

Abstract
Regulatory T cell (Treg) therapy is a promising strategy to treat inflammatory bowel disease (IBD). Data from animal models has shown that Tregs specific for intestinal antigens are more potent than polyclonal Tregs at inhibiting colitis. Flagellins, the major structural proteins of bacterial flagella, are immunogenic antigens frequently targeted in IBD subjects, leading to the hypothesis that flagellin-specific Tregs could be an effective cell therapy for IBD. We developed a novel chimeric antigen receptor (CAR) specific for flagellin derived from Escherichia coli H18 (FliC). We used this CAR to confer FliC-specificity to human Tregs and investigated their therapeutic potential. FliC-CAR Tregs were activated by recombinant FliC protein but not a control flagellin protein, demonstrating CAR specificity and functionality. In a humanized mouse model, expression of the FliC-CAR drove preferential migration to the colon and expression of the activation marker PD1. In the presence of recombinant FliC protein in vitro, FliC-CAR Tregs were significantly more suppressive than control Tregs and promoted the establishment of colon-derived epithelial cell monolayers. These results demonstrate the potential of FliC-CAR Tregs to treat IBD and more broadly show the therapeutic potential of CARs targeting microbial-derived antigens.
Keywords: Regulatory T cell, Chimeric antigen receptor, Inflammatory bowel disease, Flagellin, Treg therapy
1. INTRODUCTION
Inflammatory bowel disease (IBD) is a chronic relapsing, remitting disorder of the gastrointestinal tract that is predominantly comprised of two subtypes: ulcerative colitis (UC) and Crohn’s disease (CD). The etiology of IBD remains unclear but it is driven by a range of genetic and environmental factors that disrupt the delicate symbiotic relationship that exists between the intestinal immune system and the microbiome [1]. This loss of immune homeostasis ultimately leads to uncontrolled intestinal inflammation and tissue damage. Despite promising advances in the development of biologic agents and small-molecule drugs to treat IBD [2, 3], these conditions remain incurable and the lack of immunosuppression specificity leaves patients with an increased susceptibility to acquiring opportunistic infections and developing cancer [4, 5]. As such, novel therapeutic strategies are required.
Regulatory T cells (Treg) are a population of immunomodulatory cells that play a fundamental role in dampening undesired immune responses, particularly in the intestine [6]. Tregs employ a variety of cell contact-dependent and -independent mechanisms to suppress the action of both innate and adaptive immune cells [7]. To elicit their suppressive function, Tregs need to be stimulated via recognition of a cognate MHC-peptide complex, but once activated, they suppress cells in their local vicinity in an antigen-independent manner. This phenomenon, termed bystander or linked suppression, is key for the maintenance of intestinal homeostasis [8, 9]. In addition to their suppressive function, Tregs also act in the local environment to promote tissue repair and regeneration via a range of mechanisms such as the secretion of IL-22 [10] and amphiregulin [11].
Quantitative and qualitative deficiencies in Tregs are correlated with IBD development and progression in mice and humans [12–17]. As such, a promising strategy to restore intestinal immune homeostasis is Treg therapy. This approach is curative in mouse models of IBD [18] and has been deemed safe in a variety of clinical settings [19–22], with ongoing clinical studies investigating polyclonal Treg therapy for treating CD (NCT03185000) and UC (NCT04691232). Further data from animal models have shown that the potency of therapeutic Tregs can be significantly enhanced by using cells specific for disease-relevant antigens [23]. One approach to generate antigen-specific Tregs is to use chimeric antigen receptor (CAR) technology. CARs are synthetic fusion proteins that utilize an antibody-derived antigen-recognition domain to redirect the specificity of T cells towards desired antigens [6, 23]. This approach has been highly successful for generating human allospecific Tregs for use in a transplant setting [24–26] and has been explored in a variety of other contexts [27–30].
A group of immunogenic antigens commonly targeted in individuals with IBD are flagellins, the major structural proteins of bacterial flagella [31]. Flagellins are pathogen-associated molecular patterns that are typically engaged by Toll-like receptor (TLR)5 on both innate and adaptive immune cells [32], including Tregs [33]. However, multiple studies have found that flagellins can also be recognized as protein targets by B and T cell receptors and that individuals with IBD have a higher proportion of flagellin-specific lymphocytes than healthy controls [34–42]. As bacterial components, flagellins naturally reside on the luminal side of the intestinal barrier, thus they are predominantly only accessible and present in the lamina propria when intestinal damage is occurring [43]. This makes them ideal Treg therapy targets as cell activation would only be elicited in the context of a barrier breach. Moreover, since flagellin naturally forms oligomers [44], we hypothesized that despite not being membrane bound, their oligomeric structure would provide the minimum dimeric cross linking needed to stimulate CAR activation [45]. In this study, we developed a novel CAR to target a subset of flagellins derived from Escherichia coli H18 (FliC). We used this CAR to generate human FliC-specific Tregs and explored the potential of these cells to be used as a novel therapy for treating IBD.
2. MATERIALS AND METHODS
2.1. Subjects
Peripheral blood in the form of buffy coat products were obtained from anonymized healthy adults via Canadian Blood Services (Vancouver, BC, Canada). All blood samples were obtained with informed consent and ethical approval from the University of British Columbia Clinical and Canadian Blood Service Research Ethics Boards (H18–02553). De-identified intestinal biopsy tissue was obtained from the ascending colon of healthy subjects with informed consent at Johns Hopkins University and all methods were carried out in accordance with approved guidelines and regulations [46]. All experimental protocols were approved by the Johns Hopkins University Institutional Review Board (IRB# NA_00038329).
2.2. CAR generation
A FliC-specific hybridoma cell line, termed 5D4, was generated by immunizing BALB/c mice at the Lymphocyte Culture Center, University of Virginia [47]. This hybridoma was used to construct the FliC-CAR, using previously established protocols [24]. Briefly, total RNA from the 5D4 hybridoma cell line was extracted (Qiagen, cat#: 74134), reverse transcribed (QuantaBio, cat#: 95048–100) and the variable heavy (VH) and light chain (VL) sequences were PCR amplified (NEB, cat#: M0530L & N0447L). PCR products were purified (NEB, cat#: T1020L), cloned into a pBK-CMV vector (details on addgene.org) and Sanger sequenced. VH and VL chain sequences were then gene synthesized (Biomatik) in an scFv format that was cloned into a 3rd generation lentiviral vector upstream of in-series sequences encoding i) a 9E10 cMyc mAb epitope, ii) a human CD8α hinge, iii) a human CD28 transmembrane domain and a human CD28-CD3ζ signaling domain. CAR expression was driven by an EF1α promoter and expression of a truncated nerve growth factor receptor (ΔNGFR) reporter was driven by a minimal CMV promoter. Control cells were transduced with a previously characterized HER2-specific CAR [24] that differed from the FliC-CAR construct in the scFv domain only, or a ΔNGFR reporter without a CAR.
2.3. Cell line culture
HEK293T clone 17 cells (ATCC, cat#: CRL-11268) were cultured in IMDM supplemented with 10% fetal bovine serum, penicillin-streptomycin and GlutaMAX™ (Thermo Fisher Scientific, cat#: 12483020, 15140163, & 35050079). L cells expressing human CD32, CD58 and CD80 [48] were cultured in RPMI (Thermo Fisher Scientific, cat#: 11875–093) supplemented with bovine serum (Hyclone Laboratories Inc., cat#: SH3007203), penicillin-streptomycin and GlutaMAX™. Cells were passaged by washing with PBS, incubating with 0.05% Trypsin-EDTA (Thermo Fisher Scientific, cat#: 25300062) for 2–3 minutes and neutralizing with cell culture media. All cultures were performed at 37°C (v/v 5% CO2).
2.4. Lentivirus production
HEK293T/17 cells were co-transfected with either pCCL_FliC-CAR or pCCL_HER2-CAR and a mixture of pRSV-REV, pMDLG/pRRE, pMD2.g and pAdVAntage™ Vector (further details on addgene.org) using calcium phosphate (made in-house). Cell supernatants were harvested 45–48 hours post-transfection and lentiviral particles were concentrated by ultracentrifugation at 76,755 g. Viral titers were calculated by limiting dilution transduction of HEK293T/17 cells and virus aliquots were stored at −80°C.
2.5. Treg isolation, transduction and expansion
CD4+ T cells were enriched from peripheral blood using the Rosettesep™ Human CD4+ T Cell Enrichment Cocktail (STEMCELL Technologies, cat#: 15062) and CD25+ cells were separated using CD25 MicroBeads II (Miltenyi Biotec, cat#: 130-092-983). Naïve CD4+CD25hiCD127loCD45RA+CD45RO− Tregs and CD4+CD25−CD127+CD45RA+CD45RO− Tconvs were then purified by cell sorting. Sorted T cells were stimulated with artificial APCs (L cells expressing human CD32, CD58 and CD80; irradiated with 7500 cGy) loaded with anti-CD3 mAb (OKT3) and cultured in X-vivo™ 15 (Lonza, cat#: BEBP02–054Q) supplemented with 5% human AB serum (Wisent Bioproducts, cat#: 022210), penicillin-streptomycin, GlutaMAX™ and phenol red (Sigma-Aldrich, cat#: P3532–5G). Tregs and Tconvs were cultured with 1,000 U/mL and 100 U/mL recombinant human IL-2, respectively (Proleukin®, Prometheus Laboratories Inc., DIN: 02130181). Media and IL-2 were replenished every 2–3 days.
Cells were transduced 24 hours post-stimulation with lentivirus at a MOI of 10 viral particles per cell. After 7 days of culture, successfully transduced ΔNGFR+ cells were enriched using the EasySep™ Human CD271 Positive Selection Kit II (STEMCELL Technologies, cat#: 17849). Cells were rested overnight by culturing in the presence of 100 U/mL (Tregs) or 10 U/mL (Tconvs) IL-2 and assays were performed on day 8 of culture. All cultures were performed at 37°C (v/v 5% CO2).
2.6. Flow cytometry and cell sorting
Cells were stained using previously described protocols [49, 50] in PBS supplemented with 1% bovine serum and 5 mM EDTA (Sigma-Aldrich) with fluorescently-conjugated antibodies specific for mouse CD45 (PE-Cy7 30-F11; Thermo Fisher Scientific) and human CD45 (V500 HI30), CD4 (V500 RPA-T4, AF700 RPA-T4 or APC-R700 SK3), CD271 (PE C40–1457) GARP (BUV737 7B11 or BV711 7B11), CD8a (PE HIT8a) (all from BD Bioscience), CD45RA (PE-Cy7 HI100), CD45RO (eF450 UCHL1), FOXP3 (PE-Cy7 236A/E7), CD69 (FITC FN50), LAP (PE FNLAP), CD4 (FITC RPA-T4), CD8a (eF450 SK1) (all from Thermo Fisher Scientific), Helios (PE 22F6), PD1 (BV421 EH12.2H7, BV786 EH12.1 or BUV737 EH12.1), CD69 (Pacific Blue FN50) (all from BioLegend), CD25 (PE 4E3), CD271 (FITC ME20.4–1.H4) (all from Miltenyi Biotec), CD127 (APC-AF700 R34.34; Beckman Coulter) and cMyc (AF647 9E10; UBC AbLab). Dead cells were excluded using Fixable Viability Dye (FVD) eFluor™ 780 (ThermoFisher Scientific, cat#: 65-0865-18). Staining for intracellular markers was performed using the Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific, cat#: 00-5523-00). Cell proliferation was measured using Cell Proliferation Dye (CPD) eFluor™ 450 or 670 (Thermo Fisher Scientific, cat#: 65-0842-90 & 65-0840-90).
Cells were sorted using a MoFlow® Astrios (Beckman Coulter) or FACSAria II (BD Biosciences). Flow cytometry data were acquired using an LSRFortessa II (BD Biosciences) or CytoFLEX (Beckman Coulter) and analyzed using FlowJo X (Tree Star).
2.7. Treg-specific demethylated region (TSDR) analysis
DNA was isolated and bisulphite converted using the EZ Direct Kit (Zymo Research, cat#: D5021). The TSDR was PCR-amplified using the AllTaq PCR Core Kit (Qiagen, cat#: 203125) and the following primers: FWD: AGAAATTTGTGGGGTGGGGTAT), REV (biotinylated): ATCTACATCTAAACCCTATTATCACAACC. PCR products were run on a Q96 MD pyrosequencing system (Qiagen) using the following sequencing primer: AGAAATTTGTGGGGTGGG. Data were analyzed using Pyro Q-CpG software (Biotage) by comparing cytosine vs. thymine incorporation at 7 CpG sites in the TSDR.
2.8. Protein synthesis
BL21(DE3)pLysS bacteria (Thermo Fisher Scientific, cat#: C602003) were transformed to express recombinant FliC from enteroaggregative E. coli 042 serotype O44:H18 (GenBank accession: AF194946) or recombinant A4-Fla2 (termed Fla2) from Lachnospiraceae family bacteria A4 (GenBank accession: DQ789126) as previously described [41, 51]. Briefly, bacteria were grown in LB supplemented with 34 μg/mL chloramphenicol (Fisher Scientific, cat#: PB904), 100 μg/mL ampicillin (Fisher Scientific, cat#: PB1760) and induced with 1 mM isopropyl β-D-1-thiogalactopyranoside. The bacteria were lysed by repeated freeze/thawing and sonication. His-tagged proteins were purified using cobalt charged columns (Clontech, cat#: 635501) and eluted with 200 mM imidazole.
Purified protein preparations were depleted of endotoxins using Polymyxin B – Agarose columns (Sigma-Aldrich, cat#: P1411) and considered endotoxin-free when a negative Pyrotell lysate test (endotoxin levels < 0.03 U/mL; Associates of Cape Cod Inc., cat#: CS003) was achieved. Protein was quantified using the Pierce BCA Kit (Thermo Fisher Scientific, cat#: 23227), as per manufacturer’s protocols. Biotinylated rFliC and rFla2 were prepared using the EZ-link Sulfo-NHS-LC-biotin reagent (Thermo Fisher Scientific, cat#: 21335) according to the manufacturer’s protocol.
2.9. Bacteria lysis and flagella enrichment
042 and HS E. coli were cultured in LB overnight, pelleted by centrifugation, resuspended in PBS and blended at high speed for 1 minute in a Sorvall Omni mixer (Sorvall Products). Bacterial debris was removed by centrifugation at 3900 g for 15 minutes and supernatants were harvested and clarified by sequential 0.45 and 0.22 μm filtration. Flagella were then collected by centrifuging for 90 minutes at 100,000 g and resuspended in PBS. Protein concentrations were determined by BCA assay and flagella presence was confirmed by direct ELISA with 5D4 antibody.
2.10. Activation assays
Rested Tregs were washed and resuspended in complete X-vivo™ 15 media (described above) supplemented with 100 U/mL IL-2. For assays with plate-bound rFliC/rFla2, wells were pre-coated by incubating with 0.5, 1, 5 or 10 μg/mL rFliC or rFla2 (diluted in PBS) overnight at 4°C or for 2 hours at 37°C. Alternatively, soluble fragmented flagella were added directly to Treg cultures at concentrations of 0.5, 1, 5 or 10 μg/mL. Tregs were cultured for 24 hours at 37°C after which expression of activation markers was assessed by flow cytometry, as described above.
For cytokine secretion assays, cells were stimulated with plate-bound proteins for 48 hours. Cell culture supernatants were then harvested and analyzed for cytokine secretion using a 12-plex LEGENDplex™ HU Th Cytokine Panel (BioLegend, cat#: 741027).
For proliferation assays, rested Tregs were labelled with CPD eFluor™ 450 and cultured in wells coated with 5 μg/mL rFliC/rFla2 at 37°C for 4 days. Cell proliferation was determined by dye dilution, as measured by flow cytometry.
2.11. Suppression assays
For polyclonal assays, Tregs were labelled with CPD eFluor™ 670, serially-diluted and co-cultured with allogeneic CD3+ responder T cells (Tresp) that were labelled with CPD eFluor™ 450 and activated with anti-CD3/CD28 Dynabeads™ (1:16 bead:Tresp ratio; Thermo Fisher Scientific, cat#: 11131D). Assays were performed in complete X-vivo™ 15 media (described above) in 96-well U-bottom plates. Cells were stained and analyzed by flow cytometry after 4 days of culture. Tresp division was measured using division indices and used to calculate percent suppression (inverse of percent Tresp proliferation) relative to Tresps cultured alone.
For antigen-dependent assays, Tregs were labelled with CPD eFluor™ 670 and cultured in plates that were pre-coated with 5 μg/mL rFliC or rFla2 whilst allogeneic CD3+ Tresps were labelled with CPD eFluor™ 450 and stimulated with anti-CD3/CD28 Dynabeads™ (1:2 bead:Tresp ratio). Tregs and Tresps were cultured independently overnight in complete X-vivo™ 15 media (described above) supplemented with 100 U/mL IL-2. Dynabeads™ were then removed from the Tresps and bead-free Tresps were co-cultured with serially-diluted Tregs in complete X-vivo™ 15 media supplemented with 100 U/mL IL-2. After 3 days of co-culture, cells were stained and analyzed as described for the polyclonal suppression assay.
2.12. Human colonoid monolayers
Intestinal biopsies were used to establish adult colonic stem cell-derived epithelial cell cultures (colonoids) that were plated as monolayers on 0.4 μm Transwell® inserts (VWR, cat#: 76313–906), as previously described [46, 52]. Briefly, colonoids from Matrigel domes were dislodged using a cell scraper and incubated in Cultrex Organoid Harvesting solution (R&D Systems, cat#: 3700-100-01) at 4°C for 90 minutes. Cells were washed, treated with TrypLE Express (Thermo Fisher Scientific, cat#: 12604021) for 1 minute at 37°C, washed and resuspended in IntestiCult™ Organoid Growth Medium (OGM; STEMCELL Technologies, cat#: 06010) supplemented with 50 μg/mL gentamicin (Thermo Fisher Scientific) and 10 μM Rho kinase inhibitor Y-27632 (STEMCELL Technologies, cat#: 72304). Transwells® were pre-treated with 34 μg/mL human collagen IV (Sigma-Aldrich, cat#: C5533) and cells from 1.2 to 1.4 domes were seeded per Transwell®. Cells were incubated at 37°C (v/v 5% CO2) and media were changed every 2 days. Growth was monitored by light microscopy for confluence and transepithelial electrical resistance (TEER) was measured with a Millicell® ERS-2 Volt-Ohm meter, as previously described [46].
2.13. Colonoid barrier assay
Assays were performed when TEER measurements of colonoid monolayers reached a minimum of 500 Ω (10–12 days post-plating). Rested Tregs were resuspended in OGM supplemented with 50 μg/mL gentamicin, 10 μM Y-27632 and 100 U/mL IL-2 and added to wells of a 24-well plate that were pre-coated with 5 μg/mL rFliC or rFla2. Plates were centrifuged for 1 minute at 600 g after which colonoid monolayer-containing Transwells® were seated into the Treg-containing wells. Apical media was immediately refreshed and cells were incubated at 37°C. TEER measurements were taken 1.5 (day 0), 24 and 48 hours after the cocultures were established to monitor colonoid monolayer integrity. Probes were equilibrated in PBS and three TEER measurements were taken per well and averaged. Data is reported at % change in TEER relative to the measurements recorded on day 0.
2.14. In vivo trafficking model
CD4+ and CD8+ T cells were enriched from total PBMCs using EasySep™ Human CD4 and CD8 Positive Selection Kits II (STEMCELL Technologies, cat#: 17852 & 17853), respectively. Enriched T cells were stimulated with anti-CD3/CD28 Dynabeads® (Thermo Fisher Scientific, 3:1 bead:cell ratio) and cultured in Immunocult™ (STEMCELL Technologies, cat#: 10981) supplemented with penicillin-streptomycin. CD4+ and CD8+ T cells were cultured with 100 U/mL and 200 U/mL IL-2, respectively. Fresh media and IL-2 were replenished every 2–3 days. Cells were transduced 24 hours post-stimulation with 10 MOI lentivirus in plates that were pre-coated with 25 μg/mL RetroNectin® (Takara Bio, cat#: T100B) and blocked with 2% BSA. Transduced cells were ΔNGFR-enriched on day 7 of culture, as described above, and cultured for an additional 2 (CD4+ cells) or 3 (CD8+ cells) days before being frozen in Immunocult™ with 10% DMSO.
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratory) were bred in-house and maintained under sterile conditions. 11–12 week old male and female mice were randomly assigned to each treatment group and intravenously (tail vein) administered 2.5×106 CD4+ CAR T cells, 2.5×106 CD8+ CAR T cells and 5×106 autologous PBMCs that were thawed on the day of injection. Control mice received PBS. Thawed cells were >90% CAR+ΔNGFR+ and >70% viable (phosphatidylserine negative, Nuclear Green DCS1 negative and CytoCalcein Violet 450 positive; Apoptosis/Necrosis Detection Kit, Abcam, cat#: ab176749) at the time of injection. Human cell engraftment was tracked by weekly bleeding from the saphenous vein. 15 days post-injection, mice were anaesthetized with isoflurane and administered a single 50 μL enema of 2.5 mg dinitrobenzene sulfonic acid (DNBS; Alfa Aesar, cat#: B21430) in 50% ethanol, 3–4 cm into the colon [53, 54]. Mice were euthanized by isoflurane overdose and cervical dislocation 3 days post-enema, or earlier if pre-determined endpoint criteria were met (scoring based on weight, activity, posture, fur texture, skin integrity, signs of pain, stool consistency, dehydration and rectal bleeding).
Upon euthanasia, blood was collected by cardiac puncture and red blood cells were lysed with ammonium chloride (STEMCELL Technologies, cat#: 07850). Spleens were dissociated through 70 μm cell strainers to obtain a single cell suspension. Colon tissue was digested with collagenase VIII (Sigma-Aldrich, cat#: C2139) and lamina propria lymphocytes (LPL) were acquired using Percoll density centrifugation (GE Healthcare, cat#: 17-0891-02), as previously described [55]. Resulting lymphocytes from all tissues were stained and analyzed by flow cytometry.
All experiments were carried out in accordance with the National Research Council’s Guide for the Care and Use of Laboratory Animals.
2.15. Statistical analyses
Data was analyzed using GraphPad Prism 8 (La Jolla) and presented as mean ± SEM with the contribution of each donor shown. Statistical significance was determined using paired or unpaired two-tailed Student’s t-tests, one-way ANOVA or two-way ANOVA with the Geisser-Greenhouse correction. P-values are provided throughout where p<0.05 was considered significant.
3. RESULTS
3.1. FliC-CAR expression confers specificity for recombinant FliC
The flagellin protein FliC, derived from the enteroaggregative E. coli 042 strain (O44:H18 serotype), was selected as a model antigen as this protein is often targeted by lymphocytes in IBD patients [41]. BALB/c mice were immunized with recombinant FliC (rFliC) protein to develop a hybridoma cell line (termed 5D4) that was used to generate a FliC-specific scFv sequence [47]. This scFv sequence was cloned into a lentiviral vector immediately upstream of sequences encoding i) a cMyc 9E10 mAb epitope, ii) a CD8α hinge domain, iii) a CD28 transmembrane domain and iv), a CD28-CD3ζ signaling domain, thereby generating a second-generation CAR construct (Figure 1A). A control CAR was constructed using a similar approach by incorporating a previously characterized HER-specific scFv sequence into the same CAR backbone vector [24].
Figure 1 |. FliC-CAR-expressing cells specifically engage FliC.
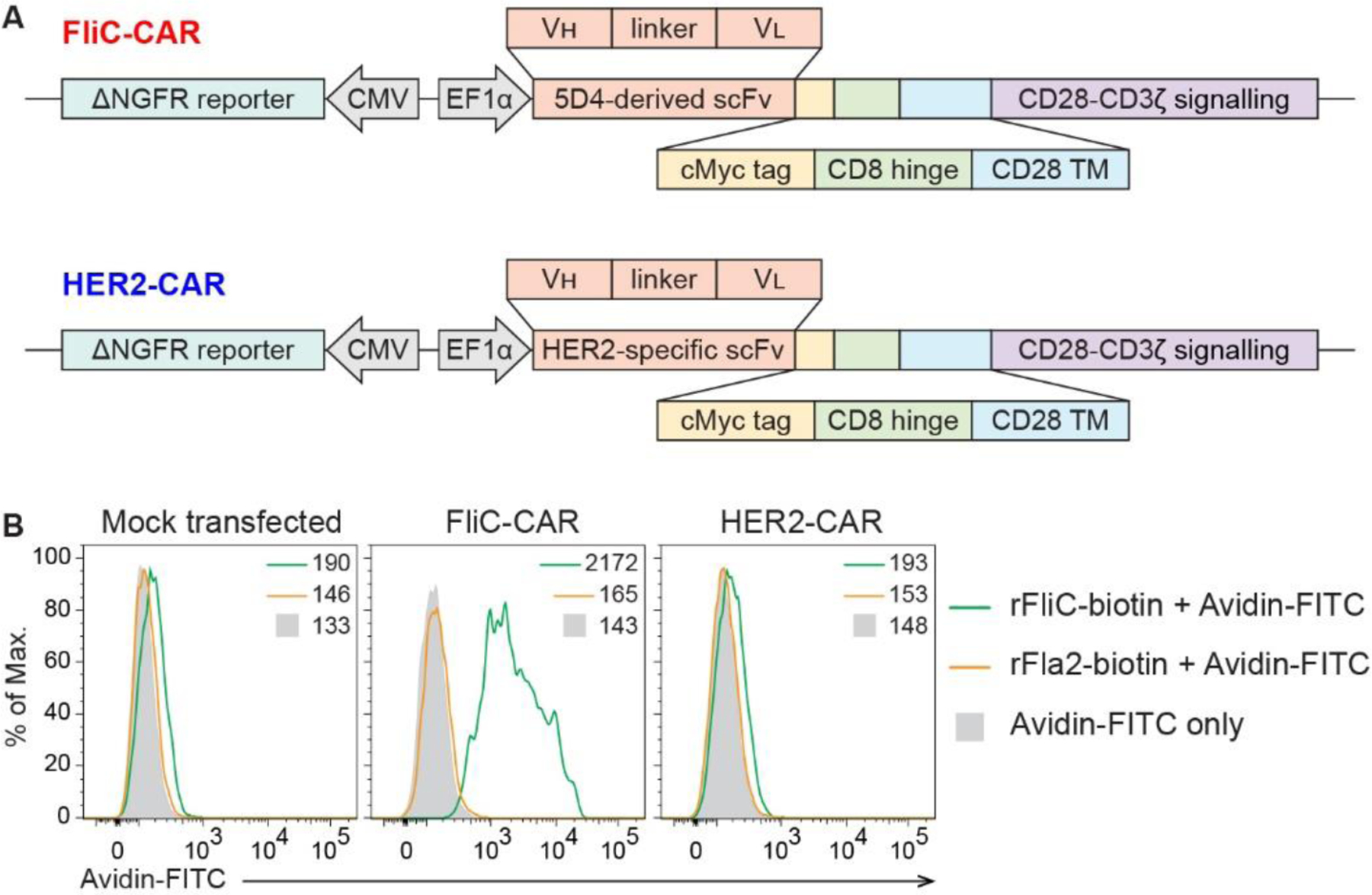
(A) Schematic diagrams of the FliC-CAR (top) and the control HER2-CAR (bottom). (B) Engagement of biotinylated rFliC and rFla2 by HEK293Ts that were transiently transfected to express the FliC-CAR or HER2-CAR. Data were acquired by flow cytometry and gated on cMyc+ΔNGFR+ cells, where possible. MFI values are provided. Representative of 2 individual experiments.
To confirm that the 5D4-derived antigen-targeting moiety maintained the ability to engage FliC when incorporated into a CAR construct, HEK293T cells were transiently transfected to express the FliC-CAR and their ability to recognize biotinylated-rFliC and avidin-FITC was assessed by flow cytometry (Figure 1B). Control cells were either mock transfected or transfected with the irrelevant HER2-CAR [24]. A phylogenetically distant recombinant flagellin protein Fla2, derived from Lachnospiraceae bacteria A4, served as a negative control antigen. rFliC binding was only observed in cells transfected with the FliC-CAR and these cells did not bind rFla2. Overall, adaptation of the 5D4 monoclonal antibody into a CAR preserved antigen specificity.
3.2. FliC-CAR Tregs maintain their phenotype and suppressive capacity
To generate human FliC-CAR Tregs, naïve CD4+CD25hiCD127loCD45RA+CD45RO− cells were sorted from the peripheral blood of healthy subjects, stimulated in a polyclonal manner, lentivirally transduced with a CAR construct after 24 hours and expanded in the presence of IL-2 for 7 days (Figure 2A). Control CD4+CD25−CD127+CD45RA+CD45RO− Tconvs were expanded in parallel, without viral transduction. Cell surface phenotypic analysis of the Tregs on day 7 showed that the cells were transduced to express their respective CARs (cMyc staining) and the ΔNGFR reporter with a high efficiency (Figure 2B). CAR+ cells were enriched as ΔNGFR+ cells to a purity of >95% (FliC-CAR: 97.3 ± 1.2%; HER2-CAR: 96.4 ± 1.2%, n=14) and rested overnight prior to functional assays.
Figure 2 |. CAR-transduced Tregs maintain their phenotype and suppressive function.
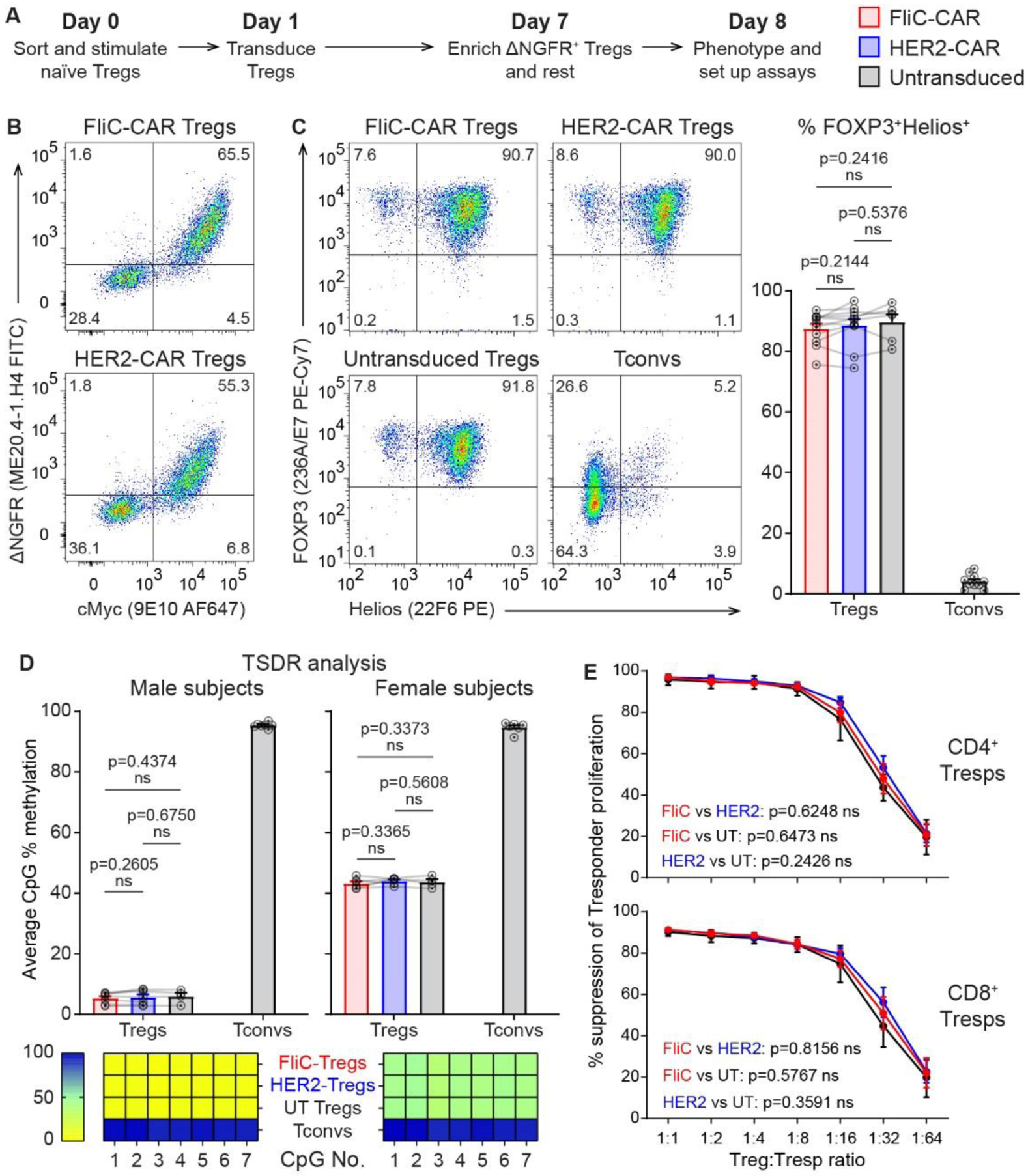
(A) Timeline for generating human CAR Tregs. (B) CAR and ΔNGFR expression before enrichment of ΔNGFR+ cells on day 7. (C) FOXP3 and Helios expression on day 8. (D) CpG methylation in the TSDR as determined by pyrosequencing on day 8. (E) Suppression of polyclonally-stimulated Tresp proliferation by CAR Tregs in the absence of CAR target antigens. Data in (B), (C) and (E) were acquired by flow cytometry, gated on live CD4+ cells. Averaged data are mean ± SEM with each connected series representing one individual. n=6–17 with data collected in 2–6 individual experiments. Statistical significance was determined using paired two-tailed Student’s t-tests (C/D) and two-way ANOVA (E) with p-values shown.
Following expansion, both the FliC-CAR and HER2-CAR Tregs maintained their expected high co-expression of FOXP3 and Helios (Figure 2C) and demethylation of the TSDR locus (Figure 2D). When activated in a polyclonal manner via anti-CD3/CD28 stimulation, FliC-CAR, HER2-CAR and untransduced Tregs were equally suppressive towards responder T cells (Figure 2E). Overall, expression of the FliC-CAR did not alter the phenotype or function of human Tregs.
3.3. FliC-CAR Tregs are activated in the presence of their target antigen
To test the function of the FliC-CAR, CAR Tregs were stimulated with the target antigen rFliC, or the negative control antigen rFla2, and expression of activation markers was measured. In the presence of rFliC, FliC-CAR Tregs significantly upregulated CD69, PD1, LAP and GARP in an antigen and dose-dependent manner; no activation was observed in the presence of rFla2 (Figure 3A–C). Conversely, HER2-CAR Tregs were not activated in the presence of either rFliC or rFla2. To exclude the possibility that this was a TLR5-mediated effect [33], CAR Tregs were stimulated with a rFliC mutant lacking the TLR5 recognition domain, termed 2H3 [56]. FliC-CAR Tregs were strongly activated by 2H3, confirming that Treg activation was mediated through the FliC-CAR and not TLR5 (Supplemental Figure 1). FliC-CAR Tregs also secreted the immunosuppressive cytokine IL-10 and proliferated in the presence of rFliC (Supplemental Figure 2). Conversely, no detectable cytokine secretion and minimal proliferation were observed by FliC-CAR Tregs in the presence of rFla2 or by HER2-CAR Tregs stimulated with either antigen.
Figure 3 |. FliC-CAR Tregs are activated in the presence of their target antigen.

FliC-CAR (red) and HER2-CAR (blue) Tregs were cultured in plates coated with varying concentrations of rFliC and rFla2 (A-C) Activation-induced expression of CD69 (A), PD1 (B), LAP and GARP (C) was measured by flow cytometry after 24 hours, gated on live CD4+ cells. (D) FliC-CAR and HER2-CAR Tregs were activated with soluble fragmented flagella from 042 or HS bacteria and expression of the indicated markers was analyzed after 24 hours. Averaged data are mean + SEM with each connected series representing one individual. n=4–6 with data collected in 2–3 individual experiments. Statistical significance was determined using one-way ANOVA with p-values shown.
In individuals with IBD, FliC-CAR Tregs would be expected to recognize flagellin as part of intact or fragmented flagella on intestinal bacteria, in addition to secreted, soluble flagellin. To confirm that FliC-CAR Tregs could be activated by natural (i.e. non-recombinant) bacterial antigens, CAR Tregs were stimulated with purified fragmented flagella obtained from 042 E. coli (from which the rFliC protein is derived) or the non-pathogenic commensal HS E. coli strain (which expresses a different flagellin) [57] and activation marker expression was assessed after 24 hours [51]. FliC-CAR Tregs significantly upregulated CD69, PD1, LAP and GARP in the presence of 042 flagella but not HS flagella whilst HER2-CAR Tregs were not activated by either flagella target (Figure 3D). Overall, these results demonstrate that the FliC-CAR is functional and that Tregs expressing this CAR are specifically activated in the presence of FliC in a TLR5-independent manner.
3.4. FliC-CAR expression in T cells promotes trafficking to the intestine
To effectively control IBD, a suitable Treg therapy should preferentially migrate to damaged intestinal tissues and respond to the presence of flagellin in the lamina propria. We and others have previously demonstrated that CARs can promote the trafficking of Tregs to tissues where their cognate antigen is expressed [25, 26, 58, 59]. To this end, we next investigated whether expression of the FliC-CAR could promote trafficking of human T cells to the inflamed intestine. In this experiment, Tconvs were used instead of Tregs due to cell number limitations. Immunodeficient NSG mice were reconstituted with a mixture of human CD4+ CAR T cells, CD8+ CAR T cells and PBMCs to aid CAR-T cell engraftment. Human CD45+ cell engraftment was measured in the peripheral blood on a weekly basis (Supplemental Figure 3). After a 15-day engraftment period, mice were administered a DNBS enema to induce intestinal inflammation and euthanized for analysis 3 days later at which point immune cells in the peripheral blood, spleen and colon were analyzed by flow cytometry (Figure 4A).
Figure 4 |. FliC-CAR T cells are activated in the intestine of humanized mice.
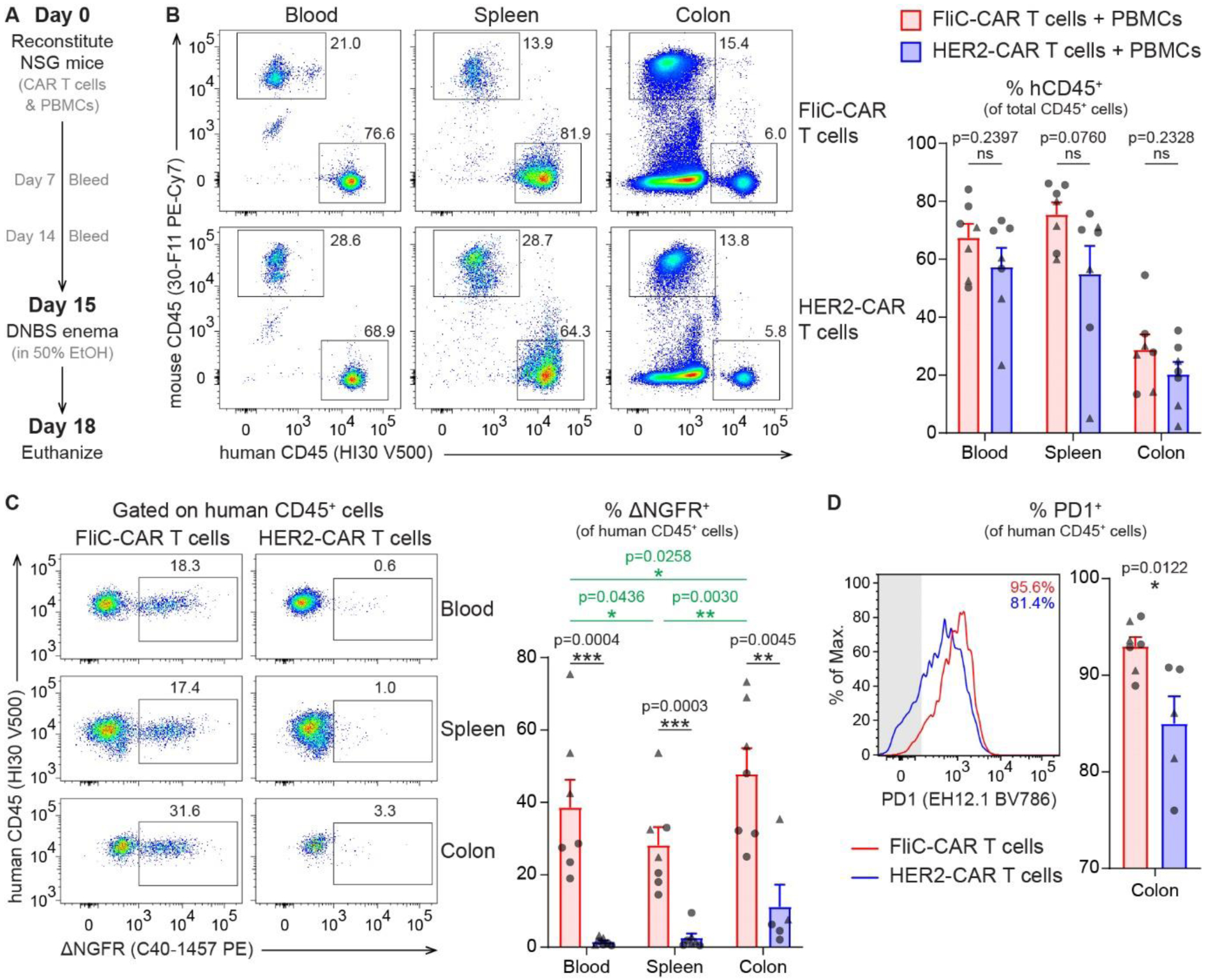
NSG mice were injected intravenously with 2.5 million CD4+ CAR T cells, 2.5 million CD8+ CAR T cells and 5 million PBMCs. After a 2-week reconstitution period, mice were administered a DNBS enema and euthanized for analysis 3 days later or when pre-established end-point criteria were met. (A) Timeline for humanized mouse model. (B) Human CD45+ cell engraftment in the peripheral blood (obtained from cardiac puncture), spleen and colon at time of euthanasia. (C) % ΔNGFR+ cells from total human CD45+ cells in the peripheral blood, spleen and colon at time of euthanasia. (D) % PD1+ cells from total human CD45+ cells in the colon at time of euthanasia. All data were acquired by flow cytometry and gated on total live cells (B) or live human CD45+ cells (C-D). Averaged data are mean + SEM with each point representing an individual mouse. n=7 per group with data collected in 2 individual experiments. Statistical significance between mice treated with FliC-CAR and HER2-CAR T cells was determined using unpaired two-tailed Student’s t-tests (black lines/text) whilst statistical significance between tissue types was determined using paired two-tailed Student’s t-tests (green lines/text) with p-values shown. Triangles and circles represent mice euthanized 2 and 3 days following a DNBS enema, respectively. For (C) and (D), data were excluded if the total number of events in the human CD45+ gate was < 500.
Mice injected with FliC-CAR and HER2-CAR T cells had a similar proportion of human CD45+ cells in each of the tissues analyzed (Figure 4B). However, within this human CD45+ population, a significantly greater proportion of cells were ΔNGFR+ in the mice that received the FliC-CAR T cells compared to mice that received HER2-CAR T cells (Figure 4C and Supplemental Figure 3). This demonstrates that the FliC-CAR T cells had a survival advantage over the HER2-CAR T cells. Further analysis of the mice injected with the FliC-CAR T cells revealed that there were significantly more ΔNGFR+ cells in the colon compared to the blood (p=0.0258) and spleen (p=0.0030), providing evidence that the FliC-CAR T cells preferentially migrate to the colon (Figure 4C). Additionally, a high proportion of the intestinal FliC-CAR T cells were PD1+, indicating that they were more activated in the colon than the HER2-CAR T cells (p=0.0122) (Figure 4D). Overall, these data show that T cells expressing the FliC-CAR preferentially home to the intestine and are activated in the inflamed colon.
3.5. FliC-CAR Tregs mediate antigen-specific immunosuppression and promote colonoid monolayer establishment
To evaluate whether expression of the FliC-CAR conferred a functional advantage to human Tregs, antigen-dependent suppression assays were performed in which the ability of the Tregs to inhibit the proliferation of polyclonally-stimulated responder T cells (Tresp) was evaluated. FliC-CAR and HER2-CAR Tregs were stimulated in wells coated with 5 μg/mL rFliC or rFla2 and allogeneic Tresps were independently stimulated using anti-CD3/CD28 beads. After 16 hours of independent culture, the anti-CD3/CD28 beads were removed from the Tresps and these cells were co-cultured for an additional 3 days with the Tregs (Figure 5A). FliC-CAR Tregs significantly inhibited the proliferation of CD4+ and CD8+ Tresps in the presence of rFliC whilst no suppression was observed in the presence of the negative control protein rFla2 (Figure 5B). Conversely, control cells including HER2-CAR Tregs, untransduced Tregs and polyclonal ΔNGFR-expressing Tregs were not suppressive in the presence of either rFliC or rFla2 (Figure 5B and Supplemental Figure 4).
Figure 5 |. FliC-CAR Tregs are suppressive and promote intestinal epithelial resistance in the presence of rFliC.
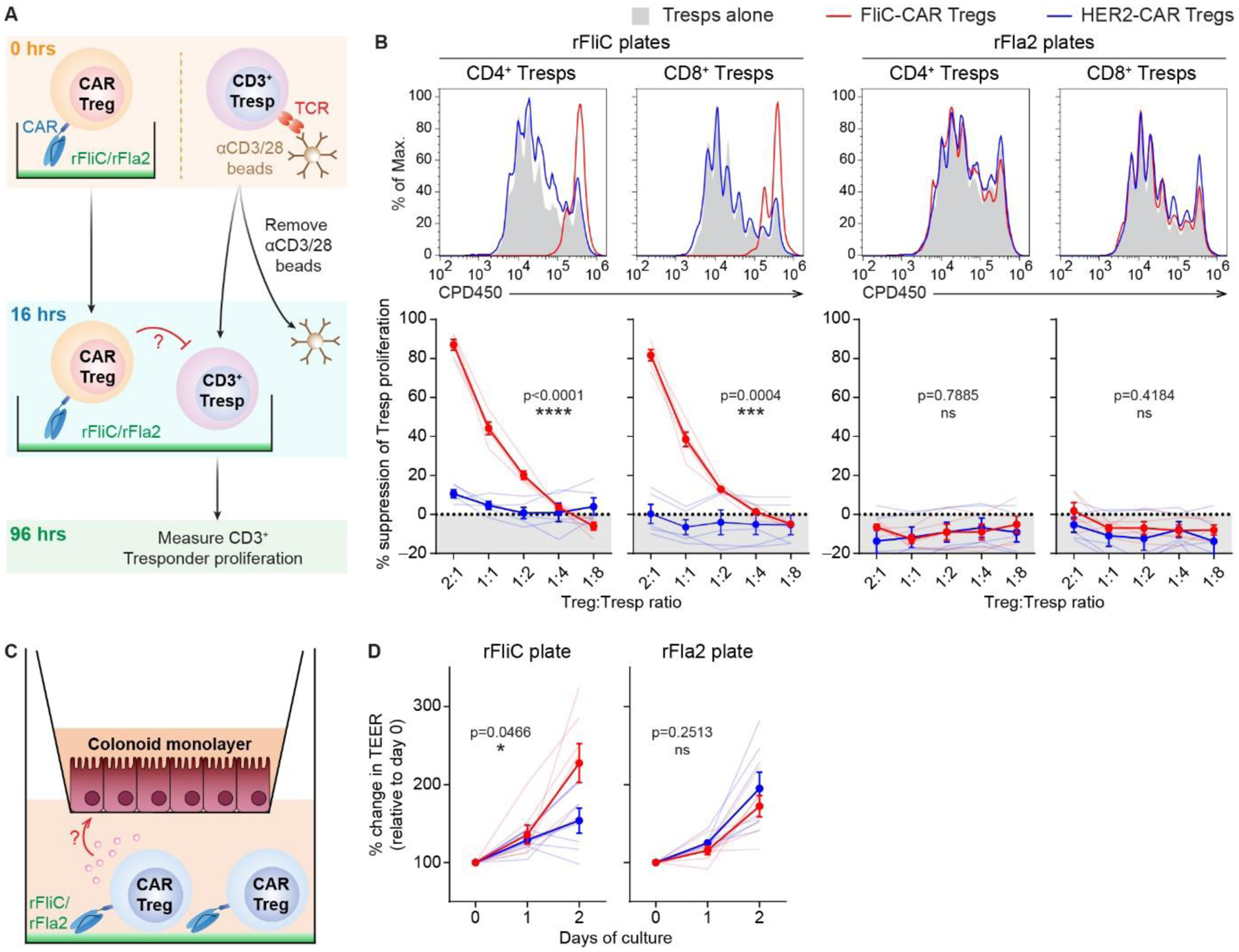
(A) Schematic diagram of suppression assay setup. CD3+ responder T cells (Tresp) were labelled with CPD eFluor™ 450 and stimulated with anti-CD3/CD28 beads. CAR Tregs were labelled with CPD eFluor™ 670 and independently cultured in plates pre-coated with 5 μg/mL rFliC or rFla2. Stimulated Tresps were harvested after 16 hours, depleted of anti-CD3/CD28 beads and added to the CAR Treg cultures. (B) CD4+ and CD8+ Tresp proliferation was measured after 4 days of culture by flow cytometry, gated on live CD4+CPDeFluor™450+CDPeFluor™670− and live CD8+CPDeFluor™450+CDPeFluor™670− cells, respectively. Representative data show Tresp proliferation following co-culture with CAR Tregs at a 2:1 Treg:Tresp ratio. % suppression of Tresp proliferation was calculated relative to Tresps that were cultured alone in the presence of 5 μg/mL plate-bound rFliC or rFla2. (C) Schematic diagram of colonoid resistance assay setup. 2D-colonoids were established on 0.4 μm Transwell® inserts and grown until a transepithelial electrical resistance (TEER) of > 500 Ω was achieved. Transwell® inserts were then transferred to plates pre-coated with 5 μg/mL rFliC or rFla2 and 1×106 Tregs were added to the basolateral compartment. (D) TEER measurements were taken 1.5 (day 0), 24 (day 1) and 48 (day 2) hours following assay setup. Data are shown as change in TEER relative to the day 0 readings. Averaged data are mean ± SEM and shown as solid lines. Faint lines represent individual subjects. n=5–7 with data collected in 2–4 individual experiments. Statistical significance was determined using two-way ANOVA with p-values shown.
As well as dampening undesired immune responses, it is well-established that Tregs also have a tissue reparative function. We have previously demonstrated that Tregs and Tr1 cells can affect epithelial cell biology and barrier function [60]. As such, we next explored whether FliC-CAR Tregs could enhance the barrier integrity of intestinal epithelial monolayers in the presence of rFliC using colonoid monolayers. Human colonic crypt-derived epithelial organoids (colonoids) were grown on Transwell® inserts that were placed into wells containing CAR Tregs stimulated with 5 μg/mL plate-bound rFliC or rFla2 (Figure 5C). In this setup, activated Tregs could secrete soluble factors that would influence the barrier integrity of human colonoid monolayers. Colonoid monolayer integrity was monitored by changes in the transepithelial electrical resistance (TEER), which increases upon the formation of tight junctions [61]. Over a two-day culture period, FliC-CAR Tregs cultured in the presence of rFliC were significantly more effective than HER2-CAR Tregs at increasing the electrical resistance of colonoid monolayers whilst no differences between these cell types was observed in the presence of rFla2. Overall, these results demonstrate that FliC-CAR expression significantly enhances the ability of human Tregs to suppress target cells and enhance colonic epithelial barrier integrity in the presence of rFliC.
4. DISCUSSION
CAR Treg therapy has emerged as a promising, and potentially curative, treatment option for a variety of inflammatory disorders, autoimmune diseases and transplant rejection [6]. Here, we adapted this strategy to generate CAR Tregs with the potential to treat IBD. Using a previously unexplored approach, we developed a novel CAR specific for a subset of flagellins and used this CAR to confer specificity to human Tregs. By targeting a microbial antigen that is only accessible in the lamina propria with intestinal barrier disruption, we hypothesized that Treg activity would be physically and temporally restricted to intestinal regions where inflammation is occurring. We found that FliC-CAR Tregs recognized and responded to soluble target flagella in a TLR5-independent manner and that expression of this CAR promoted intestinal trafficking in a humanized mouse model. FliC-CAR Tregs were also potently immunosuppressive and promoted the barrier integrity of human colonoids in the presence of their target flagellin antigen in vitro. These data not only provide support for the clinical development of flagellin-specific CAR Tregs for use in IBD, but also show the potential utility of CARs to target microbial proteins. This paradigm may also have important implications in other conditions; for example, using CAR T cells to prevent or treat colorectal cancer, which is known to be associated with specific bacterial taxa and toxic products [62, 63].
Studies using mouse Tregs have previously explored the concept of using CAR Tregs to treat colitis. In initial studies, CAR Tregs were directed towards the model antigen 2,4,6-trinitrophenol (TNP) such that they could be used to treat 2,4,6-trinitrobenzene sulphonic acid (TNBS)-induced colitis [59, 64]. Despite not targeting a disease-relevant antigen, these studies had important implications as they demonstrated that CAR Tregs exerted bystander immunosuppression and could ameliorate colitis induced by either TNBS or the unrelated hapten oxazolone. Subsequent studies investigated mouse CAR Tregs specific for carcinoembryonic antigen (CEA) [65] and found that these cells alleviated dextran sulphate sodium (DSS)-induced colitis and suppressed the development of colorectal cancer, an important complication of UC.
In our study, we used a novel approach to generate intestinal antigen-specific Tregs by directing these cells to FliC, a clinically relevant antigen that is frequently targeted by abnormal immune responses in individuals with IBD [41]. To our knowledge, this is the first example of a CAR being developed to target a clinically relevant antigen associated with IBD. Flagellins are expressed by a variety of commensal and pathogenic bacteria and are typically recognized by the innate human immune system by engaging TLR5. However, multiple studies have found that compared to healthy controls, individuals with IBD also have a higher proportion of flagellin-specific B cells [34–39] and T cells [40–42], making this an ideal target for Treg therapy. By using a CAR that specifically targeted flagellin from E. coli H18 (FliC) and not Lachnospiraceae-derived A4-Fla2, this proof-of-concept study suggests that Tregs could be tailored to function in the intestine under precise circumstances such as when inflammation is driven by certain types of pathological/immunogenic bacteria. Whilst the suitability of the FliC-CAR in a clinical setting will depend on the precise microbiota of the individual [41], the concept of targeting a luminal antigen that is predominantly accessible during active intestinal inflammation could represent a significant improvement over current drug-based therapies that deliver continuous, non-specific immunosuppression. Targeting such antigens will also prolong the lifespan of the delivered Tregs and limit the chances these cells succumbing to exhaustion as a consequence of overstimulation [66, 67].
We have previously demonstrated in a transplantation context that expression of a HLA-A2-specific CAR can drive antigen-dependent in vivo trafficking to HLA-A2+ tissues [68]. To investigate whether FliC-CAR expression promoted intestinal homing, we developed a novel humanized mouse model in which immunodeficient NSG mice were reconstituted with human immune cells and administered a DNBS hapten enema to induce intestinal inflammation, prior to the onset of xenogeneic graft-versus-host disease (xeno-GvHD) [53, 69, 70]. In this model, we found that FliC-CAR expression in human T cells promoted intestinal trafficking and that these cells exhibited evidence of colon-specific activation. However, we also observed a high level of background activation, with > 80% of the colonic HER2 CAR-T cells expressing PD1. Whilst this can be attributed to TCR-mediated xenogeneic stimulation of these cells, this is an important limitation of these experiments that should be noted. The implications of these findings are also limited by the fact that we used CAR Tconvs instead of CAR Tregs to study the trafficking bias that is driven by FliC-CAR expression.
Testing the efficacy of novel IBD therapies in a translational manner is complicated by the lack of clinically relevant humanized mouse models. It is well-established that NSG mice reconstituted with human PBMCs succumb to xeno-GvHD prior to developing spontaneous colitis [69]. Intervening with a hapten enema can promote colitis-like pathology before the onset of xeno-GvHD [53, 70] but these studies have suggested that the intestinal pathology is not driven by the engrafted human immune cells. As such, we were unable to investigate the curative potential of our CAR Tregs in vivo.
Unlike autoimmune conditions, where defects in the Treg population contribute to disease pathology [71], studies have suggested that Tregs in individuals with IBD are functional and that intestinal inflammation is instead driven by either an insufficient number of intestinal Tregs [13] or Tconvs that resist Treg-mediated suppression [7, 72, 73]. These observations directly support the rationale for seeking a Treg therapy approach to simultaneously boost the numbers and local function of Tregs in IBD. GMP-compatible Treg expansion protocols have demonstrated that therapeutically relevant numbers Tregs can be obtained using cells from individuals with IBD and that these cells are stable and functionally suppressive [74, 75].
4.1. Conclusion
CARs offer a powerful approach to significantly enhance the potency of Treg therapy products. This approach is being investigated clinically in the context of organ transplantation (NCT04817774 and NCT04838171) and promises to be an important next step for improving the efficacy of Treg therapies that are currently under clinical investigation for treating IBD (NCT03185000 and NCT04691232). We demonstrate that conferring flagellin-specificity to Treg therapy products is remarkably advantageous as it enhances the potency of these cells and promotes Tregs to only elicit their immunosuppressive and tissue reparative functions when required. This is an interesting advancement for the treatment of IBD that warrants further clinical investigation.
Supplementary Material
HIGHLIGHTS.
Chimeric antigen receptors (CAR) can be used to confer microbe specificity to Tregs
Flagellin-specific CAR Tregs preferentially home to damaged intestinal tissues
CAR Tregs elicit antigen-dependent immunosuppression
Activated CAR Tregs secrete factors that promote intestinal barrier integrity
ACKNOWLEDGEMENTS
We thank Lisa Xu for her assistance in cell sorting, Jana Gillies for performing the TSDR methylation analyses.
This work was supported by grants awarded to TSS and MKL (Crohn’s & Colitis Canada and Michael Smith Foundation for Health Research I2C-2018-2073). The authors would also like to acknowledge the Integrated Physiology Core of the Hopkins Conte Digestive Disease Basic and Translational Research Core Center (NIH NIDDK P30-DK089502).
DAB was supported by fellowships from the Canadian Institutes for Health Research (CIHR) and Michael Smith Health Research BC. JVS was supported a by 4YF from the University of British Columbia and a Vanier scholarship from CIHR. WDR was supported by a 4YF from the University of British Columbia and a Mitacs award. MKL is Canada Research Chair in Immune Engineering and receives a Scientist Salary Award from the BC Children’s Hospital Research Institute.
DECLARATION OF INTEREST
MKL received research funding from Bristol-Myers Squibb and Takeda for work unrelated to this report. TSS received research funding from Merck, Sanofi Pasteur, Rebiotix, Seres, NuBiyota, Edesa, Summit, and Pfizer for work unrelated to this project. The other authors have no commercial or financial interests to report.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].de Souza HS, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol, 2016;13:13–27. [DOI] [PubMed] [Google Scholar]
- [2].Danese S, Vuitton L, Peyrin-Biroulet L. Biologic agents for IBD: practical insights. Nat Rev Gastroenterol Hepatol, 2015;12:537–45. [DOI] [PubMed] [Google Scholar]
- [3].Hazel K, O’Connor A. Emerging treatments for inflammatory bowel disease. Ther Adv Chronic Dis, 2020;11:2040622319899297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Baumgart DC, Le Berre C. Newer Biologic and Small-Molecule Therapies for Inflammatory Bowel Disease. N Engl J Med, 2021;385:1302–15. [DOI] [PubMed] [Google Scholar]
- [5].Brunstein CG, Blazar BR, Miller JS, Cao Q, Hippen KL, McKenna DH et al. Adoptive transfer of umbilical cord blood-derived regulatory T cells and early viral reactivation. Biol Blood Marrow Transplant, 2013;19:1271–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Boardman DA, Levings MK. Emerging strategies for treating autoimmune disorders with genetically modified Treg cells. J Allergy Clin Immunol, 2022;149:1–11. [DOI] [PubMed] [Google Scholar]
- [7].Clough JN, Omer OS, Tasker S, Lord GM, Irving PM. Regulatory T-cell therapy in Crohn’s disease: challenges and advances. Gut, 2020;69:942–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dunkin D, Berin MC, Mondoulet L, Tobar S, Yeretssian G, Tordesillas L et al. Epicutaneous Tolerance Induction to a Bystander Antigen Abrogates Colitis and Ileitis in Mice. Inflamm Bowel Dis, 2017;23:1972–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jacobse J, Li J, Rings E, Samsom JN, Goettel JA. Intestinal Regulatory T Cells as Specialized Tissue-Restricted Immune Cells in Intestinal Immune Homeostasis and Disease. Front Immunol, 2021;12:716499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mizoguchi A, Yano A, Himuro H, Ezaki Y, Sadanaga T, Mizoguchi E. Clinical importance of IL-22 cascade in IBD. J Gastroenterol, 2018;53:465–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zaiss DM, van Loosdregt J, Gorlani A, Bekker CP, Grone A, Sibilia M et al. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity, 2013;38:275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity, 2008;28:546–58. [DOI] [PubMed] [Google Scholar]
- [13].Maul J, Loddenkemper C, Mundt P, Berg E, Giese T, Stallmach A et al. Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology, 2005;128:1868–78. [DOI] [PubMed] [Google Scholar]
- [14].Okou DT, Mondal K, Faubion WA, Kobrynski LJ, Denson LA, Mulle JG et al. Exome sequencing identifies a novel FOXP3 mutation in a 2-generation family with inflammatory bowel disease. J Pediatr Gastroenterol Nutr, 2014;58:561–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chabod M, Pedros C, Lamouroux L, Colacios C, Bernard I, Lagrange D et al. A spontaneous mutation of the rat Themis gene leads to impaired function of regulatory T cells linked to inflammatory bowel disease. PLoS Genet, 2012;8:e1002461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Izcue A, Hue S, Buonocore S, Arancibia-Carcamo CV, Ahern PP, Iwakura Y et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity, 2008;28:559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pedros C, Duguet F, Saoudi A, Chabod M. Disrupted regulatory T cell homeostasis in inflammatory bowel diseases. World J Gastroenterol, 2016;22:974–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mottet C, Uhlig HH, Powrie F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J Immunol, 2003;170:3939–43. [DOI] [PubMed] [Google Scholar]
- [19].MacDonald KN, Piret JM, Levings MK. Methods to manufacture regulatory T cells for cell therapy. Clin Exp Immunol, 2019;197:52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ferreira LMR, Muller YD, Bluestone JA, Tang Q. Next-generation regulatory T cell therapy. Nat Rev Drug Discov, 2019;18:749–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Raffin C, Vo LT, Bluestone JA. Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol, 2020;20:158–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Voskens C, Stoica D, Rosenberg M, Vitali F, Zundler S, Ganslmayer M et al. Autologous regulatory T-cell transfer in refractory ulcerative colitis with concomitant primary sclerosing cholangitis. Gut, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Boardman D, Maher J, Lechler R, Smyth L, Lombardi G. Antigen-specificity using chimeric antigen receptors: the future of regulatory T-cell therapy? Biochem Soc Trans, 2016;44:342–8. [DOI] [PubMed] [Google Scholar]
- [24].MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest, 2016;126:1413–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D et al. Expression of a Chimeric Antigen Receptor Specific for Donor HLA Class I Enhances the Potency of Human Regulatory T Cells in Preventing Human Skin Transplant Rejection. Am J Transplant, 2017;17:931–43. [DOI] [PubMed] [Google Scholar]
- [26].Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R et al. Prevention of Allograft Rejection by Use of Regulatory T Cells With an MHC-Specific Chimeric Antigen Receptor. Am J Transplant, 2017;17:917–30. [DOI] [PubMed] [Google Scholar]
- [27].Lee S-K, Han J, Piao H, Shin N, Jang JY, Yan J-J et al. Anti-C4d chimeric antigen receptor regulatory T cells suppressed allograft rejection in ABO-incompatible heart transplantation. Genes & Diseases, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yoon J, Schmidt A, Zhang AH, Konigs C, Kim YC, Scott DW. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood, 2017;129:238–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Imura Y, Ando M, Kondo T, Ito M, Yoshimura A. CD19-targeted CAR regulatory T cells suppress B cell pathology without GvHD. JCI Insight, 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation, 2012;9:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Alexander KL, Zhao Q, Reif M, Rosenberg AF, Mannon PJ, Duck LW et al. Human Microbiota Flagellins Drive Adaptive Immune Responses in Crohn’s Disease. Gastroenterology, 2021;161:522–35 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Caron G, Duluc D, Fremaux I, Jeannin P, David C, Gascan H et al. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J Immunol, 2005;175:1551–7. [DOI] [PubMed] [Google Scholar]
- [33].Crellin NK, Garcia RV, Hadisfar O, Allan SE, Steiner TS, Levings MK. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J Immunol, 2005;175:8051–9. [DOI] [PubMed] [Google Scholar]
- [34].Targan SR, Landers CJ, Yang H, Lodes MJ, Cong Y, Papadakis KA et al. Antibodies to CBir1 flagellin define a unique response that is associated independently with complicated Crohn’s disease. Gastroenterology, 2005;128:2020–8. [DOI] [PubMed] [Google Scholar]
- [35].Papadakis KA, Yang H, Ippoliti A, Mei L, Elson CO, Hershberg RM et al. Anti-flagellin (CBir1) phenotypic and genetic Crohn’s disease associations. Inflamm Bowel Dis, 2007;13:524–30. [DOI] [PubMed] [Google Scholar]
- [36].Lodes MJ, Cong Y, Elson CO, Mohamath R, Landers CJ, Targan SR et al. Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest, 2004;113:1296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Schoepfer AM, Schaffer T, Mueller S, Flogerzi B, Vassella E, Seibold-Schmid B et al. Phenotypic associations of Crohn’s disease with antibodies to flagellins A4-Fla2 and Fla-X, ASCA, p-ANCA, PAB, and NOD2 mutations in a Swiss Cohort. Inflamm Bowel Dis, 2009;15:1358–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Schoepfer AM, Schaffer T, Seibold-Schmid B, Muller S, Seibold F. Antibodies to flagellin indicate reactivity to bacterial antigens in IBS patients. Neurogastroenterol Motil, 2008;20:1110–8. [DOI] [PubMed] [Google Scholar]
- [39].Sitaraman SV, Klapproth JM, Moore DA 3rd, Landers C, Targan S, Williams IR et al. Elevated flagellin-specific immunoglobulins in Crohn’s disease. Am J Physiol Gastrointest Liver Physiol, 2005;288:G403–6. [DOI] [PubMed] [Google Scholar]
- [40].Wu J, Pendegraft AH, Byrne-Steele M, Yang Q, Wang C, Pan W et al. Expanded TCRbeta CDR3 clonotypes distinguish Crohn’s disease and ulcerative colitis patients. Mucosal Immunol, 2018;11:1487–95. [DOI] [PubMed] [Google Scholar]
- [41].Cook L, Lisko DJ, Wong MQ, Garcia RV, Himmel ME, Seidman EG et al. Analysis of Flagellin-Specific Adaptive Immunity Reveals Links to Dysbiosis in Patients With Inflammatory Bowel Disease. Cell Mol Gastroenterol Hepatol, 2020;9:485–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Calderon-Gomez E, Bassolas-Molina H, Mora-Buch R, Dotti I, Planell N, Esteller M et al. Commensal-Specific CD4(+) Cells From Patients With Crohn’s Disease Have a T-Helper 17 Inflammatory Profile. Gastroenterology, 2016;151:489–500 e3. [DOI] [PubMed] [Google Scholar]
- [43].Rhee SH, Im E, Riegler M, Kokkotou E, O’Brien M, Pothoulakis C. Pathophysiological role of Toll-like receptor 5 engagement by bacterial flagellin in colonic inflammation. Proc Natl Acad Sci U S A, 2005;102:13610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kumara MT, Srividya N, Muralidharan S, Tripp BC. Bioengineered flagella protein nanotubes with cysteine loops: self-assembly and manipulation in an optical trap. Nano Lett, 2006;6:2121–9. [DOI] [PubMed] [Google Scholar]
- [45].Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, Chen YY. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol, 2018;14:317–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rees WD, Telkar N, Lin DTS, Wong MQ, Poloni C, Fathi A et al. An in vitro chronic damage model impairs inflammatory and regenerative responses in human colonoid monolayers. Cell Rep, 2022;38:110283. [DOI] [PubMed] [Google Scholar]
- [47].Ivison SM, Himmel ME, Hardenberg G, Wark PA, Kifayet A, Levings MK et al. TLR5 is not required for flagellin-mediated exacerbation of DSS colitis. Inflamm Bowel Dis, 2010;16:401–9. [DOI] [PubMed] [Google Scholar]
- [48].de Waal Malefyt R, Verma S, Bejarano MT, Ranes-Goldberg M, Hill M, Spits H. CD2/LFA-3 or LFA-1/ICAM-1 but not CD28/B7 interactions can augment cytotoxicity by virus-specific CD8+ cytotoxic T lymphocytes. Eur J Immunol, 1993;23:418–24. [DOI] [PubMed] [Google Scholar]
- [49].Boardman DA, Garcia RV, Ivison SM, Bressler B, Dhar TM, Zhao Q et al. Pharmacological inhibition of RORC2 enhances human Th17-Treg stability and function. Eur J Immunol, 2020;50:1400–11. [DOI] [PubMed] [Google Scholar]
- [50].Cossarizza A, Chang HD, Radbruch A, Abrignani S, Addo R, Akdis M et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (third edition). Eur J Immunol, 2021;51:2708–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Steiner TS, Nataro JP, Poteet-Smith CE, Smith JA, Guerrant RL. Enteroaggregative Escherichia coli expresses a novel flagellin that causes IL-8 release from intestinal epithelial cells. J Clin Invest, 2000;105:1769–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Staab JF, Lemme-Dumit JM, Latanich R, Pasetti MF, Zachos NC. Co-Culture System of Human Enteroids/Colonoids with Innate Immune Cells. Curr Protoc Immunol, 2020;131:e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Goettel JA, Kotlarz D, Emani R, Canavan JB, Konnikova L, Illig D et al. Low-Dose Interleukin-2 Ameliorates Colitis in a Preclinical Humanized Mouse Model. Cell Mol Gastroenterol Hepatol, 2019;8:193–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wallace JL, Le T, Carter L, Appleyard CB, Beck PL. Hapten-induced chronic colitis in the rat: alternatives to trinitrobenzene sulfonic acid. J Pharmacol Toxicol Methods, 1995;33:237–9. [DOI] [PubMed] [Google Scholar]
- [55].Wu D, Wong MQ, Vent-Schmidt J, Boardman DA, Steiner TS, Levings MK. A method for expansion and retroviral transduction of mouse regulatory T cells. J Immunol Methods, 2021;488:112931. [DOI] [PubMed] [Google Scholar]
- [56].Donnelly MA, Steiner TS. Two nonadjacent regions in enteroaggregative Escherichia coli flagellin are required for activation of toll-like receptor 5. J Biol Chem, 2002;277:40456–61. [DOI] [PubMed] [Google Scholar]
- [57].Levine MM, Bergquist EJ, Nalin DR, Waterman DH, Hornick RB, Young CR et al. Escherichia coli strains that cause diarrhoea but do not produce heat-labile or heat-stable enterotoxins and are non-invasive. Lancet, 1978;1:1119–22. [DOI] [PubMed] [Google Scholar]
- [58].Dawson NAJ, Rosado-Sanchez I, Novakovsky GE, Fung VCW, Huang Q, McIver E et al. Functional effects of chimeric antigen receptor co-receptor signaling domains in human regulatory T cells. Sci Transl Med, 2020;12. [DOI] [PubMed] [Google Scholar]
- [59].Elinav E, Waks T, Eshhar Z. Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice. Gastroenterology, 2008;134:2014–24. [DOI] [PubMed] [Google Scholar]
- [60].Cook L, Stahl M, Nazli A, MacDonald K, Wong M, Dizzell S et al. Human Tr1 cells exhibit unique suppressive and gut reparative functions. Gastroenterol, 2019;Revisions under review. [DOI] [PubMed] [Google Scholar]
- [61].Srinivasan B, Kolli AR, Esch MB, Abaci HE, Shuler ML, Hickman JJ. TEER measurement techniques for in vitro barrier model systems. J Lab Autom, 2015;20:107–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol, 2012;10:575–82. [DOI] [PubMed] [Google Scholar]
- [63].Avril M, DePaolo RW. “Driver-passenger” bacteria and their metabolites in the pathogenesis of colorectal cancer. Gut Microbes, 2021;13:1941710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Elinav E, Adam N, Waks T, Eshhar Z. Amelioration of colitis by genetically engineered murine regulatory T cells redirected by antigen-specific chimeric receptor. Gastroenterology, 2009;136:1721–31. [DOI] [PubMed] [Google Scholar]
- [65].Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther, 2014;22:1018–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Lamarthée B, Marchal A, Charbonnier S, Blein T, Leon J, Martin E et al. Transient mTOR inhibition rescues 4–1BB CAR-Tregs from tonic signal-induced dysfunction. Nature Communications, 2021;12:6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Lamarche C, Novakovsky GE, Qi CN, Weber EW, Mackall CL, Levings MK. Repeated stimulation or tonic-signaling chimeric antigen receptors drive regulatory T cell exhaustion. bioRxiv, 2020:2020.06.27.175158. [Google Scholar]
- [68].Dawson NA, Lamarche C, Hoeppli RE, Bergqvist P, Fung VC, McIver E et al. Systematic testing and specificity mapping of alloantigen-specific chimeric antigen receptors in regulatory T cells. JCI Insight, 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ali N, Flutter B, Sanchez Rodriguez R, Sharif-Paghaleh E, Barber LD, Lombardi G et al. Xenogeneic graft-versus-host-disease in NOD-scid IL-2Rgammanull mice display a T-effector memory phenotype. PLoS One, 2012;7:e44219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Nolte T, Zadeh-Khorasani M, Safarov O, Rueff F, Gulberg V, Herbach N et al. Oxazolone and ethanol induce colitis in non-obese diabetic-severe combined immunodeficiency interleukin-2Rgamma(null) mice engrafted with human peripheral blood mononuclear cells. Clin Exp Immunol, 2013;172:349–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Selck C, Dominguez-Villar M. Antigen-Specific Regulatory T Cell Therapy in Autoimmune Diseases and Transplantation. Front Immunol, 2021;12:661875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Fantini MC, Rizzo A, Fina D, Caruso R, Sarra M, Stolfi C et al. Smad7 controls resistance of colitogenic T cells to regulatory T cell-mediated suppression. Gastroenterology, 2009;136:1308–16, e1–3. [DOI] [PubMed] [Google Scholar]
- [73].Himmel ME, Yao Y, Orban PC, Steiner TS, Levings MK. Regulatory T-cell therapy for inflammatory bowel disease: more questions than answers. Immunology, 2012;136:115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Canavan JB, Scotta C, Vossenkamper A, Goldberg R, Elder MJ, Shoval I et al. Developing in vitro expanded CD45RA+ regulatory T cells as an adoptive cell therapy for Crohn’s disease. Gut, 2016;65:584–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Wiesinger M, Stoica D, Roessner S, Lorenz C, Fischer A, Atreya R et al. Good Manufacturing Practice-Compliant Production and Lot-Release of Ex Vivo Expanded Regulatory T Cells As Basis for Treatment of Patients with Autoimmune and Inflammatory Disorders. Front Immunol, 2017;8:1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.