

Abstract
Machine and deep learning approaches can leverage the increasingly available massive datasets of protein sequences, structures, and mutational effects to predict variants with improved fitness. Many different approaches are being developed, but systematic benchmarking studies indicate that even though the specifics of the machine learning algorithms matter, the more important constraint comes from the data availability and quality utilized during training. In cases where little experimental data is available, unsupervised and self-supervised pre-training with generic protein datasets can still perform well after subsequent refinement via hybrid or transfer learning approaches. Overall, recent progress in this field has been staggering, and machine learning approaches will likely play a major role in future breakthroughs in protein biochemistry and engineering.
Keywords: mutational effect, deep learning, protein engineering, transformer, convolutional neural network, hybrid learning
1. Introduction
A long-standing question in protein biochemistry, protein engineering, and evolutionary biology is the question of where and how proteins can be mutated. Early works on this topic focused on patterns of evolutionary sequence divergence, a rich research topic whose origins trace back nearly six decades to the pioneering work by Zuckerkandl and Pauling [1]. This work was subsequently augmented with mathematical and computational approaches from condensed-matter physics, soft-condensed matter physics, and theoretical chemistry to develop models and to make quantitative predictions of the effects of mutations in proteins (see e.g. [2, 3, 4]). At the same time, experimental techniques to probe the effects of mutations have consistently improved, and today high-throughput experimental methods such as deep mutational scanning are routinely being used to systematically explore the effects of mutations empirically [5, 6].
In just the last few years, machine learning has started to complement and at times out-perform existing empirical and modeling approaches. This development has been enabled by a confluence of two separate factors, on the one hand the availability of large scale sequence and structure datasets, and on the other the insight that major advancements in computer vision and natural language processing algorithms do directly apply to problems of protein biochemistry. These powerful algorithms require an enormous amount of data, which we now have available. In fact, we have seen many of the newest algorithms developed for computer vision or natural language processing successfully applied to problems of protein biochemistry, including recurrent neural networks, Long-Short Term Memory, 1D dilated convolutions, 2D and 3D convolutions, and attention-based architectures such as transformers and graph-attention networks (Table 1).
Table 1:
Machine learning approaches commonly used to model mutational effects, and key references that have used these approaches to study mutations in proteins.
| Method | Description | Key References |
|---|---|---|
| Convolutional neural network (CNN) | A neural network architecture that processes protein structure data with convolutional filters, similar to common image processing networks. | [15, 16, 17, 18] |
| Deep generative network | A neural network architecture that learns a low-dimensional latent space that can be sampled to predict high-resolution data (such as complete protein sequences) from the latent representation. | [19, 20] |
| Gaussian Process | A Bayesian learning technique that provides a probability distribution over possible functions that fit a dataset. Because this technique includes an explicit representation of model uncertainty, it enables efficient search through protein sequence space. | [21, 22] |
| Graph neural network | A neural network architecture that represents either atoms or residues as nodes and the relationship between the atoms or residues as edges. These models learns how to update their node embeddings by aggregating information for each node based on it’s neighbors for the particular learning task. | [23, 24, 25, 26, 27] |
| Long Short-Term Memory (BiLSTM) network | A type of recurrent neural network originally developed in the context of language modeling. These models can learn positional dependencies in sequence data. | [28] |
| Support Vector Machine (SVM) | Traditional supervised machine learning method that learns boundaries in feature space separating distinct categories. One of the oldest machine learning methods applied to predicting mutational effects. | [29, 12] |
| Transformer | A very powerful neural network architecture that learns a feature embedding space and combines it with an attention mechanism to make predictions from sequence data. | [14, 30, 31, 32] |
There are four distinct problems that have seen substantial progress through the application of machine-learning tools: First, the prediction of protein structure from sequence (the folding problem, see e.g. [7]); second, the prediction of sequences that fold into a specific structure (the inverse folding problem or protein design problem, see e.g. [8]); third, the prediction of protein-ligand or protein-protein interactions (the docking problem [9, 10, 11]); fourth, the prediction of point mutations or of mutational effects [12, 13, 14]. In this review article, we will focus on the fourth problem, and we will touch occasionally on the first or second problem to the extent that they are relevant. We first introduce the fundamental machine learning approaches used, such as supervised, unsupervised, and hybrid learning. We then consider the specific problem of predicting sites primed for mutation, and we also discuss the pros and cons of using sequence or structural data. We close with a perspective of how this field may develop going forward.
2. Supervised learning
For supervised learning, we need a large dataset of mutations with known effect, and then we can train a model to predict effects on novel mutations. Because of the requirement for a large training dataset, the types of applications in which supervised learning is possible is limited. The most established one is probably prediction of stability effects (ΔΔG values).
There is a long tradition of developing methods for predicting ΔΔG values in proteins. The earliest approaches have been physics based, typically using all-atom models combined with force fields [33, 34, 35]. Simpler physics-based approaches have used statistical potentials combined with the solvent accessibility of the mutated residue [36]. In general, physics-based models require an input structure to make useful predictions.
More recently, several groups have used various machine-learning methods to substitute or augment the physics based methods. These machine-learning methods can be based entirely on sequence (e.g., [37, 38]) or can use structure as an input in some form [39, 17, 23] or combine features from both sequence and structure. Over the years, the community has explored a variety of machine learning methods and their capacity for modeling protein stability including but not limited to support vector machines [29, 12], random forests [40, 41, 12], Gaussian processes [21, 22], and dense neural networks [12], as well as ensemble models combining the different methods [39, 12]. Notably, Romero et al. [21] utilized Gaussian processes to engineer thermally stabilized P450 enzymes, producing a chimera more stable (> 8°C) than any variant obtained via directed evolution. As an example of deep learning methods adapted to this problem domain, Li et al. [17] used 3D Convolutional Neural Networks (3D CNNs) to predict ΔΔG values from a voxelized representation of the protein structure surrounding the residue of interest. This approach, originally pioneered by Torng and Altman [15], treats the protein structure like a 3D image and uses well established techniques from deep learning image processing to problems of protein biochemistry (Figure 1A). Li et al. [17] found that their approach worked well predicting stability effects. In particular, and importantly, their 3D CNN had little bias for predicting destabilizing mutations, a problem afflicting many frameworks for stability effects inference [42]
Figure 1:
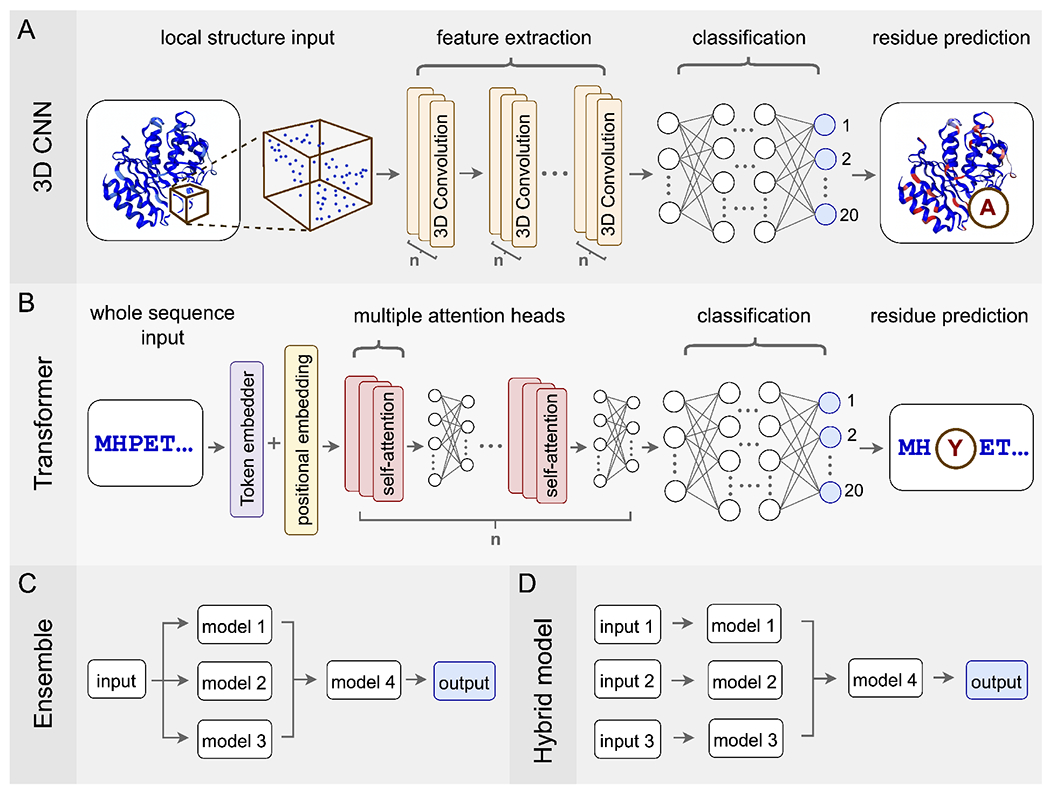
Commonly used network architectures for predicting mutations in protein sequences or structures. (A) 3D Convolutional Neural Networks (3D CNNs) take all the atoms in a subset of 3D space as input, use convolutional layers to extract features, and then use a traditional multilayer perceptron for classification. (B) Transformer models take protein sequences as input, which they process via embedding combined with the attention mechanism. The final classification occurs with a multilayer perceptron, just like for 3D CNNs. (C) Ensemble models process the same set of input data via multiple independent models, whose predictions are then combined into a final output. (D) Hybrid models are architecturally similar to ensemble models but now each of the independent models can have different input data, such as sequence data, structural data, phenotypic measurements, etc.
Mutational effects are not just limited to changes in stability. Any arbitrary phenotype we could be interested in will in general be affected by mutations. Methods that can predict such mutational effects are called variant effect predictors [43]. Examples of phenotypes of interest include but are not limited to the amount of fluorescence emitted by a fluorescent protein, the antibiotic activity of a β-lactamase, or more generally the catalytic activity of an enzyme. Predicting the effects of mutations on such phenotypes is a much more difficult problem than predicting ΔΔG, because every specific system has its own biochemistry, and we will have to develop a custom model for each system.
The advantage of supervised learning for this application is that the statistical learning approach does not require any insight or specific knowledge about the physics or biology of the system. As long as a suitable training dataset is available, we can train a predictor, often with good success. However, the number of experimental measurements required to train a good predictor can be massive. For this reason, early work in this area focused on swapping protein fragments rather than making individual point mutations [21, 44]. As large-scale datasets of mutational effects have become more available, machine-learning approaches have followed suit and have integrated these datasets into better and more fine-grained predictors [43, 45, 46].
Common sources for large-scale datasets of mutational effects include the extensive synthesis and subsequent assay of point mutants (so-called deep mutational scanning [5, 6]) as well as the synthesis of large random sequence libraries, potentially coupled with directed evolution of functional variants [47]. From a machine-learning perspective, any such dataset is useful as long as it contains a sufficiently large number of mutations that have been assayed for the phenotype of interest. Finally, manual curation of many disparate experiments into a single, consistent dataset (e.g., [48]) can also provide useful training data for variant effect predictors.
3. Unsupervised learning or zero-shot learning
Because of the requirement for large, purpose-built training datasets, supervised approaches are inherently limited. Therefore, there is substantial interest in the community to develop unsupervised approaches that can leverage general representations of protein biochemistry for specific applications without requiring application-specific training data.
Most unsupervised approaches start from sequence data, in particular multiple-sequence alignments (MSAs), and attempt to construct computational representations of the patterns encoded in these alignments. Early work in this area focused on covariation [49, 50], attempting to infer mutational effects from patterns of variation and covariation in the MSA. Subsequently, Riesselman et al. [19] demonstrated that higher-order effects can be learned by deep generative models, specifically variational autoencoders (VAEs), which can capture latent structure of arbitrary order in sequence families.
More recently, the research community has focused on applying concepts from natural language modeling to protein sequences. The goal here is to develop protein embedding models that can extract biophysically relevant quantitative features from sequence data. And as natural language modeling techniques have improved so have the protein embedding models. Bepler and Berger [28] applied a bi-directional Long Short-Term Memory (BiLSTM) network and showed they could predict global similarity of protein structures and protein secondary structure. Following this pioneering work, several groups have adapted transformer-based natural language models to learn protein embedding spaces, with considerable success [14, 30, 31, 32] (Figure 1B). For example, Rives et al. [14] demonstrated that their transformer-based ESM-1b model outcompeted alternative approaches, including LSTM networks and Hidden Markov Models, on a variety of tasks, including remote homology detection, secondary-structure prediction, and long-range contact prediction. Other network architectures inspired from language models that have shown excellent performance are deep autoregressive models [20].
Language embedding models are generally trained by taking sequence data, masking individual residues in a sequence, and then training a network to predict the masked residue. So technically speaking, these models can only predict what amino acid may reside with what likelihood at a given site in a protein sequence. However, the amino-acid likelihoods can be reinterpreted as fitness or stability effects, and this has opened the door to so-called zero-shot learning approaches where fitness effects are predicted in an unsupervised manner. This work traces back to Hopf et al. [50], who showed that sequence covariation is correlated with phenotypic measurements.
Meier et al. [51] systematically evaluated zero-shot learning for several language embedding models. They tested model predictions against an array of deep mutational scanning datasets [19], covering a wide range of different systems studied and different protein functions assayed. The general observation was that mutational effects predicted by language embedding models tend to correlate with measured mutational effects. As an interesting extension to basic language models, Hie et al. [52] parsed model predictions into components corresponding grammar (or syntax) and meaning (or semantics), where the grammaticality of a mutation is assessed by the likelihood the model assigns to that mutation at a specific site and the semantics are represented by the embedding scores. The authors were then able to show that antigenic escape mutations tended to score high on the grammar axis while also scoring high on the axis representing semantic change. In other words, these were mutations that were consistent with the protein structure while substantially changing some aspect of the protein’s biochemistry.
There are two general caveats to zero-shot language models: First, there is a wide range in model performance for different systems. Correlation coefficients range from 0.2 to 0.8. Second, it is not necessarily clear why an embedding model performs well in some cases and poorly in others, since the model has not been trained for the specific problem at hand. We do not know what specific (if any) physical quantity is being predicted by the model and whether and how it may relate to any specific phenotypic measurement.
4. Hybrid or transfer learning
From a conceptual perspective, supervised models are the appropriate approach to predicting mutational effects, and they do not suffer from the problem that it is unclear what exactly they predict. However, in practice, we rarely have sufficiently large datasets to train good supervised models. In fact, for any given protein for which a sufficiently large dataset exists we probably have already found all the interesting mutations and the machine learning approach to predicting mutations is an entirely academic exercise for this protein. In practice, prediction is useful specifically in systems where we do not have experimentally surveyed many mutations and/or where such surveys would be impractical, and consequently supervised approaches will not usually work in those settings. Unsupervised approaches, of course, don’t learn any specific fitness models and therefore are not guaranteed to make useful predictions for any specific system.
As a way of getting the best of both worlds, it is possible to devise a hybrid approach where unsupervised models are first pre-trained to learn embeddings for general protein biochemistry and/or general properties of the protein family of interest, and then fine-tuned in a supervised fashion with a small training set of measured fitness effects, also called transfer learning. In this context, we use the term “hybrid learning” to describe approaches that combine different biological input data types, such as evolutionary data from sequence alignments and phenytopic data from biochemical assays (Figure 1D), and we use the term “transfer learning” to generally represent the task of taking a pre-trained generic model and fine-tune it to a more specific dataset.
One of the first studies to demonstrate the hybrid approach was performed by Biswas et al. [53], who pretrained a recurrent neural network (RNN) model [54] to learn statistical representations of proteins and then utilized its embeddings to train a regularized linear regression and validated the final model by predicting fluorescence in GFP variants and optimizing function in TEM-1 β-lactamase. For both systems, they showed that with as little as 24 to 96 functionally assayed mutant sequences, they could train a model fine-tuned on the RNN’s embeddings to generate computational predictions that rivaled high-throughput experimental screens.
Two recent papers have performed large scale, systematic studies of hybrid learning approaches [27, 13]. Both studies highlight that the available training data may be more important than the specific network architecture or machine-learning approach taken. Hsu et al. [27] found that even relatively simple regression models supplied with assay-labeled data could outperform sophisticated embedding models without such data. Similarly, Luo et al. [13] found that combining a generic protein language model with information about the evolutionary context of a specific protein family and some measured fitness data provided superior predictive ability across approximately 50 separate high-throughput experimental datasets.
5. Predicting sites primed for mutation
In application settings where there is limited to no phenotypic data available to train or fine-tune a hybrid predictor, we are stuck with unsupervised learning approaches. However, in these scenarios, there is one additional strategy we can follow: self-supervision—where we artificially create a supervised learning task from the data itself. Here, instead of trying to predict a fitness effect of unknown meaning, we focus on predicting a masked amino acid directly, either from sequence or, more commonly, from the local microenvironment for sites all throughout the protein structure. This self-supervised approach was pioneered by Torng and Altman [15], was validated in proof-of-concept applications to blue fluorescent protein, phosphomannose isomerase, TEM-1 β lactamase [16], and has since been used to engineer highly active hydrolases for PET depolymerization [55]. Systematic benchmarking has shown that microenvironments as small as 12Å in diameter are sufficient to make predictions with over 65% accuracy [18].
The key idea in all these applications may seem counterintuitive at first. We use a neural network to predict masked wildtype residues in a known protein. Since the protein is known, the identity of any masked residues is known as well, and so most predictions of a well performing network will simply recover the correct identity of the resident residue that was masked. This in itself may not seem like a useful prediction. However, the interesting cases here are the mispredictions. Conventionally, we would consider all mispredictions to be shortcomings of the model and an opportunity for further model improvement. However, in some fraction of cases, residues will be chemically incongruent with their microenvironment and thus primed for mutation. In other words, those residues will be at odds with their immediate biochemical surroundings and can be mutated to different residues without loss of protein function. In those cases, if the network has correctly captured the relevant protein biochemistry and is not overfit to predicting the wildtype amino acid, it will confidently predict a different amino acid than the one actually present. Those “mispredictions” indicate that the wildtype amino acid is likely not the optimal amino acid and can be engineered.
This approach of trying to identify residues that are at odds with their local microenvironment has strong support from evolutionary theory, which predicts that, due to epistasis, every protein structure has some residues that are to some degree in conflict with their current surroundings and that are primed for mutation [56, 57]. In fact, evolution tends to occur at those sites. However, this approach poses an interesting challenge for training and applying a machine learning algorithm. Usually, the goal in machine learning is to get the prediction accuracy as high as possible. But, if we are interested specifically in sites where the machine-learning algorithm disagrees with the observed ground truth, then too high accuracy will be deleterious, as (in the extreme case of 100% accuracy) there won’t be any disagreements to guide protein engineering. It remains an open question how to best train models to maximize their ability to identify sites primed for mutation and predict the correct amino acid substitution.
We emphasize that in principle both sequence and structural data can be and has been used in this self-supervised approach [14, 15, 16, 18, 30]. The main distinction between self-supervised and unsupervised learning is scope: unsupervised learning refers to all machine learning techniques that attempt to learn clustering structure or embeddings from data without an annotated label while self-supervised learning is a specific type of unsupervised learning where the objective is to predict the masked portion of the input data instance where a program automatically generates the masking.
6. Pros and cons of sequence- and structure-based approaches
Some hybrid learning models notwithstanding, the majority of current machine-learning models use either exclusively sequence data or exclusively structural data as their input. There are pros and cons to both approaches. Importantly, protein sequence data is vastly more abundant than structural data, and thus it may seem practical to create machine learning models based on sequence data alone.
In particular, it has been reported that models trained on sequences can learn protein structure at the resolution of atom position [14, 32, 58], demonstrating that sequence data may implicitly contain all information required to make any predictions of interest in protein biochemistry. Moreover, sequence-based models can accept entire protein sequences as input, capturing both long- and short-range interactions within a protein. Long-range interactions in 3D space are generally excluded from training models based on structural data. Furthermore, structure-based models often represent proteins as collections of atoms fixed in space rather than a polymer of amino acids. This inaccurate representation of protein structure may undermine a protein’s flexibility and ability to adjust to novel mutations [59], a limitation that is not present in sequence-based models.
One downside of sequence data is that it does not directly provide information on physical contacts in protein structures. Although protein contacts and structure can be inferred during training, as a result, sequence-based models require a significantly larger training set and much deeper architectures than models trained on pre-solved structures [58]. This poses a challenge for model validation and testing. Sequence-based models are often trained on the entire corpus of the majority of publicly available protein sequences (e.g., UniRef100), and thus nearly any protein of interest is already present in the test set. Therefore, to what extent such models truly generalize biochemistry or rather just memorize all known sequences remains to be determined.
On the flip side, structures are richer in information than sequences and as more structures are solved with ligands, glycans, and nucleotides as co-crystals we expect to see improvement in predictions at functional residues. While protein structure data is less abundant, it allows models to learn directly from local chemistry to make predictions based on immediate biochemical features as well as the silhouette formed by immediate chemical contacts [60, 16, 18].
More interestingly, it is likely that structure-based approaches can go beyond what sequence alone can predict. Sequence-based methods de-facto only use input data from natural, evolved proteins. Such phylogenetic information has arisen via single mutational moves, and at best can be used to infer the complicated physicochemical structure of the protein as a whole. By contrast, databases of directly acquired structural information fully represent all of the atomic-level physical and chemical interactions of many different amino acids in many different proteins, all at once, and this additional and important information goes well beyond the information available in sequence databases of evolutionary phylogenies. Overall, structure-based neural networks can utilize this broader, deeper knowledge of atomic-level interactions to more fully understand the many moves that evolution has made, and to see the moves that might yet be made. By using structural information to understand and predict substitutions (including multiple substitutions) the physicochemical status of these substitutions is implicitly validated against the backdrop of structure itself, rather than against phylogenetic sequence data that merely implies structure.
The remaining limitations of models using structure as input data are currently being explored by inverse folding graph neural networks (GNNs), which combine rigid and flexible backbone features. Recent work includes geometric vector perceptron GNNs (GVP-GNNs)[8, 24, 25], structured transformer GNN [61], and protein message passing GNN (ProteinMPNNs) [26]. Where the GVP-GNNs encode geometric information for either atom-level or residue-level nodes and outperformed equivalent CNNs on the Atom3D Residue Identity benchmark [62], structured transformer GNNs utilize self-attention in an encoder-decoder architecture to capture higher-order, interaction-based dependencies that autoregressively decode masked residues. Protein-MPNN is a simplified variant of the structured transformer architecture that does away with the attention mechanism, can handle multiple protein chains at once and abstract over spatial symmetries, and has demonstrated its utility in several protein design projects by rescuing previously failed designs made with Rosetta and Alphafold.
Lastly, the ability of a structure-based model to properly learn protein biochemistry is directly dependent on the quality of experimentally-solved or computationally generated protein structures used as input data. Structures solved via crystallization methods often have low quality or display non-physiological conformations [63], and we expect this flaw to persist in computationally predicted structures since crystal structures are the primary source of training data for structure prediction models such as AlphaFold2, RosettaFold, ESMFold, or OmegaFold [7, 64, 65, 58]. Nevertheless, structure-based models are valuable tools as they can capture local protein features most critical for accurate residue prediction.
7. Looking into the future
Machine learning algorithms are fundamentally limited by the available quality and quantity of training data. As datasets continue to grow, so will the accuracy of the machine learning models trained on them. Already, for all intents and purposes, we have infinite sequence data, or rather more data than can realistically be used in training. (For example, with current compute technology it is next-to-impossible to train a machine learning model on all available microbiome datasets.) By contrast, structural data remains much more limited, and further meaningful expansions of datasets in this area should be expected.
An interesting middle ground in this context is computationally predicted structures, which are rapidly becoming more available and more high-quality. For example, DeepMind and EMBL-EBI recently released 200 million computationally predicted structures [66]. While it seems risky to train downstream models on predicted structures [8] (the downstream models may learn idiosyncracies of the predictions rather than true protein biochemistry), predicted structures can certainly be used as input to generate inferences from structure-based models. One open question for future research will be whether it is better to predict mutational effects directly from sequence or rather predict structure from sequence and then predict mutational effects using a structure-based model. Finally, we expect advancements in structure prediction that take into account conformational changes observed upon ligand binding.
Hybrid approaches integrating multiple different data sources are likely going to become increasingly important, as they can augment rather limited datasets in one domain with extensive and well understood datasets in other domains. Going forward, it may become routine to develop models that include sequence data, structural data, phenotypic data, and evolutionary data such as multiple sequence alignments and/or phylogenetic trees. Additionally, it may be possible to leverage deep learning to improve mechanistic models, for example by developing force fields that are generated by deep learning models trained on quantum chemistry data [67, 68, 69].
Finally, while much research has been focused on identifying the best architectures and training modalities to obtain well performing models, less emphasis has been placed on trying to understand the underlying biochemical principles these networks embody and abstract to make predictions. These principles can be probed with salient methods developed by the natural language processing and computer vision communities (e.g., [70]). We expect that such research will provide access to the underlying patterns (chemistries) these models are learning. Ultimately, salient methods may uncover principles of biochemistry or of protein design that have been underappreciated or undiscovered using conventional approaches. Similarly, it is possible that an understanding of how evolution has traversed phylogeny can be garnered from machine learning approaches, especially hybrid approaches that take into account both the single mutational steps available from sequence-based predictions, and the potential for multiple changes in parallel present in structure-based predictions. Based on such hybrid approaches, we may get a bird’s eye view of how evolution considers the overall issues relating to improving protein function, both the paths available, and the paths taken, and in so doing come to a deeper understanding of proteins as evolvable machines.
Funding.
This study was funded by National Institutes of Health grant R01 AI148419 and Welch Foundation grant F-1654. C.O.W. is also supported by the Blumberg Centennial Professorship in Molecular Evolution and the Reeder Centennial Fellowship in Systematic and Evolutionary Biology at The University of Texas at Austin.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest/Competing interests. D.J.D. is a cofounder of Intelligent Proteins, LLC, which uses machine learning for problems of protein engineering. A.V.K., A.D.E., and C.O.W. declare that they have no conflict of interest.
References
- [1].Zuckerkandl E, Pauling L, Evolutionary divergence and convergence in proteins, in: Bryson V, Vogel HJ (Eds.), Evolving Genes and Proteins, Academic Press, 1965, pp. 97–166. [Google Scholar]
- [2].Shakhnovich E, Protein folding thermodynamics and dynamics: Where physics, chemistry and biology meet, Chem Rev. 106 (2006) 1559–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tokuriki N, Tawfik DS, Stability effects of mutations and protein evolvability, Current Opin. Struct. Biol 19 (2009) 596–604. [DOI] [PubMed] [Google Scholar]
- [4].Serohijos AWR, Shakhnovich EI, Merging molecular mechanism and evolution: theory and computation at the interface of biophysics and evolutionary population genetics, Current Opin. Struct. Biol 26 (2014) 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Araya CL, Fowler DM, Deep mutational scanning: assessing protein function on a massive scale, Trends Biotech. 29 (2011) 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Livesey BJ, Marsh JA, Interpreting protein variant effects with computational predictors and deep mutational scanning, Dis. Model. Mech 15 (2022) dmm049510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D, Highly accurate protein structure prediction with AlphaFold, Nature 596 (2021) 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The authors developed a break-through machine-learning model that leverages evolutionary covariation in a supervised manner to accurately predict protein structures.
- [8].Hsu C, Verkuil R, Liu J, Lin Z, Hie B, Sercu T, Lerer A, Rives A, Learning inverse folding from millions of predicted structures, bioRxiv, 2022. doi: 10.1101/2022.04.10.487779. [DOI] [Google Scholar]; * This study proposed a transformer model that can predict protein sequence from a backbone structure (inverse folding problem). Importantly, the model was trained with 12 million predicted structures from AlphaFold2.
- [9].Stepniewska-Dziubinska MM, Zielenkiewicz P, Siedlecki P, Development and evaluation of a deep learning model for protein-ligand binding affinity prediction, Bioinformatics 34 (2018) 3666–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].McNutt AT, Francoeur P, Aggarwal R, Masuda T, Meli R, Ragoza M, Sunseri J, Koes DR, GNINA 1.0: molecular docking with deep learning, Journal of Cheminformatics 13 (2021) 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Stark H, Ganea O, Pattanaik L, Barzilay R, Jaakkola T, Equibind: Geometric deep learning for drug binding structure prediction, in: International Conference on Machine Learning, 2022, pp. 20503–20521. [Google Scholar]
- [12].Dehghanpoor R, Ricks E, Hursh K, Gunderson S, Farhoodi R, Haspel N, Hutchinson B, Jagodzinski F, Predicting the effect of single and multiple mutations on protein structural stability, Molecules 23 (2018) 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Luo Y, Jiang G, Yu T, Liu Y, Vo L, Ding H, Su Y, Qian WW, Zhao H, Peng J, ECNet is an evolutionary context-integrated deep learning framework for protein engineering, Nature Comm. 12 (2021) 5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rives A, Meier J, Sercu T, Goyal S, Lin Z, Liu J, Guo D, Ott M, Zitnick CL, Ma J, Fergus R, Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences, Proc. Natl. Acad. Sci. U.S.A 118 (2021) e2016239118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Torng W, Altman RB, 3D deep convolutional neural networks for amino acid environment similarity analysis, BMC Bioinf. 18 (2017) 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shroff R, Cole AW, Diaz DJ, Morrow BR, Donnell I, Annapareddy A, Gollihar J, Ellington AD, Thyer R, Discovery of novel gain-of-function mutations guided by structure-based deep learning, ACS Synth. Biol 9 (2020) 2927–2935. [DOI] [PubMed] [Google Scholar]
- [17].Li B, Yang YT, Capra JA, Gerstein MB, Predicting changes in protein thermodynamic stability upon point mutation with deep 3D convolutional neural networks, PLoS Comput. Biol 16 (2020) e1008291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kulikova AV, Diaz DJ, Loy JM, Ellington AD, Wilke CO, Learning the local landscape of protein structures with convolutional neural networks, J. Biol. Phys 47 (2021) 435–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Riesselman AJ, Ingraham JB, Marks DS, Deep generative models of genetic variation capture the effects of mutations, Nature Methods 15 (2018) 816–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shin J-E, Riesselman AJ, Kollasch AW, McMahon C, Simon E, Sander C, Manglik A, Kruse AC, Marks DS, Protein design and variant prediction using autoregressive generative models, Nature Comm. 12 (2021) 2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Romero PA, Krause A, Arnold FH, Navigating the protein fitness landscape with gaussian processes, Proc. Natl. Acad. Sci. USA 110 (2013) E193–E201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jokinen E, Heinonen M, Lähdesmäki H, mgpfusion: predicting protein stability changes with gaussian process kernel learning and data fusion, Bioinformatics 34 (2018) i274–i283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang S, Tang H, Shan P, Zuo L, ProS-GNN: predicting effects of mutations on protein stability using graph neural networks, bioRxiv, 2021. doi: 10.1101/2021.10.25.465658. [DOI] [PubMed] [Google Scholar]
- [24].Jing B, Eismann S, Suriana P, Townshend RJ, Dror R, Learning from protein structure with geometric vector perceptrons, in: 9th International Conference on Learning Representations, ICLR 2021, OpenReview.net, 2021. URL: https://openreview.net/forum?id=1YLJDvSx6J4. [Google Scholar]
- [25].Jing B, Eismann S, Soni PN, Dror RO, Equivariant graph neural networks for 3d macromolecular structure, arXiv preprint arXiv:2106.03843 (2021). [Google Scholar]; * Developed a vector-gated geometric vector perceptron (GVP) layer to act as a drop in replacement for traditional multi-layer perceptron to enable rotationally invariant message passing of vector features.
- [26].Dauparas J, Anishchenko I, Bennett N, Bai H, Ragotte RJ, Milles LF, Wicky BIM, Courbet A, de Haas RJ, Bethel N, Leung PJY, Huddy TF, Pellock S, Tischer D, Chan F, Koepnick B, Nguyen H, Kang A, Sankaran B, Bera AK, King NP, Baker D, Robust deep learning based protein sequence design using ProteinMPNN, bioRxiv, 2022. doi: 10.1101/2022.06.03.494563. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Trained a message passing graph neural network (ProteinMPNN) with an encoder-decoder architecture on the backbone coordinates of monomeric and oligomeric protein structures. They demonstrate that ProteinMPNN is able to rescue previously failed Rosetta protein design projects that involve protein-protein complex and partially deleted protein structures.
- [27].Hsu C, Nisonoff H, Fannjiang C, Listgarten J, Learning protein fitness models from evolutionary and assay-labeled data, Nature Biotechnology 40 (2022) 1114–1122. [DOI] [PubMed] [Google Scholar]
- [28].Bepler T, Berger B, Learning protein sequence embeddings using information from structure, in: 7th International Conference on Learning Representations, ICLR 2019, OpenReview.net, 2019. URL: https://openreview.net/forum?id=SygLehCqtm. [Google Scholar]
- [29].Cheng J, Randall A, Baldi P, Prediction of protein stability changes for single-site mutations using support vector machines, Proteins: Structure, Function, and Bioinformatics 62 (2006) 1125–1132. [DOI] [PubMed] [Google Scholar]
- [30].Elnaggar A, Heinzinger M, Dallago C, Rehawi G, Wang Y, Jones L, Gibbs T, Feher T, Angerer C, Steinegger M, Bhowmik D, Rost B, ProtTrans: towards cracking the language of lifes code through self-supervised deep learning and high performance computing, IEEE Transactions on Pattern Analysis and Machine Intelligence (2021). doi: 10.1109/TPAMI.2021.3095381. [DOI] [PubMed] [Google Scholar]
- [31].Marquet C, Heinzinger M, Olenyi T, Dallago C, Erckert K, Bernhofer M, Nechaev D, Rost B, Embeddings from protein language models predict conservation and variant effects, Hum. Genet (2021). doi: 10.1007/s00439-021-02411-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Brandes N, Ofer D, Peleg Y, Rappoport N, Linial M, ProteinBERT: a universal deep-learning model of protein sequence and function, Bioinformatics 38 (2022) 2102–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Guerois R, Nielsen JE, Serrano L, Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations, J. Mol. Biol 320 (2002) 369–387. [DOI] [PubMed] [Google Scholar]
- [34].Yin S, Ding F, Dokholyan NV, Modeling backbone flexibility improves protein stability estimation, Structure 15 (2007) 1567–1576. [DOI] [PubMed] [Google Scholar]
- [35].Kellogg E, Leaver-Fay A, Baker D, Role of conformational sampling in computing mutation-induced changes in protein structure and stability, Proteins: Structure, Function, and Bioinformatics 79 (2011) 830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dehouck Y, Kwasigroch JM, Gilis D, Rooman M, PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality, BMC Bioinformatics 12 (2011) 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Worth CL, Preissner R, Blundell TL, SDM—a server for predicting effects of mutations on protein stability and malfunction, Nucleic Acids Rese. 39 (2011) W215–W222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fariselli P, Martelli PL, Savojardo C, Casadio R, INPS: predicting the impact of non-synonymous variations on protein stability from sequence, Bioinformatics 31 (2015) 2816–2821. [DOI] [PubMed] [Google Scholar]
- [39].Cao H, Wang J, He L, Qi Y, Zhang JZ, DeepDDG: predicting the stability change of protein point mutations using neural networks, J. Chem. Inf. Model 59 (2019) 1508–1514. [DOI] [PubMed] [Google Scholar]
- [40].Wainreb G, Wolf L, Ashkenazy H, Dehouck Y, Ben-Tal N, Protein stability: a single recorded mutation aids in predicting the effects of other mutations in the same amino acid site, Bioinformatics 27 (2011) 3286–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Li Y, Fang J, PROTS-RF: a robust model for predicting mutation-induced protein stability changes, PLoS ONE 7 (2012) e47247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Thiltgen G, Goldstein RA, Assessing predictors of changes in protein stability upon mutation using self-consistency, PLoS ONE 7 (2012) e46084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gray VE, Hause RJ, Luebeck J, Shendure J, Fowler DM, Quantitative missense variant effect prediction using large-scale mutagenesis data, Cell Systems 6 (2018) 116–124.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bedbrook CN, Yang KK, Rice AJ, Gradinaru V, Arnold FH, Machine learning to design integral membrane channelrhodopsins for efficient eukaryotic expression and plasma membrane localization, PLoS Comput. Biol 13 (2017) e1005786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wu Z, Kan SBJ, Lewis RD, Wittmann BJ, Arnold FH, Machine learning-assisted directed protein evolution with combinatorial libraries, Proc. Natl. Acad. Sci. USA 116 (2019) 8852–8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Høie MH, Cagiada M, Frederiksen AHB, Stein A, Lindorff-Larsen K, Predicting and interpreting large-scale mutagenesis data using analyses of protein stability and conservation, Cell Reports 38 (2022) 110207. [DOI] [PubMed] [Google Scholar]; * Developed a machine learning model to predict variant effects from both structure and sequence variation. This allowed them to pinpoint which variants lose function due to loss of stability versus other mechanisms.
- [47].Cobb RE, Chao R, Zhao H, Directed evolution: Past, present and future, AIChE J. 59 (2013) 1432–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Stourac J, Dubrava J, Musil M, Horackova J, Damborsky J, Mazurenko S, Bednar D, FireProtDB: database of manually curated protein stability data, Nucleic Acids Res. 49 (2021) D319–D324. [DOI] [PMC free article] [PubMed] [Google Scholar]; * A manually curated database of experimental thermostability data for single-point mutants with a user interfaced design for training and benchmarking computational tools.
- [49].Figliuzzi M, Jacquier H, Schug A, Tenaillon O, Weigt M, Coevolutionary landscape inference and the context-dependence of mutations in beta-lactamase TEM-1, Mol. Biol. Evol 33 (2016) 268–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hopf TA, Ingraham JB, Poelwijk FJ, Schärfe CPI, Springer M, Sander C, Marks DS, Mutation effects predicted from sequence covariation, Nature Biotechnology 35 (2017) 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Meier J, Rao R, Verkuil R, Liu J, Sercu T, Rives A, Language models enable zero-shot prediction of the effects of mutations on protein function, Advances in Neural Information Processing Systems 34 (2021) 29287–29303. [Google Scholar]; * The authors trained a 650 million parameter language model on the UniRef90 dataset in an unsupervised fashion for zero-shot prediction on any protein sequence. They demonstrate their model (called ESM-1v) can predict functional effects in approximately 40 deep mutational scanning datasets.
- [52].Hie B, Zhong ED, Berger B, Bryson B, Learning the language of viral evolution and escape, Science 371 (2021) 284–288. [DOI] [PubMed] [Google Scholar]; * This study proposed a method to parse predictions from protein language models into grammatical and syntactical components, and it demonstrates that these components have distinct effects on phenotypic measurements.
- [53].Biswas S, Khimulya G, Alley EC, Esvelt KM, Church GM, Low-N protein engineering with data-efficient deep learning, Nature Methods 18 (2021) 389–396. [DOI] [PubMed] [Google Scholar]; ** The authors demonstrate the potential of transfer learning a pre-trained sequence model with as little as 24 functionally assayed mutants to build a virtual fitness landscape for virtual screening. They demonstrate how this paradigm accelerates protein engineering by engineering avGFP and TEM-1 β lactamase.
- [54].Alley EC, Khimulya G, Biswas S, AlQuraishi M, Church GM, Unified rational protein engineering with sequence-based deep representation learning, Nature Methods 16 (2019) 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lu H, Diaz DJ, Czarnecki NJ, Zhu C, Kim W, Shroff R, Acosta DJ, Alexander BR, Cole HO, Zhang Y, Lynd NA, Ellington AD, Alper HS, Machine learning-aided engineering of hydrolases for PET depolymerization, Nature 604 (2022) 662–667. [DOI] [PubMed] [Google Scholar]; * The authors leveraged a self-supervised 3D CNN model to guide the engineering of several PET-hydrolases. Their top-performing variant FAST-PETase is capable of 100 percent depolymerization of post-consumer PET packaging within a few days at moderate temperatures.
- [56].Shah P, McCandlish DM, Plotkin JB, Contingency and entrenchment in protein evolution under purifying selection, Proc. Natl. Acad. Sci. USA 112 (2015) E3226–E3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Goldstein RA, Pollock DD, Sequence entropy of folding and the absolute rate of amino acid substitutions, Nature Ecol. Evol 1 (2017) 1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lin Z, Akin H, Rao R, Hie B, Zhu Z, Lu W, dos Santos CA, Fazel-Zarandi M, Sercu T, Candido S, Rives A, Language models of protein sequences at the scale of evolution enable accurate structure prediction, bioRxiv, 2022. doi: 10.1101/2022.07.20.500902. [DOI] [Google Scholar]; ** The authors explored the learning capability of language models as they are scaled up to the point of 15 billion parameters. These massive language models are capable of predicting the three-dimensional structure of a protein at the atom level using just the protein sequence as input, removing the need to pregenerate multiple sequence alignments and in turn speeding up inference by an order of magnitude compared to AlphaFold2.
- [59].Sotomayor-Vivas C, Hernández-Lemus E, Dorantes-Gilardi R, Linking protein structural and functional change to mutation using amino acid networks., PLoS ONE 17 (2022) e0261829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wang J, Cao H, Zhang JZH, Qi Y, Computational protein design with deep learning neural networks, Scientific Reports 8 (2018) 2045–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ingraham J, Garg V, Barzilay R, Jaakkola T, Generative models for graph-based protein design, Advances in Neural Information Processing Systems 32 (2019). [Google Scholar]
- [62].Townshend RJ, Vögele M, Suriana P, Derry A, Powers A, Laloudakis Y, Balachandar S, Jing B, Anderson B, Eismann S, et al. , Atom3d: Tasks on molecules in three dimensions, arXiv preprint arXiv:2012.04035 (2020). [Google Scholar]
- [63].Domagalski MJ, Zheng H, Zimmerman MD, Dauter Z, Wlodawer A, Minor W, The quality and validation of structures from structural genomics, Methods Mol. Biol 1091 (2014) 297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, et al. , Accurate prediction of protein structures and interactions using a three-track neural network, Science 373 (2021) 871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]; * The authors trained a three-track neural network that integrates the processing of 1D sequence, 2D distance maps, and 3D coordinates to predict structures. The model is able to generate accurate protein-protein complexes from sequence information alone.
- [65].Wu R, Ding F, Wang R, Shen R, Zhang X, Luo S, Su C, Wu Z, Xie Q, Berger B, Ma J, Peng J, High-resolution de novo structure prediction from primary sequence, bioRxiv, 2022. doi: 10.1101/2022.07.21.500999. [DOI] [Google Scholar]
- [66].Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A, Žídek A, Green T, Tunyasuvunakool K, Petersen S, Jumper J, Clancy E, Green R, Vora A, Lutfi M, Figurnov M, Cowie A, Hobbs N, Kohli P, Kleywegt G, Birney E, Hassabis D, Velankar S, AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models, Nucleic Acids Res. 50 (2021) D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Devereux C, Smith JS, Huddleston KK, Barros K, Zubatyuk R, Isayev O, Roitberg AE, Extending the applicability of the ANI deep learning molecular potential to sulfur and halogens, J. Chem. Theory Comput 16 (2020) 4192–4202. [DOI] [PubMed] [Google Scholar]
- [68].Christensen AS, Sirumalla SK, Qiao Z, O’Connor MB, Smith DG, Ding F, Bygrave PJ, Anandkumar A, Welborn M, Manby FR, et al. , OrbNet Denali: a machine learning potential for biological and organic chemistry with semi-empirical cost and DFT accuracy, J. Chem. Phys 155 (2021) 204103. [DOI] [PubMed] [Google Scholar]
- [69].Jaffrelot Inizan T, Plé T, Adjoua O, Ren P, Gökcan H, Isayev O, Lagardère L, Piquemal J-P, Scalable hybrid deep neural networks/polarizable potentials biomolecular simulations including long-range effects, arXiv e-prints (2022) arXiv–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wang Q, Zhang L, Li Y, Kpalma K, Overview of deep-learning based methods for salient object detection in videos, Pattern Recognition 104 (2020) 107340. [Google Scholar]