

Abstract
The intricacies of the 3D hierarchical organization of the genome have been approached by many creative modeling studies. The specific model/simulation technique combination defines the system and phenomena that can be investigated. We present the latest modeling developments and studies of the genome, involving models ranging from nucleosome systems and small polynucleosme arrays to chromatin fibers in the kb-range, chromosomes and whole genomes, while emphasizing gene folding from first principles. Clever combinations allow the exploration of many interesting phenomena involved in gene regulation, such as nucleosome structure and dynamics, nucleosome-nucleosome stacking, polynucleosome array folding, protein regulation of chromatin architecture, mechanisms of gene folding, loop formation, compartmentalization, and structural transitions at the chromosome and genome levels. Gene level modeling with full details on nucleosome positions, epigenetic factors, and protein binding, in particular, can in principle be scaled up to model chromosomes and cells to study fundamental biological regulation.
Keywords: Genome organization, Chromatin folding, Epigenetic regulation, Gene regulation, Gene structure
1. Introduction
How our genome is structured and regulates fundamental biological processes such as transcription, replication, and repair is central for understanding and addressing human disease. While genome sequencing has revolutionized how we perceive and can study human ancestry and disease, the study of genome organization has emphasized how profoundly epigenetic processes – structural transformations of the genome – impact gene regulation. Indeed, the activation and repression of specific genes – at precise stages of the cell cycle, and in response to internal and external factors – dictates how and when basic life processes are performed.
While the problem of deciphering this epigenetic regulation of the genome may appear daunting, as so many levels and factors are involved, structural biologists and genomicists have developed powerful interrogatory techniques, both experimental and computational, to address these fascinating aspects of genome organization.
In this opinion article, we focus on computational studies that aim to scale chromatin systems from fibers to genes, the simulation approaches appropriate for these systems, and the biological applications, with insightful examples.
Gene level modeling describing how chromatin elements are defined with tailored input parameters can capture large-scale phenomena from basic physical principles, and can in principle be scaled up to model chromosomes and whole genomes to study the mechanisms that regulate gene expression associated with human disease.
2. Current View of Genome Global Organization
Genomic DNA is hierarchically organized (Figure 1) to efficiently pack 6 billion base pairs inside the diploid cell nucleus and direct biological regulation of gene expression [1, 2].
Figure 1: Hierarchical genome folding.
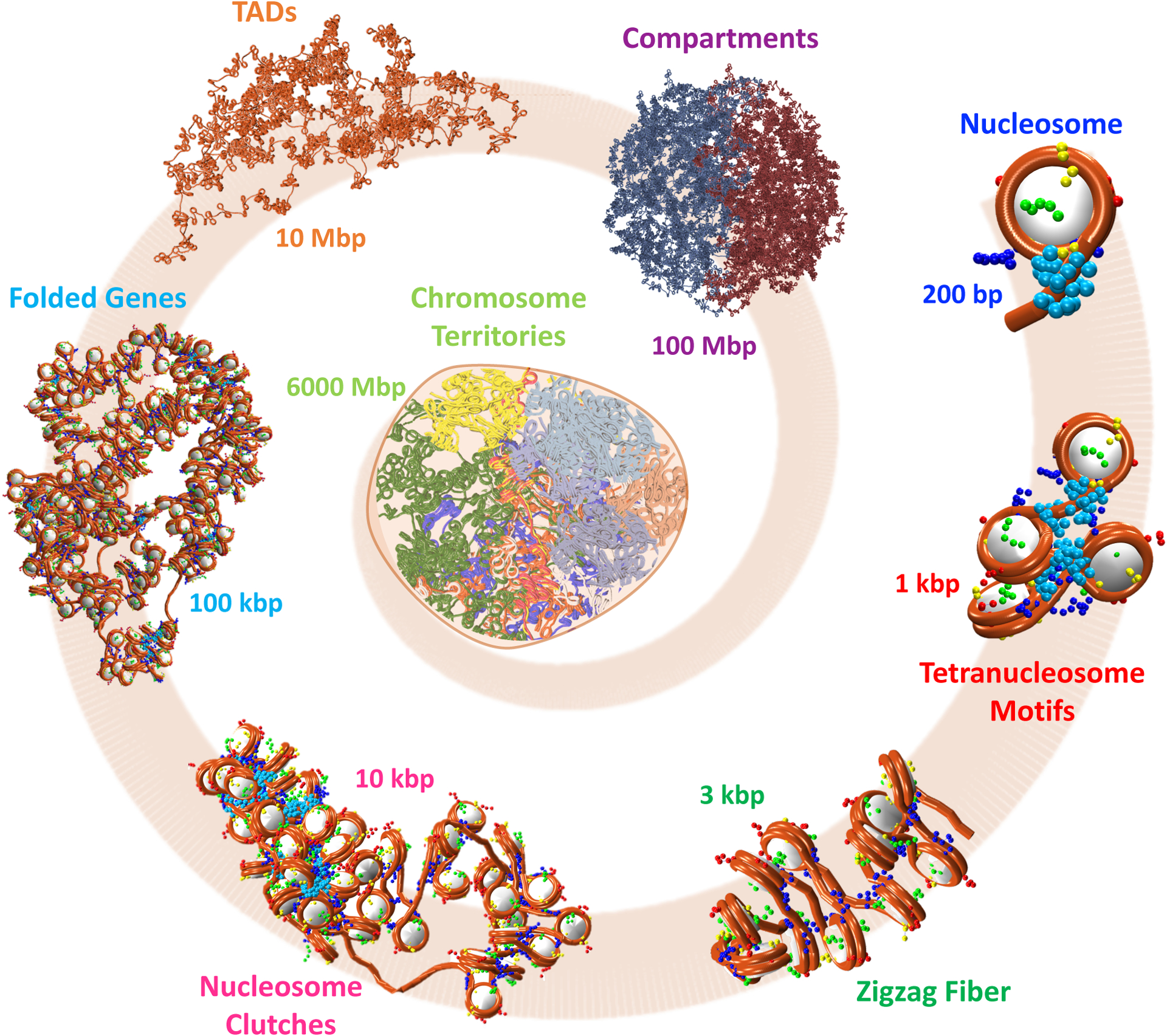
~147 bp of DNA wrap around the histone octamer to form the nucleosome, the chromatin basic building block. At the few kb level, nucleosomes fold into tetranucleosme units. The zigzag topology appears dominant in small kb arrays. Further folding through hierarchical looping creates nucleosome clutches of different sizes and levels of compaction. Genes fold while maintaining the folding features of the lower scales. Compartmentalization at the few Mb produces TADs, which further fold to form segregated compartments. Finally, chromosomes separate in territories in the cell nucleus.
Hi-C contact maps have revealed that interactions between pairs of loci across the whole eukaryotic genome dictate genome segregation into open/active and closed/inactive chromatin to form distinct Mb compartments [3]. While contacts within compartments are enriched, contacts between compartments are rare. The positions of these compartments are also cell-dependent, and dictate a large range of biological phenomena related to development, differentiation, and disease progression.
Higher resolution Hi-C maps show that at hundreds of kb scale, there is an additional level of compartmentalization involving the formation of self-interacting Topologically Associating Domains (TADs) [4, 5, 6]. Networks of loops are supported by the protein pair of cohesin and CCCTC-binding factor (CTCF) [7, 8], although cohesin is not required for the formation or maintenance of TADs at the single cell level [9]. TADs have been identified in many species across cell types, indicating that they are likely a conserved feature of genome organization. The importance of these domains in the regulation of genome expression, however, remains unclear. Overall, compartmentalization and loop formation appear to be two independent but related principle mechanisms of genome organization [10, 8].
At the sub kb level, super resolution microscopy and modeling at the nucleosome scale reveal that nucleosomes are organized in clusters or “clutches” [11, 12, 13]. Their size and compaction relates to the cell differentiation state [11, 13], and depends on epigenetic modifications [14, 12] [15]*, linker histone (LH) binding [15]*, and cohesin binding [12].
Single-molecule force spectroscopy revealed that tetranucleosomes units appear as stable secondary structure motifs in the hierarchical organization of chromatin fibers in the yeast genome [16]. Micro-C experiments combined with molecular dynamics (MD) simulations [17] show that these units organize in two basic folding motifs that favor next neighbor (i/i+2) internucleosome contacts: the α-tetrahedron and β-rhombus. While the α-tetrahedron appears between the gene start and stop sites and is related to closed chromatin, β-rhombus is associated with promoters and open chromatin. In contrast, Radiation-Induced Correlated Cleavage with sequencing (RICC-seq) experiments at the sub-kb level of the human genome reveled that i/i+2 contacts, typical of two-start zigzag fibers with stacked alternating nucleosomes, are found mostly on closed chromatin, whereas non-compact solenoid structures are found in open chromatin [18], indicating differential folding for active and repressed chromatin. Modeling and crosslinking experiments have emphasized the prevalence of zigzag architecture [19, 20].
In vitro studies have recently revealed that at high salt concentrations or presence of proteins that bind to the nucleosome, nucleosome arrays organize through liquid-liquid phase separation to form liquid droplets [21, 22]. This remains somewhat controversial as chromatin behaves as a solid at the mesoscale [23]. Similarly, in vivo experiments have provided evidence for phase separation of heterochromatin driven by repressive proteins like the heterochromatin protein 1 (HP1) [24, 25], and of actively transcribed chromatin driven by RNA Polymerase II [26] and transcription factors [27]. Thus, the compartmentalization of the genome into functionally chromatin compartments is emerging as a key regulatory feature.
While clearly genome organization is variable and heterogeneous when analyzed at the single-cell level [28], its hierarchical structure involves several distinct layers.
As we discuss below, each of these layers has been investigated by many computational studies that use models of different resolution and sampling technique to further understand genome organization and regulation. Following our overview of molecular models and techniques in the next section, we will describe associated applications in three subsequent sections organized by genome level, followed by gene folding, and thoughts regarding future directions and opportunities.
3. Methods Across the Scales
Several techniques and various multiscaling approaches are suitable for simulating the different layers of genome organization, including nucleosomes, chromatin fibers, and chromosomes. This combination of model resolution and simulation technique defines the type of systems and which phenomena can be studied (Figure 2).
Figure 2: Combination of model resolution and simulation technique for chromatin systems, each with associated system sizes and phenomena.
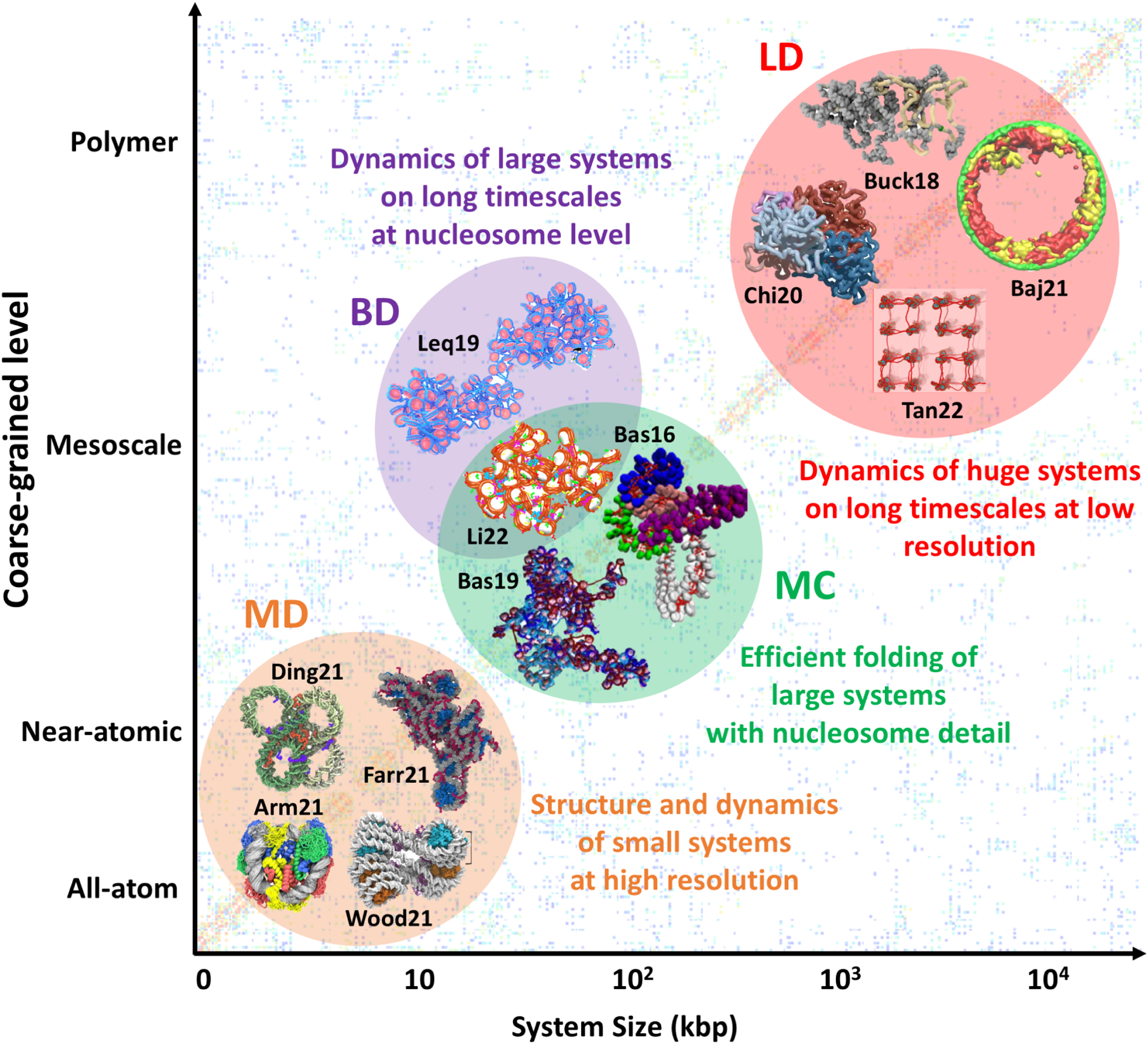
Large system sizes can be reached by reducing the number of particles (coarse-graining) with polymer models and utilizing simulation techniques like Langevin Dynamics (LD). Intermediate systems are usually studied by mesoscale models with nucleosome resolution combined with Brownian Dynamics (BD) or Monte Carlo (MC) sampling. Small systems can be studied at higher resolution with all-atom or near-atomic models by Molecular Dynamics (MD). Molecular modeling images were adapted with permission from: [42] (Ding21), [30] (Arm21), [50] (Farr21), [44] (Tan22), [59] (Baj21), and [58] (Chi20) under the Creative Commons Attribution License http://creativecommons.org/licenses/by/4.0/; [34] (Wood21) Copyright 2021 Elsevier; [146]** (Bas19); [74] (Bas16) Copyright 2016 American Chemical Society; [63]* (Li22); [61] (Leq19) under AIP Publishing.
Molecular Dynamics (MD) is a widely used technique that allows high spatial and temporal resolutions that cannot be reached by experimental methods. Its main advantage is the intense development of robust all-atom force fields and software for general-purpose MD simulations. Because it is usually combined with atomic-resolution models, it requires significant computational resources. As recently reviewed by Huertas and Cojocaru [29], all-atom MD simulations of chromatin systems are generally limited to one (~500,000 atoms) or two (~700,000 atoms) nucleosomes. The longest simulation time achieved as of this writing for a single nucleosome system is 15 μs [30]* (Arm21 in Figure 2), far from the 10 ns obtained by Bishop in 2005 for the first all-atom simulation of a nucleosome [31]. Some exceptions regarding system size are the works by Jung et al. on the GATA4 gene containing 427 nucleosomes and ~1 billion atoms [32]; Izadi et al. on a chromatin fiber of 40 nucleosomes and ~1.16 million atoms [33]; and Woods et al. on an 8-nucleosome array containing ~2.75 million atoms [34]* (Wood21 in Figure 2). However, these simulations were restricted to only 1, 0.1, and 500 ns, respectively. It remains a challenge to sample configuration space adequately for chromatin systems via all-atom MD simulations.
To overcome this issue and close the gap between the experimental and modeling time scale, enhanced sampling techniques have been applied to nucleosome and polynucleosome systems [29], including Replica Exchange, Metadynamics, Steered MD, Umbrella Sampling, Adaptively Biased MD, and Adiabatic Bias MD (see separate literature on enhanced sampling [35, 36]).
The alternative coarse-graining (CG) approaches allow longer timescales and comparison with various experimental data. Popular DNA CG models include the de Pablo three site model 3SPN (on phosphate, sugar, and nucleotide) [37] and subsequent refinements, also combined with the Wang and Takada protein CG model AICG2+ of 1 bead per residue [38], to study transcription factor binding to nucleosomes (~3,000 particles) for 10 μs [39]*; or with the Wolynes and Papoian protein CG model AWSEM of 3 beads per residue [40], to study chromatosome dynamics (~3,000 particles) for 3 μs [41]**; and with contact potentials for the amino acids derived from a tetranucleosome crystal structure, to fold tetranucleosme arrays (8,058 particles) [42]** (Ding21 in Figure 2). Similarly, Tan et al. combined it with AICG2+ and the HPS force field for discorded tails [43] to simulate a 1,024-nucleosome system (~2 million particles) [44]* (Tan22 in Figure 2).
The Pantano’s SIRAH CG force field was recently extended for protein-DNA complexes to study DNA dynamics in a tetranucleosome array (~240,000) for 5 μs [45]. Lyubartsev, Nordenskiöld, and colleagues developed a residue-based CG model for the nucleosome core particle derived from multiple all-atom MD simulations that consider the ionic environment [46].
The rigid base pair approximation by Olson and Zhurkin treats DNA’s flexibility as an harmonic oscillator with parameters extracted from experiments, accounting for sequence dependency [47]. Farr et al. recently combined this model with a residue-resolution CG model for proteins based on [48] and [49] to create a chemically-specific CG model [50]** (Farr21 in Figure 2). Similar to the multiscale used in [51] to connect all-atom to mesoscale level of chromatin, Farr et al. used two levels of coarse graining: the nucleosome all-atom model is CG into the chemically-specific model, which is then refined into a chromatin minimal model.
Eliminating the explicit representation of solvent particles in Langevin Dynamics (LD), the solvent is treated as a continuous medium surrounding the solute, leads to simulation time scales in the range of seconds. LD is generally used for simulations of polymer models in which the chromatin fiber is treated as a chain of beads connected by springs. As recently reviewed [52, 53], many polymer models to investigate chromatin structure exist. These include, the Nicodemi “strings and binders switch” (SBS) model, where chromatin is a self-avoiding polymer chain interacting with diffusing beads [54]; the Vaillant block-copolymer model, where chromatin is a self-interacting block-copolymer [55]; the Marenduzzo’s HiP-HoP [56]; and Mirny’s loop extrusion [57] models in which a loop extruding factor anchored to the chromatin fiber extrudes a progressively growing loop until barriers along the fiber are reached.
With these models, large genomic regions can be studied. For example, the HiP-HoP and SBS models were recently used with LD to study Mbp and kbp regions equivalent to 9,000 and 2,640 particles, respectively [56, 58] (Chi20 in Figure 2). Bajpai et al. used a simple polymer model in which each bead represents three nucleosomes to simulate the phase separation of a 22.4 Mbp region (37,333 particles, comparable to the X chromosome) in the nucleus (Baj21 in Figure 2) [59].
The appropriate version of LD for systems with large solvent interactions, Brownian Dynamics (BD), introduces generalized frictional interactions among the particles. Because the calculation of the frictional interactions among particles is computationally expensive, the system size is currently limited to a few thousand of particles. The group of the late Jörg Langowski first simulated 25-nucleosome arrays by BD where chromatin was treated as an array of cylindrical segments and solid spheres attached to them [60]. Recently, the de Pablo group developed the 1CPN chromatin model for BD to simulate up to 250-nucleosome arrays over 50 μs (Leq19 in Figure 2) [61]. The model treats nucleosomes with implicit tails as cylinders, linker DNA as a twistable worm-like chain, and LH as flexible beads based on the Schlick LH model [62].
BD was recently developed by the Schlick group (Li22 in Figure 2) [63]* following early work by Beard and Schlick [64] to simulate fibers with a mesoscale model of chromatin: nucleosome cores are treated as cylinders with 300 point charges distributed on its irregular surface [65], non-uniform linker DNAs are described by 1 bead per 3 nm [66], flexible histone tails are explicitly incorporated [67, 51], and flexible LHs can also be considered [62, 68]. The innovation in the new BD scheme is exploiting the computational power and parallelization of CUDA for the hydrodynamics interactions, making possible simulations of systems with up to 50 nucleosomes (~4,000 particles) [63]*.
An alternative approach to these dynamics simulation techniques is equilibrium sampling by Monte Carlo (MC), generally very efficient for surveying conformational landscapes. Early MC techniques on wormlike chains by Vologodskii and coworkers showed their efficiency for sampling supercoiled and knotted DNA [69, 70]. For chromatin, the Schlick group’s mesoscale model uses MC sampling similarly to sample kb-range fibers [71, 72]. Tailored translational and rotational moves for the nucleosome cores and linker DNA, translational moves for LH beads, regrowth moves for the histone tails, and pivot moves for short fiber segments are combined for an efficient sampling of chromatin fibers [73, 67, 62]. The largest system reached by this approach is the GATA4 gene (Bas16 in Figure 2) [74], containing 427 nucleosomes (~34,000 particles). The van Noort’s chromatin model with rigid body histone core and rigid base pair DNA model is also sampled with MC [75, 76]. During this simulation, in each MC step, a sequential replacement and evaluation of every base pair in the DNA tether is performed, and MC steps for wrapping and unwrapping of DNA are also included. Similarly, the Zhurkin and Norouzi chromatin model, which assumes fixed nucleosomes that can unwrap their DNA treated as part of a dynamic linker DNA, is also sampled with MC simulations [77, 78]. Finally, MC sampling is used in combination with Replica Exchange to simulate fibers with the Wedemann CG chromatin model that treats nucleosomes as spherocylindrical units connected by cylindrical segments for linker DNA, and considers LH implicitly [79]. Recently, this approach was used to study the organization of active and inactive chromatin by modeling ~250-nucleosome fibers [80].
Clearly, computer simulations offer an unmatched level of resolution in addition to experiments to study chromatin folding. To investigate the structure and dynamics of chromatin fibers at the Mb and chromosome scale, the most common approach is to employ coarse-grained polymer models with LD. The structure and dynamics of nucleosome arrays in kb-range lengths has been approached with coarse-grained models at the mesoscale level in combination with MC or BD simulations. Nucleosome all-atom MD simulations can offer more in-depth details at the atomistic level, although system size is significantly restricted. Thus, each genomic scale can provide insights at different resolutions, and the details learned at a certain scale can be used to build models to study the next scale. Thus, details at the nucleosome scale and all-atom resolution can be incorporated into mesoscale models for chromatin fibers, and features of chromatin fibers can be extrapolated to construct polymer models and study Mb systems.
4. Nucleosome Modeling: Revealing Structures and Dynamics at different Resolutions
The first nucleosome core particle (NCP) X-ray crystal structure at atomic resolution (2.8 Å) in 1997 [81] has been followed by many high-resolution structures that further revealed details on the mobile histone tails, variant core histones, binding of auxiliary proteins, ions, and solvent molecules [82]. These experimental anchors provided a platform for modeling NCP conformations and dynamics, including interactions among its elements, like DNA, histone tails, and linker histone (LH) (Figure 3) [29, 83].
Figure 3: Examples of recent modeling applications, from nucleosomes to chromosomes, and gene loci.
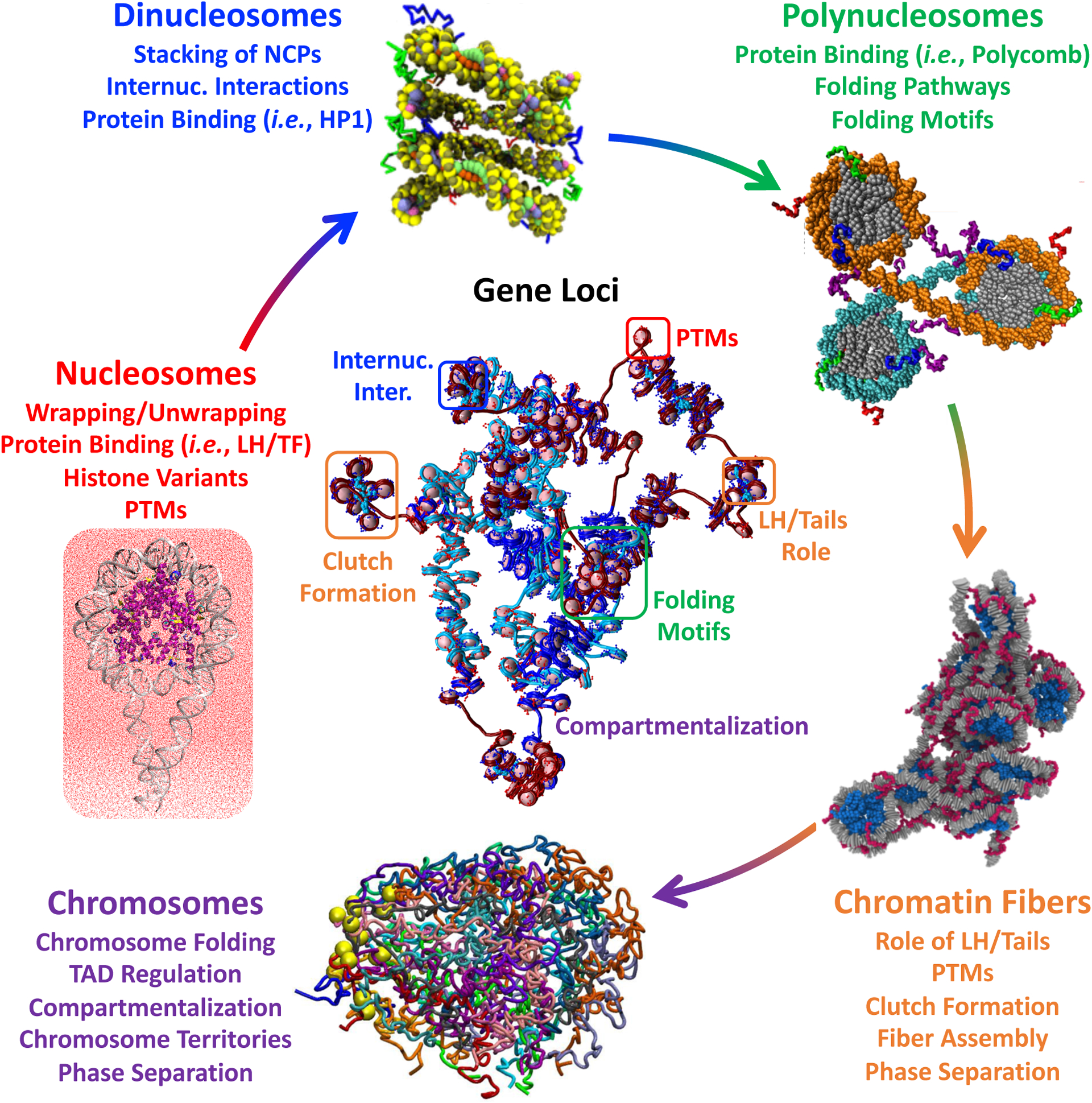
Nucleosome simulations provide insights into dynamical phenomena like wrapping and unwrapping, sliding, and breathing, the effect of LH and TF binding, and role of post translational modifications and histone variants. Dinucleosome systems are used to study regulation of internucleosme interactions and binding of repressing proteins like HP1. Polynucleosome studies reveal folding motifs and pathways, and the effect of repressive proteins like polycomb. Chromatin fibers at the kb-level reveal typologies, nucleosome clutches, role of linker DNA, LH, and histone tails, and phase separation. Modeling of chromosomes provide insights into folding and TAD formation/regulation, compartmentalization, chromosome territories, and phase separation. For gene loci, the elements and phenomena studied at each genomic scale are combined to obtain a high resolution structure. Molecular modeling images were adapted with permission from: chromatin fibers [50]** and chromosomes [142]** under Creative Commons Attribution License http://creativecommons.org/licenses/by/4.0/; dinucleosome [118] https://pubs.acs.org/doi/10.1021/acscentsci.1c00085 and under Creative Commons Attribution License https://creativecommons.org/licenses/by-nc-nd/4.0/, polynucleosome [119] Copyright 2022 Elsevier, 21 and gene loci [146]**.
Early works of nucleosomes by all-atom and CG models focused on understanding unwrapping, assembly, and sliding, as well as their sequence dependence [84]. Recently, improved models and larger simulation times have been possible. For example, Brandani et al. characterized nucleosome intermediates during nucleosome assembly [85], finding a high sequence (A/T-rich signals) and salt dependency. In agreement with previous modeling studies on nucleosome sliding [86] and unwrapping [87], genomic sequence alone likely controls nucleosome positioning.
The regulation of nucleosome dynamics by histone tails, epigenetic modifications, and histone variants have been extensively studied by molecular modeling. For example, Armeev et al. recently reported the longest all-atom MD simulation (15 μs) of the NCP (Arm21 in Figure 2) [30]*, suggesting that the unwrapping of DNA ends is mediated by H3 and H2A histone tails, and coupled to the destabilization of the DNA near the dyad. Fu et al. similarly noted that H3 tail acetylation enhances DNA mobility, which could facilitate DNA repair [88], echoing previous simulations reporting that methylation and acetylation of H3 produce a more open DNA [89, 90]. DNA methylation, instead, produces more compact nucleosomes, resistant to unwrapping, as shown by Li et al. [91]*. Histone variants also modulate NCP dynamics, as shown by Peng et al. for the H2A.B variant [92], and by Pitman et al. for heterotypic nucleosomes containing both H3 variants CENP-A and H3.3 [93]*. Epigenetic modifications and histone variants likely regulate chromatin architecture and gene expression through modulation of the NCP.
LH, with variants specific to distinct species or tissues, regulates chromatin architecture and is implicated in many functions [94]. Because of its important role, many modeling studies have investigated chromatosome structure and dynamics, as recently reviewed by Öztürk et al. [83]. Early studies performed molecular docking and modeling to understand the LH interaction with DNA and its relative position to the nucleosome [83]. Later, Wade, Cojocaru, and collaborators highlighted on/off dyad binding modes [95], conformational plasticity of the nucleosome [96], and effect of LH mutations and post translational modifications on chromatosome structure [97].
Recent modeling studies have focused on the role of LH variants and the disordered N- and C-terminal domains. Woods et al. studied chromatosomes containing the globular domain of two LH variants, Xenopus laevis generic H1 and G. gallus H1.0 [98]*. While the on-dyad binding appears enthalpically favored by both variants, especially H1.0, the off-dyad was relatively more entropically stabilized. However, the large globular domain dynamics suggested that both binding states might simultaneously occur, in agreement with what has been proposed by the Wade group [99]. Wu et al. found that the Xenopus laevis generic H1 displays disorder for both N- and C-terminal domains [41], while still resulting in a compact and rigid chromatosome. In contrast, for this same variant, Sridhar et al. suggested a disorder-to-order transition of the N-terminal domain upon nucleosome binding [100]. Finally, Zhou et al. studied chromatosomes containing the globular domain of human LH variants H1.0, H1.10, and H1.4 [101]. A more open chromatosome was found for H1.10, which highly interacts with nucleosomal DNA compared to H1.4 and H1.0, which mostly interact with linker DNA.
Overall, these various modeling studies indicate that the LH variant and binding mode lead to chromatosome heterogeneity, which is context specific. Moreover, they agree with prior work by Perišić et al. demonstrating how LH variants and binding modes affect chromatin compaction, and suggesting that combinations of on and off-dyad binding result in different levels of chromatin compaction, from relatively open to condensed arrays [68]. Earlier work by the Schlick group also revealed mechanisms of condensation by LH via DNA stem formation [62, 73, 68, 102].
Transcription factor (TF) binding to nucleosomes has recently started to be studied by molecular modeling. The Cojocaru group, reported the first simulation of a nucleosome with a TF [103]. They show that DNA local flexibility mediates Oct4 binding, and that nucleosome breathing and twisting increases with the number of TF binding sites, indicating that nucleosome dynamics facilitates TF binding. Tan et al. proposed an allosteric mechanism for the simultaneous binding of the TFs Oct4 and Sox2 [39]* in which one TF modulates the nucleosome structure to promote binding of a second TF. As recently reported by Peng et al., histone tails, in particular H3 and H4, regulate the binding of regulatory proteins to the nucleosome as they occupy the same regions in the nucleosomal or linker DNA [104]. Finally, Ishida et al. found torsional stress induced by protein binding as the mechanism regulating DNA unwrapping [105]. Thus, nucleosome stability and DNA accessibility can be tuned by the binding of regulatory proteins to control gene expression.
Despite the vast number of applications of nucleosome simulations, larger systems are needed to further understand the scaffold of genome folding. As we discuss below, coarse grained models for simulations of systems containing several nucleosomes are widely used.
5. From Dinucleosomes to Polynucleosome Arrays: The Role of Internucleosme Interactions
Favorable stacking interactions between NCPs provide the basis for chromatin fiber folding. Not many studies have focused on genome organization at this level.
Experimentally, the most interesting structural insights come from high-resolution X-ray structures of LH bound dinucleosomes [106], tetranucleosmes [107, 108], and LH bound 6-nucleosome arrays [109], as well as from Cryo-EM structures of dinucleosomes [110], trinucleosomes [111, 110], and 12-nucleosome arrays [112].
Modeling of 2- to 16-nucleosome arrays have provided insights into stacking, internucleosome interactions, and protein binding, as well as folding pathways and motifs (Figure 3). Pioneering work from the Orozco and Schlick groups used a multiscale approach to study the role of internucleosome interactions in fiber folding [51]. Their all-atom MD simulations of wild type and H4-acetylated dinucleosomes revealed that H4 and H3 tails mediate most of the internucleosome interactions, and that H4 acetylation impairs internucleosme interactions due to a decrease in tail disorder. In agreement, subsequent modeling works showed that H2A and H4 form most of the NCP-NCP stacking contacts [113, 114, 115, 116], that H4K16Ac directly reduces inter-nucleosome interactions [117], and that H4 tail and its acetylation strongly contributes to the strength and shape of the dinucleosome interaction landscape [118]. As recently suggested by Lequieu et al., besides histone tails, the linker DNA in dinucleosome systems tightly regulates the interaction landscape (Leq19 in Figure 2) [61]. Different conformations emerge depending on the linker DNA length, and while some conformations are favored by short linkers, all conformations are equally possible for medium linkers. Similarly, Kenzaki et al. showed that the folding of trinucleosomes strongly depends on the linker DNA length, finding 5 distinctive configurations [119]. These results are in line with earlier works on chromatin structure showing that variations of DNA linker lengths trigger fiber polymorphism [66, 120].
As discussed above, tetranucleosomes can arrange in α-tetrahedron and β-rhombus configurations [17]. Recently, Alvarado et al. revealed the spontaneous formation of the two α and β configurations in 4- and 16-nucleosome arrays, which appear as metastable states in the free energy surface [121]*. Similarly, Ding et al. studied the folding pathway of tetranucleosome arrays (Ding21 in Figure 2) [42]**, finding many metastable configurations, in which some shapes were similar to the α and β motifs. Woods et al. emphasized their stabilization by LH in octanucleosome arrays (Wood21 in Figure 2) [34]*.
The binding of repressive proteins to polynucleosome arrays has also been investigated by coarse grained simulations. Watanabe et al. showed that HP1α binds to two sites in adjacent nucleosomes, bridging the two nucleosomes [122]. Leicher et al. showed the simultaneous binding to nonadjacent nucleosome pairs of the Polycomb repressive complex 2 [123], demonstrating how it can bridge non contiguous chromosomal segments.
Overall, molecular modeling studies on arrays of few nucleosomes have emphasized the role of histone tails and linker DNA on internucleosome stacking and interactions, energetically favored formation of tetranucleosome units, and the regulation of dinucleosome and tetranucleosome units by protein binding. Fiber modeling at the kb level and beyond is essential for providing further details on genome organization.
6. Fiber Modeling: Understanding Genome Folding with Mesoscale and Polymer Models
Understanding chromatin 3D architecture is essential to interpret the epigenetic regulation of the genome and relate genome organization to function and human disease.
Usually, fibers of up to 100 kb are modeled at the mesoscopic level with approaches that coarse grain the chromatin elements from their atomistic structures. These are first-principle models in which the simulations aim to predict genome folding with a mechanistic basis. When systems are very large (Mb), polymer models may be preferred. These models generally aim to generate configurations that reproduce experimental contact maps rather than generating folding from first-principles, although many polymer models use only a few parameters and are more mechanistically oriented.
For recent comprehensive reviews on chromatin modeling at the mesoscale and polymer level see [124, 125, 126, 127, 128, 71]. Here, we focus on recent applications of mesoscopic and polymer models (Figure 3).
The Schlick group’s mesoscale model for MC sampling of chromatin fibers has evolved as experimental data emerged [72], and applied to simulate fibers in the kb range [71]. Early applications focused on electrostatic mechanisms of folding [64], role of tails [67], and LH and divalent ion-driven compaction [73]. Later, the model helped explain that fibers fold mostly with a zigzag topology and moderate solenoid features, producing a hybrid structure [20]. These zigzag dominant chains further fold to form higher order hierarchical loops, that are LH dependent and explain interphase and metaphase folding [19, 72, 129]. Fiber heterogeneity also emerged from non-uniform linker DNA lengths, which create fluid fibers [66, 20, 120].
In recent efforts, the role of LH on chromatin architecture, binding of antibodies [130], and nucleosome clutches were investigated. As discussed above, Perišić et al. found that combinations of LH on and off-dyad binding, and LH density produce different levels of compaction, tuning chromatin architecture [68]. Sridhar et al. found that the LH disordered C-terminal domain leads to an asymmetric and dynamical nucleosome conformation, promoting chromatin structural flexibility and long-range hierarchical loops [131]. The C-terminal domain flexibility and disorder, in particular, appeared modulated by post translational modifications, which in turn affect chromatin architecture. Yusufova et al. showed that a decrease in LH density produces a chromatin structural transition from a straight/rigid to a globular/loose structure [132], which might be involved in the upregulation of gene expression during lymphoma development. As recently revealed by Portillo-Ledesma et al., such transition occurs at an LH density ~0.5, and is tightly regulated by linker DNA length, epigenetic modifications, and salt conditions [133].
In a recent study by Portillo-Ledesma et al., the formation of nucleosome clutches, or clusters, emerged in chromatin fibers with nucleosome free regions (NFRs) [15]*, in agreement with super-resolution microscopy studies [11]. Clutch size and compaction appeared regulated by LH density and acetylation levels. Such heterogeneous clusters were also found by Bajpai et al. using a mesoscale chromatin model that considers the implicit binding of non histone proteins [134].
The role of linker DNA on chromatin architecture was recently studied using the 1CPN chromatin model by the de Pablo group (Leq19 in Figure 2) [61], the rigid base pair chromatin model by van Noort [75, 76], and the Wedemann chromatin CG modeled [80]. Overall, their results showed how the free energy of chromatin assembly and folding/unfolding mechanisms strongly depend on linker DNA length, in agreement with previous findings [66, 78].
Recently, Collepardo-Guevara and coworkers’ coarse-grained model of 125 independent 12-nucleosome chromatin arrays (300 kb) suggested that nucleosome breathing favors the liquid-liquid phase separation of chromatin due to an increase in the transient nature and heterogeneity of nucleosome–nucleosome contacts (Farr21 in Figure 2) [50]**.
Several polymer models at nucleosome resolution or coarser have been used to study larger systems, such as domains, chromosomes, or whole genomes. Wiese et al. showed with a nucleosome resolution model that nucleosome spacing in yeast strongly affects domain structure and dictates chromatin interactions and domain boundaries, being the only input parameter needed to reproduce experimental contact maps [135]. The Spakowitz nucleosome-resolution polymer model was used to predict how epigenetic marks control the 3D organization, revealing that binding of HP1 to methylated regions drives segregation of heterochromatin from euchromatin [136], and that heterochromatin preferentially positions at the nuclear periphery [137].
Polymer models with coarser resolution revealed that chromatin can exist as both a fluid or gel state, depending on the level of TAD compaction [138]**; that cell-to-cell variability on chromatin structure can be explained by phase separation [139]; and that loop extrusion and phase separation mechanisms rather than compete, co-exist to fold chromatin fibers [140]**.
Interesting developments of polymer models based on Hi-C data are the MiChroM model by Di Pierro, Wolynes, Onuchic, and collaborators [141] that uses Hi-C to incorporate active and silent chromatin types and loops positions; the Orozco’s group whole-genome 3D model [142]**, with chromosomes built as chains of beads representing a genomic region corresponding to a bin from the Hi-C map; and the Sanbonmatsu and Lee 4DHiC model [143], that uses harmonic constraints to simulate cross-linking distances. MiChroM has been applied to study the effect of condensin II on genome folding [144]**, suggesting that chromosomes separate in territories with condensin, but produce mixed centromere clustering without condensin. The Orozco group’s model was used to study the effect of DNA methylation on genome organization, finding that it increases chromatin condensation in peri-centromic regions and favors heterochromatin state [142]**.
Finally, in a breakthrough development, the group of Luthey-Schulten created a whole-cell kinetic model of a minimal cell with a reduced genome of 493 genes [145]** in which one circular chromosome of 543 kbp, treated as a self-avoiding polymer, is created from cryo-electron tomograms and 3C maps.
Clearly, these studies of chromatin fibers in the kb or Mb-range by mesoscale and polymer models provide insights into the role of LH and histone tails, formation of nucleosome clutches, fiber assembly and its regulation by nucleosome breathing, chromosome folding, phase separation, and chromosome territories, among others. However, as we discuss below, further efforts are needed to describe folding of gene loci. In particular, to understand the relationship between genome aberration and disease development, it is important to build high resolution 3D structures of gene loci that incorporate all the interacting elements and capture large-scale phenomena, such as transitions and domain formation.
7. Scaling Up to Genes
To describe how chromatin elements are defined at the gene locus level, different models and input parameters need to be tailored for these important studies to capture physical interactions between genes, promoters, and enhancers, and predict the detailed structure of gene loci and associated mechanisms.
The GATA4 gene locus was first built by a mesoscale model using 3C internucleosome contact data (Bas16 in Figure 2) [74]. Five loop restraints mimicked 3C contact data. Although a uniform DNA length of 44 bp and average LH densities of 0, 0.5, and 1 LH per nucleosome were used, the model suggested a gene repression mechanism in which hierarchical looping, produced by the combination of the 5 loops, occludes the transcription start site. This motif involves elevated long-range contacts by formation of loops of loops while maintaining local zigzag geometry, and was identified by modeling combined with crosslinking experiments [19]. Later, the GATA-4 mesoscale model was used to build the first 1 billion atom model of a gene [32].
For a more accurate gene description, the specific positions of nucleosomes, LHs, and epigenetic marks are needed. The folding of the 55-kb HOXC gene locus from the ground up was based on experimental information (Bas19 in Figure 2) [146]**. To build the HOXC system, acetylation islands were modeled based on Chip-Seq data, NFRs were identified and positioned using MNase-seq data, nucleosomes were positioned using a linker DNA length distribution obtained from chemical mapping in mouse embryonic stem cells (mESC), and LHs were placed to mimic trends seen in mESC. The simulations revealed how distinct epigenetic features cooperate to form a spontaneous contact hub that bridges promoters in the gene locus and creates two separate domains, an acetylation and LH-rich domain, emphasizing how epigenetic factors are coordinated to influence chromatin architecture. Thus, elements at the nucleosome level, such as tail acetylation and LH binding, stacking of nucleosomes, and kb-range elements like zigzag and hierarchical looping motifs, as well as nucleosome clutches converge into a complex folding (Figure 4, top) that cannot easily be generalized at all genomic scales. As commented by Di Pierro, this work advanced the field “by pushing the resolution of chromatin modeling to a level that allows us to study the inner workings of individual genes” [147]. This complex folding recapitulates the life-like folding (Figure 4, bottom). The calculated contact map resembles the experimental Micro-C map from mESC [148], showing stripes (yellow regions) arising from promoter interactions, and the contacts between the acetylation and LH-rich regions. Moreover, the 3D structure is similar to that obtained from the constrained optimization of a polymer model using experimental interparticle distances from the Micro-C map (see Figure 4 caption). However, as seen from the difference contact map (Figure 4, bottom), some structural features, like other stripes and microdomains close to the diagonal produced from loop extrusion, are not captured; this implies a more open structure compared to the one corresponding to the Micro-C map. Thus, incorporation of structural proteins like CTCF and cohesin is important for capturing all experimental factors and thus gene folding.
Figure 4: Conserved structural motifs and life-like folding by in silico gene folding.
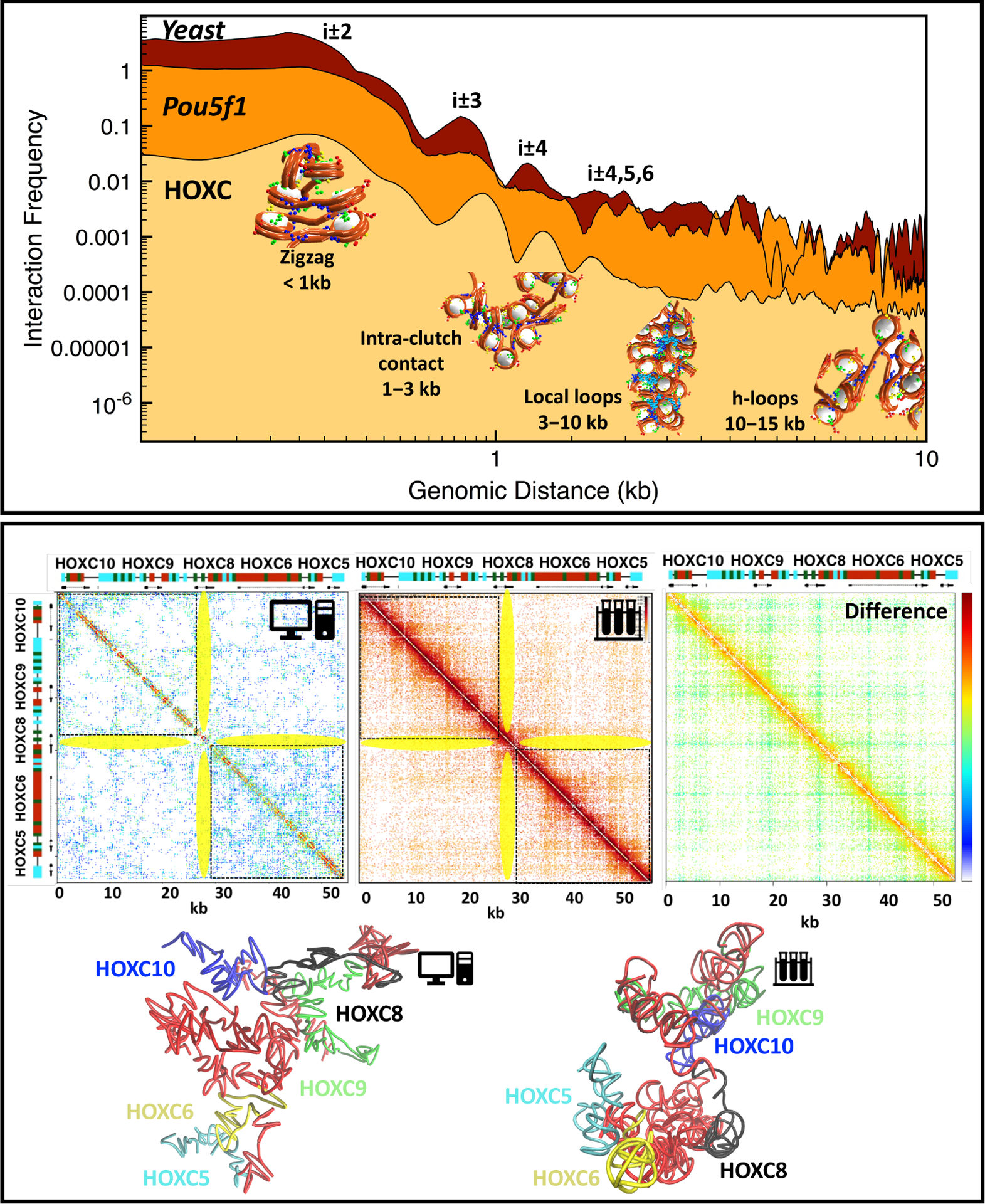
Top: Internucleosome interaction contact frequencies for HOXC [146]**, Pou5f1 [13], and yeast 30 kb genome region [149]. Structural peaks are annotated as follows: short-range contacts (<1 kb) measure next-neighbor interactions common in zigzag fibers; contacts in the 1–3 kb range arise from intra-clutch interactions; chromatin loops between neighboring clutches account for 3- to 10-kb contacts; and hierarchical loops [19] account for 10- to 15-kb contacts. Bottom, left to right: Computational contact map of the modeled HOXC from [146], experimental Micro-C map of mESC from [148], and the difference map between the computational and experimental maps. The HOXC gene locations are marked alongside the maps, with acetylation and LH-rich regions colored red and turquoise, respectively. Stripe regions are highlighted in yellow. At bottom, from left to right, are the HOXC configurations obtained in [146]** and from the Micro-C map from [148]. For the latter, we created a polymer model of 508 beads (equal to the length of the HOXC region in the Micro C map) corresponding to ~51 kb, and positioned the beads using the nucleosome and DNA beads coordinates in the initial configuration of HOXC in [146]**. Experimental Micro-C interactions from [148] were used directly to form harmonic “bonds” between connected and non connected beads, the latter 1.5 times longer, similar to Lappala et al. [143]. A cutoff was used to retain major Micro-C interactions. The22 structure was then energy minimized subject to those harmonic bond restraints.
This HOXC study served as an inspiration to model other genes. For example, the Pou5f1 gene was modeled using experimental data to study the formation and regulation of nucleosome clutches during mouse cell differentiation [13]. Similarly, MNase-seq data were used to position nucleosomes, and experimental values were used for LH density and acetylation levels, as found in mESC and neural progenitor cells. Results showed that the Pou5f1 gene folds into nucleosome clutches, with larger and more compact clutches in differentiated cells than in stem cells, recapitulating experimental results obtained at a genome-wide level [11]. Moreover, these clutch changes appeared accompanied by enhanced hierarchical looping in differentiated cells, providing a mechanistic explanation for the trends found in the experiments. Later, the differential folding in growing and quiescent or non-proliferating yeast cells was studied by modeling a 30 kb region of the yeast genome [149]. MNase-seq data were used to position nucleosomes, and Chip-seq to locate LHs and tail acetylations. Higher tail interactions of H3 and H4 with non-parental nucleosome cores were found in quiescent cells, emerging as a mechanism at the kb-range level for repressing gene expression during quiescence. Moreover, basic folding motifs, such as zigzag topology, clutches, loops, and hierarchical loops observed for HOXC, are also present in the Pou5f1 and yeast systems (Figure 4, top), underscoring common gene folding principles.
Although without nucleosome resolution, polymer models have also been used to simulate gene loci. For example, the Marenduzzo group’s polymer HiP-HoP model uses experimental ATAC-seq data for positioning transcription factors and Chip-seq data for cohesin/CTCF and epigenetic marks [56]. Application to the Pax6, globin, and SOX2 loci showed that epigenetic marks recapitulate complex genomic loci in 3D [56]. The Nicodemi SBS model was used to predict the structure of the HoxB locus containing 28 genes by defining the genomic position of CTCF binding sites and gene promoter states based on the presence of RNA polymerase [150]. Later, it was used to study the α-globin locus in embryonic stem cells (Chi20 in Figure 2) [58].
Clearly, the genome organization modeling field has advanced over the past few years. Detailed structures of gene loci are now emerging frequently. However, as shown by our comparison of the HOXC computed and experimental contact maps and structures in Figure 4, while these studies provide models for studying gene structures, chromatin interacting elements and epigenetic features must be incorporated to fully recapitulate life-like folding. For instance, the combination of DNA CpG methylation, transcription factors, CTCF/cohesin, and LH binding, as well as tail modifications, plays an important role in modulating genome architecture. In addition, inference of folding based on contact maps averaged over heterogeneous cell populations may not translate directly to single gene folding. Advances in both models and experiments and their combination will be important for moving the field forward to achieve higher resolution views of genes and genome architecture. The recent MiOS approach [151] is a promising combination of super resolution microscopy and Hi-C data with polymer and coarse grained modeling in this goal.
8. Looking Ahead
Living chromatin depends on nucleosome density and occupancy, chromatin loops of varied sizes, gene density and orientation, activating marks like histone tail acetylation and remodeling proteins like transcription factors, repressive marks like DNA methylation and protein regulators like LH and HP1, folding mechanisms, and compartmentalization.
In this perspective, we have discussed many modeling studies that aim to understand genome folding, from the nucleosome to the chromosome level, and provide insights into the mechanisms that regulate gene expression. While each study considers different chromatin elements and genomic scales, as well as employs different approaches for combining model and simulation technique to reach large system sizes and study specific biophysical phenomena, modeling chromosomes from the ground up has not yet been achieved.
How to create models that allow us to study larger systems without losing resolution is still a work in progress, although some strategies are emerging [146]** [136] [50]** [151]. It is our belief that physics-based models are necessary to provide mechanistic insights into folding and structural transitions, even though machine learning (ML) approaches may soon allow us to automatically approximate folded gene models from aggregate Hi-C maps (e.g., [141]). Given the spectacular recent success of Deep Mind’s Alpha Fold in folding the structures of millions of proteins, there is no doubt artificial intelligence and ML approaches will only increase in the near future, as we recently described in perspectives for the field of biomolecular modeling and simulation [152, 153].
Yet, as experimental techniques move toward the single cell level and nucleosome resolution, biophysical studies with new models benefiting from multidisciplinary collaborations among mathematicians, physicists, chemists, biologists, engineers, and computer scientists will drive genome research further. We thus expect many exciting developments in the near future, integrating the best of both worlds from physics-based and ML approaches, and separating aggregate cell populations from single cell structures.
Acknowledgements
This work was supported by the National Institutes of Health (NIH), National Institute of General Medical Sciences, grant MIRA R35-GM122562; National Science Foundation Rapid Award 2030377 and DMS-2151777 from the Division of Mathematical Sciences and Division of Chemistry; and Philip-Morris/Philip-Morris International, awarded to T. Schlick. We thank Jill Hoffman for preparing the Micro-C map image used in Figure 4.
Abbreviations:
- BD
Brownian Dynamics
- CG
Coarse-Graining
- Chip-seq
Chromatin immunoprecipitation sequencing
- CpG
5′-C-phosphate-G-3
- CTCF
CCCTC-binding Factor
- CUDA
Compute Unified Device Architecture
- Hi-C
High-throughput Chromosome Conformation Capture technique
- HP1
Heterochromatin Protein 1
- LD
Langevin Dynamics
- LH
Linker Histone
- MC
Monte Carlo
- MD
Molecular Dynamics
- mESC
mouse Embryonic Stem Cell
- NCP
Nucleosome Core Particle
- NFR
Nucleosome Free Region
- RICC-seq
Radiation-Induced Correlated Cleavage with sequencing
- TAD
Topologically Associating Domain
- TF
Transcription Factor
Footnotes
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Rowley MJ, Corces VG, Organizational principles of 3D genome architecture, Nat. Rev. Genet 19 (12) (2018) 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Maeshima K, Tamura S, Hansen JC, Itoh Y, Fluid-like chromatin: Toward understanding the real chromatin organization present in the cell, Curr. Opin. Cell Biol 64 (2020) 77–89. [DOI] [PubMed] [Google Scholar]
- [3].Lieberman-Aiden E, Van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J, Comprehensive mapping of long-range interactions reveals folding principles of the human genome, Science 326 (5950) (2009) 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, Van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Blüthgen N, Dekker J, Heard E, Spatial partitioning of the regulatory landscape of the X-inactivation centre, Nature 485 (7398) (2012) 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B, Topological domains in mammalian genomes identified by analysis of chromatin interactions, Nature 485 (7398) (2012) 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G, Three-dimensional folding and functional organization principles of the Drosophila genome, Cell 148 (3) (2012) 458–472. [DOI] [PubMed] [Google Scholar]
- [7].Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL, A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping, Cell 159 (7) (2014) 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rao SS, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, Huang X, Shamim MS, Shin J, Turner D, Ye Z, Omer AD, Robinson JT, Schlick T, Bernstein BE, Casellas R, Lander ES, Aiden EL, Cohesin Loss Eliminates All Loop Domains, Cell 171 (2) (2017) 305–320.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bintu B, Mateo LJ, Su J-H, Sinnott-Armstrong NA, Parker M, Kinrot S, Yamaya K, Boettiger AN, Zhuang X, Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells, Science 362 (6413) (2018) eaau1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, H Haering C, Mirny L, Spitz F, Two independent modes of chromatin organization revealed by cohesin removal., Nature 551 (7678) (2017) 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ricci MA, Manzo C, García-Parajo MF, Lakadamyali M, Cosma MP, Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo, Cell 160 (6) (2015) 1145–1158. [DOI] [PubMed] [Google Scholar]
- [12].Nozaki T, Imai R, Tanbo M, Nagashima R, Tamura S, Tani T, Joti Y, Tomita M, Hibino K, Kanemaki MT, Wendt KS, Okada Y, Nagai T, Maeshima K, Dynamic Organization of Chromatin Domains Revealed by Super-Resolution Live-Cell Imaging, Mol. Cell 67 (2) (2017) 282–293.e7. [DOI] [PubMed] [Google Scholar]
- [13].Gómez-García PA, Portillo-Ledesma S, Neguembor MV, Pesaresi M, Oweis W, Rohrlich T, Wieser S, Meshorer E, Schlick T, Cosma MP, Lakadamyali M, Mesoscale Modeling and SingleNucleosome Tracking Reveal Remodeling of Clutch Folding and Dynamics in Stem Cell Differentiation, Cell Rep 34 (2) (2021) 108614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Otterstrom J, Castells-Garcia A, Vicario C, Gomez-Garcia PA, Cosma MP, Lakadamyali M, Super-resolution microscopy reveals how histone tail acetylation affects DNA compaction within nucleosomes in vivo, Nucleic Acids Res 47 (16) (2019) 8470–8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15]*.Portillo-Ledesma S, Tsao LH, Wagley M, Lakadamyali M, Cosma MP, Schlick T, Nucleosome Clutches are Regulated by Chromatin Internal Parameters, J. Mol. Biol 433 (6) (2021) 166701, [DOI] [PMC free article] [PubMed] [Google Scholar]; The spontaneous formation of nucleosome clutches is demonstrated at the mesoscale and kb-level for chromatin fibers containing NFRs. The specific combination of NFR length, LH density, and acetylation levels defines the clutch size and compaction: shorter NFRs, higher LH density, and lower acetylation levels favor larger and compact clutches. Such nucleosome-level structures provide an additional level of epigenetic regulation.
- [16].Li W, Chen P, Yu J, Dong L, Liang D, Feng J, Yan J, Wang P-Y, Li Q, Zhang Z, Li M, Li G, FACT Remodels the Tetranucleosomal Unit of Chromatin Fibers for Gene Transcription, Mol. Cell 64 (1) (2016) 120–133. [DOI] [PubMed] [Google Scholar]
- [17].Ohno M, Ando T, Priest DG, Kumar V, Yoshida Y, Taniguchi Y, Sub-nucleosomal Genome Structure Reveals Distinct Nucleosome Folding Motifs, Cell 176 (3) (2019) 520–534.e25. [DOI] [PubMed] [Google Scholar]
- [18].Risca VI, Denny SK, Straight AF, Greenleaf WJ, Variable chromatin structure revealed by in situ spatially correlated DNA cleavage mapping, Nature 541 (7636) (2017) 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Grigoryev SA, Bascom G, Buckwalter JM, Schubert MB, Woodcock CL, Schlick T, Hierarchical Looping of Zigzag Nucleosome Chains in Metaphase Chromosomes., Proc. Natl. Acad. Sci. U.S.A 113 (5) (2016) 1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Grigoryev SA, Arya G, Correll S, Woodcock CL, Schlick T, Evidence for heteromorphic chromatin fibers from analysis of nucleosome interactions, Proc. Natl. Acad. Sci. U.S.A 106 (32) (2009) 13317–13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gibson BA, Doolittle LK, Schneider MWG, Jensen LE, Gamarra N, Henry L, Gerlich DW, Redding S, Rosen MK, Organization of Chromatin by Intrinsic and Regulated Phase Separation, Cell 179 (2) (2019) 470–484.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sanulli S, Trnka MJ, Dharmarajan V, Tibble RW, Pascal BD, Burlingame AL, Griffin PR, Gross JD, Narlikar GJ, HP1 reshapes nucleosome core to promote phase separation of heterochromatin, Nature 575 (7782) (2019) 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Strickfaden H, Tolsma TO, Sharma A, Underhill DA, Hansen JC, Hendzel MJ, Condensed Chromatin Behaves like a Solid on the Mesoscale In Vitro and in Living Cells, Cell 183 (7) (2020) 1772–1784.e13. [DOI] [PubMed] [Google Scholar]
- [24].Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, Narlikar GJ, Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin, Nature 547 (7662) (2017) 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH, Phase separation drives heterochromatin domain formation, Nature 547 (7662) (2017) 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Boehning M, Dugast-Darzacq C, Rankovic M, Hansen AS, Yu T, Marie-Nelly H, Mc-Swiggen DT, Kokic G, Dailey GM, Cramer P, Darzacq X, Zweckstetter M, RNA polymerase II clustering through carboxy-terminal domain phase separation, Nat. Struct. Mol. Biol 25 (9) (2018) 833–840. [DOI] [PubMed] [Google Scholar]
- [27].Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM, Abraham BJ, Afeyan LK, Guo YE, Rimel JK, Fant CB, Schuijers J, Lee TI, Taatjes DJ, Young RA, Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains, Cell 175 (7) (2018) 1842–1855.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Finn EH, Misteli T, Molecular basis and biological function of variability in spatial genome organization, Science 365 (6457) (2019) eaaw9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Huertas J, Cojocaru V, Breaths, twists, and turns of atomistic nucleosomes, J. Mol. Biol 433 (6) (2021) 166744. [DOI] [PubMed] [Google Scholar]
- [30]**.Armeev GA, Kniazeva AS, Komarova GA, Kirpichnikov MP, Shaytan AK, Histone dynamics mediate DNA unwrapping and sliding in nucleosomes, Nat. Commun 12 (1) (2021) 2387, [DOI] [PMC free article] [PubMed] [Google Scholar]; The longest (15 μs) all-atom MD simulation of a NCP reveals spontaneous nucleosomal DNA breathing, unwrapping, twisting, and sliding. The dynamics and plasticity of the histone octamer emerges as a key building block for these motions.
- [31].Bishop TC, Molecular dynamics simulations of a nucleosome and free DNA., J. Biomol. Struct. Dyn 22 (6) (2005) 673–686. [DOI] [PubMed] [Google Scholar]
- [32].Jung J, Nishima W, Daniels M, Bascom G, Kobayashi C, Adedoyin A, Wall M, Lappala A, Phillips D, Fischer W, Tung CS, Schlick T, Sugita Y, Sanbonmatsu KY, Scaling molecular dynamics beyond 100,000 processor cores for large-scale biophysical simulations, J. Comput. Chem 40 (21) (2019) 1919–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Izadi S, Anandakrishnan R, Onufriev AV, Implicit Solvent Model for Million-Atom Atomistic Simulations: Insights into the Organization of 30-nm Chromatin Fiber, J. Chem. Theory Comput 12 (12) (2016) 5946–5959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34]*.Woods DC, Rodríguez-Ropero F, Wereszczynski J, The Dynamic Influence of Linker Histone Saturation within the Poly-Nucleosome Array, J. Mol. Biol 433 (10) (2021) 166902, [DOI] [PMC free article] [PubMed] [Google Scholar]; A huge octa-nucleosome array with and without the globular domain of linker histoneswas simulated by all-atom MD to study fiber structures and dynamics. LH favors interactions within tetranucleosomes but not between them, indicating a role for LH in the stabilization of tetranucleosome motifs.
- [35].Bernardi RC, Melo MC, Schulten K, Enhanced sampling techniques in molecular dynamics simulations of biological systems, Biochim Biophys Acta Gen. Subj 1850 (5) (2015) 872–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yang YI, Shao Q, Zhang J, Yang L, Gao YQ, Enhanced sampling in molecular dynamics, J. Chem. Phys 151 (7) (2019) 070902. [DOI] [PubMed] [Google Scholar]
- [37].Knotts TA, Rathore N, Schwartz DC, de Pablo JJ, A coarse grain model for dna, The Journal of Chemical Physics 126 (8) (2007) 084901. [DOI] [PubMed] [Google Scholar]
- [38].Li W, Wang W, Takada S, Energy landscape views for interplays among folding, binding, and allostery of calmodulin domains, Proc. Natl. Acad. Sci. U.S.A 111 (29) (2014) 10550–10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39]*.Tan C, Takada S, Nucleosome allostery in pioneer transcription factor binding, Proc. Natl. Acad. Sci 117 (34) (2020) 20586–20596, [DOI] [PMC free article] [PubMed] [Google Scholar]; The simultaneous binding of two transcription factors (TF), Sox2 and Oct4, to the nucleosome was simulated by CG MD, revealing an allosteric mechanism in which the binding of one TF affects the nucleosomal DNA and promotes the binding of a second TF at distant sites.
- [40].Davtyan A, Schafer NP, Zheng W, Clementi C, Wolynes PG, Papoian GA, AWSEM-MD: Protein Structure Prediction Using Coarse-Grained Physical Potentials and Bioinformatically Based Local Structure Biasing, J. Phys. Chem. B 116 (29) (2012) 8494–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41]**.Wu H, Dalal Y, Papoian GA, Binding dynamics of disordered linker histone h1 with a nucleosomal particle, J. Mol. Biol 433 (6) (2021) 166881, [DOI] [PMC free article] [PubMed] [Google Scholar]; CG simulations of chromatosomes reveal that the LH N- and C-terminal domains remain disordered upon binding but condense the chromatosome structure compared to the globular domain alone. Both on- and off-dyad binding modes participate with additional asymmetric modes.
- [42]**.Ding X, Lin X, Zhang B, Stability and folding pathways of tetra-nucleosome from six-dimensional free energy surface, Nat. Commun 12 (1) (2021) 1091, [DOI] [PMC free article] [PubMed] [Google Scholar]; The stability and folding pathways of a tetra-nucleosome array is studied at near-atomistic resolution combined with meta-dynamics and temperature-accelerate MD simulations. The computed six-dimensional free energy surface as a function of internucleosome distances reveals metastable states, where some resemble the previously characterized α-tetrahedron and β-rhombus, and the global minima corresponds to the zigzag structure. These results connect modeling to reported experimental chromatin structures.
- [43].Kapcha LH, Rossky PJ, A simple atomic-level hydrophobicity scale reveals protein interfacial structure, J. Mol. Biol 426 (2) (2014) 484–498. [DOI] [PubMed] [Google Scholar]
- [44]*.Tan C, Jung J, Kobayashi C, Torre DUL, Takada S, Sugita Y, Implementation of residue-level coarse-grained models in genesis for large-scale molecular dynamics simulations, PLoS Comput. Biol 18 (4) (2022) e1009578, [DOI] [PMC free article] [PubMed] [Google Scholar]; Several residue-level CG models are implemented in the GENESIS software for large-scale simulations. Among all benchmarks, the authors test an artificial chromatin structure containing 1024 nucleosomes. Although the simulation is restricted to 1 ns, the model opens the possibility to study large-scale chromatin dynamics at near-atomistic resolution.
- [45].Brandner A, Schüller A, Melo F, Pantano S, Exploring dna dynamics within oligonucleosomes with coarse-grained simulations: Sirah force field extension for protein-dna complexes, Biochem. Biophys. Res 498 (2) (2018) 319–326. [DOI] [PubMed] [Google Scholar]
- [46].Sun T, Minhas V, Mirzoev A, Korolev N, Lyubartsev AP, Nordenskiöld L, A Bottom-Up Coarse-Grained Model for Nucleosome–Nucleosome Interactions with Explicit Ions, J. Chem. Theory Comput 18 (6) (2022) 3948–3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Olson WK, Gorin AA, Lu X, Hock LM, Zhurkin VB, Dna sequence-dependent deformability deduced from protein-dna crystal complexes, Proc. Natl. Acad. Sci. U.S.A 95 (19) (1998) 11163–11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Dignon GL, Zheng W, Kim YC, Best RB, Mittal J, Sequence determinants of protein phase behavior from a coarse-grained model, PLoS Comput. Biol 14 (1) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kim YC, Hummer G, Coarse-grained models for simulations of multiprotein complexes: Application to ubiquitin binding, J. Mol. Biol 375 (5) (2008) 1416–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50]**.Farr SE, Woods EJ, Joseph JA, Garaizar A, Collepardo-Guevara R, Nucleosome plasticity is a critical element of chromatin liquid–liquid phase separation and multivalent nucleosome interactions, Nat. Commun 12 (1) (2021) 2883, [DOI] [PMC free article] [PubMed] [Google Scholar]; A multiscale approach that involves an all-atom nucleosome model, a chemical specificCG chromatin model, and a minimal chromatin model accounts for the spontaneous nucleosome breathing. Nucleosome breathing is suggested to play a key role in the folding of disordered heterogeneous chromatin fibers and phase separation.
- [51].Collepardo-Guevara R, Portella G, Vendruscolo M, Frenkel D, Schlick T, Orozco M, Chromatin Unfolding by Epigenetic Modifications Explained by Dramatic Impairment of Internucleosome Interactions: A Multiscale Computational Study., J. Am. Chem. Soc 137 (32) (2015) 10205–10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhou R, Gao YQ, Polymer models for the mechanisms of chromatin 3d folding: review and perspective, Phys. Chem. Chem. Phys 22 (2020) 20189–20201. [DOI] [PubMed] [Google Scholar]
- [53].Fiorillo L, Bianco S, Esposito A, Conte M, Sciarretta R, Musella F, Chiariello AM, A modern challenge of polymer physics: Novel ways to study, interpret, and reconstruct chromatin structure, WIREs Comput. Mol. Sci 10 (4) (2020) e1454. [Google Scholar]
- [54].Barbieri M, Chotalia M, Fraser J, Lavitas L-M, Dostie J, Pombo A, Nicodemi M, Complexity of chromatin folding is captured by the strings and binders switch model, Proc. Natl. Acad. Sci. U.S.A 109 (40) (2012) 16173–16178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jost D, Carrivain P, Cavalli G, Vaillant C, Modeling epigenome folding: formation and dynamics of topologically associated chromatin domains, Nucleic Acids Res 42 (15) (2014) 9553–9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Buckle A, Brackley CA, Boyle S, Marenduzzo D, Gilbert N, Polymer simulations of heteromorphic chromatin predict the 3d folding of complex genomic loci, Mol. Cell 72 (4) (2018) 786–797.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny L, Formation of chromosomal domains by loop extrusion, Cell Rep 15 (9) (2016) 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chiariello AM, Bianco S, Oudelaar AM, Esposito A, Annunziatella C, Fiorillo L, Conte M, Corrado A, Prisco A, Larke MSC, Telenius JM, Sciarretta R, Musella F, Buckle VJ, Higgs DR, Hughes JR, Nicodemi M, A Dynamic Folded Hairpin Conformation Is Associated with α-Globin Activation in Erythroid Cells., Cell Rep 30 (7) (2020) 2125–2135.e5. [DOI] [PubMed] [Google Scholar]
- [59].Bajpai G, Pavlov DA, Lorber D, Volk T, Safran S, Mesoscale phase separation of chromatin in the nucleus, eLife 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ehrlich L, Munkel C, Chirico G, Langowski J, A Brownian dynamics model for the chromatin fiber., Cell Rep. Comput. Appl. Biosci. CABIOS 13 (3) (1997) 271–279. [DOI] [PubMed] [Google Scholar]
- [61].Lequieu J, Córdoba A, Moller J, De Pablo JJ, 1CPN: A coarse-grained multi-scale model of chromatin, J. Chem. Phys 150 (21) (2019) 215102. [DOI] [PubMed] [Google Scholar]
- [62].Luque A, Collepardo-Guevara R, Grigoryev S, Schlick T, Dynamic Condensation of Linker Histone C-terminal Domain Regulates Chromatin Structure., Nucleic Acids Res 42 (12) (2014) 7553–7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63]*.Li Z, Portillo-Ledesma S, Schlick T, Brownian dynamics simulations of mesoscale chromatin fibers, Biophys. J 122 (2022) 1–14, [DOI] [PMC free article] [PubMed] [Google Scholar]; An efficient Brownian Dynamics method to study the dynamics of mesoscale chromatin fibers is developed for GPU architectures by CUDA, and validated against experimental data and previous Monte Carlo simulations. The efficient implementation allows the study of up to 50-nucleosome systems in the range of the μs. Such simulations are essential to advance the study of biological processes such as gene regulation and aberrant genome-structure related diseases.
- [64].Beard DA, Schlick T, Computational Modeling Predicts the Structure and Dynamics of Chromatin Fiber, Structure 9 (2) (2001) 105–114. [DOI] [PubMed] [Google Scholar]
- [65].Zhang Q, Beard DA, Schlick T, Constructing Irregular Surfaces to Enclose Macromolecular Complexes for Mesoscale Modeling Using the Discrete Surface Charge Optimization (DiSCO) Algorithm, J. Comput. Chem 24 (16) (2003) 2063–2074. [DOI] [PubMed] [Google Scholar]
- [66].Collepardo-Guevara R, Schlick T, Chromatin fiber polymorphism triggered by variations of DNA linker lengths, Proc. Natl. Acad. Sci. U.S.A 111 (22) (2014) 8061–8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Arya G, Schlick T, Role of histone tails in chromatin folding revealed by a mesoscopic oligonucleosome model, Proc. Natl. Acad. Sci 103 (44) (2006) 16236–16241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Perisic O, Portillo-Ledesma S, Schlick T, Sensitive effect of linker histone binding mode and subtype on chromatin condensation, Nucleic Acids Res 47 (10) (2019) 4948–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Vologodskii A, Marko J, Extension of torsionally stressed dna by external force, Biophysical Journal 73 (1) (1997) 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Jian H, Vologodskii AV, Schlick T, A combined wormlike-chain and bead model for dynamic simulations of long linear DNA, Journal of Computational Physics 136 (1) (1997) 168–179. [Google Scholar]
- [71].Portillo-Ledesma S, Schlick T, Bridging chromatin structure and function over a range of experimental spatial and temporal scales by molecular modeling, WIREs Comput. Mol. Sci 10 (2) (2020) wcms.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Bascom GD, Schlick T, Mesoscale Modeling of Chromatin Fibers, in: Lavelle C, Victor J-M (Eds.), Nuclear Architecture and Dynamics, Vol. 2, Academic Press, Boston, 2017, pp. 123–147. [Google Scholar]
- [73].Arya G, Schlick T, A tale of tails: how histone tails mediate chromatin compaction in different salt and linker histone environments, J. Phys. Chem. A 113 (16) (2009) 4045–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bascom GD, Sanbonmatsu KY, Schlick T, Mesoscale Modeling Reveals Hierarchical Looping of Chromatin Fibers Near Gene Regulatory Elements, J. Phys. Chem. B 120 (33) (2016) 8642–8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].de Jong BE, Brouwer TB, Kaczmarczyk A, Visscher B, van Noort J, Rigid basepair monte carlo simulations of one-start and two-start chromatin fiber unfolding by force, Biophys. J 115 (10) (2018) 1848–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Brouwer T, Pham C, Kaczmarczyk A, de Voogd W-J, Botto M, Vizjak P, Mueller-Planitz F, van Noort J, A critical role for linker DNA in higher-order folding of chromatin fibers., Nucleic Acids Res 49 (5) (2021) 2537–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Norouzi D, Zhurkin VB, Dynamics of Chromatin Fibers: Comparison of Monte Carlo Simulations with Force Spectroscopy., Biophys. J 115 (9) (2018) 1644–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Zhurkin VB, Norouzi D, Topological polymorphism of nucleosome fibers and folding of chromatin, Biophys. J 120 (4) (2021) 577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Müller O, Kepper N, Schöpflin R, Ettig R, Rippe K, Wedemann G, Changing chromatin fiber conformation by nucleosome repositioning, Biophys. J 107 (9) (2014) 2141–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Brandstetter K, Zülske T, Ragoczy T, Hörl D, Guirao-Ortiz M, Steinek C, Barnes T, Stumberger G, Schwach J, Haugen E, Rynes E, Korber P, Stamatoyannopoulos JA, Leonhardt H, Wedemann G, Harz H, Differences in nanoscale organization of regulatory active and inactive human chromatin, Biophys. J 121 (6) (2022) 977–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ, Crystal structure of the nucleosome core particle at 2.8 Å resolution, Nature 389 (6648) (1997) 251–260. [DOI] [PubMed] [Google Scholar]
- [82].Zhou K, Gaullier G, Luger K, Nucleosome structure and dynamics are coming of age, Nat. Struct. Mol. Biol 26 (1) (2019) 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Öztürk MA, De M, Cojocaru V, Wade RC, Chromatosome Structure and Dynamics from Molecular Simulations., Annu. Rev. Phys. Chem 71 (2020) 101–119. [DOI] [PubMed] [Google Scholar]
- [84].Eslami-Mossallam B, Schiessel H, van Noort J, Nucleosome dynamics: Sequence matters, Adv. Colloid Interface Sci 232 (2016) 101–113. [DOI] [PubMed] [Google Scholar]
- [85].Brandani GB, Tan C, Takada S, The kinetic landscape of nucleosome assembly: A coarse-grained molecular dynamics study, PLoS Comput. Biol 17 (7) (2021) 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Lequieu J, Schwartz DC, de Pablo JJ, In silico evidence for sequence-dependent nucleosome sliding, Proc. Natl. Acad. Sci. U.S.A 114 (44) (2017) E9197–E9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Winogradoff D, Aksimentiev A, Molecular mechanism of spontaneous nucleosome unraveling, J. Mol. Biol 431 (2) (2019) 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Fu I, Geacintov NE, Broyde S, Molecular dynamics simulations reveal how H3K56 acetylation impacts nucleosome structure to promote DNA exposure for lesion sensing, DNA Repair 107 (2021) 103201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Li Z, Kono H, Investigating the Influence of Arginine Dimethylation on Nucleosome Dynamics Using All-Atom Simulations and Kinetic Analysis, J. Phys. Chem. B 122 (42) (2018) 9625–9634. [DOI] [PubMed] [Google Scholar]
- [90].Rajagopalan M, Balasubramanian S, Ioshikhes I, Ramaswamy A, Structural dynamics of nucleosome mediated by acetylations at H3K56 and H3K115, Eur. Biophys. J 46 (5) (2017) 471–484. [DOI] [PubMed] [Google Scholar]
- [91]*.Li S, Peng Y, Landsman D, Panchenko AR, DNA methylation cues in nucleosome geometry, stability and unwrapping, Nucleic Acids Res 50 (4) (2022) 1864–1874, [DOI] [PMC free article] [PubMed] [Google Scholar]; All-atom MD simulations of an NCP show that DNA methylation affects nucleosome dynamics and structure: more rigid nucleosome results, with an undertwistied and underwinded DNA, and increased interactions between histones and DNA. Such a possible mechanism may be involved in the repressive effect of this epigenetic modification.
- [92].Peng J, Yuan C, Hua X, Zhang Z, Molecular mechanism of histone variant H2A.B on stability and assembly of nucleosome and chromatin structures, Epigenetics Chromatin 13 (1) (2020) 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93]*.Pitman M, Dalal Y, Papoian GA, Minimal cylinder analysis reveals the mechanical properties of oncogenic nucleosomes, Biophys. J 118 (9) (2020) 2309–2318, [DOI] [PMC free article] [PubMed] [Google Scholar]; By simulating hybrid nucleosomes containing the two H3 variants CENP-A and H3.3, the Dalal and Papoian groups study how histone variants affect the mechanical properties of nucleosomes in relation to cancer development. Results show that these hybrid nucleosomes have an intermediate elasticity compared to wild type and CENP-A nucleosomes, which indicates that histone variants might regulate gene expression by tuning nucleosome flexibility and sliding.
- [94].Izzo A, Kamieniarz K, Schneider R, The histone h1 family: specific members, specific functions?, Biol Chem 389 (4) (2008) 333–343 [cited 2022-07-29]. [DOI] [PubMed] [Google Scholar]
- [95].Pachov GV, Gabdoulline RR, Wade RC, On the structure and dynamics of the complex of the nucleosome and the linker histone., Nucleic Acids Res 39 (12) (2011) 5255–5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Öztürk MA, Pachov GV, Wade RC, Cojocaru V, Conformational selection and dynamic adaptation upon linker histone binding to the nucleosome., Nucleic Acids Res 44 (14) (2016) 6599–6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Öztürk MA, Cojocaru V, Wade RC, Dependence of Chromatosome Structure on Linker Histone Sequence and Posttranslational Modification., Biophys J 114 (10) (2018) 2363–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98]*.Woods DC, Wereszczynski J, Elucidating the influence of linker histone variants on chromatosome dynamics and energetics, Nucleic Acids Res 48 (7) (2020) 3591–3604, [DOI] [PMC free article] [PubMed] [Google Scholar]; The binding of the globular domain of two LH variants to the nucleosome by all-atom MD reveals that the on-dyad binding is enthalpically stabilized in both variants, especially in one of them, and that off-dyad binding is relatively more entropically stabilized. However, the globular domain appears highly dynamic, indicating that multiple binding states may simultaneously occur.
- [99].Öztürk MA, Cojocaru V, Wade RC, Toward an Ensemble View of Chromatosome Structure: A Paradigm Shift from One to Many, Structure 26 (8) (2018) 1050–1057. [DOI] [PubMed] [Google Scholar]
- [100].Sridhar A, Orozco M, Collepardo-Guevara R, Protein disorder-to-order transition enhances the nucleosome-binding affinity of H1, Nucleic Acids Res 48 (10) (2020) 5318–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Zhou B-R, Feng H, Kale S, Fox T, Khant H, de Val N, Ghirlando R, Panchenko AR, Bai Y, Distinct Structures and Dynamics of Chromatosomes with Different Human Linker Histone Isoforms, Mol. Cell 81 (1) (2021) 166–182.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Schlick T, Hayes J, Grigoryev S, Toward convergence of experimental studies and theoretical modeling of the chromatin fiber., J. Biol. Chem 287 (8) (2012) 5183–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Huertas J, MacCarthy CM, Schöler HR, Cojocaru V, Nucleosomal DNA Dynamics Mediate Oct4 Pioneer Factor Binding., Biophys. J 118 (9) (2020) 2280–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Peng Y, Li S, Onufriev A, Landsman D, Panchenko AR, Binding of regulatory proteins to nucleosomes is modulated by dynamic histone tails, Nat. Commun 12 (1) (2021) 5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Ishida H, Kono H, Torsional stress can regulate the unwrapping of two outer half superhelical turns of nucleosomal dna, Proc. Natl. Acad. Sci. U. S. A 118 (7) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Adhireksan Z, Sharma D, Lee PL, Bao Q, Padavattan S, Shum WK, Davey GE, Davey CA, Engineering nucleosomes for generating diverse chromatin assemblies, Nucleic Acids Res 49 (9) (2021) e52–e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Schalch T, Duda S, Sargent DF, Richmond TJ, X-ray structure of a tetranucleosome and its implications for the chromatin fibre, Nature 436 (7047) (2005) 138–141. [DOI] [PubMed] [Google Scholar]
- [108].Ekundayo B, Richmond TJ, Schalch T, Capturing structural heterogeneity in chromatin fibers, J. Mol. Biol 429 (20) (2017) 3031–3042. [DOI] [PubMed] [Google Scholar]
- [109].Garcia-Saez I, Menoni H, Boopathi R, Shukla MS, Soueidan L, Noirclerc-Savoye M, Le Roy A, Skoufias DA, Bednar J, Hamiche A, Angelov D, Petosa C, Dimitrov S, Structure of an H1-Bound 6-Nucleosome Array Reveals an Untwisted Two-Start Chromatin Fiber Conformation, Mol. Cell 72 (5) (2018) 902–915.e7. [DOI] [PubMed] [Google Scholar]
- [110].Cai S, Böck D, Pilhofer M, Gan L, The in situ structures of mono-, di-, and trinucleosomes in human heterochromatin, Mol. Biol. Cell 29 (20) (2018) 2450–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Takizawa Y, Ho C-H, Tachiwana H, Matsunami H, Kobayashi W, Suzuki M, Arimura Y, Hori T, Fukagawa T, Ohi MD, Wolf M, Kurumizaka H, Cryo-em structures of centromeric tri-nucleosomes containing a central cenp-a nucleosome, Structure 28 (1) (2020) 44–53.e4. [DOI] [PubMed] [Google Scholar]
- [112].Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu RM, Zhu P, Li G, Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units, Science 344 (6182) (2014) 376–380. [DOI] [PubMed] [Google Scholar]
- [113].Saurabh S, Glaser MA, Lansac Y, Maiti PK, Atomistic Simulation of Stacked Nucleosome Core Particles: Tail Bridging, the H4 Tail, and Effect of Hydrophobic Forces, J. Phys. Chem. B 120 (12) (2016) 3048–3060. [DOI] [PubMed] [Google Scholar]
- [114].Ishida H, Kono H, H4 Tails Potentially Produce the Diversity in the Orientation of Two Nucleosomes, Biophys. J 113 (5) (2017) 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Korolev N, Lyubartsev AP, Nordenskiöld L, A systematic analysis of nucleosome core particle and nucleosome-nucleosome stacking structure, Sci. Rep 8 (1) (2018) 1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Saurabh S, Jang YH, Lansac Y, Maiti PK, Orientation Dependence of Inter-NCP Interaction: Insights into the Behavior of Liquid Crystal Phase and Chromatin Fiber Organization, J. Phys. Chem. B 124 (2) (2020) 314–323. [DOI] [PubMed] [Google Scholar]
- [117].Zhang R, Erler J, Langowski J, Histone Acetylation Regulates Chromatin Accessibility: Role of H4K16 in Inter-nucleosome Interaction, Biophys. J 112 (3) (2017) 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Moller J, Lequieu J, de Pablo JJ, The Free Energy Landscape of Internucleosome Interactions and Its Relation to Chromatin Fiber Structure, ACS Cent. Sci 5 (2) (2019) 341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Kenzaki H, Takada S, Linker dna length is a key to tri-nucleosome folding, J. Mol. Biol 433 (6) (2021) 166792. [DOI] [PubMed] [Google Scholar]
- [120].Bascom GD, Kim T, Schlick T, Kilobase Pair Chromatin Fiber Contacts Promoted by LivingSystem-Like DNA Linker Length Distributions and Nucleosome Depletion, J. Phys. Chem. B 121 (15) (2017) 3882–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121]*.Alvarado W, Moller J, Ferguson AL, de Pablo JJ, Tetranucleosome Interactions Drive Chromatin Folding, ACS Cent. Sci 7 (6) (2021) 1019–1027, [DOI] [PMC free article] [PubMed] [Google Scholar]; The folding of 4- and 16-nucleosome arrays using their 1CPN mesoscale model of chromatin combined with machine learning is studied in long time trajectories. The researchers find the spontaneus formation of the two previously characterized α-tetrahedron and β-rhombus tetranucleosome folding motifs. While the former is compact, the latter appears open and more stable, indicating that local chromatin might exist as a two-state model.
- [122].Watanabe S, Mishima Y, Shimizu M, Suetake I, Takada S, Interactions of HP1 Bound to H3K9me3 Dinucleosome by Molecular Simulations and Biochemical Assays, Biophys. J 114 (10) (2018) 2336–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Leicher R, Ge EJ, Lin X, Reynolds MJ, Xie W, Walz T, Zhang B, Muir TW, Liu S, Single-molecule and in silico dissection of the interaction between polycomb repressive complex 2 and chromatin, Proc. Natl. Acad. Sci. U.S.A 117 (48) (2020) 30465–30475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Huertas J, Woods EJ, Collepardo-Guevara R, Multiscale modelling of chromatin organisation: Resolving nucleosomes at near-atomistic resolution inside genes, Curr. Opin. Cell Biol 75 (2022) 102067. [DOI] [PubMed] [Google Scholar]
- [125].Brackey CA, Marenduzzo D, Gilbert N, Mechanistic modeling of chromatin folding to understand function, Nat. Methods 17 (8) (2020) 767–775. [DOI] [PubMed] [Google Scholar]
- [126].Moller J, de Pablo JJ, Bottom-Up Meets Top-Down: The Crossroads of Multiscale Chromatin Modeling, Biophys. J 118 (9) (2020) 2057–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Lin X, Qi Y, Latham AP, Zhang B, Multiscale modeling of genome organization with maximum entropy optimization, J. Chem. Phys 155 (1) (2021) 010901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Chiang M, Brackley CA, Marenduzzo D, Gilbert N, Predicting genome organisation and function with mechanistic modelling, Trends Genet 38 (4) (2022) 364–378. [DOI] [PubMed] [Google Scholar]
- [129].Bascom G, Schlick T, Linking Chromatin Fibers to Gene Folding by Hierarchical Looping, Biophys. J 112 (3) (2017) 434–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Myers CG, Olins DE, Olins AL, Schlick T, Mesoscale Modeling of Nucleosome-Binding Antibody PL2–6: Mono- versus Bivalent Chromatin Complexes, Biophys. J 118 (9) (2020) 2066–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Sridhar A, Farr SE, Portella G, Schlick T, Orozco M, Collepardo-Guevara R, Emergence of chromatin hierarchical loops from protein disorder and nucleosome asymmetry, Proc. Natl. Acad. Sci. U.S.A 117 (13) (2020) 7216–7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Yusufova N, Kloetgen A, Teater M, Osunsade A, Camarillo JM, Chin CR, Doane AS, Venters BJ, Portillo-Ledesma S, Conway J, Phillip JM, Elemento O, Scott DW, Béguelin W, Licht JD, Kelleher NL, Staudt LM, Skoultchi AI, Keogh MC, Apostolou E, Mason CE, Imielinski M, Schlick T, David Y, Tsirigos A, Allis CD, Soshnev AA, Cesarman E, Melnick AM, Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture, Nature 589 (7841) (2021) 299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Portillo-Ledesma S, Wagley M, Schlick T, Chromatin transitions triggered by lh density as epigenetic regulators of the genome, Nucleic Acids Res 50 (18) (2022) 10328–10342, accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Bajpai G, Padinhateeri R, Irregular chromatin: Packing density, fiber width, and occurrence of heterogeneous clusters, Biophys. J 118 (1) (2020) 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Wiese O, Marenduzzo D, Brackley CA, Nucleosome positions alone can be used to predict domains in yeast chromosomes, Proc. Natl. Acad. Sci. U.S.A 116 (35) (2019) 17307–17315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].MacPherson Q, Beltran B, Spakowitz AJ, Bottom-up modeling of chromatin segregation due to epigenetic modifications, Proc. Natl. Acad. Sci. U.S.A 115 (50) (2018) 12739–12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].MacPherson Q, Beltran B, Spakowitz AJ, Chromatin compaction leads to a preference for peripheral heterochromatin, Biophys. J 118 (6) (2020) 1479–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138]**.Salari H, Di Stefano M, Jost D, Spatial organization of chromosomes leads to heterogeneous chromatin motion and drives the liquid- or gel-like dynamical behavior of chromatin., Genome Res 32 (1) (2022) 28–43, [DOI] [PMC free article] [PubMed] [Google Scholar]; Polymer modeling of different physical states and dynamics of chromosomes suggests that TADs define the physical state of chromatin. Namely, the TAD local degree of compaction regulates the transition from an open, fluid state of chromatin to a more compact, gel state, and both states are possible.
- [139].Conte M, Fiorillo L, Bianco S, Chiariello AM, Esposito A, Nicodemi M, Polymer physics indicates chromatin folding variability across single-cells results from state degeneracy in phase separation, Nat. Commun 11 (1) (2020) 3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140]**.Conte M, Irani E, Chiariello AM, Abraham A, Bianco S, Esposito A, Nicodemi M, Loopextrusion and polymer phase-separation can co-exist at the single-molecule level to shape chromatin folding, Nat. Commun 13 (1) (2022) 4070, [DOI] [PMC free article] [PubMed] [Google Scholar]; Many polymer models that consider loop extrusion, phase separation, or both examine how these two mechanisms capture chromatin folding and whether they compete or coexist in establishing chromosome architecture in single cells. Both types of models capture the main features of single-cell chromatin conformations but the phase-separation based models better describe the segregation in globules of the genomic loci studied and the cell-to-cell structural variability. Moreover, loop-extrusion and phase-separation can coexist at the single-molecule level to determine chromatin architecture.
- [141].Di Pierro M, Zhang B, Aiden EL, Wolynes PG, Onuchic JN, Transferable model for chromosome architecture, Proc. Natl. Acad. Sci. U.S.A 113 (43) (2016) 12168–12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142]**.Buitrago D, Labrador M, Arcon JP, Lema R, Flores O, Esteve-Codina A, Blanc J, Villegas N, Bellido D, Gut M, Dans PD, Heath SC, Gut IG, Brun Heath I, Orozco M, Impact of DNA methylation on 3D genome structure, Nat. Commun 12 (1) (2021) 3243, [DOI] [PMC free article] [PubMed] [Google Scholar]; Experiments and modeling are combined to study the effect of DNA methylation on nucleosome positioning, gene expression, and genome folding. Experiments show that high levels of methylation affect the global genome structure, as well as the level of gene expression. Orozco’s group’s simulations of chromatin fibers and the whole genome further explain that methylation reduces DNA flexibility and increases chromatin condensation.
- [143].Lappala A, Wang C-Y, Kriz A, Michalk H, Tan K, Lee JT, Sanbonmatsu KY, Four-dimensional chromosome reconstruction elucidates the spatiotemporal reorganization of the mammalian X chromosome., Proc. Natl. Acad. Sci. U.S.A 118 (42) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144]**.Hoencamp C, Dudchenko O, Elbatsh AMO, Brahmachari S, Raaijmakers JA, van Schaik T, Cacciatore Ángela Sedeño, Contessoto VG, van Heesbeen RGHP, van den Broek B, Mhaskar AN, Teunissen H, Hilaire BGS, Weisz D, Omer AD, Pham M, Colaric Z, Yang Z, Rao SSP, Mitra N, Lui C, Yao, Khan R, Moroz LL, Kohn A, Leger JS, Mena A, Holcroft K, Gambetta MC, Lim F, Farley E, Stein N, Haddad A, Chauss D, Mutlu AS, Wang MC, Young ND, Hildebrandt E, Cheng HH, Knight CJ, Burnham TLU, Hovel KA, Beel AJ, Mattei P-J, Kornberg RD, Warren WC, Cary G, Gómez-Skarmeta JL, Hinman V, Lindblad-Toh K, Palma FD, Maeshima K, Multani AS, Pathak S, Nel-Themaat L, Behringer RR, Kaur P, Medema RH, van Steensel B, de Wit E, Onuchic JN, Pierro MD, Aiden EL, Rowland BD, 3D genomics across the tree of life reveals condensin ii as a determinant of architecture type, Science 372 (6545) (2021) 984–989, [DOI] [PMC free article] [PubMed] [Google Scholar]; Experiments and modeling aiming at characterizing the eukaryotic genome organization show via in situ Hi-C maps on 24 species that all species can be separated in two groups: those that show formation of chromosome territories, and those that show telomere or centromere clustering or telomere-centromere axis. Species that do not form chromosome territories lack condensin II. The Onuchic and Di Perro polymer model for chromosomes was used to better understand the effect of condensin on genome folding. Results show that condensin drives the formation of chromosomes territories.
- [145]**.Thornburg ZR, Bianchi DM, Brier TA, Gilbert BR, Earnest TM, Melo MCR, Safronova N, Sáenz JP, Cook AT, Wise KS, Hutchison CA III, Smith HO, Glass JI, Luthey-Schulten Z, Fundamental behaviors emerge from simulations of a living minimal cell, Cell 185 (2) (2022) 345–360.e28, [DOI] [PMC free article] [PubMed] [Google Scholar]; A computer model for a minimal cell with a reduced genome is developed. In this model, the physical and chemical characteristics of the cell’s nucleic acids, lipids, amino acids, and ribosomes are considered, as well as their diffusion throughout the cell. Cryo-ET data is used to reconstruct the single cell architecture containing ribosomes and chromosome configurations, then proteins, mRNA, tRNA, and degradosomes are added. These simulations allow to see how the cellular components interact and change in response to internal and external forces.
- [146]**.Bascom GD, Myers CG, Schlick T, Mesoscale modeling reveals formation of an epigenetically driven HOXC gene hub, Proc. Natl. Acad. Sci. U.S.A 116 (11) (2019) 4955–4962, [DOI] [PMC free article] [PubMed] [Google Scholar]; An in silico folding of the 55-kb HOXC gene cluster is performed from the ground up using experimental data to define gene parameters (nucleosome positions, LH and acetylation marks, etc.). The resulting structure, product of the cooperation among epigenetic elements, particularly acetylated and LH-rich regions, exhibits a complex folding in which gene promoter regions are brought close together. The study shows how natural gene marking produces a compact folded gene structure compared to gene systems containing only subcomponents of the marks. This work opened the way for nucleosome resolution models incorporating epigenetic factors to understand and predict gene folding and its regulation (commentary in [145]).
- [147].Pierro MD, Inner workings of gene folding, Proc. Natl. Acad. Sci. U.S.A 116 (11) (2019) 4774–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Hsieh T-HS, Cattoglio C, Slobodyanyuk E, Hansen AS, Rando OJ, Tjian R, Darzacq X, Resolving the 3d landscape of transcription-linked mammalian chromatin folding, Mol. Cell 78 (3) (2020) 539–553.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Swygert SG, Lin D, Portillo-Ledesma S, Lin P-Y, Hunt DR, Kao C-F, Schlick T, Noble WS, Tsukiyama T, Local chromatin fiber folding represses transcription and loop extrusion in quiescent cells, eLife 10 (2021) e72062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Barbieri M, Xie SQ, Torlai Triglia E, Chiariello AM, Bianco S, de Santiago I, Branco MR, Rueda D, Nicodemi M, Pombo A, Active and poised promoter states drive folding of the extended HoxB locus in mouse embryonic stem cells, Nat. Struct. Mol. Biol 24 (6) (2017) 515–524. [DOI] [PubMed] [Google Scholar]
- [151].Neguembor MV, Arcon JP, Buitrago D, Lema R, Walther J, Garate X, Martin L, Romero P, AlHaj Abed J, Gut M, Blanc J, Lakadamyali M, Wu C.-t., Brun Heath I, Orozco M, Dans PD, Cosma MP, MiOS, an integrated imaging and computational strategy to model gene folding with nucleosome resolution, Nature Structural & Molecular Biology 29 (10) (2022) 1011–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Schlick T, Portillo-Ledesma S, Biomolecular modeling thrives in the age of technology, Nat. Comput. Sci 1 (5) (2021) 321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Schlick T, Portillo-Ledesma S, Myers CG, Beljak L, Chen J, Dakhel S, Darling D, Ghosh S, Hall J, Jan M, Liang E, Saju S, Vohr M, Wu C, Xu Y, Xue E, Biomolecular Modeling and Simulation: A Prospering Multidisciplinary Field, Annu. Rev. Biophys 50 (2021) 267–301. [DOI] [PMC free article] [PubMed] [Google Scholar]