

Abstract
The establishment of a functional cerebral cortex depends on the proper execution of multiple developmental steps, culminating in dendritic and axonal outgrowth and the formation and maturation of synaptic connections. Dysregulation of these processes can result in improper neuronal connectivity, including that associated with various neurodevelopmental disorders. The γ-Protocadherins (γ-Pcdhs), a family of 22 distinct cell adhesion molecules that share a C-terminal cytoplasmic domain, are involved in multiple aspects of neurodevelopment including neuronal survival, dendrite arborization, and synapse development. The extent to which individual γ-Pcdh family members play unique versus common roles remains unclear. We demonstrated previously that the γ-Pcdh-C3 isoform (γC3), via its unique “variable” cytoplasmic domain (VCD), interacts in cultured cells with Axin1, a Wnt-pathway scaffold protein that regulates the differentiation and morphology of neurons. Here, we confirm that γC3 and Axin1 interact in the cortex in vivo and show that both male and female mice specifically lacking γC3 exhibit disrupted Axin1 localization to synaptic fractions, without obvious changes in dendritic spine density or morphology. However, both male and female γC3 knock-out mice exhibit severely decreased dendritic complexity of cortical pyramidal neurons that is not observed in mouse lines lacking several other γ-Pcdh isoforms. Combining knock-out with rescue constructs in cultured cortical neurons pooled from both male and female mice, we show that γC3 promotes dendritic arborization through an Axin1-dependent mechanism mediated through its VCD. Together, these data identify a novel mechanism through which γC3 uniquely regulates the formation of cortical circuitry.
SIGNIFICANCE STATEMENT The complexity of a neuron's dendritic arbor is critical for its function. We showed previously that the γ-Protocadherin (γ-Pcdh) family of 22 cell adhesion molecules promotes arborization during development; it remained unclear whether individual family members played unique roles. Here, we show that one γ-Pcdh isoform, γC3, interacts in the brain with Axin1, a scaffolding protein known to influence dendrite development. A CRISPR/Cas9-generated mutant mouse line lacking γC3 (but not lines lacking other γ-Pcdhs) exhibits severely reduced dendritic complexity of cerebral cortex neurons. Using cultured γC3 knock-out neurons and a variety of rescue constructs, we confirm that the γC3 cytoplasmic domain promotes arborization through an Axin1-dependent mechanism. Thus, γ-Pcdh isoforms are not interchangeable, but rather can play unique neurodevelopmental roles.
Keywords: cell adhesion, dendritic arborization, signaling, synapse development, synaptic maturation
Introduction
Cell-cell interactions mediated through adhesion molecules are critical for the establishment of properly functioning neuronal circuits (Sanes and Zipursky, 2020). The mammalian clustered Protocadherin (cPcdh) family, comprising over 50 adhesion molecules encoded by the Pcdha, Pcdhb, and Pcdhg gene clusters, contributes to molecular diversity at neuronal cell surfaces (Peek et al., 2017; Mountoufaris et al., 2018). Each cPcdh gene encodes 6 cadherin-like extracellular (EC) domains, a transmembrane domain, and a unique, variable cytoplasmic domain (VCD); a shared, C-terminal “constant” domain is encoded by three small exons to which each “variable” exon is spliced (Wu and Maniatis, 1999; Wu et al., 2001; Tasic et al., 2002; Wang et al., 2002a). The cPcdhs mediate homophilic trans-interactions via antiparallel EC1–4 contacts, while promiscuous cis-interactions are mediated by EC5–EC6. cPcdh interactions allow for zipper-like interfaces to form between cell surfaces, depending on the degree to which isoform repertoires match (Schreiner and Weiner, 2010; Thu et al., 2014; Nicoludis et al., 2015; Rubinstein et al., 2015; Goodman et al., 2016).
The 22-member γ-Pcdh family is critically important for multiple facets of neurodevelopment, including synapse formation/maturation (J.A. Weiner et al., 2005; Garrett and Weiner, 2009; Molumby et al., 2017; LaMassa et al., 2021; Steffen et al., 2021), dendrite self-avoidance (Lefebvre et al., 2008, 2012; Ing-Esteves et al., 2018), axon arborization (Prasad and Weiner, 2011; W.V. Chen et al., 2017), interneuron survival (Wang et al., 2002b; Prasad et al., 2008; Ing-Esteves et al., 2018; Carriere et al., 2020; Mancia Leon et al., 2020), and dendrite arborization (Garrett et al., 2012; Suo et al., 2012; Keeler et al., 2015a, b; Molumby et al., 2016). While most studies have focused on roles of the entire γ-Pcdh family, increasing evidence supports isoform-specific functions for the three “C-type” isoforms that are more broadly and highly expressed than others. γC5 interacts with the GABAA receptor (Li et al., 2012) and regulates synaptic dysfunction in mouse models of Alzheimer's disease (Li et al., 2017). γC4 specifically regulates neuronal survival in the spinal cord and retina, and is the only γ-Pcdh required for postnatal survival in mice (Garrett et al., 2019). Finally, we identified a unique interaction between the γC3 VCD and the DIX domain of Axin1 (Mah et al., 2016), a scaffolding protein first identified as important for axis formation in mouse embryos (Zeng et al., 1997). In addition to its role in Wnt signaling, Axin1 mediates key steps in neurodevelopment (Luo and Lin, 2004; Ye et al., 2015), including neurogenesis (Fang et al., 2013) and axon formation (Fang et al., 2011). Knock-down of Axin1 in cultured hippocampal neurons reduces dendritic arbor complexity, which could be rescued by overexpression of Cdc42 (Y. Chen et al., 2015). Here, we investigated whether γC3 mediates important isoform-specific roles in cortical development via Axin1.
We used CRISPR/Cas9 genome editing to create a mouse line (PcdhgC3KO) lacking the γC3 isoform because of disruption of the Pcdhgc3 variable exon, but retaining expression of the other 21 γ-Pcdhs. C3KO mice exhibit significant reductions in synapse-associated Axin1 and Cdc42 that were not associated with altered dendritic spine density or morphology. C3KO mice, but not Pcdhg mutant lines lacking a variety of other isoforms, exhibit significant reductions in the complexity of Layer V neurons in vivo and mixed cortical neurons in vitro. By re-expressing full-length or truncated γC3 in cultured C3KO neurons ± knock-down/overexpression of Axin1, we identified a mechanism by which γC3 VCD-Axin1 interactions promote dendritic arborization. Together, our data (1) confirm a specific interaction between γC3 and Axin1 in the cortex in vivo; (2) demonstrate that the previously-identified role for γ-Pcdhs in dendrite arborization is due largely to the γC3 isoform; (3) support the importance of γC3-Axin1 interactions for normal dendrite arborization in cortical neurons; and (4) reveal that the 22 γ-Pcdhs are not functionally interchangeable, but play crucial isoform-specific roles.
Materials and Methods
Animals
All experiments included both male and female animals and were conducted in accordance with the University of Iowa's Institutional Animal Care and Use Committee and National Institutes of Health guidelines. All mice were healthy, not involved in previous procedures, naive to treatment, and kept under standard housing and husbandry conditions with food and water provided ad libitum and 12/12 h light/dark cycles. All control and mutant mice were on a C57BL/6J background. The specific targeting of the Pcdhgc3 variable exon used CRISPR/Cas9 genome editing. An sgRNA (5′–GGCTTACCAGTCCGCTGCTCCGG–3′, complementary strand sequence, PAM site underlined) was used. The PAM site is in the 5th and 6th codons of the open reading frame, such that a double-stranded break and imprecise repair by nonhomologous end joining would create a small frame-shift deletion just downstream of the initiator codon. The guide (50 ng/µl) and Streptococcus pyogenes Cas9 mRNA (100 ng/µl) were microinjected into C57BL/6J zygotes, which were subsequently implanted into pseudopregnant female C57BL/6J mice. Founders were identified by PCR genotyping, and one founder carrying the 13-base pair (bp) deletion described (Fig. 1B) was bred to wild-type (WT) C57BL/6J mice for two generations to eliminate mosaicism and possible off target effects. Mice were genotyped via PCR using primers spanning the CRISPR cut site (C3KO forward: 5′–CCGGGATGAGGCAGAGACTGAA–3′ and C3KO reverse: 5′–ACTCCCACCGTTCTCCAGG–3′). The 13R1 and C4KO mutants were generated through a shotgun CRISPR/Cas9 genome editing screen performed at The Jackson Laboratory gene targeting facility and have been extensively described and characterized previously (Garrett et al., 2019). Control mice for C3KO mice were C57BL/6J wild-type littermates. Experiments with C4KO and 13R1 lines used trans-heterozygotes to avoid neonatal lethality observed in homozygotes: genotypes of experimental animals were Emx1-Cre;C4KO+/−;Pcdhgfcon3/+ and Emx1-Cre;13R1+/−;Pcdhgfcon3/+ and controls were Emx1-Cre; Pcdhgfcon3/+. Emx1-Cre mice (Gorski et al., 2002) were obtained from The Jackson Laboratory (https://www.jax.org/strain/005628), as was the Thy1-YFPH line (Feng et al., 2000) used for neuronal tracing (https://www.jax.org/strain/003782). The Pcdhgfcon3 allele has been previously described and extensively characterized in prior publications (Prasad et al., 2008; Garrett et al., 2012; Molumby et al., 2016, 2017; Steffen et al., 2021)
Figure 1.
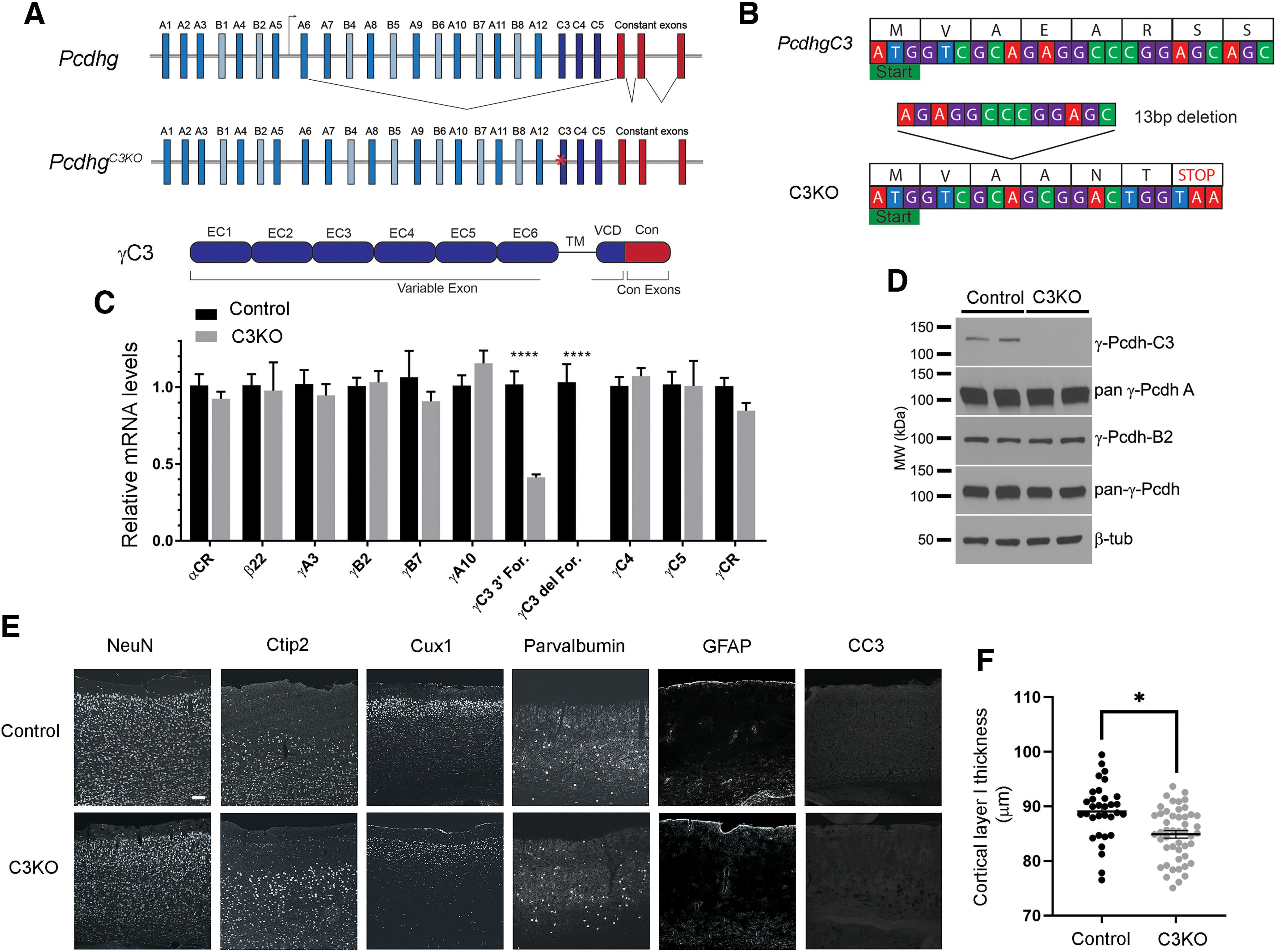
Generation of the C3KO mouse. A, Schematic representation of: the mouse Pcdhg locus, comprising 22 variable exons (γA-types: blue, γB-types: cyan, and γC-types: dark blue) which are spliced to three constant exons (red); the PcdhgC3KO locus, with frameshift deletion in the C3 variable exon indicated by a red asterisk; and the γC3 protein domain structure, with six extracellular cadherin repeats, a transmembrane domain, and a short variable cytoplasmic domain (all encoded by the variable exon) followed by a shared cytoplasmic domain (encoded by the three constant exons). B, Start of the mouse Pcdhgc3 gene with protein translation. A 13-bp deletion in the C3KO allele results in a frameshift, leading to an early stop codon. C, Quantitative PCR performed on cortical cDNA for multiple cPcdh genes shows specific loss of the γC3 isoform (no signal using a forward primer spanning the deletion; reduced transcript levels using a forward primer at the 3′ end of the variable exon), while other cPcdh and overall Pcdhg expression remain unaltered. Data are presented as relative mRNA as compared with GAPDH. ±SEM from six individual animals. A two-way ANOVA with Bonferroni post hoc test was performed to assess statistical significance. ****p < 0.0001. D, Western blottings from cortical lysates show no γC3 expression in the C3KO, while expression of other γ-Pcdhs remain largely unaltered. E, F, Cryosections of adult C3KO, and control S1 cortices were stained with multiple layer-specific and cell type-specific markers, which indicate grossly normal cell types and morphology in C3KO mice, save for a somewhat thinner Layer I, suggesting a loss of apical dendritic tufts as was seen for complete Pcdhg cluster KO mice (Garrett et al., 2012). Scale bar: 100 µm.
Plasmids
Full-length, HA-tagged γC3, γC3Δcyto, γC3Δcon, and γC3Δecto expression constructs, as well as the γC3-VCD expression construct, have been previously described (Schreiner and Weiner, 2010; Mah et al., 2016). Full-length, HA-tagged mouse Axin1, Axin1 shRNA, and scramble shRNA constructs were a kind gift from Nancy Ip and were previously described (Y. Chen et al., 2015). The zebrafish-derived CAAX-Axin1 (DIX domains, critical for γC3-Axin1 interaction (Mah et al., 2016), are ∼93% conserved between zebrafish and mouse) construct was a gift from Doug Houston previously described (Schneider et al., 2012).
Tissue preparation and immunofluorescence
Tissues from control and knock-out animals were prepared using two different methods: (1) tissue was dissected, immersed in Tissue-Tek OCT compound (Sakura Finetek) and snap frozen in isopentane cooled in a dry ice/ethanol bath; or (2) tissue was fixed in 4% paraformaldehyde (PFA) by transcardial perfusion followed by immersion overnight in the same fixative at 4°C. For cryostat sectioning, fixed tissues were sucrose sunk in 30% sucrose in PBS at 4°C, and immersed in Tissue-Tek OCT compound and frozen in isopentane cooled in a dry ice/ethanol bath. Cryostat sections were cut at 15 µm and thawed onto gelatin-subbed slides. For immunofluorescence, briefly, sections were rehydrated, blocked with 2.5% BSA, 0.1% Triton X-100 in PBS, and incubated with primary antibody overnight at 4°C in the same solution. The next day, sections were washed with PBS and incubated with fluorophore-conjugated secondary antibody for 1 h at room temperature (RT). Slides were then washed and DAPI added to the final wash to stain nuclei. FluoroGel (Electron Microscopy Sciences) was used to mount coverslips over the sections.
Image collection and analysis of dendritic spine morphology
Control and mutant mice harboring the Thy1-YFPH allele were perfused transcardially with 4% paraformaldehyde (PFA) at five weeks of age. Brains were then immersed in 4% PFA overnight. Fixed brains were then embedded in 2% agarose before being vibratome sectioned into 150-µm coronal sections. Images were collected and dendritic spine morphometrics were analyzed as previously reported (Anderson et al., 2020), with slight modifications. Basal dendritic segments (50–200 µm from the soma) of Layer V pyramidal neurons from the S1 cortex were collected using a 100× objective (N.A. of 1.4) on a Leica TCS SPE confocal microscope via the Leica LAS X software. Z-stacks were collected in the z-dimension with a step size of 0.1 µm. Z-stacks were deconvolved with Huygens Essential software using the Standard mode of the Express Deconvolution function. Images were saved as 16-bit TIFFs and opened in NeuroLucida 360 software for three-dimensional analysis. Pixel dimensions (0.07 × 0.07 × 0.10 µm) were entered to override the default x and y scaling. Dendritic segments were traced using the Rayburst Crawl user-guided method. To ensure that the tracing encompassed the whole segment, the thickness of the tracing was manually adjusted. Dendritic spines were semi-automatically detected using the following user-defined parameters. Outer range: 3 µm; minimum height: 0.3 µm; detector sensitivity: 95%; minimum count: 20 voxels. Detected spines were characterized by morphologic subtype using the following parameters: thin or mushroom spines were characterized if the head-to-neck ratio was >1.1, with spines having a head diameter >0.35 µm classified as mushroom, and otherwise were considered thin. Spines with a head-to-neck diameter ratio of <1.1 were also classified as thin if the spine length-to-neck diameter ratio was >2.5, otherwise they were classified as stubby. Classified spines were then viewed in NeuroLucida Explorer, where spine density and various morphometric details, including dendritic spine surface area and volume were obtained using the Branch Structure analysis.
Image collection and analysis of dendrite arborization
Control and mutant mice harboring the Thy1-YFPH allele were perfused transcardially with 4% paraformaldehyde (PFA) at three and six weeks of age. Brains were then immersed in 4% PFA overnight. Fixed brains were then embedded in 2% agarose before being vibratome sectioned into 200-µm coronal sections. Images were collected from Layer V pyramidal neurons from the S1 cortex using a 20× objective on a Leica TCS SPE confocal microscope via the Leica LAS X software. Z-stacks were 100 µm in the z-dimension with a step size of 0.5 µm. Individual neurons were traced using FIJI's Simple Neurite Tracer plugin, Sholl analysis was conducted for each individual neuron using the Sholl Analysis plugin. Area under the curve for each neuron was calculated and statistical significance was tested using one-way ANOVA with Bonferroni-corrected post hoc tests using Prism (GraphPad).
Layer I thickness measurements
Tissue was prepared and collected as described above for cryostat sectioning. Cryostat sections were briefly dried and then stained with DAPI. Images collected using a 10× objective from the M1/S1 region of the cortex in C3KO or WT sections were imported into FIJI. A perpendicular line from the cell body-rich Layer II to the pial surface of the brain was drawn and measured in microns. Measurements were made at six mediolateral points throughout the dorsal cortex in each brain section, and these measurements were averaged to obtain one number per section. Thirty-two to 48 brain sections were analyzed across three animals per genotype. Statistical analysis was performed using Student's t test in Prism (GraphPad).
Cortical neuronal cultures
Cultures were prepared from control or knock-out animals as described in (Garrett et al., 2012) with some modifications. In brief, cortices were isolated from postnatal day (P)0 animals, and treated with a papain enzyme solution (20 U/ml) for dissociation. The tissues were rinsed briefly in a light inhibitor solution (1 mg/ml trypsin inhibitor, 1 mg/ml BSA) and were then transferred to a heavy inhibitor solution (10 mg/ml trypsin inhibitor, 10 mg/ml BSA). The tissues were then transferred to plating media solution (Basal Medium Eagle, 5% FBS, Glutamax, N2 supplements, and 1% penicillin/streptomycin). Cells were triturated in plating media solution and plated onto German cover glass coated with Matrigel (Corning) at a density of 250,000–300,000 cells per coverslip. After 4 h, and every 2 d subsequent, media were changed to Neurobasal media supplemented with Glutamax, N21-MAX media supplement (R&D Systems) or B27-Plus (Invitrogen) and 1% penicillin/streptomycin (100 U/ml). Transfections were performed with Lipofectamine 2000 at 1 or 2 d in vitro (DIV) in Neurobasal media without antibiotics. Each coverslip was incubated with 0.5-µg total plasmid DNA or 15 pmol shRNA plasmid and 1-µl Lipofectamine 2000, mixed in OptiMEM according to manufacturer's instructions for 4 h, before being changed back to regular complete Neurobasal media. For Western blottings of cortical cultures, neurons were nucleofected using an Amaxa Nucleofector at the time of dissociation according to manufacturer's protocols for the Mouse Neuron Nucleofection kit (Lonza) and program G-013. Nucleofected neurons were plated at 1000,000 cells per coverslip and lysed for Western blottings as detailed below.
Immunoprecipitation
Cortical lysates for immunoprecipitation were prepared from control and knock-out animals at 21 d of age. Cortical tissue was homogenized in mild lysis buffer (50 mm tris-HCl, pH7.4, 150 mm NaCl, 25 mm NaF, 1% Triton X-100) using a Wheaton overhead-stirrer. Protein concentrations were calculated using the Pierce BCA protein assay, and were subsequently diluted to equal amounts before preclearing with 20 µl of Pierce Protein A/G Agarose Beads. Samples were precleared by rotating at 4°C for 30 min. Results shown are representative of three to four experiments using different cortical lysates.
Synaptosome preparations
Cortices were dissected from mice at three weeks of age. Tissues were then homogenized in Syn-Per Synaptic Protein Extraction Reagent (Thermo) supplemented with Protease and Phosphatase inhibitors using a Dounce homogenizer (1-ml Syn-Per for 100 mg of cortical tissue). Homogenized tissues were centrifuged at 1200 × g for 10 min at 4°C. The resulting supernatant was then centrifuged again at 15,000 × g for 20 min at 4°C. The supernatant (cytoplasmic fraction) was collected for analysis. The pellet (synaptosome fraction) was resuspended in more Syn-PER reagent (250 µl of Syn-Per reagent for 100 mg of starting tissue). Protein concentrations of both cytoplasmic and synaptic fractions were quantified using Pierce BCA Protein Assay. Samples were diluted to a concentration of 2 µg/µl in Laemmli buffer and boiled for 10 min at 95°C. Samples were analyzed by SDS/PAGE and Western blotting.
SDS/PAGE and Western blottings
Protein samples of equal amounts were loaded into TGX precast gels (Bio-Rad) and separated via SDS/PAGE and transferred onto nitrocellulose membranes using a TransBlot Turbo System (Bio-Rad). After protein transfer, membranes were blocked in 5% nonfat milk in Tris-buffered Saline with 0.1% Tween 20 (TBST) for at least 1 h. Membranes were then washed three times for 5 min in TBST. Primary antibodies were diluted in 5% TBST and membranes were incubated overnight at 4°C. Blots were once again washed three times for 5 min. The membrane was then incubated for at least 1 h in horseradish peroxidase (HRP)-conjugated secondary antibodies diluted into 5% nonfat milk in TBST. Signals were detected using SuperSignal West Pico or Femto Enhanced Chemiluminescent Substrates (Thermo Fisher Scientific) on a LI-COR Odyssey FC imager.
Quantitative PCR
Cortical tissue was isolated and used for RNA isolation using TRIzol reagent (ThermoFisher) according to manufacturer's protocol. Three µg of total RNA was used for cDNA synthesis using SuperScript II Reverse Transcriptase (ThermoFisher). The cDNA produced was diluted 1:10 for use with Power SYBR Green Real-Time PCR Master Mix (ThermoFisher) with the primers listed in Table 1. A Roche LightCycler instrument was used to carry out qPCR reactions.
Table 1.
List of primers used for quantitative PCR
| Primer name | Primer sequence (5′−3′) |
|---|---|
| Pcdh-αCR F | AGAGCAGGCATGCACAGC |
| Pcdh-αCR R | GACTGTTTGGGGTTGCC |
| Pcdh-β22 F | AACTATGGTAGGCAACCAGATGATC |
| Pcdh-β22 R | GAATACAGAGAGCGAAATGTGACG |
| Pcdh-γA3 F | CTCAAGATTTACTTGAAACGAAAGAAGACC |
| Pcdh-γB2 F | CAGGTACTCTTGGAGACAC |
| Pcdh-γB7 F | GGCACTGTTGGCTAGTATTTTAACTC |
| Pcdh-γA10 F | CTTTGTTAGATGATTCCAAGTGTCCTG |
| Pcdh-γC3 F | CCTGTGTTCTATAGACAGGTGTTG |
| Pcdh-γC4 F | GTCCACCCTCTGATCTTCTC |
| Pcdh-γC5 F | GTTCCCGCTCTAGTACGCTG |
| Pcdh-γA∼C5 R | CAGGTGCCAGTTTCATCACC |
| Pcdh-γCR F | CTGGCGTTTCTCTCAAGCCC |
| Pcdh-γCR R | CATGGCTTGCAGCATCTCTG |
| GAPDH F | AATGTGTCCGTCGTGGATCT |
| GAPDH R | GTTGAAGTCGCAGGAGACAA |
Retina immunostaining
Retinas were stained as described previously (Garrett et al., 2016). For whole mounts, eyes were removed and punctured with a needle, then fixed overnight by immersion in 4% PFA. Retinas were dissected from the eye and rinsed in PBS, then stained in primary antibodies diluted in 2.5% BSA with 0.5% Triton X-100 for 2–4 d at 4°C. The longer staining time was used for ChAT labeling. They were then rinsed extensively before labeling with secondary antibody overnight in the same diluent. For sectioning, fixed eyes were dissected to remove the front of the eye and the lens, cryopreserved in 30% sucrose, and frozen in Tissue-Tek OCT; 12-µm sections were cut on a cryostat, then stained on the slide with primary antibody diluted in blocking solution for 48 h at 4°C, rinsed in PBS, and labeled with secondary antibody for 1 h at room temperature.
Image analysis
For cell density, cells positive for the indicated markers were manually counted in ImageJ using the Cell Counter plugin and used to calculate cell density. Densities from two to four images per retina were averaged, and 6 retinas per genotype were compared using Student's t test.
For self-avoidance, loss of self-avoidance in starburst amacrine cells (SACs) results in larger gaps between dendrites labeled with antibodies against ChAT (Kostadinov and Sanes, 2015). This was assayed in both the OFF and ON SAC strata using two methods. First, an observer blind to genotype manually chose and measured the seven largest interdendritic areas from each image in ImageJ. These seven values were averaged per retina, and 6 retinas per genotype were compared using Student's t test. Second, images were compared using a custom web-based program and an Elo algorithm as described previously (Garrett et al., 2016). Users, blind to genotype, were instructed to compare two images and choose which had more evenly distributed dendritic coverage (i.e., smaller individual interdendritic spaces). Two random images were presented, and the user clicked on his or her choice. After choosing, a new pair of images was presented. An image's score was updated after each matchup according to the following Elo algorithm:
The value for outcome is 1 for a win and 0 for a loss, while expected is a value between 0 and 1, determined by the difference in score between the two images entering the matchup. For a matchup between images A and B, expected for image A is described by the following formula:
The image set was scored by six to eight users. Scores from each individual retina were averaged (two to four images per retina), and these retinal mean scores were used to compare across genotypes using a pairwise Wilcoxon ranked sum test.
Experimental design and statistical analysis
Statistical analysis was performed using GraphPad Prism 9 software. Data points for Western blotting graphs represent individual animals, data points for dendritic arborization graphs represent individual neurons, and data points for dendritic spine graphs represent individual dendritic segments. However, for statistical tests, the unit used was individual animals in all cases. Unpaired t test was used for comparisons between two groups. One-way ANOVA with either Bonferroni or Dunnett's post hoc test was used to correct for multiple comparisons when three or more experimental groups were compared. Pairwise Wilcox ranked sum test was used for SAC self-avoidance analysis.
Antibodies
Primary antibodies and associated information used for this study are found in Table 2. Secondary antibodies were conjugated with Alexa-488, Alexa-568, or Alexa-647 (1:500, Invitrogen) or HRP (1:1000–1:5000, Jackson ImmunoResearch).
Table 2.
Primary antibodies used in these studies
| Antibody | Host | Concentration | Company | RRID |
|---|---|---|---|---|
| Tyrosine hydroxylase | Sheep | 1:500 | Millipore | AB_11213126 |
| bNOS | Rabbit | 1:500 | Sigma-Aldrich | AB_260796 |
| Melanopsin | Rabbit | 1:1000 | Advanced Targeting Systems | AB_126799 |
| VGLUT3 | Guinea pig | 1:10,000 | Millipore | AB_2187832 |
| PKCα | Rabbit | 1:1000 | Sigma-Aldrich | AB_477345 |
| ChAT | Goat | 1:400 | Millipore | AB_2079751 |
| Glyt1 | Goat | 1:500 | Millipore | AB_90893 |
| Calbindin | Rabbit | 1:500 | Millipore | AB_2068336 |
| PKA RIIβ | Mouse | 1:500 | BD Biosciences | AB_397957 |
| Pan-γ-Pcdh N159/5 | Mouse | 1:500 | UC Davis/NIH NeuroMab | AB_2877195 |
| Pan-γ-Pcdh-A N144/32 | Mouse | 1:500 | UC Davis/NIH NeuroMab | AB_2877459 |
| γ-Pcdh-B2 N148/30 | Mouse | 1:500 | UC Davis/NIH NeuroMab | AB_2877457 |
| γ-Pcdh-C3 N174B/27 | Mouse | 1:500 | UC Davis/NIH NeuroMab | AB_2877444 |
| β-Tubulin | Mouse | 1:500 | Sigma-Aldrich | AB_477577 |
| NeuN | Mouse | 1:500 | Millipore | AB_2298772 |
| Ctip2 | Rat | 1:200 | Abcam | AB_2064130 |
| Cux1 | Rabbit | 1:100 | Santa Cruz Biotechnology | AB_2261231 |
| Parvalbumin | Mouse | 1:400 | Sigma-Aldrich | AB_477329 |
| GFAP | Mouse | 1:500 | Sigma-Aldrich | AB_477010 |
| Cleaved caspase-3 | Rabbit | 1:500 | Cell Signaling | AB_2341188 |
| Axin1 | Rabbit | 1:500 | Cell Signaling | AB_2274550 |
| Cdc42 | Mouse | 1:500 | Cytoskeleton Inc. | AB_10716593 |
| β-Actin | Mouse | 1:1000 | Cell Signaling | AB_2242334 |
| PSD-95 | Mouse | 1:1000 | Cell Signaling | AB_2721262 |
| GFP | Chicken | 1:500 | Thermo Fisher | AB_2534023 |
| HA | Rat | 1:500 | Roche | AB_390918 |
| HA | Rabbit | 1:250 | Cell Signaling | AB_1549585 |
| FoxP2 | Rabbit | 1:4000 | Abcam | AB_2107107 |
| Pax2 | Rabbit | 1:200 | Thermo Fisher | AB_2533990 |
| Neurofilaments (SMI-312) | Mouse | 1:1000 | Covance | AB_2314906 |
Results
Generation and validation of a Pcdhgc3 knock-out mouse
To investigate the role of Pcdhgc3 during cortical development, we used CRISPR/Cas9 genome editing to generate a γC3 knock-out mouse (PcdhgC3KO) by targeting the 5′ end of its variable exon (Fig. 1A). The resulting mutation generated a 13-base pair (bp) deletion introduced into Pcdhgc3 8 bp after the transcriptional start site, which created a frameshift resulting in a stop codon after 6 amino acids (Fig. 1B). PcdhgC3KO (hereafter, C3KO) mice were viable, fertile, and exhibited no outward morphologic defects. To confirm that discrete mutation of the Pcdhgc3 variable exon did not influence the expression of other cPcdh genes, we used qPCR. While we found the expected nonsense-mediated reduction of cortical Pcdhgc3 transcripts (using a forward primer in the 3′ end of the γC3 variable exon; no signal was found using a forward primer covering the deletion site), there were no significant reductions in Pcdha transcripts encoding α-Pcdhs (using primers to the α constant exons), in transcripts encoding β-Pcdh-22 (the Pcdhb cluster contains no constant exons, so we used this gene as a proxy), γA3, γA10, γB2, γB7, γC4, γC5, or in the Pcdhg cluster as a whole (using primers to the Pcdhg constant exons; Fig. 1C; ANOVA with Bonferroni's multiple comparison tests; all nonsignificant p values > 0.9999, ****p values < 0.0001, six animals per genotype). This is consistent with our prior analysis of an allelic series of Pcdhg CRISPR mutants (Garrett et al., 2019) in which expression of remaining functional variable exons was maintained. Western blotting of cortical lysates from C3KO and wild-type littermates with multiple specific monoclonal antibodies confirmed the loss of γC3 protein, with the expected maintained expression of other γ-Pcdh proteins (Fig. 1D). Histologically, the adult cerebral cortex did not display any gross disorganization, with evidence of normal neuronal density and no change in apoptosis as assessed with immunostaining for cleaved caspase-3 (Fig. 1E). However, as we found previously for mice lacking all 22 γ-Pcdh isoforms (Garrett et al., 2012), C3KO mice exhibited a small but significant reduction in the extent of the cell-sparse, dendritic process-rich Layer I (Fig. 1F; t test; *p = 0.019, three mice and 32–48 sections per genotype).
Many neuronal populations in the spinal cord and retina undergo excessive apoptosis in mice lacking all 22 γ-Pcdhs (Wang et al., 2002b; J.A. Weiner et al., 2005; Lefebvre et al., 2008; Prasad et al., 2008). In recent work, we presented evidence that the γC4 isoform was uniquely important for maintaining neuronal survival in the spinal cord and retina (Garrett et al., 2019). To confirm this, we analyzed these tissues in the C3KO mouse line. Consistent with Garrett et al. (2019), C3KO neonatal spinal cords exhibited no gross morphologic alterations with no increase in apoptotic cells or loss of particular interneuron populations (Fig. 2; t test; all ns, p = 0.14–0.44; three animals and 25–36 sections per genotype). Similarly, loss of γC3 did not cause alterations to overall retinal morphology, cellular organization, or the density of VGlut3+ or TH+ amacrine cells and melanopsin+ retinal ganglion cells [t test (Fig. 3G,T,U) or Wilcoxon (Fig. 3V); all ns, p = 0.07–0.76; six retinae per genotype]. These data support the unique, isoform-specific role of γC4 in mediating neuron survival (Garrett et al., 2019) and demonstrate that γC3 is not strictly required for grossly normal formation of the nervous system.
Figure 2.

C3KO mice do not exhibit increased apoptosis of spinal interneurons. A, Cryosections of P0 spinal cords from control and C3KO pups immunostained for the indicated markers (green) and counterstained with DAPI for nuclei (blue). Scale bar: 200 µm. No gross abnormalities of the spinal cord (previously observed in complete Pcdhg null mutants; Wang et al., 2002b; Prasad et al., 2008) were observed in C3KO mutants, nor were any alterations in the number of FoxP2-labeled ventral interneurons (B), CC3-labeled apoptotic cells (C), or Pax2-labeled dorsal and ventral interneurons (D). ns = not significant.
Figure 3.

C3KO mice do not exhibit altered cell survival or dendrite self-avoidance in the retina. Whole-mount retinas were stained for (A, B) Vglut3 to label glutamatergic amacrine cells, (C, D) tyrosine hydroxylase (TH) to label dopaminergic amacrine cells (DA cells), and (E, F) melanopsin to label intrinsically photosensitive retinal ganglion cells (ipRGCs). For all cell types, there was no significant change in cell density in adult C3KO mutants (quantified in G). Self-avoidance in starburst amacrine cells (SACs) was assayed by analyzing interdendritic space in (H, I) the OFF strata and (J, K) the ON strata. T–V, No increase in the space between SAC dendrites was observed when quantified by Elo analysis (see Materials and Methods) or manual measurement. Projections of representative cell types within the inner plexiform layer (IPL) were analyzed to assess general retinal organization. L–M, SACs (ChAT+), Vglut3+ amacrine cells and rod bipolar cells (PKCa+) targeted normally, as did (N, O) DA cells (TH+) and ipRGCs (melanopsin+), (P, Q) bnos+ GABAergic amacrine cells and Glyt1+ glycinergic amacrine cells, and (R, S) Calbindin+ cells and type 3b cone bipolar cells (PKARIIb+). The expected organization of projections, which was observed in both control and C3KO retinae, is presented in W. For all experiments, six adult retinas per genotype were analyzed. ns = not significant.
γC3 interacts with Axin1 in vivo and influences its synaptic localization
In our previous work, we established the isoform-specific ability of overexpressed, affinity-tagged γC3 to interact, via its VCD, with Axin1 and to promote its association with the membrane in HEK293 cells in vitro (Mah et al., 2016). We thus sought to establish that these proteins in fact interact in the brain in vivo. Cortical lysates were obtained from control and C3KO mice at postnatal day (P)21 and subjected to immunoprecipitation with an antibody that recognizes the shared C terminus of all 22 γ-Pcdhs (mAb N159/5). Following this, protein complexes were separated using SDS-PAGE, and Western blotted using antibodies against Axin1, γC3, and all γ-Pcdhs. Axin1 was robustly immunoprecipitated with γ-Pcdhs in wild-type lysates (Fig. 4A). Consistent with γC3 being a primary Axin1 interactor among γ-Pcdhs, comparatively little Axin1 was apparent in immunoprecipitations from C3KO cortical lysates (Fig. 4A).
Figure 4.

γC3 interacts with Axin1 in vivo and is required for normal Axin1 subcellular localization. A, Endogenous Axin1 co-immunoprecipitates with γ-Pcdhs from P21 control cortical lysates; comparatively little Axin1 is co-immunoprecipitated from cortical lysates of C3KO mice. Representative Western blot images of (B) cytoplasmic and (C) synaptic fraction preparations from P21 cortex. Quantification of Axin1 (D), CDC42 (E), and β-actin (F) intensity normalized to total protein from cytoplasmic and synaptic fractions from C3KO and control mice. Levels of all three proteins are significantly reduced in synaptic, but not cytoplasmic fractions. n = 6 animals per genotype. Unpaired t test; *p <0.05, ***p < 0.001; ns = not significant.
Considering the roles played by both Axin1 and γ-Pcdhs at the synapse, we asked whether the loss of γC3 affected Axin1's reported (Y. Chen et al., 2013) localization to synaptic fractions. We prepared synaptosomes from control and C3KO cortices at P21 and analyzed fractions by quantitative Western blotting (Fig. 4B,C). We found that C3KO samples contained decreased synaptic Axin1 without alterations to the amount of Axin1 found in the cytoplasmic fraction (Fig. 4D; t test; cytoplasmic, ns p = 0.65, synaptic, p = 0.0009; six samples per genotype). Because Axin1 is known to play an important role as a scaffolding protein at the synapse (Hirabayashi et al., 2004; Fang et al., 2011; Y. Chen et al., 2013, 2015), we hypothesized that reductions in Axin1 at the synapse might also reduce the amount of Axin1's interactors. Indeed, we also found reduced synaptic levels of Cdc42 (Fig. 4E; t test; cytoplasmic, ns p = 0.14, synaptic, p = 0.04; six samples per genotype) and β-actin (Fig. 4F; t test; cytoplasmic, ns p = 0.07, synaptic, p = 0.04; six samples per genotype). Together with our prior work (Mah et al., 2016), these data suggest that γC3 interactions with Axin1 affect its localization in neurons.
Cortical dendritic spines of mice lacking γC3 are morphologically normal
Considering the alterations to synaptic scaffolding elements we found at the synapse, we next asked whether C3KO mice exhibited alterations in dendritic spine morphology or density. We crossed C3KO mice with the Thy1-YFPH transgenic (Feng et al., 2000), in which a subset of Layer V pyramidal neurons express YFP, which fills their processes and enables analysis of morphology. Basal dendritic segments of Layer V pyramidal neurons from the S1 region of the cortex (50–200 µm from the soma) were imaged from C3KO mice and littermate controls at five weeks of age and analyzed using Neurolucida software (Fig. 5A). We found no alterations in overall dendritic spine density (Fig. 5B; t test; ns p = 0.79; at least 54 dendritic segments from at least seven mice per genotype), nor in the density of thin, mushroom, or stubby spine classes (Fig. 5C; t test; ns p = 0.40–0.98; at least 54 dendritic segments from at least seven mice per genotype). As dendritic spine measurements such as volume and surface area have been shown to correlate with synaptic strength and efficacy (Harris and Stevens, 1988, 1989; Harris et al., 1992; Grutzendler et al., 2002; Nimchinsky et al., 2002, 2004; Mizrahi and Katz, 2003; Holtmaat et al., 2005; Ashby et al., 2006), we analyzed these across spine types in both C3KO mice and controls. Again, we found no significant alteration in average spine volumes (Fig. 5D; t test; ns p = 0.50–0.87; at least 54 dendritic segments from at least seven mice per genotype) or surface areas (Fig. 5E; t test; ns p = 0.46–0.84; at least 54 dendritic segments from at least seven mice per genotype). While somewhat surprising given the altered synaptic localization of Axin1 and Cdc42 in C3KO cortex (Fig. 4B–F), this lack of effect is not inconsistent with our prior results indicating that the effect of the γ-Pcdh family on synapse density is associated primarily with their promiscuous extracellular cis-interactions with the synaptic organizers Neuroligin (Nlg)-1 and Neuroligin-2 (Molumby et al., 2017; Steffen et al., 2021). C3KO mice retain expression of 21 other γ-Pcdhs that could interact with Neuroligins and maintain normal spine density.
Figure 5.

C3KO mice show no alterations in dendritic spine density or morphology. A, Representative images showing dendritic spines of Thy1-YFPH-labeled Layer V pyramidal neurons from the S1 cortex of five-week-old mice. Scale bar: 5 µm. Quantification of overall dendritic spine density (B) and of the density of thin, mushroom, and stubby dendritic spine types (C). Quantification of average spine volume (D) and of average spine surface area (E) of all spines and of each dendritic spine class (thin, mushroom, and stubby). n ≥ 58 control dendritic segments across 7 mice, n = 54 C3KO dendritic segments across 9 mice. Error bars represent the SEM. No significant differences were observed between control and C3KO genotypes. ns = not significant.
Mice lacking γC3, but not other γ-Pcdh isoforms, exhibit decreased dendritic complexity
In addition to its role in dendritic spines, Axin1 plays a role in promoting dendritic arborization in cultured forebrain neurons (Y. Chen et al., 2015). Our prior work showed that loss of the γ-Pcdh family led to greatly reduced arborization of cortical neuron dendrites, and that both homophilic γ-Pcdh interactions and inhibition of a FAK/PKC signaling pathway by the shared constant cytoplasmic domain promotes arbor complexity (Garrett et al., 2012; Keeler et al., 2015b; Molumby et al., 2016). Because γC3 is both highly and broadly expressed compared with other γ-Pcdh isoforms (Esumi et al., 2005; Kaneko et al., 2006; Hirano et al., 2012) and because of the interaction of its unique VCD with Axin1 (Mah et al., 2016), we hypothesized that γC3 specifically contributes to the formation of dendritic arbors. The observed reduction in the extent of cortical Layer I in C3KO mice (Fig. 1F) supports this, as this layer consists largely of pyramidal neuron apical dendritic tufts.
We again bred C3KO mice to Thy1-YFPH mice to label cortical Layer V neurons and collected brains from C3KO and control littermates at three weeks (when much dendrite growth is occurring) and six weeks of age (when arbors have largely formed; Koleske, 2013). YFP-labeled Layer V neurons were imaged from 200-µm Vibratome sections, 3D stacks obtained using confocal microscopy, and arbor complexity determined using Sholl analysis. We found that pyramidal neurons lacking γC3 indeed exhibited significantly decreased dendritic complexity both at three weeks (Fig. 6A–C; t test; p = 0.001; at least 80 neurons from at least four mice per genotype) and at six weeks (Fig. 6G–I; t test; p = 0.0005; at least 80 neurons from at least four mice per genotype) of age. We also found significant reductions in other dendritic morphologic properties including number of branch points, and total dendritic length (but not in average branch length) at three weeks [t test; p = 0.001 (Fig. 6D), 0.0007 (Fig. 6E); ns, 0.07 (Fig. 6F); at least 80 neurons from at least four mice per genotype] and six weeks [t test; p=<0.0001 (Fig. 6J), 0.0009 (Fig. 6K); ns, 0.19 (Fig. 6L); at least 80 neurons from at least four mice per genotype] in C3KO mice. We showed previously that mice lacking all γ-Pcdh proteins exhibited a reduction of ∼40% in cortical Layer V dendritic arbor complexity compared with controls (Garrett et al., 2012); in comparison, our present results indicate a reduction of ∼30% in C3KO mice. This suggests that γC3 plays a particularly critical isoform-specific role in regulating dendritic complexity.
Figure 6.
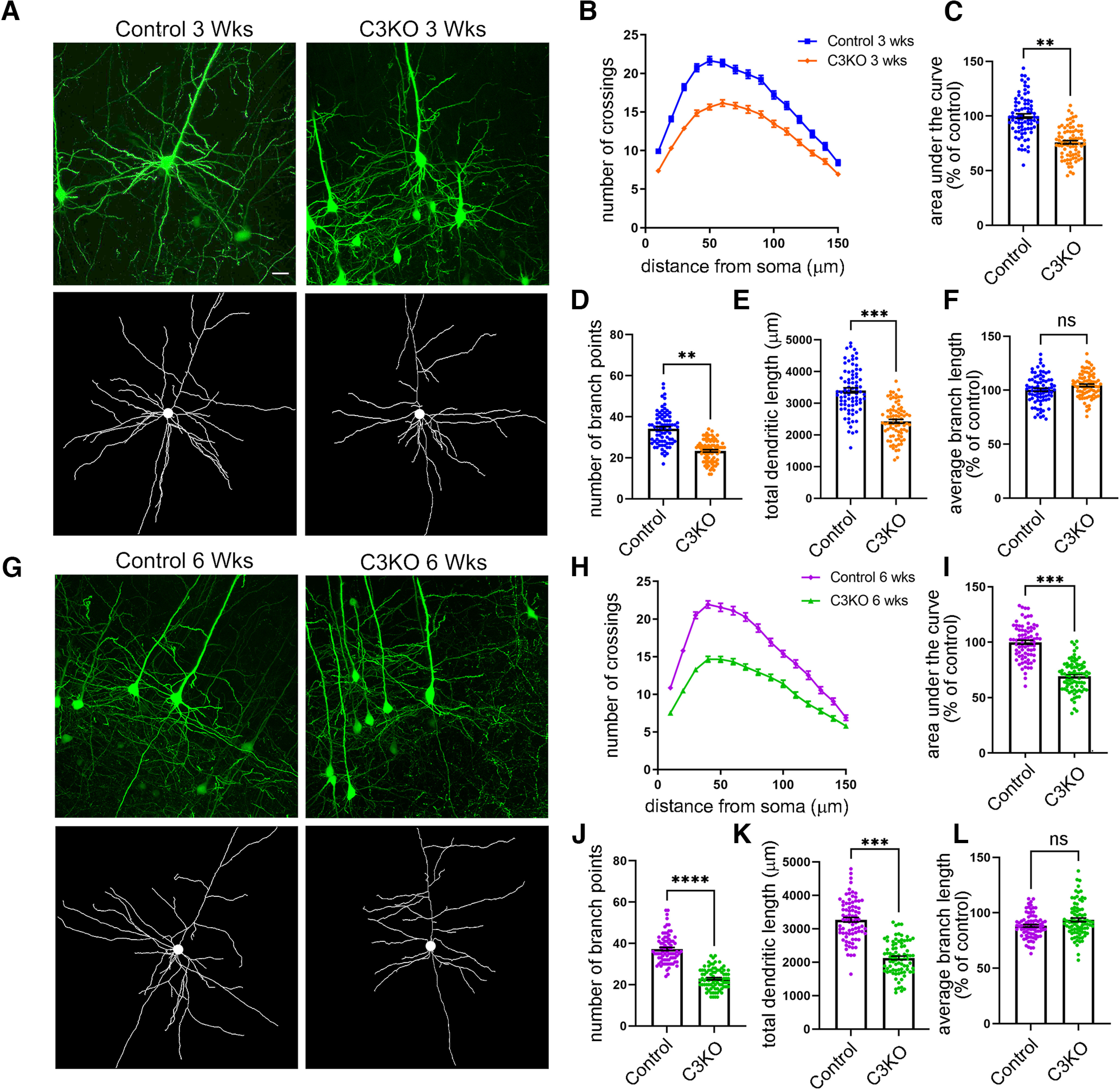
C3KO mice exhibit decreased dendritic complexity. A, Representative YFP images (top) and tracings (bottom) of Thy1-YFPH-labeled Layer V pyramidal neurons of mice three weeks of age. B, Sholl analysis graphs show dendritic crossings at spheres of increasing 10-µm intervals of mice at three weeks of age. Graphs showing area under the curve of Sholl graphs (C), average number of branch points per neuron (D), total dendritic length (E), and average branch length (F) of mice at three weeks of age. G, Representative YFP images (top) and tracings (bottom) of Thy1-YFPH-labeled Layer V pyramidal neurons of mice six weeks of age. H, Sholl analysis graphs show dendritic crossings at spheres of increasing 10-µm intervals of mice at six weeks of age. Graphs showing area under the curve of Sholl graphs (I), average number of branch points per neuron (J), total dendritic length (K), and average branch length (L) of mice at six weeks of age. Significant reductions in dendritic complexity are seen in C3KO compared with control at both ages. Scale bar: 25 µm. n = ∼80 neurons from at least 4 mice per genotype. Unpaired t test. Error bars represent the SEM; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns = not significant.
To establish that this morphologic phenotype was associated specifically with the loss of the γC3 isoform, and not simply with a reduction of the number of γ-Pcdh isoforms or total amount of protein, we analyzed dendritic complexity at six weeks of age in two additional Pcdhg mutant lines derived in our prior work (Garrett et al., 2019): line 13R1, in which nine of the 22 γ-Pcdh isoforms are absent while the other 13 (including γC3) remain expressed; and a specific C4KO line, which lacks only γC4, but retains the other 21 γ-Pcdhs (Fig. 7A). Because both of these lines lack γC4, which is the γ-Pcdh isoform uniquely critical for neuronal and postnatal organismal survival (Garrett et al., 2019), they die shortly after birth. To enable analysis of postnatal dendrite arborization, these lines were therefore bred as trans-heterozygotes over the Pcdhgfcon3 conditional locus KO allele which was excised only in the forebrain using an Emx1-Cre allele (Gorski et al., 2002; Lefebvre et al., 2008). In the resulting mice, which also carried the Thy1-YFPH allele (Feng et al., 2000), excitatory cortical neurons contained one 13R1 or C4KO allele and one Pcdhg null allele, allowing us to analyze dendrite complexity in the absence of γC4 only, or in the absence of nine isoforms (γA1, γB1, γA5, γA7, γA9, γA10, γB6, γB7, and γC4). For this experimental paradigm, it is important to note that Pcdhg heterozygous mutant neurons exhibit normal dendrite complexity (Garrett et al., 2012). For simplicity, we will use “C4KO” to refer to mice with the genotype Emx1-Cre;Pcdhgfcon3/C4KO;Thy1-YFPH and “13R1” to refer to mice with the genotype Emx1-Cre;Pcdhgfcon3/13R1;Thy1-YFPH.
Figure 7.
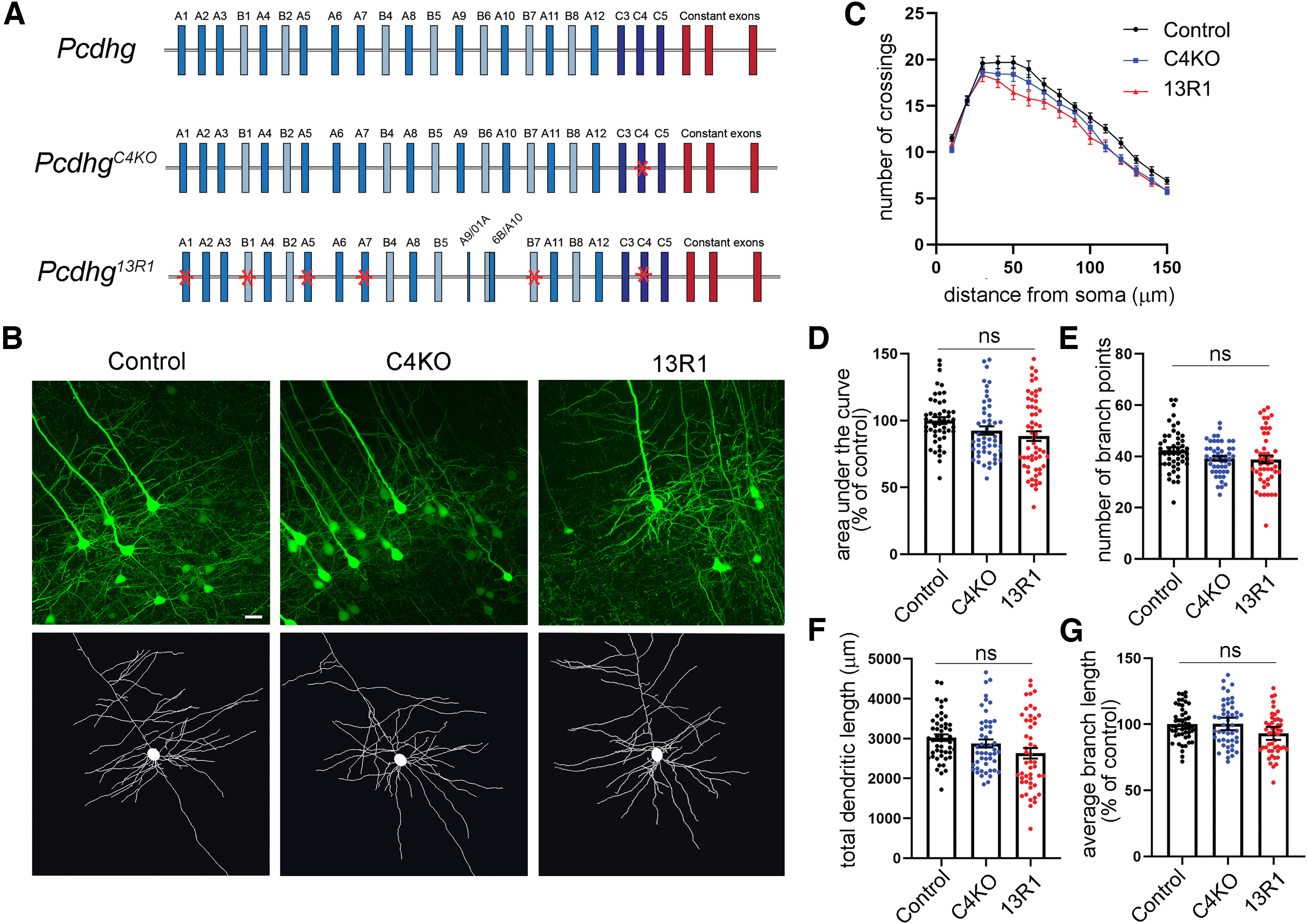
Dendritic arborization deficits found in C3KO mutants are not observed in other Pcdhg locus mutants. A, Schematic representation of the mouse Pcdhg locus (control), the PcdhgC4KO mutant mouse locus lacking only γC4, and the Pcdhg13R1 mutant locus lacking nine isoforms (mutations indicated by red asterisks). B, Representative images and tracings of Thy1-YFPH-labeled Layer V pyramidal neurons of mice six weeks of age. C, Sholl analysis graphs show dendritic crossings of spheres of increasing 10-µm intervals of mice at six weeks of age. D, Graphs showing area under the curve of Sholl graphs, (E) total branch points, (F) total dendritic length, and average branch length (G) of mice at six weeks of age. Although 13R1 neurons exhibit a slight trend toward lower complexity, neither they nor C4KO neurons were significantly different from controls on any measure. Scale bar: 25 µm. n ≥ 49 neurons per genotype and at least 3 animals from each genotype. One-way ANOVA; Dunnett's multiple comparisons test. Error bars represent the SEM; ns = not significant.
In contrast to C3KO mice, neither C4KO nor 13R1 Layer V neurons exhibited significant reductions in overall dendritic complexity, total dendritic length, average branch length, or the number of branch points (Fig. 7B–G; ANOVA; ns, p = 0.52–0.61; at least 49 neurons from at least three animals per genotype), despite a small trend toward less complexity in 13R1. This result confirms that at least some individual γ-Pcdh isoforms have distinct, nonoverlapping neuronal functions: of the isoforms tested, only γC3 is required for normal dendrite arborization (Figs. 6, 7), while only γC4 is required for neuronal and organismal survival (Garrett et al., 2019). While our analysis of these three mutant Pcdhg alleles cannot entirely preclude the possibility that a small number of other γ-Pcdh isoforms might contribute to dendritic arborization, it does provide strong evidence that γC3 plays a uniquely important role in the establishment of proper dendritic morphology in the cortex in vivo.
γC3-mediated dendritic arborization is Axin1 dependent
We next hypothesized that the dendritic arborization deficits observed in C3KO mice might be because of γC3's interaction with Axin1. To investigate this, we cultured cortical neurons from C57BL/6J WT and C3KO pups at P0 and transfected them at low efficiency with an shRNA construct previously shown to efficiently target Axin1 (Y. Chen et al., 2015) or with a control scrambled shRNA construct (Fig. 8A) along with a GFP construct to identify transfected cells. Neurons were fixed after 8 d in vitro, arbors traced using GFP fluorescence, and Sholl analysis performed to quantify dendritic complexity. First, we confirmed that C3KO cortical neurons in vitro (which include a mix of all layer specifications) exhibited a similarly substantial reduction in dendritic complexity as observed for Layer V neurons in vivo, compared with control neurons (Fig. 8B,C; ANOVA with Tukey's multiple comparison test; C3KO+Scramble shRNA vs Control+Scramble shRNA, p < 0.0001; at least 25 neurons per condition for this and all subsequent comparisons).
Figure 8.
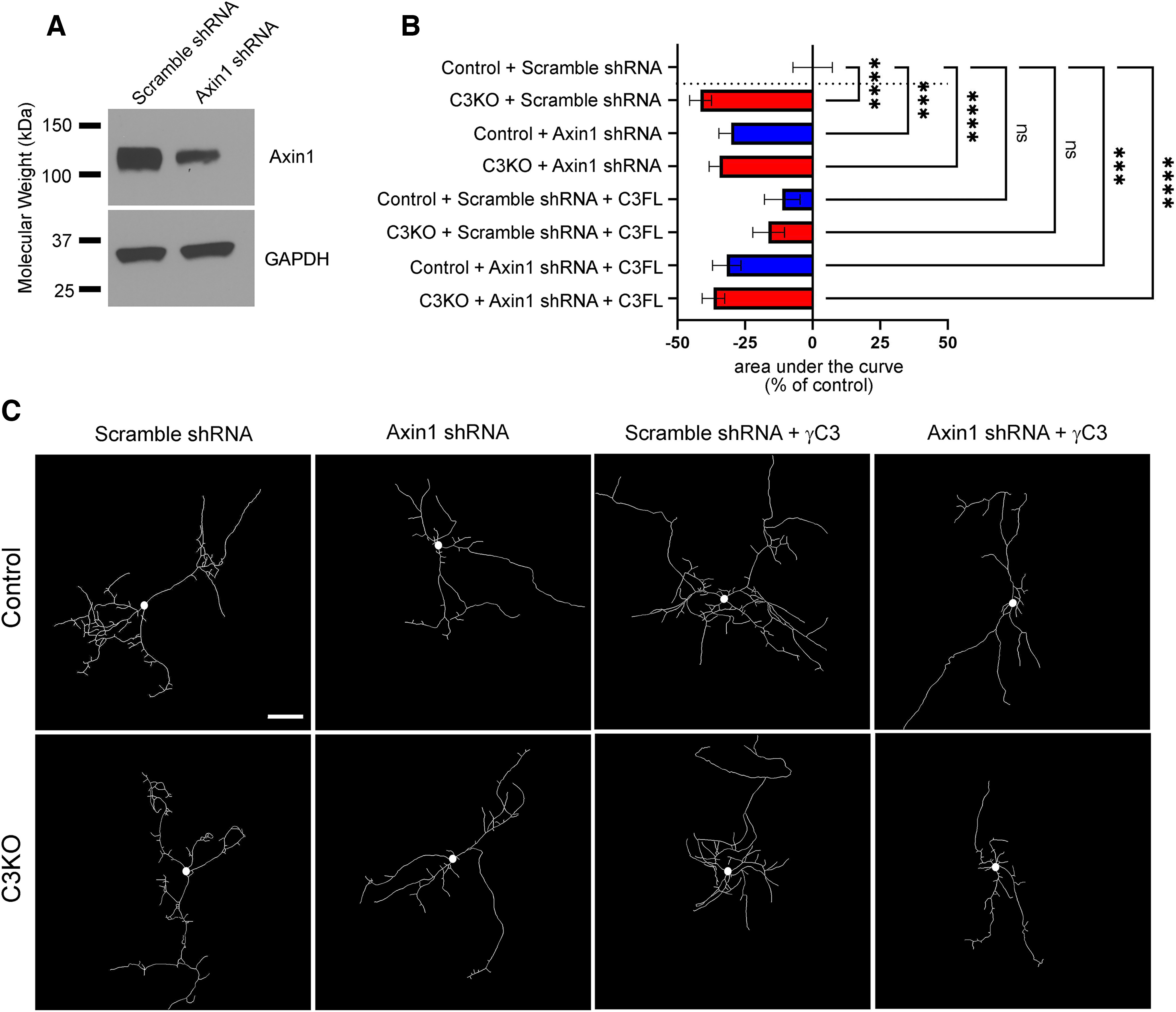
γC3-mediated dendritic arborization is dependent on Axin1. A, Cortical neurons cultured from P0 pups and nucleofected (∼50% efficiency) at plating with Axin1 shRNA (previously characterized by Chen et al., 2015) or its scrambled control. Neurons were lysed at day in vitro (DIV)8 and lysates analyzed by Western blotting using antibodies against Axin1. As expected, Axin1 levels are drastically reduced in neurons expressing the Axin1 shRNA knock-down construct. B, Quantification of area under the curve for Sholl analysis of control and C3KO cultured cortical neurons transfected with GFP, either Axin1 shRNA or the scrambled control shRNA, ± a full-length γC3 construct and analyzed at DIV8. Bars to the left indicate reduced complexity; bars to the right indicate increased complexity. C, Representative traces of neurons from each transfection condition. C3KO neurons exhibit significantly reduced dendrite complexity in culture as in vivo. Arbor complexity can be rescued by re-expression of γC3, but only if normal Axin1 levels are present. Scale bar: 50 µm. n ≥ 25 neurons per condition. One-way ANOVA; Dunnett's multiple comparisons test. Error bars represent the SEM; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns = not significant.
Next, we found that control neurons transfected with the Axin1 shRNA construct exhibited a significant decrease in dendritic complexity that was comparable to that found in C3KO cultures (Fig. 8B,C; ANOVA with Tukey's multiple comparison test; Control+Scramble shRNA vs Control+Axin1 shRNA, p = 0.0009). Importantly, C3KO neurons transfected with the Axin1 shRNA construct exhibited no further effect on arborization compared with C3KO neurons transfected with the scrambled control shRNA (Fig. 8B,C; ANOVA with Tukey's multiple comparison test; C3KO+Scramble shRNA vs C3KO+Axin1 shRNA, ns p = 0.98), consistent with γC3 and Axin1 loss affecting neurons through the same pathway. We next asked whether it was possible to rescue dendritic defects in C3KO neurons by transfecting with a construct that re-expressed full-length γC3 protein. Indeed, we found that C3KO neurons re-expressing full-length γC3 returned to levels of dendritic complexity that were statistically indistinguishable from those of control neurons (Fig. 8B,C; ANOVA with Tukey's multiple comparison tests; C3KO+Scramble shRNA+C3FL vs Control+Scramble shRNA ns, p = 0.45; C3KO+Scramble shRNA vs C3KO+Scramble shRNA+C3FL, p = 0.04). Interestingly, transfection of control neurons with the full-length γC3 construct did not increase arbor complexity, a result consistent with our prior work in vivo (Molumby et al., 2016) indicating that variation in total γ-Pcdh protein levels is not a primary driver of arborization (note also that low efficiency lipofection did not typically result in two nearby neurons both re-expressing γC3, so homophilic interactions do not come into play in this experiment; Fig. 8B,C; ANOVA with Tukey's multiple comparison tests; Control+Scramble shRNA vs Control+Scramble+C3FL shRNA ns, p = 0.81). Importantly, this rescue of C3KO neurons by re-expression of full-length γC3 was completely abrogated by Axin1 knock-down (Fig. 8B,C; ANOVA with Tukey's multiple comparison tests; C3KO+Axin1 shRNA+C3FL vs Control+Scramble shRNA p < 0.0001). Together, these data indicate that (1) reduced dendritic complexity is a general, intrinsic feature of cortical neurons lacking γC3 and (2) γC3 and Axin1 likely operate in the same signaling pathway affecting dendrite arborization.
Our previous work (Mah et al., 2016) identified the VCD of γC3 as the site of interaction with Axin1, and found that this interaction facilitates the association of Axin1 with the membrane. To confirm a link between γC3 and Axin1 signaling in the promotion of dendritic complexity, we next used a variety of deletion constructs to determine what portion of the γC3 protein was critical. In the first set of experiments, control and C3KO neurons were transfected with either (1) a C3Δecto construct (which encodes a protein lacking the 6 EC repeats, leaving only a short extracellular stub, the transmembrane domain, the VCD, and the constant cytoplasmic domain) or (2) a C3Δcyto (which encodes a protein containing only the 6 EC repeats and a transmembrane domain with a short intracellular stub, lacking both the VCD and constant cytoplasmic domain (Schreiner and Weiner, 2010; Fig. 9C) along with either the scrambled shRNA, or the Axin1 shRNA knock-down construct. We found that C3KO neurons transfected with C3Δcyto still exhibited significant dendritic arborization defects (Fig. 9A,D; ANOVA with Tukey's multiple comparison tests; C3KO+Scramble shRNA+C3Δcyto vs Control+Scramble shRNA p = 0.027; at least 25 neurons per condition for this and all subsequent comparisons), while those transfected with C3Δecto were rescued, displaying dendritic complexity indistinguishable from that of controls (Fig. 9B,D; ANOVA with Tukey's multiple comparison tests; C3KO+Scramble shRNA+C3Δecto vs Control+Scramble shRNA, ns p = 0.98). As expected from our first set of culture experiments (Fig. 8), we found that expression of the C3Δecto construct was only able to rescue dendritic arborization in C3KO neurons when normal levels of Axin1 were present: C3KO neurons transfected with both the C3Δecto and Axin1 shRNA constructs exhibited similar dendritic defects as C3KO neurons transfected with the ineffective C3Δcyto construct (Fig. 9B,D; ANOVA with Tukey's multiple comparison tests; C3KO+Axin1 shRNA+C3Δecto vs Control+Scramble shRNA p = 0.019). Again, neither the C3Δcyto nor the C3Δecto construct affected dendritic complexity significantly in control neurons that already expressed normal levels of γC3 (Fig. 9A,B,D; ANOVA with Tukey's multiple comparison tests; Control+Scramble shRNA vs Control+Scramble shRNA+C3Δcyto, ns p = 0.88; Control+Scramble shRNA vs Control+Scramble shRNA+C3Δecto, ns p = 0.98).
Figure 9.
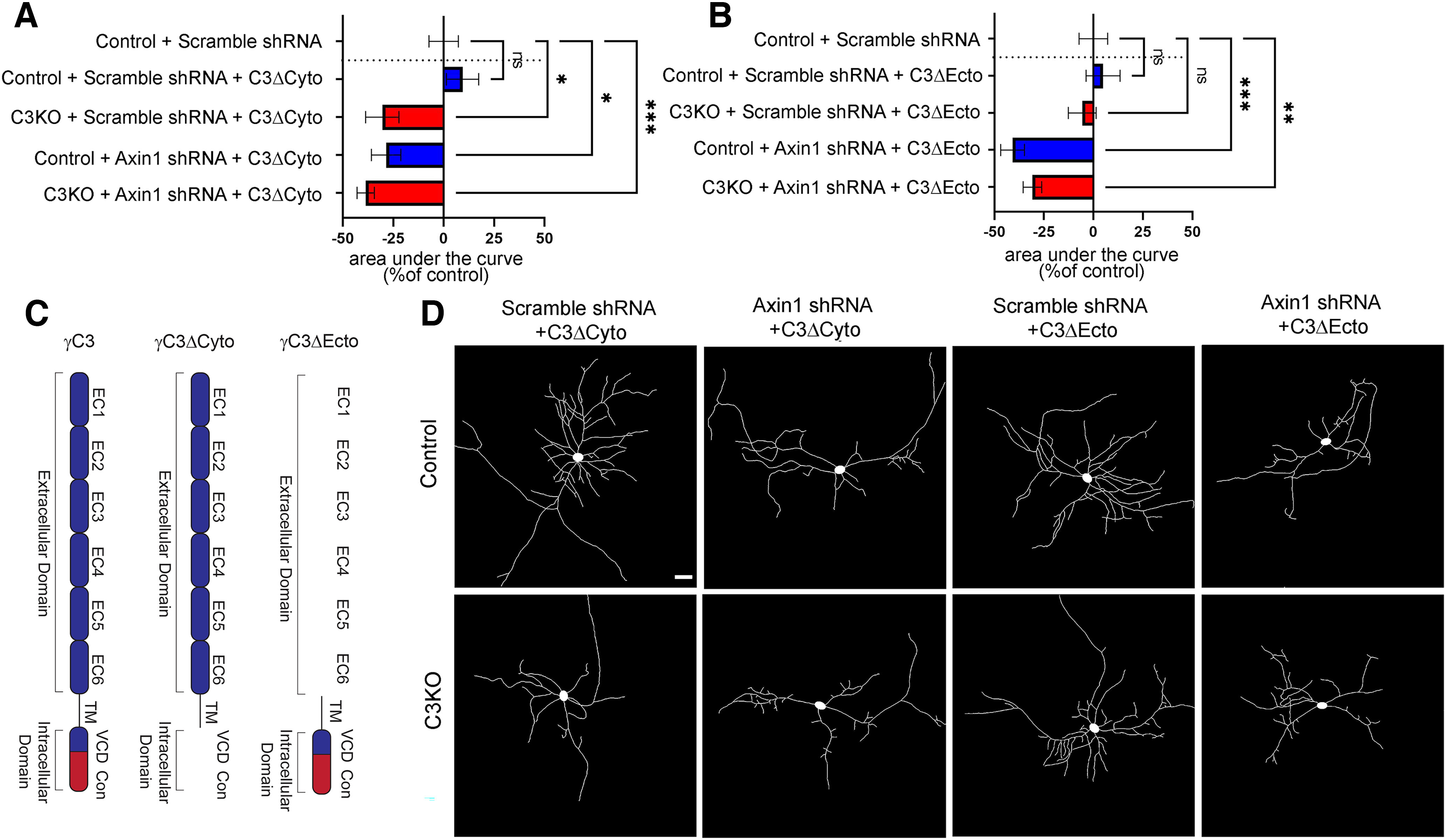
The intracellular domain of γC3 is required for Axin1-mediated dendritic arborization. A, B, Quantification of area under the curve for Sholl analysis of control and C3KO cultured cortical neurons transfected with GFP, either Axin1 shRNA or the scrambled control shRNA, and a γC3 construct lacking either the cytoplasmic (C3Δcyto; A), or the extracellular domain (C3Δecto) and analyzed at DIV8. B, Bars to the left indicate reduced complexity; bars to the right indicate increased complexity. C, Schematic depiction of γC3 deletion transfection constructs transfected. D, Representative traces of neurons from each transfection condition. Scale bar: 50 µm. n ≥ 25 neurons per condition. One-way ANOVA; Dunnett's multiple comparisons test. Error bars represent the SEM; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns = not significant.
Together, these data and those in our prior work (Mah et al., 2016) are consistent with the hypothesis that γC3 promotes dendritic arborization via interactions of its VCD with Axin1. To provide additional support for this, we performed a further set of rescue experiments in cultured C3KO neurons (Fig. 10). In addition to the already-tested C3FL and C3Δecto constructs, we transfected C3KO neurons with a C3 construct lacking only the constant domain but retaining the VCD (C3Δcon; Fig. 10A). These proteins, as well as the C3Δcyto protein discussed above (Fig. 9), localized to small puncta or patches along dendrites in transfected neurons (Fig. 10D) in a grossly similar manner. Consistent with our prior results showing that any C3 construct containing the VCD was able to bind to Axin1 and mediate C3 functions (Mah et al., 2016), we found that the C3Δcon construct significantly rescued dendritic arbor complexity when expressed in C3KO neurons, just as the C3FL and C3Δecto constructs did (Fig. 10B,C; ANOVA with Tukey's multiple comparison test; Control transfection vs C3FL, p < 0.0001; Control transfection vs C3Δecto, p < 0.0001; Control transfection vs C3Δcon, p < 0.0001; at least 29 neurons per condition for this and all subsequent comparisons). Finally, we transfected C3KO neurons with a construct expressing the isolated C3 VCD with a palmitoylation signal at its N terminus (Mah et al., 2016) to localize it to the cytoplasmic face of the plasma membrane (C3VCD; Fig. 10A); this construct also rescued arborization to the same extent (Fig. 10B,C; ANOVA with Tukey's multiple comparison tests; Control transfection vs C3-VCD, p < 0.0001). Together, these rescue experiments indicate that γC3 promotes dendritic arborization through a mechanism involving its unique VCD and Axin1.
Figure 10.
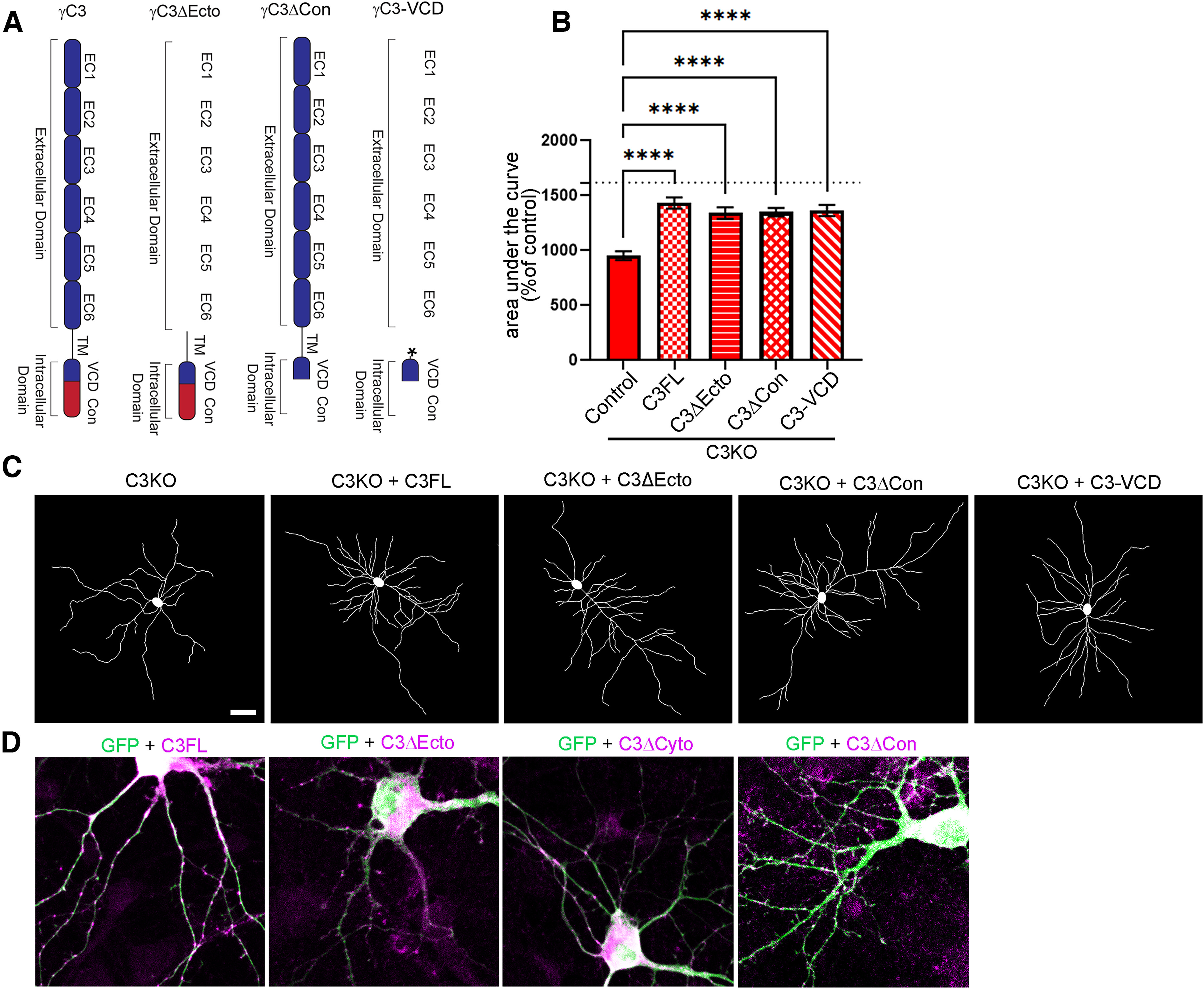
The γC3 variable cytoplasmic domain is necessary and sufficient to rescue dendritic arborization in C3KO neurons. A, Schematic depiction of γC3 deletion transfection constructs transfected. Asterisk indicates palmitoylation sequence at the N terminus of the γC3-VCD construct. B, Quantification of area under the curve for Sholl analysis of C3KO cultured cortical neurons transfected with GFP (control), a full-length (FL) γC3 construct, constructs lacking either the extracellular domain (C3Δecto) or the constant domain (C3Δcon), or a construct encoding only a palmitoylated γC3 variable cytoplasmic domain (C3-VCD) and analyzed at DIV8. All constructs significantly rescued arborization toward wild-type levels (indicated by the dashed line), indicating that the C3 VCD is necessary (Fig. 9) and sufficient. n ≥ 25 neurons per condition. One-way ANOVA; Dunnett's multiple comparisons test. Error bars represent the SEM; ****p < 0.0001. C, Representative traces of neurons from each transfection condition. D, Representative images of transfected neurons immunostained for GFP (transfected to fill neuronal processes) and the HA tag on the indicated construct. All constructs localize in a punctate or patchy manner in neuronal dendrites. Scale bar: 50 µm (C) or 10 µm (D).
Reduced dendritic complexity of C3KO neurons can be rescued by targeted Axin1 overexpression
Finally, we sought evidence that γC3's role in stabilizing Axin1 at the membrane (Mah et al., 2016) contributes to its promotion of dendritic complexity. We reasoned that if this were the case, we might be able to rescue arborization defects in C3KO neurons by overexpressing wild-type and/or membrane-targeted Axin1, along with GFP to allow for visualization. C3KO cortical neurons in vitro were transfected with a wild-type Axin1 construct, or a construct encoding Axin1 fused to a CAAX box recognized by enzymes that prenylate, and thus membrane-target, proteins. Compared with the reduced complexity of C3KO neurons expressing GFP alone (Fig. 11A), we found a substantial increase in dendritic complexity among proximal arbors for C3KO neurons transfected with wild-type Axin1, though a lack of effect in more distal processes meant that the overall area under the curve difference did not reach statistical significance (Fig. 11B,C; ANOVA with Tukey's multiple comparison tests; ns p = 0.18; at least 25 neurons per condition for this and subsequent comparisons). When C3KO neurons were transfected with the construct encoding membrane-targeted CAAX-Axin1, however, complexity was increased throughout the entire arbor, and an overall statistically significant increase was observed (Fig. 11B,C; ANOVA with Tukey's multiple comparison tests; p = 0.019). This difference was not likely due simply to higher expression levels of CAAX-Axin1 compared with wild-type Axin1, as there was no significant difference in expression level in transfected cell lines between the two constructs (Fig. 11D,E; t test; ns p = 0.48, three replicates; expression levels of the wild-type construct trended higher). Together, our data support a model in which γC3, as the primary (possibly unique) γ-Pcdh isoform regulating cortical neuron dendrite complexity, interacts with Axin1 via its VCD to stabilize it at the membrane and allow its downstream signaling pathways to promote arborization.
Figure 11.

Reduced dendritic complexity of C3KO neurons can be rescued by targeted Axin1 overexpression in vitro. A, Representative images of DIV8 C3KO neurons transfected with GFP, and either an empty vector, Axin1 overexpression construct, or membrane-targeted CAAX-tagged Axin1. B, Sholl analysis graphs show dendritic crossings of spheres of increasing 10-µm intervals of neurons analyzed at DIV8. Expression of either Axin1 or CAAX-Axin1 rescues low arbor complexity proximal to the soma; only CAAX-Axin1 maintains that rescue at more distal Sholl spheres. C, Quantification of area under the curve of Sholl analysis of transfected C3KO neurons for all conditions. A significant rescue is observed with membrane-targeted CAAX-Axin1. n ≥ 25 neurons per transfection condition. One-way ANOVA; Dunnett's multiple comparisons test. Scale bar: 25 µm. Error bars represent the SEM; *p < 0.05. D, E, Western blottings of COS7 cells transfected with the indicated constructs and blotted using Axin1 and β-tubulin (loading control) antibodies. Quantification of three replicate experiments indicates no significant difference in expression levels between the two constructs.
Discussion
Here, we tested the hypothesis that one of the 22 γ-Pcdh proteins, γC3, plays a specialized role in brain development by promoting dendrite arborization via a unique interaction with the scaffolding and signaling protein Axin1. We created a new mouse line using CRISPR/Cas9, PcdhgC3KO, in which the Pcdhgc3 variable exon was specifically disrupted, leaving the other 21 isoforms intact. C3KO mice are viable and fertile; dendritic arbor complexity of cortical neurons is, however, severely reduced in C3KO mice, and this phenotype is not recapitulated in Pcdhg mutant mouse lines lacking other γ-Pcdh isoforms. Axin1 can be co-immunoprecipitated with γC3 in cortical lysates, and C3KO mutants exhibit alterations in Axin1's subcellular localization. Reduced arbor complexity can be rescued in cultured C3KO neurons by re-expressing γC3 constructs containing its VCD, but only if Axin1 levels are normal; conversely, overexpression of membrane-targeted Axin1 can rescue C3KO neurons even in the continued absence of γC3. Together, these data support our hypothesis and–more broadly–enhance the emerging view that individual members of the clustered protocadherin family are not functionally interchangeable, but rather perform isoform-specific functions that are crucial to brain development.
Uncovering specific roles for individual clustered protocadherins
While the >50 cPcdhs have, as a group, been shown to play many crucial roles in neurodevelopment (for review, see Peek et al., 2017; Mountoufaris et al., 2018), recent studies have begun to identify isoform-specific roles mediated by the C-type cPcdhs, a highly related subfamily comprised of five proteins: αC1 and αC2 encoded within the Pcdha gene cluster, and γC3, γC4, and γC5 encoded within the Pcdhg cluster (Wu and Maniatis, 1999; Wu et al., 2001). For example, α-Pcdh-C2 has been shown to be critical for the projection of serotonergic neurons within multiple brain regions by regulating axonal tiling (W.V. Chen et al., 2017; Katori et al., 2017). Mice lacking the Pcdhgc3, Pcdhgc4, and Pcdhgc5 variable exons largely phenocopy the spinal interneuron apoptosis, Ia afferent axonal defects, and neonatal lethality first observed in mice lacking all 22 γ-Pcdhs (Wang et al., 2002b; Prasad et al., 2008; Prasad and Weiner, 2011; W.V. Chen et al., 2012). Confirming the special importance of these three Pcdhg genes, cortical interneurons lacking them undergo increased cell death (Mancia Leon et al., 2020) similar to that observed in Pcdhg-null cortical interneurons (Carriere et al., 2020).
Individual isoform-specific neurodevelopmental roles are also beginning to emerge for these Pcdhg genes. The γC5 protein can interact with the γ2 subunit of the GABAA receptor and promote its surface expression through cis cytoplasmic interactions (Li et al., 2012). The γC5 isoform has also been shown to be upregulated in APP/PS1 mice, a model for Alzheimer's disease, and this upregulation was associated with synaptic dysfunction in neurons cultured from these animals (Li et al., 2017). Using an unbiased CRISPR screen to assess the role of isoform diversity within the Pcdhg gene cluster, we showed that γC4 is the only isoform required for postnatal survival in mice, and the only isoform needed for normal neuronal survival in the spinal cord and retina (Garrett et al., 2019). Given this surprising result, it seems likely that loss of γC4 is also the crucial determinant underlying the increased cortical interneuron death observed in Pcdhg mutant mice (Carriere et al., 2020; Mancia Leon et al., 2020). Further, a recent study found that mutations in human PCDHGC4 are associated with intellectual disabilities and seizures (Iqbal et al., 2021). Consistent with this, PCDHGC4 is the only cPcdh gene predicted to be significantly constrained in humans according to the gnomAD database (Garrett et al., 2019; Karczewski et al., 2020).
In addition, there are now several studies indicating possible isoform-specific roles for γC3 in non-neuronal cells. In adenoma cells, modulation of γC3 protein levels through either overexpression or RNAi mediated knock-down was shown to regulate β-catenin/Wnt and mTOR signaling and affect apoptosis and colony formation (Dallosso et al., 2012). It has also been suggested that γC3 may be important for maintaining integrity of the blood brain barrier: when Pcdhgc3 was disrupted in cultured brain microvascular endothelial cells (BMECs), tight junction protein expression was altered and transendothelial electrical resistance was lowered (Dilling et al., 2017). Consistent with this, BMECs in which Pcdhgc3 was knocked out in vitro exhibited increased cell proliferation and migration speed in wound healing assays (Gabbert et al., 2020). As noted earlier, our previous work identified a unique and specific interaction between the VCD of γC3 and the DIX domain of Axin1; evidence suggests that γC3 competes with Dishevelled for Axin1 binding, and this results in increased Axin1 localization to the membrane in HEK293 cells (Mah et al., 2016).
γC3 regulates Axin1 localization to the synapse without altering dendritic spine morphology or density
Our data suggest a role for γC3 in stabilization of Axin1 at the synapse. Following up on our prior work using overexpressed proteins in vitro (Mah et al., 2016), we found here that endogenous γC3 and Axin1 interact in the cortex in vivo, and that mice lacking γC3 showed a significant decrease in Axin1 and Cdc42 within synaptic fractions. Despite this and Axin1's reported role in regulating dendritic spine density (Y. Chen et al., 2015), we found no significant differences in the dendritic spine density or morphology in C3KO cortex. It is important to note that in our C3KO mice, the mutation is constitutive. Thus, the entire developmental process for cortical neurons occurs in the absence of γC3. While loss of γC3 clearly reduces Axin1 and Cdc42 levels in synaptic fractions, substantial protein remains, and compensatory factors during development may act to maintain spine density and morphology. In any case, the lack of effect on spines is actually consistent with our previous findings in which we identified extracellular interactions between multiple γ-Pcdh isoforms with both Neuroligin (Nlg)-1 and Nlg-2 as the primary way in which γ-Pcdhs regulate synapse density (Molumby et al., 2017; Steffen et al., 2021). The C3KO mice have 21 remaining γ-Pcdh extracellular domains that are capable of regulating synapse density via Nlg interactions. Of course, it is possible that even in the absence of clear changes in spine density or morphology, synaptic function is abnormal in C3KO mice; future studies could be directed at assessing this possibility through electrophysiological recordings in cortical slices.
A specific role for the γC3 VCD in regulating dendritic arborization via Axin1
Binding of the γ-Pcdh constant cytoplasmic domain to focal adhesion kinase (FAK) and its homolog PYK2 inhibits these cell adhesion kinases (J. Chen et al., 2009). In a series of subsequent studies, we found that this interaction leads to decreased activity of PKC signaling, which in turn results in higher levels of hypophosphorylated (active) myristoylated alanine-rich C-kinase substrate (MARCKS), a protein that promotes actin stabilization at the membrane and thus dendritic arborization (Garrett et al., 2012; Keeler et al., 2015a). This well-supported mechanism (see also Suo et al., 2012) is presumably not specific to any of the 22 γ-Pcdh isoforms, as all share the constant domain.
Here, we have identified an additional mechanism through which a single isoform, γC3, specifically promotes dendritic arborization though its unique VCD. Given the magnitude of arborization defects observed in C3KO mice–a reduction in arbor complexity of ∼30% (Fig. 6), compared with a reduction of ∼40% in mice lacking all γ-Pcdhs (Garrett et al., 2012), it is possible that γC3 VCD interactions with Axin1 play a primary role, with constant-domain interactions with FAK/PYK2 modulating this. It is also possible that some unique aspect of γC3 VCD conformation increases the ability of its contiguous constant domain to inhibit FAK/PYK2 in a way that does not occur in other isoforms, but the ability of both the C3ΔCon and C3VCD constructs to rescue C3KO dendritic arborization (Fig. 10) suggests this is not determinative.
Interestingly, a series of studies indicates that a motif within γ-Pcdh VCDs controls their trafficking to the endolysosomal system (O'Leary et al., 2011; Shonubi et al., 2015; Phillips et al., 2017). This has been best characterized for γA3, but a number of residues are conserved in the VCDs of other γAs, γBs, and γC3, as well as α and β-Pcdhs (Shonubi et al., 2015). While our prior work (Mah et al., 2016) and the rescue of C3KO dendritic arborization by overexpression of membrane-targeted Axin1 (Fig. 11) suggests γC3 may promote branching at the plasma membrane by recruiting Axin1, it would be interesting to test whether the γC3 VCD's binding to Axin1 affects its trafficking into the endolysosomal system, or whether Axin1 signaling continues after γC3 is endocytosed. Indeed, it has been reported that endosomal Axin1 in the context of Wnt signaling can nucleate tubulin for dendritic growth in Drosophila (A.T. Weiner et al., 2020), an intriguing potential mechanism. There is also substantial evidence that γ-Pcdh homophilic interactions, including those mediated by γC3, promote dendritic arborization in cortical neurons (Molumby et al., 2016). A major outstanding question is how homophilic engagement of γ-Pcdh extracellular domains might result in altered conformations of either the VCD or the shared constant domain in such a way that their activation of arborization-promoting signaling pathways is potentiated.
Footnotes
This work was supported by National Institutes of Health Grants R01 NS055272 (to J.A.W.) and R21 NS090030 (to J.A.W. and R.W.B.). We thank Leah Fuller and Kate Miers for expert assistance with mouse colony management and members of the Weiner laboratory for helpful comments. We also thank Dr. Douglas Houston, University of Iowa Department of Biology, for sharing Axin1 plasmids.
The authors declare no competing financial interests.
References
- Anderson RM, Johnson SB, Lingg RT, Hinz DC, Romig-Martin SA, Radley JJ (2020) Evidence for similar prefrontal structural and functional alterations in male and female rats following chronic stress or glucocorticoid exposure. Cereb Cortex 30:353–370. 10.1093/cercor/bhz092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby MC, Maier SR, Nishimune A, Henley JM (2006) Lateral diffusion drives constitutive exchange of AMPA receptors at dendritic spines and is regulated by spine morphology. J Neurosci 26:7046–7055. 10.1523/JNEUROSCI.1235-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriere CH, Wang WX, Sing AD, Fekete A, Jones BE, Yee Y, Ellegood J, Maganti H, Awofala L, Marocha J, Aziz A, Wang LY, Lerch JP, Lefebvre JL (2020) The gamma-protocadherins regulate the survival of GABAergic interneurons during developmental cell death. J Neurosci 40:8652–8668. 10.1523/JNEUROSCI.1636-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Lu Y, Meng S, Han MH, Lin C, Wang X (2009) Alpha- and gamma-protocadherins negatively regulate PYK2. J Biol Chem 284:2880–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WV, Alvarez FJ, Lefebvre JL, Friedman B, Nwakeze C, Geiman E, Smith C, Thu CA, Tapia JC, Tasic B, Sanes JR, Maniatis T (2012) Functional significance of isoform diversification in the protocadherin gamma gene cluster. Neuron 75:402–409. 10.1016/j.neuron.2012.06.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WV, Nwakeze CL, Denny CA, O'Keeffe S, Rieger MA, Mountoufaris G, Kirner A, Dougherty JD, Hen R, Wu Q, Maniatis T (2017) Pcdhαc2 is required for axonal tiling and assembly of serotonergic circuitries in mice. Science 356:406–411. 10.1126/science.aal3231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Fu AK, Ip NY (2013) Axin: an emerging key scaffold at the synapse. IUBMB Life 65:685–691. [DOI] [PubMed] [Google Scholar]
- Chen Y, Liang Z, Fei E, Chen Y, Zhou X, Fang W, Fu WY, Fu AK, Ip NY (2015) Axin regulates dendritic spine morphogenesis through Cdc42-dependent signaling. PLoS One 10:e0133115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallosso AR, Øster B, Greenhough A, Thorsen K, Curry TJ, Owen C, Hancock AL, Szemes M, Paraskeva C, Frank M, Andersen CL, Malik K (2012) Long-range epigenetic silencing of chromosome 5q31 protocadherins is involved in early and late stages of colorectal tumorigenesis through modulation of oncogenic pathways. Oncogene 31:4409–4419. 10.1038/onc.2011.609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilling C, Roewer N, Förster CY, Burek M (2017) Multiple protocadherins are expressed in brain microvascular endothelial cells and might play a role in tight junction protein regulation. J Cereb Blood Flow Metab 37:3391–3400. 10.1177/0271678X16688706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esumi S, Kakazu N, Taguchi Y, Hirayama T, Sasaki A, Hirabayashi T, Koide T, Kitsukawa T, Hamada S, Yagi T (2005) Monoallelic yet combinatorial expression of variable exons of the protocadherin-alpha gene cluster in single neurons. Nat Genet 37:171–176. 10.1038/ng1500 [DOI] [PubMed] [Google Scholar]
- Fang WQ, Ip JP, Li R, Ng YP, Lin SC, Chen Y, Fu AK, Ip NY (2011) Cdk5-mediated phosphorylation of Axin directs axon formation during cerebral cortex development. J Neurosci 31:13613–13624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang WQ, Chen WW, Fu AK, Ip NY (2013) Axin directs the amplification and differentiation of intermediate progenitors in the developing cerebral cortex. Neuron 79:665–679. 10.1016/j.neuron.2013.06.017 [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR (2000) Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28:41–51. 10.1016/s0896-6273(00)00084-2 [DOI] [PubMed] [Google Scholar]
- Gabbert L, Dilling C, Meybohm P, Burek M (2020) Deletion of protocadherin gamma C3 induces phenotypic and functional changes in brain microvascular endothelial cells in vitro. Front Pharmacol 11:590144. 10.3389/fphar.2020.590144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett AM, Weiner JA (2009) Control of CNS synapse development by γ-protocadherin-mediated astrocyte-neuron contact. J Neurosci 29:11723–11731. 10.1523/JNEUROSCI.2818-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett AM, Schreiner D, Lobas MA, Weiner JA (2012) γ-Protocadherins control cortical dendrite arborization by regulating the activity of a FAK/PKC/MARCKS signaling pathway. Neuron 74:269–276. 10.1016/j.neuron.2012.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett AM, Tadenev ALD, Hammond YT, Fuerst PG, Burgess RW (2016) Replacing the PDZ-interacting C-termini of DSCAM and DSCAML1 with epitope tags causes different phenotypic severity in different cell populations. Elife 5:e16144. 10.7554/eLife.16144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett AM, Bosch PJ, Steffen DM, Fuller LC, Marcucci CG, Koch AA, Bais P, Weiner JA, Burgess RW (2019) CRISPR/Cas9 interrogation of the mouse Pcdhg gene cluster reveals a crucial isoform-specific role for Pcdhgc4. PLoS Genet 15:e1008554. 10.1371/journal.pgen.1008554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman KM, Rubinstein R, Thu CA, Bahna F, Mannepalli S, Ahlsén G, Rittenhouse C, Maniatis T, Honig B, Shapiro L (2016) Structural basis of diverse homophilic recognition by clustered α- and β-protocadherins. Neuron 90:709–723. 10.1016/j.neuron.2016.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR (2002) Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci 22:6309–6314. 10.1523/JNEUROSCI.22-15-06309.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grutzendler J, Kasthuri N, Gan WB (2002) Long-term dendritic spine stability in the adult cortex. Nature 420:812–816. 10.1038/nature01276 [DOI] [PubMed] [Google Scholar]
- Harris KM, Stevens JK (1988) Dendritic spines of rat cerebellar Purkinje cells: serial electron microscopy with reference to their biophysical characteristics. J Neurosci 8:4455–4469. 10.1523/JNEUROSCI.08-12-04455.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Stevens JK (1989) Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci 9:2982–2997. 10.1523/JNEUROSCI.09-08-02982.1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B (1992) Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci 12:2685–2705. 10.1523/JNEUROSCI.12-07-02685.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi S, Nishimura W, Iida J, Kansaku A, Kishida S, Kikuchi A, Tanaka N, Hata Y (2004) Synaptic scaffolding molecule interacts with axin. J Neurochem 90:332–339. 10.1111/j.1471-4159.2004.02497.x [DOI] [PubMed] [Google Scholar]
- Hirano K, Kaneko R, Izawa T, Kawaguchi M, Kitsukawa T, Yagi T (2012) Single-neuron diversity generated by Protocadherin-beta cluster in mouse central and peripheral nervous systems. Front Mol Neurosci 5:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K (2005) Transient and persistent dendritic spines in the neocortex in vivo. Neuron 45:279–291. 10.1016/j.neuron.2005.01.003 [DOI] [PubMed] [Google Scholar]
- Ing-Esteves S, Kostadinov D, Marocha J, Sing AD, Joseph KS, Laboulaye MA, Sanes JR, Lefebvre JL (2018) Combinatorial effects of alpha- and gamma-protocadherins on neuronal survival and dendritic self-avoidance. J Neurosci 38:2713–2729. 10.1523/JNEUROSCI.3035-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal M, et al. (2021) Biallelic variants in PCDHGC4 cause a novel neurodevelopmental syndrome with progressive microcephaly, seizures, and joint anomalies. Genet Med 23:2138–2149. 10.1038/s41436-021-01260-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko R, Kato H, Kawamura Y, Esumi S, Hirayama T, Hirabayashi T, Yagi T (2006) Allelic gene regulation of Pcdh-alpha and Pcdh-gamma clusters involving both monoallelic and biallelic expression in single Purkinje cells. J Biol Chem 281:30551–30560. 10.1074/jbc.M605677200 [DOI] [PubMed] [Google Scholar]
- Karczewski KJ, et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katori S, Noguchi-Katori Y, Okayama A, Kawamura Y, Luo W, Sakimura K, Hirabayashi T, Iwasato T, Yagi T (2017) Protocadherin-αC2 is required for diffuse projections of serotonergic axons. Sci Rep 7:15908. 10.1038/s41598-017-16120-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeler AB, Molumby MJ, Weiner JA (2015a) Protocadherins branch out: multiple roles in dendrite development. Cell Adh Migr 9:214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeler AB, Schreiner D, Weiner JA (2015b) Protein kinase C phosphorylation of a gamma-protocadherin C-terminal lipid binding domain regulates focal adhesion kinase inhibition and dendrite arborization. J Biol Chem 290:20674–20686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koleske AJ (2013) Molecular mechanisms of dendrite stability. Nat Rev Neurosci 14:536–550. 10.1038/nrn3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostadinov D, Sanes JR (2015) Protocadherin-dependent dendritic self-avoidance regulates neural connectivity and circuit function. Elife 4:e08964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMassa N, Sverdlov H, Mambetalieva A, Shapiro S, Bucaro M, Fernandez-Monreal M, Phillips GR (2021) Gamma-protocadherin localization at the synapse is associated with parameters of synaptic maturation. J Comp Neurol 529:2407–2417. 10.1002/cne.25102 [DOI] [PubMed] [Google Scholar]
- Lefebvre JL, Zhang Y, Meister M, Wang X, Sanes JR (2008) Gamma-protocadherins regulate neuronal survival but are dispensable for circuit formation in retina. Development 135:4141–4151. 10.1242/dev.027912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre JL, Kostadinov D, Chen WV, Maniatis T, Sanes JR (2012) Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature 488:517–521. 10.1038/nature11305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xiao H, Chiou TT, Jin H, Bonhomme B, Miralles CP, Pinal N, Ali R, Chen WV, Maniatis T, De Blas AL (2012) Molecular and functional interaction between protocadherin-γC5 and GABAA receptors. J Neurosci 32:11780–11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chen Z, Gao Y, Pan G, Zheng H, Zhang Y, Xu H, Bu G, Zheng H (2017) Synaptic adhesion molecule Pcdh-γC5 mediates synaptic dysfunction in Alzheimer's disease. J Neurosci 37:9259–9268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Lin SC (2004) Axin: a master scaffold for multiple signaling pathways. Neurosignals 13:99–113. 10.1159/000076563 [DOI] [PubMed] [Google Scholar]
- Mah KM, Houston DW, Weiner JA (2016) The γ-protocadherin-C3 isoform inhibits canonical Wnt signalling by binding to and stabilizing Axin1 at the membrane. Sci Rep 6:31665. 10.1038/srep31665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancia Leon WR, Spatazza J, Rakela B, Chatterjee A, Pande V, Maniatis T, Hasenstaub AR, Stryker MP, Alvarez-Buylla A (2020) Clustered gamma-protocadherins regulate cortical interneuron programmed cell death. Elife 9:e55374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrahi A, Katz LC (2003) Dendritic stability in the adult olfactory bulb. Nat Neurosci 6:1201–1207. 10.1038/nn1133 [DOI] [PubMed] [Google Scholar]
- Molumby MJ, Keeler AB, Weiner JA (2016) Homophilic protocadherin cell-cell interactions promote dendrite complexity. Cell Rep 15:1037–1050. 10.1016/j.celrep.2016.03.093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molumby MJ, Anderson RM, Newbold DJ, Koblesky NK, Garrett AM, Schreiner D, Radley JJ, Weiner JA (2017) γ-Protocadherins interact with neuroligin-1 and negatively regulate dendritic spine morphogenesis. Cell Rep 18:2702–2714. 10.1016/j.celrep.2017.02.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountoufaris G, Canzio D, Nwakeze CL, Chen WV, Maniatis T (2018) Writing, reading, and translating the clustered protocadherin cell surface recognition code for neural circuit assembly. Annu Rev Cell Dev Biol 34:471–493. 10.1146/annurev-cellbio-100616-060701 [DOI] [PubMed] [Google Scholar]
- Nicoludis JM, Lau SY, Schärfe CP, Marks DS, Weihofen WA, Gaudet R (2015) Structure and sequence analyses of clustered protocadherins reveal antiparallel interactions that mediate homophilic specificity. Structure 23:2087–2098. 10.1016/j.str.2015.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K (2002) Structure and function of dendritic spines. Annu Rev Physiol 64:313–353. 10.1146/annurev.physiol.64.081501.160008 [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Yasuda R, Oertner TG, Svoboda K (2004) The number of glutamate receptors opened by synaptic stimulation in single hippocampal spines. J Neurosci 24:2054–2064. 10.1523/JNEUROSCI.5066-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary R, Reilly JE, Hanson HH, Kang S, Lou N, Phillips GR (2011) A variable cytoplasmic domain segment is necessary for g-protocadherin trafficking and tubulation in the endosome/lysosome pathway. Mol Biol Cell 22:4362–4372. 10.1091/mbc.E11-04-0283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek SL, Mah KM, Weiner JA (2017) Regulation of neural circuit formation by protocadherins. Cell Mol Life Sci 74:4133–4157. 10.1007/s00018-017-2572-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips GR, LaMassa N, Nie YM (2017) Clustered protocadherin trafficking. Semin Cell Dev Biol 69:131–139. 10.1016/j.semcdb.2017.05.001 [DOI] [PubMed] [Google Scholar]
- Prasad T, Weiner JA (2011) Direct and indirect regulation of spinal cord Ia afferent terminal formation by the gamma-protocadherins. Front Mol Neurosci 4:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad T, Wang X, Gray PA, Weiner JA (2008) A differential developmental pattern of spinal interneuron apoptosis during synaptogenesis: insights from genetic analyses of the protocadherin-gamma gene cluster. Development 135:4153–4164. 10.1242/dev.026807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein R, Thu CA, Goodman KM, Wolcott HN, Bahna F, Mannepalli S, Ahlsen G, Chevee M, Halim A, Clausen H, Maniatis T, Shapiro L, Honig B (2015) Molecular logic of neuronal self-recognition through protocadherin domain interactions. Cell 163:629–642. 10.1016/j.cell.2015.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Zipursky SL (2020) Synaptic specificity, recognition molecules, and assembly of neural circuits. Cell 181:1434–1435. 10.1016/j.cell.2020.05.046 [DOI] [PubMed] [Google Scholar]
- Schneider PN, Slusarski DC, Houston DW (2012) Differential role of Axin RGS domain function in Wnt signaling during anteroposterior patterning and maternal axis formation. PLoS One 7:e44096. 10.1371/journal.pone.0044096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner D, Weiner JA (2010) Combinatorial homophilic interaction between gamma-protocadherin multimers greatly expands the molecular diversity of cell adhesion. Proc Natl Acad Sci U S A 107:14893–14898. 10.1073/pnas.1004526107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonubi A, Roman C, Phillips GR (2015) The clustered protocadherin endolysosomal trafficking motif mediates cytoplasmic association. BMC Cell Biol 16:28. 10.1186/s12860-015-0074-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen DM, Ferri SL, Marcucci CG, Blocklinger KL, Molumby MJ, Abel T, Weiner JA (2021) The γ-protocadherins interact physically and functionally with neuroligin-2 to negatively regulate inhibitory synapse density and are required for normal social interaction. Mol Neurobiol 58:2574–2589. 10.1007/s12035-020-02263-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suo L, Lu H, Ying G, Capecchi MR, Wu Q (2012) Protocadherin clusters and cell adhesion kinase regulate dendrite complexity through Rho GTPase. J Mol Cell Biol 4:362–376. [DOI] [PubMed] [Google Scholar]
- Tasic B, Nabholz CE, Baldwin KK, Kim Y, Rueckert EH, Ribich SA, Cramer P, Wu Q, Axel R, Maniatis T (2002) Promoter choice determines splice site selection in protocadherin alpha and gamma pre-mRNA splicing. Mol Cell 10:21–33. 10.1016/s1097-2765(02)00578-6 [DOI] [PubMed] [Google Scholar]
- Thu CA, Chen WV, Rubinstein R, Chevee M, Wolcott HN, Felsovalyi KO, Tapia JC, Shapiro L, Honig B, Maniatis T (2014) Single-cell identity generated by combinatorial homophilic interactions between α, β, and γ protocadherins. Cell 158:1045–1059. 10.1016/j.cell.2014.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su H, Bradley A (2002a) Molecular mechanisms governing Pcdh-gamma gene expression: evidence for a multiple promoter and cis-alternative splicing model. Genes Dev 16:1890–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Weiner JA, Levi S, Craig AM, Bradley A, Sanes JR (2002b) Gamma protocadherins are required for survival of spinal interneurons. Neuron 36:843–854. [DOI] [PubMed] [Google Scholar]
- Weiner JA, Wang X, Tapia JC, Sanes JR (2005) Gamma protocadherins are required for synaptic development in the spinal cord. Proc Natl Acad Sci U S A 102:8–14. 10.1073/pnas.0407931101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner AT, Seebold DY, Torres-Guitierrez P, Folker C, Swope RD, Kothe GO, Stoltz JG, Zalenski MK, Kozlowski C, Barbera DJ, Patel MA, Thyagarajan P, Shorey M, Nye DMR, Keegan M, Behari K, Song S, Axelrod JD, Rolls MM (2020) Endosomal Wnt signaling proteins control microtubule nucleation in dendrites. PLoS Biol 18:e3000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Maniatis T (1999) A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell 97:779–790. 10.1016/s0092-8674(00)80789-8 [DOI] [PubMed] [Google Scholar]
- Wu Q, Zhang T, Cheng JF, Kim Y, Grimwood J, Schmutz J, Dickson M, Noonan JP, Zhang MQ, Myers RM, Maniatis T (2001) Comparative DNA sequence analysis of mouse and human protocadherin gene clusters. Genome Res 11:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye T, Fu AK, Ip NY (2015) Emerging roles of Axin in cerebral cortical development. Front Cell Neurosci 9:217. 10.3389/fncel.2015.00217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Fagotto F, Zhang T, Hsu W, Vasicek TJ, Perry WL 3rd, Lee JJ, Tilghman SM, Gumbiner BM, Costantini F (1997) The mouse Fused locus encodes Axin, an inhibitor of the Wnt signaling pathway that regulates embryonic axis formation. Cell 90:181–192. 10.1016/s0092-8674(00)80324-4 [DOI] [PubMed] [Google Scholar]