

Abstract
Objectives: In this study, we aimed to identify the metabolic genes associated with the metastasis and immunotherapy resistance of oral squamous cell carcinoma (OSCC) and to construct a metabolic gene-related predictive model for the prognosis of OSCC. Methods: RNA-seq data were download from The Cancer Genome Atlas (TCGA). Weighted gene co-expression network analysis (WGCNA) was applied to identify the modules related to EMT, stemness, and checkpoint signatures in OSCC. Univariate Cox and the least absolute shrinkage and selection operator (LASSO) methods were used to construct the metabolic gene signature. Furthermore, the scRNA-seq data were obtained from Gene Expression Omnibus (GEO) database and analyzed using “Seurat” and “CopyKAT” packages. Results: The risk prediction model was constructed using the 12 metabolic-related gene signature. Based on this model, risk score of each sample was calculated and used to divide the samples into low- and high-risk groups. Our model was effective as the risk score was significantly associated with clinical features and genetic mutations. Meanwhile, we found that lipid metabolism, glycolysis, amino acid metabolism, and drug metabolism differed between high- and low-risk groups. Pathways associated with malignant tumor and immunosuppression were enriched in high-risk group. Furthermore, low-risk group showed a more activated immune status and was predicted to have better response to immunotherapy. Finally, through single-cell transcriptome analysis, we assessed the expression of these 12 genes in tumor and non-tumor cells and verified the existence of two clusters of tumor cells with different degrees of malignancy at the cellular level. Conclusions: Our study demonstrates the clinical significance of metabolic related gene signature for the treatment of OSCC and suggests potential therapeutic targets and pathways for OSCC.
Keywords: Oral squamous cell carcinoma, WGCNA, metabolism, metastasis, immunotherapy resistance
Introduction
Oral squamous cell carcinoma (OSCC) is the most fatal oral disease and the most common head and neck malignancy. Its incidence is increasing with approximately 400,000 new cases and 180,000 deaths annually [1]. OSCC originates from the mucosal epithelium of the oral cavity and pharynx, and the risk factors for OSCC include smoking, excessive alcohol consumption, betel nut chewing, and human papillomavirus (HPV) exposure [2,3]. Although early OSCC can achieve a good prognosis through surgical resection, most OSCC patients do not exhibit symptoms at the early stage but in advanced stages at the time of diagnosis. Furthermore, OSCC also have high recurrence rates [4]; hence, the survival rate of OSCC is still not significantly improved even with the improved treatment strategies [5,6].
Lymph node metastasis occurs in more than half of the patients with OSCC and is closely related to the prognosis of OSCC [7,8]. An important factor attributing to lymph node and distant metastasis is epithelial-mesenchymal transition (EMT), a genetic and epigenetically regulated biological process in which an epithelial cell transforms into a mesenchymal state [9-11]. Tumor cells with EMT are characterized by changes in cytoskeleton, decreased adhesion to their surroundings, enhanced migration, and apoptotic resistance [12,13]. Many studies have demonstrated that EMT activation is closely interconnected with the generation of cancer stem cells (CSCs) in various types of carcinomas [14]. EMT activation also enables tumor cells to escape immune killing by increasing the expression of immune checkpoint genes and acquire resistance to chemotherapy and immunotherapy [15-18], which are also characteristics of CSCs. Anti-PD1 monoclonal antibodies, such as pembrolizumab and nivolumab, have been used as first-line treatment for malignant OSCC [19,20], however, many patients do not respond to immunotherapy. Studies have shown that tumor cells resistant to chemotherapy, targeted therapy, or immunotherapy, commonly exhibit activated EMT [21].
EMT is a dynamic and reversible process that involves extensive crosstalk with metabolic reprogramming, which is regulated by EMT transcription factors (EMT-TFs) and epigenetic modifying enzymes that use metabolites as cofactors and substrates. Moreover, changes in the migration and invasion of tumor cells require metabolic reprogramming to provide energy [22]. Transforming Growth Factor β (TGF-β) and hypoxia can activate EMT in tumor cells as well as induce glycolysis and fatty acid oxidation (FAO) pathways [23,24]. Glutamine metabolism and mitochondrial metabolism are also changed during EMT, and the expression levels of SLC2A1/3, HK2, CD36, and GLS1/2 involved in the above metabolic processes are altered [25-27]. These extensive changes in metabolic processes are essential for EMT and constitute potential metabolic targets for inhibiting EMT.
Although EMT activation has been known in the tumor cells of patients with OSCC, contributing to the poor prognosis of OSCC [28], the interaction between metabolic pathways and EMT in OSCC remains unclear. Investigating this link is essential for improving immunotherapy outcomes in patients with OSCC and providing metabolically targeted therapies. Therefore, in this study, we used TCGA database to identify the most relevant metabolic gene modules for EMT, stemness, and checkpoint signatures in OSCC. We constructed a metabolism-related prognostic signature containing 12 genes using least absolute shrinkage and selection operator (LASSO) regression. Furthermore, we showed the association between the risk score derived from the signature and the clinical features and immune landscape. Finally, we used single-cell RNA sequencing (scRNA-seq) data from OSCC patients to explore the expression of these 12 genes in tumor and non-tumor cells. We divided tumor cells into two clusters based on copy number variation (CNV) to explain the heterogeneity of tumor cells in terms of risk metabolic gene.
Materials and methods
Study design and data collection
The workflow diagram of this study was presented in Figure S1. First, RNA-seq data, clinical information, somatic mutations, and CNVs data were downloaded from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/). In total, 330 patients with OSCC were screened from the TCGA-HNSC cohort. Next, ScRNA-seq data were downloaded from the GEO database (GSE103322, http://www.ncbi.nlm.nih.gov/geo/) which was provided by Bradley et al. [29], and contained data of 5902 cells from 18 OSCC patients. R4.1.2 software was used for data preprocessing.
Co-expression network construction and identification of signature-related modules
We obtained 2119 genes related to “Metabolism” pathways (R-HSA-1430728) from the Reactome database using the “ReactomePA” package [30]. Among these genes, the EMT signature- [31], stemness signature- [32], and checkpoint signature- [33] related genes were downloaded and subjected to single-sample gene set enrichment analysis (ssGSEA) to calculate the scores of EMT, stemness, and checkpoint. The “WGCNA” package was applied to construct a co-expression network of metabolic genes. The matrix of adjacencies was built based on the Pearson’s correlation value between paired genes. An unsigned scale-free co-expression network was constructed based on a soft thresholding power of β = 4. Then, “TOMsimilarity” and “cutreeDynamic” were used to construct the metabolic gene co-expression module, and the merge height cut was set at 0.4 to merge modules. The correlation between module eigengenes (MEs) and signatures was tested using Pearson’s correlation. The adjacency threshold was set at 0.05 to select hub genes from the MEs of interest. The “iRegulon” plug in Cytoscape was used to identify the transcription factors of hub genes (FDR < 0.001).
Construction of the prognosis signature
We used genes with an adjacency threshold greater than 0.05 in significant MEs for the univariate Cox regression. LASSO regression was used to screen out the genes most related to survival and to calculate their coefficients. Finally, the risk score was calculated for patients in TCGA data using the formula below, and validation was performed in the GEO dataset (GSE41613).
Functional and pathway enrichment analyses
Functional enrichment analysis of the hub genes was performed using Metascape (http://metascape.org) [34]. The software GSEA (http://www.gsea-msigdb.org/) and the C2, C6, and C7 gene sets were downloaded to analyze different pathways in the high- and low-risk groups. Differential expression gene (DEG) analysis was performed by “limma3.50.3” package using the expression matrix of patients in the top 25% and the bottom 25% of the risk score, and genes with |logFC| > 1, P < 0.05 were screened for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment. Only the gene sets with P < 0.05 were considered significant.
Metabolic pathways, immune signatures, and cell types enrichment analysis
Metabolic gene sets, involving 90 human metabolic pathways, were downloaded from the KEGG pathway database (https://www.genome.jp/kegg/pathway.html) [35]. Immune signatures were identified based on previously published studies [33,36,37]. Cell type characterization and composition estimation were performed using CIBERSORT (http://cibersort.stanford.edu) [38]. Package “ggplot2” was used to display the immune cell composition and immune signature of different groups. Function “mantel_test” was used to identify the relationship between the 19 signatures and the risk score, and the results were shown by “ggcor” package. The relationship among different types of immune cells was analyzed by Spearman correlation and graphed by “corrplot” package. In addition, we calculated the hazard ratio (HR) of 22 types of immune cells by using “coxph” function in “survival” package.
Evaluation of immune-checkpoint blockade (ICB) response and drug sensitivity prediction
The ICB response signature was adopted from published research, and ssGSEA was used to estimate the ICB response score. Since the KMplot tool (http://kmplot.com/analysis) contains immunotherapy data for multiple tumors, we selected anti-PD1 treatment data and pretreatment samples to obtain the survival curves of high- and low-risk patients. Furthermore, we used “pRRophetic” package to calculate the half-maximal inhibitory concentration (IC50) of 5-Fluorouracil, Cetuximab and Cisplatin according to “cgp2016” dataset.
Single cell RNA-seq analysis
ScRNA-seq data of 5902 cells were first subjected to quality control using the “Seurat” package, and the data with nFeature > 2000, nCount < 20000, and percent.mt < 15 were filtered for further analysis, yielding a total of 5746 cells. The data were normalized using log2 (TPM/10+1), and t-distributed stochastic neighbor embedding (tSNE) was used for dimensionality reduction and cluster classification. Furthermore, CopyKAT (Copynumber Karyotyping of Tumors), a package designed to identify genomic copy number for scRNA-seq data, was used to distinguish tumor cells by recognizing aneuploid cells, and we set win.size at 25 & KS.cut at 0.2. Then, tumor cells were divided into two clones by calculating Euclidean distance with CopyKAT [39].
Results
Identification of significant genes related to EMT, stemness, and checkpoints
We obtained RNA-seq data of 546 HNSC samples from the TCGA database and screened 330 oral cancer samples from the lip, gingiva, tongue, oral cavity, and pharynx. The detailed patient information was provided in Table S1. Since EMT is a key factor contributing to the metastasis of cancers [40], and regulates not only tumor cell migration but also the generation of cancer stem cells as well as the expression of checkpoint genes [41], in this study, we calculated the EMT, stemness, and checkpoint signature scores of 330 HNSC samples using ssGSEA (Table S2). A total of 2119 metabolic-related genes were obtained from the Reactome database. We chose β = 4 (scale-free R2 = 0.92) as the soft threshold power to build a scale-free network (Figure S2A and S2B). According to the expression of these genes, we divided them into seven modules by average linkage clustering (Figure S2C-E). We then displayed a heatmap of the correlation between modules and signatures, showing a significant positive relationship among the green module, EMT, and checkpoint signatures (Figure 1A). As invasive metastasis is an inefficient process in which only a small portion of EMT tumor cells can survive immune surveillance [42], to obtain genes with greater clinical value for immune therapy, we also selected from the brown module genes that negatively correlated with the checkpoint for subsequent analysis.
Figure 1.

Module-trait associations and identification of significant genes in green and brown modules. A. The correlation between modules and signatures. Each cell contains the corresponding correlation and P value. B. Function enrichment of genes in green and brown module by Metascape. C. The results of 259 genes in green and brown modules and 21 genes were significantly associated with the survival of patients.
According to topological overlap above 0.05 in MEs, there were 165 hub genes in the green module and 94 hub genes in the brown module. We used “iRegulon” to predict the transcription factors (TFs) of genes in green and brown modules (FDR < 0.001). The top 5 TFs for green modules were SPL1, SOX21, CTCF, CEBPA, ELF2, while the top 5 TFs for brown modules were FOXO1, TCF12, EP300, MSRB3, NR4A1 (Figure S3). Upon importing these genes into Metascape for analysis, hub genes in green module were significantly related to glycosaminoglycan metabolism and chondroitin sulfate/dermatan sulfate metabolism pathways (Figure 1B) which are mainly involved the composition of the extracellular matrix and likely to facilitate the activation of EMT process. Moreover, the enrichment of vitamin and lipid metabolic pathways in the green module support the rapid energy supply for the malignant tumor cells. The hub genes in brown module were enriched in the pathways associated with oxidative stress (Figure 1B), such as NRF2 which is activated under oxidative stress conditions and can reduce inflammatory damage by stimulating the expression of antioxidants and anti-inflammatory cytokines. Furthermore, 259 genes expressed in TCGA OSCC cohort were analyzed for its association with overall survival (OS) by univariate Cox regression, and the results showed that 21 genes were closely related to the prognosis of patients (P < 0.05) (Figure 1C).
Construction of a metabolism-related prognostic signature in OSCC
Using the LASSO regression algorithm, an immune-related metabolic signature including 12 genes for prognosis prediction was constructed (Figure 2A, 2B), and the coefficients of the 12 genes were shown in Table S3. The 330 OSCC patients were divided into high- (n = 165) and low-risk groups (n = 165) using the median risk score as a threshold, and the mortality rate of patients increased with the risk score (Figure 2C) as the OSCC patients in low-risk group showed better survival rates than those in high-risk group (Figure 2E). Gene expression patterns were shown in Figure 2D. Furthermore, the area under the curve (AUC) of the prognostic signature for 1-, 3-, and 5-year survival was 0.66, 0.74, and 0.71, respectively, which was further validated in GSE41613 cohort (Figure 2F, 2G), indicating the reliability of this model.
Figure 2.
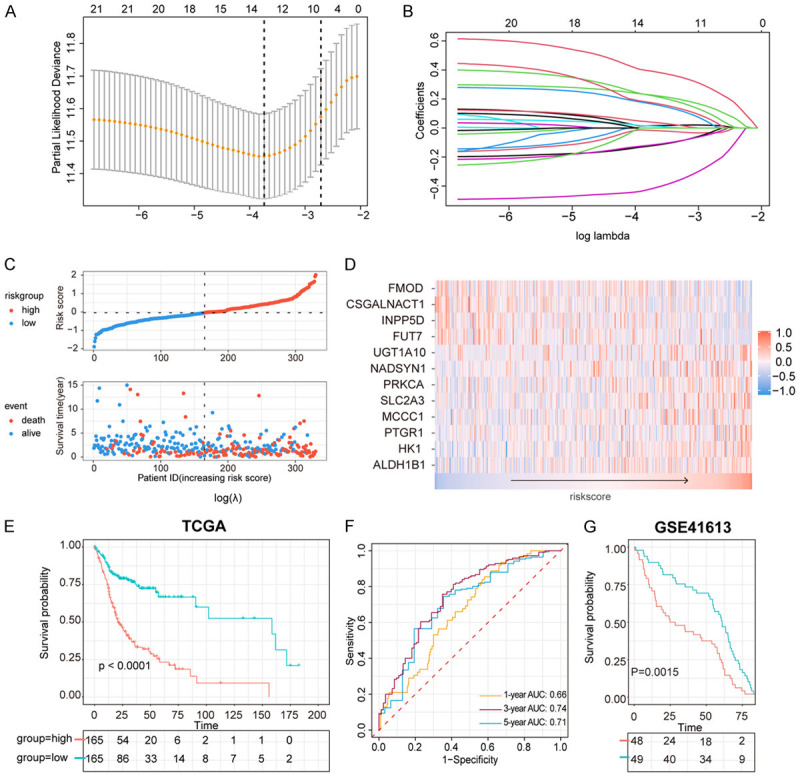
Construction of metabolic prognostic signature. A. Selection of the tuning parameter (lambda) in the LASSO model. B. LASSO coefficient profiles of 12 genes in metabolic signature. C. Distribution of risk score in patients, and scatterplot of OS events with risk score. D. Heatmap of 12 genes in metabolic signature with the increasing risk score. E. OS between high-risk group and low risk group in TCGA cohort. F. Survival-dependent ROC curve of metabolic signature at 1, 3, 5 years in TCGA cohort. G. Validation of metabolic signature in GEO41613.
Association of the risk score with clinical features and somatic mutations in OSCC
Figure 3A showed the distribution of the relevant patient characteristics and the risk scores. To investigate the relationship between the risk score and the clinical features, we grouped patients according to TNM staging system and found that the risk score of the metabolic signature increased with increased T and N stages, suggesting that the signature was reliable (Figure 3B). However, because most patients were classified as M0, the risk score did not show a significant difference in the M classification.
Figure 3.

The relationship between risk score and clinical features and comparison of somatic mutations and CNV in different risk group. A. Distribution of clinical features with the increasing risk score. B. Comparison of risk score in TNM stage and tumor stage. C. The overall TMB in different risk group. D. The somatic mutations (upper) and CNV (lower) in high-risk group and low-risk group.
Moreover, we analyzed the somatic mutations and CNV data downloaded from TCGA database. Patients in high-risk group had higher tumor mutation burden (TMB) (Figure 3C) and exhibited a higher frequency of TP53 mutations than the patients in the low-risk group (Figure 3D). Similarly, the high- and low-risk groups harbored different CNV alteration sites, and more CNVs in both amplification (AMP) and deletion (DEL) were observed in the high-risk group (Figure 3D). For example, AP_9:3q28, a large amplification of TP63, was observed in the high-risk group, but not in the low-risk group.
Metabolic signature was an independent factor of a survival predictive nomogram
We used univariate and multivariate Cox regression analyses to explore the relationship between the clinical characteristics and the metabolism-related risk scores. We found that patient’s age (hazard ratio [HR] = 1.02) and metabolism-related risk score (HR = 2.70) were two independent prognostic factors (P < 0.05) (Figure 4A, 4B). Furthermore, the age and the risk score were used to construct a nomogram to estimate the patient’s 1-, 3-, and 5-year survival (Figure 4C). The calibration curve for predicting patient survival indicated that the predicted OS was close to the actual OS (Figure 4D). Moreover, the ROC curve demonstrated that the three-index combined model was more accurate than each index alone (Figure 4E). The C-index value of the model combining age and risk score was 0.721.
Figure 4.

The metabolic signature was an independent factor for prognosis of the survival of OSCC patients. A. Univariate Cox regression analysis of clinical features and risk score with the OS of patients. B. Multivariate Cox regression analysis of risk score and age with the OS of patients. C. Nomogram predictions for survival predictions at 1, 3, 5 years. D. The calibration plot for the nomogram at 1, 3, 5 years. E. ROC curve for the predictions by age, risk score and the combine of them.
Identification of different metabolic pathways and functional enrichment analysis
To further investigate the changes in metabolism between the two risk groups, we calculated the scores of all samples in 90 KEGG metabolic pathways using ssGSEA and then performed a t-test to screen the pathways with p-values less than 0.01 (Figure 5A; Table S4). We found that eight of the 31 metabolic pathways with significant difference between risk groups were related to lipid metabolism and were enriched in the low-risk group. The pathways associated with glucose metabolism were enriched in the high-risk group, including the glycolysis/gluconeogenesis pathway. Notably, many redox-related pathways were enriched in the high-risk group, including ascorbate and aldarate metabolism, glutathione metabolism, and cytochrome P450-mediated xenobiotic metabolism, which are related to the resistance of adverse environments. In addition, the enrichment of amino acid synthesis and drug metabolism pathways in the high-risk group was in consistent with high malignancy.
Figure 5.

The ssGSEA score of metabolic pathways in different group and the DEGs analysis by GSEA, GO, KEGG pathway enrichment. A. Heatmap of 31 different metabolism pathways in high-risk and low-risk group (P < 0.01). B. GO enrichment of DEGs. C. KEGG enrichment of DEGs. D. GSEA enrichment of DEGs.
Subsequently, we quadrupled the patients by risk score and took the highest and lowest quartiles for differential gene expression analysis. The “limma3.50.3” package was used to identify the DEGs among these two groups (high risk vs low risk), and 371 genes were obtained, with 281 genes upregulated and 90 genes downregulated in the high-risk group. GO pathway enrichment analysis showed that the upregulated genes were enriched in chromosome segregation and nuclear division, while genes with decreased expression were enriched in steroid metabolic process, positive regulation of cellular catabolic process, actin filament organization, regulation of stress-activated mitogen-activated protein kinase (MAPK) cascade, secondary alcohol metabolism process, and response to hypoxia (Figure 5B). KEGG enrichment analysis showed that DNA replication, cell cycle, and mismatch repair pathways were enriched in the high-risk group, while the AMPK and Hippo pathways were enriched in the low-risk group (Figure 5C). Taken together, we found that more genetic mutations, cell cycle and metabolic disorders existed in the high-risk group, whereases more stable cytoskeleton and stronger cell adhesion were in the low-risk group. In addition, we analyzed the DEGs by using GSEA software with three gene sets: C2 (curated gene sets), C6 (oncogenic signature gene sets), and C7 (immunologic signature gene sets) (Figure 5D). The results showed that pathways relevant to immune response, such as chemokine receptors, T lymphocytes, and CD40 signaling pathways, were enriched in the low-risk group, while pathways related to tumor malignancy and proliferation were enriched in the high-risk group. In the C6 gene sets, some tumor suppressor gene-related signaling pathways such as P53 and PTEN, and some oncogenic pathways such as NOTCH1 and KRAS were normal in the low-risk group but were mutated in the high-risk group. Higher MYC expression was also noted in the high-risk group, indicating that tumors in the high-risk group were more malignant. In addition, the C7 gene sets were associated with immune pathways, where the high-risk group was enriched with more immunosuppressive pathways, including PD1 ligation, Treg, and IL6-induced M2 polarization of macrophages, while the low-risk group was enriched with CD8+ T cells and interferon (IFN) pathways.
Distinct immune landscape and ICB efficiency between different risk groups
To further investigate the immune characteristics of the different risk groups, 22 types of immune cells were analyzed using CIBERSORT. We ranked the samples with increasing number of CD8+ T cells in the immune cell heatmap and demonstrated that the area of CD8+ T cells (blue) was larger in low-risk patients (Figure 6A). To better show the differences in cell type between the high- and low-risk groups, we used box plots to present the results. As shown in Figure 6B, the high-risk group was enriched with M2 macrophages which are recognized as immunosuppressive cells. M0 Macrophages were also enriched in the high-risk group although the difference from the low-risk group was not significant. Interestingly, Figure 6B also indicated that CD8+ T cells, dendritic cells, B cells, T follicular helper cells (Tfh), and Tγδ were enriched in the low-risk group. Some studies have shown that B cells and CD4+ T cells contribute to the formation of tertiary lymphoid structures (TLSs) in EMT. Crosstalk between immune cells in TLSs could promote a series of immune processes including antigen presentation, maintenance of T cell states, and the formation of memory T cells; the interplay network was shown in Figure 6C. Given that DEGs were enriched in the CD40 signaling pathway in GSEA enrichment analysis, we investigated CD40 and CD40LG expression in high- and low-risk groups and found that CD40LG expression was higher in the low-risk group, which might promote the activation of T cells and the killing of CD40+ tumor cells (Figure 6D). Furthermore, we collected gene sets that represented immune molecules and processes (Table S5) and analyzed them using ssGSEA. The data suggested that the low-risk group provided a microenvironment more suitable for T cell co-stimulation; thus, cytolytic activity and type I and II IFN responses were stronger in the low-risk group than in the high-risk group, which further enhanced the antitumor effect and increased the MHC presentation on tumor cells (Figure 6E). Moreover, the low-risk group had higher checkpoint scores due to many factors, such as increased T-cell infiltration and better T-cell activation, suggesting that patients in this group might benefit from ICB treatment. The expression levels of the seven checkpoint genes were presented in Figure S4A. To further validate our ssGSEA results, we performed ssGSEA again using the dataset from Thomas Powles et al. [36] and conducted a correlation analysis with each signature (Figure 6F). The results showed that the risk score was associated with EMT, antigen presentation, CD8+ T effector cells, and DNA repair as well as angiogenesis. More importantly, the metabolic risk score could be used as an indicator to predict the efficacy of immunotherapy. We compared the therapeutic response to anti-PD-1 between the two risk groups via the ICB response score and revealed that the low-risk group had a better response to ICB therapy (Figure 6G). Consistently, data from KMplot dataset which contains information on patients with anti-PD1 mAb treatment showed that the low-risk group had higher survival rate than the high-risk group (Figure 6H). We also used pRRophetic package to evaluate the IC50 of three first-line drugs: 5-Fluorouracil (Figure 6I), Cetuximab (Figure S4B), and Cisplatin (Figure S4C), and the results suggested that 5-Fluorouracil might not have therapeutic effect in patients at the high-risk group.
Figure 6.

The immune landscape in different risk groups and the relationship between rick score and drug response. A. The heatmap of 22 types of immune cells estimated by CIBERDORT. B. The boxplots of 22 types of immune cells estimated by CIBERDORT. C. The crosstalk between 22 types of immune cells. D. CD40 and CD40LG expression of different risk groups. E. The ssGASE score of immune processes in different risk groups. F. The correlation of risk score and 19 signatures provided by Thomas Powles et al. G. The ICB-Response score in high- and low-risk groups. H. Kaplan-Meier survival of patients grouped by metabolic signature in KM plot’s anti-PD1 therapy array. I. The evaluated IC50 of 5-Fluorouracil. *P < 0.05, **P < 0.01, ***P < 0.001.
Single-cell sequencing analysis reveals the expression of 12 genes on different cell clusters
The published scRNA sequencing dataset (GSE136103) from the GEO database contains 5902 cells from 18 patients. After filtering, we used 5746 cells for subsequent analysis (Figure S5A-C). Eighteen clusters were generated using the tSNE algorithm (Figure S5D). Based on the expression signatures of these cells (Table S6), we classified them into eight clusters: B cells, CD4+ T cells, CD8+ NK cells, dendritic cells, endothelial cells, tumor cells, fibroblasts, macrophages, and mast cells (Figures S5E, 7A). Our results were consistent with the results from dataset provided by Bradley et al. [29]. We also used EPCAM to identify malignant cell clusters (Figure 7B). Furthermore, we performed single cell CNV analysis by using the “CopyKAT” package, which discriminated malignant cells from normal cells. The results showed that out of the 5746 cells, 2318 cells were malignant cells, while the other 3428 cells were normal cells (Figure S6). In addition, the malignant cells belonged to the same group as the tumor cell population we previously identified. Furthermore, we divided the tumor cells into two clusters (1654 cells in cluster 1 and 664 cells in cluster 2) based on CNV (Figures S7, 7C) and found that EPCAM was expressed at significantly higher levels in cluster 2 compared to cluster 1. It is well known that EPCAM is a biomarker of cancer stem cells and is involved in EMT. In addition, cells in cluster 2 overexpressed SOX2, another marker of cancer stem cells (Figure 7D). Combined the CNV mutation data with the expression of EPCAM and SOX2, we speculated that tumors in cluster 2 were more malignant than that in cluster 1.
Figure 7.
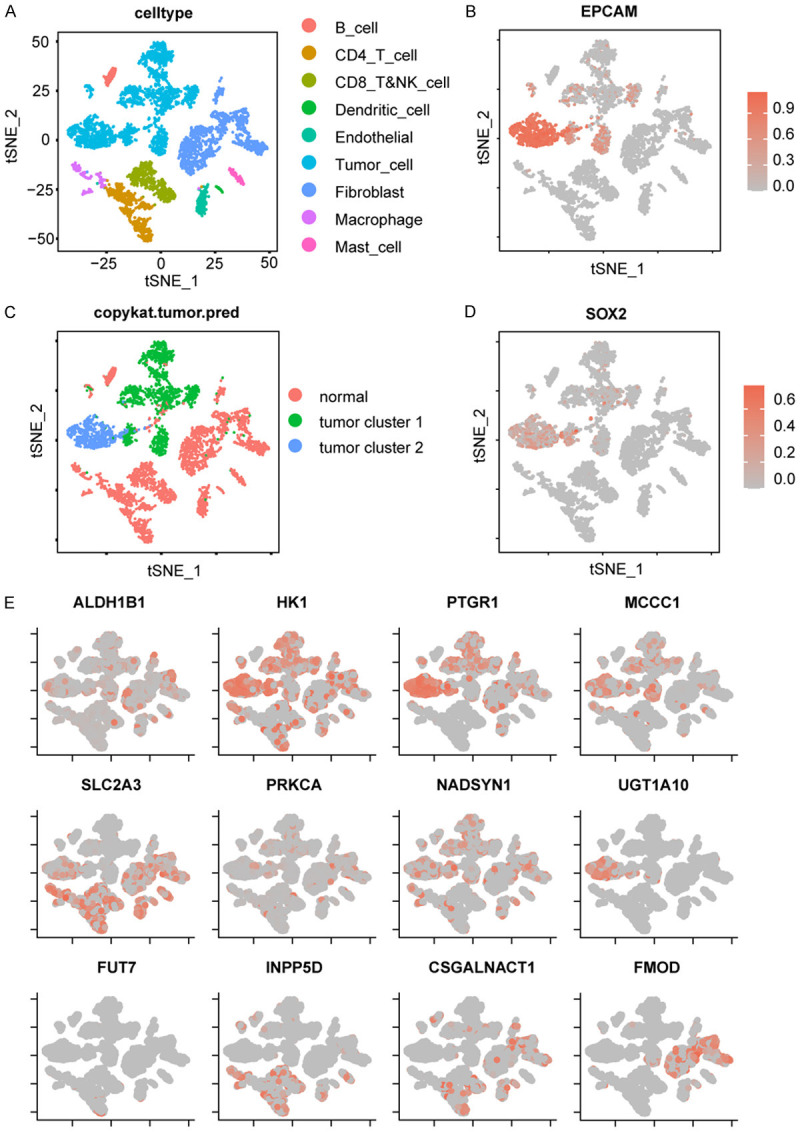
Single cell sequencing revealed the specific expression of the 12 genes in 8 cell types. A. Cell types were identified by marker genes. B. The expression of EPCAM in each cell type. C. Tumor cells were divided into 2 clusters by CopyKAT. D. The expression of SOX2 in each cell type. E. Expression profile of the 12 genes in each cell type.
Among the 12 genes we used to construct metabolism-related signature, HK1, PTGR1, MCCC1, UGT1A10, and NADSYN1 were highly expressed in tumor cells, while SLC2A3, INPP5D, and CSGALNACT1 were highly expressed in immune cells and fibroblasts. In addition, SLC2A3 was also expressed in cluster 2 tumor cells, while FOMD was expressed only in fibroblasts (Figure 7E). SLC2A3, also known as GLUT3, has the highest affinity for glucose among the four glucose transporters and tends to be highly expressed in malignant tumors to meet their metabolic requirements. Furthermore, PTGR1 and UGT1A10 expression was upregulated in tumors of cluster 2 compared to tumors in cluster 1. In contrast, INPP5D, CSGALNACT1, and FOMD were only expressed in non-tumor cells and had negative coefficients with the risk score. Together, our analysis on the gene expression profile in each cell type provides a useful reference for the development of therapeutic drugs.
Discussion
Various studies have focused on the EMT process in OSCC to investigate the mechanisms by which OSCC metastasizes. EMT activation results in the acquisition of stem cell properties, aggressive mobility, and immune resistance. EMT is usually associated with metabolic reprogramming, which is widely implemented by malignant cells to meet their needs for rapid proliferation and mobility in harsh tumor microenvironments. In this study, we used WGCNA, a systematic biology method, to identify the metabolic genes related to EMT, stemness, and checkpoints. We identified a green module which had the most positive correlation with EMT and checkpoints, but not with stemness, which might be due to the existence of the multiple states of EMT, each exhibiting distinct cancer stem cells marker [43]. We also selected the brown module related to the checkpoint signature for subsequent analysis. The biological functions of the green module were related to extracellular matrix synthesis. It has been reported that glycosaminoglycan can suppress the activation of T cells and dendritic cells [44], and that abundant ECM can impede immune cell infiltration, leading to the immune escape of tumor cells in OSCC. In addition, the oxidative stress-related pathways were also enriched in the module, suggesting the important role in the induction of EMT. One important molecule in the oxidative stress-related pathways was NRF2 which can promote the transcription of several genes involved in the formation of NADPH to help scavenge ROS [45].
We also constructed a metabolic signature consisting of 12 genes for predicting OSCC prognosis by comprehensive bioinformatic analysis of the green and brown modules. We verified that the risk score and age could be used as independent prognostic factors. OSCC patients with high-risk scores had poor outcomes. Furthermore, the risk score also correlated with other common malignant features. For example, high-risk score was associated with a more severe TNM stage and a higher TMB. A high mutation rate of TP53 was found in OSCC patients, especially in the high-risk group. The CNV patterns differed between the two risk groups, with the high-risk group showing more genomic amplifications of several genes, including TP63. Nevertheless, whether TP63 acts as a tumor suppressive or tumor promoting factor is not clear [46-48], and our results showed that TP63 amplification was associated with more malignant OSCC.
The high- and low-risk groups presented different patterns of metabolic pathways. Glycolysis, amino acid synthesis, and glutamine metabolism pathways were upregulated in the high-risk group. Similarly, drug metabolism and CYP450 pathway were upregulated, suggesting that the high-risk group were more resistant to chemotherapy. Interestingly, the lipid metabolism and arachidonic acid metabolism pathways were enriched in the low-risk group, as these pathways are generally considered to be positively correlated with malignant tumors. Furthermore, GO pathway analysis revealed that the MAPK pathway was enriched in the low-risk group. Although activating mutations in MAPK are common in tumors and are generally considered to be oncogenic [49,50], recent studies suggest that the role of MAPK pathway mutations may be different in HNSCC, which was supported by our results. Other evidence also supports this notion. For example, multiple MAPK pathway activating mutations found in HNSCC could significantly suppress p-ErbB3 expression and promote CD8+ T cell infiltration, which are associated with better survival [51]. Furthermore, previous studies have shown that arachidonic acid and other fatty acids can activate members of the MAPK superfamily [52,53], which explains why lipid and arachidonic acid metabolism were active in the low-risk OSCC group. We also found the differential expression of some classical tumor-associated pathways, including TP53, PTEN, MYC, KRAS, and E2F1, between the high- and low-risk groups.
In this study, we showed that the risk score was associated with the immune landscape of OSCC patients. First, the population of CD8+ T cells was expanded in the low-risk group, and other factors, including HLA, cytolytic activity, and co-stimulation, also indicated that the low-risk group had a more active immune environment. In the low-risk group, T cells exhibited higher CD40L expression, but tumor cells exhibited lower CD40 expression, suggesting the possibility of applying CD40LG signal in the design of chimeric antigen receptor T cell (CAR-T) for an improved OSCC therapy. Another significant finding from our analysis was that the risk score was reliable in predicting the response to immunotherapy, with a lower risk score indicating a higher ICB-response score and a better survival outcome. The predicted IC50 results showed that the high-risk score predicted resistance to 5-fluorouracil but not to cetuximab and cisplatin, suggesting cetuximab and cisplatin as more efficient therapeutic drug in treating malignant OSCC.
Finally, scRNA-seq data were used to further explain the signature we constructed. The tumors in cluster 2 expressed high levels of EPCAM and SOX2 which are markers of malignant tumor cells. Among the 12 signature genes, PTGR1, UTG1A10 and SLC2A3, were differentially expressed between the two tumor clusters, indicating that these genes could be potential therapeutic targets in the treatment of refractory OSCC. Indeed, inhibitors of PTGR1 and UTG1A10, including indomethacin (PTGR1 inhibitor) and sorafenib (UTG1A10 inhibitor), have been used clinical in other cancer types [54,55]. As for SLC2A3, the molecular basis for its inhibition by exofacial inhibitors has been reported [56], which could serve as potential therapeutic target. On the other hand, Hexokinase 1 (HK1), the first rate-limiting enzyme in glycolysis and a closely related enzyme to cancer progression, is expressed in both tumor and non-tumor cells. It has been reported that HK1 is upregulated when T cells are activated [57], which would probably interfere with our predictions using the risk score. Therefore, the risk score would be more suitable for the assessment of patients prior to treatment. Notably, another two genes in the 12 gene signature, CSGALNACT1 and FMOD were associated with the metabolism of the extracellular matrix, suggesting that the immune state of the EMT can be improved by regulating the extracellular matrix to achieve good therapeutic outcomes in OSCC.
In conclusion, we constructed a metabolic gene co-expression network for OSCC and identified metabolic genes most relevant to EMT, stemness, and checkpoint signatures. A prognostic metabolic signature was constructed and validated. The risk score derived from the signature was closely related to the clinical features and could be used to predict the response to immune therapy. Importantly, some potential targets identified from the analyzing metabolic characteristics in different risk groups may help develop clinical treatment plans.
Nevertheless, there were some limitations in this work. First, datasets of large immunotherapy, especially in OSCC, should be used to verify our signature. Second, in vitro and in vivo experiments should be carried out to validate these potential targets and pathways. Finally, our research only studied the relationship between metabolism and OSCC metastasis at the transcriptional level, and we look forward to a multi-omics study to reveal more details about the role of metabolism in OSCC metastasis.
Acknowledgements
We thank all researchers sharing the data used in this work. Supported by the projects named as Health Commission of Hubei Province scientific research project [grant number WJ2021M166], and Supporting project of medical science and technology innovation platform of Zhongnan Hospital of Wuhan University [grant number PTXM2021014].
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Hashibe M, Brennan P, Benhamou S, Castellsague X, Chen C, Curado MP, Dal Maso L, Daudt AW, Fabianova E, Fernandez L, Wünsch-Filho V, Franceschi S, Hayes RB, Herrero R, Koifman S, La Vecchia C, Lazarus P, Levi F, Mates D, Matos E, Menezes A, Muscat J, Eluf-Neto J, Olshan AF, Rudnai P, Schwartz SM, Smith E, Sturgis EM, Szeszenia-Dabrowska N, Talamini R, Wei Q, Winn DM, Zaridze D, Zatonski W, Zhang ZF, Berthiller J, Boffetta P. Alcohol drinking in never users of tobacco, cigarette smoking in never drinkers, and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. J Natl Cancer Inst. 2007;99:777–789. doi: 10.1093/jnci/djk179. [DOI] [PubMed] [Google Scholar]
- 3.Blot WJ, McLaughlin JK, Winn DM, Austin DF, Greenberg RS, Preston-Martin S, Bernstein L, Schoenberg JB, Stemhagen A, Fraumeni JF Jr. Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res. 1988;48:3282–3287. [PubMed] [Google Scholar]
- 4.Zittel S, Moratin J, Horn D, Metzger K, Ristow O, Engel M, Mrosek J, Freier K, Hoffmann J, Freudlsperger C. Clinical outcome and prognostic factors in recurrent oral squamous cell carcinoma after primary surgical treatment: a retrospective study. Clin Oral Investig. 2022;26:2055–2064. doi: 10.1007/s00784-021-04186-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geum DH, Roh YC, Yoon SY, Kim HG, Lee JH, Song JM, Lee JY, Hwang DS, Kim YD, Shin SH, Chung IK, Kim UK. The impact factors on 5-year survival rate in patients operated with oral cancer. J Korean Assoc Oral Maxillofac Surg. 2013;39:207–216. doi: 10.5125/jkaoms.2013.39.5.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Cruz AK, Vaish R, Dhar H. Oral cancers: current status. Oral Oncol. 2018;87:64–69. doi: 10.1016/j.oraloncology.2018.10.013. [DOI] [PubMed] [Google Scholar]
- 7.Ozdek A, Sarac S, Akyol MU, Unal OF, Sungur A. Histopathological predictors of occult lymph node metastases in supraglottic squamous cell carcinomas. Eur Arch Otorhinolaryngol. 2000;257:389–392. doi: 10.1007/s004050000231. [DOI] [PubMed] [Google Scholar]
- 8.Roberts TJ, Colevas AD, Hara W, Holsinger FC, Oakley-Girvan I, Divi V. Number of positive nodes is superior to the lymph node ratio and American Joint Committee on Cancer N staging for the prognosis of surgically treated head and neck squamous cell carcinomas. Cancer. 2016;122:1388–1397. doi: 10.1002/cncr.29932. [DOI] [PubMed] [Google Scholar]
- 9.Zeng YT, Liu XF, Yang WT, Zheng PS. REX1 promotes EMT-induced cell metastasis by activating the JAK2/STAT3-signaling pathway by targeting SOCS1 in cervical cancer. Oncogene. 2019;38:6940–6957. doi: 10.1038/s41388-019-0906-3. [DOI] [PubMed] [Google Scholar]
- 10.Gumireddy K, Li A, Gimotty PA, Klein-Szanto AJ, Showe LC, Katsaros D, Coukos G, Zhang L, Huang Q. KLF17 is a negative regulator of epithelial-mesenchymal transition and metastasis in breast cancer. Nat Cell Biol. 2009;11:1297–1304. doi: 10.1038/ncb1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 12.Saxena M, Stephens MA, Pathak H, Rangarajan A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011;2:e179. doi: 10.1038/cddis.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20:69–84. doi: 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- 14.Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol. 2012;22:396–403. doi: 10.1016/j.semcancer.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL, Weinberg RA. Epithelial-to-mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res. 2017;77:3982–3989. doi: 10.1158/0008-5472.CAN-16-3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chockley PJ, Chen J, Chen G, Beer DG, Standiford TJ, Keshamouni VG. Epithelial-mesenchymal transition leads to NK cell-mediated metastasis-specific immunosurveillance in lung cancer. J Clin Invest. 2018;128:1384–1396. doi: 10.1172/JCI97611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nilsson MB, Sun H, Robichaux J, Pfeifer M, McDermott U, Travers J, Diao L, Xi Y, Tong P, Shen L, Hofstad M, Kawakami M, Le X, Liu X, Fan Y, Poteete A, Hu L, Negrao MV, Tran H, Dmitrovsky E, Peng D, Gibbons DL, Wang J, Heymach JV. A YAP/FOXM1 axis mediates EMT-associated EGFR inhibitor resistance and increased expression of spindle assembly checkpoint components. Sci Transl Med. 2020;12:eaaz4589. doi: 10.1126/scitranslmed.aaz4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paz-Ares L, Ciuleanu TE, Cobo M, Schenker M, Zurawski B, Menezes J, Richardet E, Bennouna J, Felip E, Juan-Vidal O, Alexandru A, Sakai H, Lingua A, Salman P, Souquet PJ, De Marchi P, Martin C, Pérol M, Scherpereel A, Lu S, John T, Carbone DP, Meadows-Shropshire S, Agrawal S, Oukessou A, Yan J, Reck M. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22:198–211. doi: 10.1016/S1470-2045(20)30641-0. [DOI] [PubMed] [Google Scholar]
- 20.Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, Harrington KJ, Kasper S, Vokes EE, Even C, Worden F, Saba NF, Docampo LCI, Haddad R, Rordorf T, Kiyota N, Tahara M, Lynch M, Jayaprakash V, Li L, Gillison ML. Nivolumab vs investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck: 2-year long-term survival update of CheckMate 141 with analyses by tumor PD-L1 expression. Oral Oncol. 2018;81:45–51. doi: 10.1016/j.oraloncology.2018.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611–629. doi: 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sciacovelli M, Frezza C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017;284:3132–3144. doi: 10.1111/febs.14090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19:1438–1449. doi: 10.1038/nm.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corbet C, Bastien E, Santiago de Jesus JP, Dierge E, Martherus R, Vander Linden C, Doix B, Degavre C, Guilbaud C, Petit L, Michiels C, Dessy C, Larondelle Y, Feron O. TGFβ2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat Commun. 2020;11:454. doi: 10.1038/s41467-019-14262-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson M, Marayati R, Moffitt R, Yeh JJ. Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget. 2017;8:56081–56094. doi: 10.18632/oncotarget.9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park JH, Vithayathil S, Kumar S, Sung PL, Dobrolecki LE, Putluri V, Bhat VB, Bhowmik SK, Gupta V, Arora K, Wu D, Tsouko E, Zhang Y, Maity S, Donti TR, Graham BH, Frigo DE, Coarfa C, Yotnda P, Putluri N, Sreekumar A, Lewis MT, Creighton CJ, Wong LC, Kaipparettu BA. Fatty acid oxidation-driven Src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 2016;14:2154–2165. doi: 10.1016/j.celrep.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guerra F, Guaragnella N, Arbini AA, Bucci C, Giannattasio S, Moro L. Mitochondrial dysfunction: a novel potential driver of epithelial-to-mesenchymal transition in cancer. Front Oncol. 2017;7:295. doi: 10.3389/fonc.2017.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Jiang C, Li N, Wang F, Xu Y, Shen Z, Yang L, Li Z, He C. The circEPSTI1/miR-942-5p/LTBP2 axis regulates the progression of OSCC in the background of OSF via EMT and the PI3K/Akt/mTOR pathway. Cell Death Dis. 2020;11:682. doi: 10.1038/s41419-020-02851-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS, Deschler DG, Varvares MA, Mylvaganam R, Rozenblatt-Rosen O, Rocco JW, Faquin WC, Lin DT, Regev A, Bernstein BE. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171:1611–1624. e24. doi: 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu G, He QY. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol Biosyst. 2016;12:477–479. doi: 10.1039/c5mb00663e. [DOI] [PubMed] [Google Scholar]
- 31.Alonso SR, Tracey L, Ortiz P, Pérez-Gómez B, Palacios J, Pollán M, Linares J, Serrano S, Sáez-Castillo AI, Sánchez L, Pajares R, Sánchez-Aguilera A, Artiga MJ, Piris MA, Rodríguez-Peralto JL. A high-throughput study in melanoma identifies epithelial-mesenchymal transition as a major determinant of metastasis. Cancer Res. 2007;67:3450–3460. doi: 10.1158/0008-5472.CAN-06-3481. [DOI] [PubMed] [Google Scholar]
- 32.Miranda A, Hamilton PT, Zhang AW, Pattnaik S, Becht E, Mezheyeuski A, Bruun J, Micke P, de Reynies A, Nelson BH. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc Natl Acad Sci U S A. 2019;116:9020–9029. doi: 10.1073/pnas.1818210116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao B, Liu L, Li A, Xiang C, Wang P, Li H, Xiao T. Identification and verification of immune-related gene prognostic signature based on ssgsea for osteosarcoma. Front Oncol. 2020;10:607622. doi: 10.3389/fonc.2020.607622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xing L, Guo M, Zhang X, Zhang X, Liu F. A transcriptional metabolic gene-set based prognostic signature is associated with clinical and mutational features in head and neck squamous cell carcinoma. J Cancer Res Clin Oncol. 2020;146:621–630. doi: 10.1007/s00432-020-03155-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, Jhunjhunwala S, Banchereau R, Yang Y, Guan Y, Chalouni C, Ziai J, Şenbabaoğlu Y, Santoro S, Sheinson D, Hung J, Giltnane JM, Pierce AA, Mesh K, Lianoglou S, Riegler J, Carano RAD, Eriksson P, Höglund M, Somarriba L, Halligan DL, van der Heijden MS, Loriot Y, Rosenberg JE, Fong L, Mellman I, Chen DS, Green M, Derleth C, Fine GD, Hegde PS, Bourgon R, Powles T. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martín-Algarra S, Mandal R, Sharfman WH, Bhatia S, Hwu WJ, Gajewski TF, Slingluff CL Jr, Chowell D, Kendall SM, Chang H, Shah R, Kuo F, Morris LGT, Sidhom JW, Schneck JP, Horak CE, Weinhold N, Chan TA. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171:934–949. e16. doi: 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao R, Bai S, Henderson YC, Lin Y, Schalck A, Yan Y, Kumar T, Hu M, Sei E, Davis A, Wang F, Shaitelman SF, Wang JR, Chen K, Moulder S, Lai SY, Navin NE. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat Biotechnol. 2021;39:599–608. doi: 10.1038/s41587-020-00795-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mittal V. Epithelial mesenchymal transition in tumor metastasis. Annu Rev Pathol. 2018;13:395–412. doi: 10.1146/annurev-pathol-020117-043854. [DOI] [PubMed] [Google Scholar]
- 41.Jiang Y, Zhan H. Communication between EMT and PD-L1 signaling: new insights into tumor immune evasion. Cancer Lett. 2020;468:72–81. doi: 10.1016/j.canlet.2019.10.013. [DOI] [PubMed] [Google Scholar]
- 42.Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, Groom AC. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998;153:865–873. doi: 10.1016/S0002-9440(10)65628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang CY, Yaswen P, Goga A, Werb Z. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015;526:131–135. doi: 10.1038/nature15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.du Souich P, García AG, Vergés J, Montell E. Immunomodulatory and anti-inflammatory effects of chondroitin sulphate. J Cell Mol Med. 2009;13:1451–1463. doi: 10.1111/j.1582-4934.2009.00826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu S, Lu H, Bai Y. Nrf2 in cancers: a double-edged sword. Cancer Med. 2019;8:2252–2267. doi: 10.1002/cam4.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Melino G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ. 2011;18:1487–1499. doi: 10.1038/cdd.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu D, Jiang XH, Jiang YH, Ding WC, Zhang CL, Shen H, Wang XL, Ma D, Hu Z, Wang H. Amplification and overexpression of TP63 and MYC as biomarkers for transition of cervical intraepithelial neoplasia to cervical cancer. Int J Gynecol Cancer. 2014;24:643–648. doi: 10.1097/IGC.0000000000000122. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe H, Ma Q, Peng S, Adelmant G, Swain D, Song W, Fox C, Francis JM, Pedamallu CS, DeLuca DS, Brooks AN, Wang S, Que J, Rustgi AK, Wong KK, Ligon KL, Liu XS, Marto JA, Meyerson M, Bass AJ. SOX2 and p63 colocalize at genetic loci in squamous cell carcinomas. J Clin Invest. 2014;124:1636–1645. doi: 10.1172/JCI71545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hiraide T, Ikegami K, Sakaguchi T, Morita Y, Hayasaka T, Masaki N, Waki M, Sugiyama E, Shinriki S, Takeda M, Shibasaki Y, Miyazaki S, Kikuchi H, Okuyama H, Inoue M, Setou M, Konno H. Accumulation of arachidonic acid-containing phosphatidylinositol at the outer edge of colorectal cancer. Sci Rep. 2016;6:29935. doi: 10.1038/srep29935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong SH, Avis I, Vos MD, Martínez A, Treston AM, Mulshine JL. Relationship of arachidonic acid metabolizing enzyme expression in epithelial cancer cell lines to the growth effect of selective biochemical inhibitors. Cancer Res. 1999;59:2223–2228. [PubMed] [Google Scholar]
- 51.Ngan HL, Liu Y, Fong AY, Poon PHY, Yeung CK, Chan SSM, Lau A, Piao W, Li H, Tse JSW, Lo KW, Chan SM, Su YX, Chan JYK, Lau CW, Mills GB, Grandis JR, Lui VWY. MAPK pathway mutations in head and neck cancer affect immune microenvironments and ErbB3 signaling. Life Sci Alliance. 2020;3:e201900545. doi: 10.26508/lsa.201900545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cussac D, Schaak S, Denis C, Paris H. alpha 2B-adrenergic receptor activates MAPK via a pathway involving arachidonic acid metabolism, matrix metalloproteinases, and epidermal growth factor receptor transactivation. J Biol Chem. 2002;277:19882–19888. doi: 10.1074/jbc.M110142200. [DOI] [PubMed] [Google Scholar]
- 53.Gijón MA, Leslie CC. Regulation of arachidonic acid release and cytosolic phospholipase A2 activation. J Leukoc Biol. 1999;65:330–336. doi: 10.1002/jlb.65.3.330. [DOI] [PubMed] [Google Scholar]
- 54.Wang X, Yin G, Zhang W, Song K, Zhang L, Guo Z. Prostaglandin reductase 1 as a potential therapeutic target for cancer therapy. Front Pharmacol. 2021;12:717730. doi: 10.3389/fphar.2021.717730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu YJ, Zheng B, Wang HY, Chen L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. 2017;38:614–622. doi: 10.1038/aps.2017.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang N, Zhang S, Yuan Y, Xu H, Defossa E, Matter H, Besenius M, Derdau V, Dreyer M, Halland N, He KH, Petry S, Podeschwa M, Tennagels N, Jiang X, Yan N. Molecular basis for inhibiting human glucose transporters by exofacial inhibitors. Nat Commun. 2022;13:2632. doi: 10.1038/s41467-022-30326-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Araujo L, Khim P, Mkhikian H, Mortales CL, Demetriou M. Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. Elife. 2017;6:e21330. doi: 10.7554/eLife.21330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.