

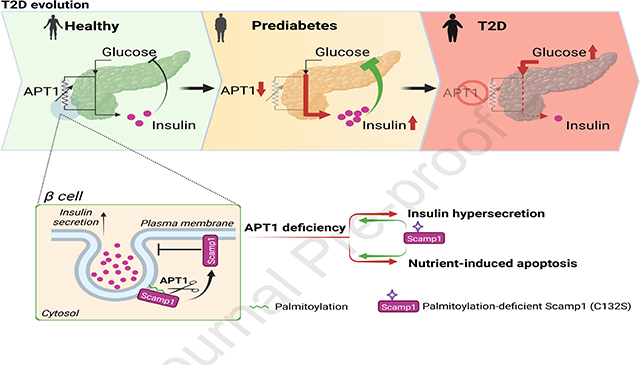
SUMMARY
Hyperinsulinemia often precedes type 2 diabetes. Palmitoylation, implicated in exocytosis, is reversed by acyl-protein thioesterase 1 (APT1). APT1 biology was altered in pancreatic islets from humans with type 2 diabetes, and APT1 knockdown in nondiabetic islets caused insulin hypersecretion. APT1 knockout mice had islet autonomous increased glucose-stimulated insulin secretion that was associated with prolonged insulin granule fusion. Using palmitoylation proteomics, we identified Scamp1 as an APT1 substrate that localized to insulin secretory granules. Scamp1 knockdown caused insulin hypersecretion. Expression of a mutated Scamp1 incapable of being palmitoylated in APT1 deficient cells rescued insulin hypersecretion and nutrient induced apoptosis. High fat fed islet specific APT1 knockout mice and global APT1 deficient db/db mice showed increased β-cell failure. These findings suggest that APT1 is regulated in human islets, and that APT1 deficiency causes insulin hypersecretion leading to β-cell failure, modeling the evolution of some forms of human type 2 diabetes.
eTOC blurb
The depalmitoylation enzyme acyl-protein thioesterase 1 (APT1) is altered in pancreatic islets from people with type 2 diabetes and in nondiabetic islets exposed to hyperglycemia. APT1 deficiency in mice increases insulin secretion that promotes β-cell failure in the setting of metabolic stress, suggesting involvement of palmitoylation in diabetes pathogenesis.
Graphical Abstract
INTRODUCTION
Type 2 diabetes is a lethal multisystem disease defined by elevated levels of circulating glucose. Hyperglycemia in type 2 diabetes results from β-cell dysfunction usually developing in the context of insulin resistance.1,2 Hyperinsulinemia is common in type 2 diabetes, and hyperglycemia may represent the failure of β-cells to compensate for peripheral insulin resistance. The mechanisms responsible for the evolution of type 2 diabetes remain obscure, and the disease has a spectrum of presentations3 that encompass variable risks for diabetes complications and death.4 Some of these presentations are characterized by insulin hypersecretion in the absence of insulin resistance long before circulating glucose levels reach levels diagnostic of diabetes.5,6 The etiology of this hypersecretion is unknown, and it is unknown whether increased insulin secretion contributes to β-cell exhaustion or the progression of insulin resistance and diabetes phenotypes.7
Nutrient excess leading to tissue accumulation of lipids is common in type 2 diabetes, and elevated levels of the fatty acid palmitate are associated with hyperinsulinemia and β-cell dysfunction.8 Nutrient toxicity, sometimes called glucolipotoxicity, is poorly understood and the role of fatty acids in diabetes pathogenesis is controversial.9,10 Palmitoylation, the modification of protein structure through the formation of a thioester bond between the ester moiety of palmitate and the sulfhydryl group on cysteine residues, affects at least 4000 human proteins11 and has been neglected as a potential contributor to metabolic disease.
Palmitoylation is driven by a large family of DHHC acyltransferases and reversed by a small family of acyl-protein thioesterases (APTs). Glucose increases the number of palmitoylated proteins in cells that model pancreatic β-cells,12 suggesting that fuel flow might coordinate metabolic function through palmitoylation.
APT1 is a dominant mediator of depalmitoylation,13 the process of removing palmitate from cysteine residues. APT1 enzyme activity is decreased in diabetic endothelial cells, exposure of normal endothelial cells to hyperglycemia decreases APT1 enzyme activity, and genetic APT1 deficiency mimics diabetic vascular disease,14 suggesting that palmitoylation/depalmitoylation cycling may be important in diabetes pathophysiology. Since many components of insulin secretion and insulin action are palmitoylated,15 we tested the hypothesis that palmitoylation affects β-cell function in type 2 diabetes. Our findings are consistent with a central role for dynamic palmitoylation in preventing progression to type 2 diabetes.
RESULTS
APT1 Expression, Enzyme Activity, and Effect on Insulin Secretion in Human Islets
Palmitoylation mediates synaptic vesicle fusion in neurons through many of the same proteins involved in insulin secretion,16 and APT1 affects function and intracellular trafficking in a neuronal disease model.17 We studied APT1 in human pancreatic islets. In islets (see Table S1A for demographics) from 7 nondiabetic subjects (mean Hb A1c 5.1%) and 4 diabetic subjects (mean Hb A1c 7.0%), APT1 mRNA was 48% higher in the setting of diabetes (p = 0.0160, Figure 1A). In human endothelial cells, hyperglycemia increases APT1 mRNA but decreases APT1 enzyme activity.14 Hyperglycemia also decreased APT1 enzyme activity in human islets. We assayed activity with a probe (DPP3) bearing a thioester that fluoresces when this bond is hydrolyzed by depalmitoylases.18 Using samples different from those utilized for APT1 expression, human islets (see Table S1B for demographics) were cultured in 5.5 mM or 25 mM glucose for 48 h followed by incubation with the depalmitoylation probe, FACS separation, and quantification of emission fluorescence. Figure 1B shows the gating strategy and DPP3 histogram for whole live cells, islet cells (DAPI−Hpi2+), and β-cells (DAPI−Hpi2+CD26−NTPDase3+). Enzyme activity is expressed as the % change in fluorescent signal (arbitrary units) in cells treated with 25 mM glucose as compared to 5.5 mM glucose. High glucose decreased depalmitoylation activity in islets from three different nondiabetic human donors (Figure 1C).
Figure 1. APT1 expression, enzyme activity, and effect of APT1 deficiency on insulin secretion in human islets.
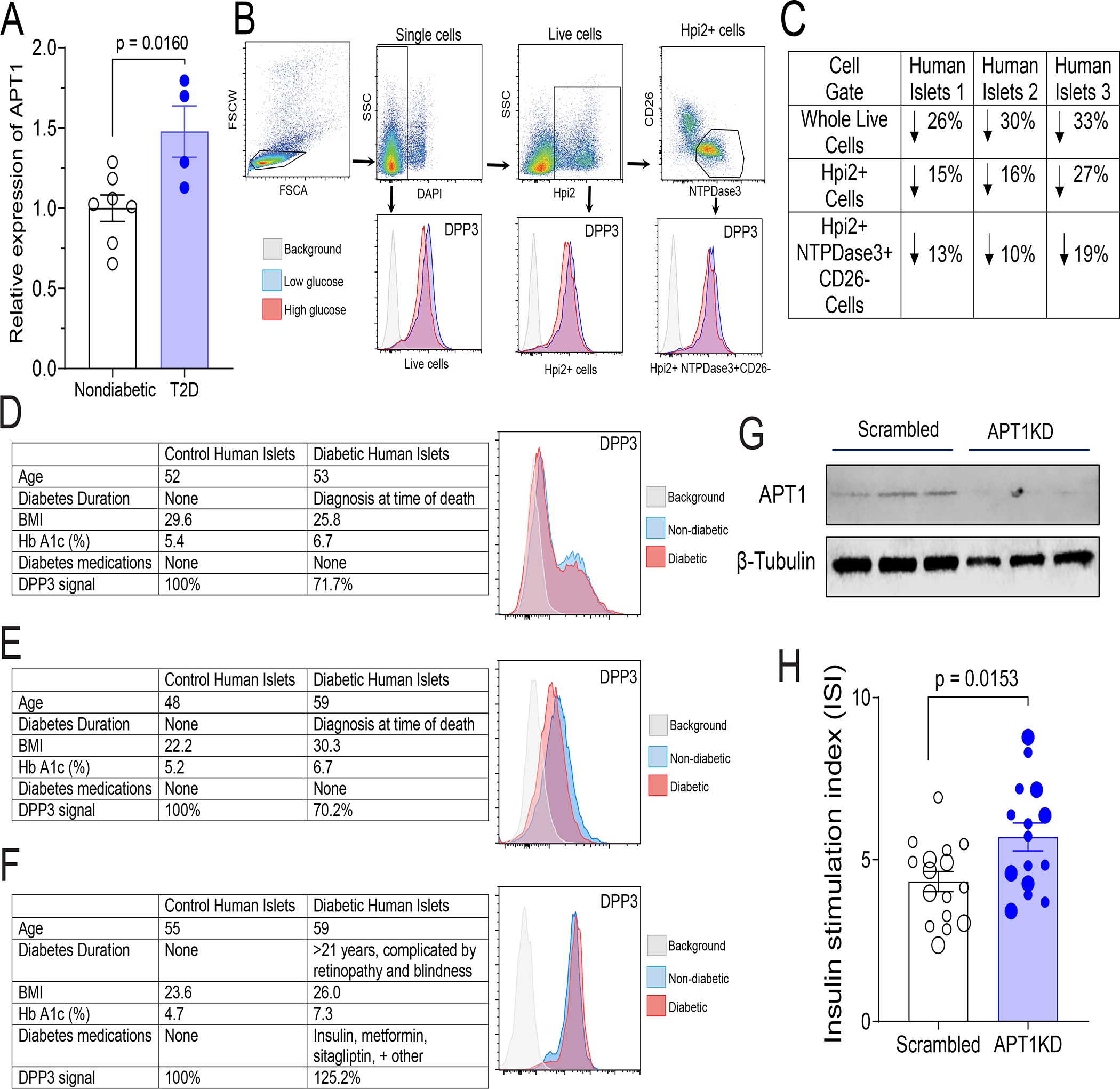
(A) APT1 mRNA in islets from control (nondiabetic) and type 2 diabetes (T2D) humans. Donor demographics are provided in Table S1A. (B,C) Hyperglycemia decreases APT1 enzyme activity in nondiabetic human islets. APT1 activity was assayed using fluorescent depalmitoylation probe DPP3 in human islets treated with low glucose (5.5 mM) or high glucose (25 mM) for 48 h. FACS gating strategy is shown in top of panel B and the DPP3 histogram for each gate is shown in the bottom of panel B. Whole live cells: single+ DAPI−; Islet cells: single+DAPI−Hpi2+; β-cells: single+DAPI−Hpi2+CD26−NTPDase3+. Panel C shows the activity results for three independent assays using islets from three different donors. Donor demographics are provided in Table S1B. (D,E,F) Comparison of APT1 enzyme activity in human diabetic and nondiabetic islets. Panels D (donor IDs HP-20021-01, HP-20019-01T2D) and E (donor IDs HP-21124-01, HP-21129-01T2D) used islets from donors with no prior history of diabetes; Panel F (donor IDs HP-20268-01, HP-20259-01T2D) used islets from a donor with a long history of diabetes and its complications. (G,H) APT1 knockdown in nondiabetic human islets increases insulin secretion. Western blots to verify knockdown are shown for three culture aliquots in panel G. Insulin stimulation index (insulin after 16.7 mM glucose as compared to 3 mM glucose) in multiple aliquots of islets from two different normal donors indicated by different size symbols. Donor demographics are provided in Table S1C.
We also measured human islet APT1 enzyme activity under the relatively uncommon circumstance of obtaining tissues from diabetic and nondiabetic humans at the same time. In two separate comparisons (Figure 1D, 1E) performed 16 months apart using tissues not confounded by known complications of longstanding diabetes, β-cell APT1 enzyme activity was decreased by ~30% in freshly isolated cells from two male subjects with diabetes (Hb A1c 6.7%, 6.7%, taking no mediations for diabetes since the diagnosis for each was made post-mortem) as compared to nondiabetic male subjects (Hb A1c 5.4%, 5.2%). In another comparison (Figure 1F) using tissue from a patient with longstanding diabetes with severe complications, β-cell yield was low and APT1 enzyme activity was increased by ~25% in male diabetic cells (Hb A1c 7.3%, multiple diabetes medications) as compared to male nondiabetic cells (Hb A1c 4.7%).
Using islets from two nondiabetic humans (see Table S1C for demographics), APT1 was knocked down (average knockdown 77%) followed by separation into multiple culture replicates and assessment of insulin secretion. Validation of the knockdown by Western blotting for APT1 protein is shown in Figure 1G. APT1 knockdown increased glucose-stimulated insulin secretion (p = 0.0153) in nondiabetic human islets (Figure 1H with two different sizes of symbols denoting replicates from two different human donors).
Glucose Metabolism in Two Mouse Models of APT1 Deficiency
Global APT1 deficient mice (APT1 global KO) in the C57BL/6J background generated as described14 were compared with littermates without APT1 deficiency. Both sexes were studied. Male and female chow fed APT1 global KO mice had enhanced glucose tolerance (Figure S1A), no difference in insulin sensitivity by insulin tolerance tests (Figure S1B), and increased secretion of both insulin (Figure S1C) and C-peptide (Figure S1D) during glucose tolerance tests. We generated islet specific APT1 knockout mice (APT1 islet KO) by crossing floxed Lypla1 (the gene encoding APT1 protein) animals with tamoxifen-inducible Pdx1PBCreER™ mice.19 Male chow fed APT1 islet KO mice had enhanced glucose tolerance (Figure S1E), no difference in insulin sensitivity by insulin tolerance tests (Figure S1F), and increased secretion of both insulin (Figure S1G) and C-peptide (Figure S1H) during glucose tolerance tests. Female chow fed APT1 islet KO mice also had increased secretion of insulin during glucose tolerance tests (Figure S2A), but their phenotype was milder than in males and not consistently associated with enhanced glucose tolerance (Figure S2B). APT1 message was absent in global APT 1 KO islets (Figure S2C). After treatment with tamoxifen, APT1 message in islets from APT1 islet KO mice was decreased by ~50% as compared to islets from floxed animals without Cre (Figure S2D), a value similar to the extent of β-cell recombination reported in this Cre line.19 Body weight and fasting plasma values for the global APT1 deficient animals and their controls as well as the islet specific APT1 KO mice and their controls are shown in Table S2A. Cre expression alone may affect islet function, but glucose tolerance (Figure S1I) and insulin levels during glucose tolerance tests (Figure S1J) were the same in tamoxifen-treated non-floxed mice in the presence and absence of Cre. Unlike mice with global APT1 deficiency (APT1 global KO), APT1 global heterozygote mice had glucose tolerance and insulin levels during glucose tolerance tests that were not different from their controls (Figure S1K,L). APT1 is homologous to a related protein with similar function, APT2, but APT2 global KO mice generated as described14 showed glucose tolerance and insulin levels during glucose tolerance tests that were not different from their controls (Figure S2E,F).
Characterization of APT1 deficient Islets
We compared β-cell area for chow fed APT1 global KO mice with enhanced glucose tolerance and their controls. Representative images of islets for both genotypes are shown in Figure 2A with insulin in green, glucagon in red, and nuclei in blue. Histomorphometry of 152 islets from controls and 160 islets from APT1 global KO mice is presented in Figure 2B with total values presented on the left and frequency distribution by area presented on the right. β-cell area per islet did not differ by genotype. Analyses of β-cell area expressed as a percentage of pancreatic area showed no difference between APT1 global KO mice and controls on chow diet (Figure 2C). There was also no difference in pancreatic islet insulin content between APT1 global KO and control mice (Figure 2D).
Figure 2. Isolated islets from APT1 deficient mice have increased glucose-stimulated insulin secretion.
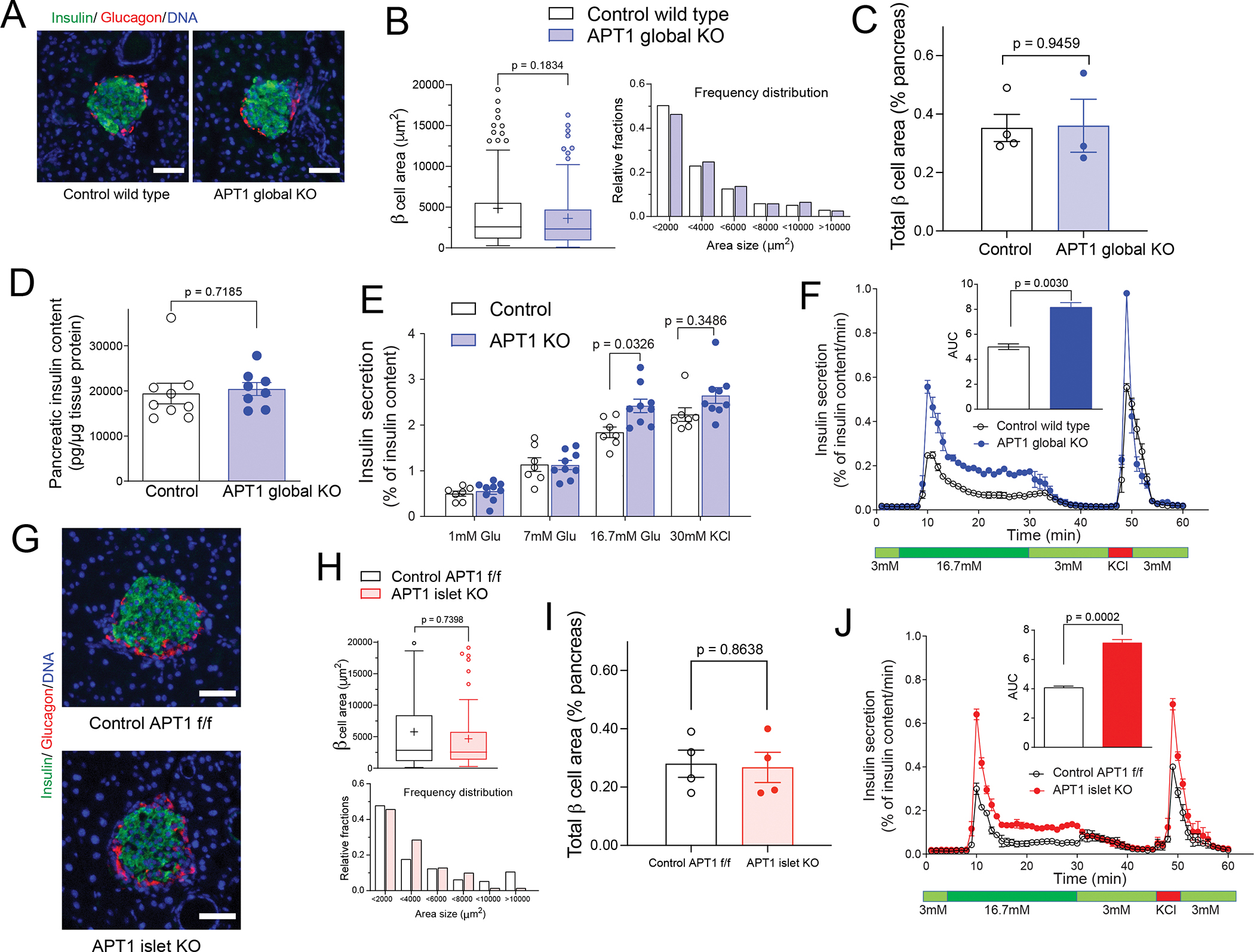
(A,B) Islet histomorphometry is unaffected in chow fed APT1 global KO mice. (C) Analysis of β-cell area as % of pancreatic area in chow fed APT1 global KO mice. (D) Pancreatic insulin content in control and APT1 global KO mice. (E) Static glucose-stimulated insulin secretion in control and global APT1 KO islets. (F) Dynamic glucose-stimulated insulin secretion in control and global APT1 KO islets performed by perifusion. (G,H) Islet histomorphometry is unaffected in chow fed APT1 islet KO mice. (I) Analysis of β-cell area as % of pancreatic area in chow fed APT1 islet KO mice. (J) Dynamic glucose-stimulated insulin secretion in control and islet APT1 KO islets performed by perifusion. Scale=50 μm.
Static glucose-stimulated insulin secretion was increased in APT1 global KO islets as compared to islets from control littermates incubated in 16.7 mM glucose (Figure 2E), but basal insulin secretion did not differ by genotype. Dynamic glucose-stimulated insulin secretion showed increased biphasic release of insulin in APT1 global KO islets compared to control islets that was rapidly extinguished when the ambient glucose concentration was decreased in the perifusion chamber (Figure 2F). Basal secretion did not differ by genotype under dynamic conditions.
Representative images of islets for chow fed APT1 islet KO mice and their floxed controls are shown in Figure 2G. Histomorphometry of 129 islets from controls and 79 islets from APT1 islet KO mice showed no difference in β-cell area per islet by genotype (Figure 2H). Analyses of β-cell area expressed as a percentage of pancreatic area showed no difference between APT1 islet KO mice and controls on chow diet (Figure 2I). As with islets from APT1 global KO mice, perifused islets from APT1 islet KO mice had increased glucose-stimulated biphasic release of insulin compared to control islets with no effect on basal insulin secretion (Figure 2J).
Glucose-stimulated insulin secretion requires glucose metabolism that leads to closure of KATP channels, membrane depolarization, opening of calcium channels, and increased intracellular calcium that mediates the release of insulin granules. Calcium dynamics measured by Fluo-4 imaging showed that calcium signals were the same in isolated islets from control and APT1 global KO islets in 2 mM glucose and following stimulation with 16.7 mM glucose (Figure 3A). There was also no difference in the number of insulin stained granules docked at the plasma membrane between control and APT1 KO islets in low or high glucose (Figure 3B). Collectively, these findings suggest that insulin hypersecretion with APT1 deficiency is mediated downstream of depolarization dependent calcium influx and the insulin biosynthetic apparatus.
Figure 3. Calcium imaging, docked granules, cortical actin, and kinetics of granule fusion.
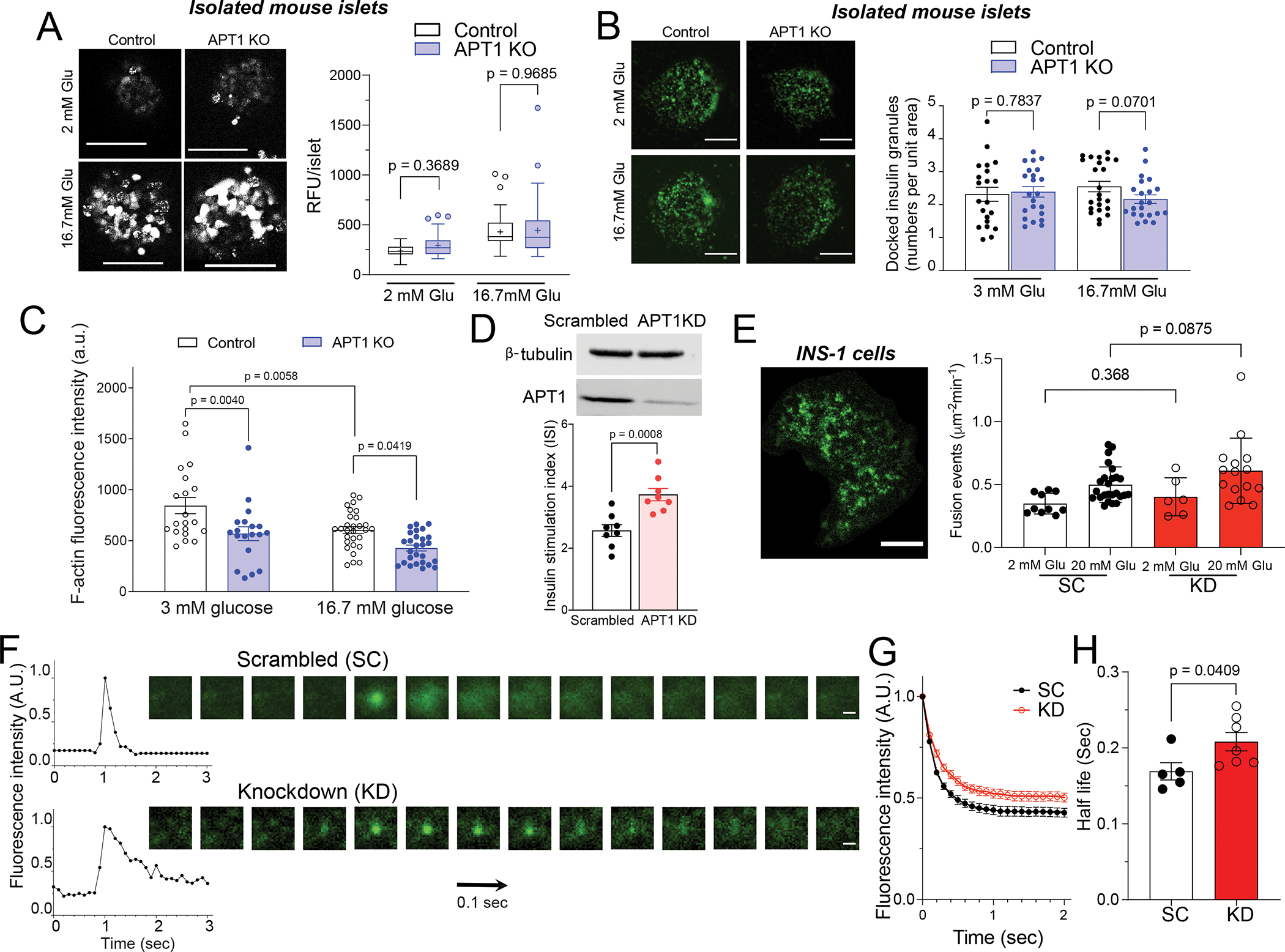
(A) Fluo-4 live cell imaging of isolated islets in the presence of 2 mM and 16.7 mM glucose. Scale=100 μm. (B) Docked insulin granule analysis by TIRF imaging in the presence of 3 mM and 16.7 mM glucose. Scale=5 μm. (C) Dispersed islets from global APT1 knockout and control mice were isolated and F-actin was imaged in cells incubated in 3 mM or 16.7 mM glucose. P values represent comparisons by two-way ANOVA. (D) Western blotting to validate the knockdown of APT1 in INS-1 832/13 cells (top panel) and increased glucose-stimulated insulin secretion in APT1 knockdown cells (bottom panel). (E) VAMP2-pHluorin imaging of insulin granules in an INS-1 cell by TIRF (left panel, Scale=5 μm) and fusion events in scrambled (SC) and APT1 knockdown (KD) INS-1 cells (right panel). (F) Intensity of isolated granule fusion events (left) and video frames of representative fusion events imaged by TIRF (right). Scale=0.5 μm. (G) Decay curve of peak intensity during fusion events for scrambled (SC) and APT1 knockdown (KD) cells. (H) Half-life of granule fusion for scrambled (SC) and APT1 knockdown (KD) cells.
Normal biphasic insulin release requires glucose dependent localized F-actin remodeling,20–22 and we confirmed this finding by demonstrating decreased F-actin intensity after glucose treatment in islets isolated from control mice (Figure 3C). F-actin intensity was significantly lower in APT1 KO islets compared to control islets in the setting of either 3 mM or 16.7 mM glucose (Figure 3C). Since we found no difference in docked insulin granules with APT1 deficiency, these findings suggest that enhanced glucose-dependent insulin secretion in APT1 KO islets could be mediated by F-actin-dependent events involved in the process of granule fusion.
To assess granule fusion, we studied insulinoma INS-1 832/13 cells transfected with VAMP2-pHluorin, a pH sensitive secretory granule marker that demonstrates increased fluorescence (unquenching) when exposed to extracellular pH. APT1 knockdown in INS-1 cells, validated by Western blotting (Figure 3D top panel), increased glucose-stimulated insulin secretion (Figure 3D bottom panel). INS-1 cells were transfected with VAMP2-pHluorin and imaged by TIRF microscopy to show multiple membrane fusion events (Figure 3E left panel) that were quantified. There was no significant difference in the number of vesicle fusion events in controls and knockouts (Figure 3E right panel). The kinetics of individual fusion events were characterized in control (scrambled) and APT1 knockdown cells (representative events shown in Figure 3F). The decay of fluorescence intensity was delayed in the APT1 deficient cells (Figure 3G), indicating an increased half-life for granule release with APT1 deficiency (KD vs SC in Figure 3G,H). These findings are consistent with altered granule trafficking as a potential mediator of insulin hypersecretion with APT1 deficiency.
Scamp1 is an APT1 Substrate that Partially Localizes to Insulin Secretory Granules
We used palmitoylation proteomics to identify potential APT1 substrates in islets. Islets isolated from 3 global APT1 KO and 3 control mice were subjected to resin-assisted capture (RAC) to select for palmitoylated proteins followed by labeling with tandem mass tags and mass spectrometry multiplexing according to the strategy shown in Figure S3A. Tubulin α1c and Ckap4 are known palmitoylated proteins, and increased palmitoylation was confirmed by RAC assays in APT1 knockdown INS-1 cells (Figure S3C). A heatmap of 390 palmitoylated proteins detected from control and APT1 KO islets is presented in Figure S3B, and the volcano plot for this analysis is shown in Figure 4A. Under high stringency (threshold 1.5 fold change, ≥2 peptides, moderated p value ≤0.005), only three proteins were identified as having increased palmitoylation in APT1 KO as compared to control mice (Table S2B; see Table S3 for all proteins): Rpl17, Rpl27a, and Scamp1 (secretory carrier membrane protein 1), a protein implicated in exocytosis.23,24 Scamp1 palmitoylation was increased in APT1 knockdown cells compared to control cells as determined by RAC assays (Figure 4B), and Scamp1 palmitoylation was verified by click chemistry using Myc-tagged Scamp1 and palmitic acid alkyne (Figure 4C).
Figure 4. Scamp1 is a palmitoylated protein that partially localizes to insulin secretory granules.
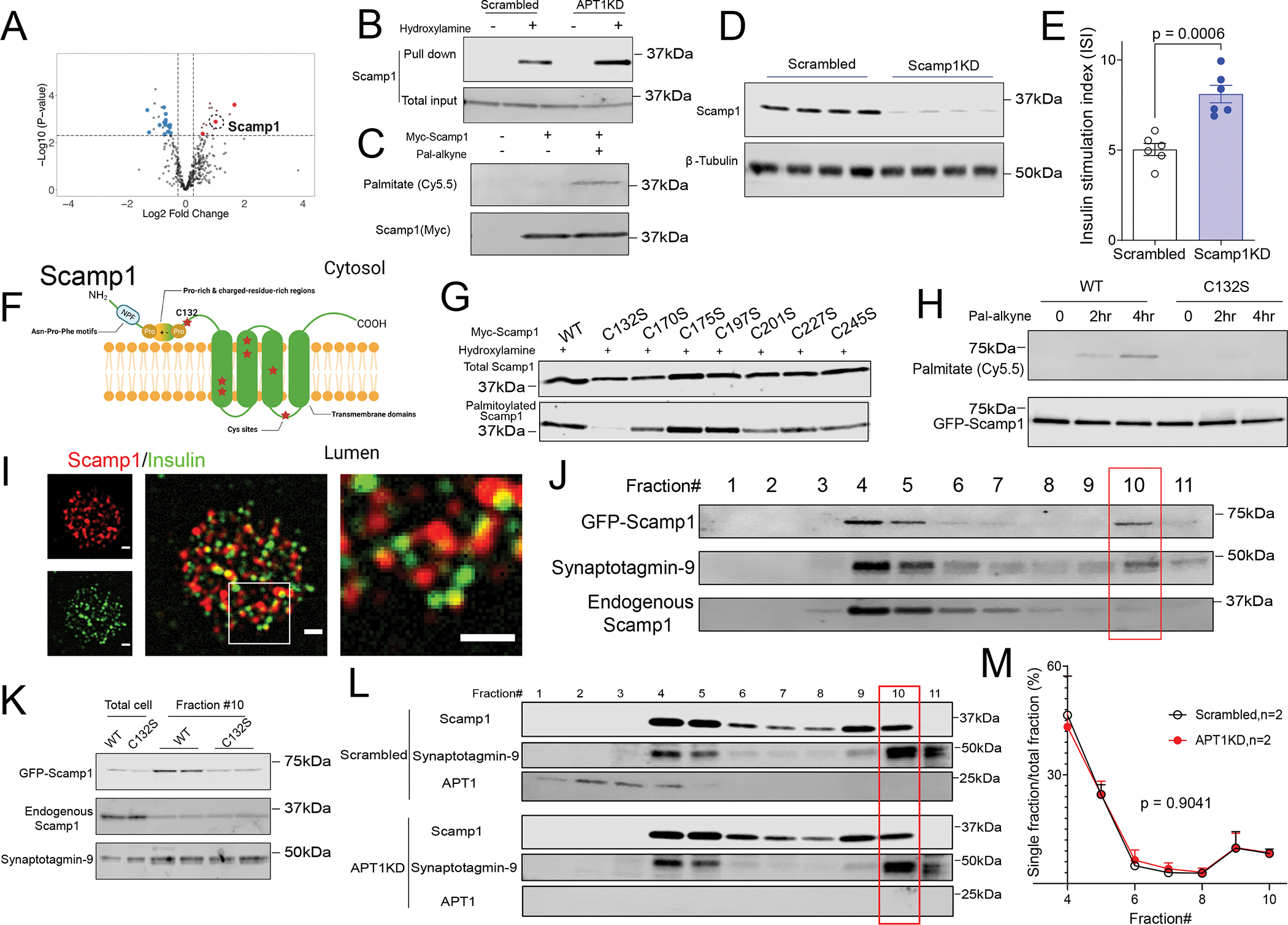
(A) Volcano plot of palmitoylation screen to identify proteins with increased palmitoylation in APT1 deficient islets. Large symbols indicate ≥ 2 peptides; small symbols < 2 peptides. (B,C) Validation of Scamp1 as a palmitoylated protein and substrate for APT1 by RAC assay (B) and click chemistry (C). (D) Western blotting to validate the knockdown of Scamp1 in INS-1 cells. (E) Increased glucose-stimulated insulin secretion in Scamp1 knockdown cells. (F) Diagram of the transmembrane vesicle protein Scamp1 with cysteine residues denoted by red stars. (G) RAC assay of wild type and mutant Scamp1 proteins with individual cysteine to serine mutations at each of the cysteine residues in Scamp1. (H) Click chemistry assay for palmitoylation in wild type and C132S Scamp1. (I) Co-localization of Scamp1 (red) and insulin (green) granules in dispersed mouse islets by TIRF. The box in the merged image of the middle panel is shown at higher magnification in the right panel. Scale=1 μm. (J) Scamp1 localization in dense core secretory granules of INS-1 cells. (K) Decreased accumulation of the palmitoylation defective C132S Scamp1 mutant in dense core granules (fraction #10). (L) Western blotting of Scamp1 in various fractions of control/Scrambled and APTKD INS-1 cells. (M) Quantification of Scamp1 fractions including dense core secretory granules (fraction #10) in INS-1 cells from two independent experiments.
When Scamp1 was knocked down in INS-1 832/13 cells (Figure 4D), glucose-dependent insulin secretion was increased (Figure 4E), the same response seen in APT1 knockdown human islets (Figure 1H), APT1 knockdown INS-1 cells (Figure 3D) and in isolated islets from two models of mice with APT1 deficiency (Figures 2F, 2J). These observations suggest that Scamp1 palmitoylation is involved in insulin secretion. To pursue this possibility, we identified the Scamp1 palmitoylation site. Conventional reversible palmitoylation involves thiols in cysteine residues. Scamp1, belonging to a family of vesicular trafficking proteins characterized by 4 central transmembrane regions,25 has 7 cysteines represented by red stars in Figure 4F. Each cysteine was mutated to serine and all were assayed for palmitoylation in assays specific for S-palmitoylation (Figure 4G). The mutation of Cys132, the only cysteine in one of the cytosolic domains of Scamp1 (and thus expected to be accessible to APT1), abolished palmitoylation (Figure 4G). The C132S mutant had decreased modification by palmitic acid alkyne in click chemistry assays as compared to wild type Scamp1 (Figure 4H).
Scamp1 (red) and insulin (green) were detected in fixed, dispersed islets from control mice (Figure 4I, left panels), and merged images revealed partial co-localization of Scamp 1 with insulin (Figure 4I, middle panel with box expanded at higher magnification image in right panel). GFP tagged (green) Scamp1 and mCherry tagged (red) NPY (an insulin granule marker) were detected in INS-1 cells (Figure S4, left panels), and merged images also revealed partial co-localization of Scamp1 with NPY (Figure S4, middle panel with box expanded at higher magnification in right panel). Iodixanol density gradient purification of insulin granules from INS-1 cells26 showed that GFP tagged wild type Scamp1 and Synaptotagmin-9, one of the calcium binding proteins responsible for glucose-stimulated insulin secretion,27 were found in insulin secretory granules (Figure 4J, fraction #10). However, the C132S Scamp1 mutant was not enriched in insulin secretory granules (Figure 4K). This finding suggests that depalmitoylated Scamp1 trafficking is distinct from wild type Scamp1 trafficking, implicating differentially palmitoylated forms of Scamp1 in insulin secretion. To assess native Scamp1 content in secretory granules in APT1 deficiency, we compared density gradients in control and APT1KD cells (Figure 4L). Scamp1 did not differ in fractions (including dense core secretory granules, fraction #10) from APT1 replete and deficient cells (Figure 4M).
Islet Specific APT1 Deficiency Unexpectedly Predisposes Mice to β-cell Failure
Chow fed islet specific APT1 knockout mice (APT1 islet KO) had normal β-cell area measurements (Figure 2H,I) and enhanced glucose tolerance due to increased insulin secretion (Figure S1E–H), raising the possibility that these animals would be protected from experimental diabetes. Unexpectedly, we found the opposite phenotype after feeding a high fat diet.
After 12 weeks of high fat diet, there was no difference in body weight between control and APT1 islet KO mice (Figure 5A). Surprisingly, APT1 islet KO mice had impaired glucose tolerance compared to controls (Figure 5B) in the absence of a genotype specific effect on insulin tolerance (Figure 5C). Impaired glucose tolerance in the knockouts was associated with decreased insulin:glucose ratio (Figure 5D) and decreased C-peptide (Figure 5E) at 15 min after administration of glucose. β-cell area per islet was decreased in the knockouts compared to controls by histomorphometry (Figure 5F,G) and by analyses of β-cell area expressed as percentage of pancreatic area (Figure 5H) consistent with more pronounced β-cell failure in the knockouts.
Figure 5. Islet specific APT1 deficiency promotes β-cell failure induced by feeding a high fat diet.
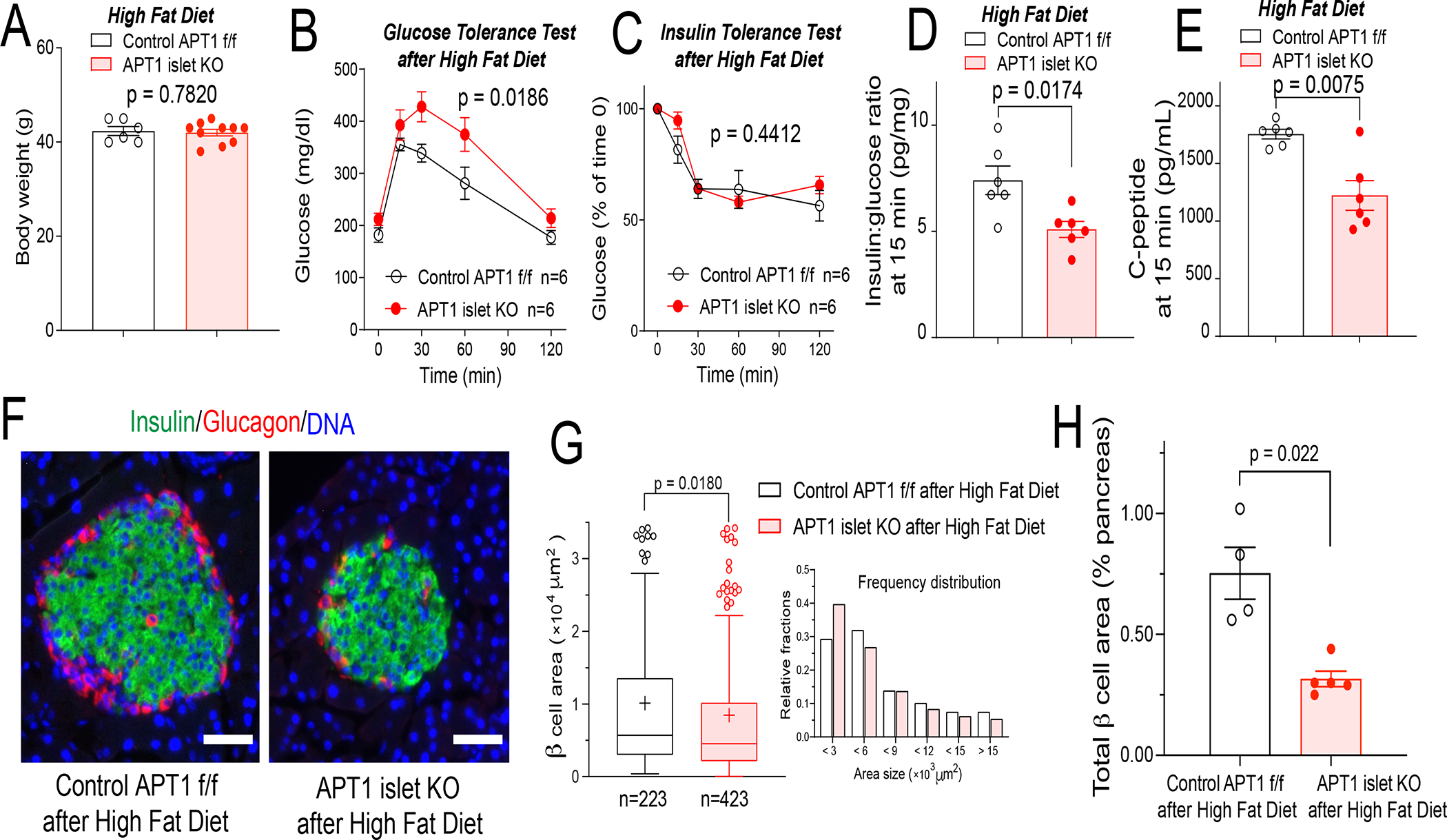
Male APT1 islet KO mice with improved glucose tolerance due to increased insulin secretion (Figure S1 E–H) were placed on a high fat diet for 12 weeks. (A) Body weight of APT1 islet KO and control mice after 12 weeks on high fat diet. (B) Impaired glucose tolerance in APT1 islet KO mice after high fat diet. (C) No effect on insulin sensitivity by insulin tolerance testing in APT1 islet KO mice compared to control mice after high fat diet. (D,E) Decreased insulin secretion in APT1 islet KO mice as shown by decreased insulin:glucose ratio (D) and decreased C-peptide (E) 15 min after glucose administration. (F) Islet images after high fat diet. Insulin (green), glucagon (red), DAPI (blue). Scale=50 μm. (G) Histomorphometry of islets after high fat diet. (H) Analysis of β-cell area as % of pancreatic area in high fat fed APT1 islet KO mice.
Since the APT1 islet KO mice may target a small portion of α-cells19 in addition to β-cells, it is possible that these mice could exhibit aberrant glucagon secretion. There was no significant difference in fasting glucagon levels between control and APT1 islet KO male mice on a chow (Figure S5A) or high fat diet (Figure S5B). We also addressed the same concern in our global KO model. There was no significant difference in glucagon levels between APT1 global KO mice and controls of both sexes (Figure S5C). For the high fat diet APT1 islet KO mice with more pronounced β-cell failure in Figure 5, there was no significant difference in β-cell proliferation or apoptosis (Figure S5D,E).
Palmitoylation Defective Scamp1 Rescues Insulin Hypersecretion and Nutrient Induced Apoptosis in APT1 Deficient Cells
Co-localization of the APT1 substrate Scamp1 with insulin in secretory granules is impaired with the use of a mutant, C132S Scamp1, incapable of normal palmitoylation (Figure 4I,J,K). When wild type Scamp1 and C132S Scamp1 were expressed at levels ~10-fold greater than endogenous Scamp1 (Figure S6A) in native INS-1 cells (with normal APT1 expression), both forms of Scamp1 suppressed glucose-stimulated insulin secretion (Figure S6B) with C132S having the greater effect. One interpretation of these results is that the depalmitoylated form of Scamp1 restrains insulin secretion. A fraction of exogenously expressed wild type Scamp1 would be depalmitoylated due to the presence of APT1 activity, and at high levels of expression, this depalmitoylated form would be sufficiently abundant to affect insulin secretion. C132S Scamp1 would be predicted to be exclusively depalmitoylated, accounting for a greater effect on insulin secretion. Wild type and C132S Scamp1 expression levels were titrated in multiple experiments to achieve levels equal to or less than levels of endogenous Scamp1 (Figure 6A) in INS-1 cells with APT1 activity (Scrambled) or without APT1 activity (APT1KD). Expression of wild type Scamp1 did not prevent hypersecretion of insulin induced by APT1 deficiency, but C132S Scamp1 rescued hypersecretion in APT1 deficient cells (Figure 6B), suggesting that modification of Scamp1 palmitoylation status by APT1 activity is physiologically relevant to glucose dependent insulin secretion.
Figure 6. Palmitoylation defective Scamp1 (C132S) rescues increased insulin secretion and nutrient induced apoptosis in APT1 knockdown INS-1 cells.
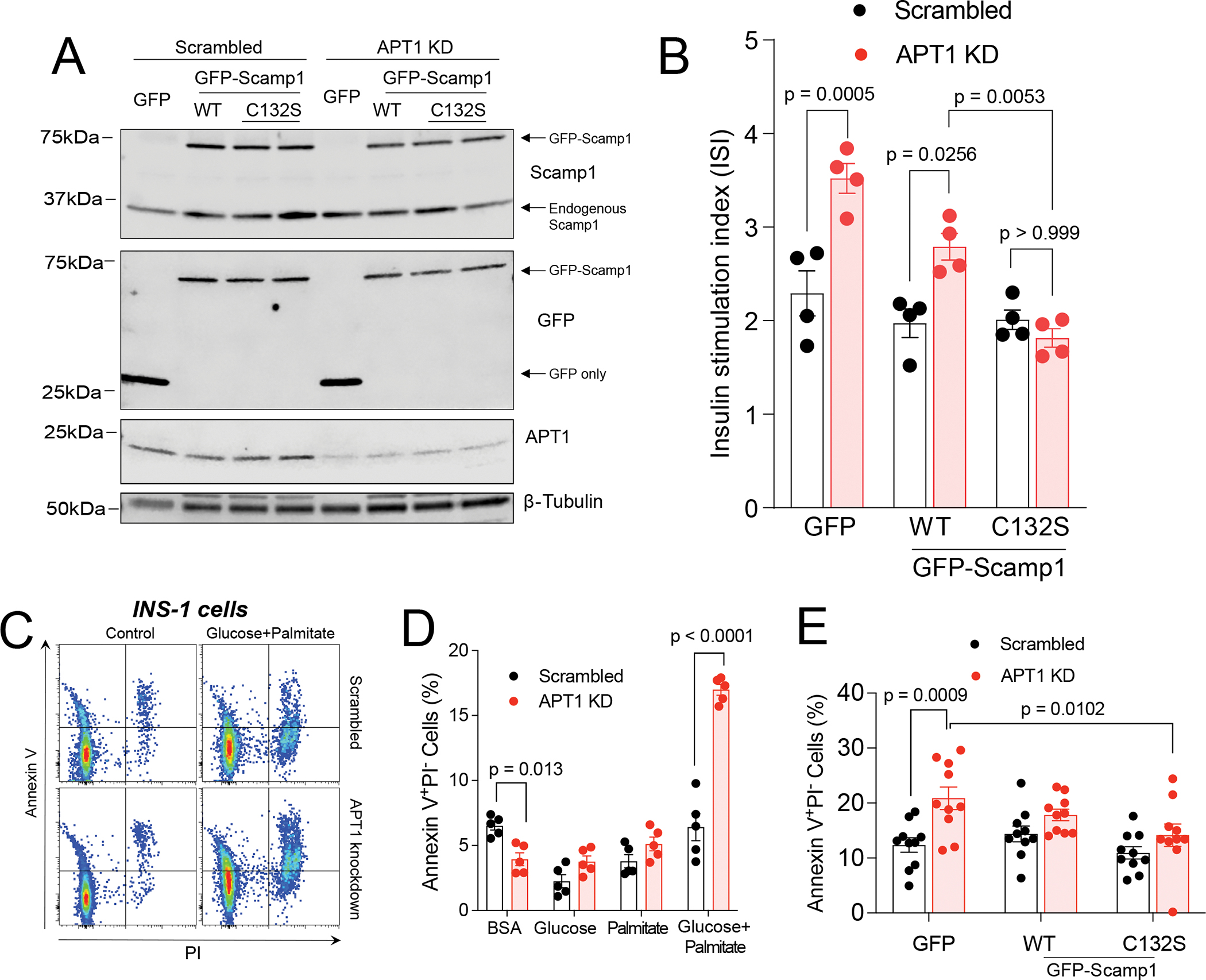
(A) Expression of wild type and C132S mutant Scamp1 in control (scrambled) and APT1 knockdown INS-1 cells at levels comparable to endogenous levels of Scamp1. (B) Glucose-stimulated insulin secretion in control and APT1 knockdown cells with expression of GFP only, GFP-wild type Scamp1, and GFP-C132S Scamp1. (C,D) APT1-deficient INS-1 cells have increased susceptibility to apoptosis. FACS analysis for Annexin V and PI in the presence and absence of high glucose (25 mM) + palmitate (0.5 mM) complexed with BSA (C) and quantitation of Annexin V data after exposure to different media for 24 h (D). (E) INS-1 cells treated with Scrambled or APT1 knockdown shRNA were transduced with GFP alone, WT Scamp1 or C132S Scamp1 for 48 h, then treated with high glucose (25 mM) + palmitate (0.5 mM) complexed with BSA for 24 h followed by FACS assay for apoptosis.
Nutrient excess may promote apoptosis to induce β-cell failure in APT1 deficient mice with insulin hypersecretion, but β-cell apoptosis can be difficult to detect in the pancreas.28 Like APT1 deficient mice, APT1 knockdown INS-1 cells have increased glucose induced insulin secretion (Figure 3D). In cells treated with BSA only, there were significantly fewer annexin V positive cells in the setting of APT1 knockdown as compared to cells with normal APT1 activity (Figure 6C,D), suggesting that APT1 knockdown alone does not induce cell death. The combination of 25 mM glucose (“Glucose”) and 0.5 mM palmitate (“Palmitate”) for 24 h significantly increased apoptosis in APT1 knockdown as compared to APT1 replete cells (Figure 6C,D), suggesting that APT1 knockdown predisposes to the loss of insulin secreting cells in the setting of glucolipotoxicity. C132S Scamp1 rescued apoptosis in APT1 knockdown cells (Figure 6E), suggesting that modification of Scamp1 palmitoylation status by APT1 activity is physiologically relevant to nutrient induced apoptosis. Treatment with 25 mM glucose and 0.5 mM palmitate for 24 h increased abundance of two ER stress markers, thioredoxin-interacting protein (Txnip) and the active form of X-box binding protein 1 (XBP-1s), but the effects were not different between control/Scrambled and APT1 knockdown INS-1 cells (Figure S6C).
APT1 Deficiency in db/db Mice Causes Insulin Hypersecretion Followed by β-cell Failure
To determine if the β-cell failure phenotype could be confirmed in another model of type 2 diabetes, db/db mice (BKS.Cg-Dock7m++ Leprdb/J) with (APT1 KO db/db) or without (control db/db) global APT1 deficiency were characterized over time.
At 1 month of age, APT1 KO animals as compared to controls had enhanced glucose tolerance (Figure 7A), and increased secretion of both insulin (Figure 7B) and C-peptide (Figure 7C) during glucose tolerance tests. By 2 months of age, fasting glucose levels were higher in both genotypes consistent with the progression of diabetes in the db/db model, enhanced glucose tolerance in the APT1 KO mice was lost (Figure 7D), and there was no difference in insulin secretion or C-peptide (Figure 7E,F) between genotypes. By 4 months of age, fasting glucose levels exceeded 500 mg/dl in both groups. APT1 KO animals at this age had impaired glucose tolerance compared to controls (Figure 7G), and decreased secretion of both insulin (Figure 7H) and C-peptide (Figure 7I) during glucose tolerance tests. Representative images of islets after development of pronounced glucose intolerance at 4–5 months of age are shown in Figure 7J. Analyses of β-cell area expressed as percentage of pancreatic area was not different between genotypes at 6–7 weeks of age (Figure 7K), and was numerically but not significantly decreased at 4–5 months of age (Figure 7L). β-cell area per islet by histomorphometry was significantly decreased in the knockouts at 4 months of age (Figure 7M) compared to controls, consistent with more pronounced β-cell failure. Between APT1 KO and controls in this db/db model, there were no significant differences in β-cell proliferation at 6–7 weeks of age or 4–5 months of age (Figure S7A,B). There were also no significant differences in apoptosis at 6–7 weeks of age or 4–5 months of age (Figure S7C,D).
Figure 7. APT1 deficiency promotes β-cell failure over time in db/db mice.
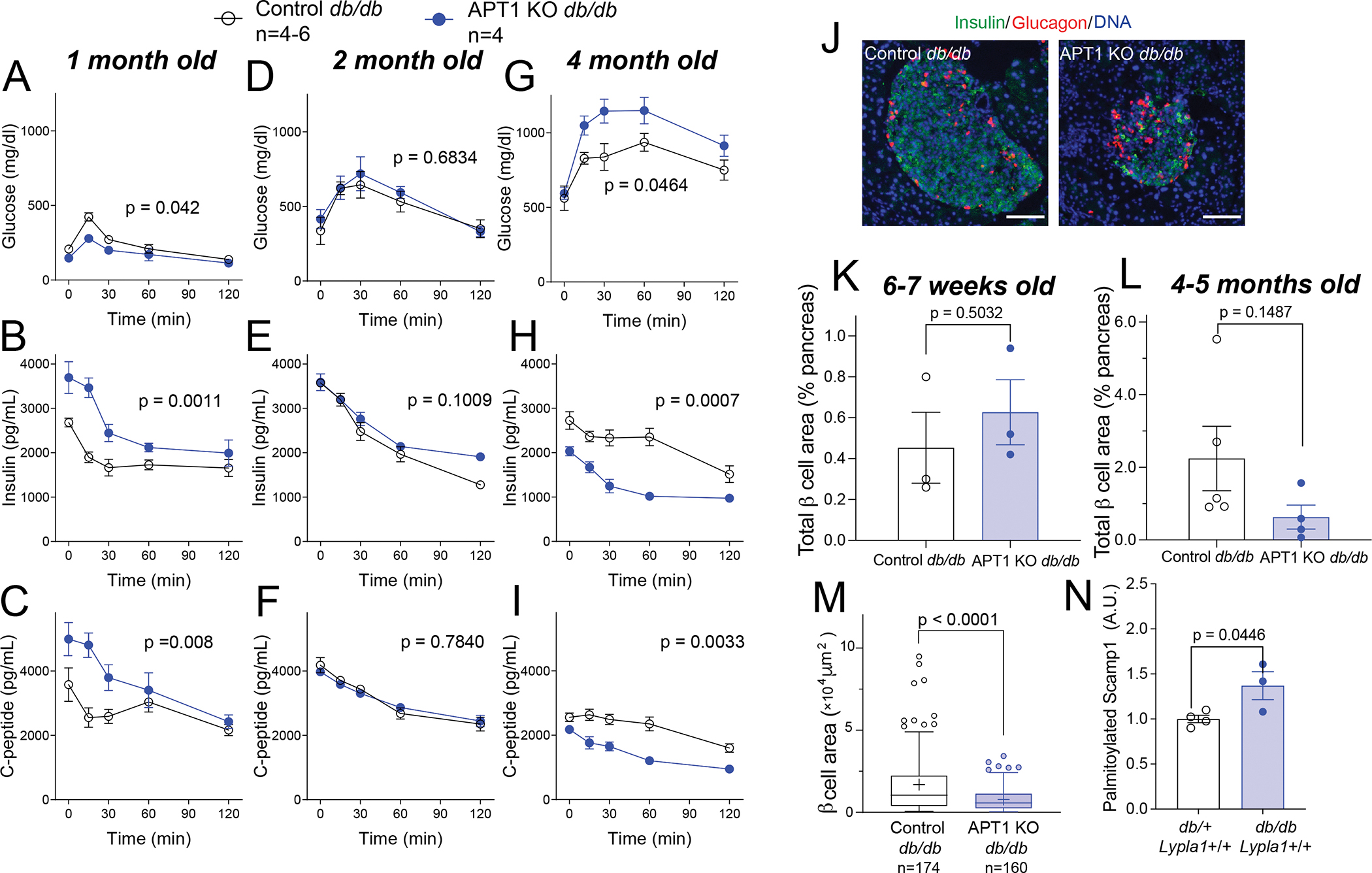
(A,B,C) Male and female db/db mice with APT1 deficiency at 1 month of age have enhanced glucose tolerance (A), and increased secretion of insulin (B) and C-peptide (C). (D,E,F) No difference in glucose tolerance, insulin secretion or C-peptide secretion by genotype at 2 months of age. (G,H,I) db/db mice with APT1 deficiency at 4 months of age have impaired glucose tolerance (G), and decreased secretion of insulin (H) and C-peptide (I). (J) Images of islets after development of more pronounced glucose intolerance in the APT1 deficient animals. (K) Analysis of β-cell area as % of pancreatic area in APT1 KO db/db and control db/db mice at 6–7 weeks of age and at (L) 4–5 months of age. (M) Islet histomorphometry of APT1 KO and control db/db mice at 4 months of age. (N) Palmitoylated Scamp1 in isolated islets of db/db mice without APT1 deficiency and control db/+ mice without APT1 deficiency as determined by RAC assay. Scale=100 μm.
In mice without defects in Lypla1 (the gene encoding APT1), Scamp1 palmitoylation determined by RAC assay was increased in islets from db/db as compared to islets from control db/+ mice (Figure 7N), suggesting that β-cell failure in db/db mice may be related to impaired removal of palmitate from Scamp1.
DISCUSSION
Diabetes occurs when insulin production by β-cells is insufficient to control blood glucose. Human genome-wide association studies have identified hundreds of type 2 diabetes signals,29 and those associated with coding variants are usually implicated in pancreatic islet function. However, individual signals account for a small percentage of disease risk, and gene-gene interactions do not increase this small effect.30 Most type 2 diabetes risk is related to obesity,31 consistent with the notion that nutrient excess associated with obesity interacts with many genes to cause diabetes. How nutrient excess enhances β-cell death or dedifferentiation is unknown.9,10 APT1 enzyme activity, a mediator of the posttranslational process of palmitoylation dynamics, is suppressed by hyperglycemia,14 and here we show that disruption of APT1 causes insulin hypersecretion that can progress to insulin deficiency.
APT1 deficiency in chow fed mice increased insulin secretion in the absence of an effect on insulin sensitivity. Isolated islets showed normal basal insulin secretion but increased glucose-stimulated biphasic insulin secretion. APT1 deficiency did not affect insulin content, insulin granule docking, or membrane excitability, but altered insulin granule trafficking to prolong fusion events. Palmitoylation proteomics identified increased palmitoylation of the transmembrane vesicle protein Scamp1 in APT1 deficient islets, Scamp1 deficiency caused increased insulin secretion, Scamp1 was associated with insulin secretory granules, and expression of a palmitoylation-deficient mutant Scamp1 rescued insulin hypersecretion as well as nutrient induced apoptosis in APT1 deficiency. APT1 deficient mice subjected to the nutrient excess of a high fat diet developed glucose intolerance, insulin deficiency, and β-cell loss in the absence of an effect on insulin sensitivity. APT1 deficient mice subjected to nutrient excess modeled by db/db mice showed enhanced glucose tolerance due to insulin hypersecretion that evolved to insulin deficiency and β-cell loss. Together, these findings suggest that defective depalmitoylation, which can result from nutrient excess, causes insulin hypersecretion leading to β-cell exhaustion.
Scamp1 appears to be involved in the insulin hypersecretion phenotype. Implicating Scamp1 in insulin granule biology is novel, and consistent with previous reports of the role of this protein in vesicle biology. Secretory carrier-associated membrane proteins (Scamps), present in all eukaryotic cells, exist as multiple co-localized isoforms implicated in post-Golgi trafficking. Scamp1 and 2 regulate exocytosis of secretory granules in mast cells and neuroendocrine (PC12) cells.23,25,32,33 In PC12 cells, Scamp1 specifically regulates the closure of fusion pores during late stages of exocytosis serving to limit the extent of compound exocytosis.24 These effects mirror current observations in INS-1 cells. Scamp1 deficiency promotes insulin secretion, and APT1 deficiency promotes insulin secretion in the setting of increased palmitoylation of Scamp1 that appears to be due to prolongation of granule fusion. Complete vesicle fusion is energetically favorable and likely responsible for signals in control INS-1 cells (SC in Figure 3E,F). Increased Scamp1 palmitoylation caused by APT1 deficiency may interfere with complete fusion, favoring prolonged vesicular kiss-and-run or cavicapture events allowing increased insulin release (KD in Figure 3F). Expression of a palmitoylation-deficient Scamp1 mutant decreases its association with secretory granules (Figure 4K) and corrects insulin hypersecretion (Figure 6B), suggesting that the depalmitoylated form of Scamp1 (generated by APT1) regulates insulin secretory granule fusion. Knockdown of Scamp1 increases insulin secretion (Figure 4E), likely caused by decreased availability of depalmitoylated Scamp1 required for normal fusion.
Little is known about the potential roles of APT1 and Scamp1 in human type 2 diabetes progression. A single-cell RNA-seq study reported that Scamp1-AS is an anticorrelated gene for exocytosis in human β-cells.34 Scamp1 was found to be upregulated in β-cells from patients with type 2 diabetes as compared with non-diabetic β-cells.35 We are unaware of APT1 and Scamp1 variants directly associated with diabetes in human GWAS studies, which might be expected since APT1 enzyme activity and Scamp1 palmitoylation are posttranscriptional processes potentially modulated by nutrient status.
Human islet data suggest that depalmitoylation may impact diabetes in people. APT1 mRNA was increased in islets from people with type 2 diabetes as compared to people without diabetes (Figure 1A) while APT1 enzyme activity was decreased in normal human islets incubated with high glucose (Figure 1C). The identical effect occurs in human endothelial cells, namely, increased APT1 message in the setting of decreased APT1 enzyme activity caused by hyperglycemia,14 suggesting that impaired depalmitoylation prompts a compensatory increase in APT1 mRNA. Hyperglycemia decreased APT1 enzyme activity in vitro using islets from three nondiabetic human donors, and endogenous hyperglycemia was associated with decreased APT1 enzyme activity in two donors with undiagnosed diabetes not confounded by the chronicity of disease or diabetes medications. Knockdown of APT1 in normal human islets increased glucose-stimulated insulin secretion (Figure 1H). These results raise the possibility that impaired function of APT1 could participate in insulin hypersecretion predisposing to diabetes in humans. Our detection of increased APT1 enzyme activity in islets from a patient with chronic β-cell failure is confounded by diabetes complications and multiple diabetes medications, but since decreased APT1 promotes β-cell dysfunction, it is plausible that increased APT1 in advanced disease might represent a compensatory process to improve insulin secretory homeostasis.
Hyperinsulinemia has been proposed to promote obesity and insulin resistance in the evolution of β-cell failure 36, but body weight and insulin sensitivity were unaffected in our mice with APT1 deficiency and insulin hypersecretion. Multiple clinical phenotypes may lead to a type 2 diabetes diagnosis,3,4 but obesity can be associated with increased insulin secretion in the absence of insulin resistance,37 and insulin hypersecretion independent of insulin resistance can predict type 2 diabetes.5,6
Nutrient excess is associated with β-cell dysfunction, but specific mechanisms underlying this process are scarce.9,10 APT1 represents a discrete target for modification by the metabolic milieu that could be mediating the nutrient toxicity presumed to be responsible for obesity-associated diabetes.
Limitations of Study
There are limitations to our work. In human islet studies, we used glucose at 25 mM, a concentration that is not physiologic but can be seen in poorly controlled diabetes. It is not clear if lesser degrees of hyperglycemia affect APT1 to regulate insulin secretion or contribute to diabetes pathophysiology. Lentiviral knockdown of APT1 in intact human islets increased insulin secretion, but we do not know how many β-cells were transduced or the extent of knockdown in β-cells. Since APT1 is expressed in other cells including endothelial cells, APT1 knockdown in cell types other than β-cells may contribute to our findings in human islets. We focused on Scamp1 as a putative mediator of the effects of APT1 deficiency in β-cells, but other palmitoylated proteins are also likely to be involved in diabetes progression. Two additional candidates identified in our palmitoylation proteomic screen, Rpl17 and Rpl27a, are proteins found in ribosomes, an organelle important for insulin hypersecretion. We confirmed statistically decreased β-cell area expressed as a percentage of pancreatic area in high fat fed APT1 islet KO mice, but the numerical decrease in β-cell area expressed as a percentage of pancreatic area in APT1 KO db/db mice did not reach statistical significance in part due to small sample sizes. We did not detect β-cell apoptosis or altered proliferation in our animal models of β-cell failure, perhaps because our imaging was performed at time points that missed relevant pathologic events such as cell death. We demonstrated physiologically relevant findings in human islets from people with and without diabetes, but pursued mechanisms using a rat insulinoma cell line, INS-1 832/13. Finally, APT1 deficiency promotes both insulin hypersecretion and nutrient induced apoptosis, but the mechanistic link between these processes is unknown. Additional work to address these issues might include study of human cell lines, tissue specific modulation of Scamp1, and detailed characterization of the cell biology of insulin hypersecretion-induced β-cell failure over time.
STAR ★ METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Clay F Semenkovich (csemenko@wustl.edu).
Materials availability
Plasmids generated in this study are available from the lead contact upon request.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. Source data and Western blot images for the figures in the manuscript are available as Data S1: Sources, Related to Figures 1–7 and S1–S7.
This paper does not report original code.
Any additional information required to analyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human pancreatic islets
Human pancreatic islets for research from deidentified organ donors were obtained from Prodo Laboratories Inc. following the instructions of the Integrated Islet Distribution Program (IIDP), and studied with the approval of the Washington University Human Research Protection Office (HRPO). Table S1 A–C summarizes donor information.
Mouse models
Animal protocols were approved by the Washington University Institutional Animal Care and Use Committee (IACUC) and complied with the NIH Guide for the Care and Use of Laboratory Animals. APT1 global deficient mice (APT1 global KO) in the C57BL/6J background and floxed Lypla1 mice were generated as described.14 Islet-specific APT1 knockout mice (APT1 islet-KO) were produced by crossing floxed animals with tamoxifen-inducible Pdx1PBCreER™ mice.19 Adult mice were administered tamoxifen (75 mg/kg i.p.) for five consecutive days. APT1 global KO mice were crossed with db/+ (BKS.Cg-Dock7m++ Leprdb/J) animals followed by intercrossing the progeny to yield db/db mice with and without APT1 deficiency. APT2 global knockout mice (APT2KO) were generated by the trans-NIH Knock-Out Mouse Project (KOMP) and obtained from the KOMP Repository (Project ID: CSD34805). Mice were housed in a specific pathogen-free facility with a 12 h light-dark cycle and fed with commercial chow or high-fat diet with 42% of calories as fat (Envigo Cat. No. TD 88137). Both sexes were metabolically phenotyped, and detailed studies of islet physiology were further pursued in both genders for global APT1KO mice, or males only for the islet APT1 KO model due to the stronger phenotype in males as compared to females with APT1 deficiency.
Cell lines
In this study, viruses were generated in 293T cells that were purchased from ATCC (Cat No. CRL-3216). INS-1 832/13 cells38 used for mechanistic studies were obtained from the Metabolic Tissue Function Core of the Washington University Diabetes Research Center (DRC).
METHOD DETAILS
APT1 enzyme activity assay
For determination of the effects of hyperglycemia on APT1 enzyme activity, ~6000 islet equivalents (IEQ) were separated into two equal aliquots and cultured in CMRL 1066 complete media (10% FBS, 1% Pen/Strep, 1% Sodium pyruvate, 0.1 mM MEM-non-essential amino acids in CMRL 1066 basal media) as islet culture media containing 5.5 mM or 25 mM glucose for 48 hours. Islet cells were then dispersed with Accutase (Sigma, Cat. No. A6964), stained for Hpi2 (clone HIC1-2B4.2B, Novus, Cat. No. NBP1-18946PECY7), CD26 (clone BA5b, Biolegend, Cat. No. 302705) and NTPDase 3 (J. Sevigny, Cat. No. hN3-B3s),39 followed by incubation with DAPI and the depalmitoylation probe DPP318 during flow cytometry acquisition. The emission fluorescence was acquired by digital FACScan, and data analyzed in FlowJo_V10.8.
Mouse islet isolation
Mouse islet isolations were performed essentially by the procedure described by Carter et al.40 Briefly, in anesthetized mice, the common bile duct was clamped off near the junction with the small intestine and cannulated. The pancreas was perfused with 4.5 mL of 1.4 mg/mL collagenase P (Roche, Cat. No. 11213857001) solution (10 mM HEPES, 1 mM MgCl2 and 5 mM glucose in Hank’s Balanced Salt Solution without CaCl2, MgCl2, NaHCO3 and phenol) and then removed from the mouse and kept with 1 mL of collagenase P solution on ice. Pancreatic digestion was performed in 10 mL glass tubes at 37°C for 10 min, and facilitated with hand-shaking for approximately 15 s to complete the separation of tissue. The reaction was stopped by dilution with 8 mL wash buffer (1 mM CaCl2.2H2O, 10 mM HEPES, 1 mM MgCl2 and 5 mM glucose in Hank’s Balanced Salt Solution without CaCl2, MgCl2, NaHCO3 and phenol) on ice, and the digested pancreas was transferred into a 50 mL Falcon tube for washing three times with wash buffer. After the final wash, the pancreas suspension was filtered through a 70 μm cell strainer, and the filtrate maintained in complete RPMI (Gibco, Cat. No. 11875-085) media (10% FBS, 2% Pen/Strep and 2 mM GlutaMax in RPMI 1640 basal media). Individual islets were then picked up manually under the stereomicroscope, and cultured in RPMI 1640 complete media overnight for further experiments.
Quantitative RT-PCR analysis
Total RNA from mouse and human islets was extracted using an RNeasy micro kit (Qiagen, Cat. No. 74004) and reverse transcribed using an iScript cDNA synthesis kit (Bio-Rad, Cat. No. 1708890). PCR reactions were performed with an Applied Biosystems StepOnePlus Real-Time PCR System (Thermofisher) using the SYBR Green PCR Master Mix assay (SYBR® Premix Ex Taq™, Takara, Cat. No. RR420A).
Gene knockdown in INS-1 cells or human islets
Two approaches were used for knockdown experiments, including siRNA (for APT1 or Scamp1 in INS-1 cells) and lentiviral-transduced shRNA (for APT1 in INS-1 cells or human islets). For siRNA experiments in INS-1 cells, cells were seeded at a density of 5 × 105 cells/cm2 in a 12 well plate or 2 × 106 cells/cm2 in a 6 well plate for overnight culture in RPMI 1640 complete media. DharmaFECT 1 Transfection Reagent (Horizon Discovery, Cat. No. T-2001-02) was used to transfect the non-targeting siRNA control (NTC, Horizon Discovery, Cat. No. D-001810-10-20) or the ON-TARGETplus SMARTpool siRNA (Horizon Discovery, Cat. No. L-089651-02-0005 for APT1, Cat. No. L-086797-02-0010 for Scamp1). Cells were then studied 72 h later. For shRNA experiments, lentiviral vectors were obtained from commercial sources (pGFP-C-shLenti system for rat INS-1 cells from Origene, Cat. No. TL709612, or MISSION shRNA system for human islets from Sigma), and virus was generated in 293T cells by transfection of scrambled control or target shRNA expression vectors with packaging helper plasmids psPAX2 and pMD2.G. Lentiviruses were then concentrated from culture supernatants by Lenti-X™ concentrator (Takara, Cat. No. 631231) and titers were determined by Lenti-X™ qRT-PCR titration kit (Takara, Cat. No. 631235). For APT1 knockdown in INS-1 cells, cells were seeded at a density of 5 × 105 cells/cm2 in a 12 well plate or 2 × 106 cells/cm2 in a 6 well plate for overnight culture in RPMI 1640 complete media. Cells were transduced with Scrambled shRNA or shRNA lentivirus encoding for Lypla1 with 4 μg/mL polybrene (Hexadimethrine bromide, Sigma, Cat. No. H9268-10G) for 24 h. Cells were harvested 4–5 days post transduction for further experiments. For APT1 knockdown in human islets, human islets were cultured in CMRL 1066 complete media for 24 h upon arrival. Size-matched human islets (30 islets/replicate) were infected with either LYPLA1 expression shRNA lentivirus or scrambled shRNA at an Infectious Units (IFU) per cell of 50 or 100 with 4 μg/mL polybrene in a 35 mm Petri dish for 24 h. Western blotting was performed four days later for APT1 knockdown confirmation.
Human islet static GSIS assay
Multiple aliquots of human islets after treatment under scrambled or APT1 knockdown conditions were subjected to static glucose-stimulated insulin secretion (GSIS) assay. For static GSIS measurement, human islets were precultured with 1 mL Krebs-Ringer bicarbonate HEPES buffer (KRBH) buffer (128 mM NaCl, 5 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4. 7H2O, 5 mM NaHCO3, 2.5 mM CaCl2.2H2O and 10 mM HEPES) supplemented with 5.5 mM glucose and 0.1% BSA for 1 h for equilibration. Then the islets were incubated with KRBH buffer for 1 h at 5.5 mM glucose and an additional 1 h at 25 mM glucose. At the end of each incubation, the media were collected and were assayed for human insulin content (Alpco, Cat. No. 80-INSHU-E01.1).
Mouse metabolic characterization
Glucose tolerance (GTT) and insulin tolerance tests (ITT) were performed in mice after a 6 h fast as described.41 Glucose was injected at 1 g/kg i.p. and insulin at 0.75 U/kg i.p. C-peptide and insulin were measured by ELISA using reagents from Crystal Chem (Cat. No. 90050 or 90080). Fasting chemistries were measured using reagents from FUJIFILM Wako Chemicals (glucose, Cat. No. 99703001; NEFA, Cat. No. 99934691), or Thermofisher (triglycerides, Cat. No. TR22421; cholesterol, Cat. No. TR13421). Basal glucagon levels were measured in mice after an overnight fast using kit from Mercodia Inc (Cat. No. 10-1271-01) by the Translational Diagnostics Core of the Washington University Diabetes Research Center.
Histological analysis
Mouse pancreas was fixed with 4% paraformaldehyde for 24 h, followed by cryopreservation with gradient sucrose solutions (15% and then 30%) overnight prior to OCT embedding. Frozen sections (cut at 10 μm thickness by Leica CM1850 Cryostat) were permeabilized with 0.1% Triton X-100 and blocked in 5% goat serum in PBS, followed by incubation with primary antibody overnight at 4°C, and subsequently secondary antibody at room temperature for 1 h. Nuclei were counterstained with DAPI. Images of insulin staining for whole pancreas were captured by automated fluorescence slide scaner (Zeiss AxioScan 7), and other images were obtained using an epifluorescence microscope (Leica DMI4000B). For histological evaluation of islets, immunofluorescence imaging was performed for insulin (recombinant anti-insulin antibody from Abcam, Cat. No. ab181547; clone ICBTACLS from Thermofisher, Cat. No. 53-9769-82; or guinea pig polyclonal antibody from Abcam, Cat. No. ab7842) and glucagon (polyclonal, Abcam, Cat. No. ab133195). For measuring pancreatic β-cell proliferation, frozen sections were stained with anti-Ki67 antibody (Clone SolA15 from Thermofisher, Cat. No.14-5698-80). Apoptotic nuclei were stained by TUNEL assay with an in Situ cell death detection kit (Sigma-Aldrich, Cat. No.11684795910) as described in the manual. Images were analyzed with Fiji-ImageJ software. Morphometry of the beta cell area fraction was analyzed in 10 sections that were ~150 μm apart for each mouse.
Mouse islet static GSIS and dynamic GSIS
For evaluation of insulin secretion, overnight cultured isolated mouse islets were stimulated for insulin secretion under static or dynamic conditions. Under static conditions, islets were matched for size and preincubated with 1 mM glucose for 1 h, then incubated with varying glucose concentrations (1 mM, 7 mM, or 16.7 mM) or 30 mM KCl for 1 h followed by assaying the media for mouse insulin content as described for static GSIS for human islets. Under dynamic conditions, cultured mouse islets were studied using a multi-chamber perifusion system (Biorep Technologies, PERI-4.2). Each chamber was simultaneously perifused with KRBH buffer in the presence of 3 mM glucose, 16.7 mM glucose, or 30 mM KCl as indicated. Islets were perifused at 100 μL/min, solutions were collected each minute, and insulin production was quantified utilizing an insulin ELISA Kit (Crystal Chem, Cat. No. 90080) according to the manufacturer’s specifications.
Mouse islet calcium imaging
For calcium imaging, isolated islets from control or APT1 global deficient mice were incubated at room temperature for 30 min with 4 μM Fluo-4 AM (Thermofisher, Cat. No. F14201) prior to live cell imaging performed with an LSM880 two-photon inverted microscope (Carl Zeiss Inc.) using a heated stage-top incubator (Pecon GmbH). Relative fluorescence was captured after exposure to 2 mM or 16.7 mM glucose for multiple islets from each group, and images were analyzed using Fiji-imageJ software.
Dispersed mouse islet immunofluorescence
For measurement of docked insulin granules, dispersed islet cells from control or APT1 global deficient mice were fixed with 4% paraformaldehyde for 10 min, then monolayer cells were permeabilised using 0.1% Triton-X 100, and blocked with 5% goat serum in PBS at room temperature for 1 h. Then cells were stained for insulin (guinea pig polyclonal antibody from Abcam, Cat. No. ab7842) overnight at 4°C. TIRF images were then acquired by a Nikon TIRF system in the Cell and Tissue Imaging Core of the Washington Univesity Diabetes Research Center and granule density was counted by Fiji-ImageJ. For the assessment of cortical F-actin, Alexa Fluor 594 conjugated phalloidin (Thermofisher, Cat. No. A12381) was used to stain dispersed islet cells and images were acquired by a Nikon A1plus laser scan confocal microscope. Cell borders were delineated and fluoresence intensity of F-actin was quantitated after subtraction of background (Fiji-ImageJ).
Acyl-resin assisted capture (RAC) assay
Palmitoylated proteins were purified by RAC as described 42. Mouse islets or INS-1 cells were homogenized with lysis buffer containing 2% Triton-X-100 and 0.2 mM HDSF (hexadecylsulfonyl fluoride from Santa Cruz, Cat. No. sc-221708). After protein concentrations were determined by BCA protein assay kits (Thermofisher, Cat. No. 23227), the same amounts of protein lysates were incubated in 2.5% SDS with 0.1% MMTS (S-methyl methanethiosulfonate from Sigma, Cat. No. 64306) to block free sulfhydryl groups. Proteins were then precipitated, washed, and resuspended in binding buffer (100 mM HEPES, 1 mM EDTA, 1% SDS with protease Inhibitor) followed by thiopropyl-sepharose bead (GE healthcare, Cat. No.17-0420-01) pull-down. Incubation with neutral hydroxylamine (0.8 M, pH 7.4, Alfa Aesar, Cat. No. B22202) was used to specifically cleave the thioester linkage in palmitoylated proteins, and enriched palmitoylated proteins were eluted with 50 mM DTT (dithiothreitol). Eluted proteins were then denatured and followed by Western blotting with antibodies to proteins of interest including Scamp1 antibody (Thermofisher, Cat. No. PA1-739).
Palmitoylation proteomics
Proteomic screening was performed essentially as described.14 Palmitoylation-enriched samples were first obtained by acyl-RAC from APT1 global KO mouse islets. Trypsin-digested peptides were labeled with tandem mass tag reagents (TMT10plex™ Isobaric Label Reagent Set from Thermofisher, Cat. No. 90110), suspended in formic acid, and loaded onto a nano-LC column. Eluted peptides were subjected to MS1 scans and selected ions subjected to MS2 scans, then MS raw data were converted to peak lists and analyzed using Mascot search engine with specification for known protein modifications. Peptide matches were filtered at 1% FDR and identities containing >=2 Occam’s razor peptides were accepted. Relative quantification analyses were performed using R, and heat-map intensities were created with the R package: pheatmap. Assessing significance for differences between control and APT1 global KO islets was performed by t-test.
Scamp1 cysteine site mutagenesis
An expression plasmid encoding Myc-tagged wild type mouse Scamp1 cDNA was obtained from Origene Technologies (Myc-Scamp1, Cat. No. MR205042). Mutation of individual cysteine residues (C132S, C170S, C175S, C197S, C201S, C227S or C245S) in Scamp1 was performed by a modified site-directed mutagenesis PCR method using primer pairs containing non-overlapping sequences43 with SequalPrep™ Long PCR Kit (Thermo Fisher Scientific, Cat. No. A10498). The primer pairs are provided in Table S4. These constructs were used in 293T cell transfection experiments for palmitoylation assays.
Cloning of Scamp1 expression plasmids
For constitutive expression and imaging detection, GFP-WT-Scamp1 and GFP-C132S-Scamp1 were generated. The cDNA inserts from Myc-WT-Scamp1 or Myc-C132S-Scamp1 were subcloned into an mGFP lentiviral expression vector.14 The following primers were used for PCR cloning of the insert: forward- TCTAACAATTGTATGTCGGATTTTGACAGCA; reverse - GTACCTCTAGATTACATCTGGTTACCCTTGAAG. The restriction enzymes used were MfeI/XbaI and EcoRI/XbaI, for the vector and inserts, respectively. The plasmids were confirmed by Sanger sequencing.
Click chemistry for Scamp1 palmitoylation
Scamp1 palmitoylation was confirmed by click chemistry44 in 293T cells overexpressing Myc-Scamp1 or GFP-Scamp. Forty-eight hours post transfection, cells were incubated with media containing 20 μM palmitic acid alkyne (Cayman Chemical, Cat. No. 13266) for various times. Cells were then lysed, and solubilized protein fractions were isolated. The same amounts of protein lysates were immunoprecipitated with Myc-trap or GFP-trap magnetic agarose (Chromotek, Cat. No. ytma-10, Cat. No. gtma-10). Immunoprecipitates were then click labeled on beads with labeling buffer (PBS, pH=7.4, 1 mM CuSO4, 1 mM TCEP, 0.1 mM TBTA, and 40 μM azide-Cy5.5 from Click Chemistry Tools) at room temperature for 1 h. Labeled proteins were blotted for Scamp1 using Myc primary antibody (clone 9E10, Santa Cruz, Cat. No. sc-40) and IRDye-680CW conjugated secondary antibody (Licor, Cat. No. 925-68070). Resultant blots were scanned by a dual color fluorescence Licor imager.
Fluorescence imaging of Scamp1 granules
For mouse islets, dispersed islet cells from control mice were fixed and stained for insulin and Scamp1 (Thermofisher, Cat. No. PA1-739) as described above. For INS-1 cells, the cells were co-transfected with GFP-Scamp1 and mCherry-NPY (Addgene, Cat. No. 67156). TIRF images were acquired by a Nikon TIRF system as described above.
Insulin granule fusion analyses
VAMP2-pHluorin45 was transfected into INS-1 cells after siRNA knockdown treatment. Twenty four h later, cells were subjected to TIRF live cell imaging to quantify fusion events as described.46 On the day of the live cell imaging experiment, cells were preincubated with KRBH buffer containing 2 mM glucose in an incubator and then placed in a metal chamber mounted on a heated stage and kept at 37°C throughout the imaging experiments. Time lapse images were acquired by a Nikon TIRF system equipped with the PerfectFocus stability mechanism at 100 ms exposure and readout rate 540 MHz from an Andor Zyla 4.2 Megapixel sCMOS camera. To analyze fusion events, background normalized image sequences were first processed by Trackmate plugin in Fiji-ImageJ.47 The statistics of all identified spots were extracted. The data were then calculated by custom script in Python to generate time series intensity curves for each spot track including the preceding and following frames. The spot tracks were finally compiled for visualization within Fiji-Image J for manual confirmation. Individual fusion events were defined by the rapid appearance and disappearance of VAMP2-pHluorin fluoresence labeled vesicles. Kinetics of fusion decay curve for identified fusion events were extracted and normalized by the peak signal.
Dense core granule purification
Dense core secretory granules (DCVs) were purified from INS-1 cells as described.26 Cells were scraped in homogenate medium containing 0.26 M sucrose, 0.2 mM EDTA and 5 mM MOPS (Sigma, Cat. No. M1442-100ML) and then homogenized by passing through a 29-G needle 10 times. Cells were then subjected to sequential centrifugation steps (1500 rpm for 10 min and 5900 rpm for 15 min) at 4° C to remove cell nuclei and heavy membranes. The supernantants were ultracentrifuged in discontinuous gradients (0, 14.5%, and 30%) of iodixanol (Sigma, Cat. No. D1556) at 190,000 g for 5 h. A total of 11 gradient fractions were collected. Fraction #10 represents DCVs. Gradient fractions were analyzed by Western blotting for the presence of GFP-Scamp1, synaptotagmin-9 (polycolonal antibody from Synaptic systems, Cat. No. 105053), and endogenous Scamp1.
Apoptosis detection in INS-1 cells
INS-1 cells subjected to stable knockdown of APT1 by lentiviral shRNA were incubated in control media (5 mM glucose and 0.5% BSA), high glucose (25 mM glucose), palmitate (0.5 mM palmitate complexed with BSA), or a combination of high glucose and palmitate for 24 h. Then apoptotic cells were detected by using an APC Annexin V Apoptosis Detection Kit (Biolegend, Cat. No. 640932) according to the manufacturer’s instruction. Briefly, cells were collected, washed with PBS and re-suspended in FACS buffer (4% FBS and 2 mM EDTA in PBS). The cells were stained with APC/Annexin V and propidium iodide (PI) for 15 min at room temperature in the dark, prior to the addition of Annexin V binding buffer. Flow cytometry analysis was performed on the cell suspensions and the data obtained were analyzed using FlowJo_V10.8 software. In rescue experiments, INS-1 cells were subjected to stable knockdown of APT1 by lentiviral shRNA. The cells were subsequently transduced with GFP alone, GFP-WT Scamp1, or GFP-C132S-Scamp1 by lentivirus for 48 h. Then cells were incubated in high glucose and palmitate/BSA for 24 h followed by staining and FACS analyses using the same procedure as described above.
ER stress marker Western blotting
INS-1 cells with stable knockdown of APT1 by lentiviral shRNA were precultured with 5 mM glucose for 16 h, then cells were incubated in control media (5 mM glucose and 0.5% BSA), or in a stress media with the combination of high glucose (25 mM glucose) and palmitate (0.5 mM palmitate complexed with BSA) for 24 h. Then cells were harvested for Western blotting for ER stress markers. Briefly, cells were washed with PBS, lysed in buffers supplemented with protease inhibitors and phosphatase inhibitors for protein quantification. The protein samples were electrophoresed on SDS-PAGE gels and transferred to PVDF membranes before being blocked with blocking buffer (Licor, Cat. No. 927-60001) for 1 h. The membrane was incubated with primary antibodies, including anti-Txnip (MBL International, Cat. No. K0205-3), anti-XBP-1s (Cell Signaling Technology, Cat. No. 40435), anti-β tubulin (Cell Signaling Technology, Cat. No. 2146) and anti-APT1 (Abcam, Cat. No. ab91606) at 4°C overnight. Corresponding secondary antibodies were incubated for 1 h at room temperature and the proteins were visualized using Licor Odyssey FC Imaging system and quantitatively analyzed using Licor Image Studio Lite Ver 5.2 software.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed in Prism 9 software (GraphPad Software, La Jolla, CA) using unpaired t test (with Welch’s correction where appropriate), or two-way ANOVA (matched values over different time points with Bonferroni’s correction for multiple comparisons). Normal distribution was analyzed by Shapiro-Wilk test and Kolmogorov-Smirnov test. Data are presented as pooled data (n refers to numbers or sample numbers as indicated in figure legends) as mean ± sem, and p<0.05 was considered significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HPi2 antibody (HIC1-2B4.2B) | Novus Biologicals | Cat# NBP1-18946PECY7; RRID:AB_1625453 |
| PE anti-human CD26 antibody | BioLegend | Cat# 302705, RRID:AB_314289 |
| Mouse monoclonal anti-NTPDase 3 | ectonucleotidases antibodies | Cat# hN3-B3S, RRID:AB_2752250 |
| Mouse monoclonal anti-Insulin | Thermo Fisher Scientific | Cat# 53-9769-80, RRID:AB_2574468 |
| Guinea pig polyclonal anti-Insulin | Abcam | Cat# ab7842, RRID:AB_306130 |
| Rabbit polyclonal anti-Glucagon | Abcam | Cat# ab133195, RRID:AB_11156483 |
| Rat monoclonal anti-Ki67 | Thermo Fisher Scientific | Cat# 14-5698-80, RRID:AB_10853185 |
| Mouse monoclonal anti-Myc | Santa Cruz Biotechnology | Cat# sc-40, RRID:AB_627268 |
| Rabbit monoclonal anti-Lypla1 | Abcam | Cat# ab91606, RRID:AB_10565192 |
| Rabbit polyclonal anti-Scamp1 | Thermo Fisher Scientific | Cat# PA1-739, RRID:AB_2184624 |
| Rabbit polyclonal anti-Synaptotagmin-9 | Synaptic Systems | Cat# 105 053, RRID:AB_2199639 |
| Mouse monoclonal anti-Txnip | MBL International | Cat# K0205-3, RRID:AB_592934) |
| Rabbit monoclonal anti-XBP-1s | Cell Signaling Technology | Cat# 40435, RRID:AB_2891025 |
| Mouse monoclonal anti-GFP | Thermo Fisher Scientific | Cat# MA5-15256, RRID:AB_10979281 |
| Rabbit monoclonal anti-β-Tubulin | Cell Signaling Technology | Cat# 2146, RRID:AB_2210545 |
| Mouse monoclonal anti-Tubulin α1c | Santa Cruz Biotechnology | Cat# sc-134239, RRID:AB_2210215 |
| Human Fc block | BD Biosciences | Cat# 564220, RRID:AB_2869554 |
| Rabbit polyclonal anti-CKAP4 | Proteintech | Cat# 16686-1-AP, RRID:AB_2276275 |
| Bacterial and virus strains | ||
| MISSION shRNA Bacterial Glycerol Stock for human APT1 | Sigma | Cat# SHCLNG-NM_006330 |
| Biological samples | ||
| Human islets | Prodo labs | https://prodolabs.com/ |
| Chemicals, peptides, and recombinant proteins | ||
| Accutase®solution | Sigma | Cat# A6964 |
| Collagenase P | Roche | Cat# 11213857001 |
| Hexadimethrine bromide (Polybrene) | Sigma | Cat# H9268-10G |
| Fluo-4 AM | Thermo Fisher Scientific | Cat# F14201 |
| Alexa Fluor 594 phalloidin | Thermo Fisher Scientific | Cat# A12381; RRID:AB_2315633 |
| Transfection Reagent | Horizon Discovery | Cat# T-2001-502 02 |
| Hexadecylsulfonyl fluoride (HDSF) | Santa Cruz Biotechnology | Cat# sc-221708 |
| S-methyl methanethiosulfonate (MMTS) | Sigma | Cat# 64306 |
| Palmitic acid alkyne | Cayman Chemical | Cat# 13266 |
| Iodixanol | Sigma | Cat# D1556 |
| MOPS solution | Sigma | Cat# M1442 |
| Intercept blocking buffer | Licor | Cat# 927-60001 |
| Critical commercial assays | ||
| Human Insulin ELISA kit | Alpco | Cat# 80-INSHU-E01.1 |
| Rat Insulin ELISA kit | Alpco | Cat# 80-INSRT-E01 |
| Crystal Chem ultrasensitive mouse Insulin kit | Crystal Chem Inc | Cat# 90080 |
| Crystal Chem ultrasensitive mouse C-peptide kit | Crystal Chem Inc | Cat# 90050 |
| RNeasy micro kit | Qiagen | Cat# 74004 |
| Mercodia Glucagon ELISA | Mercodia Inc | Cat#10-1271-01 |
| iScript cDNA synthesis kit | Bio-Rad | Cat# 1708890 |
| SYBR® Premix Ex Taq™ | TAKARA BIO | Cat# RR420A |
| Glucose assay kit | FUJIFILM Wako Chemicals | Cat# 99703001 |
| Triglycerides assay kit | Thermo Fisher Scientific | Cat# TR22421 |
| Cholesterol assay kit | Thermo Fisher Scientific | Cat# TR13421 |
| Lenti-X™ concentrator | TAKARA BIO | Cat# 631231 |
| Lenti-X™ qRT-PCR titration kit | TAKARA BIO | Cat# 631235 |
| SequalPrep™ Long PCR Kit | Thermo Fisher Scientific | Cat# A10498 |
| BCA protein assay kits | Thermo Fisher Scientific | Cat# 23227 |
| TMT10plex™ Isobaric Label Reagent set | Thermo Fisher Scientific | Cat# 90110 |
| APC Annexin V Apoptosis Detection Kit | Biolegend | Cat# 640932 |
| In Situ Cell Death Detection Kit | Sigma | Cat# 12156792910 |
| Deposited data | ||
| Mass spectrometry analysis of differential palmitoylation | PRIDE | Accession PXD038554 DOI 10.6019/PXD038554 |
| Source data and Western blot images for manuscript figures | This paper | Data S1: Sources |
| Experimental models: Cell lines | ||
| 293T | ATCC | Cat# CRL-3216; RRID:CVCL_0063 |
| INS-1 832/13 | Hohmeier et al., 2000 | https://wolframsyndrome.wustl.edu/urano-lab |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J floxed Lypla1 | Wei et al., 2020 | N/A |
| Mouse: Lypla2tm1a(KOMP)Mbp | KOMP Repository | Project ID: CSD34805; RRID:IMSR_KOMP:CSD34805-1a-Mbp |
| Mouse: BKS.Cg-Dock7m +/+ Leprdb/J | The Jackson Laboratory | JAX: 000642; RRID:IMSR_JAX:000642 |
| Mouse: Pdx1PB-CreER™ mice | Zhang et al., 2005 | https://endocrinology.wustl.edu/items/remedi-lab/ |
| Oligonucleotides | ||
| RT-PCR primer: hLypla1-forward CCTTTGCAGGTATCAGAAGTTCA |
Wei et al., 2020 | N/A |
| RT-PCR primer: hLypla1-reverse GCTGCCTGTTTAATCCCAGAT |
Wei et al., 2020 | N/A |
| RT-PCR primer: hTBP-forward CCACTCACAGACTCTCACAAC | This paper | N/A |
| RT-PCR primer: hTBP-reverse CTGCGGTACAATCCCAGAACT |
This paper | N/A |
| Rat APT1 siRNA (Smartpool) | Horizon Discovery | Cat# L-089651-02-0005 |
| Rat Scamp1 siRNA (Smartpool) | Horizon Discovery | Cat# L-086797-02-0010 |
| RT-PCR primer: mLypla1-forward CAGGTATCAAAAGTCCCCACATC |
This paper | N/A |
| RT-PCR primer: mLypla1-reverse GGTTTCTGCTGCCTGTTTAATTC |
This paper | N/A |
| RT-PCR primer: mTBP-forward AGAACAATCCAGACTAGCAGCA |
This paper | N/A |
| RT-PCR primer: mTBP-Reverse GGGAACTTCACATCACAGCTC |
This paper | N/A |
| Recombinant DNA | ||
| GFP-WT-Scamp1 | This paper | N/A |
| GFP-C132S-Scamp1 | This paper | N/A |
| Scrambled-shRNA Plasmid | Origene Technologies | Cat# TL709612-D |
| Rat Lypla1 shRNA Plasmid | Origene Technologies | Cat# TL709612-B |
| Mouse Scamp1Tagged ORF Clone | Origene Technologies | Cat# MR205042 |
| mCherry-NPY | Barg et al., 2010 | Addgene # 67156 |
| VAMP2-pHluorin | Miesenböck et al., 1998 | https://pistonlab.wustl.edu/piston-lab-members/ |
| Software and algorithms | ||
| Fiji-ImageJ | National Inst. Of Health | https://imagej.nih.gov/ij/ |
| Fiji plugin: Trackmate | Tinevez et al., 2017 | https://imagej.net/TrackMate |
| Python 3 | Python Software Foundation | http://www.python.org |
| Image Studio Lite Ver 5.2 | Licor | https://www.licor.com/bio/image-studio/ |
| FlowJo_V10.8 | Flowjo LLC. | https://www.flowjo.com/ |
| Prism 9 | Graphpad software | https://www.graphpad.com/scientific-software/prism/ |
| Mascot | Perkins et al., 1999 | http://www.matrixscience.com/ |
| R | R Core Team (2018) | https://www.R-project.org/ |
| R package: mixtools:: normalmixEM | Tatiana Benaglia, Didier Chauveau, David R. Hunter, Derek Young (2009) | http://www.jstatsoft.org/v32/i06/ |
| Other | ||
| High fat diet | Envigo | Cat# TD88137 |
| CMRL 1066 media | Corning | Cat# 99-603-CV |
| RPMI 1640 | Gibco | Cat# 11875-085 |
| Thiopropyl sepharose bead | GE healthcare | Cat# 17-0420-01 |
| Myc-trap magnetic agarose | Chromotek | Cat# ytma-10 |
| GFP-trap magnetic agarose | Chromotek | Cat# gtma-10 |
Highlights.
Acyl-protein thioesterase 1 (APT1) shows altered expression in human pancreatic islets
APT1 deficiency causes insulin hypersecretion
Deficiency of the APT1 substrate Scamp1 causes insulin hypersecretion
APT1 deficiency promotes β-cell failure in high fat fed mice and db/db mice
ACKNOWLEDGMENTS
This work was supported by the Washington University Centene Personalized Medicine Initiative (P19-00559), the Washington University Diabetes Research Center (DK020579), and the following NIH grants: DK101392, DK056341, DK115972, DK123301, DK112921, GM119840, T32 HL125241, F30 DK131830, HL157154. G.D. is supported by China Scholarship Council (CSC) grant 201608420067. S.M. is supported by Manpei Suzuki Diabetes Foundation and JSPS Overseas Research Fellowships. We thank Jeff Millman, Leonardo Velaszco-Cruz, Kristina Maxwell, Punn Augsornworawat, Cris Brown (all at Washington University) and Richard O’Brien (Vanderbilt University) for advice.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors have no financial interests to declare. The lead contact is a member of the Cell Metabolism advisory board.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Porte D Jr. (1991). Banting lecture 1990. Beta-cells in type II diabetes mellitus. Diabetes 40, 166–180. 10.2337/diab.40.2.166. [DOI] [PubMed] [Google Scholar]
- 2.Kahn SE (2003). The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 46, 3–19. 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- 3.Tura A, Grespan E, Gobl CS, Koivula RW, Franks PW, Pearson ER, Walker M, Forgie IM, Giordano GN, Pavo I, et al. (2021). Profiles of Glucose Metabolism in Different Prediabetes Phenotypes, Classified by Fasting Glycemia, 2-Hour OGTT, Glycated Hemoglobin, and 1-Hour OGTT: An IMI DIRECT Study. Diabetes 70, 2092–2106. 10.2337/db21-0227. [DOI] [PubMed] [Google Scholar]
- 4.Wagner R, Heni M, Tabak AG, Machann J, Schick F, Randrianarisoa E, Hrabe de Angelis M, Birkenfeld AL, Stefan N, Peter A, et al. (2021). Pathophysiology-based subphenotyping of individuals at elevated risk for type 2 diabetes. Nat Med 27, 49–57. 10.1038/s41591-020-1116-9. [DOI] [PubMed] [Google Scholar]
- 5.Weyer C, Hanson RL, Tataranni PA, Bogardus C, and Pratley RE (2000). A high fasting plasma insulin concentration predicts type 2 diabetes independent of insulin resistance: evidence for a pathogenic role of relative hyperinsulinemia. Diabetes 49, 2094–2101. 10.2337/diabetes.49.12.2094. [DOI] [PubMed] [Google Scholar]
- 6.Trico D, Natali A, Arslanian S, Mari A, and Ferrannini E (2018). Identification, pathophysiology, and clinical implications of primary insulin hypersecretion in nondiabetic adults and adolescents. JCI Insight 3. 10.1172/jci.insight.124912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esser N, Utzschneider KM, and Kahn SE (2020). Early beta cell dysfunction vs insulin hypersecretion as the primary event in the pathogenesis of dysglycaemia. Diabetologia 63, 2007–2021. 10.1007/s00125-020-05245-x. [DOI] [PubMed] [Google Scholar]
- 8.Staaf J, Ubhayasekera SJ, Sargsyan E, Chowdhury A, Kristinsson H, Manell H, Bergquist J, Forslund A, and Bergsten P (2016). Initial hyperinsulinemia and subsequent beta-cell dysfunction is associated with elevated palmitate levels. Pediatr Res 80, 267–274. 10.1038/pr.2016.80. [DOI] [PubMed] [Google Scholar]
- 9.Weir GC (2020). Glucolipotoxicity, beta-Cells, and Diabetes: The Emperor Has No Clothes. Diabetes 69, 273–278. 10.2337/db19-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prentki M, Peyot ML, Masiello P, and Madiraju SRM (2020). Nutrient-Induced Metabolic Stress, Adaptation, Detoxification, and Toxicity in the Pancreatic beta-Cell. Diabetes 69, 279–290. 10.2337/dbi19-0014. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, and Yang W (2021). Proteome-Scale Analysis of Protein S-Acylation Comes of Age. J Proteome Res 20, 14–26. 10.1021/acs.jproteome.0c00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdel-Ghany M, Sharp GW, and Straub SG (2010). Glucose stimulation of protein acylation in the pancreatic beta-cell. Life Sci 87, 667–671. 10.1016/j.lfs.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smotrys JE, and Linder ME (2004). Palmitoylation of intracellular signaling proteins: regulation and function. Annu Rev Biochem 73, 559–587. 10.1146/annurev.biochem.73.011303.073954. [DOI] [PubMed] [Google Scholar]
- 14.Wei X, Adak S, Zayed M, Yin L, Feng C, Speck SL, Kathayat RS, Zhang Q, Dickinson BC, and Semenkovich CF (2020). Endothelial Palmitoylation Cycling Coordinates Vessel Remodeling in Peripheral Artery Disease. Circ Res 127, 249–265. 10.1161/CIRCRESAHA.120.316752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chamberlain LH, Shipston MJ, and Gould GW (2021). Regulatory effects of protein S-acylation on insulin secretion and insulin action. Open Biol 11, 210017. 10.1098/rsob.210017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prescott GR, Gorleku OA, Greaves J, and Chamberlain LH (2009). Palmitoylation of the synaptic vesicle fusion machinery. J Neurochem 110, 1135–1149. 10.1111/j.1471-4159.2009.06205.x. [DOI] [PubMed] [Google Scholar]
- 17.Virlogeux A, Scaramuzzino C, Lenoir S, Carpentier R, Louessard M, Genoux A, Lino P, Hinckelmann MV, Perrier AL, Humbert S, and Saudou F (2021). Increasing brain palmitoylation rescues behavior and neuropathology in Huntington disease mice. Sci Adv 7. 10.1126/sciadv.abb0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kathayat RS, Elvira PD, and Dickinson BC (2017). A fluorescent probe for cysteine depalmitoylation reveals dynamic APT signaling. Nat Chem Biol 13, 150–152. 10.1038/nchembio.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Fujitani Y, Wright CV, and Gannon M (2005). Efficient recombination in pancreatic islets by a tamoxifen-inducible Cre-recombinase. Genesis 42, 210–217. 10.1002/gene.20137. [DOI] [PubMed] [Google Scholar]
- 20.Kalwat MA, and Thurmond DC (2013). Signaling mechanisms of glucose-induced F-actin remodeling in pancreatic islet beta cells. Exp Mol Med 45, e37. 10.1038/emm.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Houtz J, Borden P, Ceasrine A, Minichiello L, and Kuruvilla R (2016). Neurotrophin Signaling Is Required for Glucose-Induced Insulin Secretion. Dev Cell 39, 329–345. 10.1016/j.devcel.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang B, Lin H, Li X, Lu W, Kim JB, Xu A, and Cheng KKY (2020). The adaptor protein APPL2 controls glucose-stimulated insulin secretion via F-actin remodeling in pancreatic beta-cells. Proc Natl Acad Sci U S A 117, 28307–28315. 10.1073/pnas.2016997117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandez-Chacon R, Alvarez de Toledo G, Hammer RE, and Sudhof TC (1999). Analysis of SCAMP1 function in secretory vesicle exocytosis by means of gene targeting in mice. J Biol Chem 274, 32551–32554. 10.1074/jbc.274.46.32551. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, and Castle D (2011). Regulation of fusion pore closure and compound exocytosis in neuroendocrine PC12 cells by SCAMP1. Traffic 12, 600–614. 10.1111/j.1600-0854.2011.01170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernandez-Chacon R, and Sudhof TC (2000). Novel SCAMPs lacking NPF repeats: ubiquitous and synaptic vesicle-specific forms implicate SCAMPs in multiple membrane-trafficking functions. J Neurosci 20, 7941–7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kreutzberger AJB, Kiessling V, Doyle CA, Schenk N, Upchurch CM, Elmer-Dixon M, Ward AE, Preobraschenski J, Hussein SS, Tomaka W, et al. (2020). Distinct insulin granule subpopulations implicated in the secretory pathology of diabetes types 1 and 2. Elife 9. 10.7554/eLife.62506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gauthier BR, and Wollheim CB (2008). Synaptotagmins bind calcium to release insulin. Am J Physiol Endocrinol Metab 295, E1279–1286. 10.1152/ajpendo.90568.2008. [DOI] [PubMed] [Google Scholar]
- 28.Lee JH, Mellado-Gil JM, Bahn YJ, Pathy SM, Zhang YE, and Rane SG (2020). Protection from beta-cell apoptosis by inhibition of TGF-beta/Smad3 signaling. Cell Death Dis 11, 184. 10.1038/s41419-020-2365-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, et al. (2018). Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 50, 1505–1513. 10.1038/s41588-018-0241-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nag A, McCarthy MI, and Mahajan A (2020). Large-Scale Analyses Provide No Evidence for Gene-Gene Interactions Influencing Type 2 Diabetes Risk. Diabetes 69, 2518–2522. 10.2337/db20-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schnurr TM, Jakupovic H, Carrasquilla GD, Angquist L, Grarup N, Sorensen TIA, Tjonneland A, Overvad K, Pedersen O, Hansen T, and Kilpelainen TO (2020). Obesity, unfavourable lifestyle and genetic risk of type 2 diabetes: a case-cohort study. Diabetologia 63, 1324–1332. 10.1007/s00125-020-05140-5. [DOI] [PubMed] [Google Scholar]
- 32.Guo Z, Liu L, Cafiso D, and Castle D (2002). Perturbation of a very late step of regulated exocytosis by a secretory carrier membrane protein (SCAMP2)-derived peptide. J Biol Chem 277, 35357–35363. 10.1074/jbc.M202259200. [DOI] [PubMed] [Google Scholar]
- 33.Liu L, Guo Z, Tieu Q, Castle A, and Castle D (2002). Role of secretory carrier membrane protein SCAMP2 in granule exocytosis. Mol Biol Cell 13, 4266–4278. 10.1091/mbc.e02-03-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Camunas-Soler J, Dai XQ, Hang Y, Bautista A, Lyon J, Suzuki K, Kim SK, Quake SR, and MacDonald PE (2020). Patch-Seq Links Single-Cell Transcriptomes to Human Islet Dysfunction in Diabetes. Cell Metab 31, 1017–1031 e1014. 10.1016/j.cmet.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Avrahami D, Wang YJ, Schug J, Feleke E, Gao L, Liu C, Consortium H, Naji A, Glaser B, and Kaestner KH (2020). Single-cell transcriptomics of human islet ontogeny defines the molecular basis of beta-cell dedifferentiation in T2D. Mol Metab 42, 101057. 10.1016/j.molmet.2020.101057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erion K, and Corkey BE (2018). beta-Cell Failure or beta-Cell Abuse? Front Endocrinol (Lausanne) 9, 532. 10.3389/fendo.2018.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Vliet S, Koh HE, Patterson BW, Yoshino M, LaForest R, Gropler RJ, Klein S, and Mittendorfer B (2020). Obesity Is Associated With Increased Basal and Postprandial beta-Cell Insulin Secretion Even in the Absence of Insulin Resistance. Diabetes 69, 2112–2119. 10.2337/db20-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, and Newgard CB (2000). Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and - independent glucose-stimulated insulin secretion. Diabetes 49, 424–430. 10.2337/diabetes.49.3.424. [DOI] [PubMed] [Google Scholar]
- 39.Saunders DC, Brissova M, Phillips N, Shrestha S, Walker JT, Aramandla R, Poffenberger G, Flaherty DK, Weller KP, Pelletier J, et al. (2019). Ectonucleoside Triphosphate Diphosphohydrolase-3 Antibody Targets Adult Human Pancreatic beta Cells for In Vitro and In Vivo Analysis. Cell Metab 29, 745–754 e744. 10.1016/j.cmet.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carter JD, Dula SB, Corbin KL, Wu R, and Nunemaker CS (2009). A practical guide to rodent islet isolation and assessment. Biol Proced Online 11, 3–31. 10.1007/s12575-009-9021-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei X, Song H, Yin L, Rizzo MG, Sidhu R, Covey DF, Ory DS, and Semenkovich CF (2016). Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature 539, 294–298. 10.1038/nature20117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forrester MT, Hess DT, Thompson JW, Hultman R, Moseley MA, Stamler JS, and Casey PJ (2011). Site-specific analysis of protein S-acylation by resin-assisted capture. J Lipid Res 52, 393–398. 10.1194/jlr.D011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu H, and Naismith JH (2008). An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol 8, 91. 10.1186/1472-6750-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin BR, Wang C, Adibekian A, Tully SE, and Cravatt BF (2011). Global profiling of dynamic protein palmitoylation. Nat Methods 9, 84–89. 10.1038/nmeth.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miesenbock G, De Angelis DA, and Rothman JE (1998). Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394, 192–195. 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 46.Xu Y, Toomre DK, Bogan JS, and Hao M (2017). Excess cholesterol inhibits glucose-stimulated fusion pore dynamics in insulin exocytosis. J Cell Mol Med 21, 2950–2962. 10.1111/jcmm.13207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, and Eliceiri KW (2017). TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90. 10.1016/j.ymeth.2016.09.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. Source data and Western blot images for the figures in the manuscript are available as Data S1: Sources, Related to Figures 1–7 and S1–S7.
This paper does not report original code.
Any additional information required to analyze the data reported in this paper is available from the lead contact upon request.