

Abstract
Cerebral creatine deficiency syndromes (CCDS) are a rare group of inherited metabolic disorders (IMDs) that often present with nonspecific findings including global developmental delay (GDD), intellectual disability (ID), seizures, hypotonia, and behavioral differences. Creatine transporter (CRTR) deficiency is the most common CCDS, exhibiting X-linked inheritance and an estimated prevalence as high as 2.6% in individuals with neurodevelopmental disorders. Here, we present a 20-month-old boy with worsening failure to thrive (FTT) and GDD admitted for evaluation. He was found to have persistently low serum creatinine levels and a family history notable for a mother with learning disabilities and a maternal male cousin with GDD. Urine analyses revealed a marked elevation of creatine and elevated creatine:creatinine ratio suggestive of CRTR deficiency. Molecular genetic testing of SLC6A8 identified a maternally inherited hemizygous variant and brain magnetic resonance spectroscopy (MRS) showed diffusely diminished creatine peaks, further supporting the diagnosis of CRTR deficiency. The proband was started on creatine, arginine, and glycine supplementation and has demonstrated improved development. This case highlights that CRTR deficiency should be considered in all patients presenting with FTT and abnormal neurodevelopmental features, particularly if creatinine levels are low on serum chemistry studies. The nonspecific presentation of this condition in males and females likely has resulted in CRTR deficiency being underdiagnosed. There are existing therapies for individuals affected with CRTR deficiency and other CCDS, highlighting the importance of early diagnosis and intervention for affected individuals.
Keywords: creatine transporter deficiency, failure to thrive, developmental delay, intellectual disability, neurodevelopmental disorder, newborn screening, case report
Background
Isolated failure to thrive (FTT) is often not genetic in origin; however, there are scenarios that should raise a clinician’s suspicion for a possible genetic condition. These include cases of FTT that are congenital, persistent, severe, require hospital admission, and/or occur in combination with other clinical features. Additional features particularly suggestive of a genetic cause include developmental delays, seizures, abnormal fat distribution, dysmorphic features, and abnormalities on chemistry studies. Obtaining a detailed family history can also be helpful to determine if a Mendelian (e.g., autosomal dominant, autosomal recessive, or X-linked) or mitochondrial inheritance pattern is present.
Genetic conditions associated with FTT can be divided into 2 categories: (1) inherited metabolic disorders (IMDs; also known as inborn errors of metabolism) and (2) non-metabolic genetic conditions. Many IMDs can be treated with dietary modification and/or metabolic supplementation, highlighting the importance of a timely and accurate diagnosis. While state newborn screening (NBS) evaluates for several IMDs, most IMDs are not included in NBS, including cerebral creatine deficiency syndromes (CCDS) such as creatine transporter (CRTR) deficiency.1,2
Cerebral creatine deficiency syndromes are a rare group of IMDs which include X-linked CRTR deficiency and 2 autosomal recessive creatine biosynthesis disorders: guanidinoacetate methyltransferase (GAMT) deficiency and L-arginine:glycine amidinotransferase (AGAT) deficiency. These disorders often present with nonspecific features including global developmental delay (GDD), intellectual disability (ID), seizures, hypotonia, and behavioral differences.3,4 Self-injurious behavior can be a feature in either CRTR or GAMT deficiency, whereas pyramidal and extrapyramidal findings are most associated with GAMT deficiency.4 Abnormal values for urine guanidinoacetate, creatine, and creatinine are common among all 3 conditions; however, the pattern varies for each specific disorder. Elevations in urine creatine and creatine:creatinine are specific to CRTR deficiency, while marked elevation and reduction of guanidinoacetate are most consistent with GAMT deficiency and AGAT deficiency, respectively (Table 1).4,5
Table 1.
Features of Cerebral Creatine Deficiency Syndromes (CCDS).
| Disorder | Gene (inheritance) | Clinical features | Urine guanidinoacetate | Urine creatine | Urine creatine: creatinine |
|---|---|---|---|---|---|
| L-arginine:glycine amidinotransferase (AGAT) deficiency | GATM (AR) | Onset 3 weeks to 25 years; speech-predominant global developmental delay, intellectual disability, autistic behavior, hypotonia, and myopathy | Decreased | Decreased | Normal |
| Guanidinoacetate methyltransferase (GAMT) deficiency | GAMT (AR) | Onset 3 months to 3 years; speech-predominant global developmental delay, intellectual disability, autistic behavior, aggression, self-injurious behavior, seizures, movement disorders, and basal ganglia findings | Increased | Decreased | Normal |
| Creatine transporter (CRTR) deficiency | SLC6A8 (XLR); findings may be less severe or absent in heterozygous females | Onset <3 years; speech-predominant global developmental delay, intellectual disability, autistic behavior, seizures, and hypotonia | Normal | Increased | Increased |
Abbreviations: AGAT, L-arginine:glycine amidinotransferase; GATM, glycine amidinotransferase, mitochondrial; GAMT, guanidinoacetate methyltransferase; CRTR, creatine transporter; AR, autosomal recessive; XLR, X-linked recessive.
Creatine transporter deficiency is the most common of the 3 CCDS, exhibiting X-linked inheritance and accounting for approximately 64% to 72% of CCDS.3 Hemizygous males are typically more severely affected, whereas heterozygous females typically present with a variable phenotype ranging from asymptomatic to mild ID; however, females can be as severely affected as males, a phenomenon that appears unrelated to X-inactivation.6 The prevalence of CRTR deficiency among males with ID is estimated to be between 0.4% and 1.4%,7 with one study reporting a prevalence of 2.64% within a cohort of males and females with neurodevelopmental disorders who underwent screening for a CCDS.8 Developmental delay is the most common feature in males with CRTR deficiency, present in 80% of males at the time of initial presentation, with 8% of males presenting with FTT.7
Creatine supplementation with oral creatine-monohydrate is a shared therapy among all the CCDS in addition to supportive therapy of associated features. Creatine transporter deficiency therapy includes high-dose oral creatine supplementation (400 mg/kg/day) in an effort to use any residual transporter activity with the goal of increasing cerebral creatine. In addition, large doses of creatine precursors, L-arginine (400 mg/kg/day) and/or glycine (150 mg/kg/day), can be given orally to promote creatine synthesis.4,9 Response to combined supplementation of creatine, arginine, and glycine varies among individuals but has been shown to improve clinical outcomes (e.g. seizure frequency, psychiatric and behavioral features, cognition).10 In some cases, increases in cerebral creatine on magnetic resonance spectroscopy (MRS) have been observed after starting combined supplementation,10 whereas high-dose creatine alone does not appear to have the same clinical or radiological effects.11 Clinical improvement with combined supplementation appears to be high in women and men with milder features, most likely due to residual CRTR function that results in a milder phenotype as well as increased response to therapy.10,12
Here we describe a 20-month-old boy found to have CRTR deficiency who presented with GDD and worsening FTT requiring hospital admission and was found to have persistently low serum creatinine levels. He was ultimately diagnosed with CRTR deficiency and started on creatine, arginine, and glycine supplementation with demonstrated developmental improvement. This case illustrates a common but nonspecific presentation of CRTR deficiency, demonstrating the importance of including disorders of creatine metabolism on the differential diagnosis in patients presenting with FTT and neurodevelopmental features.
Case Presentation
A 20-month-old boy with GDD was transferred for worsening FTT after inability to demonstrate adequate caloric intake and weight gain during a 3-day admission at an outside hospital. Perinatal history was notable for being small for gestational age at 39 weeks’ gestation (birth weight 2.33 kg, second percentile, Z = −2.2; birth length 49.5 cm, 32nd percentile, Z = −0.5; head circumference unavailable). He was born to a gravida 1, para 0, woman, and NBS at birth was unremarkable. Developmental history was significant for sitting at 6 months and cruising at 12 months, and at 20-months of age, he was unable to walk independently or use spoken words to communicate. There was no history of diarrhea, constipation, recurrent emesis, or seizures. Physical exam was notable for weight 7.4 kg (<third percentile, Z-score = −3.8), height 66 cm (<third percentile, Z-score = −6.5), head circumference 45 cm (<third percentile, Z-score = −2.3), and nondysmorphic features.
Prior imaging studies included a normal renal ultrasound and echocardiogram; a bone-age study at 20 months of age revealed a bone-age of 14 months. Laboratory studies at 18 months of age included normal thyroid-stimulating hormone (TSH) 1.4 µIU/mL (0.2-5.0), free T4 1.27 ng/dL (0.5-1.76), random cortisol 7.6 µg/dL (>2), insulin-like growth factor 1 (IGF-1) 38 ng/mL (30-167), and lead <2 µg/dL (<5). Previous genetic testing included a normal karyotype and chromosomal microarray. On admission, a comprehensive metabolic panel was normal except for a low creatinine of <0.06 mg/dL (0.18-0.35) and elevated blood urea nitrogen of 26 mg/dL (5–18). A nasal-gastric tube was placed to increase caloric intake and Medical Genetics was consulted due to concern for an underlying genetic condition.
A 3-generation pedigree was suggestive of maternal inheritance of an X-linked or mitochondrial disorder, notable for a family history of learning disabilities in the proband’s mother and a maternal male first cousin with GDD; there was no family history of seizures. Given the finding of persistently low serum creatinine, a urine creatine disorder panel was ordered and revealed mild elevation of guanidinoacetate (254 mmol/mol cr; reference 0-162 mmol/mol cr), marked elevation of creatine (6022 mmol/mol cr; reference 0-1706 mmol/mol cr), low-normal creatinine (546 nmol/mL; reference 0-14 833 nmol/mL), and elevated creatine: creatinine ratio (11, reference <2.5). These biochemical laboratory results were highly suggestive of CRTR deficiency (Table 1), an X-linked condition, and supported a focused genetic testing strategy as opposed to a broader testing approach (e.g. large gene panel or exome sequencing). Full-gene sequencing with deletion/duplication analysis of SLC6A8, the gene associated with CRTR deficiency, identified a novel, maternally inherited hemizygous variant of uncertain significance (VUS; NM_005629.4: c.191_193del (p. Ser64del)). Brain MRS showed diffusely diminished creatine peaks across the sampled regions, further supporting the diagnosis of CRTR deficiency (Figure 1). The patient was started on creatine, arginine, and glycine supplementation, and maternal relatives were referred to Biochemical Genetics for outpatient evaluation and treatment. At 40 months of age and approximately a year after placement of a gastric tube and initiation of creatine, arginine, and glycine supplementation, there is documented evidence of improved development and weight gain (weight 13.3 kg, 14th percentile). He is an active and playful 3-year-old, currently no longer requiring or receiving physical or occupational therapy. His verbal communication is limited to 5 to 10 words, though he is able sign and express his wants/needs with demonstrated receptive language and comprehension as he awaits speech therapy placement. Notably, he follows with neurology biannually and there continues to be no evidence of seizure-like activity; no electroencephalogram or repeat MRS has been performed to date. The proband’s affected mother and presumably affected maternal relatives have not presented to care for further evaluation.
Figure 1.
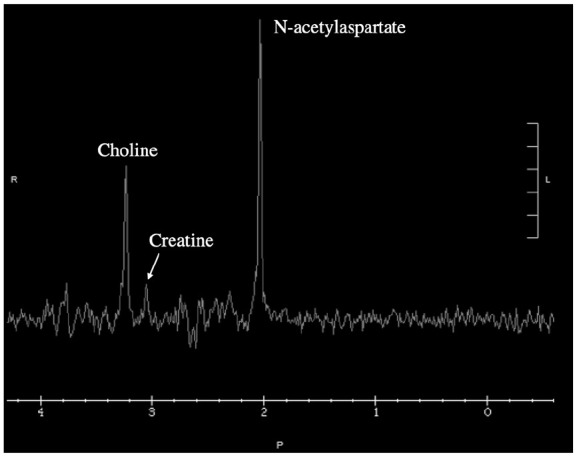
Magnetic resonance spectroscopy (MRS) of white matter in an individual with creatine transporter (CRTR) deficiency prior to starting creatine supplementation. Creatine peak at 3.0 ppm is severely reduced from normal (typically larger than choline peak at 3.2 ppm in unaffected individuals).
Discussion and Conclusions
It is important to consider genetic conditions and IMDs in patients presenting with FTT to provide patients and families appropriate diagnostic, prognostic, therapeutic, and recurrence risk counseling. If possible, early consultation with a medical geneticist is warranted to guide further evaluation and recommend treatment when available. Nonmetabolic genetic conditions on the differential diagnosis for FTT include Russell-Silver syndrome (the most common genetic cause of FTT after trisomy 21 and notable for relative macrocephaly and triangular facial appearance), Prader-Willi syndrome (poor feeding until 18–36 months with subsequent paradoxical hyperphagia), chromosome abnormalities, and monogenic ID disorders with FTT secondary to behavioral features.13 Teratogen exposures such as congenital infections14,15 and in utero ethanol exposure,16 while not genetic in nature, should also be considered.
In a child with FTT, additional features particularly suggestive of an IMD include abnormalities on chemistry panels that are long-term or worsen in times of stress (e.g., elevated anion gap, hypoglycemia, hyperammonemia, elevated lactate, and low creatinine), seizures, developmental delay (often secondary to a prior metabolic insult), and abnormal fat distribution. The IMD differential diagnosis is broad and includes disorders of amino acid metabolism, fatty acid oxidation disorders, organic acidurias, urea cycle disorders, and mitochondrial disorders. Disorders of creatinine metabolism and congenital disorders of glycosylation should also be considered as clinical features associated with these IMDs are often nonspecific and demonstrate significant interindividual variability. Biochemical laboratory studies are useful to evaluate for an IMD in a patient with FTT with or without additional features; they can also be of particular value in interpreting the clinical significance of a VUS identified by molecular genetic testing by providing functional evidence for or against pathogenicity. For this reason, biochemical laboratory testing in consultation with a medical geneticist can help ensure correct interpretation of test results and appropriate recommendations for further evaluation.
Healthcare providers should be aware of FTT as an early diagnostic clue for CRTR deficiency, and our proband’s clinical presentation provides support for this consideration in the work-up of this chief complaint. In a study of 101 males with CRTR deficiency, gastrointestinal problems were a presenting feature in 16% with 8% presenting with FTT.7 Miller et al17 evaluated 20 boys with molecularly confirmed CRTR deficiency (genotypes not available) and found parents reported vomiting or a significant feeding issue prior to 12 months of age in over half of cases.
While the SLC6A8 variant identified in our proband and his mother (NM_005629.4: c.191_193del (p. Ser64del)) was classified as a VUS, several other disease-causing single amino acid (3 bp) deletions have been reported in this gene.11,18,19 The serine residue at amino acid position 64 is located within the first of 12 SLC6A8 transmembrane (TM) helices, specifically localized within the ligand-binding site.19,20 The TM1 helix, together with TM2, 6, and 7, constitute the dynamic bundle domain, allowing creatine to enter the transporter and to be released.20 Similarly to missense variants in SLC6A8 that confer residual transporter activity and possibly a milder phenotype,7 this variant may also represent a hypomorphic allele that results in a milder phenotype and possible increased response to therapy. Despite the current classification of the variant identified in our proband, the diagnosis of CRTR deficiency was made based on the constellation of clinical, laboratory, radiological, and family history findings supporting a diagnosis of CRTR deficiency, as well as response to therapeutic intervention.
Given that CRTR deficiency and other CCDS produce a relatively nonspecific phenotype and require nonroutine yet specific biochemical evaluation to diagnose, it is likely CCDS are underdiagnosed, particularly CRTR deficiency which is more common.21 The concept of treatable IMDs presenting with seemingly nonspecific clinical features has been recognized, prompting development of clinical protocols such as the Treatable Intellectual Disability Endeavor (TIDE) in British Columbia.22 With a shortage of medical geneticists in an era of increased access to genetic testing, general pediatricians and other providers need to be provided tools and resources to systematically broaden their differential for indications such as FTT, GDD, ID, hypotonia, and seizures. It is important to note that CCDS are treatable9 and thus, early diagnosis is of significant clinical value as it allows for timely therapeutic intervention which can result in improved clinical outcomes, particularly in symptomatic females.23,24 In addition, there is meaningful benefit to providing families of children with an IMD a diagnosis and prognosis, as well as comprehensive genetic counseling as early as possible.
Newborn screening programs in the United States currently evaluate newborns for several IMDs; however, the screening varies by state and furthermore, most IMDs are not included in NBS.1,2 Thus a history of a negative NBS does not eliminate the possibility of an IMD in any child. With therapy available for CCDS, these disorders have been nominated for inclusion on the NBS Recommended Uniform Screening Panel24,25; however, currently, only Utah, Michigan, and New York include screening for GAMT deficiency.24 Newborn screening for GAMT deficiency in these states has identified affected newborns, allowing initiation of therapy by 2 weeks of age with subsequent report of normal growth and development.23 In New York, the identification of a newborn affected with GAMT deficiency not only enabled timely diagnosis and therapeutic intervention for the infant proband, but also lead to the diagnosis and initiation of therapy in the proband’s 6-year-old sibling who had developmental delay, absent speech, and hypotonia of previously unknown cause.23 As broad molecular genetic testing becomes more accessible, it is likely more individuals with CRTR deficiency and other CCDS will be diagnosed; however, diagnoses made through this approach will likely occur after the onset of clinical manifestations.
In conclusion, CRTR deficiency should be considered in all individuals presenting with FTT and abnormal neurodevelopmental features, particularly if creatinine level is low. If seizures are present, the suspicion for an IMD, including CRTR deficiency, should be further heightened, but a lack of seizures should not exclude this disorder from one’s differential diagnosis. Laboratory evaluation for CRTR deficiency and other CCDS includes the measurement and calculated ratios of urine guanidinoacetate, creatine, and creatinine; if obtaining brain imaging, consider including brain MRS for measurement of creatine and other metabolites. The immense value and benefit of providing families of children with an IMD, such as CRTR deficiency, a diagnosis, prognosis, therapeutic options, and comprehensive genetic counseling as early as possible is immeasurable.
Acknowledgments
The authors thank the individuals and families mentioned in this report, members of the Stanford Biochemical Genetics Laboratory who assisted with this diagnosis, and Dr. Bryan Lanzman for providing radiologic expertise.
Footnotes
Authors’ Contributions: C.G.T. drafted the manuscript, obtained, analyzed, and interpreted the patient data. M.J.P. developed the table and figures, obtained, analyzed, and interpreted the patient data. K.P.C.O. analyzed and interpreted the patient data and provided expertise content knowledge. D.R.M. obtained, analyzed, and interpreted the patient data and provided expertise content knowledge. All authors read and approved the final manuscript.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics Approval: Our institution does not require ethical approval for reporting individual cases or case series.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed Consent for Publication: Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.
ORCID iD: Christina G. Tise  https://orcid.org/0000-0002-6227-7895
https://orcid.org/0000-0002-6227-7895
References
- 1. Watson MS, Lloyd-Puryear MA, Mann MY, et al. Main report. Genet Med. 2006;8:1S-252S. [Google Scholar]
- 2. Health Resources & Services Administration. Recommended uniform screening panel. Published 2017. Accessed February 3, 2021. https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/index.html
- 3. Mercimek-Andrews S, Salomons GS. Creatine deficiency disorders. In: Adam MP, Everman DB, Mirzaa GM, et al. eds. GeneReviews ®. Seattle, WA: University of Washington; 1993. Accessed October 6, 2022. http://www.ncbi.nlm.nih.gov/books/NBK3794/ [PubMed] [Google Scholar]
- 4. Stöckler-Ipsiroglou S, Mercimek-Mahmutoglu S, Salomons GS. Creatine deficiency syndromes. In: Saudubray JM, Baumgartner MR, Walter J, eds. Inborn metabolic Diseases: Diagnosis and Treatment. Berlin: Springer:243-248. [Google Scholar]
- 5. Mercimek-Andrews S, Salomons GS. Creatine deficiency syndromes. In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews ®. Seattle, WA: University of Washington; 1993. Accessed May 6, 2021. http://www.ncbi.nlm.nih.gov/books/NBK3794/ [PubMed] [Google Scholar]
- 6. van de Kamp JM, Mancini GMS, Pouwels PJW, et al. Clinical features and X-inactivation in females heterozygous for creatine transporter defect. Clin Genet. 2011;79(3):264-272. [DOI] [PubMed] [Google Scholar]
- 7. van de Kamp JM, Betsalel OT, Mercimek-Mahmutoglu S, et al. Phenotype and genotype in 101 males with X-linked creatine transporter deficiency. J Med Genet. 2013;50:463-472. [DOI] [PubMed] [Google Scholar]
- 8. Bahl S, Cordeiro D, MacNeil L, et al. Urine creatine metabolite panel as a screening test in neurodevelopmental disorders. Orphanet J Rare Dis. 2020;15:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. UpToDate. Congenital disorders of creatine synthesis and transport. Date unknown. Accessed September 18, 2021. https://www.uptodate.com/contents/congenital-disorders-of-creatine-synthesis-and-transport?search=creatine%20deficiency&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1#H1472866778
- 10. Dunbar M, Jaggumantri S, Sargent M, et al. Treatment of X-linked creatine transporter (SLC6A8) deficiency: systematic review of the literature and three new cases. Mol Genet Metab. 2014;112(4):259-274. [DOI] [PubMed] [Google Scholar]
- 11. Shi K, Zhao H, Xu S, et al. Treatment efficacy of high-dose creatine supplementation in a child with creatine transporter (SLC6A8) deficiency. Mol Genet Genomic Med. 2021;9(4):e1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bruun TUJ, Sidky S, Bandeira AO, et al. Treatment outcome of creatine transporter deficiency: international retrospective cohort study. Metab Brain Dis. 2018;33(3):875-884. [DOI] [PubMed] [Google Scholar]
- 13. Rabago J, Marra K, Allmendinger N, et al. The clinical geneticist and the evaluation of failure to thrive versus failure to feed. Am J Med Genet C Semin Med Genet. 2015;169(4):337-348. [DOI] [PubMed] [Google Scholar]
- 14. Natsheh J, Abu-Libdeh B, Abu-Libdeh A. Growth hormone deficiency in congenital toxoplasmosis. JCS. 2018;08:e43-e45. [Google Scholar]
- 15. Congenital CMV Outcomes; National CMV Foundation. https://www.nationalcmv.org/overview/outcomes
- 16. Amos-Kroohs RM, Fink BA, Smith CJ, et al. Abnormal eating behaviors are common in children with fetal alcohol spectrum disorders. J Pediatr. 2016;169:194-200.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miller JS, Thomas RP, Bennett A, et al. Early indicators of creatine transporter deficiency. J Pediatr. 2019;206:283-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. National Institutes of Health. ClinVar (VCV000011698.14). Date unknown. Accessed February 1, 2022. https://www.ncbi.nlm.nih.gov/clinvar/variation/11698/?new_evidence=false
- 19. van de Kamp JM, Mancini GM, Salomons GS. X-linked creatine transporter deficiency: clinical aspects and pathophysiology. J Inherit Metab Dis. 2014;37(5):715-733. [DOI] [PubMed] [Google Scholar]
- 20. Colas C, Banci G, Martini R, et al. Studies of structural determinants of substrate binding in the creatine transporter (CreaT, SLC6A8) using molecular models. Sci Rep. 2020;10:6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sharer JD, Bodamer O, Longo N, et al. Laboratory diagnosis of creatine deficiency syndromes: a technical standard and guideline of the American College of Medical Genetics and Genomics. Genet Med. 2017;19(2):256-263. [DOI] [PubMed] [Google Scholar]
- 22. van Karnebeek CD, Stockler-Ipsiroglu S. Early identification of treatable inborn errors of metabolism in children with intellectual disability: the Treatable Intellectual Disability Endeavor protocol in British Columbia. Paediatr Child Health. 2014;19(9):469-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hart K, Rohrwasser A, Wallis H, et al. Prospective identification by neonatal screening of patients with guanidinoacetate methyltransferase deficiency. Mol Genet Metab. 2021;134(1-2):60-64. [DOI] [PubMed] [Google Scholar]
- 24. Association for Creatine Deficiencies. Newborn screening. Date unknown. Accessed January 29, 2022. https://creatineinfo.org/nbs/
- 25. Health Resources & Services Administration. Previously nominated conditions. Published 2017. Accessed January 29, 2022. https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/previous-nominations.html