

Abstract
Hepatocellular carcinoma (HCC) is one of the most malignant tumors worldwide and HCC patients often develop drug resisitene. Long non-coding RNAs (LncRNAs) are closely related to cell cycle, growth, development, differentiation, and apoptosis. Abnormally expressed lncRNAs have been proved to mediate drug resistance in tumor cells. However, the effect of LIMT on drug resistance has not been explored in HCC. In this study, we explored the effect of long non-coding RNA LIMT on drug resistance and its underlying mechanism in hepatocellular carcinoma (HCC). Our results showed that LncRNA LINC01089 (LIMT) expression is downregulated in 78.57% (44/56) of 56 HCC tumor tissue samples. LIMT expression is also downregulated in HCC cells compared with that in normal liver LO2 cells. Inhibition of LIMT increases the resistance to sorafenib and promotes cell invasion via regulation of epithelial to mesenchymal transition (EMT) in HCC. StarBase V3.0 was used to predict the potential binding site of miR-665 in LIMT. Furthermore, miR-665 participates in sorafenib resistance and also regulates the level of EMT-related proteins in HCC cells. A rescue experiment demonstrated that silencing of LIMT eliminats the inhibitory effect of the miR-665 inhibitor on sorafenib resistance in HCC cells. Taken together, our findings revealed that downregulation of LIMT increases the resistance of HCC to sorafenib via miR-665 and EMT. Therefore, LIMT, which serves as a therapeutically effective target, will provide new hope for the treatment of HCC.
Keywords: LncRNA LIMT, sorafenib, chemoresistance, EMT, HCC
Introduction
Hepatocellular carcinoma (HCC) is one of the most pernicious tumors worldwide [1]. Because of the high rate of recurrence and metastasis, more and more patients are diagnosed with HCC every year and also with a poor prognosis [2]. Sorafenib, which acts as a multiple kinase inhibitor, is the first-line drug for the treatment of HCC in many countries [3]. Although sorafenib can extend HCC patients’ survival time, its curative effect is short because of the development of drug-resistant cells. Some patients with HCC are not only resistant to the long-term effects of sorafenib, but also show resistance in the early stage [4]. Thus, it is urgent to reveal the detailed mechanisms of sorafenib resistance in HCC.
Long non-coding RNAs (lncRNAs> 200 nucleotides) are closely related to a series of physiological activities, such as cell cycle, growth, development, differentiation, and apoptosis. Several abnormally expressed lncRNAs have been proved to mediate drug resistance in tumor cells. For example, inhibition of HOTAIR could decrease drug resistance in nonsmall-cell lung cancer (NSCLC) by regulating the expression of Unc-51 like autophagy activating kinase 1 (ULK1) [5]. Mechanistically, lncRNAs and microRNAs (miRNAs) can interact to mediate drug resistance and cell invasion in various cancer cells. For instance, silencing of SNHG16 was shown to increase chemotherapy resistance to sorafenib in HCC by upregulating miR-140-5p [6]. In addition, drug resistance was inhibited following transfection with a small interfering RNA (siRNA) targeting FOXD2-AS1 via the miR-98-5p/cytoplasmic polyadenylation element binding protein 4 (CPEB4) axis and the regulation of cell proliferation and invasion [7]. LncRNA LINC01089 (LIMT) functions as a metastasis inhibitory lncRNA, which was first discovered to be inhibited by epidermal growth factor (EGF) in breast cancer, and is related to cell migration and metastasis in different cancers [ 8, 9]. However, the effect of LIMT on drug resistance has not been explored so far in HCC.
The process of epithelial to mesenchymal transition (EMT) accompanied by epithelial cells is rendered into cells containing mesenchymal phenotypes via special steps [ 10, 11]. EMT is related to a great diversity of biological processes, such as embryonic development, tissue reconstruction, chronic inflammation, cancer metastasis, and drug resistance [ 12, 13]. Cancer cells generally acquire multi-drug resistance during EMT. Alternatively, EMT is closely associated with chemo-resistance to cisplatin and doxorubicin in HCC [ 14– 16]. In addition, evidence shows that EMT and its accompanying molecules are the key determinants of resistance to sorafenib therapy in HCC. Therefore, analysis of the molecular mechanism underlying EMT might help to improve drug resistance to achieve an anti-tumor effect.
This study intended to reveal the involvement of LIMT in sorafenib resistance and the potential underlying mechanism. It provides important evidence that LIMT may be a novel therapeutic target and a biomarker to predict the effect of sorafenib therapy in HCC.
Materials and Methods
Cell culture and the HCC human tissues
The human HCC cells (Huh7, Hep3B, and SNU449) were purchased from ATCC (Manassas, USA) and cultured separately in Dulbecco’s modified Eagle’s medium (DMEM), minimal essential medium (MEM), and Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco, Carlsbad, USA) supplemented with 10% FBS (Sigma, St Louis, USA) under a suitable environment (5% CO 2 at 37°C). The tumor tissue samples of HCC patients were obtained from Tongde Hospital of Zhejiang Province. All patients provided written informed consent and the study protocol was approved by the Clinical Research Ethics Committee of Tongde Hospital of Zhejiang Province.
Cell viability
Cell viability in different treatment groups was determined by Cell Counting Kit-8 (CCK-8) assay. HCC cell suspension (5×10 3 cells) was inoculated into 96-well plates (100 μl/well) and cultured at 37°C with 5% CO 2 until the cells adhered. Next, 10 μL of CCK-8 solution (Dojindo, Tokyo, Japan) was add into each well and incubated at 37°C for 2 h. Thereafter, the optical density (OD) value of each well was detected at 450 nm with a microplate reader.
Transfection
LIMT siRNAs (LIMT siRNA1:LIMT siRNA2:LIMT siRNA3=1:1:1), miR-665 siRNA, miR-665 mimics, miR-665 inhibitor and their corresponding negative controls were designed and manufactured by GenePharma (Shanghai, China). Transfections were performed using lipofectamine 2000 (Invitrogen, Carlsbad, USA) according to the manufacturer’s protocol. The sequences are shown in Table 1.
Table1 The siRNA sequences used in this study
|
Name |
Sequence |
|
LIMT-homo-177 |
Sense: 5′-CCAUUCAUGUCAGCAGUUATT-3′; Antisense: 5′-UAACUGCUGACAUGAAUGGTT-3′ |
|
LIMT-homo-295 |
Sense: 5′-GCAGAACGUGAGGGUGUAATT-3′ Antisense: 5′-UUACACCCUCACGUUCUGCTT-3′ |
|
LIMT-homo-888 |
Sense: 5′-GCUUCCAACCUCCAUUGCATT-3′; Antisense: 5′-UGCAAUGGAGGUUGGAAGCTT-3′ |
|
miR-665 mimic |
Sense: 5′-ACCAGGAGGCUGAGGCCCCU-3′; Antisense: 5′-GGGCCUCAGCCUCCUGGUUU-3′ |
|
miR-665 inhibitor |
5′-AGGGGCCUCAGCCUCCUGGU-3′; |
Western blot analysis
The concentrations of extracted protein from different treatment groups were measured by bicinchoninic acid (BCA) protein assay. Before separation by SDS-PAGE, equal amounts of protein (40 μg) were denatured by heating with loading buffer at 100°C. The separated proteins on the gel were electro-transferred onto polyvinylidene fluoride (PVDF) membranes. After being blocked with 5% nonfat dry milk in TBS-T buffer for 2 h at 37°C, the membranes were incubated with indicated primary antibodies, including anti-fibronectin 1 (FN-1) antibody, anti-E-cadherin antibody, anti-vimentin antibody and anti-GAPDH antibody (1:1000 dilution; Abcam, Cambridge, UK), overnight at 4°C. Then the membranes were washed and incubated with the corresponding HRP-conjugated secondary antibodies (1:2000 dilution; Cell Signaling Technology, Beverly, USA). The immunoreactive protein bands were visualized using an ECL kit (GE Healthcare, Piscataway, USA). GAPDH was used as the loading control.
qRT-PCR analysis
Total RNA was extracted from HCC cells using Trizol reagent (Invitrogen) and then reverse transcribed to cDNA using a Reverse Transcription kit (TaKaRa, Tokyo, Japan). Utilizing SYBR Premix Ex Taq (TaKaRa), qRT-PCR was run and analyzed on the Applied Biosystems Real-time PCR System (Foster City, USA). GAPDH and U6were used as reference controls for the lncRNA, mRNA, and miRNA. The primers used were as follows: LIMT forward primer: 5′-CGAATGGACAATCTTTCCTTCTGTC-3′, reverse primer: 5′-GCTAGAGGTTGAGGGCCTGAGT-3′; and miR-665 forward primer: 5′-ACCAGGAGGCTGAGGC-3′, reverse primer: 5′-GAACATGTCTGCGTATCTC-3′; GAPDH forward primer: 5′-CGGAGTCAACGGATTTGGTCGTAT-3′ reverse primer: 5′-AGCCTTCTCCATGGTGGTGAAGAC-3′; and U6 forward primer: 5′-GCTTCGGCAGCACATATACTAAAAT-3′, reverse primer: 5′-CGCTTCACGAATTTGCGTGTCAT-3′.
Dual-luciferase reporter assays
Reporter plasmids containing the LIMT-3′ UTR, which contained the mutant (MUT) or wild-type (WT) binding sequence for miR-665, were constructed by GenePharma. The plasmids were then transiently co-transfected into cells with luciferase reporter vectors together with the miR-665-mimic, miR-665 inhibitor, or control using Lipofectamine 2000. Forty-eight hours after transfection, the relative luciferase activity of the WT or MUT LIMT-3′-UTR was measured using a dual-luciferase reporter assay kit (Promega, Madison, USA).
Ethynyl deoxyuridine (EdU) assay
Cell proliferation was determined using a Click-iT ® EdU Imaging kit according to the instructions of the manufacturer (Invitrogen). Briefly, HCC cells were seeded in 96-well plates at a density of 3×10 3 cells/well in growth media. The medium was replaced by serum-free medium to synchronize the cells. After 24 h of incubation, the serum-free medium was replaced by growth media with the indicated concentrations and cuktured for 48 h. After incubation with 20 μM EdU for 3 h, Cell nuclei were stained with Hoechst 33342 (Invitrogen) at a concentration of 5 μg/mL for 30 min. The percentage of positive cells in five random fields of view per slide was determined under an inverted fluorescence microscope (Olympus, Tokyo, Japan) and expressed relative to that in the untreated control cells.
Cell cycle and apoptosis analysis
Cell cycle and apoptosis analysis were performed by flow cytometry using Cell Cycle Assay kit-PI/RNase Staining and Annexin V/FITC Apoptosis Detection kit (Roche, Basel, Switzerland) according to manufacturer’s instructions. Cell cycle analysis was carried out by Cell Cycle Assay Kit-PI/RNase Staining. Briefly, the cell density was adjusted to 2×10 6 cells/mL, and 1 mL of cell suspension was added into a 1.5-mL microtube and centrifuged at 1000 g for 3 min. Then the supernatant was discarded and 1 mL of 70% ethanol (–20°C) was added to the cell pellet to disperse the cells by votexing and standing at 4°C for 2 h. After centrifugation at 1000 g for 3 min, the ethanol was removed and 1 mL PBS buffer was added to wash the cells. After centrifugation at 1000 g for 3 min, the supernatant was discard and 0.5 mL of Working Solution (500 μL assay buffer, containing 25 μL PI solution and 2.5 μL RNase solution) was added. After vortexing and incubation for 30 min at 37°C in the dark, the mixture was further incubated for 30 min at 4°C in the dark, followed by vortexing and filtering through a nylon mesh to remove cell clumps in the sample. Finally, cell cycle was detected by flow cytometry.
Cell apoptosis analysis was carried out using Annexin V/FITC Apoptosis Detection kit. Briefly, HCC cells (treated as above) were harvested by trypsinization, rinsed with ice-cold phosphate PBS, and centrifuged to remove the supernatant. Then, the cells were resuspended in 100 μL 1× binding buffer and incubated with Annexin V-FITC for 15 min in the dark at room temperature. Finally, a flow cytometer was used to determine the number of apoptotic cells from B2 and B4 quadrants.
Transwell assay
HCC cell suspension (100 μL of 2.5×10 4 cells/μL) was added into the upper Transwell chamber (Corning Costar, New York, USA) coated with Matrigel (BD, Franklin Lakes, USA). After 48 h of incubation, the membranes were fixed with methanol for 30 min and stained with 0.1% crystal violet for 20 min. Then the infiltrated cells were counted and photographed in five random fields under a microscope (Olympus) at a magnification of 100×.
Nude mouse xenograft model
Female BALB/c nu/ nu mice (4–5 weeks old) were purchased from Shanghai SLAC Laboratory Animal Co., Ltd (Shanghai, China). Huh7 cells were subcutaneously injected into the left hip of three mice. After the tumor was formed, a small section (1 mm 3) of tumor tissue was inoculated into the experimental group nude mice. After 10 days, the tumors had a diameter of 0.5 cm and reached a volume of ~50–100 mm 3. The mice were randomly divided into four groups ( n=3 per group): control, sorafenib group, LIMT plasmid group, and LIMT plasmid + sorafenib group. LIMT plasmid was injected intratumorally four times from day 0 to day 14, while sorafenib was injected into the tail vein once every two days for two weeks. Tumor volumes were recorded every two days and body weight was measured daily. The tumor volume (mm 3) was calculated using the formula: Volume=(length×width 2/2). The mice were sacrificed humanely on day 15 after treatment, and the resected tumors were weighed and subject to subsequent analysis.
TUNEL analysis
The apoptosis in paraffin-embedded mouse tissue sections (5-mm) was determined by an In situ cell death detection kit (Roche, Basel, Switzerland). The apoptotic cells were observed under a light microscope (Olympus). The assay was independently repeated three times. The positive rates were measured using IPP 6.0 software (Media Cybernetics, Bethesda, USA).
Statistical analysis
GraphPad software 8 (GraphPad Inc., San Diego, USA) was used for calculating statistical comparisons. Data were shown as the mean±SD and analyzed by two-tailed Student’s t-test. A value of P≤0.05 was considered to be significantly different. StarBase 3 ( http://starbase.sysu.edu.cn/index.php) was used to analyze the relationship between LIMT and miR-665.
Results
The level of LIMT is associated with sorafenib resistance
To verify the relationship between the expression level of LIMT and sorafenib resistance, qRT-PCR was performed to detect LIMT expression in 56 pairs of HCC tumor tissues and matched adjacent tissues. The results showed that LIMT expression was downregulated in 78.57% (44/56) HCC tumor tissue ( Figure 1 A,B). We also investigated the relationship between LIMT expression level and clinical characteristics in the 33 HCC cancer cases to determine whether LIMT expression is related to clinical features. The results showed that low expression of LIMT in 33 HCC patients was closely related to large tumor size ( P=0.0270), TNM stage ( P=0.0173), and HBsAg ( P=0.0221). However, LIMT expression level was not correlated with other parameters, such as patient age, sex, alpha-fetoprotein (AFP), and portal vein tumor thrombus ( Table 2). Next, the expression of LIMT was examined in HCC cells and normal liver epithelial LO2 cells, which showed that LIMT expression was reduced in HCC cells ( Figure 1C). Furthermore, we found that the SNU449 cell line, with the lowest LIMT expression, was most resistant to sorafenib. The IC 50 values for sorafenib in HCC cells are presented in Figure 1D,E.
Figure1 .

The level of LIMT is assiciated with sorafenib resistance(A,B) LIMT expression was detected in 56 pairs of HCC tumor tissues and matched adjacent tissues. **P<0.01. (C) LIMT expression was determined in HCC cells and normal liver LO2 cells. *P<0.05, **P<0.01, and ***P<0.001. (D,E) Cell viability was examined by CCK-8 assay after treatment with sorafenib, and the IC50 values are represented by a bar chart.
Table2 Correlations between LIMT and clinical characteristics of 33 HCC cancer patients
|
Clinical parameter |
n=33 |
High |
Low |
P-value |
|
|
Sex |
Female |
9 |
6 |
3 |
0.1340 |
|
Male |
24 |
9 |
15 |
|
|
|
Age (year) |
<60 |
19 |
10 |
9 |
0.5787 |
|
≥60 |
14 |
6 |
8 |
|
|
|
Stage |
I–II |
19 |
12 |
7 |
0.0173* |
|
III–IV |
14 |
3 |
11 |
|
|
|
Tumor size (cm 3) |
<3 |
13 |
9 |
4 |
0.0270* |
|
≥3 |
20 |
6 |
14 |
|
|
|
AFP |
<20 |
16 |
8 |
8 |
0.6109 |
|
≥20 |
17 |
7 |
10 |
|
|
|
Portal vein tumor thrombus |
Yes |
7 |
4 |
3 |
0.8764 |
|
No |
26 |
14 |
12 |
|
|
|
HBsAg |
Yes |
16 |
4 |
12 |
0.0221* |
|
No |
17 |
11 |
6 |
|
AFP, alpha-fetoprotein.
Interference with LIMT enhances sorafenib resistance and promotes invasion by promoting EMT
To further reveal the chemoresistance effect of LIMT on sorafenib, HCC cells were transfected with LIMT siRNAs (LIMT siRNA1:LIMT siRNA2:LIMT siRNA3=1:1:1) and observed the changes of cell viability and proliferation. As shown in Figure 2, LIMT silencing increased the resistance to sorafenib by enhancing cell viability and proliferation, while LIMT plasmid treatment decreased this effect in HCC cells ( Figure 2A,B and Supplementary Figure S1A). Transwell analysis was then used to determine the invasion ability of HCC cells. Compared with cells treated with sorafenib alone, the number of HCC cells that invaded the Matrigel layer was increased after transfection with LIMT siRNAs ( Figure 2C). EMT was also found to play an important role in HCC resistance to sorafenib and cell invasion in a variety of cancers [ 17– 19]. Western blot analysis of the EMT-related proteins, including FN-1, E-cadherin, and Vimentin, showed that LIMT silencing promoted the sorafenib-induced reduction in E-cadherin, whereas it upregulated sorafenib-induced increase in the expressions of FN-1, Vimentin and Twist 1 ( Figure 2D). The phenotypic changes was shown in Supplementary Figure S3A. Furthermore, flow cytometry analysis showed that sorafenib treatment could increase cell apoptosis and enhance the rate of G2/S. However, LIMT siRNA combined with sorafenib could reduce cell apoptosis and the rate of G2/S compared with those in the sorafenib group ( Supplementary Figure S1B,C). The cell interference efficiency of LIMT was shown in Supplementary Figure S2A–C.
Figure2 .

Interference with LIMT enhances sorafenib resistance and promotes invasion via regulation of EMT(A,B) CCK-8 and EdU determination of the cell viability and proliferation of HCC cells following transfection with LIMT siRNAs (LIMT siRNA 1, LIMT siRNA 2, LIMT siRNA 3) and combined with or without sorafenib. **P<0.01 and ***P<0.001. (C) Transwell invasion analysis of HCC cells transfected with LIMT siRNA combined with sorafenib. *P<0.05, **P<0.01, and ***P<0.001. (D) Western blot analysis of FN-1, E-cadherin, Vimentin and Twist 1 expression levels in HCC cells.
LIMT negatively regulates miR-665 expression
To further explore the mechanisms underlying the effect of LIMT, the potential miRNAs binding site in LIMTwas identified from multidimensional sequencing data using StarBase V3.0 and the reporter plasmids containing the LIMT-3′UTR were further constructed, which contained the mutant (MUT) or wild-type (WT) binding sequences to verify the interaction between LIMT and miR-665 by Dual-Luciferase Reporter Assays ( Figure 3A,B). Moreover, we detected the levels of miR-665, E-cadherin and Vimentin in HCC tumor tissues and matched adjacent tissues, and the results revealed that E-cadherin was donw-regulated, and the expressions of miR-665 and Vimentin were increased in HCC tumor tissues ( Figure 3C), indicating that LIMT was negative-correlated with the level of miR-665 ( Figure 3C), which is consistant with results from starBase v3.0 ( Supplementary Figure S2D). The level of miR-665 was significantly increased after LIMT siRNA transfection, while its expression was downregulated after treatment with LIMT plasmid ( Figure 3D). Transfection with miR-665 mimics reduced LIMT level, while the miR-665 inhibitor upregulated LIMT level ( Figure 3E). These data suggest that LIMT level is inversely correlated with the miR-665 level in HCC cells.
Figure3 .

LIMT is negatively correlated with miR-665 expression(A) StarBase V3.0 was used to predict the potential binding site for miR-665 in LIMT. (B) Relative luciferase activity of HCC cells co-transfected with WT-LIMT or Mut-LIMT and NC, or miR-665 mimics. *P<0.05. (C) The levels of miR-665 and EMT-related mRNAs were detected in HCC tumor tissues and matched adjacent tissues. The correlation between LIMT and miR-665 (**P<0.01 and ***P<0.001). (D) The level of miR-665 in HCC cells treating with LIMT siRNA or the LIMT overexpressing plasmid was determined by qRT-PCR. **P<0.01 and ***P<0.001. (E) LIMT expression was detected after incubation with miR-665 mimics or inhibitors. **P<0.01 and ***P<0.001.
miR-665 regulates resistance to sorafenib
To investigate the functions of miR-665 in HCC cells, we also examined resistance to sorafenib and cell invasion after treatment with miR-665 mimics, inhibitor, with or without sorafenib. The results showed that miR-665 mimics increased sorafenib resistance, whereas the miR-665 inhibitor enhanced sorafenib sensitivity ( Figure 4A). EdU analysis also confirmed these results ( Figure 4B). The number of HCC cells passing through the membrane was increased after miR-665 mimic treatment, indicating that miR-665 mimics could enhance the invasion capability of HCC cells ( Figure 4C). To reveal whether miR-665 participates in the EMT process, we determined the levels of EMT-related proteins. Compared with the sorafenib group, miR-665 mimics combined with sorafenib could downregulate E-cadherin level and increase FN-1, Vimentin, Twist 1 expressions. However, miR-665 inhibitor plus sorafenib could reverse these effects ( Figure 4D). The phenotypic changes are shown in Supplementary Figure S3B.
Figure4 .

MiR-665 regulates sorafenib resistance in HCC cells(A,B) CCK-8 assay and EdU analysis were used to determin cell viability and proliferation of HCC cells exposed to sorafenib following miR-665 mimics or inhibitors treatment. **P<0.01 and ***P<0.001. (C) The invasion ability of HCC cells transfected with miR-665 mimics or inhibitors combined with sorafenib was confirmed by Transwell invasion analysis. *P<0.05, **P<0.01, and ***P<0.001. (D) Western blot analysis of FN-1, E-cadherin, Vimentin and Twist 1 levels in HCC cells.
LIMT regulates sorafenib resistance through regulating miR-665 and EMT
To verify whether LIMT plays the role in sorafenib resistance via the regulation of miR-665 and EMT, we transfected the miR-665 inhibitor into HCC cells along with the LIMT siRNAs, followed by treatment with sorafenib to perform a rescue experiment. As expected, the results showed that the LIMT siRNA inhibited the increase of miR-665 inhibitor-induced sorafenib sensitivity, and showed no significant difference between the control group and silencing of LIMT plus the miR-665 inhibitor group ( Figure 5A –C). MiR-665 inhibitor combined with LIMT siRNA could reduce the invasion ability compared with LIMT siRNA treatment alone ( Figure 5D). Western blot analysis also confirmed that miR-665 inhibitor combined with LIMTsilencing could inhibit EMT compared with LIMT siRNA treatment alone ( Figure 5E). These results indicated that inhibition of LIMT might enhance chemotherapy resistance to sorafenib by upregulating the effect of miR-665.
Figure5 .

LIMT could regulate sorafenib resistance through regulating miR-665 and EMT(A–C) HCC cells were treated with the LIMT siRNA alone or combined with miR-665 inhibitor, following treatment with sorafenib, and cell viability was examined by CCK-8 assay. (D) The rate of invasive HCC cells treated with LIMT siRNA alone or combined with miR-665 inhibitor following treatment with sorafenib were determined by Transwell invasion analysis. **P<0.01 and ***P<0.001. (E) The levels of FN-1, E-cadherin, Vimentin and Twist 1 proteins were determined by western blot analysis.
LIMT over-expression suppresses tumor growth in vivo
To explore the effect of LIMT on sorafenib resistance in vivo, tumor nude mouse model was established and treated with normal saline alone, sorafenib alone, LIMT over-expression alone, or sorafenib plus LIMT over-expression. The results showed that the tumor volume was significantly reduced in the sorafenib plus LIMT over-expression group ( Figure 6A,B). TUNEL analysis revealed that sorafenib plus LIMT over-expression treatment could enhance tumor cell apoptosis compared with that in other groups ( Figure 6C). Furthermore, the expressions of E-cadherin, FN-1 and Vimentin were determined, showing that compared with the sorafenib group, the level of E-cadherin was upregulated, and the expression levels of FN-1 and Vimentin were downregulated in the sorafenib plus LIMT over-expression group ( Figure 6D). These data indicated that up-regulation of LIMT could inhibit tumor growth in vivo.
Figure6 .

LIMT over-expression could suppress tumor growth in vivo (A,B) Growth curves of xenograft tumors and the tumor size treated with control, sorafenib, LIMT plasmid, or sorafenib plus LIMT plasmid (n = 3 per group). (C) TUNEL was used to analyze cell apoptosis in the treatment groups. (D) RT-qPCR was used to analyze the mRNA expression levels of E-cadherin, Vimentin and FN-1.
Discussion
Sorafenib resistance and distant metastasis are closely correlated with the poor prognosis in HCC patients. So, it is very important to reveal the underlying molecular mechanisms and explore novel therapeutic targets. Accumulated evidence indicates that lncRNAs regulate gene expression related to various processes, such as cell apoptosis, proliferation, and migration, suggesting that they may play a carcinogenic or tumor-suppressive role in cancers [20]. Furthermore, lncRNAs are involved in sorafenib resistance in various cancers [21]. LIMT, located on chromosome 12, was determined to be downregulated in cancers [ 9, 22]. It had been reported that over-expression of LIMT could inhibit the proliferation, migration, and invasion of NSCLC cells by regulating the miR-152-3p/PTEN axis [23]. Furthermore, it has inhibitory effects on cell migration and metastasis in a variety of cancer cells, such as gastric cancer, colorectal cancer, breast cancer, and cervical cancer [ 9, 22, 24, 25]. However, the role of LIMT in drug resistance has not been explored so far in HCC. Here, for the first time we explored the effect of LIMT on chemotherapy resistance in HCC. Our results verified that the level of LIMT was downregulated in HCC tumor tissue and HCC cells, implying that LIMT might play a role in suppressing cancers. As expected, knockdown of LIMT significantly increased sorafenib resistance and promoted cell invasion in HCC cells.
Evidence shows that some lncRNAs bind competitively to targeted miRNAs, thus acting as sponges of miRNA molecules. Silencing SNHG16 inhibitor increased chemotherapy resistance by upregulating miR-140-5p in HCC [6]. Knockdown of HOTAIRenhanced sorafenib resistance by upregulating miR-217 in HCC [26]. Our study verified that downregulation of LIMT promoted sorafenib resistance by promoting EMT. The regulatory relationship between lncRNAs and miRNAs is related to many cell biological processes [ 27, 28]. Thus, we further investigated the relationship between LIMT and miRNAs. We found that LIMT interacts directly with miR-665 to reduce its apparent level. Furthermore, several miRNAs have been proved to participate in sorafenib resistance in HCC. Upregulation of miR-223 could promote sorafenib resistance through regulation of FBW7 (encoding F-box and WD repeat domain containing 7) expression in HCC [29]. MiR-221 participates in sorafenib resistance [30]. Increasing evidence suggests that miR-665 is a tumor suppressor or cancer-promoting miRNA, which is expressed in tumor cells. For instance, miR-665 was reported to be a tumor-promoting factor in HCC. High miR-665 expression promotes cell metastasis, growth, and EMT of HCC and NSCLC cells [ 31– 33]. Consistent with these previous reports, in this study we found that miR-665 mimics could enhance sorafenib resistance and invasion capability of HCC cells. Moreover, LIMT silencing abolished the effect of the miR-665 inhibitor on sorafenib resistance in HCC cells. Thus, LIMT could regulate HCC cell sorafenib resistance.
Recently, EMT has been found to be closely associated with the progress of chemoresistance, and reversing the formation of EMT could reduce chemotherapy resistance in various cancers, indicating that EMT is an effective therapeutic target for cancer metastasis and chemotherapy resistance [ 34, 35]. It has been reported that TGF-β1/ROCK signaling pathway is involved in vasculogenic mimicry formation by inducing EMT [36]. For instance, a previous study demonstrated that H19 could promote paclitaxel resistance by adjusting miR-340 expression and EMT in breast cancer cells [37]. Upregulation of miR-206 could inhibit cell proliferation, migration and promote apoptosis in HCC cells by modulating cMET expression [38]. Over-expression of miR-4458 could also inhibit the migration, invasion, and EMT of HCC cells by suppressing the TGF-β signaling [39]. Our study also revealed that inhibition of LIMT could affect sorafenib resistance and promote cell invasion by regulating EMT. EMT-induced drug resistance in cancer cells involves many typical signaling pathways, such as Wnt/β-catenin, nuclear factor kappa B (NF-κB), and phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/protein kinase B (AKT) [ 40– 42]. Therefore, the relationship between the LIMT and EMT-related signaling pathways still needs further investigation in the future.
In summary, the present study revealed that regulation of LIMT is a mechanism involved in the resistance to sorafenib directly through regulating miR-665 and EMT in HCC cells ( Figure 7). Our results demonstrated that down-regulation of LIMTcould enhance sorafenib resistance and promote EMT by regulation of miR-665, suggesting that LIMT might serve as a molecular target for sorafenib resistance detection treatment and prognosis in HCC by regulating the miR-665/EMT axis.
Figure7 .
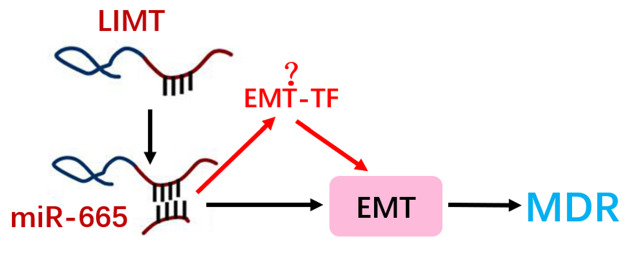
The mechanism of the LIMT in regulating resistance to sorafenib of HCC cellsSchematic diagram of the regulatory mechanism of the LIMT/miR-665/EMT axis in regulating resistance to sorafenib of HCC cells.
Supporting information
Supplementary Data
Supplementary data is available at Acta Biochimica et Biophysica Sinicaonline.
COMPETING INTERESTS
The authors declare that they have no conflict of interest.
Funding Statement
This work was supported by the grants from the Medical and Health Research Project of Zhejiang Province (No. 2018KY126 to L.Y.), Wenzhou Municipal Science and Technology Bureau (No. Y20170095 to L.Y.), and Zhejiang Provincial Natural Science Foundation of China (No. LQ19H280008 to X.Z.).
References
- 1.Gupta M, Chandan K, Sarwat M. Role of microRNA and long non-coding RNA in hepatocellular carcinoma. Curr Pharm Des. . 2020;26:415–428. doi: 10.2174/1381612826666200115093835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei L, Wang X, Lv L, Liu J, Xing H, Song Y, Xie M, et al. The emerging role of microRNAs and long noncoding RNAs in drug resistance of hepatocellular carcinoma. Mol Cancer. . 2019;18:147. doi: 10.1186/s12943-019-1086-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keating GM. Sorafenib: a review in hepatocellular carcinoma. Targ Oncol. . 2017;12:243–253. doi: 10.1007/s11523-017-0484-7. [DOI] [PubMed] [Google Scholar]
- 4.Mendez-Blanco C, Fondevila F, Garcia-Palomo A, et al. Sorafenib resistance in hepatocarcinoma: role of hypoxia-inducible factors. Exp Mol Med. 2018, 50: 1-9 . [DOI] [PMC free article] [PubMed]
- 5.Yang Y, Jiang C, Yang Y, Guo L, Huang J, Liu X, Wu C, et al. Silencing of LncRNA-HOTAIR decreases drug resistance of non-small cell lung cancer cells by inactivating autophagy via suppressing the phosphorylation of ULK1. Biochem Biophys Res Commun. . 2018;497:1003–1010. doi: 10.1016/j.bbrc.2018.02.141. [DOI] [PubMed] [Google Scholar]
- 6.Ye J, Zhang R, Du X, Chai W, Zhou Q. Long noncoding RNA SNHG16 induces sorafenib resistance in hepatocellular carcinoma cells through sponging miR-140-5p. Onco Targets Ther. . 2019;12:415–422. doi: 10.2147/OTT.S175176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu N, Wang X, Di Z, Xiong J, Ma Y, Yan Y’, Qian Y, et al. Silencing lncRNA FOXD2-AS1 inhibits proliferation, migration, invasion and drug resistance of drug-resistant glioma cells and promotes their apoptosis via microRNA-98-5p/CPEB4 axis. Aging. . 2019;11:10266–10283. doi: 10.18632/aging.102455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sas-Chen A, Aure MR, Leibovich L, Carvalho S, Enuka Y, Körner C, Polycarpou-Schwarz M, et al. LIMT is a novel metastasis inhibiting lnc RNA suppressed by EGF and downregulated in aggressive breast cancer . EMBO Mol Med. . 2016;8:1052–1064. doi: 10.15252/emmm.201606198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li M, Guo X. LINC01089 blocks the proliferation and metastasis of colorectal cancer cells via regulating miR-27b-3p/HOXA10 axis. Onco Targets Ther. . 2020;13:8251–8260. doi: 10.2147/OTT.S256148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saitoh M. Involvement of partial EMT in cancer progression. J Biochem. . 2018;164:257–264. doi: 10.1093/jb/mvy047. [DOI] [PubMed] [Google Scholar]
- 11.Du B, Shim JS. Targeting epithelial–mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules. . 2016;21:965. doi: 10.3390/molecules21070965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aleksakhina SN, Kashyap A, Imyanitov EN. Mechanisms of acquired tumor drug resistance. BioChim Biophys Acta (BBA) - Rev Cancer. . 2019;1872:188310. doi: 10.1016/j.bbcan.2019.188310. [DOI] [PubMed] [Google Scholar]
- 13.Mittal V. Epithelial mesenchymal transition in tumor metastasis. Annu Rev Pathol Mech Dis. . 2018;13:395–412. doi: 10.1146/annurev-pathol-020117-043854. [DOI] [PubMed] [Google Scholar]
- 14.Zhang X, Chen J, Jiang S, He S, Bai Y, Zhu L, Ma R, et al. N-Acetyltransferase 10 enhances doxorubicin resistance in human hepatocellular carcinoma cell lines by promoting the epithelial-to-mesenchymal transition. Oxid Med Cell Longev. . 2019;2019:1–14. doi: 10.1155/2019/7561879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Lu Y, Zhang C, Huang D, Wu W, Zhang Y, Shen J, et al. FSCN‑1 increases doxorubicin resistance in hepatocellular carcinoma through promotion of epithelial-mesenchymal transition. Int J Oncol. . 2018;52:1455. doi: 10.3892/ijo.2018.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bao Y, Zhang Y, Lu Y, Guo H, Dong Z, Chen Q, Zhang X, et al. Overexpression of microRNA-9 enhances cisplatin sensitivity in hepatocellular carcinoma by regulating EIF5A2-mediated epithelial-mesenchymal transition. Int J Biol Sci. . 2020;16:827–837. doi: 10.7150/ijbs.32460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu N, Zhang X, Bao Y, Yu H, Jia D, Ma C. Down‐regulation of GAS5 ameliorates myocardial ischaemia/reperfusion injury via the miR‐335/ROCK1/AKT/GSK‐3β axis. J Cell Mol Med. . 2019;23:8420–8431. doi: 10.1111/jcmm.14724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M, Duan L, Li Y, Liu B. Long noncoding RNA/circular noncoding RNA–miRNA–mRNA axes in cardiovascular diseases. Life Sci. . 2019;233:116440. doi: 10.1016/j.lfs.2019.04.066. [DOI] [PubMed] [Google Scholar]
- 19.Ma J, Zeng S, Zhang Y, Deng G, Qu Y, Guo C, Yin L, et al. BMP4 promotes oxaliplatin resistance by an induction of epithelial-mesenchymal transition via MEK1/ERK/ELK1 signaling in hepatocellular carcinoma. Cancer Lett. . 2017;411:117–129. doi: 10.1016/j.canlet.2017.09.041. [DOI] [PubMed] [Google Scholar]
- 20.Sanchez Calle A, Kawamura Y, Yamamoto Y, Takeshita F, Ochiya T. Emerging roles of long non-coding RNA in cancer. Cancer Sci. . 2018;109:2093–2100. doi: 10.1111/cas.13642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai Y, Feng B, Abudoureyimu M, Zhi Y, Zhou H, Wang T, Chu X, et al. Non-coding RNAs: emerging regulators of sorafenib resistance in hepatocellular carcinoma. Front Oncol. . 2019;9:1156. doi: 10.3389/fonc.2019.01156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan H, Qin Y, Zeng B, Feng Y, Li Y, Xiang T, Ren G. Long noncoding RNA LINC01089 predicts clinical prognosis and inhibits cell proliferation and invasion through the Wnt/β-catenin signaling pathway in breast cancer. Onco Targets Ther. . 2019;12:4883–4895. doi: 10.2147/OTT.S208830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang H, Zhang H, Li X, Huang S, Guo Q, Geng D. LINC01089 functions as a ceRNA for miR-152-3p to inhibit non‐small lung cancer progression through regulating PTEN. Cancer Cell Int. . 2021;21:143. doi: 10.1186/s12935-021-01846-7. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Guo X, Li M. LINC01089 is a tumor-suppressive lncRNA in gastric cancer and it regulates miR-27a-3p/TET1 axis. Cancer Cell Int. . 2020;20:507. doi: 10.1186/s12935-020-01561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li S, Han Y, Liang X, Zhao M. LINC01089 inhibits the progression of cervical cancer via inhibiting miR‐27a‐3p and increasing BTG2. J Gene Med. . 2021;23:e3280. doi: 10.1002/jgm.3280. [DOI] [PubMed] [Google Scholar]
- 26.Tang X, Zhang W, Ye Y, Li H, Cheng L, Zhang M, Zheng S, et al. LncRNA HOTAIR contributes to sorafenib resistance through suppressing miR-217 in hepatic carcinoma. Biomed Res Int. . 2020;2020:1–10. doi: 10.1155/2020/9515071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L, Cho KB, Li Y, Tao G, Xie Z, Guo B. Long Noncoding RNA (lncRNA)-mediated competing endogenous RNA networks provide novel potential biomarkers and therapeutic targets for colorectal cancer. Int J Mol Sci. . 2019;20:5758. doi: 10.3390/ijms20225758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luan X, Wang Y. LncRNA XLOC_006390 facilitates cervical cancer tumorigenesis and metastasis as a ceRNA against miR-331-3p and miR-338-3p. J Gynecol Oncol. . 2018;29:e95. doi: 10.3802/jgo.2018.29.e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang X, Yang W, Shu Z, Shen X, Zhang W, Cen C, Cao L, et al. MicroRNA‑223 promotes hepatocellular carcinoma cell resistance to sorafenib by targeting FBW7. Oncol Rep. . 2019;41:1231. doi: 10.3892/or.2018.6908. [DOI] [PubMed] [Google Scholar]
- 30.Fornari F, Pollutri D, Patrizi C, La Bella T, Marinelli S, Casadei Gardini A, Marisi G, et al. In hepatocellular carcinoma miR-221 modulates sorafenib resistance through inhibition of caspase-3–mediated apoptosis. Clin Cancer Res. . 2017;23:3953–3965. doi: 10.1158/1078-0432.CCR-16-1464. [DOI] [PubMed] [Google Scholar]
- 31.Hu Y, Yang C, Yang S, Cheng F, Rao J, Wang X. miR-665 promotes hepatocellular carcinoma cell migration, invasion, and proliferation by decreasing Hippo signaling through targeting PTPRB. Cell Death Dis. . 2018;9:954. doi: 10.1038/s41419-018-0978-y. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Qu Z, Wu J, Wu J, Ji A, Qiang G, Jiang Y, Jiang C, et al. Exosomal miR-665 as a novel minimally invasive biomarker for hepatocellular carcinoma diagnosis and prognosis. Oncotarget. . 2017;8:80666–80678. doi: 10.18632/oncotarget.20881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xia J, Li D, Zhu X, Xia W, Qi Z, Li G, Xu Q. Upregulated miR‑665 expression independently predicts poor prognosis of lung cancer and facilitates tumor cell proliferation, migration and invasion. Oncol Lett. . 2020;19:3578. doi: 10.3892/ol.2020.11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Erin N, Grahovac J, Brozovic A, Efferth T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resistance Updates. . 2020;53:100715. doi: 10.1016/j.drup.2020.100715. [DOI] [PubMed] [Google Scholar]
- 35.Bakir B, Chiarella AM, Pitarresi JR, Rustgi AK. EMT, MET, plasticity, and tumor metastasis. Trends Cell Biol. . 2020;30:764–776. doi: 10.1016/j.tcb.2020.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, Zhang J, Zhou H, Liu G, Li Q. Rho kinase mediates transforming growth factor-β1-induced vasculogenic mimicry formation: involvement of the epithelial–mesenchymal transition and cancer stemness activity. Acta Biochim Biophys Sin. . 2020;52:411–420. doi: 10.1093/abbs/gmaa014. [DOI] [PubMed] [Google Scholar]
- 37.Yan L, Yang S, Yue CX, Wei XY, Peng W, Dong ZY, Xu HN, et al. Long noncoding RNA H19 acts as a miR ‐340‐3p sponge to promote epithelial‐mesenchymal transition by regulating YWHAZ expression in paclitaxel‐resistant breast cancer cells . Environ Toxicol. . 2020;35:1015–1028. doi: 10.1002/tox.22938. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, Tai Q, Zhang J, Kang J, Gao F, Zhong F, Cai L, et al. MiRNA-206 inhibits hepatocellular carcinoma cell proliferation and migration but promotes apoptosis by modulating cMET expression. Acta Biochim Biophys Sin. . 2019;51:243–253. doi: 10.1093/abbs/gmy119. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Shi K, Liu H, Chen W, Luo Y, Wei X, Wu Z. miR-4458 inhibits the epithelial–mesenchymal transition of hepatocellular carcinoma cells by suppressing the TGF-β signaling pathway via targeting TGFBR1. Acta Biochim Biophys Sin. . 2020;52:554–562. doi: 10.1093/abbs/gmaa029. [DOI] [PubMed] [Google Scholar]
- 40.Xu J, Liu D, Niu H, Zhu G, Xu Y, Ye D, Li J, et al. Resveratrol reverses Doxorubicin resistance by inhibiting epithelial-mesenchymal transition (EMT) through modulating PTEN/Akt signaling pathway in gastric cancer. J Exp Clin Cancer Res. . 2017;36:19. doi: 10.1186/s13046-016-0487-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun XH, Chang X, Wang Y, Xu B, Cao X. Oroxylin A suppresses the cell proliferation, migration, and EMT via NF- κ B signaling pathway in human breast cancer cells . Biomed Res Int. . 2019;2019:1–10. doi: 10.1155/2019/9241769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang S, Liu Y, Li MY, Ng CSH, Yang SL, Wang S, Zou C, et al. FOXP3 promotes tumor growth and metastasis by activating Wnt/β-catenin signaling pathway and EMT in non-small cell lung cancer. Mol Cancer. . 2017;16:124. doi: 10.1186/s12943-017-0700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.