubated in blocking solution for 1 h and incubated with the primary antibody directed against the respective protein or GAPDH for loading control. Densitometric analysis of immunoreactive bands for each protein was conducted using Syngene's GenTools software. Protein levels were normalized to GAPDH. Fold change was calculated by normalization to a saline-treated control group for each protein.
Hippocampal slice preparation and electrophysiology
After one week of acclimation, nine-week-old mice were randomly assigned to one of four treatment groups [saline-saline (Sal-Sal); saline-ketamine (Sal-Ket); MK-801-saline (MK-Sal); MK-801-ketamine (MK-Ket)] and, at the same time daily (9:30 A.M.), animals were treated with either saline or 0.1 mg/kg MK-801 followed by either saline or 10 mg/kg ketamine 10 min later. One mouse within each cage received each of the four treatments. Animals were anesthetized with isoflurane and killed 24 h posttreatment.
Hippocampal slice preparation and electrophysiology experiments were conducted generally as previously described (Preston et al., 2019; Brown et al., 2021). Briefly, brains were removed and rapidly submerged in oxygenated (95% O2/5% CO2), ice-cold dissection artificial CSF (ACSF; 120 mm NaCl, 3 mm KCl, 4 mm MgCl2, 1 mm NaH2PO4, 26 mm NaHCO3, and 10 mm glucose). The brain was mounted on its dorsal surface and sectioned along the horizontal plane with a vibratome to acquire 400-µm slices containing the hippocampus. The hippocampus was subdissected free from the rest of the slice and the CA3 subfield was removed. These slices were then quickly placed in a humidified holding chamber at room temperature (20–22°C). Following 90 min of recovery, slices were transferred to a submersion-type chamber and continuously perfused (1.5 ml/min; Ismatec Reglo ICC Digital Pump; Cole-Parmer) with oxygenated (95% O2/5% CO2) ACSF (120 mm NaCl, 3 mm KCl, 1.5 mm MgCl2, 1 mm NaH2PO4, 2.5 mm CaCl2, 26 mm NaHCO3, and 10 mm glucose) during recording experiments. Schaffer collateral fibers were stimulated by placing a bipolar electrode (100 µs in duration at 0.05 Hz; FHC) in the stratum radiatum of the CA1 subfield and an ACSF-filled glass recording pipette (3–5 MΩ; World Precision Instruments) recorded a field EPSP (fEPSP) in the same layer of CA1.
The stimulus intensity was then modified to elicit 35% of the maximal fEPSP slope and paired-pulse fEPSPs (50-ms interpulse interval) were recorded each min for 5 min. Following paired-pulse recordings, individual stimulus pulses were applied every 20 s for 10 min and baseline fEPSP responses were monitored. After recording baseline responses for 10 min, a high-frequency stimulation (HFS) protocol (4 × 100 Hz/1-s train at 20-s intervals) induced long-term potentiation, and fEPSP responses were monitored for the subsequent 60 min.
Data analysis
Experimental groups were executed and analyzed by an experimenter blind to treatment groups. Sample sizes were based on our prior experience using the same paradigms. Distance traveled, immobility time, and escape failures following administration of different doses of the GluN2A-selective NMDAR positive allosteric modulator GNE-5729, as well as escape failures (learned helplessness) and discrimination index (novel object recognition) after administration of different doses of ketamine were analyzed via one-way ANOVA. Effects of NMDAR antagonists on the antidepressant-like behavioral (forced-swim test and learned helplessness), biochemical (western blots), or novel object recognition effects of ketamine or the other putative rapid-acting antidepressant drugs were analyzed using two-way ANOVA, with factors “pretreatment” × “treatment.” Social defeat results were analyzed using a repeated measures three-way ANOVA, with factors “pretreatment” × “treatment” × “experimental phase” (repeated factor). ANOVAs were followed by Holm–Sidak's multiple comparisons post hoc test when significance was reached (i.e., p < 0.05). Electrophysiology data were digitized at 10 kHz, filtered at 3 kHz, and analyzed with pCLAMP 10.7 software (Molecular Devices). Slices with an average baseline fEPSP slope value from 1–5 min that exhibited >10% variation compared with 6–10 min were excluded from the analysis. Between-group comparisons were analyzed via two-way analysis of variance (factor A: treatment; factor B: pretreatment) and treatment means were separated via Holm–Šídák post hoc comparisons. fEPSP values were normalized to the average fEPSP slope response recorded during the last 5 min of baseline. Individual normalized slope responses represent the average normalized slope value recorded at 20-s intervals successively over a 1 min period. LTP magnitude was calculated by averaging the normalized fEPSP slope values 56–60 min after HFS. An average of four slices were collected per animal, which were averaged to determine responses for slices from a given animal. Reported n-values indicate the number of mice assessed. All statistical analysis and graphic production were completed using GraphPad Prism version 9.2 (GraphPad Software Inc.). An α level of 0.05 was used as the criterion for statistical significance. All data are presented as mean ± SEM. Statistical outliers (based on priori criteria) were determined and removed from the dataset using the ROUT method (Motulsky and Brown, 2006), provided by GraphPad Prism; parameter used: Q = 1%. Out of a total of 846 mice used, 13 mice were excluded from the analyses based on the outlier ROUT method identification. For a detailed description on the exact statistical analyses used see Table 1.
Table 1.
Statistical Analyses
| Figure/statistical test | Number of mice (as appear in graph) | Factorial effects | Interaction effect | |||||
|---|---|---|---|---|---|---|---|---|
| Overall effects for Figure 1 | ||||||||
| Factor “treatment” | ||||||||
| 1 A | One-way ANOVA | n = 7, 7, 7, 6, 7, 7 | F(5,35) = 4.10; | p = 0.005 | ||||
| Factor “treatment” | Factor “CSDS phase” | Factor “treatment” × “CSDS phase” | ||||||
| 1 B | Two-way RM ANOVA | n = 8, 6, 7 | F(2,19) = 1.288; | p = 0.299 | F(2,38) = 84.300; | p < 0.0001 | F(4,38) = 2.737; | p = 0.043 |
| Overall effects for Figure 2 | ||||||||
| Factor “pretreatment” | Factor “treatment” | Factor “pretreatment” × “treatment” | ||||||
| 2 A | Two-way ANOVA | n = 13, 13, 13, 14 | F(1,48) = 3.333; | p = 0.074 | F(1,48) = 5.134; | p = 0.028 | F(1,48) = 1.742; | p = 0.193 |
| 2 B | Two-way ANOVA | n = 16, 16, 17, 17 | F(1,62) = 2.900; | p = 0.094 | F(1,62) = 4.625; | p = 0.035 | F(1,62) = 5.317; | p = 0.025 |
| Factor “treatment” × “phase” | Factor “pretreatment” × “treatment” | Factor “pretreatment” × “treatment” × “phase” | ||||||
| 2 C | Three-way RM ANOVA | n = 6, 6, 7, 6 | F(2,42) = 2.316; | p = 0.111 | F(1,21) = 6.452; | p = 0.019 | F(2,42) = 3.278; | p = 0.048 |
| Factor “pretreatment” | Factor “treatment” | Factor “pretreatment” × “treatment” | ||||||
| 2 D | Two-way ANOVA | n = 8, 9, 9, 9 | F(1,31) = 4.109; | p = 0.051 | F(1,31) = 3.230; | p = 0.082 | F(1,31) = 7.313; | p = 0.011 |
| Factor “co-treatment” | Factor “treatment” | Factor “pretreatment” × “treatment” | ||||||
| 2 E | Two-way ANOVA | n = 7, 9, 8, 8 | F(1,28) = 10.020; | p = 0.004 | F(1,28) = 7.631; | p = 0.010 | F(1,28) = 7.606; | p = 0.010 |
| Overall effects for Figure 3 | ||||||||
| Factor “pretreatment” | Factor “treatment” | Factor “pretreatment” × “treatment” | ||||||
| 3 A | Two-way ANOVA | n = 8, 9, 8, 9 | F(1,30) = 6.804; | p = 0.014 | F(1,30) = 43.040; | p < 0.0001 | F(1,30) = 6.202; | p = 0.019 |
| 3 B | Two-way ANOVA | n = 8, 9, 9, 9 | F(1,31) = 5.795; | p = 0.022 | F(1,31) = 5.261; | p = 0.029 | F(1,31) = 9.041; | p = 0.005 |
| 3 C | Two-way ANOVA | n = 8, 8, 9, 8 | F(1,29) = 6.209; | p = 0.019 | F(1,29) = 3.499; | p = 0.072 | F(1,29) = 6.077; | p = 0.020 |
| 3 D | Two-way ANOVA | n = 11, 8, 11, 10 | F(1,36) = 11.060; | p = 0.002 | F(1,36) = 44.570; | p < 0.0001 | F(1,36) = 19.760; | p < 0.0001 |
| 3 E | Two-way ANOVA | n = 8, 10, 7, 10 | F(1,35) = 5.753; | p = 0.022 | F(1,35) = 6.958; | p = 0.012 | F(1,35) = 3.737; | p = 0.061 |
| 3 F | Two-way ANOVA | n = 8, 7, 10, 8 | F(1,29) = 9.865; | p = 0.004 | F(1,29) = 7.547; | p = 0.010 | F(1,29) = 11.540; | p = 0.002 |
| Overall effects for Figure 4 | ||||||||
| Factor “treatment” | ||||||||
| 4 A | One-way ANOVA | n = 8, 8, 8 | F(2,21) = 15.28; | p < 0.0001 | ||||
| Factor “pretreatment” | Factor “treatment” | Factor “pretreatment” × “treatment” | ||||||
| 4 B | Two-way ANOVA | n = 8, 7, 7, 8 | F(1,26) = 16.410; | p < 0.001 | F(1,26) = 7.356; | p = 0.012 | F(1,26) = 5.937; | p = 0.022 |
| 4 D | Two-way ANOVA | n = 10, 10, 10, 10 | F(1,36) = 4.468; | p = 0.042 | F(1,36) = 1.963; | p = 0.170 | F(1,36) = 3.944; | p = 0.054 |
| 4 E | Two-way ANOVA | n = 10, 10, 10, 10 | F(1,36) = 1.253; | p = 0.270 | F(1,36) = 4.389; | p = 0.043 | F(1,36) = 2.729; | p = 0.107 |
| 4 H | Two-way ANOVA | n = 8, 5, 5, 6 | F(1,20) = 11.630; | p = 0.003 | F(1,20) = 4.616; | p = 0.044 | F(1,20) = 6.246; | p = 0.021 |
| Overall effects for Figure 5 | ||||||||
| Factor “pretreatment” | Factor “treatment” | Factor “pretreatment” × “treatment” | ||||||
| 5 A | Two-way ANOVA | n = 6, 7, 7, 7, 7, 7 | F(2,35) = 1.320; | p = 0.280 | F(1,35) = 7.287; | p = 0.011 | F(2,35) = 4.796; | p = 0.014 |
| Factor “treatment” | ||||||||
| 5 B | One-way ANOVA | n = 6, 6, 6, 7, 7 | F(4,27) = 2.469; | p = 0.069 | ||||
| 5 C | One-way ANOVA | n = 8, 8, 8, 8, 8 | F(4,35) = 2.469; | p = 0.088 | ||||
| 5 D | One-way ANOVA | n = 9, 8, 8, 8, 7, 8 | F(5,42) = 2.110; | p = 0.083 | ||||
Results
For full details on the statistical tests used, F values, and n numbers for each of the experiments, see Table 1.
High doses of ketamine do not exert antidepressant-relevant behavioral actions
We assessed the effects of different doses of ketamine to reverse escape failures following learned helplessness induced by inescapable shocks. This model is sensitive to single administration of (R,S)-ketamine, but not to traditional antidepressants, when tested 24 h following drug exposure (see Ramaker and Dulawa, 2017). Mice were given injections of different doses of ketamine (1–100 mg/kg) and tested 24 h later for reversal of learned helplessness (Fig. 1A). Ketamine administration significantly reduced escape failures of susceptible mice at the dose of 10 mg/kg and indicated a trend to reduce escape failures at the dose of 30 mg/kg (p = 0.068). Doses of 1, 3, and 100 mg/kg were ineffective, with 100 mg/kg completely preventing the antidepressant-like actions of ketamine compared with the dose of 10 mg/kg.
Figure 1.

High doses of ketamine do not elicit antidepressant-relevant behavioral actions in mice. Mice received an injection of vehicle or different doses of racemic ketamine and were assessed for antidepressant-like responses 24 h later. A, Ketamine at the dose of 10 mg/kg significantly decreased escape failures in helpless mice, whereas the doses of 1, 3, 30, and 100 mg/kg did not exert significant antidepressant-relevant reductions in escape failures in the learned helplessness paradigm. B, Similarly, ketamine at the dose of 10 mg/kg reversed the decrease in sucrose preference of mice that underwent chronic social defeat stress whereas the high dose of 100 mg/kg did not elicit such an antidepressant-related response. Mice were tested for sucrose versus water preference during the 24-h period following drug administration. Data are the mean ± SEM; *p < 0.05, ***p < 0.001 as indicated by Holm–Šídák post hoc comparisons. See Table 1 for complete details on the statistical analyses and precise group sizes. CSDS, chronic social defeat stress; KET, racemic ketamine; SAL, saline; Treat, treatment.
To further assess whether ketamine is antidepressant at high doses, we tested the effects of ketamine to reverse sucrose preference deficits induced by 10 d of social defeat stress. Either 10 or 100 mg/kg ketamine, or vehicle, was administered to mice that exhibited <55% sucrose preference following chronic social defeat stress, and sucrose preference was assessed over a 24-h period after drug administration (Fig. 1B). While ketamine at the dose of 10 mg/kg reversed the deficit in sucrose preference, the dose of 100 mg/kg was ineffective in ameliorating sucrose preference deficits in chronically stressed, susceptible mice.
NMDAR activity mediates antidepressant-relevant behavioral effects of ketamine
To assess the role of NMDAR activity on ketamine's antidepressant-like behavioral actions, we gave mice an injection of the readily brain penetrant NMDAR channel blocker MK-801 (0.1 mg/kg), followed by the administration of ketamine 10 min later, and tested these mice for reversal of learned helplessness behavior 24 h after administration of ketamine (Fig. 2A). Ketamine's antidepressant-relevant actions were absent in mice that received MK-801 before ketamine (Fig. 2A). Similarly, pretreatment with the competitive NMDAR blocker (±)-CPP (which crosses the blood-brain barrier and exerts centrally-mediated behavioral actions; Lehmann et al., 1987) completely prevented the effect of ketamine on escape failures in helpless mice (Fig. 2B). Pretreatment with MK-801 also prevented ketamine's actions on sucrose preference in socially-defeated susceptible mice (Fig. 2C) and immobility time in the forced-swim test (Fig. 2D). As in all the other tests, the forced-swimming procedure was conducted 24 h after drug administration, a time point long after any locomotor or anesthetic effects of ketamine disappear.
Figure 2.

Blocking NMDAR activity prevents the antidepressant-relevant behavioral effects of ketamine. A, Mice received an injection of vehicle or the NMDAR channel blocker MK-801 (0.1 mg/kg) and 10 min later they received an injection of vehicle or racemic ketamine (10 mg/kg). MK-801 pretreatment prevented the antidepressant-relevant behavioral actions of ketamine in the learned helplessness paradigm. B, Similarly, pretreatment with the competitive NMDAR blocker (±)-CPP (10 mg/kg) blocked ketamine's antidepressant-relevant actions in the learned helplessness paradigm. C, MK-801 (0.1 mg/kg) pretreatment also prevented the anti-anhedonic actions of ketamine in mice that underwent chronic social defeat stress, as measured by the sucrose preference test. D, MK-801 (0.03 mg/kg) pretreatment prevented ketamine's actions on immobility time in the forced-swim test. E, Co-administration of subeffective doses of ketamine with the NMDAR positive modulator rapastinel induced a synergistic reduction of escape failures in helpless mice in the learned helplessness paradigm. In all paradigms, mice were tested 24 h following drug administration. Data are the mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001 as indicated by Holm–Šídák post hoc comparisons. See Table 1 for complete details on the statistical analyses and precise group sizes. CSDS, chronic social defeat stress; KET, racemic ketamine; SAL, saline; Treat, treatment.
These results might indicate that NMDAR activity is necessary for the antidepressant-like behavioral actions of ketamine, or they could also simply indicate that a higher degree of NMDAR inhibition generally prevents rapid antidepressant activity. We thus tested the hypothesis that ketamine's antidepressant-like action(s) converge with positive allosteric modulators of the NMDAR, via co-administration of subeffective doses of ketamine (3 mg/kg) with a 1 mg/kg subeffective dose of GLYX-13 (rapastinel), which was determined by preliminary dose–response experiments. Rapastinel is an NMDAR positive allosteric modulator with EC50 effects on NMDAR subunits as measured in vitro of 9.8 pm (GluN2A), 9.9 nm (GluN2B), 2.2 pm (GluN2C), and 1.7 pm (GluN2D; Donello et al., 2019). An earlier study revealed that the antidepressant-related effects of rapastinel, as well as the pharmacologically-related NMDAR positive allosteric modulator NYX-2925, were blocked with prior administration of the competitive NMDAR antagonist CPP (Burgdorf et al., 2015; Khan et al., 2018). Rapastinel at the dose of 1 mg/kg was ineffective to induce any antidepressant-relevant behavioral actions on its own, but the combination with the low (also subeffective) dose of ketamine induced a synergistic effect to reverse escape failures 24 h following administration, suggesting that NMDAR activation possibly contributes to the antidepressant-like effects of ketamine (Fig. 2E).
Blocking NMDAR activity prevents the antidepressant-relevant behavioral effects of rapid-acting antidepressant drugs with distinct mechanisms
It is possible that our results could indicate only that excessive NMDAR inhibition [arising from a combined administration of ketamine and other NMDAR antagonists (i.e., MK-801 or CPP)] prevents the rapid antidepressant behavioral effects of ketamine. However, these data could also provide a novel mechanism underlying ketamine's rapid antidepressant action involving NMDAR activation. In order to examine the critical role of NMDAR activation as a general mechanism underlying rapid-acting antidepressant efficacy, we assessed whether pretreatment with the NMDAR inhibitor MK-801 before administration of other preclinically characterized, putative rapid-acting antidepressants also prevents their antidepressant-relevant behavioral actions.
(2R,6R)-HNK is a metabolite of ketamine that has been shown to exert ketamine-like antidepressant biobehavioral actions (Zanos et al., 2016, 2019; Pham et al., 2018; Fukumoto et al., 2019; Lumsden et al., 2019; Aleksandrova et al., 2020; Riggs et al., 2020), without blocking NMDAR function at relevant concentrations (Suzuki et al., 2017; Zanos et al., 2017b; Lumsden et al., 2019). As above, pretreatment with MK-801 at a dose of 0.1 mg/kg completely prevented the antidepressant-like actions of (2R,6R)-HNK tested in the forced-swim test 24 h after administration (Fig. 3A), whereas pretreatment with MK-801 at 0.03 mg/kg did not (Fig. 3B). The finding that (2R,6R)-HNK's effects to change forced swimming behavior required a higher dose of MK-801 compared with that required to reverse ketamine's effects may be reflecting that (2R,6R)-HNK itself is not an NMDAR antagonist at the brain concentrations elicited by the dose used (Suzuki et al., 2017; Zanos et al., 2017b; Lumsden et al., 2019).
Figure 3.
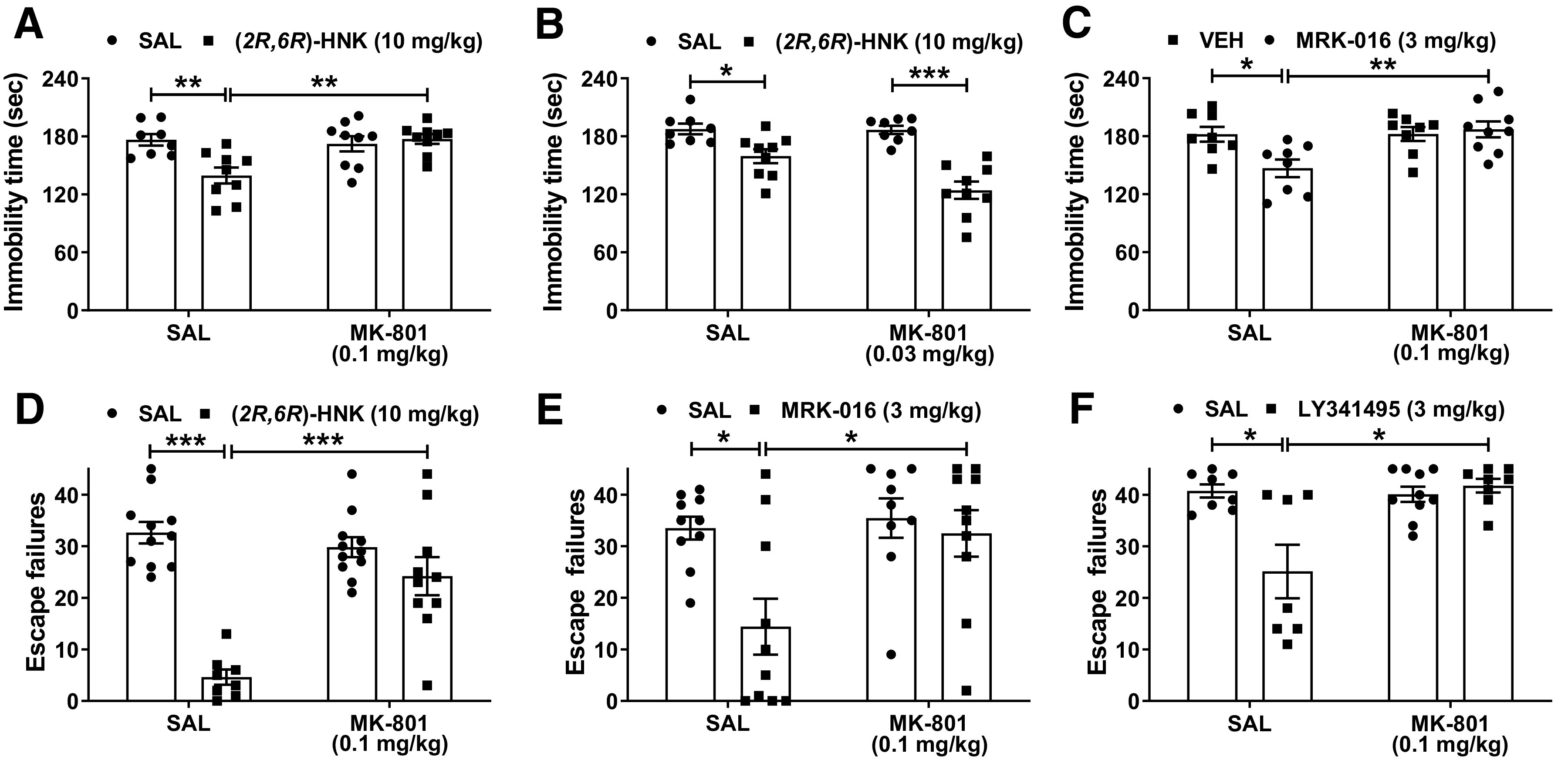
Blocking NMDAR activity prevents the antidepressant-relevant behavioral effects of distinct rapid-acting antidepressant compounds. A, B, Mice received an injection of vehicle or the NMDAR channel blocker MK-801 and 10 min later were given an additional injection of vehicle or ketamine's metabolite (2R,6R)-hydroxynorketamine (HNK; 10 mg/kg) and tested in the forced swim test 24 h later. While (A) the dose of 0.1 mg/kg MK-801 completely prevented the antidepressant-like behavioral actions of (2R,6R)-HNK, (B) 0.03 mg/kg MK-801 did not prevent (2R,6R)-HNK's actions to decrease immobility time in the forced-swim test. MK-801 pretreatment (0.1 mg/kg) prevented the antidepressant-relevant behavioral actions of (C) the negative allosteric modulator of GABAA receptors containing α5 subunits (GABA-NAM) MRK-016 in the forced-swim test. MK-801 pretreatment (0.1 mg/kg) prevented the antidepressant-like effects of (D) (2R,6R)-HNK, (E) MRK-016, and (F) the mGlu2/3 receptor antagonist LY341495 in the learned helplessness paradigm 24 h following drug administration. Data are the mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001 as indicated by Holm–Šídák post hoc comparisons. See Table 1 for complete details on the statistical analyses and precise group sizes. KET, racemic ketamine; SAL, saline.
Negative allosteric modulators of GABAA receptors containing α5 subunits (GABA-NAM), which promote glutamate release and enhanced excitatory glutamatergic transmission, have been shown to exert ketamine-like rapid and sustained antidepressant-relevant actions in rodent tests (Fischell et al., 2015; Zanos et al., 2017a; Xiong et al., 2018; Troppoli et al., 2021). As we found with ketamine and (2R,6R)-HNK, pretreatment with MK-801 (0.1 mg/kg) prevented the anti-immobility actions of the GABA-NAM MRK-016 in the forced-swim test (Fig. 3C). We also found that MK-801 pretreatment (0.1 mg/kg) prevented the reversal of helpless behavior following inescapable shock induced by both (2R,6R)-HNK (Fig. 3D) and MRK-016 24 h after their administration (Fig. 3E). mGlu2/3 receptor antagonists have been shown to exert rapid antidepressant-relevant behavioral actions in rodent tests (Chaki et al., 2004; Yoshimizu et al., 2006; Bespalov et al., 2008; Witkin et al., 2016), likely through increasing glutamatergic neurotransmission in mood-regulating synapses. Similar to these earlier reports we found that the mGlu2/3 receptor antagonist, LY341495, reversed helpless behavior in the learned helplessness test. Pretreatment with MK-801 (0.1 mg/kg) prevented this effect (Fig. 3F). Together, these findings suggest a shared necessity of NMDAR activation for the behavioral antidepressant effects of putative rapid-acting antidepressants with distinct mechanisms of action.
Role for NMDAR-mediated hippocampal plasticity in the effects of ketamine
Ketamine and its (2R,6R)-HNK metabolite have been established to enhance SC-CA1 synaptic strength (Autry et al., 2011; Zanos et al., 2016; Aleksandrova et al., 2020; Riggs et al., 2020, 2022). Performance in novel object recognition task is mediated, in part, by plasticity of SC-CA1 synapses, and is NMDAR activity dependent (Clarke et al., 2010; Warburton et al., 2013; Fig. 4A). We confirmed the earlier finding that ketamine increases performance in the novel object recognition task (Papp et al., 2017; Willner et al., 2019; Aleksandrova et al., 2020). The administration of ketamine was performed 24 h before testing to avoid any ataxic and anesthetic effects of the drug. Specifically, ketamine (10 mg/kg) increased the discrimination index in the novel object recognition test compared with saline-treated mice in a 1 h recognition memory task (Fig. 4A). In contrast, the high dose of ketamine (100 mg/kg) impaired short-term object recognition memory compared with controls (Fig. 4A).
Figure 4.
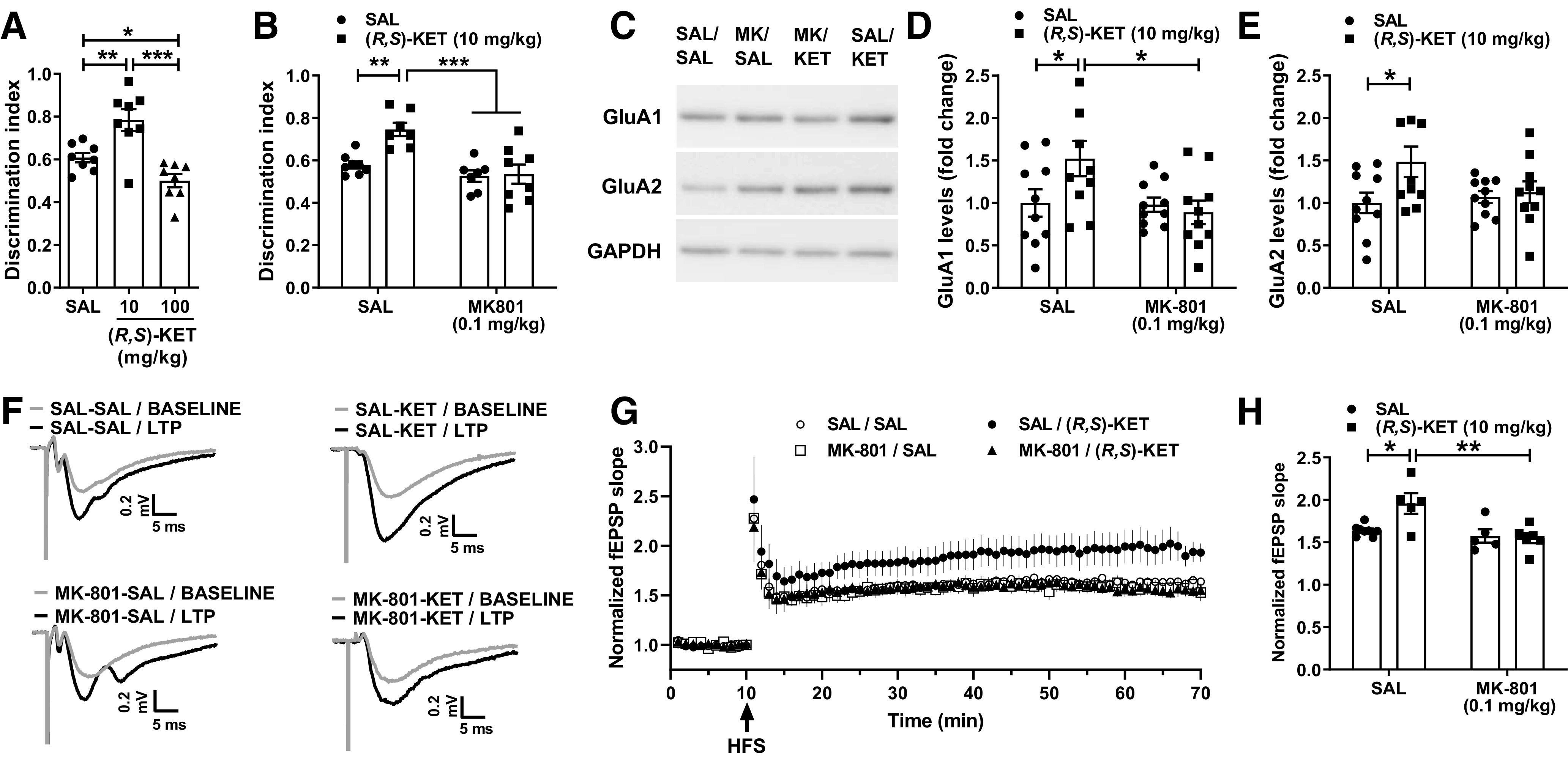
Blocking NMDAR activity prevents the procognitive and synaptic actions of ketamine. A, Mice received an injection of vehicle or different doses of racemic ketamine and were tested for short-term novel object recognition memory 24 h after drug administration. Ketamine (10 mg/kg) enhanced the discrimination index, indicative of a procognitive effect, whereas a higher dose of ketamine (100 mg/kg) impaired object recognition memory. B, Administration of the NMDAR channel blocker MK-801 (0.1 mg/kg) 10 min before ketamine (10 mg/kg) prevented the procognitive effect of ketamine in the novel object recognition test. C, Representative western blot images for GluA1 and GluA2 AMPAR subunits from hippocampal synaptoneurosomes. D, E, Pretreatment with MK-801 prevented the ketamine-induced enhancement of synaptoneurosomal levels of GluA1 and GluA2 AMPAR subunits. F, Traces composed of representative sweeps from 5 min pre-tetanus (gray) and 56–60 min post-tetanus (black) from SAL-SAL, SAL-KET, MK-801-SAL, and MK-801-KET treatment groups. G, H, Pretreatment with MK-801 prevented the metaplastic effect of ketamine on long-term potentiation magnitude at the SC-CA1 synapse. Data are the mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001 as indicated by Holm–Šídák post hoc comparisons. See Table 1 for complete details on the statistical analyses and precise group sizes. HFS, high-frequency stimulation; KET, racemic ketamine; LTP, long-term potentiation; MK, MK-801; SAL, saline.
To assess the role of NMDAR activity on the procognitive effects of ketamine in the novel object recognition test we gave mice an injection of MK-801 (0.1 mg/kg), followed by the administration of ketamine 10 min later, and tested these mice in the novel object recognition task 24 h after administration of ketamine (Fig. 4B). Ketamine administration increased the time spent with the novel object compared with saline-treated controls (Fig. 4B); however, ketamine's procognitive actions were absent in mice that received MK-801 before ketamine (Fig. 4B; p = 0.847 compared with the respective controls). Similar to high-dose ketamine, the MK-801 treatment resulted in an overall effect to decrease novel object recognition performance (Table 1).
Studies have reported that expression of the AMPA receptor GluA1 and GluA2 subunits in the hippocampus is increased 24 h after administration of ketamine to rodents, consistent with the finding that enhanced AMPA receptor activity underlies ketamine's sustained antidepressant-relevant behavioral effects (Maeng et al., 2008; Li et al., 2010; Autry et al., 2011; Zanos et al., 2016). We administered saline or the NMDAR channel blocker MK-801 (0.1 mg/kg), followed by the administration of ketamine 10 min later, and collected hippocampi from these mice at 24 h. While ketamine administration induced an enhancement of both GluA1 (Fig. 4C,D) and GluA2 (Fig. 4) AMPAR subunits, MK-801 pretreatment abolished this effect of ketamine for both the GluA1 (p = 0.682) and GluA2 (p = 0.756) subunits (Fig. 4C–E).
Ketamine administration to rodents has been found to induce a metaplastic effect on ex vivo SC-CA1 synaptic activity, resulting in enhanced long-term potentiation (LTP; Burgdorf et al., 2013; Graef et al., 2015; Widman et al., 2018). We administered vehicle or ketamine (10 mg/kg) 10 min after administration of vehicle or MK-801 (0.1 mg/kg). Ketamine treatment induced a metaplastic effect on LTP magnitude 24 h after ketamine treatment, an effect that was blocked by MK-801 pretreatment (Fig. 4F,G). Post hoc comparisons revealed a significant increase in LTP magnitude in SAL-KET-treated mice compared with SAL-SAL controls (Fig. 4H). Pretreatment with MK-801 prevented the ketamine-induced enhancement of LTP magnitude (Fig. 4H). These data suggest that the metaplastic effect of ketamine on LTP magnitude at the SC-CA1 synapse in mouse hippocampal slices requires NMDAR activation.
GluN2A activity mediates antidepressant actions of ketamine
A convergent enhancement in excitatory neurotransmission in brain areas including prefrontal cortex and hippocampus, has been characterized as a key mechanism that underlies ketamine's rapid antidepressant action (Thompson, 2023). Generally, synaptic NMDARs primarily contain GluN2A subunits, which mediate long-term synaptic plasticity and display faster kinetics, whereas GluN2B receptor subunits are expressed extrasynaptically and display slower kinetics (Traynelis et al., 2010; Paoletti et al., 2013). Furthermore, the positive modulator rapastinel that we found augmented the effects of ketamine (Fig. 2E) has reported selectivity to GluN2A compared with GluN2B (Donello et al., 2019). Therefore, we hypothesized a primary role of GluN2A-containing receptors in ketamine's actions described above. We administered saline or the GluN2A subunit-selective NMDAR antagonist PEAQX (at the doses of either 5 or 30 mg/kg), followed by the administration of ketamine 10 min later, and assessed the reversal of helpless behaviors 24 h later. Pretreatment with PEAQX at the doses of 5 and 30 mg/kg completely prevented the antidepressant-like actions of ketamine to reverse learned helplessness (Fig. 5A).
Figure 5.

The antidepressant-like actions of ketamine are mediated through GluN2A activity. A, Mice received an injection of vehicle or the GluN2A-selective NMDAR negative allosteric modulator PEAQX (5 or 30 mg/kg) followed by an injection of vehicle or ketamine (10 mg/kg) 10 min later, and then were tested for reversal of helpless behavior 24 h later. PEAQX pretreatment, at both doses administered, prevented the antidepressant-relevant actions of ketamine to decrease escape failures of helpless mice. B, The GluN2A-selective NMDAR positive allosteric modulator GNE-5729 induced a decrease in locomotor activity of mice in the open-field test only at the highest dose administered (3 mg/kg). C, In the forced-swim test, GNE-5729 at the dose of 3 mg/kg significantly reduced immobility time of mice, indicative of an antidepressant response. D, Similarly, the dose of 1 mg/kg of GNE-5729 significantly reduced escape failures of helpless mice. Data are the mean ± SEM; *p < 0.05, ***p < 0.001 as indicated by Holm–Šídák post hoc comparisons. See Table 1 for complete details on the statistical analyses and precise group sizes. KET, ketamine; SAL, saline; VEH, vehicle.
As GluN2A-specific blockade prevented the antidepressant-like effects of ketamine, we then tested whether the GluN2A NMDAR positive modulator GNE-5729 may produce antidepressant-like behavioral effects comparable to those of ketamine. The effect of GNE-5729 at doses ranging from 0.1–3 mg/kg on the locomotor activity of mice was evaluated, where we found that only the dose of 3 mg/kg reduced distance traveled compared with vehicle-treated mice (Fig. 5B, inset). The doses of 0.1, 0.3, and 1 mg/kg GNE-5729 did not alter the locomotion of mice compared with the controls (Fig. 5B). We next assessed the ability of GNE-5729 to induce antidepressant-like behaviors. GNE-5729 (3 mg/kg) induced a significant decrease in immobility time of mice in the forced-swim test compared with the control mice tested 24 h after administration (Fig. 5C), indicative of the antidepressant-like efficacy of this GluN2A NMDAR positive allosteric modulator. In the learned helplessness paradigm, GNE-5729 at the dose of 1 mg/kg also significantly decreased escape failures of helpless mice compared with vehicle-treated controls when tested 24 h after administration (Fig. 5D). None of the other doses changed immobility time or escape failures in the forced-swim and learned helplessness tests, respectively (Fig. 5C,D). Collectively, these results indicate that GluN2A activation is necessary and sufficient to induce behavioral ketamine-like effects.
Discussion
We hypothesize that the antidepressant effects of ketamine are mediated by the induction of NMDAR activation-dependent synaptic plasticity. Specifically, increases in synchronous neuronal activation and glutamate release lead to acute activation of NMDARs and the induction of persistent changes in synaptic strength and plasticity that underlie maintained therapeutic actions after the drug is no longer present. Such an NMDAR activation mechanism converges with increased glutamate release probability, either through ketamine disinhibition via acting on interneurons or hydroxynorketamine's direct action, resulting in postsynaptic AMPAR activation, membrane depolarization release of NMDAR magnesium block, NMDAR activation, calcium influx and consequential postsynaptic plasticity including upregulation of synaptic AMPARs. We tested the role of NMDAR activation in the antidepressant actions of ketamine 24 h following administration of the drug (where no relevant brain concentrations of ketamine or its metabolites are present), confirming that ketamine exerts an inverted U-shaped dose response in previously unassessed behavioral outcomes. Our findings suggest that NMDAR inhibition induced by higher doses of ketamine prevents antidepressant-related effects of ketamine that are triggered by lower doses. New findings include that (1) ketamine's persistent antidepressant-like behavioral actions are blocked following acute pretreatment with either a competitive (CPP) or channel blocking (MK-801) NMDAR antagonist, (2) an otherwise ineffective low dose of ketamine acts synergistically with an NMDAR positive allosteric modulator to exert a persistent antidepressant-like behavioral effect, (3) the antidepressant-like actions of other rapid-acting antidepressant drug classes in development (that do not block the NMDAR themselves) are similarly blocked by NMDAR inhibition, (4) ketamine-induced up-regulation of hippocampal synaptic AMPAR subunits GluA1 and GluA2 are blocked by acute NMDAR inhibition, (5) ketamine-induced metaplasticity measured ex vivo using electrically-stimulated LTP is blocked by NMDAR inhibition, (6) ketamine-induced antidepressant-relevant behavioral actions are blocked by a GluN2A-preferring NMDAR antagonist, and (7) pharmacological activation of the NMDAR subunit GluN2A is sufficient to exert antidepressant-like behavioral effects.
In humans, the typical dose/route of ketamine administration used in clinical antidepressant studies is 0.5 mg/kg administered during a 40-min intravenous infusion. 1.0 mg/kg can be also effective, while a lower dose appears to be generally less effective (Fava et al., 2020). However, there are no controlled studies to date that have compared these to higher doses. Previous preclinical studies have demonstrated that antidepressant-like effects following administration of ketamine are consistently observed in animal experiments when subanesthetic doses are administered, but these actions are absent at higher doses near or within the range used for anesthesia (Duncan et al., 1998; Li et al., 2010; Chowdhury et al., 2012, 2017; Zanos et al., 2016; Miller et al., 2018; Hibicke et al., 2020; Kim and Monteggia, 2020). To our knowledge, all previous reports have used the forced swimming test rodent behavioral assay. Our current results add to these reports, further showing a U-shaped dose response in the reversal of learned helplessness. We also found that 10 mg/kg ketamine effectively reversed the deleterious effects of chronic social defeat stress on anhedonia as measured by the sucrose preference test, whereas the dose of 100 mg/kg was ineffective. We note that at the 24 h time point of testing, ketamine and its metabolites are eliminated from mice (below detectable limits by 2 h), and any anesthetic or other side effects of ketamine have been absent for over 23 h (Zanos et al., 2018b).
We also found that ketamine's persistent antidepressant-like behavioral actions are blocked following pretreatment with other NMDAR antagonists. In particular, we demonstrated that preadministration of the noncompetitive antagonist MK-801 blocks ketamine's antidepressant-like effects, as measured in the forced swimming test, learned helplessness test, and recovery of sucrose preference following chronic social defeat stress. Similarly, pretreatment with the competitive NMDAR antagonist CPP also blocked ketamine's effects to reverse learned helplessness. We conclude that NMDAR activation is required for ketamine's ability to induce the antidepressant-relevant behavioral responses to ketamine. We also conclude that the failure of higher doses of ketamine to induce antidepressant-like behavioral responses is because of the block of some critical fraction of NMDARs that is needed for the induction of antidepressant-relevant actions.
Our studies identified that the antidepressant-like actions of other putative rapid-acting antidepressant drugs are blocked by NMDAR inhibition. Specifically, we found that the antidepressant-like behavioral effects of ketamine's metabolite (2R,6R)-HNK and the GABA-NAM MRK-016 in the forced swimming and learned helplessness tests, as well as the antidepressant-like effects of the mGlu2/3 receptor antagonist LY341495 in the learned helplessness test, were prevented by preadministration with MK-801. We note that prevention of (2R,6R)-HNK's effects to change forced swimming behavior (Fig. 3A,B) required a higher dose of MK-801 compared with that required to reverse ketamine's effects (Fig. 2D), potentially reflecting the fact that (2R,6R)-HNK is not an NMDAR antagonist at the doses used here (Suzuki et al., 2017; Zanos et al., 2017b; Lumsden et al., 2019). We have previously shown that antidepressant-relevant doses of (2R,6R)-HNK increase the probability of glutamate release independent of NMDAR inhibition, potentially via a mechanism convergent with mGlu2 receptor signaling (Zanos et al., 2016, 2019; Riggs et al., 2020, 2022). MRK-016 is an α5-selective GABA-NAM that exerts rapid antidepressant-like and anti-anhedonic actions in addition to restoring stress-weakened synapses in the hippocampus, presumably by disinhibition of excitatory activity (Fischell et al., 2015; Zanos et al., 2017a; Xiong et al., 2018; Troppoli et al., 2021). Consistent with this, we found that ketamine acts synergistically with the NMDAR positive allosteric modulator rapastinel to exert an antidepressant-like behavioral effect. These results support the conclusions that rapid-acting antidepressant compounds share a common downstream NMDAR-activation dependent effector mechanism, despite their wide range of independent upstream targets.
Canonical SC-CA1 LTP, mediated by NMDAR activation, is maintained by upregulation of postsynaptic GluA1 and GluA2 (Huganir and Nicoll, 2013). The novel object recognition task is established to involve SC-CA1 potentiation and to be NMDAR activity dependent (Clarke et al., 2010; Warburton et al., 2013). The function of this hippocampal synapse may also have relevance to the cognitive deficits observed in depression and may be reversed by ketamine (see Gill et al., 2021). Here, we demonstrated that a 10 mg/kg dose of ketamine improved novel object discrimination. Furthermore, the effectiveness of 10 mg/kg ketamine in enhancing novel object discrimination was blocked by preadministration with MK-801. Consistent with a ketamine-mediated activation of NMDAR signaling, we identified that ketamine-induced up-regulation of AMPAR subunits GluA1 and GluA2 in a synaptoneurosome preparation from the hippocampus 24 h after administration, was blocked by the prior administration of MK-801. These data are consistent with previous observations that antidepressant-relevant doses of ketamine produce effects on plasticity-mediated cellular signaling pathways that are not observed at higher doses (Duncan et al., 1998; Li et al., 2010; Chowdhury et al., 2012, 2017; Kim and Monteggia, 2020).
In vivo administration of ketamine has been reported to enhance ex vivo SC-CA1 LTP (Burgdorf et al., 2013; Graef et al., 2015; Widman et al., 2018). Similarly, the NMDAR PAMs rapastinel and NYX-295 induce a similar metaplasticity (Burgdorf et al., 2013, 2015; Khan et al., 2018). Ketamine and its metabolite (2R,6R)-HNK ameliorate impaired SC-CA1 LTP measured in anesthetized Wistar-Kyoto rats 3.5 h after drug injection (Aleksandrova et al., 2020). Bath application of ketamine has also been reported to induce enhanced synaptic potentiation in hippocampal slices obtained from mice treated with ketamine 7 d before slice collection (Kim et al., 2021). Here, we report that the metaplastic effect of ketamine on ex vivo electrically-stimulated LTP assessed 24 h following administration is blocked by the prior administration of MK-801. While it is unclear whether SC-CA1 function is directly related to ketamine's antidepressant actions, or whether such changes are generally representative of ketamine-induced plasticity of excitatory synapses (Thompson, 2023), these findings suggest that ketamine induces NMDAR-activation-dependent process that enhances LTP induction. We note that while concentrations in the range of 1 and 20 μm ketamine are reported to enhance SC-CA1 synaptic strength in hippocampal slices, a higher concentration of 100 μm does not have this effect, which can also be blocked by APV (Kim and Monteggia, 2020; Izumi et al., 2022). Ketamine (50uM) also blocks LTP induced in nucleus accumbens slices, which may explain a lack of ketamine in vivo dopamine-dependent plasticity in this region (Simmler et al., 2022).
Finally, we report that pharmacological activation of NMDAR subunit GluN2A with GNE-5729 is sufficient to exert antidepressant-like behavioral effects when tested in both the forced swimming and learned helplessness assays of antidepressant efficacy (maximally effective at 3 and 1 mg/kg, respectively). We also show that pretreatment with the GluN2A antagonist PEAQX prevents the antidepressant-like effects of ketamine in the learned helplessness assay, suggesting that ketamine requires, and potentially acts downstream via, GluN2A activation to exert its antidepressant behavioral actions.
Our results support targeting NMDAR-dependent LTP-like synaptic potentiation as an effective antidepressant strategy. Our findings implicate ketamine-induced SC-CA1 synaptic to be NMDAR-activation dependent, but it remains to be determined whether such changes are necessary, or unique, to this synapse. A focus on the strength of inhibition of NMDAR signaling to develop future treatments of depression may be counterproductively resulting in drugs that prevent increases in NMDAR-activation-dependent increases in synaptic strength necessary for efficacy.
Footnotes
This research was supported by the National Institutes of Health Grant MH107615, United States Department of Veterans Affairs Merit Awards 1I01BX004062 and 101BX003631, an Allergan Pharmaceuticals Investigator-Initiated Research Grant (T.D.G.), the National Institutes of Health Grant MH086828 (to S.M.T.), and the Brain and Research Foundation Young Investigator Grant National Alliance for Research on Schizophrenia and Depression Grant 26826 (to P.Z.). We thank Ms. Thatchana Rajasekar for her technical assistance in analyzing the novel object recognition experiments. We also thank Dr. Patrick Morris who characterized the GluN2A PAM, GNE-5729.
T.D.G. has received research funding from Roche Pharmaceuticals and served as a consultant for FSV7 LLC, during the preceding three years. C.A.Z. is listed as a co-inventor on a patent for the use of ketamine in major depression and suicidal ideation. P.Z., C.A.Z., and T.D.G. are listed as co-authors in patents and patent applications related to the pharmacology and use of (2R,6R)-HNK in the treatment of depression, anxiety, anhedonia, suicidal ideation, and posttraumatic stress disorders. S.M.T. is listed as an inventor on a patent for the use of GABA-NAMs for treatment of depression. All other authors declare no competing financial interests. C.A.Z. has assigned his patent rights to the Unites States Government but will share a percentage of any royalties that may be received. P.Z., S.M.T., and T.D.G. have assigned patent rights to the University of Maryland, Baltimore, MD, but will share a percentage of any royalties that may be received by the University of Maryland.
References
- Aleksandrova LR, Wang YT, Phillips AG (2020) Ketamine and its metabolite, (2R,6R)-HNK, restore hippocampal LTP and long-term spatial memory in the Wistar-Kyoto rat model of depression. Mol Brain 13:92. 10.1186/s13041-020-00627-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anis NA, Berry SC, Burton NR, Lodge D (1983) The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methyl-aspartate. Br J Pharmacol 79:565–575. 10.1111/j.1476-5381.1983.tb11031.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, Monteggia LM (2011) NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475:91–95. 10.1038/nature10130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH (2000) Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 47:351–354. 10.1016/s0006-3223(99)00230-9 [DOI] [PubMed] [Google Scholar]
- Bespalov AY, van Gaalen MM, Sukhotina IA, Wicke K, Mezler M, Schoemaker H, Gross G (2008) Behavioral characterization of the mGlu group II/III receptor antagonist, LY-341495, in animal models of anxiety and depression. Eur J Pharmacol 592:96–102. 10.1016/j.ejphar.2008.06.089 [DOI] [PubMed] [Google Scholar]
- Bevins RA, Besheer J (2006) Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study 'recognition memory.' Nat Protoc 1:1306–1311. 10.1038/nprot.2006.205 [DOI] [PubMed] [Google Scholar]
- Brown KA, Carpenter JM, Preston CJ, Ludwig HD, Clay KB, Harn DA, Norberg T, Wagner JJ, Filipov NM (2021) Lacto-N-fucopentaose-III ameliorates acute and persisting hippocampal synaptic plasticity and transmission deficits in a Gulf War Illness mouse model. Life Sci 279:119707. 10.1016/j.lfs.2021.119707 [DOI] [PubMed] [Google Scholar]
- Burgdorf J, Zhang XL, Nicholson KL, Balster RL, Leander JD, Stanton PK, Gross AL, Kroes RA, Moskal JR (2013) GLYX-13, a NMDA receptor glycine-site functional partial agonist, induces antidepressant-like effects without ketamine-like side effects. Neuropsychopharmacology 38:729–742. 10.1038/npp.2012.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgdorf J, Zhang XL, Weiss C, Gross A, Boikess SR, Kroes RA, Khan MA, Burch RM, Rex CS, Disterhoft JF, Stanton PK, Moskal JR (2015) The long-lasting antidepressant effects of rapastinel (GLYX-13) are associated with a metaplasticity process in the medial prefrontal cortex and hippocampus. Neuroscience 308:202–211. 10.1016/j.neuroscience.2015.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerecor (2016) Cerecor reports top-line data from CERC-301 phase 2 study for major depressive disorder. Rockville: Cerecor, Inc. [Google Scholar]
- Chaki S, Yoshikawa R, Hirota S, Shimazaki T, Maeda M, Kawashima N, Yoshimizu T, Yasuhara A, Sakagami K, Okuyama S, Nakanishi S, Nakazato A (2004) MGS0039: a potent and selective group II metabotropic glutamate receptor antagonist with antidepressant-like activity. Neuropharmacology 46:457–467. 10.1016/j.neuropharm.2003.10.009 [DOI] [PubMed] [Google Scholar]
- Chowdhury GM, Behar KL, Cho W, Thomas MA, Rothman DL, Sanacora G (2012) ¹H-[¹³C]-nuclear magnetic resonance spectroscopy measures of ketamine's effect on amino acid neurotransmitter metabolism. Biol Psychiatry 71:1022–1025. 10.1016/j.biopsych.2011.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury GM, Zhang J, Thomas M, Banasr M, Ma X, Pittman B, Bristow L, Schaeffer E, Duman RS, Rothman DL, Behar KL, Sanacora G (2017) Transiently increased glutamate cycling in rat PFC is associated with rapid onset of antidepressant-like effects. Mol Psychiatry 22:120–126. 10.1038/mp.2016.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke JR, Cammarota M, Gruart A, Izquierdo I, Delgado-García JM (2010) Plastic modifications induced by object recognition memory processing. Proc Natl Acad Sci U S A 107:2652–2657. 10.1073/pnas.0915059107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly EJ, Singh JB, Fedgchin M, Cooper K, Lim P, Shelton RC, Thase ME, Winokur A, Van Nueten L, Manji H, Drevets WC (2018) Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry 75:139–148. 10.1001/jamapsychiatry.2017.3739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donello JE, Banerjee P, Li YX, Guo YX, Yoshitake T, Zhang XL, Miry O, Kehr J, Stanton PK, Gross AL, Burgdorf JS, Kroes RA, Moskal JR (2019) Positive N-methyl-D-aspartate receptor modulation by rapastinel promotes rapid and sustained antidepressant-like effects. Int J Neuropsychopharmacol 22:247–259. 10.1093/ijnp/pyy101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS (2014) Pathophysiology of depression and innovative treatments: remodeling glutamatergic synaptic connections. Dialogues Clin Neurosci 16:11–27. 10.31887/DCNS.2014.16.1/rduman [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan GE, Moy SS, Knapp DJ, Mueller RA, Breese GR (1998) Metabolic mapping of the rat brain after subanesthetic doses of ketamine: potential relevance to schizophrenia. Brain Res 787:181–190. 10.1016/s0006-8993(97)01390-5 [DOI] [PubMed] [Google Scholar]
- Fava M, Freeman MP, Flynn M, Judge H, Hoeppner BB, Cusin C, Ionescu DF, Mathew SJ, Chang LC, Iosifescu DV, Murrough J, Debattista C, Schatzberg AF, Trivedi MH, Jha MK, Sanacora G, Wilkinson ST, Papakostas GI (2020) Double-blind, placebo-controlled, dose-ranging trial of intravenous ketamine as adjunctive therapy in treatment-resistant depression (TRD). Mol Psychiatry 25:1592–1603. 10.1038/s41380-018-0256-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischell J, Van Dyke AM, Kvarta MD, LeGates TA, Thompson SM (2015) Rapid antidepressant action and restoration of excitatory synaptic strength after chronic stress by negative modulators of alpha5-containing GABAA receptors. Neuropsychopharmacology 40:2499–2509. 10.1038/npp.2015.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto K, Fogaça MV, Liu RJ, Duman C, Kato T, Li XY, Duman RS (2019) Activity-dependent brain-derived neurotrophic factor signaling is required for the antidepressant actions of (2R,6R)-hydroxynorketamine. Proc Natl Acad Sci U S A 116:297–302. 10.1073/pnas.1814709116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiou P, et al. (2022) Experimenters' sex modulates mouse behaviors and neural responses to ketamine via corticotropin releasing factor. Nat Neurosci 25:1191–1200. 10.1038/s41593-022-01146-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill H, Gill B, Rodrigues NB, Lipsitz O, Rosenblat JD, El-Halabi S, Nasri F, Mansur RB, Lee Y, McIntyre RS (2021) The effects of ketamine on cognition in treatment-resistant depression: a systematic review and priority avenues for future research. Neurosci Biobehav Rev 120:78–85. 10.1016/j.neubiorev.2020.11.020 [DOI] [PubMed] [Google Scholar]
- Gould TD, Zarate CA Jr, Thompson SM (2019) Molecular pharmacology and neurobiology of rapid-acting antidepressants. Annu Rev Pharmacol Toxicol 59:213–236. 10.1146/annurev-pharmtox-010617-052811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef JD, Newberry K, Newton A, Pieschl R, Shields E, Luan FN, Simmermacher J, Luchetti D, Schaeffer E, Li YW, Kiss L, Bristow LJ (2015) Effect of acute NR2B antagonist treatment on long-term potentiation in the rat hippocampus. Brain Res 1609:31–39. 10.1016/j.brainres.2015.03.019 [DOI] [PubMed] [Google Scholar]
- Hess EM, Riggs LM, Michaelides M, Gould TD (2022) Mechanisms of ketamine and its metabolites as antidepressants. Biochem Pharmacol 197:114892. 10.1016/j.bcp.2021.114892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibicke M, Landry AN, Kramer HM, Talman ZK, Nichols CD (2020) Psychedelics, but not ketamine, produce persistent antidepressant-like effects in a rodent experimental system for the study of depression. ACS Chem Neurosci 11:864–871. 10.1021/acschemneuro.9b00493 [DOI] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA (2013) AMPARs and synaptic plasticity: the last 25 years. Neuron 80:704–717. 10.1016/j.neuron.2013.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim L, Diaz Granados N, Jolkovsky L, Brutsche N, Luckenbaugh D, Herring WJ, Potter WZ, Zarate CAJ (2012) A randomized, placebo-controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. J Clin Psychopharmacol 32:551–557. 10.1097/JCP.0b013e31825d70d6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Hsu FF, Conway CR, Nagele P, Mennerick SJ, Zorumski CF (2022) Nitrous oxide, a rapid antidepressant, has ketamine-like effects on excitatory transmission in adult hippocampus. Biol Psychiatry 92:964–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Monteggia LM (2020) Targeting homeostatic synaptic plasticity for treatment of mood disorders. Neuron 106:715–726. 10.1016/j.neuron.2020.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Monteggia LM (2023) Rapid homeostatic plasticity and neuropsychiatric therapeutics. Neuropsychopharmacology 48:54–60. 10.1038/s41386-022-01411-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Houck DR, Gross AL, Zhang XL, Cearley C, Madsen TM, Kroes RA, Stanton PK, Burgdorf J, Moskal JR (2018) NYX-2925 is a novel NMDA receptor-specific spirocyclic-β-lactam that modulates synaptic plasticity processes associated with learning and memory. Int J Neuropsychopharmacol 21:242–254. 10.1093/ijnp/pyx096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Monteggia LM (2020) Increasing doses of ketamine curtail antidepressant responses and suppress associated synaptic signaling pathways. Behav Brain Res 380:112378. 10.1016/j.bbr.2019.112378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Autry AE, Na ES, Adachi M, Björkholm C, Kavalali ET, Monteggia LM (2021) Sustained effects of rapidly acting antidepressants require BDNF-dependent MeCP2 phosphorylation. Nat Neurosci 24:1100–1109. 10.1038/s41593-021-00868-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Abdallah CG, Sanacora G, Charney DS, Duman RS (2019) Ketamine: a paradigm shift for depression research and treatment. Neuron 101:774–778. 10.1016/j.neuron.2019.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann J, Schneider J, McPherson S, Murphy DE, Bernard P, Tsai C, Bennett DA, Pastor G, Steel DJ, Boehm C (1987) CPP, a selective N-methyl-D-aspartate (NMDA)-type receptor antagonist: characterization in vitro and in vivo. J Pharmacol Exp Ther 240:737–746. [PubMed] [Google Scholar]
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS (2010) mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329:959–964. 10.1126/science.1190287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumsden EW, et al. (2019) Antidepressant-relevant concentrations of the ketamine metabolite (2R,6R)-hydroxynorketamine do not block NMDA receptor function. Proc Natl Acad Sci U S A 116:5160–5169. 10.1073/pnas.1816071116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeng S, Zarate CA Jr, Du J, Schloesser RJ, McCammon J, Chen G, Manji HK (2008) Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry 63:349–352. 10.1016/j.biopsych.2007.05.028 [DOI] [PubMed] [Google Scholar]
- Miller OH, Grabole N, Wells I, Hall BJ (2018) Genome-wide translating mRNA analysis following ketamine reveals novel targets for antidepressant treatment. bioRxiv 254904. [Google Scholar]
- Moghaddam B, Adams B, Verma A, Daly D (1997) Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci 17:2921–2927. 10.1523/JNEUROSCI.17-08-02921.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motulsky HJ, Brown RE (2006) Detecting outliers when fitting data with nonlinear regression - a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics 7:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrough JW, Perez AM, Pillemer S, Stern J, Parides MK, aan het Rot M, Collins KA, Mathew SJ, Charney DS, Iosifescu DV (2013) Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biol Psychiatry 74:250–256. 10.1016/j.biopsych.2012.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14:383–400. 10.1038/nrn3504 [DOI] [PubMed] [Google Scholar]
- Papp M, Gruca P, Lason-Tyburkiewicz M, Willner P (2017) Antidepressant, anxiolytic and procognitive effects of subacute and chronic ketamine in the chronic mild stress model of depression. Behav Pharmacol 28:1–8. 10.1097/FBP.0000000000000259 [DOI] [PubMed] [Google Scholar]
- Park LT, Kadriu B, Gould TD, Zanos P, Greenstein D, Evans JW, Yuan P, Farmer CA, Oppenheimer M, George JM, Adeojo LW, Snodgrass HR, Smith MA, Henter ID, Machado-Vieira R, Mannes AJ, Zarate CA (2020) A randomized trial of the N-methyl-d-aspartate receptor glycine site antagonist prodrug 4-chlorokynurenine in treatment-resistant depression. Int J Neuropsychopharmacol 23:417–425. 10.1093/ijnp/pyaa025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson B, Fraser H, Wang C, Marcus R (2015) A randomized, double- blind, placebo-controlled, sequential parallel study of CERC-301 in the adjunctive treatment of subjects with severe depression and recent active suicidal ideation despite antidepressant treatment. In: National network of depression centers annual conference. Ann Arbor: Michigan. [Google Scholar]
- Pham TH, Defaix C, Xu X, Deng SX, Fabresse N, Alvarez JC, Landry DW, Brachman RA, Denny CA, Gardier AM (2018) Common neurotransmission recruited in (R,S)-ketamine and (2R,6R)-hydroxynorketamine-induced sustained antidepressant-like effects. Biol Psychiatry 84:e3–e6. 10.1016/j.biopsych.2017.10.020 [DOI] [PubMed] [Google Scholar]
- Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW (2008) An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol 28:631–637. 10.1097/JCP.0b013e31818a6cea [DOI] [PubMed] [Google Scholar]
- Preston CJ, Brown KA, Wagner JJ (2019) Cocaine conditioning induces persisting changes in ventral hippocampus synaptic transmission, long-term potentiation, and radial arm maze performance in the mouse. Neuropharmacology 150:27–37. 10.1016/j.neuropharm.2019.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaker MJ, Dulawa SC (2017) Identifying fast-onset antidepressants using rodent models. Mol Psychiatry 22:656–665. 10.1038/mp.2017.36 [DOI] [PubMed] [Google Scholar]
- Riggs LM, Aracava Y, Zanos P, Fischell J, Albuquerque EX, Pereira EFR, Thompson SM, Gould TD (2020) (2R,6R)-hydroxynorketamine rapidly potentiates hippocampal glutamatergic transmission through a synapse-specific presynaptic mechanism. Neuropsychopharmacology 45:426–436. 10.1038/s41386-019-0443-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs LM, Thompson SM, Gould TD (2022) (2R,6R)-hydroxynorketamine rapidly potentiates optically-evoked Schaffer collateral synaptic activity. Neuropharmacology 214:109153. 10.1016/j.neuropharm.2022.109153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Smith MA, Pathak S, Su HL, Boeijinga PH, McCarthy DJ, Quirk MC (2014) Lanicemine: a low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Mol Psychiatry 19:978–985. 10.1038/mp.2013.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Johnson MR, Khan A, Atkinson SD, Riesenberg RR, Schronen JP, Burke MA, Zajecka JM, Barra L, Su HL, Posener JA, Bui KH, Quirk MC, Piser TM, Mathew SJ, Pathak S (2017) Adjunctive lanicemine (AZD6765) in patients with major depressive disorder and history of inadequate response to antidepressants: a randomized, placebo-controlled study. Neuropsychopharmacology 42:844–853. 10.1038/npp.2016.224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmler LD, Li Y, Hadjas LC, Hiver A, van Zessen R, Lüscher C (2022) Dual action of ketamine confines addiction liability. Nature 608:368–373. 10.1038/s41586-022-04993-7 [DOI] [PubMed] [Google Scholar]
- Smith EG, Deligiannidis KM, Ulbricht CM, Landolin CS, Patel JK, Rothschild AJ (2013) Antidepressant augmentation using the N-methyl-D-aspartate antagonist memantine: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 74:966–973. 10.4088/JCP.12m08252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Nosyreva E, Hunt KW, Kavalali ET, Monteggia LM (2017) Effects of a ketamine metabolite on synaptic NMDAR function. Nature 546:E1–E3. 10.1038/nature22084 [DOI] [PubMed] [Google Scholar]
- Thompson SM (2023) Plasticity of synapses and reward circuit function in the genesis and treatment of depression. Neuropsychopharmacology 48:90–103. 10.1038/s41386-022-01422-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496. 10.1124/pr.109.002451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troppoli TA, Zanos P, Georgiou P, Gould TD, Rudolph U, Thompson SM (2021) Negative allosteric modulation of gamma-aminobutyric acid a receptors at alpha5 subunit-containing benzodiazepine sites reverses stress-induced anhedonia and weakened synaptic function in mice. Biol Psychiatry 92:216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton EC, Barker GR, Brown MW (2013) Investigations into the involvement of NMDA mechanisms in recognition memory. Neuropharmacology 74:41–47. 10.1016/j.neuropharm.2013.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener N, Nagel J, Gross R, Chambon C, Greco S, Pietraszek M, Gravius A, Danysz W (2011) Evaluation of brain pharmacokinetics of (+)MK-801 in relation to behaviour. Neurosci Lett 503:68–72. 10.1016/j.neulet.2011.08.012 [DOI] [PubMed] [Google Scholar]
- Widman AJ, Stewart AE, Erb EM, Gardner E, McMahon LL (2018) Intravascular ketamine increases theta-burst but not high frequency tetanus induced LTP at CA3-CA1 synapses within three hours and devoid of an increase in spine density. Front Synaptic Neurosci 10:8. 10.3389/fnsyn.2018.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willner P, Gruca P, Lason M, Tota-Glowczyk K, Litwa E, Niemczyk M, Papp M (2019) Validation of chronic mild stress in the Wistar-Kyoto rat as an animal model of treatment-resistant depression. Behav Pharmacol 30:239–250. 10.1097/FBP.0000000000000431 [DOI] [PubMed] [Google Scholar]
- Witkin JM, Monn JA, Schoepp DD, Li X, Overshiner C, Mitchell SN, Carter G, Johnson B, Rasmussen K, Rorick-Kehn LM (2016) The rapidly acting antidepressant ketamine and the mGlu2/3 receptor antagonist LY341495 rapidly engage dopaminergic mood circuits. J Pharmacol Exp Ther 358:71–82. 10.1124/jpet.116.233627 [DOI] [PubMed] [Google Scholar]
- Wulf HA, Browne CA, Zarate CA, Lucki I (2022) Mediation of the behavioral effects of ketamine and (2R,6R)-hydroxynorketamine in mice by kappa opioid receptors. Psychopharmacology (Berl) 239:2309–2316. 10.1007/s00213-022-06118-4 [DOI] [PubMed] [Google Scholar]
- Xiong Z, Zhang K, Ishima T, Ren Q, Chang L, Chen J, Hashimoto K (2018) Comparison of rapid and long-lasting antidepressant effects of negative modulators of α5-containing GABAA receptors and (R)ketamine in a chronic social defeat stress model. Pharmacol Biochem Behav 175:139–145. 10.1016/j.pbb.2018.10.005 [DOI] [PubMed] [Google Scholar]
- Yoshimizu T, Shimazaki T, Ito A, Chaki S (2006) An mGluR2/3 antagonist, MGS0039, exerts antidepressant and anxiolytic effects in behavioral models in rats. Psychopharmacology (Berl) 186:587–593. 10.1007/s00213-006-0390-7 [DOI] [PubMed] [Google Scholar]
- Zanos P, Piantadosi SC, Wu HQ, Pribut HJ, Dell MJ, Can A, Snodgrass HR, Zarate CA Jr, Schwarcz R, Gould TD (2015) The prodrug 4-chlorokynurenine causes ketamine-like antidepressant effects, but not side effects, by NMDA/glycineB-site inhibition. J Pharmacol Exp Ther 355:76–85. 10.1124/jpet.115.225664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, Alkondon M, Yuan P, Pribut HJ, Singh NS, Dossou KS, Fang Y, Huang XP, Mayo CL, Wainer IW, Albuquerque EX, Thompson SM, Thomas CJ, Zarate CA Jr, Gould TD (2016) NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533:481–486. 10.1038/nature17998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P, Nelson ME, Highland JN, Krimmel SR, Georgiou P, Gould TD, Thompson SM (2017a) A negative allosteric modulator for alpha5 subunit-containing GABA receptors exerts a rapid and persistent antidepressant-like action without the side effects of the NMDA receptor antagonist ketamine in mice. eNeuro 4:ENEURO.0285-16.2017. 10.1523/ENEURO.0285-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, Alkondon M, Yuan P, Pribut HJ, Singh NS, Dossou KSS, Fang Y, Huang XP, Mayo CL, Albuquerque EX, Thompson SM, Thomas CJ, Zarate CA, Gould TD (2017b) Zanos et al. reply. Nature 546:E4–E5. 10.1038/nature22085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P, Thompson SM, Duman RS, Zarate CA Jr, Gould TD (2018a) Convergent mechanisms underlying rapid antidepressant action. CNS Drugs 32:197–227. 10.1007/s40263-018-0492-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P, Moaddel R, Morris PJ, Riggs LM, Highland JN, Georgiou P, Pereira EFR, Albuquerque EX, Thomas CJ, Zarate CA Jr, Gould TD (2018b) Ketamine and ketamine metabolite pharmacology: insights into therapeutic mechanisms. Pharmacol Rev 70:621–660. 10.1124/pr.117.015198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P, Highland JN, Stewart BW, Georgiou P, Jenne CE, Lovett J, Morris PJ, Thomas CJ, Moaddel R, Zarate CA Jr, Gould TD (2019) (2R,6R)-hydroxynorketamine exerts mGlu2 receptor-dependent antidepressant actions. Proc Natl Acad Sci U S A 116:6441–6450. 10.1073/pnas.1819540116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA Jr, Singh JB, Quiroz JA, De Jesus G, Denicoff KK, Luckenbaugh DA, Manji HK, Charney DS (2006a) A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J Psychiatry 163:153–155. 10.1176/appi.ajp.163.1.153 [DOI] [PubMed] [Google Scholar]
- Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK (2006b) A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63:856–864. 10.1001/archpsyc.63.8.856 [DOI] [PubMed] [Google Scholar]
- Zarate CA Jr, Mathews D, Ibrahim L, Chaves JF, Marquardt C, Ukoh I, Jolkovsky L, Brutsche NE, Smith MA, Luckenbaugh DA (2013) A randomized trial of a low-trapping nonselective N-methyl-D-aspartate channel blocker in major depression. Biol Psychiatry 74:257–264. 10.1016/j.biopsych.2012.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Hillhouse TM, Anderson PM, Koppenhaver PO, Kegen TN, Manicka SG, Lane JT, Pottanat E, Van Fossen M, Rice R, Porter JH (2021) Opioid receptor system contributes to the acute and sustained antidepressant-like effects, but not the hyperactivity motor effects of ketamine in mice. Pharmacol Biochem Behav 208:173228. 10.1016/j.pbb.2021.173228 [DOI] [PubMed] [Google Scholar]