

Abstract
Substrate side chain conformation impacts reactivity during glycosylation and glycoside hydrolysis and is restricted by many glycosidases and glycosyltransferases during catalysis. We show that the side chains of gluco and manno iminosugars can be restricted to predominant conformations by strategic installation of a methyl group. Glycosidase inhibition studies reveal that iminosugars with the gauche,gauche side chain conformations are 6‐ to 10‐fold more potent than isosteric compounds with the gauche,trans conformation; a manno‐configured iminosugar with the gauche,gauche conformation is a 27‐fold better inhibitor than 1‐deoxymannojirimycin. The results are discussed in terms of the energetic benefits of preorganization, particularly when in synergy with favorable hydrophobic interactions. The demonstration that inhibitor side chain preorganization can favorably impact glycosidase inhibition paves the way for improved inhibitor design through conformational preorganization.
Keywords: Glycosidase, Iminosugar, Inhibitor, Methyl Group, Side Chain Conformation
The first experimental demonstration that preorganization of inhibitor side chain conformation can favorably impact glycosidase inhibition is reported. By strategic installation of a methyl group at C6, a manno‐configured iminosugar with the gauche,gauche side chain conformation was found to be a 27‐fold better inhibitor than 1‐deoxymannojirimycin.
The biosynthesis and degradation of glycosides are catalyzed by two classes of enzymes: glycosyltransferases (GTs), which install glycosidic bonds, and glycoside hydrolases (GHs), or glycosidases, which cleave them. Inhibition of GTs or GHs has applications in the treatment of many diseases, including bacterial infection, viral infection, diabetes, cancer and genetic disorders.[ 1 , 2 ] Inhibitor design is often based on mimicry of the substrate conformation and/or of the positively charged transition state.[ 3 , 4 , 5 , 6 ] This is illustrated by the drugs: miglitol (Figure 1), a GH inhibitor used to treat type II diabetes by targeting intestinal α‐glucosidases, is an iminosugar whose conjugate acid mimics the positive charge at the transition state for the enzymatic glucoside hydrolysis. [3] Zanamivir, a drug used to treat influenza by inhibiting a neuraminidase, on the other hand was designed to mimic the conformation of the natural substrate at the transition state for hydrolysis. [3] Finally, 4′‐deaza‐1′‐aza‐2′‐deoxy‐9‐methylene Immucillin‐G (DADMe‐Immucillin‐G), a purine nucleoside phosphorylase inhibitor with potential for the treatment for malaria, is a transition state analog that, like miglitol, is protonated and positively charged when bound to the enzyme. [4]
Figure 1.
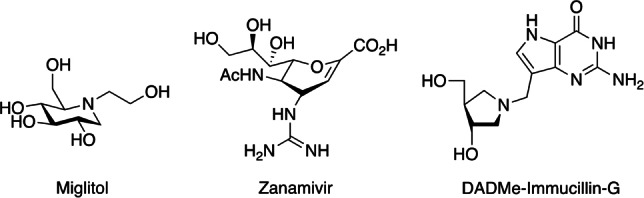
Structures of miglitol, zanamivir, and DADMe‐Immucillin‐G. Ac, acetyl.
In chemical glycosylation and glycoside hydrolysis, the reactivity of glycosidic bond‐forming or bond‐breaking events is influenced by the carbohydrate side chain conformation: gauche,gauche (gg), gauche,trans (gt), or trans,gauche (tg).[ 7 , 8 , 9 , 10 ] The relative population of these conformers in aqueous solution is configuration‐dependent with D‐glucopyranosides and D‐mannopyranosides being ≈50 : 50 mixtures of gg and gt conformers at equilibrium (Scheme 1).[ 11 , 12 ]
Scheme 1.
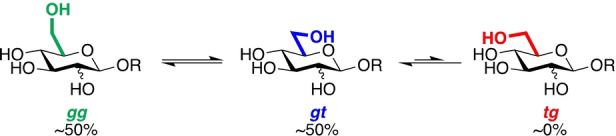
Approximate aqueous solution populations of three staggered conformations of D‐glucopyranosides (equatorial OH at C2) and D‐mannopyranosides (axial OH at C2).
Seminal studies by Bols and co‐workers demonstrated the influence of side chain conformation on glycoside reactivity by evaluation of spontaneous hydrolysis at pH 6.5 of a series of conformationally locked dinitrophenyl glucopyranosides (Figure 2). [8] The rate of glycoside hydrolysis was found to be gg>gt>tg, as corroborated in the galacto series by our laboratory. [9] In addition, our recent findings have revealed that the influence of side chain conformation on reactivity is exploited by nature, where many GHs and GTs bind their ligands with specific side chain conformations.[ 13 , 14 ] For example, >80 % of glucosidases restrict the side chain of glucose in their active sites to the gg conformation: considering the ≈50 : 50 unbound population of gg:gt conformers, these enzymes appear to have evolved to take the advantage of the substrate side chain conformation to gain additional transition state stabilization. [13] Davies and co‐workers studied the GH family 1 β‐glucosidase from Thermotoga maritima (TmGH1) and found that the conformationally rigid castanospermine 1 (Figure 3) is a four‐fold better inhibitor than 1‐deoxynojirimycin 2, whose side chain adopts the gg conformation in complex with TmGH1 (Figure 4a),[ 5 , 15 , 16 ] suggesting that nature has already benefitted from the concept of side chain conformational control to evolve improved inhibitors. [13] Collectively, these studies suggest that preorganization of inhibitor side chain conformation can be used to influence their activity.
Figure 2.
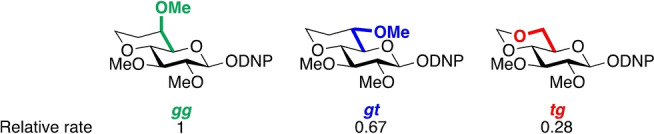
Relative rates of spontaneous hydrolysis of dinitrophenyl glycosides. DNP, 2,4‐dinitrophenyl.
Figure 3.
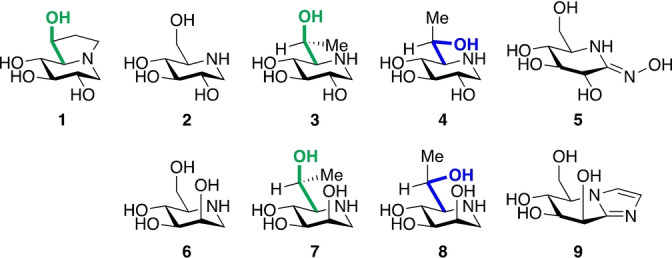
Structures of castanospermine 1, 1‐deoxynojirimycin 2, glucohydroximolactam 5, 1‐deoxymannojirimycin 6, mannoimidazole 9, and the targeted inhibitors 3, 4, 7 and 8.
Figure 4.
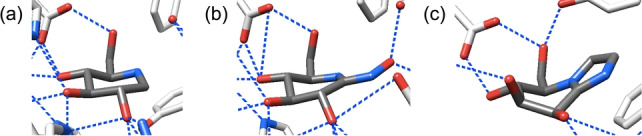
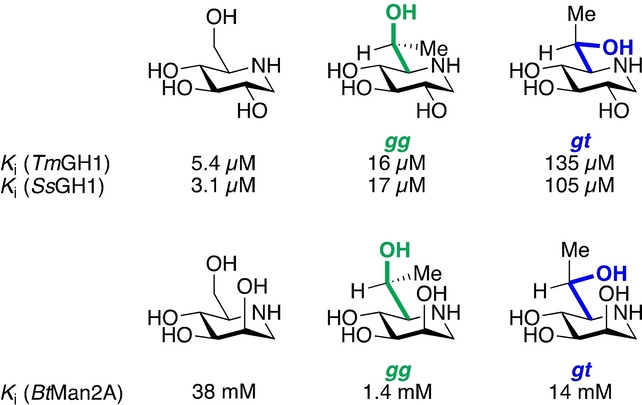
Partial X‐ray crystal structures of a) TmGH1 in complex with 2 (PDB ID: 2 J77), b) SsGH1 in complex with 5 (PDB ID: 1UWU), and c) BtMan2A in complex with 9 (PDB ID: 2VMF).
Following established principles for the restriction of side chain conformation,[ 17 , 18 ] to investigate whether side chain preorganization can influence inhibitor potency, we designed two derivatives (3 and 4, Figure 3) of 1‐deoxynojirimycin 2 carrying a methyl group at C6. In addition to TmGH1, whose crystal structure in complex with 2 is known (Figure 4a), [5] we also selected a GH family 1 β‐glycosidase from Sulfolobus solfataricus (SsGH1) for our studies on these compounds: X‐ray studies (Figure 4b) indicate that glucohydroximolactam‐type inhibitor 5 adopts the gg side chain conformation when bound to this enzyme.[ 19 , 20 ] Additionally, we designed two manno‐configured derivatives (7 and 8) of 1‐deoxymannojirimycin 6, and a GH family 2 β‐mannosidase from Bacteroides thetaiotaomicron (BtMan2A) was selected for studies on this series of compounds: the crystal structure of the mannoimidazole 9:BtMan2A complex reveals the side chain of 9 to be bound in the gg conformation (Figure 4c). [21] Compound 3 with the L‐glycero‐D‐gluco configuration and compound 7 with the L‐glycero‐D‐manno configuration, whose side chains were predicted to predominantly adopt the gg conformation,[ 17 , 18 ] were anticipated to be better inhibitors of the respective glycosidases than their respective isomers 4 and 8 with the gt conformation of their side chains.
The synthesis of compounds 2–4 commenced with known[ 22 , 23 ] intermediate 10 (Scheme 2). Selective tritylation of O6, followed by protection of O4 with the p‐methoxybenzyl (PMB) group provided the fully protected sugar 11 in 98 % yield. Thioglycoside 11 was hydrolyzed to hemiacetal 12 in 90 % yield using N‐iodosuccinimide (NIS) in wet acetone. Reduction of 12 with lithium aluminum hydride afforded diol 13 in 89 % yield. Swern oxidation of 13, followed by double reductive amination [24] afforded iminosugar 14 in 62 % yield. The basic amine in 14 was protected with a benzyl group to form intermediate 15, and then selective cleavage of the trityl ether with a 1 : 1 mixture of acetic acid and ethanol at 80 °C provided alcohol 16 in 60 % yield over two steps. Parikh‐Doering oxidation [25] of 16 and immediate treatment with methyl magnesium chloride in THF at 0 °C furnished the C6‐methyl‐substituted products 17 and 18 as an inseparable 2 : 1 mixture in 80 % yield over two steps. Cleavage of the PMB ethers in 17 and 18 was achieved with trifluoroacetic acid (TFA) in dichloromethane to give diol 19 in 51 % isolated yield and isomer 20 in 26 % yield. To determine the configuration at C6, compounds 19 and 20 were converted to rigid bicyclic derivatives 21 in 72 % yield and 22 in 78 % yield, respectively, by installation of the 4,6‐O‐di‐tert‐butylsilylene acetal group. Bicyclic compound 21 displayed a 3 J H5,H6 of 5.0 Hz and nuclear Overhauser effect (NOE) correlations of H4 with the methyl group, a tert‐butyl group and H2, indicative of an axial methyl group and the L‐glycero‐D‐gluco configuration. Compound 22 had a 3 J H5,H6 of 9.5 Hz and NOE correlations of H4 with H6, H2 and a tert‐butyl group, indicating an equatorial methyl group and the D‐glycero‐D‐gluco configuration. Diols 19 and 20 were subjected to hydrogenolysis to provide iminosugars 3 and 4 as hydrochloride (HCl) salts in quantitative yields. The L‐glycero‐D‐gluco isomer 3 had an NOE correlation between the methyl group and H5 and a 3 J H5,H6 of 2.3 Hz consistent with the predicted predominant gg conformation, while the D‐glycero‐D‐gluco isomer 4 had an NOE correlation between the methyl group and H4 and a 3 J H5,H6 of 3.3 Hz indicative of the anticipated predominant gt conformation. The HCl salts of 3 and 4 were also converted to the free bases, whose side chain conformations were determined in the same manner to be predominantly gg and gt, respectively (Figure S1). In parallel, intermediate 15 was treated with TFA to form diol 23, which was debenzylated to afford 1‐deoxynojirimycin 2 [26] in 72 % yield.
Scheme 2.

Synthesis of gluco iminosugars 2–4. Bn, benzyl; Tol, p‐tolyl; Trt, triphenylmethyl; DMAP, 4‐(dimethylamino)pyridine; PMB, p‐methoxybenzyl; TBAI, tetrabutylammonium iodide; DMF, N,N‐dimethylformamide; NIS, N‐iodosuccinimide; DMSO, dimethyl sulfoxide; MS, molecular sieves; DIPEA, N,N‐diisopropylethylamine; TFA, trifluoroacetic acid; Tf, trifluoromethanesulfonyl.
The synthesis and establishment of relative configuration and side chain conformation of manno iminosugars 6–8 were accomplished analogously, starting with known [27] diol 24 (Scheme 3). Alcohol 30 was oxidized and then reacted with methyl magnesium chloride in THF at 0 °C to give the separable C6‐methyl‐substituted products 31 and 32. [28] Compound 31 was used to generate iminosugar 7 as an HCl salt via deprotection of the PMB group and hydrogenolysis in 86 % yield over two steps, while compound 32 was converted to the HCl salt of iminosugar 8 in 72 % yield. As expected, the L‐glycero‐D‐manno iminosugar 7 showed an NOE correlation between the methyl group and H5 and a 3 J H5,H6 of 3.1 Hz, consistent with the predominant gg conformation, while the D‐glycero‐D‐manno isomer 8 had an NOE correlation between the methyl group and H4 and a 3 J H5,H6 of 3.5 Hz, indicative of the gt side chain conformation. The HCl salts of 7 and 8 were also converted to their conjugate bases, whose side chain conformations were assigned analogously as gg and gt, respectively (Figure S2). Finally, intermediate 29 was used to prepare 1‐deoxymannojirimycin 6 [29] in 62 % yield.
Scheme 3.

Synthesis of manno iminosugars 6–8. NBS, N‐bromosuccinimide.
We evaluated the inhibition potency of these six compounds against the selected glycosidases by determining inhibition constants (K i) of gluco iminosugars 2–4 for TmGH1 and SsGH1 (Table 1) and of manno compounds 6–8 for BtMan2A (Table 2). Gratifyingly, a consistent pattern emerged according to which iminosugars 3 and 7, with the predominant gg side chain conformation, are between six‐ and ten‐fold better inhibitors than the isosteric 4 and 8, respectively, with the enforced gt conformation validating the design hypotheses that side chain conformation can be used to influence activity and that the gg conformation is optimal. We attribute the weaker activity of the gluco isomer 3 with the gg conformation relative to the unrestricted 1‐deoxynojirimycin 2 against the two glucosidase enzymes (Table 1) to an unfavorable steric or hydrophobic interaction of the conformation‐controlling methyl group with the active site. In contrast, the manno isomer 7 with the gg conformation is a significantly better inhibitor of the mannosidase tested (Table 2) than 1‐deoxymannojirimycin 6 itself, reflecting a synergistic combination of the optimal side chain conformation with a favorable hydrophobic or other interaction of the added methyl group with the active site.
Table 1.
Inhibition constants (K i) of inhibitors 2–4 for TmGH1 and SsGH1.
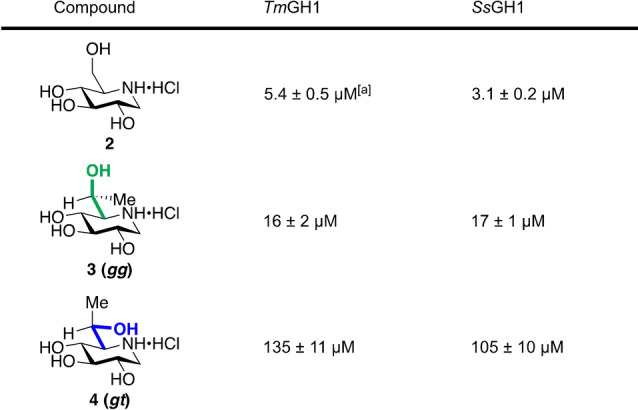
[a] Reported K i=3.8 μM with 2,4‐dinitrophenyl β‐D‐glucopyranoside as a substrate. [15]
Table 2.
Inhibition constants (K i) of inhibitors 6–8 for BtMan2A.
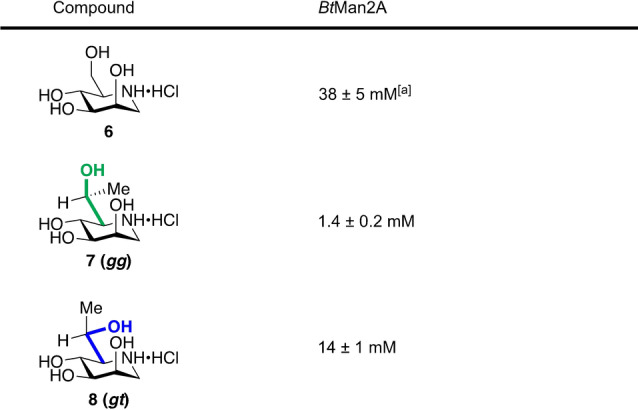
[a] Reported K i=33 mM with 2,4‐dinitrophenyl β‐D‐mannopyranoside as a substrate. [21]
The weaker potency of the isomers 4 and 8, whose side chains are restricted to the gt conformation, likely arises from the greater penalty that must be paid for the enzymes to force their side chains to adopt their higher energy gg conformations, and/or from a reduced contribution to binding by their ground state gt conformations. The manno isomer 8 is nevertheless a stronger inhibitor than 1‐deoxymannojirimycin 6, indicating that the methyl group in 8 must afford a favorable interaction, possibly a CH‐π interaction with Trp656, with the glycosidase active site that more than offsets the effect of the less favorable nature of the gt side chain conformation in this compound. Overall, these results are fully consistent with i) the design hypotheses, with the imposed gg conformation affording better inhibitors than the gt conformation in each case studied, and ii) the well‐appreciated and wide‐ranging influence of methyl groups in medicinal chemistry that is maximized when conformational restriction and hydrophobic effects are in synergy.[ 30 , 31 , 32 ]
In summary, we have synthesized and evaluated gluco iminosugars 2–4 as inhibitors of TmGH1 and SsGH1 and manno iminosugars 6–8 as inhibitors of BtMan2A. Inhibition studies with these three glycosidases consistently showed that compounds 3 and 7 with the gg side chain conformation are better inhibitors than the isosteric compounds 4 and 8, respectively, with the gt side chain conformation. Moreover, preorganization of the side chain into the gg conformation by an ideally located methyl group gives the manno‐configured iminosugar 7 27‐fold greater potency over 1‐deoxymannojirimycin 6 itself, underlining the benefits to be gained from the synergy of conformational restriction and hydrophobic interactions, consistent with well‐established principles in medicinal chemistry.[ 30 , 31 , 32 ] We anticipate this concept of conformational preorganization will be beneficial for the design of future inhibitors.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgments
Financial support was provided by the NIH (GM62160). We thank Bhargavi M. Boruah and Chin Huang (University of Georgia) for helpful discussions on the glycosidase inhibition and Prof. Breeanna Urbanowicz's laboratory (University of Georgia) for sharing their microplate reader. Jarvis Hill (University of Georgia) is thanked for critical reading of the manuscript. Graphics for X‐ray crystal structures were generated using UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from the NIH (P41‐GM103311).
Tseng P.-S., Ande C., Moremen K. W., Crich D., Angew. Chem. Int. Ed. 2023, 62, e202217809; Angew. Chem. 2023, 135, e202217809.
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1. Asano N., Glycobiology 2003, 13, 93R–104R. [DOI] [PubMed] [Google Scholar]
- 2. Tedaldi L., Wagner G. K., MedChemComm 2014, 5, 1106–1125. [Google Scholar]
- 3. Gloster T. M., Davies G. J., Org. Biomol. Chem. 2010, 8, 305–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schramm V. L., Acc. Chem. Res. 2015, 48, 1032–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gloster T. M., Meloncelli P., Stick R. V., Zechel D., Vasella A., Davies G. J., J. Am. Chem. Soc. 2007, 129, 2345–2354. [DOI] [PubMed] [Google Scholar]
- 6. González-Cuesta M., Sidhu P., Ashmus R. A., Males A., Proceviat C., Madden Z., Rogalski J. C., Busmann J. A., Foster L. J., García Fernández J. M., Davies G. J., Ortiz Mellet C., Vocadlo D. J., J. Am. Chem. Soc. 2022, 144, 832–844. [DOI] [PubMed] [Google Scholar]
- 7.The three staggered conformations of the hydroxymethyl group in sugars are described as gauche,gauche (gg), gauche,trans (gt) and trans,gauche (tg) based on the relationship of the C6−O6 bond to the C5−O5 and C5−C4 bonds, respectively.
- 8. Jensen H. H., Nordstrøm L. U., Bols M., J. Am. Chem. Soc. 2004, 126, 9205–9213. [DOI] [PubMed] [Google Scholar]
- 9. Moumé-Pymbock M., Furukawa T., Mondal S., Crich D., J. Am. Chem. Soc. 2013, 135, 14249–14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kancharla P. K., Crich D., J. Am. Chem. Soc. 2013, 135, 18999–19007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bock K., Duus J. Ø., J. Carbohydr. Chem. 1994, 13, 513–543. [Google Scholar]
- 12. Amarasekara H., Dharuman S., Kato T., Crich D., J. Org. Chem. 2018, 83, 881–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Quirke J. C. K., Crich D., J. Am. Chem. Soc. 2020, 142, 16965–16973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Quirke J. C. K., Crich D., ACS Catal. 2021, 11, 5069–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zechel D. L., Boraston A. B., Gloster T., Boraston C. M., Macdonald J. M., Tilbrook D. M. G., Stick R. V., Davies G. J., J. Am. Chem. Soc. 2003, 125, 14313–14323. [DOI] [PubMed] [Google Scholar]
- 16. Gloster T. M., Madsen R., Davies G. J., ChemBioChem 2006, 7, 738–742. [DOI] [PubMed] [Google Scholar]
- 17. Pirrone M. G., Gysin M., Haldimann K., Hobbie S. N., Vasella A., Crich D., J. Org. Chem. 2020, 85, 16043–16059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Upadhyaya K., Bagul R. S., Crich D., J. Org. Chem. 2021, 86, 12199–12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gloster T. M., Roberts S., Ducros V. M., Perugino G., Rossi M., Hoos R., Moracci M., Vasella A., Davies G. J., Biochemistry 2004, 43, 6101–6109. [DOI] [PubMed] [Google Scholar]
- 20. Gloster T. M., Roberts S., Perugino G., Rossi M., Moracci M., Panday N., Terinek M., Vasella A., Davies G. J., Biochemistry 2006, 45, 11879–11884. [DOI] [PubMed] [Google Scholar]
- 21. Tailford L. N., Offen W. A., Smith N. L., Dumon C., Morland C., Gratien J., Heck M.-P., Stick R. V., Blériot Y., Vasella A., Gilbert H. J., Davies G. J., Nat. Chem. Biol. 2008, 4, 306–312. [DOI] [PubMed] [Google Scholar]
- 22. Zou X., Qin C., Pereira C. L., Tian G., Hu J., Seeberger P. H., Yin J., Chem. Eur. J. 2018, 24, 2868–2872. [DOI] [PubMed] [Google Scholar]
- 23. Senf D., Ruprecht C., de Kruijff G. H. M., Simonetti S. O., Schuhmacher F., Seeberger P. H., Pfrengle F., Chem. Eur. J. 2017, 23, 3197–3205. [DOI] [PubMed] [Google Scholar]
- 24. Wennekes T., van den Berg R. J. B. H. N., Donker W., van der Marel G. A., Strijland A., Aerts J. M. F. G., Overkleeft H. S., J. Org. Chem. 2007, 72, 1088–1097. [DOI] [PubMed] [Google Scholar]
- 25. Parikh J. R., Doering W. v. E., J. Am. Chem. Soc. 1967, 89, 5505–5507. [Google Scholar]
- 26. De Angelis M., Sappino C., Mandic E., D'Alessio M., De Dominicis M. G., Sannino S., Primitivo L., Mencarelli P., Ricelli A., Righi G., Tetrahedron 2021, 79, 131837. [Google Scholar]
- 27. Walvoort M. T. C., de Witte W., van Dijk J., Dinkelaar J., Lodder G., Overkleeft H. S., Codée J. D. C., van der Marel G. A., Org. Lett. 2011, 13, 4360–4363. [DOI] [PubMed] [Google Scholar]
- 28.Intriguingly but beyond the scope of this paper, the selectivity for the Grignard reaction in the manno series (31 and 32, L-glycero:D-glycero=1 : 3) was reproducibly opposite to that for the gluco series (17 and 18, L-glycero:D-glycero=2 : 1).
- 29. Stauffert F., Lepage M., Pichon M., Hazelard D., Bodlenner A., Compain P., Synthesis 2016, 48, 1177–1180. [Google Scholar]
- 30. Barreiro E. J., Kümmerle A. E., Fraga C. A. M., Chem. Rev. 2011, 111, 5215–5246. [DOI] [PubMed] [Google Scholar]
- 31. Leung C. S., Leung S. S. F., Tirado-Rives J., Jorgensen W. L., J. Med. Chem. 2012, 55, 4489–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.For an important example of the importance of a methyl group (threonine vs. serine) in the structure and properties of glycoproteins, see: Corzana F., Busto J. H., Jiménez-Osés G., García de Luis M., Asensio J. L., Jiménez-Barbero J., Peregrina J. M., Avenoza A., J. Am. Chem. Soc. 2007, 129, 9458–9467. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.