

Summary
Apoptotic cell (AC) clearance (efferocytosis) is performed by phagocytes, such as macrophages, that inhabit harsh physiological environments. Here, we find macrophages display enhanced efferocytosis under prolonged (chronic) physiological hypoxia, characterized by increased internalization and accelerated degradation of ACs. Transcriptional and translational analysis revealed that chronic physiological hypoxia induces two distinct but complimentary states. The first, ‘primed’ state consists of concomitant transcription and translation of metabolic programs in AC-naïve macrophages that persist during efferocytosis. The second, ‘poised’ state consists of transcription, but not translation, of phagocyte function programs in AC-naïve macrophages that are translated during efferocytosis. Mechanistically, macrophages efficiently flux glucose into a noncanonical pentose phosphate pathway (PPP) loop to enhance NADPH production. PPP-derived NADPH directly supports enhanced efferocytosis under physiological hypoxia by ensuring phagolysosomal maturation and redox homeostasis. Thus, macrophages residing under physiological hypoxia adopt states that support cell fitness and ensure performance of essential homeostatic functions rapidly and safely.
eTOC
Tissues pose distinct challenges that macrophages must overcome, including limited nutrient availability (glucose, oxygen), to perform essential functions such as apoptotic cell clearance (efferocytosis). Here, Wang et al. find that macrophages undergo novel metabolic adaptations to prolonged physiological hypoxia that directly support rapid, efficient efferocytosis.
Graphical Abstract
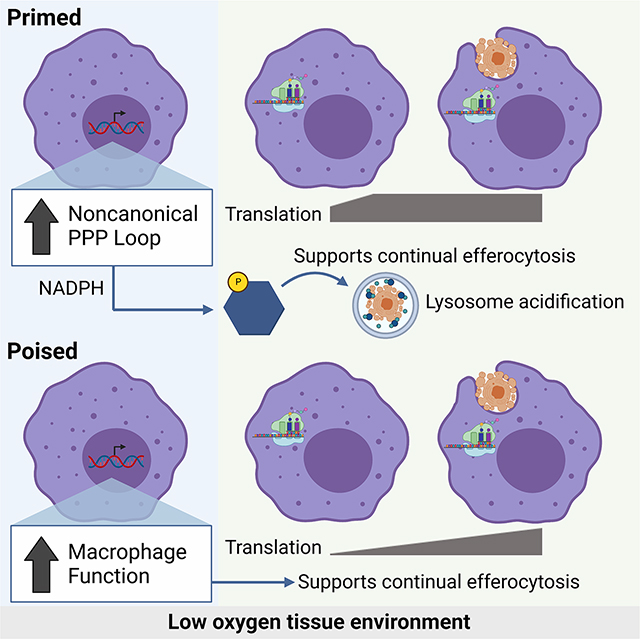
Introduction
Efferocytosis, the phagocytic clearance of apoptotic cells (ACs), is indispensable for organismal homeostasis1–3. Efferocytosis occurs in all major tissues and organs, ensuring that disparate processes such as barrier epithelial cell recycling, removal of spent neutrophils and red blood cells, elimination of apoptotic neurons, and clearance of negatively-selected thymocytes are executed rapidly and safely4–6. These clearance processes are performed by various types of phagocytes, especially by tissue-resident macrophages (TRMs7,8). At homeostasis, each tissue exhibits a substantial rate of cellular turnover, collectively amounting to ~1–2% body mass each day9. As the body’s main professional phagocyte, TRMs shoulder the brunt of this massive burden10.
TRMs are generally long-lived cells that seed a tissue early during development and often reside in a tissue throughout the organism’s lifespan11–14. It is now clear that TRMs adapt to their unique and often harsh tissue environment to perform their core functions11,13,15, including residing in tissues with a relative dearth of oxygen availability and exposure to tissue-specific debris and metabolites. For instance, the tissues that feature the most cell turnover (bone marrow, spleen, thymus9,16,17 are also tissues with the lowest oxygen availability (~1% O2, termed physiological hypoxia18–20). TRMs, then, must both adapt to the tissue environment in which they reside and cope with the continuous influx of internalized biological material. Recent work has illustrated mechanisms by which phagocytes sense and respond to internalized ACs21–25, however, how tissue environment factors, such as oxygen availability, inform the ability to perform efferocytosis remains unknown.
Here, we made the striking discovery that exposure to prolonged (‘chronic’) physiological hypoxia, similar to that experienced by several TRM populations, resulted in increased internalization and accelerated degradation of ACs. We found that chronic exposure to physiological hypoxia induced two distinct but complimentary states. One state, which we term ‘primed’, generally consists of simultaneous transcriptional and translational induction or suppression of metabolic programs in AC-naïve macrophages that remain induced or suppressed during efferocytosis. The other state, which we term ‘poised’, generally consists of transcription, without concomitant translation, of phagocytic functional programs in AC-naïve macrophages that are instead translated during efferocytosis. Importantly, we discovered that both states are necessary for enhanced continual efferocytosis. Subsequent exploration of primed state metabolic programs revealed that macrophages exposed to chronic physiological hypoxia switch to the efficient utilization of glucose to generate NADPH via a noncanonical pentose phosphate pathway (PPP) loop that features recycling of PPP-derived intermediates back through the oxidative PPP. The PPP-dependent generation of NADPH prior to efferocytosis served to both support enhanced internalization and degradation of ACs via phagolysosomal maturation and protect phagocytes from runaway oxidative stress. These studies reveal that local tissue environment, in this case physiological hypoxia, programs distinct states in macrophages that simultaneously support cell fitness and ensure the ability to perform critical homeostatic functions, such as efferocytosis.
Results
Efferocytosis is enhanced under prolonged (‘chronic’) physiological hypoxia
To address the effect of prolonged (‘chronic’) physiological hypoxia on efferocytosis, we optimized a system and protocols to continually manipulate and culture primary professional phagocytes (macrophages) in low (1%) oxygen. Strikingly, chronic physiological hypoxia-conditioned macrophages internalized significantly more ACs than both macrophages cultured under atmospheric (‘standard’) oxygen levels (21%) and macrophages exposed to ‘acute’ (3h) hypoxia (Figure 1A). Chronic physiological hypoxia-conditioned macrophages internalized significantly more ACs irrespective of the size of the target cell, the type of target cell, the method of cell death induction, or the transformation status of the target cell (Figure 1B; Figure S1A–D). Furthermore, macrophages internalized significantly more ACs regardless of whether they were initially differentiated in standard oxygen or under physiological hypoxia prior to conditioning (Figure 1C), and this enhanced efferocytosis capacity was lost in chronic physiological hypoxia-conditioned macrophages when transferred back to standard oxygen conditions (Figure S1E).
Figure 1: Efferocytosis is enhanced under prolonged (‘chronic’) physiological hypoxia.
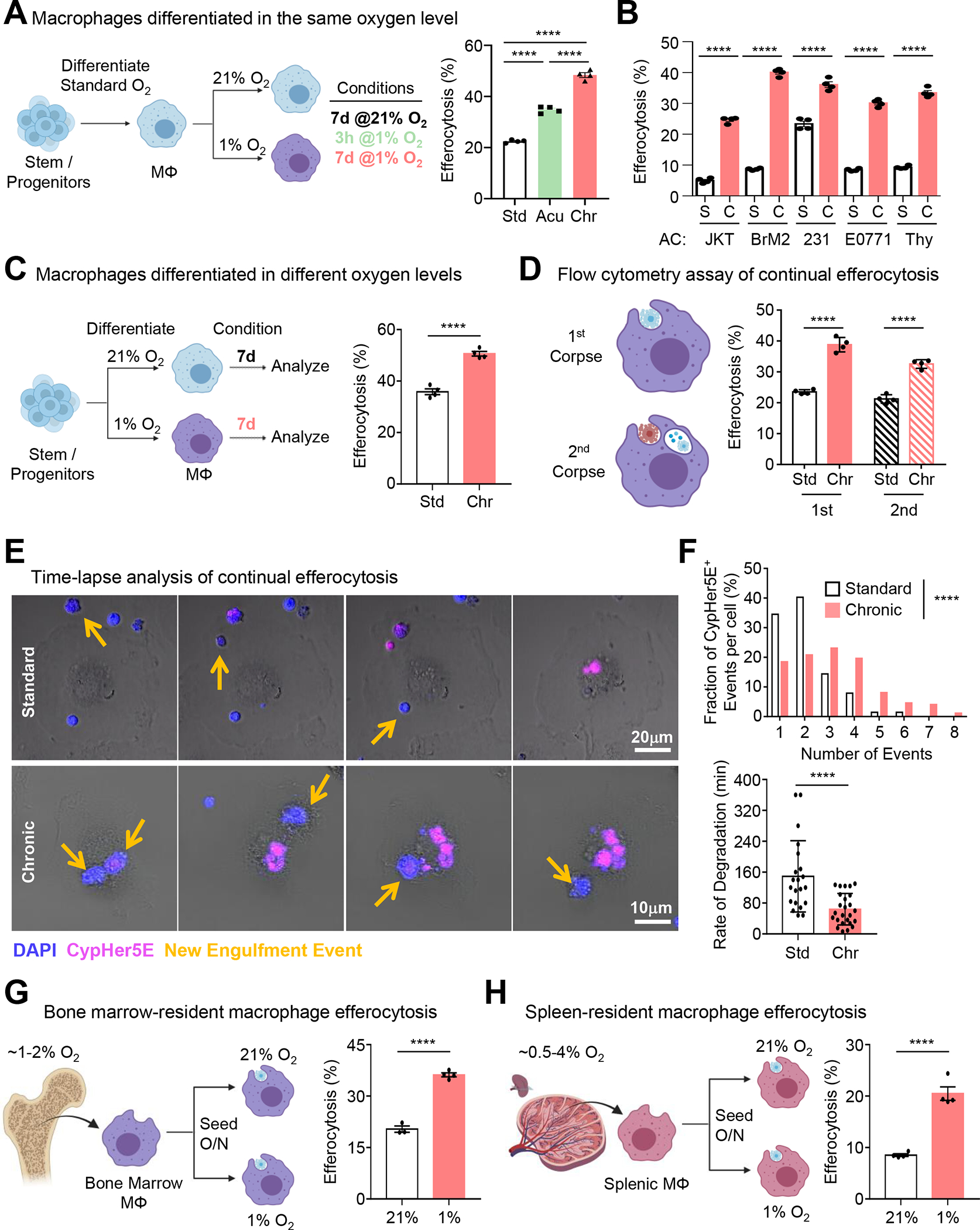
(A) Conditioned macrophages (left) were co-cultured with CypHer5E-labeled apoptotic MDA-MB-231s for 1h, then assessed via flow cytometry. Data are from four independent experiments, shown as mean ± SEM.
(B) Experiments performed as in (A), with the following targets: Jurkats, BrM2s, MDA-MD-231s, E0771s, and thymocytes. Data are from four independent experiments, shown as mean ± SEM.
(C) Conditioned macrophages (left) were co-cultured with CypHer5E-labeled apoptotic MDA-MB-231s for 1h, then assessed via flow cytometry. Data are from three independent experiments, shown as mean ± SEM.
(D) Experiments performed as in (A) but used to assess continual efferocytosis. Data are from three independent experiments, shown as mean ± SEM.
(E, F) Experiments performed as in (A), but imaged via time-lapse confocal microscopy. Yellow arrows indicate newly internalized ACs. (F) Quantification of CypHer5E+ events (top) and rate of degradation of internalized ACs (bottom). For quantification, 115 efferocytotic macrophages from 8 standard oxygen scenes and 155 efferocytotic macrophages from 6 chronic hypoxia scenes were analyzed. Data were binned as number of events per-cell and presented as a fraction of 100%. For analysis of degradation rate, 21 (standard oxygen) and 25 (chronic hypoxia) efferocytotic macrophages were analyzed. Degradation time = time to shrink internalized AC 50% after acidification (CypHer5E+).
(G, H) Bone marrow (G) or splenic (H) macrophages were isolated and seeded at either 1% or 21% oxygen overnight, then co-cultured with CypHer5E-labeled apoptotic MDA-MD-231 cells for 1h. Efferocytosis was assessed via flow cytometry. Data are from four independent experiments, shown as mean ± SEM.
Significance was determined by Student’s t-test in C,F-H, and by one-way ANOVA in A,B,D,F, ****p < .0001.
Professional phagocytes are often responsible for internalization of ACs in quick succession, a phenomenon known as ‘continual efferocytosis’, which protects against the manifestation of inflammatory disease, such as atherosclerosis23,24,26,27. Interestingly, chronic physiological hypoxia-conditioned macrophages also exhibited significantly higher levels of continual efferocytosis (Figure 1D). Despite the increased proportion of macrophages internalize more ACs on a population level, our flow cytometric analysis of the geometric mean fluorescence intensity of AC uptake (a surrogate of per-cell uptake) was similar between conditions (Figure S1F), suggesting that either 1) individual macrophages do not take up more ACs, or 2) chronic physiological hypoxia-conditioned macrophages internalize more ACs and digest internalized ACs quicker on a per-cell basis. To test these hypotheses, we performed time-lapse confocal microscopy of macrophages conditioned in standard oxygen or chronic physiological hypoxia. Indeed, chronic physiological hypoxia-conditioned macrophages not only internalized significantly more ACs on a per-cell basis but also degraded internalized ACs significantly faster (Figure 1E and 1F).
Multiple tissue-resident macrophage populations reside under physiological hypoxia. For instance, bone marrow-resident macrophages and macrophage subsets in the spleen chronically experience oxygen levels as low as ~1%19. To test the in vivo relevance of physiological hypoxia on efferocytosis, we isolated tissue-resident macrophages (TRMs) from either the bone marrow or spleen and seeded them in either standard or 1% oxygen (Figure 1G and 1H). Consistent with our in vitro conditioning studies, we found that both bone marrow and splenic TRMs exhibited higher efferocytosis when maintained in 1% oxygen compared to standard oxygen (Figure 1G and 1H). Collectively, our data suggest that professional phagocytes better internalize and degrade potentially dangerous ACs under prolonged physiological hypoxia.
Characterization of macrophages under chronic physiological hypoxia
To investigate how macrophages adapt to chronic physiological hypoxia, we performed RNA sequencing (RNAseq) of primary macrophages cultured in standard oxygen (21%), exposed to acute hypoxia (1% for 3h), and exposed to prolonged physiological hypoxia (1% for 7d; Figure 2A). Analysis of chronic physiological hypoxia-conditioned macrophages revealed several differentially expressed transcriptional programs (Figure 2B). For instance, we observed significant downregulation of lipid and mitochondrial metabolism programs and upregulation of carbohydrate metabolism and hypoxia-responsive programs (Figure 2B), broadly consistent with a previous study of cancerous cells cultured under chronic hypoxia28. We observed several programs not previously associated with physiological hypoxia. For instance, we observed downregulation of metabolic programs (e.g., amino acid metabolism, insulin signaling), cell biological processes (e.g., endocytosis, vesicular transport), and immunity (e.g., pro-inflammatory, antigen presentation) (Figure 2B; Figure S2A and S2B). Contrarily, we observed upregulation of homeostatic macrophage function programs (e.g., phagocytosis, wound healing), cell biological processes (e.g., lysosome biology, cell adhesion), and pro-tumorigenic programs (Figure 2B; Figure S2A and S2B). Although the majority of the chronic physiological hypoxia-specific program was induced only after prolonged hypoxia, a fraction of the program was modestly, albeit non-significantly, induced in response to acute hypoxia (Chronic vs. Standard Only; Figure 2C). Because these genes significantly increase after prolonged physiological hypoxia, we consider them part of the chronic physiological hypoxia-induced program.
Figure 2: Characterization of macrophages under chronic physiological hypoxia.
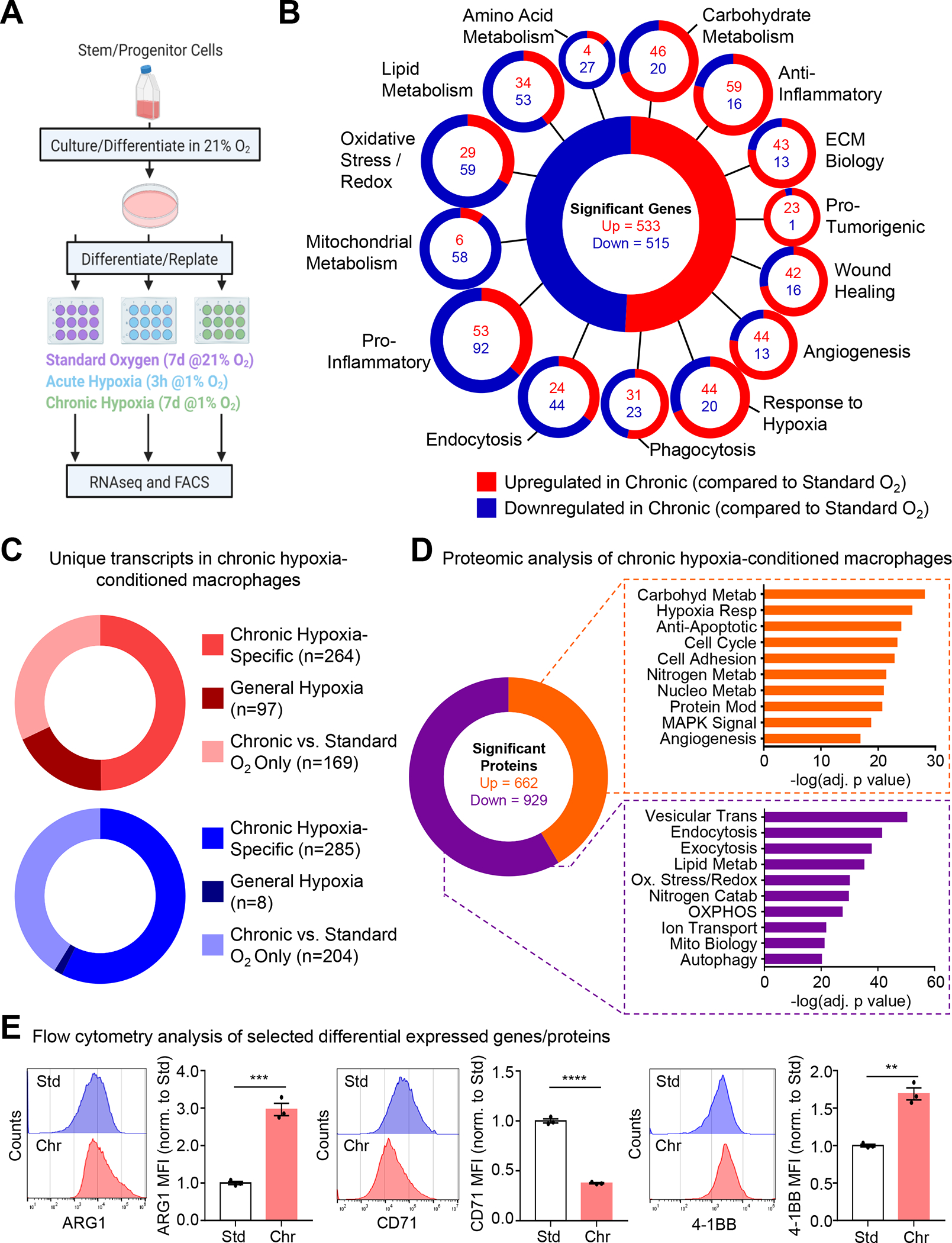
(A, B) (A) Schematic of method used for RNA sequencing analysis. (B) Shown are classifications of differentially expressed genes according to known or putative function. Three independent experiments were analyzed for each condition. ECM, extracellular matrix.
(C) Analysis of data from (A) to identify unique transcripts in chronic hypoxia-conditioned macrophages compared to standard oxygen and acute hypoxia (‘Chronic Hypoxia-Specific’), differentially regulated under acute hypoxia and maintained during chronic conditioning (‘General Hypoxia’), or differentially regulated in chronic hypoxia-conditioned macrophages compared to standard oxygen (‘Chronic vs. Standard O2 Only’).
(D) Similar to (A) but instead analyzed via TMT-labeled mass spectrometry. Shown are differentially expressed proteins classified according to known or putative function. Six independent experiments were analyzed for each condition. MAPK, mitogen-activated protein kinase. OXPHOS, oxidative phosphorylation. Pathway significance was determined using Fisher’s Exact Tests.
(E) Representative flow cytometry histograms and normalized mean fluorescence intensity (MFI) from analysis of macrophages conditioned as in (A). Data represent three independent experiments, shown as mean ± SEM. Significance was determined by Student’s t-test. **p < 0.01. ***p < 0.001. ****p < .0001.
In parallel, we performed proteomic analysis of chronic physiological hypoxia-conditioned macrophages. Our analysis revealed several differentially regulated programs that overlap with programs identified via RNAseq, especially programs involved in cellular metabolism (Figure 2D). We validated representative targets fitting into one of three criteria: 1) a target upregulated in both RNAseq and proteomics analysis, Arginase 1 (ARG1, gene symbol Arg1), 2) a target downregulated in both RNAseq and proteomics analysis, transferrin receptor protein 1 (CD71, gene symbol Tfrc), and 3) a target upregulated in RNAseq but not detected via proteomics, 4–1BB (CD137, gene symbol Tnfrs9) (Figure 2E). Taken together, these data indicate that macrophages induce unique gene and protein programs in response to chronic physiological hypoxia.
Chronic physiological hypoxia induces states both primed and poised for efferocytosis
Beyond differentially-regulated genes that we failed to detect signal for in our proteomics analysis, we also observed that a plurality of genes differentially regulated under chronic hypoxia are detected but remain unchanged at the protein level (Figure S3A). One hypothesis is that macrophages in low oxygen environments accumulate function-specific mRNAs that allow for rapid protein synthesis during the execution of canonical functions. To test this, we used a strategy for performing mass spectrometry analysis of proteins in efferocytotic macrophages while excluding contaminating peptides from internalized ACs. Specifically, we labeled live target cells with 13C-lysine prior to use in efferocytosis assay (Figure 3A and Figure S3B). Analysis of the core chronic physiological hypoxia-induced mRNA program revealed two distinct patterns of protein expression. First, we observed transcriptional programs that were also significantly differentially expressed at the protein level in AC-naïve macrophages, which remained differentially expressed in efferocytotic macrophages (‘Primed’ Programs, Figure 3A; Figure S4, PFKL/Pfkl in Figure 3B). This cluster consisted primarily of cellular metabolism programs. Second, we observed programs differentially regulated at the mRNA level but not the protein level in AC-naïve macrophages, that were subsequently differentially regulated at the protein level in efferocytotic macrophages (‘Poised’ Programs, Figure 3A; Figure S4, SAMD9L/Samd9l in Figure 3B). This cluster consisted primarily of macrophage function programs, including phagocytosis. Thus, chronic physiological hypoxia induces two distinct but complementary states in macrophages. The first ‘primed’ state responds immediately by coupling transcription and translation to prime AC-naïve macrophages for efferocytosis. The second ‘poised’ state features transcriptional changes without concomitant translation in AC-naïve macrophages, instead poising AC-naïve macrophages with function-specific transcripts that are subsequently translated during efferocytosis.
Fig 3: Chronic hypoxia induces states both primed and poised for efferocytosis.
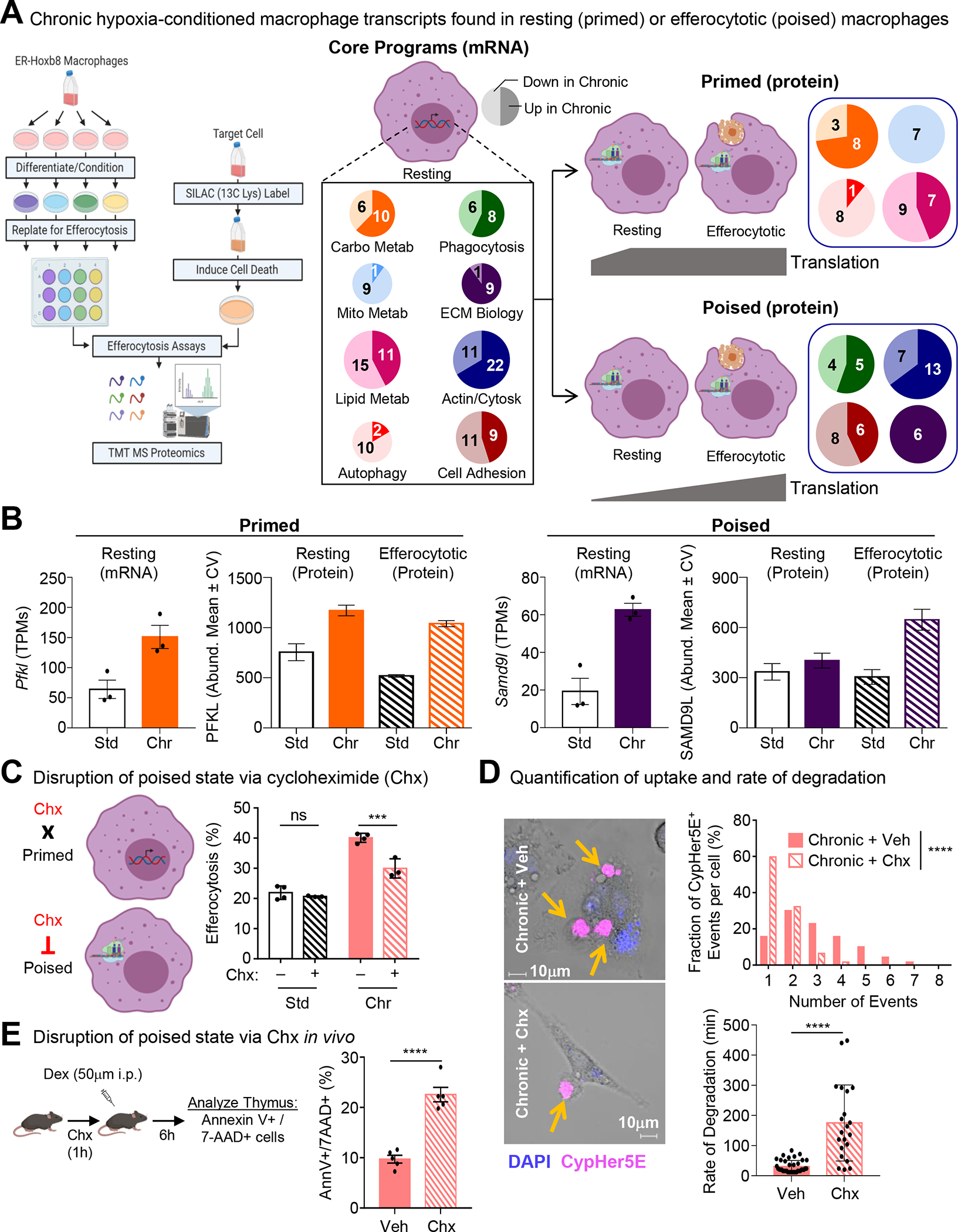
(A) Similar to Figure 1A, except macrophages were co-cultured with apoptotic MDA-MB-231s labeled with 13C-Lysine then analyzed via mass spectrometry. Four independent experiments were analyzed for each condition. (Middle/Right) Differentially regulated mRNA transcripts (Core Programs; Middle) were either concomitantly differentially regulated at the protein level in AC-naïve macrophages (Primed; Top Right) or not differentially regulated in AC-naïve macrophages but instead were differentially regulated in efferocytotic macrophages (Poised; Bottom Right). Numbers represent genes/proteins differentially regulated, with light shading representing downregulated and dark shading representing upregulated.
(B) Shown are representative transcripts/proteins from each state observed in Figure 3A. PFKL/Pfkl, ATP-dependent 6-phosphofructokinase, liver-type. SAMD9L/Samd9l, Sterile alpha motif domain-containing protein 9-like.
(C) (Left) Schematic illustrating the differential effect of cycloheximide (Chx) treatment on ‘primed’ versus ‘poised’ programs. (Right) Experiments performed as in Figure 1A, but with the addition of cycloheximide treatment. Data represent three independent experiments, shown as mean ± SEM.
(D) Experiments performed and analyzed as in Figure 1E. Shown are representative images (Left), quantification of CypHer5E+ events per-cell (Top Right), and analysis of degradation rate (Bottom Right). For quantification, 70 efferocytotic macrophages from 5 vehicle-treated scenes and 62 efferocytotic macrophages from 7 Chx-treated scenes were analyzed. For degradation rate, 38 (vehicle-treated) and 21 (Chx-treated) efferocytotic macrophages were analyzed. Data shown as mean ± SEM.
(E) Schematic of experimental design (Left) and summary plot (Right) of AnnexinV+ 7-AAD+ thymocytes 6h post-dexamethasone (Dex) injection in vehicle (n=5) or cycloheximide (Chx)-treated mice (n=5). Data shown as mean ± SEM.
Significance was determined by Student’s t-test in D,E, and by one-way ANOVA in C,D, ***p < .001, ****p < .0001. ns = not significant.
Our discovery of ‘poised’ state programs raised the question, what purpose does enhanced transcription of core functional genes without concomitant protein synthesis serve? One hypothesis is that translation of ‘poised’ state programs is necessary to support continuous uptake and degradation of ACs observed in chronic physiological hypoxia-conditioned macrophages. To test this, we took advantage of the properties of cycloheximide (Chx) to temporally-disrupt translation with minimal effect on transcription (Figure 3C). Treatment of standard oxygen-conditioned macrophages with Chx immediately prior to culture with ACs did not affect efferocytosis (Figure 3C). Contrarily, treatment of chronic physiological hypoxia-conditioned macrophages with Chx immediately prior to culture with ACs resulted in a significant decrease in efferocytosis (Figure 3C). Furthermore, we tested if disrupting translation affects per-cell internalization and AC degradation rate. We observed that chronic physiological hypoxia-conditioned macrophages treated with Chx internalized significantly fewer ACs on a per-cell basis (Figure 3D, top graph). Additionally, temporally disrupted translation resulted in a slower rate of AC degradation (Figure 3D, bottom graph), suggesting that translation of the ‘poised’ program is essential for rapid, continuous efferocytosis observed in chronic physiological hypoxia-conditioned macrophages.
Finally, we sought to determine if the ‘poised’ program is important for efferocytosis in vivo. Many of the tissues where macrophages perform high efferocytosis burden feature physiological hypoxia8. For instance, macrophages in the thymus are responsible for clearing millions of apoptotic thymocytes daily16,17 despite the thymus featuring physiological hypoxia (10mmHg, or ~1%, O229). Taking advantage of its in vivo activity, we treated mice with Chx or vehicle prior to the induction of thymocyte apoptosis using dexamethasone (Figure 3E). Similar to our in vitro findings, mice treated with Chx exhibited significantly decreased clearance of apoptotic thymocytes (Figure 3E). Thus, the ‘poised’ program observed in chronic physiological hypoxia-conditioned macrophages is necessary for efficient continual efferocytosis in vitro and in vivo.
Mapping the metabolic pathways unique to chronic hypoxia-conditioned macrophages
We further explored macrophage metabolism under chronic physiological hypoxia. We first performed a network analysis of the differentially regulated metabolic programs in ‘primed’ macrophages (Figure 4A, Figure S4A). Our analysis revealed that the broader metabolic programs clustered into specific metabolic pathways. For instance, carbohydrate metabolism clustered into three categories: gluconeogenesis/ glucose transport, one carbon/aldehyde metabolism, and pentose phosphate pathway (PPP; Figure 4A). Consistent with our RNAseq and proteomics analyses, we observed significant differences in glucose uptake by macrophages conditioned in hypoxia (Figure 4B). However, contrary to our expectations, we observed that chronic physiological hypoxia-conditioned macrophages consume significantly less glucose from media than macrophages in standard oxygen (Figure 4B, left graph). Furthermore, chronic physiological hypoxia-conditioned macrophages secrete less lactate (Figure 4B, right graph) and contain less ATP (Figure 4C).
Figure 4: Metabolic pathway use in chronic physiological hypoxia-conditioned macrophages.
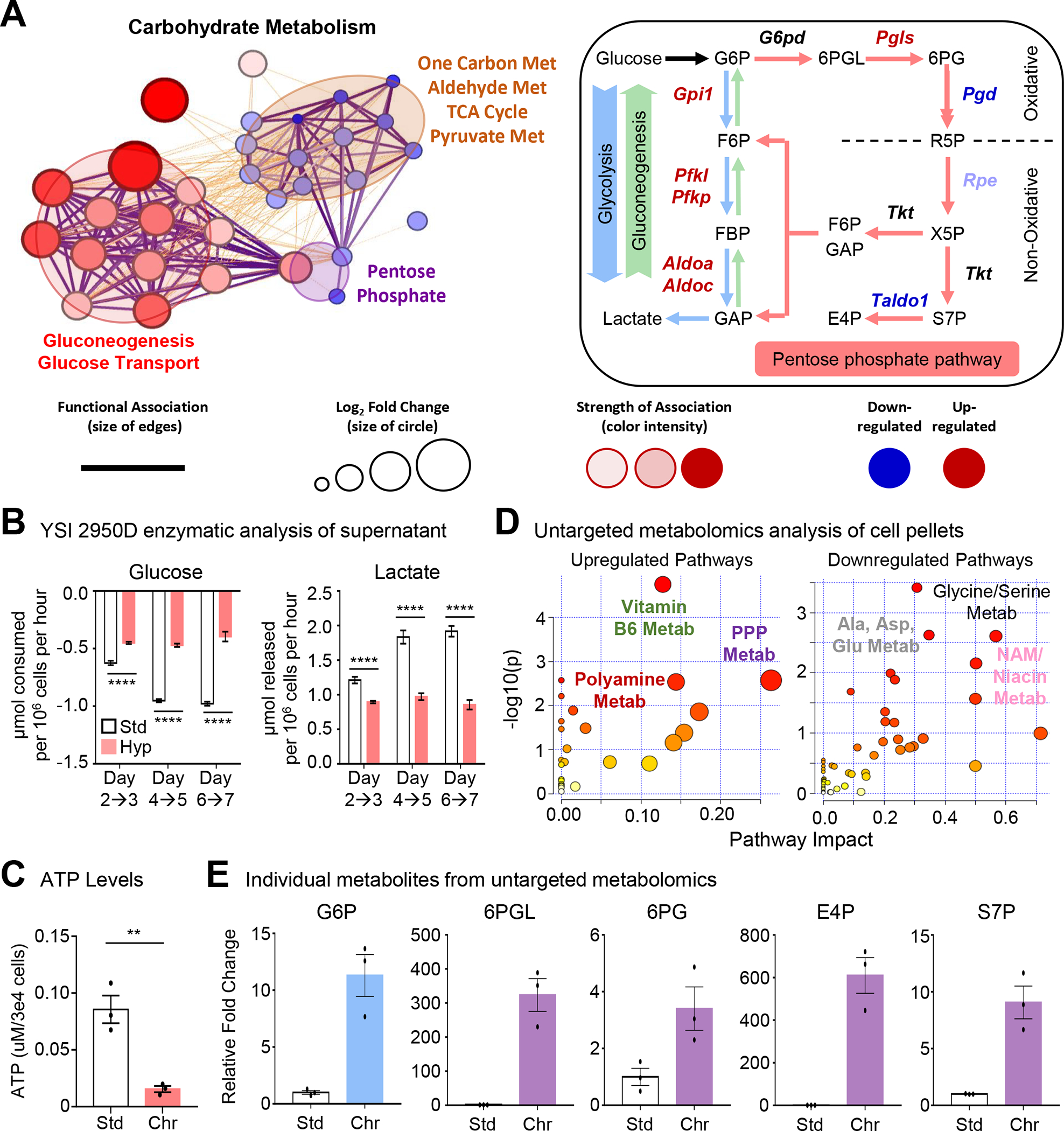
(A) (Left) Differentially regulated metabolic genes (Figure 2) are represented using network analysis. (Right) Schematic of routes of glucose use. Genes in red are significantly upregulated whereas genes in blue are downregulated.
(B) Macrophages cultured as in Figure 1A but with glucose and lactate media levels in the media analyzed. Data is from two independent experiments, with four biological replicates per experiment. Data are shown as mean ± SEM.
(C) Macrophages cultured as in Figure 1A but analyzed for cellular ATP levels. Data is from three independent experiments, shown as mean ± SEM.
(D) Macrophages cultured as in Figure 1A then isolated for untargeted metabolomic analysis. (Left) upregulated and (Right) downregulated pathways in chronic hypoxia-conditioned macrophages are shown. Three independent experiments were performed for each condition.
(E) Shown are representative metabolites from Figure 4C. Data are shown as mean ± SEM. All metabolites are significant, p < .0001. G6P, glucose 6-phosphate. 6PGL, 6-phosphogluconolactone. 6PG, 6-phosphogluconate. E4P, erythrose 4-phosphate. S7P, sedoheptulose 7-phosphate.
Significance was determined by Student’s t-test in C, and by one-way ANOVA in B, **p < .01, ****p < .0001.
Our findings that chronic physiological hypoxia-conditioned macrophages consume and utilize less glucose was surprising given previous studies30–32. To further elucidate the metabolic state of chronic physiological hypoxia-conditioned macrophages, we performed untargeted metabolomics analysis. Chronic physiological hypoxia-conditioned macrophages exhibited a different metabolite profile than macrophages from either standard or acute hypoxia conditions (Figure S4B). Our analysis revealed significant upregulation of PPP, polyamine, and vitamin B6 metabolism (Figure 4D and 4E; Figure S4C). On the other hand, we observed downregulation of alanine/aspartate/glutamine, glycine/serine (one carbon), and niacin/nicotinamide (vitamin B3) metabolism (Figure 4D and 4E; Figure S4C). We also observed decreased TCA cycle intermediates (Figure S4C), consistent with our observation that chronic physiological hypoxia-conditioned macrophages downregulate mitochondrial metabolism transcriptionally (Figure S4A). Analysis of individual metabolites revealed relative enrichment of key PPP and upper glycolysis intermediates but downregulated/unchanged downstream glycolysis intermediates (Figure 4D; Figure S4C). Thus, chronic physiological hypoxia-conditioned macrophages induce or suppress distinct metabolic programs, including increased PPP activity, despite decreased glucose uptake/lactate secretion and ATP synthesis.
Macrophages adapt to chronic physiological hypoxia by co-opting a noncanonical pentose phosphate loop to ensure redox homeostasis
Beyond ATP generated via glycolysis, glucose also contributes to redox homeostasis and nucleotide synthesis by shunting the intermediate glucose 6-phosphate (G6P) into the PPP (Figure 5A). Our finding that chronic physiological hypoxia-conditioned macrophages consume less glucose, secrete less lactate, contain less ATP, and accumulate fewer TCA cycle intermediates but accumulate PPP intermediates suggests that macrophages are using glucose in an unconventional way. One possibility is that chronic physiological hypoxia-conditioned macrophages accumulate PPP intermediates because of efficient shunting of glucose into the PPP. To test this hypothesis, we performed 1,2-13C-glucose tracing of standard oxygen- and chronic physiological hypoxia-conditioned macrophages, which allows us to track intermediates stemming from glucose uptake (Figure 5A33). In chronic physiological hypoxia-conditioned macrophages, we observed increased fractional enrichment and relative abundance of several intermediates that had cycled through the PPP (Figure 5A; Figure S5A). Contrarily, we observed decreased fractional enrichment and relative abundance of the terminal glycolysis intermediates phosphoenolpyruvate (PEP) and lactate as well as decreased contribution of glucose to the glucose-alanine and TCA cycles in chronic physiological hypoxia-conditioned macrophages (Figure S5A). Importantly, we detected significantly less M+1 and M+2 lactate in conditioned media from chronic physiological hypoxia-conditioned macrophages using carbon nuclear magnetic resonance (Figure 5B), suggesting that chronic physiological hypoxia-conditioned macrophages utilize less glucose for energy generation via glycolysis, instead efficiently shunting glucose into the PPP.
Figure 5: Macrophages co-opt noncanonical pentose phosphate loop for redox homeostasis.
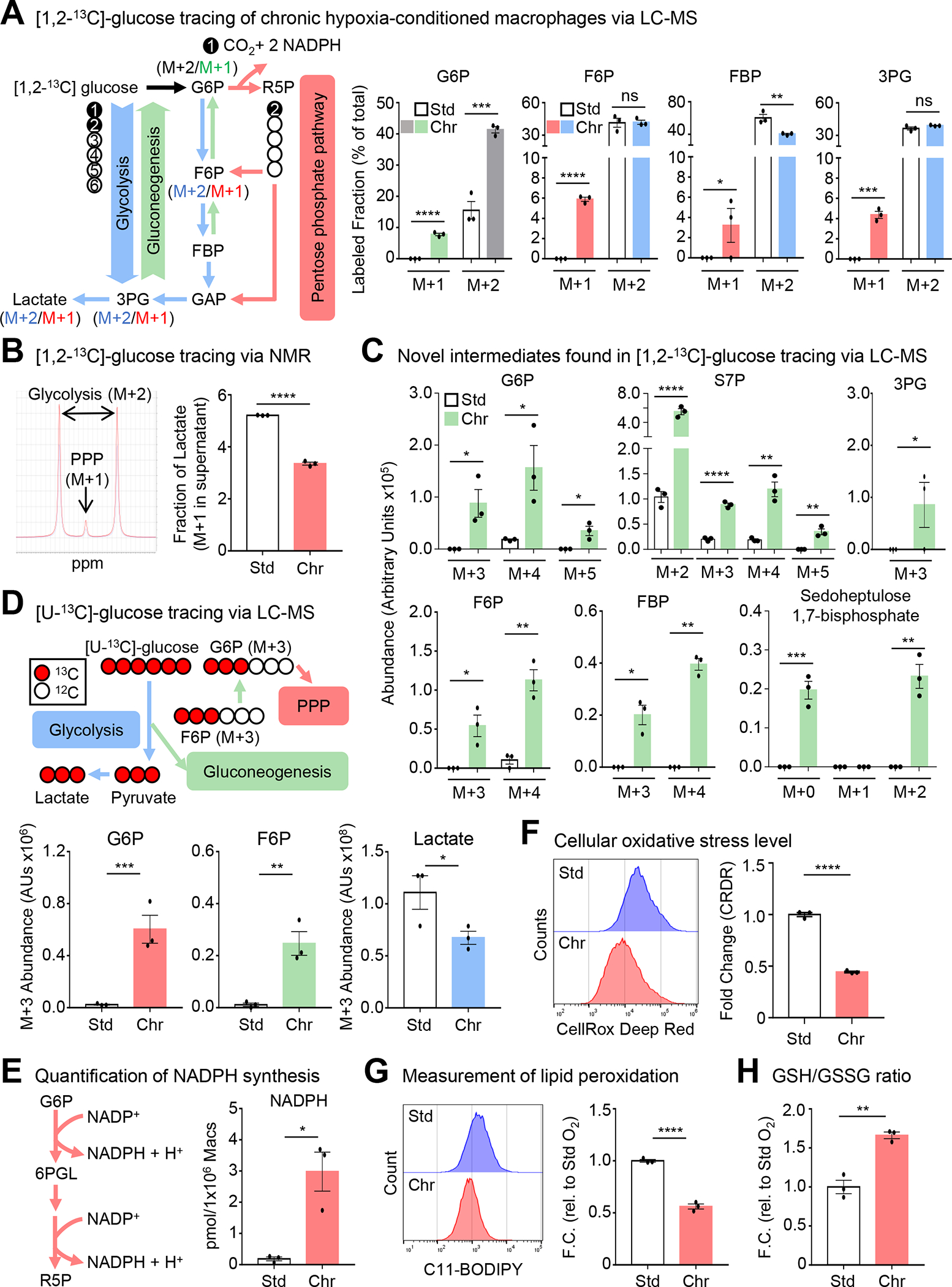
(A) (Left) Schematic for isotopologue analysis of [1,2-13C]-glucose tracing experiments. (Right) Macrophages conditioned as in Figure 1A, but cultured with media containing [1,2-13C]-glucose for 16h then analyzed via LC-MS. Shown is the fractional enrichment of the indicated isotopologues. Three independent experiments were performed for each condition. Data are shown as mean ± SEM. G6P, glucose 6-phosphate. F6P, fructose 6-phosphate. FBP, fructose 1,6-bisphosphate. 3PG, 3-phosphoglyceric acid.
(B) (Left) Macrophages conditioned as in Figure 1A. Representative peaks for PPP- and glycolysis-derived lactate in media analyzed via NMR. (Right) Cumulative data of the M+1 lactate fraction. Data are from four biological replicates, shown as mean ± SEM.
(C) Analysis of experiments from Figure 5A. Data shown as mean ± SEM. G6P, glucose 6-phosphate. S7P, sedoheptulose 7-phosphate. 3PG, 3-phosphoglyceric acid. F6P, fructose 6-phosphate. FBP, fructose 1,6-bisphosphate.
(D) (Top) Schematic for [U-13C]-glucose flux analysis. (Bottom) Similar to Figure 5A, except using [U-13C]-glucose. Shown is the relative abundance of the indicated isotopologues. Data are from three independent experiments, shown as mean ± SEM. G6P, glucose 6-phosphate. F6P, fructose 6-phosphate.
(E) (Left) Schematic of NADPH synthesis via PPP. (Right) Macrophages conditioned as in Figure 5A, followed by NADPH quantification. Data are from three independent experiments, shown as mean ± SEM.
(F) Macrophages conditioned as in Figure 5A. Oxidative stress levels were measured using CellRox Deep Red. Shown are representative flow cytometry plots (Left) and summary plots (Right). Data are representative of three independent experiments, shown as mean ± SEM.
(G) Experiments performed as in Figure 5A. Lipid peroxidation was measured using C11-BODIPY 581/591. Shown are representative flow cytometry plots (Left) and summary plots (Right). Data are representative of three independent experiments, shown as mean ± SEM.
(H) Experiments performed as in Figure 5A, then analyzed for total glutathione, reduced glutathione (GSH), and oxidized glutathione (GSSG). Data are from four independent experiments, shown as mean ± SEM.
Significance was determined by Student’s t-test in B,D-H, and by one-way ANOVA in A,C, *p < .05, **p < .01, ***p < .001, ****p < .0001. ns = not significant.
As part of our 1,2-13C-glucose tracing studies, we observed intermediates with 13C labeling beyond the first and second position (Figure 5C; Figure S5B). For instance, we observed increased fractional enrichment and relative abundance of M+3, M+4, and M+5 isotopologues of G6P in chronic physiological hypoxia-conditioned macrophages (Figure 5C; Figure S5B). The presence of these isotopologues could only arise if intermediates generated during glycolysis or PPP cycled back to G6P, through a process resembling gluconeogenesis in the liver. Unlike conventional gluconeogenesis, however, we did not detect newly synthesized glucose. Instead, together with G6P, we observe increased fractional enrichment and relative abundance of newly synthesized glycolytic intermediates (Figure 5C). Additionally, we observed increased relative abundance and fractional enrichment of newly synthesized isotopologues of S7P (Figure 5C; Figure S5B). In parallel, we performed uniformly-labeled (U)-13C-glucose tracing of standard oxygen- and chronic physiological hypoxia-conditioned macrophages to directly test glucose carbon recycling and turnover (Figure 5D). Similar to our 1,2-13C-glucose tracing studies, we observed less fractional enrichment and relative abundance of the glycolysis-generated M+3 lactate isotopologue in chronic physiological hypoxia conditioned macrophages (Figure 5D; Figure S5C). Furthermore, chronic physiological hypoxia-conditioned macrophages exhibited increased fractional enrichment and relative abundance of newly-synthesized M+3 G6P and F6P isotopologues (Figure 5D; Figure S5C). Chronic physiological hypoxia-conditioned macrophages also produced less alanine and decreased contribution to the TCA cycle, irrespective of route (via M+2 citrate or M+3 oxaloacetate, Figure S5D and S5E). Finally, glucose that enters the PPP can provide the five-carbon nucleoside sugar necessary for de novo nucleotide synthesis. We observed decreased de novo synthesis of both inosine monophosphate (IMP) and guanosine monophosphate (GMP; Figure S6F) in chronic physiological hypoxia-conditioned macrophages, suggesting that the adaptation macrophages undergo in response to physiological hypoxia includes a specific and efficient repurposing of glucose for the generation of NADPH.
Although the conventional PPP is important for inflammatory macrophage function, this often coincides with enhanced glycolysis, decreased G6P, and increased TCA cycle intermediates34–36. Contrarily, we observe increased PPP absent the other features present in inflammatory macrophages, suggesting that chronic physiological hypoxia-conditioned macrophages co-opt an unconventional PPP. In support of this, we detected M+0 and M+2 sedoheptulose 1,7-bisphosphate (SBP) only in chronic physiological hypoxia-conditioned macrophages (Figure 5C). SBP is an unusual metabolite, previously identified as a novel PPP intermediate in the liver (L-type PPP37–39). Interestingly, a recent report found that SBP accumulates in the hepatoma HepG2 line in response to oxidative stress37, suggesting that cells upregulate the noncanonical PPP to cope with contexts that pose a danger to redox homeostasis. Indeed, chronic physiological hypoxia-conditioned macrophages exhibited increased synthesis of NADPH and a decreased NADP+/NADPH ratio (Figure 5E, Figure S7A). Furthermore, macrophages exhibit lower cellular oxidative stress (Figure 5F) and lipid peroxidation (Figure 5G), further supporting the notion that macrophages adapt to physiological hypoxia by efficiently enhancing reductive potential and actively maintaining redox homeostasis.
Our RNAseq and proteomic analyses suggested that macrophages respond to chronic physiological hypoxia by regulating mitochondrial metabolism and biology programs (Figure 2 and 3A; Figure S2, S3C, and S5A), which are major regulators of redox state40. Subsequent analysis of chronic physiological hypoxia-conditioned macrophages revealed decreased mitochondrial mass and potential (Figure S7B and S7C) and decreased mitochondrial superoxide levels (Figure S6D), suggesting that macrophages limit mitochondrial processes that produce reactive species when exposed to prolonged physiological hypoxia. Additionally, we observed an increased ratio of reduced glutathione (GSH) to oxidized glutathione (GSSG) (Figure 5H) despite decreased glutathione synthesis (Figure S7E) in chronic physiological hypoxia-conditioned macrophages relative to standard oxygen-conditioned macrophages. Collectively, our data indicate that macrophages adapt to chronic physiological hypoxia by preferentially shunting glucose into a noncanonical PPP loop that enhances reductive potential, including increased NADPH generation, to maintain low levels of cellular oxidative stress.
The noncanonical pentose phosphate pathway loop supports continual efferocytosis
We hypothesized that induction of ‘primed’ programs in macrophages exposed to prolonged physiological hypoxia directly support ‘poised’ functional programs, such as efferocytosis. We tested whether induction of noncanonical PPP activity, one of the chronic physiological hypoxia-induced ‘primed’ programs, directly supports efferocytosis. Specifically, we used multiple strategies to target the PPP specifically during efferocytosis (Figure S7F). First, use of the noncompetitive inhibitor of glucose-6-phosphate dehydrogenase (G6PD), G6PDi1, resulted in decreased efferocytosis by chronic physiological hypoxia-conditioned macrophages but not macrophages cultured in standard oxygen (Figure S7G, left graph). Second, treatment with the small molecule inhibitor 6-aminonicotinamide (6-AN), to target G6PD activity, also resulted in significantly decreased efferocytosis in chronic physiological hypoxia-conditioned macrophages (Figure S7G, center graph). Third, to test the contribution of conversion of fructose 1,6-bisphosphate to fructose 6-phosphate, we targeted fructose 1,6-bisphosphatase-1 (FBP1) activity using the selective inhibitor FBPi. Consistent with our tracing studies suggesting that PPP intermediates cycle back to G6P, we found that FBP1 inhibition led to decreased efferocytosis exclusively in chronic physiological hypoxia-conditioned macrophages (Figure S7G, right graph). Collectively, our combination of approaches temporally targeting components of the noncanonical PPP loop suggest that macrophages depend on this loop for efficient efferocytosis under prolonged physiological hypoxia.
We next queried whether PPP activity supports continual efferocytosis. Temporal targeting of G6PD activity not only suppressed internalization of the first AC but also reduced internalization of subsequent ACs (Figure 6A). Continual efferocytosis depends on a phagocytes ability to rapidly mature the phagolysosome and degrade ACs, as observed in chronic physiological hypoxia-conditioned macrophages (Figure 1E and 1F). Indeed, time-lapse confocal microscopy revealed three key findings. First, inhibition of G6PD activity resulted in fewer macrophages internalizing ACs (Figure 6B). Second, macrophages internalize significantly fewer ACs on a per-cell basis when G6PD activity was inhibited (Figure 6B, top graph). Third, inhibition of G6PD activity resulted in a slower rate of AC degradation (Figure 6B, bottom graph), essentially reversing the ‘primed’ adaptations macrophages undergo during prolonged physiological hypoxia. Finally, we sought to determine if temporal perturbation of the ‘primed’ program affects efferocytosis in vivo. We treated mice with 6-AN or vehicle prior to the induction of thymocyte apoptosis using dexamethasone (Figure 6C). Similar to our in vitro findings and two recent independent studies41,42, mice treated with 6-AN exhibited significantly decreased clearance of apoptotic thymocytes (Figure 6C).
Figure 6: Noncanonical pentose phosphate loop supports continual efferocytosis.
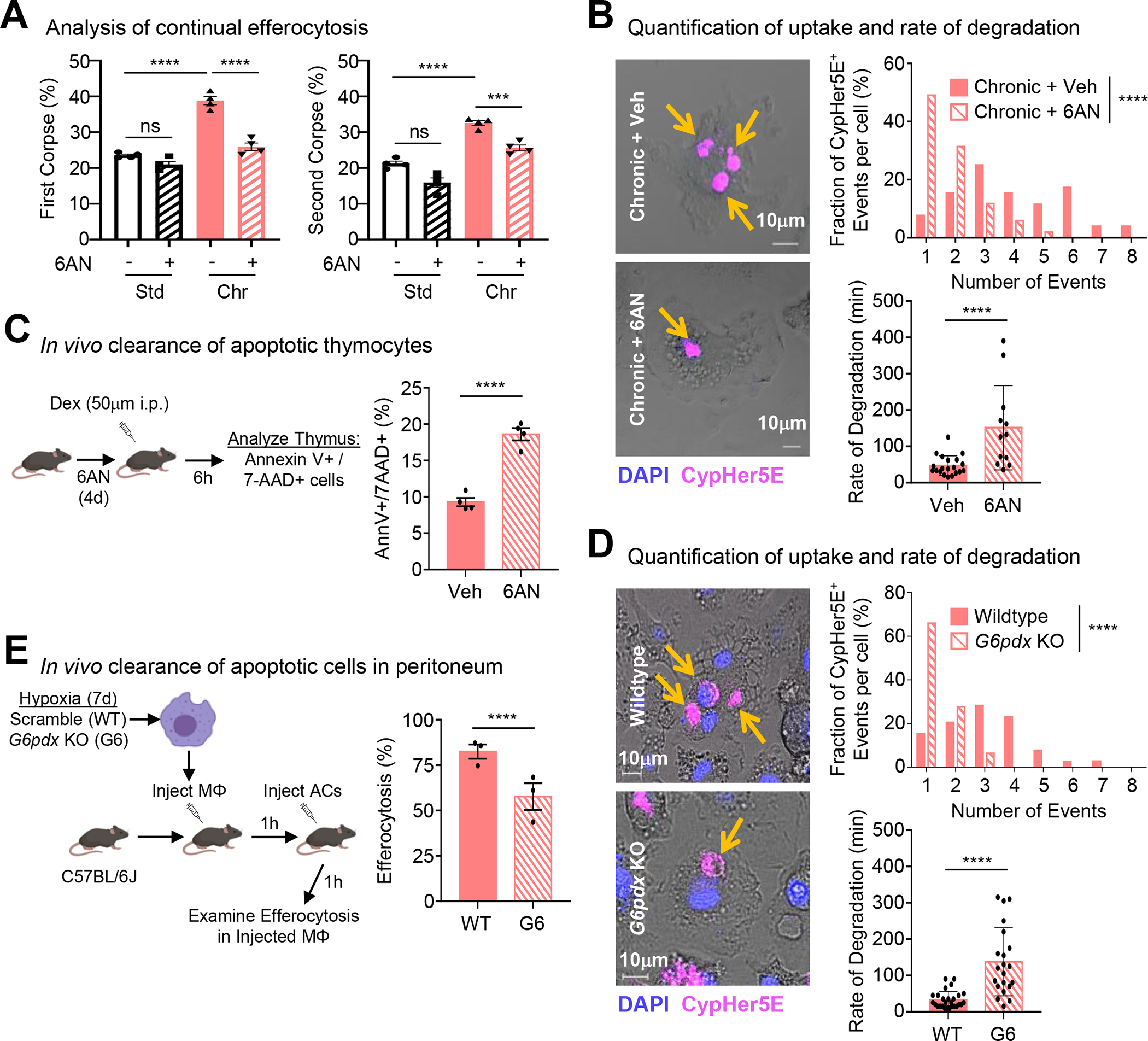
(A) Continual efferocytosis experiments performed as in Figure 1D. Data represent three independent experiments, shown as mean ± SEM.
(B) Experiments performed and analyzed as in Figure 1E. Shown are representative images (Left), quantification of CypHer5E+ events per-cell (Top Right), and analysis of degradation rate (Bottom Right). For quantification, 53 efferocytotic macrophages from 5 vehicle-treated scenes and 51 efferocytotic macrophages from 7 6AN-treated scenes were analyzed. For degradation rate, 21 (vehicle-treated) and 13 (6AN-treated) efferocytotic macrophages were analyzed. Data shown as mean ± SEM.
(C) Schematic of experimental design (Left) and summary plot (Right) of AnnexinV+ 7-AAD+ thymocytes 6h post-dexamethasone (Dex) injection in vehicle (n=4) or 6AN-treated mice (n=4). Data shown as mean ± SEM.
(D) Experiments performed and analyzed as in Figure 1E except using G6PDX-deficient (G6) macrophages. Shown are representative images (Left), quantification of CypHer5E+ events (Top Right), and analysis of degradation rate (Bottom Right). For quantification, 39 efferocytotic macrophages from 5 WT scenes and 47 efferocytotic macrophages from 7 G6pdx-deficient scenes were analyzed. For degradation rate, 27 (WT) and 21 (G6pdx-deficient) efferocytotic macrophages were analyzed. Data shown as mean ± SEM.
(E) (Left) Schematic of experimental design. (Right) Summary plot of efferocytosis rate by GFP+ CD11b+ F4/80+ macrophages in WT mice (n=3) and G6pdx-deficient mice (n=3). Data shown as mean ± SEM.
Significance was determined by Student’s t-test in B-E, and by one-way ANOVA in A,B,D, ***p < .001, ****p < .0001. ns = not significant.
We next sought to determine if genetic targeting of G6pdx, the mouse isoform of G6PD, affects efferocytosis by chronic physiological hypoxia-conditioned macrophages. Similar to our results with the small molecule 6-AN, targeting of G6pdx (Figure S7H) revealed several striking findings. First, macrophages lacking G6pdx internalized significantly fewer ACs on a per-cell basis (Figure 6D, top graph). Second, macrophages lacking G6pdx degraded ACs at a slower rate (Figure 6D, bottom graph), essentially reversing the ‘primed’ adaptations macrophages make under prolonged physiological hypoxia. This decreased efferocytosis capacity was due to G6PDX deficiency directly, as introduction of a G6pdx ORF significantly rescued efferocytosis capacity (Figure S7I). Finally, G6pdx-deficient macrophages internalized significantly fewer ACs than control macrophages in vivo (Figure 6E). Thus, the ‘primed’ program, in particular the noncanonical PPP loop, is necessary for efficient continual efferocytosis in vitro and in vivo.
Noncanonical pentose phosphate loop supports phagolysosomal maturation and prevents redox crisis in efferocytotic macrophages
Phagolysosomal maturation and subsequent AC degradation requires NADPH oxidase activity43 but if not buffered against, can lead to redox crisis featuring runaway lysosomal acidification and oxidative stress44,45. Macrophages under standard oxygen produce minimal NADPH (Figure 7A45). On the other hand, we observed significant PPP-dependent production of NADPH (Figure 7A). Although NADPH production remained abundant during efferocytosis by chronic physiological hypoxia-conditioned macrophages (Figure 7B), NADPH pools were further depleted in efferocytotic macrophages with temporally disrupted PPP activity (Figure 7B) and decreased G6pdx levels (Figure 7C). Thus, chronic physiological hypoxia-conditioned macrophages boost PPP-dependent NADPH production prior to efferocytosis which remains active during efferocytosis.
Figure 7: Noncanonical pentose phosphate loop prevents redox crisis in efferocytotic macrophages.

(A) Experiments performed as in Figure 1A, with inclusion of 6AN. Data are from three independent experiments, shown as mean ± SEM.
(B) Experiments performed as in Figure 7A. Data are from three independent experiments, shown as mean ± SEM.
(C) Experiments performed as in Figure 7A except using G6PDX-deficient (G6) macrophages. Data are from three independent experiments, shown as mean ± SEM.
(D, E) Experiments performed as in Figure 7A using 6AN-treated macrophages (D) or G6PDX-deficient (G6) macrophages (E). Lysosomal acidification was measured in efferocytotic macrophages (CypHer5E+). Shown are representative flow cytometry plots (Left) and MFI (Right) from three independent experiments. Data are shown as mean ± SEM.
(F, G) Experiments performed as in Figure 7A using 6AN-treated macrophages (F) or G6PDX-deficient (G6) macrophages (G). Cellular ROS was measured in efferocytotic macrophages (CypHer5E+). Shown are representative flow cytometry plots (Left) and MFI (Right) from three independent experiments. Data are shown as mean ± SEM.
Significance was determined by Student’s t-test in C, and by one-way ANOVA in A,B,D-G, *p < .05, **p < .01, ***p < .001, ****p < .0001. ns = not significant.
Our observation that chronic physiological hypoxia-conditioned macrophages increase PPP-dependent NADPH production both prior to and during efferocytosis raises the hypothesis that the noncanonical PPP loop, a core ‘primed’ program, directly supports efferocytosis, a core ‘poised’ program. To test this hypothesis, we sought to determine if temporal perturbation or genetic disruption of the PPP also leads to runaway lysosomal acidification and oxidative stress in efferocytotic macrophages. In AC-naïve macrophages, inhibition of PPP activity resulted in a modest increase in lysosomal acidity (Figure 7D; Figure S7J). On the other hand, efferocytotic macrophages with perturbed PPP activity displayed significantly higher lysosomal acidity than untreated AC-naïve and efferocytotic macrophages (Figure 7D; Figure S7J). We observed similar results in chronic physiological hypoxia-conditioned macrophages with genetic disruption of G6pdx (Figure 7E). Finally, we tested the hypothesis that disrupted PPP activity leads to increased oxidative stress in efferocytotic macrophages. Indeed, we observed that temporal perturbation of PPP activity resulted in significantly increased lipid peroxidation (Figure S7K) and cellular oxidative stress (Figure 7F; Figure S7L) in chronic physiological hypoxia-conditioned efferocytotic macrophages. Importantly, genetic disruption of G6pdx also resulted in higher oxidative stress in chronic physiological hypoxia-conditioned efferocytotic macrophages (Figure 7G). Thus, our data suggest that PPP-mediated NADPH production, a core ‘primed’ program, supports enhanced efferocytosis, a core ‘poised’ program, by providing the NADPH necessary for both rapid phagolysosomal maturation and protection from excessive lysosomal acidification and oxidative stress under settings of prolonged physiological hypoxia.
Discussion
Professional phagocytes, such as macrophages, reside in tissues throughout the body in limited nutrient environments often for long periods of time13,15. This is especially true for oxygen, which ranges between 38 mmHg (~5%) and 10 mmHg (~1%, physiological hypoxia18). Despite the relative dearth of oxygen, phagocytes remain responsible for removing millions of ACs daily19, a process that is metabolically demanding and potentially perilous10. How phagocytes adapt to continuous inhabitance in physiological hypoxic environments and how such adaptations inform efferocytosis remains unexplored. Here, we made the striking observation that macrophages conditioned in prolonged physiological hypoxia (~1% O2) in vitro or in vivo are better phagocytes, both internalizing more ACs and degrading internalized ACs faster. As part of an adaptive response to prolonged physiological hypoxia, macrophages induce two distinct states that support efficient efferocytosis. The first state, which we term ‘primed’, is characterized by concomitant changes in mRNA and protein programs, especially metabolic programs, in AC-naïve macrophages that remain induced during efferocytosis. The second state, which we term ‘poised’, is characterized by transcription, but not translation, of phagocyte function programs (e.g., phagocytosis programs) in AC-naïve macrophages that are subsequently translated during efferocytosis. We found that both primed and poised state programs support enhanced continual efferocytosis by macrophages residing under chronic physiological hypoxia. Importantly, we found that this ‘primed’ state, in particular induction of a noncanonical pentose phosphate pathway loop that generates abundant NADPH, directly supports enhanced, continual efferocytosis (a core ‘poised’ state program). Based on our findings, we propose that phagocytes that reside in physiological hypoxic environments, such as highly phagocytic macrophages in the bone marrow or spleen9,19, adopt distinct states that both ensure their own fitness and their readiness to perform core homeostatic functions.
Previous analysis of phagocytosis in different oxygen levels has produced mixed findings. For instance, past studies have suggested that acute hypoxia enhances internalization of opsonized targets46, bacteria47,48, and ACs19, whereas other studies have suggested that acute hypoxia has no effect on phagocytosis48–50 or decreases clearance of ACs51, and that macrophages in hypoxic regions may produce less of the anti-inflammatory mediatory IL-10 typically produced during efferocytosis25. Much of the contradiction might relate to variations in how hypoxia was induced/maintain or for how long cells were cultured in hypoxia, typically ranging from 3h to 24h. Indeed, significant cellular changes can occur, including degradation of HIF proteins and HIF-dependent transcriptional programs, as quickly as 24h after introduction to hypoxia28,52. Although we observed a modest increase in population-level efferocytosis in macrophages exposed to acute physiological hypoxia, these cells did not exhibit enhanced lysosomal degradation. Instead, the most dramatic differences in both uptake and degradation of ACs were observed in macrophages conditioned in chronic physiological hypoxia. It will be interesting to further interrogate the importance of prolonged physiological hypoxia, including identified ‘primed’ and ‘poised’ programs, especially considering phagocytes residing in physiologically hypoxic tissues are often responsible for clearing biological material beyond conventional ACs8,10.
Despite the growing body of work detailing how cells undergo metabolic adaptation to chronic hypoxia, understanding how such changes inform cellular function remains largely unexplored. We move beyond a phenomenological understanding of cellular metabolic adaption to chronic physiological hypoxia by mechanistically linking the changes in macrophage metabolism to enhanced AC uptake and degradation. Unexpectedly, we found that macrophages under prolonged physiological hypoxia limit glucose flux to most major pathways including anaerobic glycolysis and the TCA cycle. Instead, macrophages shunt glucose into a noncanonical PPP loop involving repeated cycling of PPP intermediates back to G6P via a process resembling gluconeogenesis. We found that this PPP loop is necessary for generation of NADPH in AC-naïve macrophages and appears to provide the NADPH necessary for appropriate phagolysosomal maturation and AC degradation. The importance of the PPP for efficient efferocytosis was also recently independently demonstrated using the small molecule 6-AN in vitro and in the thymus in vivo41,42. Our results suggest that it is this noncanonical PPP loop, and not the canonical PPP per se, that is required in tissue-resident macrophages residing in physiological hypoxic tissues. Collectively, we speculate that the adaptations observed in our study represent normal physiology of tissue-resident macrophages residing in physiological hypoxic tissues.
Limitations of the study
A series of important questions arise from our work related to our observation that chronic physiological hypoxia seems to induce both ‘primed’ and ‘poised’ states in macrophages. For instance, what macrophage functions, beyond efferocytosis, are ‘poised’ under chronic physiological hypoxia? Efferocytosis is generally a core macrophage function shared across tissue-resident macrophage populations, but are there functions that are informed by a combination of oxygen availability and tissue-specific factors (e.g., presence of heme)? Finally, how are these poised programs regulated, for example, are they maintained by ribosomal stalling or increased chromatin accessibility/transcription? Future studies are required to determine the answers to these questions and could provide important clues on how macrophage function in specific environments to maintain tissue homeostasis.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Justin S. A. Perry (perryj@mskcc.org).
Materials availability
This study did not generate new reagents or materials.
Data and code availability
mRNA sequencing raw data files have been deposited in NCBI Gene Expression Omnibus (GEO) archive (accession GSE192969). An Excel file with plotted data points and a pdf image with uncropped western blots are in Data S1.
No new code was used in the preparation of this manuscript.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Animals were housed at the Memorial Sloan Kettering Cancer Center (MSKCC) animal facility under specific pathogen free (SPF) conditions on a 12-hour light/dark cycle under ambient conditions with ad libitum access to food and water. All studies were approved by the Sloan Kettering Institute (SKI) Institutional Animal Care and Use Committee (IACUC).
Cell Lines and Primary Cells
Primary ER-Hoxb8 myeloid progenitors, Jurkat T lymphoma cells, MDA-MB-231 breast cancer cells, BrM2 metastatic breast cancer cells, and E0771 breast cancer cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) or Roswell Park Memorial Institute Medium (RPMI) supplemented with lot-tested 10% fetal bovine serum (FBS), 1mM L-glutamine, penicillin, and streptomycin. Cells were cultured at 37°C in a humidified 5% CO2 standard oxygen (21%) or hypoxic (1%) cell incubator.
METHOD DETAILS
Standard Oxygen and Hypoxia Conditioning
To model standard oxygen, acute hypoxia, and chronic hypoxia environments, we optimized protocols using the BioSpherix Xvivo X3 system, which allows us to dynamically regulate CO2 and O2 (down to 0.1%) in four different chambers without exposing cells to atmosphere. Primary macrophage progenitors immortalized with a modified estrogen receptorHoxb8 fusion (ER-Hoxb8) were generated as previously reported53 from C57BL/6J mice. Progenitors were maintained in RPMI 1640 containing 5% heat-inactivated fetal bovine serum, 5% P885L (GM-CSF producing)-conditioned media, and 0.5μM β-estradiol (Sigma) in standard oxygen (~21% O2). To differentiate into primary macrophages, progenitors were first washed three times with cold PBS to remove β-estradiol. Progenitors were then cultured in a-MEM containing 5% heat-inactivated fetal bovine serum, 1% PenicillinStreptomycin-Glutamine (100X), and 10% L929 (M-CSF producing)-conditioned media. On day 6 of differentiation, mature macrophages were replated in non-TC treated plates and cultured in either standard oxygen or hypoxia (1% O2) for 7 days prior to use in efferocytosis assays. For some experiments, progenitors were instead cultured in standard oxygen or hypoxia for 7 days prior to differentiation and use in efferocytosis assays. In all cases, media was replenished every other day and cells were incubated @37°C.
Induction of Apoptosis
For thymocyte phagocytosis, thymi from 6-week-old mice were harvested and crushed between frosted slides to release thymocytes. Cells were then treated with dexamethasone (50̼M) for 4 hours prior to downstream use. Jurkat T lymphoma, MDA-MB-231, BrM2, and E0771 cells, were induced to undergo apoptosis by treatment with 150mJ cm−2 (Jurkat cells) or 650mJ cm−2 ultraviolet C irradiation (Stratalinker) at ~5×106 cell density per 10cm non-TC treated dish. Then, cells were incubated for 4h, 8h, 16h, or 24h before downstream use. Apoptosis was confirmed via Annexin V/7-AAD staining to be greater than 70% Annexin V+ single-positive (Figure S1A).
In Vitro Efferocytosis Assays
Apoptotic cells were stained with either 1μM CypHer5E (Cytiva) or 50μM TAMRA-SE (ThermoFisher) in serum-free HBSS for 45min. Then, cells were washed by incubating in serum-containing assay media for an additional 25min. Apoptotic cells were co-cultured with macrophages at a 1:1 phagocyte to target ratio. All efferocytosis assays included four technical replicates for each condition with at least three biological replicates. For all pharmacological studies, phagocytes were pre-incubated with the compounds listed below or vehicle (DMSO) for 16h or 6d prior to addition of apoptotic cells. Macrophages were treated with the following: Cycloheximide (Chx; Sigma, 100μM), 6-AN (MedChemExpress, 100μM), G6PDi-1 (Cayman Chemical, 50μM), or FBP1i (Cayman Chemical, 25μM). Macrophages were examined via microscopy to ensure no gross morphological changes or cell death had occurred due to drug treatment. Macrophages and apoptotic cells were co-cultured for 1h. Apoptotic cells were subsequently removed via three washes with cold assay media and phagocytes were harvested using a cell scraper (Biotium) and assessed by flow cytometry. For continual efferocytosis, the first round of apoptotic cells was labeled with CFSE (ThermoFisher) and co-cultured with macrophages at a 1:1 ratio for 1h. Then, apoptotic cells were washed away using cold assay media. Macrophages were subsequently rested for 1.5h prior to co-culture with a second round of apoptotic cells that were labeled with CypHer5E. Continual efferocytosis was assessed by flow cytometry as CFSE+ (1st AC uptake) and CFSE+ CypHer5E+ (2nd AC uptake) macrophages. flow cytometry analysis was performed using an Attune NxT Flow Cytometer (ThermoFisher) and analyzed using FlowJo 10.7.1.
Time-lapse Confocal Microscopy
Equipment.
Experiments to evaluate per-cell uptake and degradation were performed on a Zeiss Axio Observer.Z1 7 inverted fluorescence microscope equipped with a Zeiss 20x PlanApo (0.8 NA) objective, a 6-channel, 7-laser LSM 980 (405nm, 445nm, 488nm, 514nm, 561nm, 594nm, 639nm), and a Airyscan 2 multiplex detector. The imaging stage is fully encased in a black-out environmental chamber and the Z PIEZO stage is fully encapsulated with a heating/gas-controlled insert, together allowing us to control the temperature, humidity, CO2, and O2. Time-lapse experiments were acquired using Zen Blue software (Zeiss).
Experiment.
Prior to imaging, cells were allowed to acclimate for at least 10min. After selection of scenes (see below), apoptotic cells were added to phagocyte-containing chamber slides and scenes were rapidly focused prior to the beginning of data collection. For each condition/experiment, at least 8 regions (‘scenes’) were selected and imaged. Scenes were selected if they featured at least 10 cells per region, and when experimental manipulations were performed (e.g., 6-AN treatment), regions were selected that featured similar cell numbers between vehicle and treatment conditions. Scenes were imaged every 4min for a minimum of 8h per experiment, which generally equates to ~0.5μs per pixel or 5s per scene. Time-lapse experiments were performed in Multiplex CO-8Y mode which allows for gentle confocal-resolution imaging at a high frame rate. Specifically, we set the pinhole to 2.42 Airy units (57μm) with the image scaling (per pixel) set at 0.414μm × 0.414μm with 2x averaging. Focusing was ensured using the Definite Focus 2 module with a two-part strategy that was first based on the initial focus of individual scenes and then subsequently updated after each round of images. Analysis, including generation of scale bars, was performed using Zen Blue or Fiji.
Ex Vivo Efferocytosis
To perform ex vivo efferocytosis, tissues were isolated from 7-week-old female C57BL/6 mice. To isolate bone marrow-resident macrophages (BMMs), bone marrow was flushed from bones using a 25G needle with α-MEM media containing FBS in a hypoxia chamber set to 1% oxygen. Red blood cells were lysed, and remaining bone marrow cells were plated in a non-TC treated 6-well plate in BMDM media and cultured overnight in either hypoxia (1%) or standard (21%) oxygen. Floating cells were removed and fresh BMDM media was added. BMMs were then co-cultured with CypHer5E-labeled apoptotic MDA-MD-231 cells at a 1:1 ratio for 1h. Macrophages were isolated, stained with CD11b and F4/80, and analyzed via flow cytometry. To isolate splenic macrophages, spleens were minced into small pieces in a hypoxia chamber set to 1% oxygen and digested for 20min at 37C. Cell suspensions were strained using a 70μm cell strainer. Red blood cells were lysed, and remaining splenic cells were plated and treated identical to bone marrow-resident macrophages as described above.
In Vivo Efferocytosis
To perform in vivo efferocytosis, we performed two models: dexamethasone (Dex)-induced apoptotic thymocyte removal and peritoneal apoptotic T cell removal. For Dex-induced clearance, six- to eight-week-old mice were injected i.p. with 300μl PBS containing 250μg dexamethasone (50μM; Sigma) with or without cycloheximide (5mg/kg) or 6-AN (3mg/kg) dissolved in EtOH. 6h after injection, thymi were harvested from mice and the numbers of thymocytes with annexin V staining only (apoptotic) versus annexin V/7-AAD double positive cells (secondarily necrotic) were assessed via flow cytometry. For the peritoneal clearance model, Cas9+ macrophages bearing either a scramble control guide or guides targeting G6pdx where initially conditioned in physiological hypoxia (1% O2) for 7d. Conditioned macrophages were subsequently i.p. injected 1h prior to injection of apoptotic cells. Then, mice were injected with 1e6 CypHer5E-labeled apoptotic Jurkat T cells and allowed to rest. Mice were subsequently euthanized 1h post-injection. Peritoneal lavage was collected in 8ml cold PBS. Collected lavage was centrifuged and cells were blocked with CD16/32 (clone 24G2) prior to staining with CD11b (eBioscience, 48-0112-82, clone M1/70, 1:750) and F4/80 (eBioscience, 12-4801-82, clone BM8, 1:750) for 30min at 4°C. Efferocytosis was analyzed by quantifying CypHer5E+ events within the CD11b+ F4/80high macrophages via flow cytometry.
RNA Sequencing
For bulk RNA sequencing, 4.5×105 macrophages were conditioned for 7d in standard O2 (~21%) and either immediately collected or pulsed with 3h of 1% O2 (acute) and then collected. In parallel, 4.5×105 macrophages were conditioned for 7d in 1% O2 (chronic) then collected. Collected samples were subsequently collected, washed with cold PBS, and lysed on ice with beta-mercaptoethanol-containing lysis buffer (Buffer RA, Machery-Nagel) for downstream RNA isolation. Total RNA was isolated using the NucleoSpin RNA isolation kit with on-column rDNase digestion (Machery-Nagel) and mRNA libraries were generated by polyA capture and reverse transcription of cDNA. Libraries were then sequenced at 150bp (paired-end) reads with ~20 million reads per sample using an Illumina NovaSeq 6000 sequencer.
Proteomics
For 13C SILAC labeling, target cells were grown in lysine- and arginine-deficient DMEM with 10% dialyzed FBS, 1% PSQ, and 2mM L-glutamine, supplemented with ‘heavy’ 13C6-lysine and ‘light’ 12C6-arginine (100mg/L; Cambridge Isotope Laboratories). After 8–10 passages, incorporation of 13C6-lysine-labeled amino acids into proteins was verified via LC-MS/MS to be >99%. Heavy isotope-labeled target cells were expanded in 13C6-lysine media and induced to undergo apoptosis. Macrophages were first conditioned in standard oxygen or hypoxia (see above), and then cultured with or without apoptotic cells for 1h. Apoptotic cells were subsequently removed using two cold assay media washes and two cold PBS washes. Macrophages were then removed using a cell scraper, pelleted to remove any residual liquid, and immediately snap frozen on dry ice.
Sample Preparation.
Cell pellets were lysed with 200–300μL buffer containing 8M urea and 200mM EPPS (pH at 8.5) with protease inhibitor (Roche) and phosphatase inhibitor cocktails 2 and 3 (Sigma). Lysates were aspirated 2x on ice, followed by water sonication for 2min @4°C. Benzonase (Millipore) was added to a concentration of 50u/mL and incubated on ice for 15min. Samples were centrifuged at 14,000g for 10 min (@4°C) and the supernatant was subsequently extracted. The Pierce bicinchoninic acid (BCA) protein concentration assay was used to determine protein concentration. Protein disulfide bonds were reduced with 5mM tris (2-carboxyethyl) phosphine (@RT, 30min), then alkylated with 10mM iodoacetamide (@RT, 30min, in the dark). The reaction was quenched with 10mM dithiothreitol (@RT, 15min). Equivalent volumes of lysate aliquots were taken for each sample (100–200μg in each sample) and diluted to approximately 100μL with lysis buffer. Samples were subjected to methanol for 5s, then chloroform was added for an additional 5s prior to centrifugation and precipitation54. Pellets were reconstituted in 100μL of 200mM EPPS buffer and digested with Lys-C (1:100 enzyme-to-protein ratio) and incubated @37°C for 4h. Trypsin was then added (1:100 enzyme-to-protein ratio) and digested @37°C overnight. Anhydrous acetonitrile was then added to make a final volume of 30% ACN.
TMT Labeling.
Samples were TMT-labeled as described54. Briefly, samples were TMT-tagged by adding 10μL (28μg/μL) of TMTPro reagent for each sample and incubated for 1h @RT. A ratio check was performed by taking a 2μL aliquot from each sample and desalted by the StageTip method55 to confirm labeling efficiency. TMT-tags were then quenched with hydroxylamine to a final concentration of 0.3% for 15min @RT. Samples were pooled in their entirety, then dried via vacuum-centrifugation. Dried samples were reconstituted in 1mL of 3% ACN/1% TFA, desalted using a 100mg tC18 SepPak (Waters), and lyophilized overnight. Lyophilized peptides were dried using vacuum-centrifugation and reconstituted in 1mL of 2% ACN/25mM ABC. Peptides were fractionated into 48 fractions. Next, an Ultimate 3000 HPLC (Dionex) coupled to an Ultimate 3000 Fraction Collector using a Waters XBridge BEH130 C18 column (3.5μm × 4.6mm × 250mm) was operated at 1mL/min. The Buffers A, B, and C used below consisted of 100% water, 100% ACN, and 25mM ABC, respectively. The fractionation gradient operated as follows: 1% B to 5% B in 1min, 5% B to 35% B in 61min, 35% B to 60% B in 5min, 60% B to 70% B in 3min, 70% B to 1% B in 10min, with 10% C the entire gradient to maintain pH. The 48 fractions were then concatenated to 12 fractions (i.e., fractions 1, 13, 25, 37 were pooled, followed by fractions 2, 14, 26, 38, etc.) so that every 12th fraction was used to pool. Pooled fractions were vacuum-centrifuged then reconstituted in 1% ACN/0.1% FA for LCMS/MS.
LC-MS/MS.
Total fractions were analyzed by LC-MS/MS using a NanoAcquity (Waters) with a 50cm EASY-Spray Column (PepMap RSLC, C18, 2μm, 100Å, 75μm I.D.) heated to 60°C coupled to a Orbitrap Eclipse Tribrid Mass Spectrometer (Thermo Fisher Scientific). Peptides were separated by direct inject at a flow rate of 300nL/min using a gradient of 5 to 30% acetonitrile (0.1% FA) in water (0.1% FA) over 3h then to 50% ACN in 30min and analyzed by SPS-MS3. MS1 scans were acquired over a range of m/z 375–1500, 120K resolution, AGC target (4.0e5), and maximum IT of 50ms. MS2 scans were acquired on MS1 scans of charge 2–7 using an isolation of 0.7m/z, collision induced dissociation with activation of 32%, turbo scan and max IT of 50ms. MS3 scans were acquired using specific precursor selection (SPS) of 10 isolation notches, m/z range 100–1000, 50K resolution, AGC target (1.0e5), HCD activation of 45%, and max IT of 150ms. The dynamic exclusion was set at 60s.
TMT Data Analysis.
Raw data files were processed using Proteome Discoverer (PD) version 2.4.1.15 (Thermo Scientific). For each of the TMT experiments, raw files from all fractions were merged and searched with the SEQUEST HT search engine with a UniProt protein database downloaded on 2019/01/09 (176,945 entries). The precursor and fragment mass tolerances were 10ppm and 0.6Da respectively. A maximum of two trypsin missed cleavages were permitted. Searches used a reversed sequence decoy strategy to control peptide false discovery rate (FDR) and 1% FDR was set as threshold for identification.
Analysis of Nutrient Consumption and Secretion via YSI
Macrophages (2.5×106) were cultured in either standard oxygen or hypoxia (1% O2) and conditioned for a total of 7 days. Media was changed at day 1, day 3, and day 5 prior to a 24h period of conditioning. At the end of the 24h period (denoted as day 2–3, day 4–5, and day 6–7 periods), media was collected, spun down to remove cell debris, and immediately snap frozen for downstream analysis via a 2950D Biochemistry Analyzer (YSI Life Sciences) to determine glucose and lactate concentrations. Absolute rates of consumption (glucose) or secretion (lactate) were calculated first by subtracting the concentration observed in control (cell-free) media incubated in parallel, then normalized to cell number and volume of media. These experiments were performed independently at least two times.
Untargeted Metabolomics
Macrophages (2.5×106) were conditioned in standard oxygen or hypoxia for 7d. To protect from experimental artifact, all samples were collected in the same oxygen environment that they were conditioned in. Plates containing cells were placed on ice and washed with cold PBS three times, then moved to dry ice and ice cold 80% methanol was added. Cells were subsequently scraped and transferred to a cold centrifuge tube on dry ice. The cell-methanol slurries were then vortexed for 1min, allowed to rest on dry ice for 5min, then vortexed an additional 1min to create cell lysates. Lysates were centrifuged for 20min at 14,000g in a refrigerated centrifuge set to 4°C to remove debris. Supernatants were transferred to clean tubes and lyophilized in the absence of heat and dissolved in water. Targeted LC/MS analyses were performed on a Q Exactive Orbitrap mass spectrometer (Thermo Scientific) coupled to a Vanquish UPLC system (Thermo Scientific). The Q Exactive operated in polarity-switching mode. A Sequant ZIC-HILIC column (2.1mm i.d. × 150mm, Merck) was used for separation of metabolites. Flow rate was set at 150μL/min. Buffers consisted of 100% acetonitrile for mobile B, and 0.1% NH4OH/20mM CH3COONH4 in water for mobile A. Gradient ran from 85% to 30% B in 20min followed by a wash with 30% B and re-equilibration at 85% B. MS data were processed using Compound Discoverer (Thermo Scientific). An in-house metabolite library as well as Chemspider were searched for metabolite identification. Three levels of metabolite identification were reported: 1) identified compounds: definitive identification based on the mass (within 5ppm) and retention time of authentic chemical standards; 2) putatively annotated compounds by searching ChemSpider (mass tolerance 5ppm); 3) compounds with predicted chemical composition based on mass. Relative metabolite quantitation was performed based on peak area for each metabolite. Hierarchical clustering analysis and principal component analysis (PCA) were performed by Compound Discoverer (Thermo Scientific).
13C-Metabolic Flux Analysis
Experiments were performed similar to those outlined in Untargeted Metabolomics with the following modification. On day 5, glucose-deficient RPMI containing 1 g/L [1,2-13C]-glucose or [U-13C]-glucose (both from Cambridge Isotope Laboratories), 10% L929-conditioned media, 1% PSQ, and 10% FBS was conditioned in either standard oxygen or hypoxia for 16–24h. On day 6, conditioned macrophages were washed with PBS twice and then cultured with heavy isotope-containing media for 16h. Metabolites were extracted using the same protocol detailed in Untargeted Metabolomics. LC/MS analyses were performed on a Q Exactive Orbitrap mass spectrometer (Thermo Scientific) coupled to a Vanquish UPLC system (Thermo Scientific). The Q Exactive operated in polarity-switching mode. A Sequant ZIC-HILIC column (2.1mm i.d. × 150mm, Merck) was used for separation of metabolites. Flow rate was set at 150μL/min. Buffers consisted of 100% acetonitrile for mobile B, and 0.1% NH4OH/20mM CH3COONH4 in water for mobile A. Gradient ran from 85% to 30% B in 20min followed by a wash with 30% B and re-equilibration at 85% B. Data analysis was done using El-MAVEN (v0.12.0). Metabolites and their 13C isotopologues were identified on the basis of exact mass within 5ppm and standard retention times. Relative metabolite quantitation was performed based on peak area for each metabolite.
Analysis of Mitochondrial Properties
For analysis of mitochondrial superoxide levels, conditioned macrophages were labeled with the live cell-permeant fluorogenic probe MitoSOX Red (ThermoFisher). MitoSOX Red is targeted to the mitochondria and is oxidized by superoxide but not reactive oxygen or nitrogen species, inducing an increase in fluorescence which we quantified using flow cytometry. To estimate mitochondrial shape and content, conditioned macrophages were labeled with both MitoTracker Green (MTG) and MitoTracker Deep Red (MTDR, both ThermoFisher). MTG localizes to mitochondria independent of mitochondrial membrane potential whereas MTDR labeling of mitochondria is dependent on mitochondrial membrane potential. Combined, the two fluorogenic probes were used to estimate potential and quantity (via flow cytometry).
NADP+/NADPH, ATP, and GSH/GSSG Measurement
4×106 or 2.5×104 (for GSH/GSSG) macrophages were seeded for conditioning and subsequent NADP+/NADPH, ATP, or GSH/GSSG measurement. In some experiments, macrophages were treated with 6-AN (100nM) or vehicle (DMSO) beginning one day after replating. On day 7, cells were collected, processed, and the ratio of NADP+/NADPH or GSH/GSSG was measure using the NADP/NADPH Quantitation Colorimetric Kit (BioVision) or the GSH/GSSG-Glo Assay (Promega), respectively, or ATP was quantitated using the Luminescent ATP Detection Assay Kit (Abcam), following the manufacturer’s protocol.
Measurement of Redox State and Lysosomal Acidity
Lipid peroxidation and cellular ROS was determined by labeling of phagocytes using the reagent BODIPY 581/591 C11 and CellROX Deep Red (both ThermoFisher), respectively, followed by flow cytometric analysis. Lysosomal acidity was determined by labeling of phagocytes using the reagent Lysosensor Green DND-189 (ThermoFisher). Labeling was performed according to manufacturer instructions. For analysis of redox state or lysosomal acidity in efferocytotic cells, phagocytes were first gated on CypHer5E+.
CRISPR/Cas9 Deletion or siRNA knockdown of SLCs
Stable, individual clones of Cas9-expressing Hoxb8 macrophages were generated using bone marrow from Cas9 mice (JAX Strain #026179) and a lentiviral transduction protocol adapted from Fang Zhang and colleagues. G6pdx was disrupted using the Zhang lab lentiGuide-Puro sgRNA plasmid with a verified guide for G6pdx. lentiGuide-Puro was a gift from Feng Zhang (Addgene plasmid # 52963). Guide RNAs targeting G6pdx and scramble control were generated using the following oligo pairs:
G6pdx:
5’---CACCGACCTGAAGATACCTTCATTG-−-3’
5’---AAACCAATGAAGGTATCTTCAGGTC-−-3’
Scramble Control:
5’--- CACCGCACTCACATCGCTACATCA-−-3’
3’--- AAACTGATGTAGCGATGTGAGTGC-−-5’.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were executed using GraphPad Prism 7, SPSS v.22, and R v.4.0.5. We determined statistical significance, depending on the structure of the data, via unpaired two-tailed Student’s t-test, nonparametric Mann–Whitney U-test, one-way or two-way ANOVA, or Fisher’s exact test. The statistical test used for each experiment can be found in the corresponding figure legend. R v.4.0.5 was used for graphical and statistical analyses and the R package DESeq2 was used for differential gene expression analysis of transcriptomic and proteomic analyses. All genes were curated according to a previously described approach21. Briefly, complementary analyses of biological pathways were performed by comparing significantly differentially expressed genes or proteins and cross-referencing them with the Molecular Signatures Database (MSigDB). Gephi (v.0.9.1 https://gephi.org/) was used to perform standard network clustering analyses. The “Link Communities” algorithm for biological network analysis was used to calculate edges and nodes56. The Yifan Hu layout algorithm was used to determine network structure. All biologically independent samples were included and used for statistical and graphical analyses. No data was excluded from this manuscript and can be found online under Data S1 - Source Data. Sample sizes were not predetermined using statistical methods. RNA sequencing data is deposited under NCBI GEO accession GSE192969.
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| β-actin | SCBT | sc-47778 |
| G6PDX | Abcam | ab91034 |
| CD16/32 (FcR block) | BioxCell | BP0307 |
| CD11b | eBioscience | 48-0112-82 |
| F4/80 | eBioscience | 12-4801-82 |
| Annexin V | Biolegend | 640912 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Thymocytes | C57BL/6J mice | |
| Bone marrow macrophages | C57BL/6J mice | |
| Splenic macrophages | C57BL/6J mice | |
| Chemicals, peptides, and recombinant proteins | ||
| CypHer5E NHS Ester | Cytiva | PA15405 |
| 5(6)-TAMRA, SE | ThermoFisher | C1171 |
| Cycloheximide | Sigma | C4859 |
| 6-AN | MedChemExpress | HY-W010342 |
| G6PDi-1 | Cayman Chemicals | 31484 |
| FBP1i | Cayman Chemicals | 18860 |
| CFSE | ThermoFisher | C34554 |
| Dexamethasone | Sigma | D4902 |
| 7-AAD | Biolegend | 420403 |
| 13C6-lysine | Cambridge Isotopes | 201740-81-0 |
| 12C6-arginine | Cambridge Isotopes | 1119-34-2 |
| [1,2-13C]-glucose | Cambridge Isotopes | 138079-87-5 |
| [U-13C]-glucose | Cambridge Isotopes | 110187-42-3 |
| MitoSOX Red | ThermoFisher | M36008 |
| MitoTracker Green | ThermoFisher | M7514 |
| MitoTracker Red | ThermoFisher | M22425 |
| BODIPY 581/591 C11 | ThermoFisher | D3861 |
| CellRox Deep Red | ThermoFisher | C10491 |
| Lysosensor Green DND-189 | ThermoFisher | L7535 |
| Critical commercial assays | ||
| NADP/NADPH Quantitation Colorimetric Kit | BioVision | K347 |
| GSH/GSSG-Glo Assay | Promega | V6611 |
| Luminescent ATP Detection Assay Kit | Abcam | ab113849 |
| Deposited data | ||
| mRNA sequencing data | NCBI GEO | GSE192969 |
| Data S1 – Source Data | ||
| Experimental models: Cell lines | ||
| Jurkat T lymphoma | ATCC | TIB-152 |
| MDA-MB-231 | MSKCC | |
| BrM2 | MSKCC | |
| E0771 | CH3 Biosystems | 94A001 |
| Experimental models: Organisms/strains | ||
| C57BL6/J mice | JAX | 000664 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| Mouse G6pdx ORF | OriGene | MR208269 |
| Software and algorithms | ||
| GraphPad Prism 7 | ||
| SPSS v.22 | ||
| R v.4.0.5 | ||
| Gephi v.0.9.1 | ||
| Other | ||
LIFE SCIENCE TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Snail | Cell Signaling Technology | Cat#3879S; RRID: AB_2255011 |
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 |
| Rabbit polyclonal anti-BMAL1 | This paper | N/A |
| Bacterial and virus strains | ||
| pAAV-hSyn-DIO-hM3D(Gq)-mCherry | Krashes et al.1 | Addgene AAV5; 44361-AAV5 |
| AAV5-EF1a-DIO-hChR2(H134R)-EYFP | Hope Center Viral Vectors Core | N/A |
| Cowpox virus Brighton Red | BEI Resources | NR-88 |
| Zika-SMGC-1, GENBANK: KX266255 | Isolated from patient (Wang et al.2) | N/A |
| Staphylococcus aureus | ATCC | ATCC 29213 |
| Streptococcus pyogenes: M1 serotype strain: strain SF370; M1 GAS | ATCC | ATCC 700294 |
| Biological samples | ||
| Healthy adult BA9 brain tissue | University of Maryland Brain & Tissue Bank; http://medschool.umaryland.edu/btbank/ | Cat#UMB1455 |
| Human hippocampal brain blocks | New York Brain Bank | http://nybb.hs.columbia.edu/ |
| Patient-derived xenografts (PDX) | Children’s Oncology Group Cell Culture and Xenograft Repository | http://cogcell.org/ |
| Chemicals, peptides, and recombinant proteins | ||
| MK-2206 AKT inhibitor | Selleck Chemicals | S1078; CAS: 1032350-13-2 |
| SB-505124 | Sigma-Aldrich | S4696; CAS: 694433-59-5 (free base) |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124-87-8 |
| Human TGF-β | R&D | 240-B; GenPept: P01137 |
| Activated S6K1 | Millipore | Cat#14-486 |
| GST-BMAL1 | Novus | Cat#H00000406-P01 |
| Critical commercial assays | ||
| EasyTag EXPRESS 35S Protein Labeling Kit | PerkinElmer | NEG772014MC |
| CaspaseGlo 3/7 | Promega | G8090 |
| TruSeq ChIP Sample Prep Kit | Illumina | IP-202-1012 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE63473 |
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Nanog STILT inference | This paper; Mendeley Data | http://dx.doi.org/10.17632/wx6s4mj7s8.2 |
| Affinity-based mass spectrometry performed with 57 genes | This paper; Mendeley Data | Table S8; http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Experimental models: Cell lines | ||
| Hamster: CHO cells | ATCC | CRL-11268 |
| D. melanogaster: Cell line S2: S2-DRSC | Laboratory of Norbert Perrimon | FlyBase: FBtc0000181 |
| Human: Passage 40 H9 ES cells | MSKCC stem cell core facility | N/A |
| Human: HUES 8 hESC line (NIH approval number NIHhESC-09-0021) | HSCI iPS Core | hES Cell Line: HUES-8 |
| Experimental models: Organisms/strains | ||
| C. elegans: Strain BC4011: srl-1(s2500) II; dpy-18(e364) III; unc-46(e177)rol-3(s1040) V. | Caenorhabditis Genetics Center | WB Strain: BC4011; WormBase: WBVar00241916 |
| D. melanogaster: RNAi of Sxl. y[1] sc[*] v[1]; P{TRiP.HMS00609}attP2 | Bloomington Drosophila Stock Center | BDSC:34393; FlyBase: FBtp0064874 |
| S. cerevisiae: Strain background: W303 | ATCC | ATTC: 208353 |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J | The Jackson Laboratory | JAX: 006494 |
| Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J | The Jackson Laboratory | RRID: IMSR_JAX:008471 |
| Zebrafish: Tg(Shha:GFP)t10: t10Tg | Neumann and Nuesslein-Volhard3 | ZFIN: ZDB-GENO-060207-1 |
| Arabidopsis: 35S::PIF4-YFP, BZR1-CFP | Wang et al.4 | N/A |
| Arabidopsis: JYB1021.2: pS24(AT5G58010)::cS24:GFP(-G):NOS #1 | NASC | NASC ID: N70450 |
| Oligonucleotides | ||
| siRNA targeting sequence: PIP5K I alpha #1: ACACAGUACUCAGUUGAUA | This paper | N/A |
| Primers for XX, see Table SX | This paper | N/A |
| Primer: GFP/YFP/CFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC | This paper | N/A |
| Morpholino: MO-pax2a GGTCTGCTTTGCAGTGAATATCCAT | Gene Tools | ZFIN: ZDB-MRPHLNO-061106-5 |
| ACTB (hs01060665_g1) | Life Technologies | Cat#4331182 |
| RNA sequence: hnRNPA1_ligand: UAGGGACUUAGGGUUCUCUCUAGGGACUUAGGGUUCUCUCUAGGGA | This paper | N/A |
| Recombinant DNA | ||
| pLVX-Tight-Puro (TetOn) | Clonetech | Cat#632162 |
| Plasmid: GFP-Nito | This paper | N/A |
| cDNA GH111110 | Drosophila Genomics Resource Center | DGRC:5666; FlyBase:FBcl0130415 |
| AAV2/1-hsyn-GCaMP6- WPRE | Chen et al.5 | N/A |
| Mouse raptor: pLKO mouse shRNA 1 raptor | Thoreen et al.6 | Addgene Plasmid #21339 |
| Software and algorithms | ||
| ImageJ | Schneider et al.7 | https://imagej.nih.gov/ij/ |
| Bowtie2 | Langmead and Salzberg8 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al.9 | http://samtools.sourceforge.net/ |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al.10 | https://github.com/ChristophRau/wMICA |
| ICS algorithm | This paper; Mendeley Data | http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Other | ||
| Sequence data, analyses, and resources related to the ultra-deep sequencing of the AML31 tumor, relapse, and matched normal | This paper | http://aml31.genome.wustl.edu |
| Resource website for the AML31 publication | This paper | https://github.com/chrisamiller/aml31SuppSite |
PHYSICAL SCIENCE TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| QD605 streptavidin conjugated quantum dot | Thermo Fisher Scientific | Cat#Q10101MP |
| Platinum black | Sigma-Aldrich | Cat#205915 |
| Sodium formate BioUltra, ≥99.0% (NT) | Sigma-Aldrich | Cat#71359 |
| Chloramphenicol | Sigma-Aldrich | Cat#C0378 |
| Carbon dioxide (13C, 99%) (<2% 18O) | Cambridge Isotope Laboratories | CLM-185-5 |
| Poly(vinylidene fluoride-co-hexafluoropropylene) | Sigma-Aldrich | 427179 |
| PTFE Hydrophilic Membrane Filters, 0.22 μm, 90 mm | Scientificfilters.com/Tisch Scientific | SF13842 |
| Critical commercial assays | ||
| Folic Acid (FA) ELISA kit | Alpha Diagnostic International | Cat# 0365-0B9 |
| TMT10plex Isobaric Label Reagent Set | Thermo Fisher | A37725 |
| Surface Plasmon Resonance CM5 kit | GE Healthcare | Cat#29104988 |
| NanoBRET Target Engagement K-5 kit | Promega | Cat#N2500 |
| Deposited data | ||
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Structure of compound 5 | This paper; Cambridge Crystallographic Data Center | CCDC: 2016466 |
| Code for constraints-based modeling and analysis of autotrophic E. coli | This paper | https://gitlab.com/elad.noor/sloppy/tree/master/rubisco |
| Software and algorithms | ||
| Gaussian09 | Frish et al.1 | https://gaussian.com |
| Python version 2.7 | Python Software Foundation | https://www.python.org |
| ChemDraw Professional 18.0 | PerkinElmer | https://www.perkinelmer.com/category/chemdraw |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al.2 | https://github.com/ChristophRau/wMICA |
| Other | ||
| DASGIP MX4/4 Gas Mixing Module for 4 Vessels with a Mass Flow Controller | Eppendorf | Cat#76DGMX44 |
| Agilent 1200 series HPLC | Agilent Technologies | https://www.agilent.com/en/products/liquid-chromatography |
| PHI Quantera II XPS | ULVAC-PHI, Inc. | https://www.ulvac-phi.com/en/products/xps/phi-quantera-ii/ |
Highlights.
Chronic physiological hypoxia enhances macrophage apoptotic cell uptake and degradation
Chronic physiological hypoxia induces both primed and poised states in macrophages
Both primed and poised state programs directly support enhanced continual efferocytosis
A noncanonical PPP loop supports enhanced efferocytosis and maintains redox homeostasis
Acknowledgements
We thank members of the Perry laboratory and co-authors for edits and discussions related to this manuscript. We thank Justin Cross and the Donald B. and Catherine C. Marron Cancer Metabolism Center at MSKCC for help with YSI experiments. This work was supported by grants to J.S.A.P. from the NIH (NCI 5R00CA237728; NIGMS 1DP2GM146337), a Parker Institute for Cancer Immunotherapy Career Development Award, a V Foundation Scholars Award, a MSKCC Functional Genomics Institute pilot award, a grant to K.R.K. from the NIH (NCI 1R01CA248364), and MSKCC Cancer Center Support Grant P30CA008748. Some figure panels were created using the commercial version of BioRender.
Footnotes
Declaration of Interests
K.R.K. Serves on the scientific advisory board of NVision Imaging Technologies. J.S.A.P. and K.R.K holds patents related to imaging and modulation of cellular metabolism.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boada-Romero E, Martinez J, Heckmann BL, and Green DR (2020). The clearance of dead cells by efferocytosis. Nature Reviews Molecular Cell Biology 21, 398–414. 10.1038/s41580-020-0232-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doran AC, Yurdagul A, and Tabas I (2020). Efferocytosis in health and disease. Nature Reviews Immunology 20, 254–267. 10.1038/s41577-019-0240-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rothlin CV, Hille TD, and Ghosh S (2020). Determining the effector response to cell death. Nat Rev Immunol. 10.1038/s41577-020-00456-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elliott Michael R., and Ravichandran Kodi S. (2016). The Dynamics of Apoptotic Cell Clearance. Developmental Cell 38, 147–160. 10.1016/j.devcel.2016.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henson PM (2017). Cell Removal: Efferocytosis. Annual Review of Cell and Developmental Biology 33, 127–144. 10.1146/annurev-cellbio-111315-125315. [DOI] [PubMed] [Google Scholar]
- 6.Morioka S, Maueröder C, and Ravichandran KS (2019). Living on the Edge: Efferocytosis at the Interface of Homeostasis and Pathology. Immunity 50, 1149–1162. 10.1016/j.immuni.2019.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Penberthy KK, Lysiak JJ, and Ravichandran KS (2018). Rethinking Phagocytes: Clues from the Retina and Testes. Trends Cell Biol 28, 317–327. 10.1016/j.tcb.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zago G, Saavedra PHV, Keshari KR, and Perry JSA (2021). Immunometabolism of Tissue-Resident Macrophages – An Appraisal of the Current Knowledge and Cutting-Edge Methods and Technologies. Frontiers in Immunology 12. 10.3389/fimmu.2021.665782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sender R, and Milo R (2021). The distribution of cellular turnover in the human body. Nature Medicine 27, 45–48. 10.1038/s41591-020-01182-9. [DOI] [PubMed] [Google Scholar]
- 10.Trzeciak A, Wang Y-T, and Perry JSA (2021). First we eat, then we do everything else: The dynamic metabolic regulation of efferocytosis. Cell Metabolism. 10.1016/j.cmet.2021.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amit I, Winter DR, and Jung S (2016). The role of the local environment and epigenetics in shaping macrophage identity and their effect on tissue homeostasis. Nature Immunology 17, 18–25. 10.1038/ni.3325. [DOI] [PubMed] [Google Scholar]
- 12.Guilliams M, and Svedberg FR (2021). Does tissue imprinting restrict macrophage plasticity? Nature Immunology 22, 118–127. 10.1038/s41590-020-00849-2. [DOI] [PubMed] [Google Scholar]
- 13.Guilliams M, Thierry GR, Bonnardel J, and Bajenoff M (2020). Establishment and Maintenance of the Macrophage Niche. Immunity 52, 434–451. 10.1016/j.immuni.2020.02.015. [DOI] [PubMed] [Google Scholar]
- 14.Okabe Y, and Medzhitov R (2016). Tissue biology perspective on macrophages. Nature Immunology 17, 9–17. 10.1038/ni.3320. [DOI] [PubMed] [Google Scholar]
- 15.Blériot C, Chakarov S, and Ginhoux F (2020). Determinants of Resident Tissue Macrophage Identity and Function. Immunity 52, 957–970. 10.1016/j.immuni.2020.05.014. [DOI] [PubMed] [Google Scholar]
- 16.Breed ER, Watanabe M, and Hogquist KA (2019). Measuring Thymic Clonal Deletion at the Population Level. Journal of immunology (Baltimore, Md. : 1950) 202, 3226–3233. 10.4049/jimmunol.1900191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stritesky GL, Xing Y, Erickson JR, Kalekar LA, Wang X, Mueller DL, Jameson SC, and Hogquist KA (2013). Murine thymic selection quantified using a unique method to capture deleted T cells. Proceedings of the National Academy of Sciences of the United States of America 110, 4679–4684. 10.1073/pnas.1217532110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, and Kieda C (2011). Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med 15, 1239–1253. 10.1111/j.1582-4934.2011.01258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norris PC, Libreros S, and Serhan CN (2019). Resolution metabolomes activated by hypoxic environment. 5, eaax4895. doi: 10.1126/sciadv.aax4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trayhurn P (2019). Oxygen-A Critical, but Overlooked, Nutrient. Front Nutr 6, 10–10. 10.3389/fnut.2019.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morioka S, Perry JSA, Raymond MH, Medina CB, Zhu Y, Zhao L, Serbulea V, Onengut-Gumuscu S, Leitinger N, Kucenas S, et al. (2018). Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature 563, 714–718. 10.1038/s41586-018-0735-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perry JSA, Morioka S, Medina CB, Iker Etchegaray J, Barron B, Raymond MH, Lucas CD, Onengut-Gumuscu S, Delpire E, and Ravichandran KS (2019). Interpreting an apoptotic corpse as anti-inflammatory involves a chloride sensing pathway. Nature Cell Biology 21, 1532–1543. 10.1038/s41556-019-0431-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Subramanian M, Yurdagul A, Barbosa-Lorenzi VC, Cai B, de Juan-Sanz J, Ryan TA, Nomura M, Maxfield FR, and Tabas I (2017). Mitochondrial Fission Promotes the Continued Clearance of Apoptotic Cells by Macrophages. Cell 171, 331–345.e322. 10.1016/j.cell.2017.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yurdagul A, Subramanian M, Wang X, Crown SB, Ilkayeva OR, Darville L, Kolluru GK, Rymond CC, Gerlach BD, Zheng Z, et al. (2020). Macrophage Metabolism of Apoptotic Cell-Derived Arginine Promotes Continual Efferocytosis and Resolution of Injury. Cell Metabolism 31, 518–533.e510. 10.1016/j.cmet.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang S, Weinberg S, DeBerge M, Gainullina A, Schipma M, Kinchen JM, Ben-Sahra I, Gius DR, Yvan-Charvet L, Chandel NS, et al. (2019). Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair. Cell Metabolism 29, 443–456.e445. 10.1016/j.cmet.2018.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park D, Han CZ, Elliott MR, Kinchen JM, Trampont PC, Das S, Collins S, Lysiak JJ, Hoehn KL, and Ravichandran KS (2011). Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature 477, 220–224. 10.1038/nature10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Viaud M, Ivanov S, Vujic N, Duta-Mare M, Aira L-E, Barouillet T, Garcia E, Orange F, Dugail I, Hainault I, et al. (2018). Lysosomal Cholesterol Hydrolysis Couples Efferocytosis to Anti-Inflammatory Oxysterol Production. Circulation research 122, 1369–1384. doi: 10.1161/CIRCRESAHA.117.312333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jain IH, Calvo SE, Markhard AL, Skinner OS, To T-L, Ast T, and Mootha VK (2020). Genetic Screen for Cell Fitness in High or Low Oxygen Highlights Mitochondrial and Lipid Metabolism. Cell 181, 716–727.e711. 10.1016/j.cell.2020.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hale LP, Braun RD, Gwinn WM, Greer PK, and Dewhirst MW (2002). Hypoxia in the thymus: role of oxygen tension in thymocyte survival. 282, H1467–H1477. 10.1152/ajpheart.00682.2001. [DOI] [PubMed] [Google Scholar]
- 30.Denko NC (2008). Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nature Reviews Cancer 8, 705–713. 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 31.Sadiku P, and Walmsley SR (2019). Hypoxia and the regulation of myeloid cell metabolic imprinting: consequences for the inflammatory response. EMBO Rep 20, e47388. 10.15252/embr.201847388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor CT, and Colgan SP (2017). Regulation of immunity and inflammation by hypoxia in immunological niches. Nature Reviews Immunology 17, 774–785. 10.1038/nri.2017.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jang C, Chen L, and Rabinowitz JD (2018). Metabolomics and Isotope Tracing. Cell 173, 822–837. 10.1016/j.cell.2018.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Artyomov MN, and Van den Bossche J (2020). Immunometabolism in the Single-Cell Era. Cell Metabolism 32, 710–725. 10.1016/j.cmet.2020.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makowski L, Chaib M, and Rathmell JC (2020). Immunometabolism: From basic mechanisms to translation. 295, 5–14. 10.1111/imr.12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryan DG, and O’Neill LAJ (2020). Krebs Cycle Reborn in Macrophage Immunometabolism. Annual review of immunology 38, 289–313. 10.1146/annurev-immunol-081619-104850. [DOI] [PubMed] [Google Scholar]
- 37.Cheng M-L, Lin J-F, Huang C-Y, Li G-J, Shih L-M, Chiu DT-Y, and Ho H-Y (2019). Sedoheptulose-1,7-bisphospate Accumulation and Metabolic Anomalies in Hepatoma Cells Exposed to Oxidative Stress. Oxidative Medicine and Cellular Longevity 2019, 5913635. 10.1155/2019/5913635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Longenecker JP, and Williams JF (1980). Quantitative measurement of the L-type pentose phosphate cycle with [2–14C]glucose and [5–14C]glucose in isolated hepatocytes. The Biochemical journal 188, 859–865. 10.1042/bj1880859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams JF, Blackmore PF, and Arora KK (1985). The significance of sedoheptulose 1,7-bisphosphate in the metabolism and regulation of the pentose pathway in liver. Biochemistry international 11, 599–610. [PubMed] [Google Scholar]
- 40.Muri J, and Kopf M (2020). Redox regulation of immunometabolism. Nature Reviews Immunology. 10.1038/s41577-020-00478-8. [DOI] [PubMed] [Google Scholar]
- 41.Tsai TL, Zhou TA, Hsieh YT, Wang JC, Cheng HK, Huang CH, Tsai PY, Fan HH, Feng HK, Huang YC, et al. (2022). Multiomics reveal the central role of pentose phosphate pathway in resident thymic macrophages to cope with efferocytosis-associated stress. Cell Rep 40, 111065. 10.1016/j.celrep.2022.111065. [DOI] [PubMed] [Google Scholar]
- 42.He D, Mao Q, Jia J, Wang Z, Liu Y, Liu T, Luo B, and Zhang Z (2021). Pentose Phosphate Pathway Regulates Tolerogenic Apoptotic Cell Clearance and Immune Tolerance. Front Immunol 12, 797091. 10.3389/fimmu.2021.797091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bagaitkar J, Huang J, Zeng MY, Pech NK, Monlish DA, Perez-Zapata LJ, Miralda I, Schuettpelz LG, and Dinauer MC (2018). NADPH oxidase activation regulates apoptotic neutrophil clearance by murine macrophages. Blood 131, 2367–2378. 10.1182/blood-2017-09-809004 %J Blood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mantegazza AR, Savina A, Vermeulen M, Pérez L, Geffner J, Hermine O, Rosenzweig SD, Faure F, and Amigorena S (2008). NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood 112, 4712–4722. 10.1182/blood-2008-01-134791 %J Blood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yvan-Charvet L, Pagler TA, Seimon TA, Thorp E, Welch CL, Witztum JL, Tabas I, and Tall AR (2010). ABCA1 and ABCG1 Protect Against Oxidative Stress–Induced Macrophage Apoptosis During Efferocytosis. 106, 1861–1869. doi: 10.1161/CIRCRESAHA.110.217281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Acosta-Iborra B, Elorza A, Olazabal IM, Martín-Cofreces NB, Martin-Puig S, Miró M, Calzada MJ, Aragonés J, Sánchez-Madrid F, and Landázuri MO (2009). Macrophage oxygen sensing modulates antigen presentation and phagocytic functions involving IFN-gamma production through the HIF-1 alpha transcription factor. Journal of immunology (Baltimore, Md. : 1950) 182, 3155–3164. 10.4049/jimmunol.0801710. [DOI] [PubMed] [Google Scholar]
- 47.Anand RJ, Gribar SC, Li J, Kohler JW, Branca MF, Dubowski T, Sodhi CP, and Hackam DJ (2007). Hypoxia causes an increase in phagocytosis by macrophages in a HIF-1α-dependent manner. 82, 1257–1265. 10.1189/jlb.0307195. [DOI] [PubMed] [Google Scholar]
- 48.Fritzenwanger M, Jung C, Goebel B, Lauten A, and Figulla HR (2011). Impact of short-term systemic hypoxia on phagocytosis, cytokine production, and transcription factor activation in peripheral blood cells. Mediators Inflamm 2011, 429501–429501. 10.1155/2011/429501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dehn S, DeBerge M, Yeap X-Y, Yvan-Charvet L, Fang D, Eltzschig HK, Miller SD, and Thorp EB (2016). HIF-2α in Resting Macrophages Tempers Mitochondrial Reactive Oxygen Species To Selectively Repress MARCO-Dependent Phagocytosis. 197, 3639–3649. 10.4049/jimmunol.1600402 %J The Journal of Immunology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin N, Shay JES, Xie H, Lee DSM, Skuli N, Tang Q, Zhou Z, Azzam A, Meng H, Wang H, et al. (2018). Myeloid Cell Hypoxia-Inducible Factors Promote Resolution of Inflammation in Experimental Colitis. Frontiers in Immunology 9. 10.3389/fimmu.2018.02565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marsch E, Theelen TL, Demandt JA, Jeurissen M, van Gink M, Verjans R, Janssen A, Cleutjens JP, Meex SJ, Donners MM, et al. (2014). Reversal of hypoxia in murine atherosclerosis prevents necrotic core expansion by enhancing efferocytosis. Arteriosclerosis, thrombosis, and vascular biology 34, 2545–2553. 10.1161/atvbaha.114.304023. [DOI] [PubMed] [Google Scholar]
- 52.Ginouvès A, Ilc K, Macías N, Pouysségur J, and Berra E (2008). PHDs overactivation during chronic hypoxia “desensitizes” HIFα and protects cells from necrosis. 105, 4745–4750. 10.1073/pnas.0705680105 %J Proceedings of the National Academy of Sciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang GG, Calvo KR, Pasillas MP, Sykes DB, Häcker H, and Kamps MP (2006). Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nature Methods 3, 287. 10.1038/nmeth865 https://www.nature.com/articles/nmeth865#supplementary-information. [DOI] [PubMed] [Google Scholar]
- 54.Navarrete-Perea J, Yu Q, Gygi SP, and Paulo JA (2018). Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. Journal of Proteome Research 17, 2226–2236. 10.1021/acs.jproteome.8b00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature Protocols 2, 1896–1906. 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- 56.Ahn Y-Y, Bagrow JP, and Lehmann S (2010). Link communities reveal multiscale complexity in networks. Nature 466, 761–764. 10.1038/nature09182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
mRNA sequencing raw data files have been deposited in NCBI Gene Expression Omnibus (GEO) archive (accession GSE192969). An Excel file with plotted data points and a pdf image with uncropped western blots are in Data S1.
No new code was used in the preparation of this manuscript.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.