

Summary
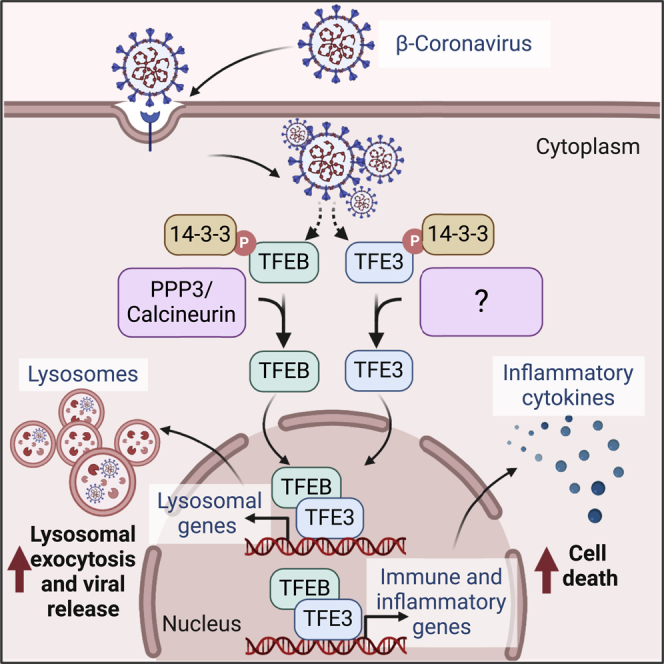
Beta-coronaviruses have emerged as a severe threat to global health. Undercovering the interplay between host and beta-coronaviruses is essential for understanding disease pathogenesis and developing efficient treatments. Here we report that the transcription factors TFEB and TFE3 translocate from the cytosol to the nucleus in response to beta-coronavirus infection by a mechanism that requires activation of calcineurin phosphatase. In the nucleus, TFEB and TFE3 bind to the promoter of multiple lysosomal and immune genes. Accordingly, MHV-induced upregulation of immune regulators is significantly decreased in TFEB/TFE3-depleted cells. Conversely, over-expression of either TFEB or TFE3 is sufficient to increase expression of several cytokines and chemokines. The reduced immune response observed in the absence of TFEB and TFE3 results in increased cellular survival of infected cells but also in reduced lysosomal exocytosis and decreased viral infectivity. These results suggest a central role of TFEB and TFE3 in cellular response to beta-coronavirus infection.
Subject areas: Virology, Molecular biology
Graphical abstract
Highlights
-
•
TFEB and TFE3 translocate to the nucleus in response to beta-coronavirus infection
-
•
TFEB and TFE3 induce expression of multiple immune genes upon viral infection
-
•
MHV-induced cell death is diminished in TFEB/TFE3-depleted cells
-
•
TFEB and TFE3 facilitate virus egress from cells by promoting lysosomal exocytosis
Virology; Molecular biology
Introduction
The transcription factors TFEB and TFE3 regulate energy homeostasis and cellular response to a wide variety of stress conditions, including nutrient deprivation, oxidative stress, organelle damage, and pathogens.1,2 TFEB and TFE3 remain sequestered in the cytosol under normal conditions. This retention is mediated by the mechanistic target of rapamycin complex 1 (mTORC1)-dependent phosphorylation of TFEB at serine-211 (S211) and TFE3 at serine-321 (S321) and the consequent binding of the transcription factors to 14-3-3.3,4,5 Following stress, inactivation of mTORC1 and/or activation of specific phosphatases result in TFEB-S211 and TFE3-S321 dephosphorylation, dissociation of the TFEB/TFE3-14-3-3 complex, and translocation to the nucleus.3,4,5,6,7,8 Active TFEB and TFE3 bind to the promoter of hundreds of genes inducing expression of critical regulators of lysosomal biogenesis, autophagy, metabolism, and cellular response to stress, thus allowing restoration of cellular homeostasis.9,10,11
Recent evidence has pointed out an important role of TFEB and TFE3 in innate immunity and inflammation, in particular orchestrating antibacterial responses. Activation of TFEB in response to Mycobacterium tuberculosis infection induces autophagy, limiting bacterial growth in bone marrow-derived macrophages (BMDMs).12 A separate study showed that preventing TFEB activation in mice inhibits the expansion of the lysosomal compartment induced by Salmonella typhimurium, resulting in increased frequency of liver necrosis and decreased survival rates.13 TFEB and TFE3 also influence immune responses by directly controlling expression of several cytokines and chemokines,14,15,16,17 promoting differentiation of macrophages into pro-inflammatory states,18,19 and modulating migration of dendritic cells.20 The antibacterial function of these transcription factors is well established in Caenorhabditis elegans where depletion of HLH-30, the ortholog of TFEB and TFE3 in worms, causes increased susceptibility to Staphylococcus aureus.14,21,22,23
The role of TFEB and TFE3 in antiviral response is less understood but includes some intriguing examples. Recognition of HIV by Toll-like receptor 8 (TLR8) induces TFEB nuclear translocation, initially resulting in autophagy activation. At later infection times, however, the HIV accesory protein Nef causes TFEB retention in the cytosol, leading to persistent viral infection.24 Furthermore, the TFEB activator Flubendazole increases autophagy, decreasing HIV infection and transfer from dendritic to CD4+ T cells.25 The role of TFEB in Coxsackievirus B3 (CVB3) infection is more controversial. Under caloric-restriction conditions, activation of TFEB increases susceptibility to CVB3, resulting in accelerated pancreatic pathology.26 In contrast, another study has described that cleavage of TFEB by the CVB3 proteinase 3C prevents its transcriptional activity allowing increased virus survival.27
Coronaviruses (CoVs) are a group of enveloped, single-strand RNA viruses that cause respiratory, gastrointestinal, hepatic, and neurological diseases in mammalian and avian species. The beta-coronavirus genus includes the highly pathologic Middle East respiratory syndrome coronavirus (MERS-CoV) and the severe acute respiratory syndrome coronaviruses SARS-CoV and SARS-CoV-2, which have caused devastating outbreaks and pose a severe threat to global public health.28,29 Accumulating evidence has shown that SARS-CoV-2 manipulates several steps of the autophagic/lysosomal pathway, and these alterations have important consequences for different aspects of the viral life cycle, including infection, replication, and secretion.30,31,32,33 In particular, it was described that newly assembled SARS-CoV-2 particles are delivered to lysosomes, and the virus promotes lysosomal exocytosis to facilitate its release from cells.30,31
In this study we found that beta-coronavirus induced a robust and persistent activation of TFEB and TFE3. Translocation of the transcription factors to the nucleus was required for efficient expression of multiple immune regulators and contributed to viral infection-induced cell death. TFEB and TFE3 activation also increased lysosomal exocytosis, enhancing virus egress. Our results indicate that TFEB and TFE3 play a crucial role in the interface between host and beta-coronavirus.
Results
MHV induces TFEB and TFE3 nuclear translocation
In this study we used mouse hepatitis virus (MHV) strain A59 (further referred to as MHV) as a model for beta-coronavirus infection. One advantage of this system is that the work can be performed under Biosafety Level 2 (BSL-2) conditions by infecting HeLa cells carrying the receptor for MHV (HeLa-mCC1a) (Figure S1A). It is well established that infection of MHV in mice results in pathogenesis that includes acute pneumonia, severe lung injury, and neurological symptoms, and it is considered a surrogate model for SARS-CoV and SARS-CoV-2.34 Likewise, Hela-mCC1a cells are a well-accepted system to study MHV infection.30,35,36,37,38,39,40
We infected HeLa-mCC1a cells with MHV at a high MOI and analyzed the distribution of viral particles in a time course manner using an antibody (J1.3) that primarily recognizes the viral M protein present in assembled viral particles.30 In agreement with previous studies, we found co-localization of viral particles with ER and trans-Golgi network (TGN) markers at early infection stages (Figures S1B and S1C), followed by a progressively increased co-localization with lysosomal markers at later infection times (11-14 hours post infection) (Figure S1D).30,41,42
Next, we asked whether the delivery of viral particles to lysosomes might affect lysosomal signaling or lysosomal stress pathways. As seen in Figures 1A, S1E, and S1F, MHV did not cause noticeable changes in mTORC1 activity, as assessed by measuring the phosphorylation status of the mTORC1 targets p70 S6 kinase and 4EBP1. As control, we confirmed mTORC1 inactivation in HeLa-mCC1a cells in response to starvation (Figure 1A). Recent evidence has suggested that the transcription factor STAT3 functions as a sensor of lysosomal stress by increasing its translocation to the nucleus in response to lysosomal cargo overload or defective lysosomal protease activity.43 However, we did not detect changes in STAT3 phosphorylation or intracellular distribution in response to MHV (Figures 1A and S1G–S1I). In contrast, we observed robust and persistent activation of the transcription factors TFEB and TFE3 in response to MHV infection. Whereas TFEB remained retained in the cytosol at early infection times (between 0 and 11 h p.i.), a significant nuclear accumulation was observed at 14 h p.i., with further activation at 16 and 24 h p.i. (Figures 1B–1D). Activation of TFE3 was a little bit faster, with over 80% of infected cells showing significant nuclear accumulation at 14 h p.i. (Figures S1J, 1C and 1E).
Figure 1.

The transcription factors TFEB and TFE3 translocate to the nucleus in response to MHV infection
(A) Immunoblot analysis of protein lysates from HeLa-mCC1a cells treated with DMSO (0 h), infected with MHV for 8 h, 11 h, and 14 h, or incubated in EBSS medium for 4 h (STV). Immunoblots are representative of at least three independent experiments.
(B) Representative immunofluorescence images of MHV (Mj1.3) (red) and endogenous TFEB (green). Cells were stained with DAPI (blue). n = 3 independent experiments. Scale bars, 20 μm.
(C) Representative immunofluorescence images of HeLa-mCC1a cells treated with DMSO (0 h) or infected with MHV for 16 h and 24 h. Endogenous TFEB and TFE3 are shown in green, MHV M protein in red, and DAPI staining in blue. n = 3 independent experiments. Scale bars, 20 μm.
(D) Quantification of the percentage of infected cells with nuclear TFEB using confocal microscopy from (C). n = 600 cells per condition from 3 independent experiments.
(E) Quantification of the percentage of infected cells with nuclear TFE3 using confocal microscopy from (C). n = 600 cells per condition from 3 independent experiments.
(F) Quantification of the p-S211-TFEB/total TFEB ratio represented as fold increase. n = 3 independent experiments.
(G) Quantification of the p-S321-TFE3/total TFE3 ratio represented as fold increase. n = 3 independent experiments.
(H) Calu-3 cells were infected with SARS-CoV-2 for the indicated times. Endogenous TFEB and TFE3 are shown in green and DAPI staining in blue. Scale bars, 20 μm.
(I) Representative immunoblot analysis of protein lysates from HeLa-mCC1a cells infected with MHV for the indicated times.
(J and K) Quantification of immunoblot data shown in (I). n = 3 independent experiments.
(L) Representative western blot of TFEB and TFE3 levels in HeLa-mCC1a cells treated with either non-target or DCAF7 siRNAs for 72 h followed by infection with MHV for 16 h.
(M) Quantification of immunoblot data shown in (L). n = 3 independent experiments. Statistical analysis with one-way ANOVA followed by Dunnett’s multiple comparison post-test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Data represent mean ± SEM.
It is well established that mTOR-mediated phosphorylation of TFEB at serine-211 and TFE3 at serine-321 is required for binding of these transcription factors to 14-3-3 and consequent retention in the cytosol.3,4,5 Dephosphorylation of TFEB-S211 and TFE3-S321 is, therefore, a prerequisite for their translocation to the nucleus. Consistently, we found a significant reduction in TFEB-S211 and TFE3-S321 phosphorylation at late times of MHV infection (Figures 1F and 1G). Virus-mediated TFEB and TFE3 activation was also confirmed by subcellular fractionation analysis (Figure S1K). Furthermore, infection of Calu-3 human lung epithelial cells with SARS-CoV-2 resulted in TFEB and TFE3 nuclear accumulation at late infection times (Figure 1H), suggesting a general role of these transcription factors in cellular response against beta-coronavirus.
Viruses often target specific cellular transcription factors for degradation with the goal to evade immune response and increase viral propagation. Interestingly, we found a time-dependent reduction in TFEB and TFE3 protein levels following viral infection. Whereas the amount of TFEB and TFE3 remained constant up to 11 h p.i., the protein levels progressively decreased at 16 and 24 h p.i. (Figures 1I–1K). This fall occurred without a concomitant reduction in TFEB and TFE3 mRNA levels (Figure S1L). Similar observations were recently reported in human airway cells infected with human coronavirus OC43 and 229E.44 In this case, coronavirus infection induced PAK2-dependent TFEB phosphorylation, priming the transcription factor for recognition by the E3 ubiquitin ligase DDB1-and CUL4-associated factor 7 (DCAF7) and causing TFEB proteasomal degradation. In agreement with these observations, we found that depletion of DCAF7 with specific small interfering RNAs (siRNAs) prevented MHV-induced TFEB and TFE3 degradation (Figures 1L and 1M). The efficiency of DCAF7 depletion was assessed by qPCR (Figure S1L). Our results indicate that MHV triggers post-translational degradation of TFEB and TFE3, thus suggesting a potential role of these transcription factors in antiviral host defense.
Calcineurin promotes TFEB activation in response to MHV
Several studies have established the contribution of different phosphatases and kinases to TFEB/TFE3 activation. These include the phosphatases PP2A8 and PPP3/calcineurin,7 as well as the AMP-activated protein kinase (AMPK),22 protein kinase C (PKC), and protein kinase D (PKD).21 To further explore the mechanisms involved in TFEB/TFE3 activation following MHV infection, we determined the effect of different inhibitors on TFEB/TFE3 nuclear translocation. HeLa-mCC1a cells were seeded in optical-bottom 96-well plastic plates, infected with MHV for 2 h, and then treated with calcium chelators (BAPTA-AM) or inhibitors against PPP3/calcineurin (FK506), PKC (BIM-IV), PKD (CRT-0066101), and AMPK (Compound C) for up to 16 h. Microscopy images were acquired with CellVoyager CV7000, and the accumulation of TFEB in the nucleus was measured using Columbus software. Interestingly, we observed that the PPP3/calcineurin inhibitor FK506 almost completely prevented TFEB activation, while PKC inhibition had a partial effect (Figures 2A and 2B). The phosphatase activity of PPP3/calcineurin is activated by intracellular calcium. Accordingly, incubation with the calcium chelator BAPTA-AM very significantly reduced TFEB nuclear translocation (Figures 2A and 2B).
Figure 2.

Calcineurin regulates MHV-induced TFEB nuclear translocation
(A) Representative images of the endogenous TFEB translocation assay obtained by confocal automated microscopy. HeLa-mCC1a cells were treated with MHV for 16 h. After 2 h of MHV infection, cells were treated with FK506 (PPP3/calcineurin inhibitor, 10 μM), BAPTA-AM (Ca2+ chelator, 10 μM), BIM IV (PKC inhibitor, 1 μm), CRT-0066101 (PKD inhibitor, 2.5 μM), and compound C (AMPK inhibitor, 2.5 μM). n = 3 independent experiments. Scale bars, 50 μm.
(B) Quantification of average intensity of nuclear endogenous TFEB fluorescence. Automated imaging analysis included at least 10,000 cells per condition. n = 3 independent experiments.
(C) Representative immunofluorescence images of endogenous TFEB (green) in HeLa-mCC1a cells treated with either non-target or PPP3/calcineurin catalytic subunits A and B siRNAs for 72 h followed by infection with MHV for 16 h. Cells were stained with DAPI (blue) n = 3 independent experiments. Scale bars, 20 μm.
(D) Immunoblot analysis of protein lysates from HeLa-mCC1a treated with either non-target or PPP3/calcineurin catalytic subunits A and B siRNAs for 72 h followed by infection with MHV for 16 h n = 3 independent experiments.
(E) Quantification of percentage of infected cells with nuclear TFEB using confocal microscopy from (C). n = 600 cells per condition from 3 independent experiments.
(F) Representative immunofluorescence images of endogenous TFEB (green) in HeLa-mCC1a cells treated with either non-target or PPP2 catalytic subunits A and B siRNAs for 72 h followed by infection with MHV for 16 h. Cells were stained with DAPI (blue) n = 3 independent experiments. Scale bars, 20 μm
(G) Quantification of percentage of infected cells with nuclear TFEB using confocal microscopy from (F). n = 300 cells per condition from 3 independent experiments. Statistical analysis with one-way ANOVA followed by Dunnett’s multiple comparison post-test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Data represent mean ± SEM.
It is important to note that treatment with either FK506 or BAPTA-AM did not affect viral replication (Figure S2A). To further confirm these results, we used specific siRNAs to deplete the two PPP3/calcineurin catalytic subunits (PPP3CA and PPP3CB) and found that MHV-induced TFEB nuclear translocation was significantly dismissed when compared with control cells (Figures 2C–2E). In contrast, depletion of the PP2A catalytic subunits did not affect TFEB activation in response to coronavirus (Figures 2F and 2G). Unexpectedly, we observed that none of these treatments prevented virus-induced TFE3 activation (Figures S2B–S2F). These data suggest distinctive mechanisms of activation that may explain the different kinetics of TFEB and TFE3 nuclear translocation in response to MHV. Thus, TFE3 would activate at earlier infection times by a mechanism that does not affect TFEB, while TFEB nuclear translocation likely requires an increase in intracellular calcium levels and the consequent activation of PPP3/calcineurin.
Upregulation of immune genes is diminished in TFEB/TFE3-depleted cells
To investigate the contribution of TFEB and TFE3 to host antiviral response, we performed RNA sequencing (RNA-seq) analysis in HeLa-mCC1a cells treated with either control or TFEB/TFE3 siRNAs after 24 h infection with MHV (Figure 3A). The comparative transcriptome analysis between uninfected and MHV-infected control cells indicated that 11,722 genes were differentially expressed between the two conditions, 5,967 upregulated and 5,755 downregulated (Figure 3B and Table S1). A recent study investigating the transcriptional response to SARS-CoV-2 infection identified a set of 25 signature genes (12 upregulated and 13 downregulated) that represent the host response to infection.45 Interestingly, 11 of the 12 upregulated genes and 9 of the 13 downregulated genes were also up- or downregulated by MHV, respectively, further supporting the validity of our experimental conditions to understand SARS-CoV-2 host transcriptional response (Figure S3A).
Figure 3.

TFEB and TFE3 depletion reduces the immune response induced by MHV infection
(A) Principal-component analysis of genes with q-value <0.05 reveals distinct clustering of siControl- and siTFEB/TFE3-treated cells upon 24 h MHV infection.
(B) Volcano plot indicating distribution of genes significantly up- (red) and downregulated (green) in control cells versus cells infected with MHV for 24 h. Cutoffs indicate genes with q-value <0.05.
(C) Enriched gene ontology (GO) terms in the ‘MSigDB Hallmark’ category of differentially increased expression genes between control conditions and treated with MHV for 24 h.
(D) Heatmap of differentially expressed immune response genes from RNA-Seq analysis of siControl versus siTFEB/3-treated cells in response to MHV infection.
(E) Relative quantitative RT-PCR analysis of the mRNA expression of immune genes in HeLa-mCC1a cells treated with either control or TFEB/3 siRNAs upon incubation with MHV for 0 h, 6 h, 16 h, and 24 h n = 3 independent experiments. Statistical analysis with one-way ANOVA followed by Dunnett’s multiple comparison post-test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Data represent mean ± SEM.
MSigDM Hallmark Pathway gene ontology analysis of those genes most upregulated by the virus (shrunkenLog2FC > 1, padjusted value < 0.05) revealed that among the most relevant categories were many related to immune and inflammatory response (Figure 3C). Notably, the MHV-induced upregulation of numerous immune genes was significantly impaired by depletion of TFEB and TFE3 (Figure 3D, Tables S2 and S3). These included several SARS-CoV-2 signature genes, such as TNFAIP3, PTX3, PPP1R15A, IL1A, and CXCL2 (Figure 3D), suggesting that TFEB and TFE3 play an important role in cellular response against coronavirus infection.
To confirm our RNA-seq data, we analyzed expression of several immune genes at different times of MHV infection by qPCR. As seen in Figure 3E, upregulation of several critical immune regulators was reduced in TFEB/TFE3-depleted cells. Efficient TFEB and TFE3 depletion was verified both by qPCR (Figure 3E) and immunoblot (Figures S3B and S3C). Expression of the virus M protein at different infection times was also measured by qPCR to confirm that TFEB/TFE3 depletion did not affect virus replication (Figure 3E). Quantification of cytokine and chemokine protein levels by cytokine blot arrays further confirmed that the production of several cytokines in response to MHV was severely impaired in TFEB/TFE3-depleted cells (Figures S3D and S3E).
It is important to note that the most significant differences in cytokine expression between control and TFEB/TFE3-depleted cells were observed at late infection times. This is consistent with the lack of significant TFEB and TFE3 activation before 14 h p.i. (Figures 1D and 1E). We hypothesized that early cytokine production might be regulated by other transcription factors, such as NFkB. Accordingly, we observed a robust induction of NFkB activity in response to MHV infection at 1 and 3 h p.i., as assessed by increased phosphorylation of NFkB-p65 and IκBα (Figures S3F–S3H). Strong NFkB responses have also been described in cells infected with SARS-CoV-2.46 MHV infection also led to engagement of the interferon response, as shown by increased STAT1 phosphorylation after 3 h of incubation with the virus (Figures S3F and S3I).
Collectively, these results suggest an important role for TFEB and TFE3 in cellular response against beta-coronavirus, in particular by maintaining elevated cytokine production at late infection times.
TFEB and TFE3 mediate transcription of multiple immune regulators
To better understand the specific contribution of TFEB and TFE3 to the cellular transcriptional response against coronavirus, we performed chromatin immunoprecipitation sequencing (ChIP-seq) analysis in HeLa-mCC1a cells infected with MHV for 14 h and assessed promoter occupancy by endogenous TFE3. As expected, given the well-known role of these transcription factors in lysosomal biogenesis and autophagy regulation, we observed interaction of TFE3 with the promoter of multiple lysosomal and autophagic genes (Figure S4A). Furthermore, the increased autophagy activation caused by MHV infection was significantly reduced in TFEB/TFE3-depleted cells (Figures S4B and S4C). In addition, we detected TFE3 binding to the promoter of several important immune regulators. As an example, the peaks corresponding to some of these genes, including ILF3, B2M, IL24, SIRT1, SH2D5, and TNFAIP3 are shown in Figure 4A. We also found recruitment of TFE3 to the promoter of genes that, while not considered prototypical immune genes, have recently been shown to play an important role in SARS-CoV-2 infection. These include the nucleoporin NUP188,47 the translation initiation factor EIF4G1,48 the calcium ion channel ORAI1,49 and the SUMO1-ligase SAE150 (Figure 4A).
Figure 4.

TFEB and TFE3 induce expression of multiple immune genes
(A) Schematic representations of the TFE3 binding region in the promoter of several immune genes in HeLa-mCC1a cells infected with MHV for 14 h. The transcription start site is indicated as TSS.
(B) Relative quantitative RT-PCR analysis of the mRNA levels of the indicated immune genes in HeLa-mCC1a cells infected with either control adenovirus (Ad-null) or adenovirus expressing TFE3-321A (Ad-TFE3-S321A)for 40 h n = 3 independent experiments.
(C) Relative quantitative RT-PCR analysis of the mRNA levels of the indicated immune genes in HeLa-mCC1a cells infected with either control adenovirus (Ad-null) or adenovirus expressing TFEB-S211A (Ad-TFEB-S211A) for 40 h n = 3 independent experiments. Statistical analysis with Student’s t-tests. ∗p < 0.05, ∗∗p < 0.01,∗∗∗p < 0.001. Data represent mean ± SEM.
To further corroborate our ChIP-seq results, we over-expressed recombinant TFE3 in HeLa-mCC1a cells and assessed the expression of several potential TFE3-targeted immune genes by qPCR. As seen in Figure 4B, over-expression of TFE3 was sufficient to induce a strong mRNA upregulation of many critical regulators of innate immune response, including TRIM25, IL1A, B2M, SIRT1, GEM, RELB, IRF9, IL24, SH2D5, TNFAIP3, CXCL2, and FOXO3. Similar results were observed following over-expression of recombinant TFEB in HeLa-mCC1a cells (Figure 4C). Altogether, our results indicate that TFE3 and TFEB play an important role in host response to coronavirus by directly regulating expression of key immune genes.
MHV-mediated TFEB and TFE3 activation leads to apoptosis
It is well established that beta-coronavirus can induce cell death via a variety of signaling pathways, including apoptosis, necrosis, and pyroptosis.51,52 To assess the effect of MHV on cell viability, MHV-infected cells were stained with Annexin V/7-AAD and analyzed by flow cytometry. As seen in Figures 5A and 5B, MHV caused a robust increase in apoptosis after 24 h infection, while the number of necrotic or necrotic/late apoptotic cells did not change significantly. Accordingly, we detected cleavage of caspase-3/7/9, as well as the caspase-3 target PARP1, upon infection, and this activation was blocked by the caspase pan-inhibitor Z-VAD(OH)-FMK (Figures S5A–S5E). Furthermore, incubation with Z-VAD(OH)-FMK at 2 h following MHV infection completely prevented cell death (Figures S5F and S5G). Notably, depletion of TFEB and TFE3 led to reduced levels of apoptotic cells (Figures 5A and 5B), suggesting that the decreased expression of inflammatory cytokines observed in TFEB/TFE3-depleted cells may have an important impact on cell viability. Cleavage of caspase-3 and PARP1 was also significantly reduced in the absence of TFEB and TFE3 (Figures 5C–5E).
Figure 5.

TFEB and TFE3 modulate cell viability in response to viral infection
(A) Representative flow cytometry plots of HeLa-mCC1a cells treated with either siRNA Control (siControl) or siRNA against TFEB and TFE3 (siTFEB/TFE3) and infected with MHV for 16 and 24 h. Annexin V+ are apoptotic cells, 7-AAD + are necrotic cells, and Annexin V+/7-AAD + are late apoptotic and necrotic cells.
(B) Quantification of the population of apoptotic cells (Annexin V+), necrotic cells (7-AAD+), and late apoptotic and necrotic cells (Annexin+/7-AAD+) from (A). n = 3 independent experiments.
(C) Representative western blot of PARP1, Caspase-3, TFEB, and TFE3 from HeLa-mCC1a cells treated with siControl or siTFEB/TFE3 and then infected with MHV for 16 and 24 h n = 3 independent experiments.
(D and E) Quantification of immunoblot data shown in (C). n = 3 independent experiments.
(F) Quantification of LDH levels released to the extracellular media in HeLa-mCC1a cells treated with MHV for 16 h or 24 h. Cells were previously transfected with siRNA control (siControl) and siRNA against TFEB and TFE3 (siTFEB/3). In addition, cells were treated with a pan-caspase inhibitor Z-VAD after 2 h of MHV infection when indicated. n = 4 independent experiments. Statistical analyzes were performed with two-way ANOVA with Sıdak’s. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Values are mean ± SEM.
As an additional way to monitor cell death, we measured the release of the cytosolic enzyme lactate dehydrogenase (LDH) into the extracellular media, which can be used as an indicator of plasma membrane integrity. In agreement with our previous data, we found a clear increase in LDH release at 24 h p.i. that was abolished by either TFEB/TFE3 depletion or incubation with caspase inhibitors (Figure 5F).
TFEB and TFE3 are required for MHV-induced lysosomal exocytosis
We previously described that over-expression of either TFEB or TFE3 causes fusion of lysosomes with the plasma membrane.5,53 In addition, Ghosh and colleagues recently reported that MHV utilizes lysosomes to egress from cells and this has important consequences for antigen presentation and viral spread.30 Therefore, we hypothesized that TFEB/TFE3 activation may contribute to MHV-induced lysosomal exocytosis. Confocal microscopy analysis confirmed increased levels of the lysosomal protein LAMP1 at the plasma membrane in cells infected with MHV at 16 and 24 h p.i. (Figures 6A and 6B). Incubation with the calcium ionophore agent, ionomycin, was used as a positive control of lysosomal exocytosis (Figures S6A and S6B). As predicted, lysosomal exocytosis was almost completely blocked upon TFEB and TFE3 depletion both at 16 and 24 h p.i. (Figures 6A and 6B). To further corroborate these data, we measured the presence of lysosomal enzymes in the extracellular medium. As seen in Figure 6C, we observed a modest but significant increase in β-hexosaminidase activity in the extracellular medium in MHV-infected cells, while this increase was reduced by TFEB/TFE3 depletion.
Figure 6.

TFEB and TFE3 facilitate virus exit from cells and contribute to viral infectivity
(A and B) HeLa-mCC1a cells were transfected with either non-target siRNAs (siControl) or siRNAs against TFEB and TFE3 (siTFEB/3) for 72 h and then infected with MHV for 16 h or 24 h. Representative immunofluorescence images showing surface LAMP1 levels. n = 3 independent experiments. Scale bars, 20 μm (B) Quantification of surface LAMP1 levels from (A). n = 30 cells per condition from 3 independent experiments.
(C) Extracellular β-Hexosaminidase activity assay from siControl- or siTFEB/TFE3-treated HeLa-mCC1a cells following infection with MHV for 24 h n = 3 independent experiments.
(D) Relative quantitative RT-PCR analysis of the MHV mRNA levels in the extracellular medium of HeLa-mCC1a transfected with non-target (siControl) or siRNA against TFEB and TFE3 (siTFEB/3) for 72 h and infected with MHV for 16 h or 24 h n = 3 independent experiments.
(E) HeLa-mCC1a cells were transfected with non-target (siControl) or siRNA against TFEB and TFE3 (siTFEB/3) for 72 h and then treated with MHV for 16 h or 24 h. Then Supernatants were collected and reinoculated into new HeLa-mCC1a cells. After 48–72 h TCID50/mL was calculated. n = 3 independent experiments.
(F) Model depicting the proposed role of TFEB and TFE3 in cellular response to beta-coronavirus. Statistical analysis with one-way ANOVA followed by Dunnett’s multiple comparison post-test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Data represent mean ± SEM.
Next, we collected supernatants from MHV-infected control and TFEB/TFE3-depleted cells at 16 and 24 h p.i. and measured MHV RNA by qPCR. Notably, we found that TFEB/TFE3-depleted cells showed decreased viral genome release compared with siControl-treated cells (Figure 6D). It is important to keep in mind that our LDH assays did not detect changes in membrane permeability at 16 h p.i. (Figure 5E), suggesting that viral egress occurs in the absence of cell lysis at this infection time and that the reduced extracellular MHV levels observed in TFEB/TFE3-depleted cells are likely due to diminished lysosomal exocytosis. In contrast, both reduced lysosomal exocytosis and cell death may contribute to the lowered viral exit found in TFEB/TFE3-depleted cells at 24 h p.i. Furthermore, when we performed plaque assays to determine viral titer, we found that significantly reduced number of colonies were formed by the supernatant from TFEB/TFE3-depleted cells compared with control cells (Figures 6E and S6C), indicating that the diminished lysosomal exocytosis observed in the absence of the transcription factors may have a direct impact on viral infectivity.
Altogether, our data indicate that the TFEB/TFE3 axis is part of the cellular network implicated in detecting, controlling, and eradicating beta-coronavirus (Figure 6F). The ability of MHV to evade (TFEB/TFE3 degradation) and utilize (lysosomal exocytosis) this system for its own benefit represents a clear example of viral hijacking of the host stress response pathways.
Discussion
Coronavirus infection can result in acute respiratory distress syndrome and multiple organ failure. Identifying the transcription factors regulating host defense mechanisms is therefore essential for understanding host-pathogen interactions and developing therapeutic measurements.
In this study we describe that MHV, a well-accepted surrogate model for SARS-CoV and SARS-CoV-2, induces activation of the transcription factors TFEB and TFE3. Translocation of TFEB and TFE3 from the cytosol to the nucleus occurs at late infection times, and it is consistent with the delayed kinetics of activation previously observed in lipopolysaccharide (LPS)-stimulated macrophages.15 The virus did not seem to cause noticeable changes in mTORC1 activity, at least as assessed by monitoring the phosphorylation status of the mTORC1 targets p70 S6 kinase and 4EBP1, but TFEB nuclear translocation was inhibited by depletion of the phosphatase PPP3/calcineurin or incubation with BAPTA-AM. This suggests that increased intracellular calcium levels, which occur upon virus-induced ER stress54 or lysosomal damage,31,32 may contribute to TFEB activation. Accordingly, we and others have previously described that release of Ca2+ from either ER or lysosomes results in TFEB and TFE3 activation.7,55
A recent report has shown that expression of the SARS-CoV-2 accessory protein ORF3a induces TFEB nuclear translocation. However, expression of TFEB target genes was not observed under these conditions,32 pointing to the limitation of expressing individual viral proteins versus infection with whole viral particles. Furthermore, these data suggest that additional viral or virus-induced proteins may be required to ensure TFEB/TFE3 transcriptional competence. While we recognize that studying coronavirus infection in HeLa cells may also hold some limitations, it is important to note that this system has been extensively used to characterize SARS-CoV-2 infection. For example, ACE2-expressing wild types (HeLa-ACE2) were used in one of the first studies identifying SARS-CoV-2 infectivity in humans,56 as well as in many others characterizing SARS-CoV-2 genome stability,57 internalization,58 assembly,59 and mechanisms of infection and host response.60,61 Finally, our experiments in Calu-3 cells confirmed TFEB and TFE3 activation in lung cells in response to SARS-CoV-2, further suggesting a role of these transcription factors in cellular defense against human coronavirus.
Infection-associated immune responses are central to the pathogenesis of beta-coronavirus. As expected, transcriptome analysis of MHV-infected cells revealed a robust upregulation in the expression of numerous immune regulators, including pro-inflammatory cytokines and chemokines. Expression of many of these immune modulators was significantly reduced upon TFEB and TFE3 depletion, further corroborating previous studies suggesting a key role of these transcription factors in cellular defense against pathogens. Furthermore, ChIP-seq analysis revealed direct binding of TFE3 to the promoter of numerous immune regulators, indicating that the contribution of TFEB and TFE3 to immune response does not solely rely on their ability to modulate the lysosomal and autophagy pathways but also rely on their capacity to directly induce cytokine/chemokine expression. Among the TFEB and TFE3 immune direct targets newly identified in this study, we found cytokines (IL24), chemokines (CXCL2), proteins implicated in major histocompatibility complex (MHC)-I antigen presentation (B2M), regulators of viral replication (ILF3), and transcription factors involved in inflammatory response (LITAF and FOXO3), suggesting a multifactorial role of TFEB and TFE3 in antiviral response.
Beta-coronavirus pathology is caused both by direct cytotoxin effect of the virus and aberrant host immune response. Excessive elevated cytokine and chemokine levels (cytokine storm) play a central role in severity and lethality in SARS-CoV-2 infection.62,63 It has been suggested that blocking cytokine-mediated inflammatory cell death signaling pathways may limit tissue damage and benefit patients with COVID-19.51 Importantly, our study shows that the reduced expression of immune mediators observed in TFEB/TFE3-depleted cells leads to decreased virus-induced cell death, suggesting that activation of these transcription factors may contribute to the immunopathology of beta-coronavirus. It is important to keep in mind that TFEB and TFE3 might also promote cell death by directly increasing expression of proapoptotic proteins, as it has been described under certain stress conditions.55,64
Pathogens usually interfere with the function of those transcription factors that are involved in immune defense. It is therefore not surprising that MHV induces proteasomal-mediated degradation of TFEB and TFE3 as a way to limit immune response and cell death. However, the timing of this regulation may hold important information about the virus dependency of specific cellular pathways. Whereas autophagy and lysosomes play key roles in host antiviral defenses by promoting virus degradation, at least a fraction of beta-coronavirus is delivered to lysosomes to be released from cells upon induction of lysosomal exocytosis. It is therefore plausible that the virus initially allows TFEB and TFE3 activation in order to take advantage of their ability to modulate lysosomal function and promote fusion of lysosomes with the plasma membrane. At later infection times, however, virus-dependent activation of specific E3 ligases, such as DCAF7, would prevent an efficient immune response.
The overall contribution of TFEB and TFE3 to host response against beta-coronavirus is likely complex and context dependent. The ability of these transcription factors to increase expression of inflammatory cytokines suggests that they are essential components of the host immune response. However, excessive or prolonged TFEB/TFE3 activation has the potential to contribute to cytokine storm and tissue damage. Furthermore, while the TFEB/TFE3-mediated regulation of the autophagic and lysosomal pathways may contribute to virus degradation in some cell types, their ability to induce lysosomal exocytosis may also help promoting viral dissemination. Summarizing, TFEB and TFE3 play an important role in the cellular response against beta-coronavirus, suggesting that modulation of their activity might be a promising target for antiviral treatments.
Limitations of the study
There are limitations to our study: the mechanism underlying TFE3 activation has not been elucidated. In addition, the source of Ca2+ release leading to calcineurin activation (e.g., plasma membrane, ER, or lysosomes) should be further explored. Finally, our study was performed in culture cells; future studies will be required to confirm the contribution of TFEB and TFE3 to the immune response against beta-coronavirus in vivo.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Caspase-3 (D3R6Y) Rabbit mAb | Cell Signaling Technology | Cat# 9664; RRID:AB_2798429 |
| Cleaved Caspase-3 (Asp175) (5A1E) Rabbit mAb | Cell Signaling Technology | Cat# 9664, RRID:AB_2070042 |
| PARP Antibody | Cell Signaling Technology | Cat# 9542, RRID:AB_2160739 |
| Cleaved PARP (Asp214) (D64E10) XP Rabbit mAb | Cell Signaling Technology | Cat# 5625, RRID:AB_10699459 |
| Caspase-9 (C9) Mouse mAb | Cell Signaling Technology | Cat# 9508, RRID:AB_2068620 |
| Cleaved Caspase-9 (Asp330) (E5Z7N) Rabbit mAb | Cell Signaling Technology | Cat# 52873, RRID:AB_2799423 |
| Caspase-7 (D2Q3L) Rabbit Antibody | Cell Signaling Technology | Cat# 12827, RRID:AB_2687912 |
| Cleaved Caspase-7 (Asp198) (D6H1) Rabbit mAb | Cell Signaling Technology | Cat# 8438; RRID:AB_11178377 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074, RRID:AB_2099233 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7076, RRID:AB_330924 |
| TFE3 Antibody | Cell Signaling Technology | Cat# 14779, RRID:AB_2687582 |
| TFEB Antibody | Cell Signaling Technology | Cat# 4240, RRID:AB_11220225 |
| GAPDH Monoclonal Antibody (6C5) | Thermo Fisher Scientific | Cat# AM4300, RRID:AB_2536381 |
| Goat anti-Rabbit IgG (Heavy Chain), Superclonal Recombinant Secondary Antibody, Alexa Fluor™ 488 | Thermo Fisher Scientific | Cat# A27034, RRID:AB_2536097 |
| Goat anti-Rabbit IgG (Heavy Chain), Superclonal Recombinant Secondary Antibody, Alexa Fluor™ 555 | Thermo Fisher Scientific | Cat# A27039, RRID:AB_2536100 |
| LAMP-1 (human) | DSHB | Cat# H4A3, RRID:AB_2296838 |
| p70 S6 Kinase Antibody | Cell Signaling Technology | Cat# 9202, RRID:AB_331676 |
| Phospho-p70 S6 Kinase (Thr389) Antibody | Cell Signaling Technology | Cat# 9205, RRID:AB_330944 |
| Phospho-4E-BP1 (Ser65) Antibody | Cell Signaling Technology | Cat# 9451, RRID:AB_330947 |
| 4E-BP1 (53H11) Rabbit mAb | Cell Signaling Technology | Cat# 9644, RRID:AB_2097841 |
| Phospho-Stat3 (Tyr705) (D3A7) XP Rabbit mAb | Cell Signaling Technology | Cat# 9145, RRID:AB_2491009 |
| Stat3 (D3Z2G) Rabbit mAb #12640 | Cell Signaling Technology | Cat# 12640, RRID:AB_2629499 |
| Calnexin (C5C9) Rabbit mAb | Cell Signaling Technology | Cat# 2679, RRID:AB_2228381 |
| Pan-Calcineurin A Antibody | Cell Signaling Technology | Cat# 2614, RRID:AB_2168458 |
| Phospho-TFEB (Ser211) (E9S8N) Rabbit mAb | Cell Signaling Technology | Cat# 37681, RRID:AB_2799117 |
| Phospho-Stat3 (Ser727) | Cell Signaling Technology | Cat# 34911, RRID:AB_2737598 |
| Histone H3 (D1H2) XP Rabbit mAb | Cell Signaling Technology | Cat# 4499, RRID:AB_10544537 |
| NFAT1 (D43B1) XP Rabbit mAb | Cell Signaling Technology | Cat# 5861, RRID:AB_10834808 |
| Rabbit anti-TFEB Antibody, Affinity Purified | Bethyl | Cat# A303-673A, RRID:AB_1120475 |
| Anti-TFE3 antibody produced in rabbit | Sigma-Aldrich | Cat# HPA023881, RRID:AB_1857931 |
| Anti-phospho S321 TFE3 | Martina et al., 201655 | N/A |
| Anti-MHV(MJ1.3) | Ghosh et al., 202030 | N/A |
| Rabbit Anti-Human TGN46 Polyclonal antibody, Unconjugated | Bio-Rad | Cat# AHP1586, RRID:AB_2303333 |
| Anti-p18 INK, C-Terminal antibody produced in rabbit | Sigma-Aldrich | Cat# SAB4500079, RRID:AB_10742707 |
| NF-κB p65 (D14E12) XP® Rabbit mAb #8242 | Cell Signaling Technology | Cat# 8242, RRID:AB_10859369 |
| Phospho-NF-κB p65 (Ser536) (93H1) Rabbit mAb #3033 | Cell Signaling Technology | Cat# 3033, RRID:AB_331284 |
| Phospho-IκBα (Ser32) (14D4) Rabbit mAb #2859 | Cell Signaling Technology | Cat# 2859, RRID:AB_561111 |
| IκBα (L35A5) Mouse mAb (Amino-terminal Antigen) #4814 | Cell Signaling Technology | Cat# 4814, RRID:AB_390781 |
| Stat1 (D1K9Y) Rabbit mAb #14994 | Cell Signaling Technology | Cat# 14994, RRID:AB_2737027 |
| Phospho-Stat1 (Tyr701) (58D6) Rabbit mAb #9167 | Cell Signaling Technology | Cat# 9167, RRID:AB_561284 |
| CHOP (L63F7) Mouse mAb #2895 | Cell Signaling Technology | Cat# 2895, RRID:AB_2089254 |
| ATF-4 (D4B8) Rabbit mAb #11815 | Cell Signaling Technology | Cat# 11815, RRID:AB_2616025 |
| LC3B Antibody #2775 | Cell Signaling Technology | Cat# 2775, RRID:AB_915950 |
| Bacterial and virus strains | ||
| Murine Coronavirus Mouse Hepatitis Virus A-59 (MHV-A59) | Ghosh et al., 202030 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Dorsomorphin dihydrochloride | Tocris | Cat# 3093 |
| FK 506 | Tocris | Cat# 3631 |
| BAPTA AM | Tocris | Cat# 2787 |
| Torin 1 | Tocris | Cat# 4247 |
| Z-VAD(OH)-FMK (Caspase Inhibitor VI) | Tocris | Cat# S8102 |
| CRT0066101 (hydrochloride) | Cayman Chemical Company | Cat# 15337 |
| Bisindolylmaleimide IV | Cayman Chemical Company | Cat# 13299 |
| Hoechst 33342 Solution (20 mM) | Thermo Fisher Scientific | Cat# 62249 |
| HCS CellMask™ Deep Red Stain | Thermo Fisher Scientific | Cat# H32721 |
| Gelatin from cold water fish skin | Sigma | Cat# G7041 |
| Ionomycin, Calcium Salt | Cell Signaling Technology | Cat# 9995S |
| Crystal Violet | Fisher Scientific | Cat# C581-25 |
| Critical commercial assays | ||
| LDH Cytotoxicity Assay Kit | Cell Signaling Technology | Cat# 37291 |
| eBioscience™ Annexin V Apoptosis Detection Kits | Thermo Fisher Scientific | Cat# 88-8006-72 |
| PureLink™ RNA Mini Kit | Thermo Fisher Scientific | Cat# 12183018A |
| Beta Hexosaminidase Activity Assay Kit | Cell Biolabs | Cat# MET-5095 |
| Proteome Profiler Human Cytokine Array Kit | R&D Systems | Cat# ARY005B |
| Deposited data | ||
| ChIP-Seq | This paper | GEO: GSE210675 |
| RNA-Seq | This paper | GEO: GSE210676 |
| Experimental models: Cell lines | ||
| HeLa-mCC1a | Ghosh et al., 202030 | N/A |
| Calu-3 | ATCC | Cat#HTB-55 |
| Oligonucleotides | ||
| See Table S4 for a list of oligonucleotides | N/A | N/A |
| Software and algorithms | ||
| Columbus 2.9.1.532 | PerkinElmer | https://www.perkinelmer.com/product/image-data-storage-and-analysis-system-columbus |
| FlowJo 10.8.1 | Becton, Dickinson & Company | https://www.flowjo.com/ |
| GraphPad Prism 9.4.1 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Fiji | Schneider et al., 201265 | https://imagej.net/Fiji |
| SoftMax Pro Software | Molecular Devices | https://www.moleculardevices.com/ |
| ShinyGO 0.76 | Ge et al., 202066 | http://bioinformatics.sdstate.edu/go/ |
| QuantStudio 12K Software v1.5 | Thermo Fisher Scientific | https://www.thermofisher.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Rosa Puertollano (puertolr@mail.nih.gov).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Cell lines and cell culture
HeLa-mCC1a cells were cultured in DMEM GlutaMAX (Thermo Fisher Scientific, Cat# 10569044), supplemented with 10% (v/v) FBS (Thermo Fisher Scientific, Cat# 10438034), 1% (v/v) penicillin-streptomycin (Thermo Fisher Scientific, Cat# 15140122) at 37°C in 5% CO2.
Method details
Virus infection
HeLa-mCC1a cells were incubated for 2 h with MHV-A59, washed, and kept in DMEM GlutaMAX, supplemented with 10% (v/v) FBS, 1% (v/v) penicillin-streptomycin at 37°C in 5% CO2 for the indicated periods of time. Calu-3 cells (ATCC HTB-55) were cultivated in Eagle’s minimum essential medium containing 10% fetal calf serum (FCS), 2 mM L-glutamine, and non-essential amino acids. Cells were grown on coverslips and incubated overnight at 37 °C and 5% CO2 in a humified atmosphere. The medium was removed, and the cells were infected with SARS-CoV-2 D614G (4.7e5 TCID50/mL). Mock samples were treated with medium only. One hour post infection (p.i.), the cells were washed with PBS (PBS), and fresh medium was added. After 1, 6, 12, and 24 h p.i., the medium was removed, and cells were fixed and processed for immunofluorescence.
SDS-PAGE/western blot
To obtain total lysates, HeLa-mCC1a cells were washed with cold PBS 1X and then RIPA Lysis Buffer (Thermo Fisher Scientific, Cat# 87788) with Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, Cat# 78440) was added. Lysates were incubated for 20 min on ice and centrifuged at 14,000 x g for 30 min at 4°C. Subsequently, the proteins were quantified by the bicinchoninic acid method (Thermo Fisher Scientific, Cat# 23235). 30 μg of proteins were incubated at 70°C for 10 min in presence of NuPAGE LDS Sample Buffer (Thermo Fisher Scientific, Cat# NP0007) and NuPAGE Sample Reducing Agent (Thermo Fisher Scientific, Cat# NP0009), separated by SDS-PAGE and blotting onto nitrocellulose membranes (Thermo Fisher Scientific, Cat# LC2000). Membranes were blocked in presence of milk/TBS 5% for 1 h with shaking and then incubated with primary antibody overnight with shaking at 4°C. Incubation with HRP-conjugated secondary antibody (1:5000) was performed for 1 h at room temperature with shaking. Detection was performed with SuperSignal West Femto (Thermo Fisher Scientific, Cat# 34096) using the C300 gel imaging system for chemiluminescence (Azure Biosystem). Fiji was used for quantification by densitometry.
Array cytokines
HeLa-mCC1a cells were incubated for 2 h with MHV-A59, washed, and kept in DMEM GlutaMAX, supplemented with 10% (v/v) FBS, 1% (v/v) penicillin-streptomycin at 37°C in 5% CO2 for 24 h. The cell culture medium obtained was used to be analyzed with Proteome Profiler Human Cytokine Array Kit (R&D Systems, Cat# ARY005B) according to the manufacturer’s instructions.
Subcellular fractionation
Cells were washed, scraped gently, and centrifuged. Pellet was incubated with buffer A (20 mM Tris-HCL 7.6; 0.1 mM EDTA; 2 mM MgCl2; Protease and Phosphatase Inhibitor Cocktail) for 2 min at room temperature followed by 10 min on ice. Then NP-40 1% was added and samples were passes through a 20G syringe 3 times. After this, samples were centrifuged 500 x g for 3 min at 4°C. Supernatants, which correspond to cytoplasmic fractions, were stored at −80°C. Cell pellets ware washed 3 times with buffer A + 1% NP-40, treated with buffer B (20 mM HEPES pH 7.9; 400 mM NaCl; 2.5% (V/V) Glycerol; 1 mM EDTA; 0.5 mM DTT; Protease and Phosphatase Inhibitor Cocktail), incubated in liquid nitrogen, and then at 37°C. Samples were incubated on ice for 20 min, centrifuged at 20.000 x g for 20 min. Supernatant, that corresponded to nuclear fraction, was stored at −80°C.
Real-time PCR
Total RNA was obtained with the PureLink RNA Mini Kit (Thermo Fisher Scientific). Reverse transcriptase reactions were carried out with SuperScript III First-Strand Synthesis Super-Mix for qRT-PCR (Thermo Fisher Scientific) after treatment with TURBO DNA-free Kit (Thermo Fisher Scientific) at 37°C for 30 min. Real-time PCR was performed with PowerUp SYBR Green Master Mix and run on the QuantStudio 12K Flex real-time PCR machine (Thermo Fisher Scientific). Relative expression was normalized against GAPDH expression levels. RNA expression was calculated with normalized expression (2−dCt) using the Ct method. For each gene, 3 technical replicates and 3 biological samples were analyzed. Primers used in this study are listed in Table S4.
RNAi
ON-TARGETplus Human siRNA for TFEB and TFE3, DCAF7, PPP2CA/B, PPP3CA/B or ON-TARGETplus Non-targeting Control Pool (Horizon Discovery) was mixed with Opti-MEM I Reduced Serum Medium (Thermo Fisher Scientific, Cat# 31985062) and Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific, Cat# 13778075). Next, the mixture was added to the cells with a final concentration of 100 nM. After 72 h, the cells were used for the treatments.
RNAseq
HeLa-mCC1a cells were incubated for 2 h with MHV-A59, washed, and kept in DMEM GlutaMAX, supplemented with 10% (v/v) FBS, 1% (v/v) penicillin-streptomycin at 37°C in 5% CO2 for 24 h. Washed cell pellets were snap-frozen on dry ice and then processed for RNA-seq assay by Active Motif, Inc. Total RNA was isolated from cells using the Qiagen RNeasy Mini Kit (Qiagen, Cat# 74104). For each sample, 0.5 ng of total RNA was then used in Illumina’s TruSeq Stranded mRNA Library kit (Illumina, Cat# 20020594). Libraries were sequenced on Illumina NextSeq 500 as paired-end 42-nt reads. Sequence reads were analyzed with the STAR alignment – DESeq2 software pipeline. The generated RNA-Seq data of HeLa-mCC1a can be downloaded at GEO (Accession GSE210676).
ChIP-seq
HeLa-mCC1a cells were incubated for 2 h with MHV-A59, washed, and kept in DMEM GlutaMAX, supplemented with 10% (v/v) FBS, 1% (v/v) penicillin-streptomycin at 37°C in 5% CO2 for 14 h. Also, HeLa-mCC1a cells were treated for 4 h with EBSS medium. Then the cell was crosslinked with 1/10 volume of freshly prepared Formaldehyde Solution (described in the “RIME” section) to the existing media of cells and agitated for 15 min at room temperature. Fixation was stopped by adding ½0 vol Glycine to the existing media. Cells were washed by transferring to a 15 mL conical tube and centrifuged at 800 x g for 10 min at 4°C. Supernatants were removed and cells were resuspended with cold PBS containing 0.5% Igepal CA-630. Cell pellets were resuspended and washed one more time with cold PBS containing 0.5% Igepal CA-630 and 1 mM PMSF. Cells were centrifuged again to pellet and snap-frozen on dry ice and then processed for Chip-seq assay by Active Motif, Inc. The generated ChIP-Seq data of HeLa-mCC1a can be downloaded at GEO (Accession GSE210675).
LDH assay and β-hexosaminidase
Relative LDH levels were measured in cell culture media. For this, the media were centrifuged for 5 min at 500 x g at room temperature and analyzed in 96 plates using LDH Cytotoxicity Assay Kit (Cell Signaling Technology, Cat# 37291) or Beta Hexosaminidase Activity Assay Kit (Cell Biolabs, Cat# MET-5095) in SpectraMax iD3 (Molecular Devices).
TCID50/mL determination
Extracellular medium collected from infected cell cultures was serially diluted and dilutions used to inoculate multiple cell cultures for up to 72 h. Cytopathic changes in the cells were tabulated to calculate TCID50/mL values, as described.67 Briefly, supernatant (media) from MHV infected obtained and inoculated to HeLa-mCC1a cells seeded at 4 × 104 cells/well in 96-well format. 8 serial dilution of the media was prepared in DMEM (10−1 to 10−11) and added in triplicates or quadruplicates for statistical significance. Cells were left in 37°C incubator for up to 72 h. Thereafter the media was discarded and each well received 100 mL of Crystal Violet solution (25% crystal violet supplemented with 20% ethanol) and incubated for 15 min. The solution was discarded, each well was washed twice with water and the image of the 96-well plate was captured. Dilution that showed 50% cell death (i.e., where 50% of the cells in dish were left behind, stained with Crystal Violet) was used for calculating TCID50/mL.
Flow cytometry analysis
HeLa-mCC1a were treated with Cellstripper (Corning, Cat# 25-056-CI) for 15 min at 37°C. Subsequently, the cells were treated with eBioscience Annexin V Apoptosis Detection Kits (Thermo Fisher Scientific, Cat# 88-8006-72) according to the manufacturer’s instructions. Cells were analyzed by flow cytometry using a BD Fortessa cytometer. The percentage of differentially labeled cells was quantified using FlowJo 10.8.1.
Immunofluorescence confocal microscopy
HeLa-mCC1a cells were seeded in coverslips in 24-well culture plates. After treatment, cells were washed with PBS and fixed in 4% paraformaldehyde in PBS for 20 min. Cells were then permeabilized in 0.2% Triton X-100 in PBS for 10 min and, after two washes, cells were incubated in 3% BSA in PBS for 30 min at room temperature. Cells were incubated with primary antibodies at 4°C overnight, washed four times with PBS and incubated with secondary antibodies (Life Technologies) at room temperature for 1h.
For LAMP1 surface staining analysis, HeLa-mCC1a were treated with anti-Lamp1 antibody (H4A3) in DMEM GlutaMAX supplemented with 10 mM HEPES and 1% BSA for 20 min at 4°C, fixed with PFA 4% for 20 min, washed once in PBS, and incubated with secondary antibody for 1h at room temperature. Cells were then washed and incubated with Hoechst 33,342 20 mM (1:5000) for 15 min at room temperature. Samples were examined under a Zeiss LSM 510 confocal microscope and analyzed and quantified with the ImageJ software.
High-throughput imaging
HeLa mCC1a cells were seeded in 96-well plates (Greiner Bio-One 655,090) at a density of 60,000 cells/cm2. Cells were incubated for 2 h with MHV-A59, washed, and treated with drugs until 16 h p.i. Subsequently, cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with TX-100 1% for 30 min and blocked with 3% fish gelatin for 1 h at room temperature. The cells were incubated with anti-TFEB or anti-TFE3 1:150 over night at 4°C and with Alexa Fluor 488 secondary antibodies 1:1000 for 1 h at room temperature. To stain the membrane and nucleus, HCS CellMask Deep Red Stain (1:50,000) and Hoechst 33,342 20 mM (1:5000) were used for 15 min at room temperature, respectively. Images were acquired with Cell_Voyager (CV7000) in confocal mode using 405/488/640 nm filters, 20× objective (N.A. = 0.75), and laser-based autofocus. 16 images (20× magnification) were acquired per well and then analyzed with Columbus 2.9.1.532 software. Cells were segmented by cell nucleus (blue color) and the intensity of green fluorescence was measured to identify the cells with nuclear TFEB. The script was designed using Torin 1 (0.3 μM) as a positive control and without anti-TFEB or anti-TFE3 as a negative control.
Quantification and statistical analysis
Statistical analyzes were performed with One-way ANOVA with Dunnett’s as a post-test. two-way ANOVA with Sıdak’s as a post-test. Also, Student’s t-tests were performed to compare two populations. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Data represent mean ± SEM.
Acknowledgments
This work was supported by the Intramural Research Program of the NIH, National Heart, Lung, and Blood Institute (NHLBI) (ZIA HL006151). We thank Drs. Philip McCoy, Maria-Lopez Ocasio, and Pradeep Dagur (Flow Cytometry Core, NHLBI) for their assistance in the Annexin V apoptosis analysis. High-throughput imaging work was performed at the High-Throughput Imaging Facility (HiTIF)/Center for Cancer Research/National Cancer Institute/NIH. Graphics in Figures S1A, 2A, S2B, and 6F and graphical abstract were created with BioRender.com
Author contributions
P.S.C., P.J.T., and E.J. were involved in the experimental strategy, performed the experiments, analyzed the data, and participated in the preparation of the manuscript; S.G. provided reagents and valuable suggestions; N.A. reviewed the manuscript and provided reagents and valuable suggestions; R.P. designed the research, analyzed the data, supervised the project, and wrote the manuscript. All the authors reviewed the manuscript.
Declaration of interests
The authors declare no competing interest.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location.
Published: February 9, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106169.
Supplemental information
Data and code availability
-
•
RNA-Seq and ChIP-Seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Raben N., Puertollano R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu. Rev. Cell Dev. Biol. 2016;32:255–278. doi: 10.1146/annurev-cellbio-111315-125407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tan A., Prasad R., Lee C., Jho E.H. Past, present, and future perspectives of transcription factor EB (TFEB): mechanisms of regulation and association with disease. Cell Death Differ. 2022;29:1433–1449. doi: 10.1038/s41418-022-01028-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martina J.A., Chen Y., Gucek M., Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914. doi: 10.4161/auto.19653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roczniak-Ferguson A., Petit C.S., Froehlich F., Qian S., Ky J., Angarola B., Walther T.C., Ferguson S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012;5:ra42. doi: 10.1126/scisignal.2002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martina J.A., Diab H.I., Lishu L., Jeong-A L., Patange S., Raben N., Puertollano R. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal. 2014;7:ra9. doi: 10.1126/scisignal.2004754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Settembre C., Zoncu R., Medina D.L., Vetrini F., Erdin S., Erdin S., Huynh T., Ferron M., Karsenty G., Vellard M.C., et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Medina D.L., Di Paola S., Peluso I., Armani A., De Stefani D., Venditti R., Montefusco S., Scotto-Rosato A., Prezioso C., Forrester A., et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015;17:288–299. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martina J.A., Puertollano R. Protein phosphatase 2A stimulates activation of TFEB and TFE3 transcription factors in response to oxidative stress. J. Biol. Chem. 2018;293:12525–12534. doi: 10.1074/jbc.RA118.003471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sardiello M., Palmieri M., di Ronza A., Medina D.L., Valenza M., Gennarino V.A., Di Malta C., Donaudy F., Embrione V., Polishchuk R.S., et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 10.Settembre C., Di Malta C., Polito V.A., Garcia Arencibia M., Vetrini F., Erdin S., Erdin S.U., Huynh T., Medina D., Colella P., et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Markby G.R., Sakamoto K. Transcription factor EB and TFE3: new metabolic coordinators mediating adaptive responses to exercise in skeletal muscle? Am. J. Physiol. Endocrinol. Metab. 2020;319:E763–E768. doi: 10.1152/ajpendo.00339.2020. [DOI] [PubMed] [Google Scholar]
- 12.Kim Y.S., Lee H.M., Kim J.K., Yang C.S., Kim T.S., Jung M., Jin H.S., Kim S., Jang J., Oh G.T., et al. PPAR-Alpha activation mediates innate host defense through induction of TFEB and lipid catabolism. J. Immunol. 2017;198:3283–3295. doi: 10.4049/jimmunol.1601920. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Z., Chen C., Yang F., Zeng Y.X., Sun P., Liu P., Li X. Itaconate is a lysosomal inducer that promotes antibacterial innate immunity. Mol. Cell. 2022;82:2844–2857.e10. doi: 10.1016/j.molcel.2022.05.009. [DOI] [PubMed] [Google Scholar]
- 14.Visvikis O., Ihuegbu N., Labed S.A., Luhachack L.G., Alves A.M.F., Wollenberg A.C., Stuart L.M., Stormo G.D., Irazoqui J.E. Innate host defense requires TFEB-mediated transcription of cytoprotective and antimicrobial genes. Immunity. 2014;40:896–909. doi: 10.1016/j.immuni.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pastore N., Brady O.A., Diab H.I., Martina J.A., Sun L., Huynh T., Lim J.A., Zare H., Raben N., Ballabio A., Puertollano R. TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy. 2016;12:1240–1258. doi: 10.1080/15548627.2016.1179405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tseng H.H.L., Vong C.T., Kwan Y.W., Lee S.M.Y., Hoi M.P.M. Lysosomal Ca(2+) signaling regulates high glucose-mediated interleukin-1beta secretion via transcription factor EB in human monocytic cells. Front. Immunol. 2017;8:1161. doi: 10.3389/fimmu.2017.01161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayama Y., Kimura T., Takeda Y., Nada S., Koyama S., Takamatsu H., Kang S., Ito D., Maeda Y., Nishide M., et al. Lysosomal protein Lamtor1 controls innate immune responses via nuclear translocation of transcription factor EB. J. Immunol. 2018;200:3790–3800. doi: 10.4049/jimmunol.1701283. [DOI] [PubMed] [Google Scholar]
- 18.Fang L., Hodge J., Saaoud F., Wang J., Iwanowycz S., Wang Y., Hui Y., Evans T.D., Razani B., Fan D. Transcriptional factor EB regulates macrophage polarization in the tumor microenvironment. OncoImmunology. 2017;6:e1312042. doi: 10.1080/2162402X.2017.1312042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen D., Xie J., Fiskesund R., Dong W., Liang X., Lv J., Jin X., Liu J., Mo S., Zhang T., et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat. Commun. 2018;9:873. doi: 10.1038/s41467-018-03225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bretou M., Sáez P.J., Sanséau D., Maurin M., Lankar D., Chabaud M., Spampanato C., Malbec O., Barbier L., Muallem S., et al. Lysosome signaling controls the migration of dendritic cells. Sci. Immunol. 2017;2:eaak9573. doi: 10.1126/sciimmunol.aak9573. [DOI] [PubMed] [Google Scholar]
- 21.Najibi M., Labed S.A., Visvikis O., Irazoqui J.E. An evolutionarily conserved PLC-PKD-TFEB pathway for host defense. Cell Rep. 2016;15:1728–1742. doi: 10.1016/j.celrep.2016.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El-Houjeiri L., Possik E., Vijayaraghavan T., Paquette M., Martina J.A., Kazan J.M., Ma E.H., Jones R., Blanchette P., Puertollano R., Pause A. The transcription factors TFEB and TFE3 link the FLCN-AMPK signaling Axis to innate immune response and pathogen resistance. Cell Rep. 2019;26:3613–3628.e6. doi: 10.1016/j.celrep.2019.02.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martina J.A., Guerrero-Gómez D., Gómez-Orte E., Antonio Bárcena J., Cabello J., Miranda-Vizuete A., Puertollano R. A conserved cysteine-based redox mechanism sustains TFEB/HLH-30 activity under persistent stress. EMBO J. 2021;40:e105793. doi: 10.15252/embj.2020105793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campbell G.R., Rawat P., Bruckman R.S., Spector S.A. Human immunodeficiency virus type 1 Nef inhibits autophagy through transcription factor EB sequestration. PLoS Pathog. 2015;11:e1005018. doi: 10.1371/journal.ppat.1005018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chauhan S., Ahmed Z., Bradfute S.B., Arko-Mensah J., Mandell M.A., Won Choi S., Kimura T., Blanchet F., Waller A., Mudd M.H., et al. Pharmaceutical screen identifies novel target processes for activation of autophagy with a broad translational potential. Nat. Commun. 2015;6:8620. doi: 10.1038/ncomms9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alirezaei M., Flynn C.T., Garcia S.D., Kimura T., Whitton J.L. A food-responsive switch modulates TFEB and autophagy, and determines susceptibility to coxsackievirus infection and pancreatitis. Autophagy. 2021;17:402–419. doi: 10.1080/15548627.2020.1720425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohamud Y., Tang H., Xue Y.C., Liu H., Ng C.S., Bahreyni A., Luo H. Coxsackievirus B3 targets TFEB to disrupt lysosomal function. Autophagy. 2021;17:3924–3938. doi: 10.1080/15548627.2021.1896925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fung T.S., Liu D.X. Human coronavirus: host-pathogen interaction. Annu. Rev. Microbiol. 2019;73:529–557. doi: 10.1146/annurev-micro-020518-115759. [DOI] [PubMed] [Google Scholar]
- 29.Li G., Fan Y., Lai Y., Han T., Li Z., Zhou P., Pan P., Wang W., Hu D., Liu X., et al. Coronavirus infections and immune responses. J. Med. Virol. 2020;92:424–432. doi: 10.1002/jmv.25685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghosh S., Dellibovi-Ragheb T.A., Kerviel A., Pak E., Qiu Q., Fisher M., Takvorian P.M., Bleck C., Hsu V.W., Fehr A.R., et al. Beta-coronaviruses use lysosomes for egress instead of the biosynthetic secretory pathway. Cell. 2020;183:1520–1535.e14. doi: 10.1016/j.cell.2020.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen D., Zheng Q., Sun L., Ji M., Li Y., Deng H., Zhang H. ORF3a of SARS-CoV-2 promotes lysosomal exocytosis-mediated viral egress. Dev. Cell. 2021;56:3250–3263.e5. doi: 10.1016/j.devcel.2021.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miao G., Zhao H., Li Y., Ji M., Chen Y., Shi Y., Bi Y., Wang P., Zhang H. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell. 2021;56:427–442.e5. doi: 10.1016/j.devcel.2020.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y., Sun H., Pei R., Mao B., Zhao Z., Li H., Lin Y., Lu K. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discov. 2021;7:31. doi: 10.1038/s41421-021-00268-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korner R.W., Majjouti M., Alcazar M.A.A., Mahabir E. Of mice and men: The coronavirus MHV and mouse models as a translational approach to understand SARS-CoV-2. Viruses. 2020;12 doi: 10.3390/v12080880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bedell K., Buchaklian A.H., Perlman S. Efficacy of an automated multiple emitter whole-room ultraviolet-C disinfection system against coronaviruses MHV and MERS-CoV. Infect. Control Hosp. Epidemiol. 2016;37:598–599. doi: 10.1017/ice.2015.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burkard C., Verheije M.H., Wicht O., van Kasteren S.I., van Kuppeveld F.J., Haagmans B.L., Pelkmans L., Rottier P.J.M., Bosch B.J., de Haan C.A.M. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog. 2014;10:e1004502. doi: 10.1371/journal.ppat.1004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ulasli M., Gurses S.A., Bayraktar R., Yumrutas O., Oztuzcu S., Igci M., Igci Y.Z., Cakmak E.A., Arslan A. The effects of Nigella sativa (Ns), Anthemis hyalina (Ah) and Citrus sinensis (Cs) extracts on the replication of coronavirus and the expression of TRP genes family. Mol. Biol. Rep. 2014;41:1703–1711. doi: 10.1007/s11033-014-3019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thorp E.B., Gallagher T.M. Requirements for CEACAMs and cholesterol during murine coronavirus cell entry. J. Virol. 2004;78:2682–2692. doi: 10.1128/jvi.78.6.2682-2692.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gosert R., Kanjanahaluethai A., Egger D., Bienz K., Baker S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 2002;76:3697–3708. doi: 10.1128/jvi.76.8.3697-3708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rao P.V., Gallagher T.M. Intracellular complexes of viral spike and cellular receptor accumulate during cytopathic murine coronavirus infections. J. Virol. 1998;72:3278–3288. doi: 10.1128/JVI.72.4.3278-3288.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tooze J., Tooze S.A., Fuller S.D. Sorting of progeny coronavirus from condensed secretory proteins at the exit from the trans-Golgi network of AtT20 cells. J. Cell Biol. 1987;105:1215–1226. doi: 10.1083/jcb.105.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ujike M., Taguchi F. Incorporation of spike and membrane glycoproteins into coronavirus virions. Viruses. 2015;7:1700–1725. doi: 10.3390/v7041700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martínez-Fábregas J., Prescott A., van Kasteren S., Pedrioli D.L., McLean I., Moles A., Reinheckel T., Poli V., Watts C. Lysosomal protease deficiency or substrate overload induces an oxidative-stress mediated STAT3-dependent pathway of lysosomal homeostasis. Nat. Commun. 2018;9:5343. doi: 10.1038/s41467-018-07741-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Y., Lear T., Larsen M., Lin B., Cao Q., Alfaras I., Kennerdell J., Salminen L., Camarco D., Lockwood K., et al. Modulation of lysosomal function as a therapeutic approach for coronaviral infections. Res. Sq. 2021 doi: 10.21203/rs.3.rs-419305/v1. [DOI] [Google Scholar]
- 45.Li Y., Duche A., Sayer M.R., Roosan D., Khalafalla F.G., Ostrom R.S., Totonchy J., Roosan M.R. SARS-CoV-2 early infection signature identified potential key infection mechanisms and drug targets. BMC Genom. 2021;22:125. doi: 10.1186/s12864-021-07433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nilsson-Payant B.E., Uhl S., Grimont A., Doane A.S., Cohen P., Patel R.S., Higgins C.A., Acklin J.A., Bram Y., Chandar V., et al. The NF-kappaB transcriptional footprint is essential for SARS-CoV-2 replication. J. Virol. 2021;95:e0125721. doi: 10.1128/JVI.01257-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen Q., Wang Y.E., Palazzo A.F. Crosstalk between nucleocytoplasmic trafficking and the innate immune response to viral infection. J. Biol. Chem. 2021;297:100856. doi: 10.1016/j.jbc.2021.100856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmidt N., Lareau C.A., Keshishian H., Ganskih S., Schneider C., Hennig T., Melanson R., Werner S., Wei Y., Zimmer M., et al. The SARS-CoV-2 RNA-protein interactome in infected human cells. Nat. Microbiol. 2021;6:339–353. doi: 10.1038/s41564-020-00846-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu B., Ramaiah A., Garcia G., Jr., Hasiakos S., Arumugaswami V., Srikanth S. ORAI1 limits SARS-CoV-2 infection by regulating tonic type I IFN signaling. J. Immunol. 2022;208:74–84. doi: 10.4049/jimmunol.2100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ibrahim I.H., Ellakwa D.E.S. SUMO pathway, blood coagulation and oxidative stress in SARS-CoV-2 infection. Biochem. Biophys. Rep. 2021;26:100938. doi: 10.1016/j.bbrep.2021.100938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karki R., Sharma B.R., Tuladhar S., Williams E.P., Zalduondo L., Samir P., Zheng M., Sundaram B., Banoth B., Malireddi R.K.S., et al. Synergism of TNF-alpha and IFN-gamma triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell. 2021;184:149–168.e17. doi: 10.1016/j.cell.2020.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng M., Williams E.P., Malireddi R.K.S., Karki R., Banoth B., Burton A., Webby R., Channappanavar R., Jonsson C.B., Kanneganti T.D. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J. Biol. Chem. 2020;295:14040–14052. doi: 10.1074/jbc.RA120.015036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Medina D.L., Fraldi A., Bouche V., Annunziata F., Mansueto G., Spampanato C., Puri C., Pignata A., Martina J.A., Sardiello M., et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell. 2011;21:421–430. doi: 10.1016/j.devcel.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Versteeg G.A., van de Nes P.S., Bredenbeek P.J., Spaan W.J.M. The coronavirus spike protein induces endoplasmic reticulum stress and upregulation of intracellular chemokine mRNA concentrations. J. Virol. 2007;81:10981–10990. doi: 10.1128/JVI.01033-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martina J.A., Diab H.I., Brady O.A., Puertollano R. TFEB and TFE3 are novel components of the integrated stress response. EMBO J. 2016;35:479–495. doi: 10.15252/embj.201593428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wakida H., Kawata K., Yamaji Y., Hattori E., Tsuchiya T., Wada Y., Ozaki H., Akimitsu N. Stability of RNA sequences derived from the coronavirus genome in human cells. Biochem. Biophys. Res. Commun. 2020;527:993–999. doi: 10.1016/j.bbrc.2020.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ou X., Liu Y., Lei X., Li P., Mi D., Ren L., Guo L., Guo R., Chen T., Hu J., et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020;11:1620. doi: 10.1038/s41467-020-15562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumar B., Hawkins G.M., Kicmal T., Qing E., Timm E., Gallagher T. Assembly and entry of severe acute respiratory syndrome coronavirus 2 (SARS-CoV2): evaluation using virus-like particles. Cells. 2021;10 doi: 10.3390/cells10040853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sui C., Xiao T., Zhang S., Zeng H., Zheng Y., Liu B., Xu G., Gao C., Zhang Z. SARS-CoV-2 NSP13 inhibits type I IFN production by degradation of TBK1 via p62-dependent selective autophagy. J. Immunol. 2022;208:753–761. doi: 10.4049/jimmunol.2100684. [DOI] [PubMed] [Google Scholar]
- 61.Shi G., Chiramel A.I., Li T., Lai K.K., Kenney A.D., Zani A., Eddy A.C., Majdoul S., Zhang L., Dempsey T., et al. Rapalogs downmodulate intrinsic immunity and promote cell entry of SARS-CoV-2. J. Clin. Invest. 2022;132:e160766. doi: 10.1172/JCI160766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jose R.J., Manuel A. COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir. Med. 2020;8:e46–e47. doi: 10.1016/S2213-2600(20)30216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mehta P., McAuley D.F., Brown M., Sanchez E., Tattersall R.S., Manson J.J., HLH Across Speciality Collaboration UK COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–1034. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang C.B., Liu J., Tong B.C.K., Wang Z.Y., Zhu Z., Su C.F., Sreenivasmurthy S.G., Wu J.X., Iyaswamy A., Krishnamoorthi S., et al. TFEB, a master regulator of autophagy and biogenesis, unexpectedly promotes apoptosis in response to the cyclopentenone prostaglandin 15d-PGJ2. Acta Pharmacol. Sin. 2022;43:1251–1263. doi: 10.1038/s41401-021-00711-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ge S.X., Jung D., Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics. 2020;36:2628–2629. doi: 10.1093/bioinformatics/btz931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Leibowitz J., Kaufman G., Liu P. Coronaviruses: propagation, quantification, storage, and construction of recombinant mouse hepatitis virus. Curr. Protoc. Microbiol. 2011;Chapter 15 doi: 10.1002/9780471729259.mc15e01s21. Unit 15E 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA-Seq and ChIP-Seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.