

Abstract

In this work, we explored a microwave-assisted glycolysis process to chemically recycle rigid polyurethane (PU) foam waste to obtain a single-phase product with suitable physio-chemical properties as a secondary raw material for the preparation of new rigid PU products. Such an approach was compared to a conventionally heated (ConvH) process, analyzing the performances of different catalysts. The use of microwaves allowed a 94% decrease in the reaction time scale of rigid PU depolymerization, with a concurrent 45% reduction in energy expense. By using a PU/diethylene glycol mass ratio of 1.5, best performances were obtained with a 30 mmol/100gPU potassium acetate concentration, both in terms of the product viscosity and aromatic amine byproduct content. The glycolysis products recovered were employed in substitution to virgin polyol for rigid PU foam preparation, showing improved compressive strength and comparable thermal insulation properties up to a 30% content with respect to the traditional non-recycled counterpart.
1. Introduction
Polyurethane (PU) is the sixth most used synthetic polymer all over the world. Despite the negative impact on the PU market turnover, which decreased from 65B$ to 45B$ in the 2018–2020 period, the improvement in the pandemic situation and the material key role as an insulating material to preserve the cold chain during vaccine delivery1 have increased the trust in the future increase of its production with an expected compound annual growth rate of 5.6% in the 2021–2028 forecast period. In 2021, the market size value was 59B$.2 PU demand comes mainly from the construction sector, driven by the increasing investments for the development of energy-efficient buildings for both commercial and residential purposes. Other important applications are in the automotive, furniture, and footwear sectors and in the electronic industry. The widespread success of this material can be attributed to its unique set of properties which provide excellent performances in terms of durability, flexibility, lightweightness, and optimal thermal insulation, combined with low production and processing costs. However, as a direct consequence of such commercial success, a growing amount of PU waste has been disposed of in landfills over the last decades due to the lack of suitable and economically viable recycling solutions. Such waste includes not only post-consumer products but also scraps from slab stock manufacturing for thermal insulation (up to 10% of total PU foam production). From this perspective, in the light of the new European Union policy to promote the transition toward a circular economy, there is a growing interest toward the development of new recycling strategies. Landfilling is indeed the least desirable solution among the different expanded PU end-of-life scenarios3,4 due to its low density, which generates useless costs and waste landing, while incineration, despite the possible derived energy recovery, is problematic for the development of toxic gases with combustion.5,6 Therefore, the two possible options left for PU recovery are physical and chemical recycle. Although physical recycle is the more straightforward solution, the products obtained with this approach usually have not only mediocre properties but also often different and lower-value applications.7 On the other hand, chemical recycle through solvolysis is believed to be the cornerstone of PU recycling to overcome many of the limitations of the other solutions.8,9 With this approach, the material depolymerization is promoted by the reaction of the urethane bonds with a suitable cleaving agent, allowing the recovery of raw materials and the production of new PU products. Many different thermo-chemical routes can be employed for PU depolymerization9,10 and have been investigated over the years such as hydrolysis,11,12 aminolysis,13,14 glycolysis,15−17 phosphorolysis,18,19 and acidolysis20,21 (Figure 1). Among them, especially glycolysis has shown promising potential for industrial upscaling.22−27 Glycolysis is, in effect, a transesterification reaction between the ester part of the urethane group of the PU waste and the hydroxyl groups of a glycolytic agent to produce mixed structure polyols comprising the original glycols and isocyanate. The actual sets of reaction involved in this process are, however, more complex due to the specific structural composition of the polymer backbone and the presence of possible side reactions. Indeed, in the PU network, ureic units are also present and subjected to glycolysis,28 producing an amine with the isocyanate substituent. Since derivatives of diphenylmethane-4,4′-diisocyanate (MDI) are the most used isocyanates for PU preparation, methylenedianiline (MDA, also known as diaminodiphenylmethane) represents the typical byproduct.29 MDA can also be produced by hydrolysis, due to the presence of residual humidity, and pyrolysis reactions in case of high reaction temperatures. On the other hand, if the PU is derived from polyester polyols, the cleavage agent can induce the transesterification of the ester groups in the polymer structure as well, while ureic groups and ester groups of the polyols can be subjected to hydrolysis.16,17 From this point of view, there are still some challenges to face in order to recover high-value products from glycolysis, hindering the economic and environmental sustainability of the process in comparison with virgin PU production, such as to reduce reaction byproducts, increase the recycled content in new PU products, and decrease the high energy demand and long reaction times.
Figure 1.

Possible pathways for PU chemical recycle (ϕ- is the substituent derived from the isocyanate polymer, R- is the substituent derived from the virgin polyol, and R′-and R″- are derived from the glycolytic agent).
A possible strategy to overcome some of these issues is provided by the use of alternative heating mechanisms to provide the heat required to sustain the process. In this context, over the last few years, several emerging techniques have attracted the attention of the research community due to their considerable process intensification potential. Among them, microwave (MW) heating has been established as an efficient strategy for both organic synthesis and extraction processes. Indeed, since initial experiments back in 1986,30,31 MW-assisted technologies have been rapidly developed, ensuring high levels of controllability and reproducibility which have expanded their applications in several chemistry and engineering fields on different scales.32 Through a controlled irradiation, MWs can indeed be absorbed by suitable substances and readily converted into heat. Such a mechanism is capable of producing a volumetric heating effect far more efficient than the surface conduction of traditional heating elements.
In recent days, MWs have also been tested in the plastic recycling field. Several authors have focused their attention on the recycling of polyethylene terephthalate (PET). Hoang et al.33 used zinc sulfate to lead to PET glycolysis. Myren et al.34 compared the chemical and electrochemical pathways for polyester recycling. López-Fonseca et al.35 evaluated the use of phase transfer catalysts in PET hydrolysis. Achilias and Karayannidis36 used manganese acetate as a transesterification catalyst, while Alnaqbi et al.37 worked with ionic liquids. Chen et al.38 recovered the depolymerization kinetics. Finally, Zeltins et al.39 depolymerized PET through an MW-assisted process and used the reaction product together with renewable rapeseed oil to produce new PU rigid foams.
Very few authors have worked on MW-assisted glycolysis of PU. Nikje et al.40−42 investigated this process applied to flexible foams using high-functionality glycols such as glycerin. They proved the feasibility of an MW-assisted reaction that is capable of reducing the reaction time by up to 30 min. They studied the effect of pentaerythritol (PER) up to 30% (considered a green medium) at low foam-to-solvent ratios and very active catalysts, such as sodium and potassium hydroxide, to cleave the foam. PER increases the viscosity of the lower phase and the reaction time. The best PER concentration is found to be 20%. The obtained upper phase can be used in the preparation of new foams; however, the bottom phase is greasy and unusable in PU formulations. In these studies, they studied the effect of the operative temperature.
In another work,43 they recycled a rigid foam to obtain a single-phase product by varying the output power of the MW reactor. They showed that a PU to solvent ratio of 0.5 is suitable to obtain a good product capable of substituting virgin polyols up to 40% in new PU formulations. However, they have not measured any mechanical performance of the new foam. They also performed MW-assisted hydroglycolysis reactions44 to assess the effect of the water content on depolymerization performances. It was found that the higher the water content, the lower the viscosity of the product and, therefore, the higher the degree of depolymerization. However, they did not produce new foams with these products.
Kiss et al.45 compared the atmospheric glycolysis of flexible PU foam with MW-assisted and autoclave-led glycolysis, finding that the latter is the most efficient as it allows the recycling of a larger amount of the waste material. They produced new flexible foams by substituting the virgin polyol with up to 5% of recycled polyol. Finally, Grdadolnik et al.46 applied acidolysis obtaining a polyol with carboxylic functionalities from flexible PU foam. They used MWs to shorten the reaction time, and they obtained an improvement in mechanical performances of the flexible foam produced with the recycled polyol. Foams showed a higher modulus and stress at 40% compression and a higher compression set, indicating a strong effect of carboxyl-terminated polyol due to esterification.
In this work, we carried out an MW-assisted glycolysis to recycle a PU foam waste and obtained a single-phase product that can be used as a secondary raw material to produce new rigid PU foams. The MW process was compared with a conventionally heated (ConvH) process to assess its feasibility. A higher waste to solvent ratio was used in order to ensure a noticeable substitution percentage of the glycolysis product in the new prepared foam.
2. Materials and Methods
2.1. Materials
Scrap rigid PU wastes derived from the cutting stage of rigid foams for insulating panels were kindly provided in the powder form by an external company. The material is a rigid water-blown PU with an apparent density of 40 kg/m3. Its composition is based on a formulation comprising polymeric MDI (pMDI) and a mixture of two different polyols, a trifunctional polyether and a bifunctional polyester.
For the glycolysis experiments, diethylene glycol (DEG, Sigma-Aldrich) was used as the solvolysis agent, while the different catalysts employed were potassium acetate (KOAc), tin (II) 2-ethylhexanoate or stannous octoate (SnOct2) (both from Sigma-Aldrich), and monoethanolamine (MEA), provided by Evonik Industries AG. For the characterization tests, methanol, high-performance liquid chromatography (HPLC)-grade tetrahydrofuran (THF), and potassium hydroxide were employed both as eluents for the analytical techniques and in sample preparation, while N-methyl-2-pyrrolidone, acetic anhydride, and 4-dimethylaminopyridine were used in hydroxyl value (HV) determination. All these chemicals were reagent grade and purchased from Sigma-Aldrich.
The raw materials for the preparation of new foams were
isocyanate: Desmodur 44V20L (pMDI, Covestro AG, Leverkusen, Germany), NCO % = 30.5–32.5, viscosity at 25 °C = 160–240 mPa·s
polyol: the same mixture of the PU waste was used. 50% of Alcupol C-5710 (polyether triol, Repsol S.A., Madrid, Spain), hydroxyl value, HV = 570 mgKOH/g, viscosity at 25 °C = 700 mPa·s and 50% of Adicrol FD 315 (aromatic DEG-based, Nord Composites UK Ltd, Wednesbury, UK), HV = 300–330 mgKOH/g, viscosity at 25 °C = 2000–3000 mPa·s
catalysts: Niax A1 [bis(2-dimethylaminoethyl)ether, Momentive, Waterford (NY), USA] and N,N-dimethylcyclohexylamine [Sigma-Aldrich, St. Louis (MO), USA], respectively, as gel and blow catalysts while Kosmos45 (potassium acetate, Evonik Industries AG, Essen, Germany) as the trimerization catalyst
surfactant: Tegostab B8460 (polyether polydimethylsiloxane, Evonik Industries AG, Essen, Germany)
blowing agent: deionized water
2.2. Equipment
2.2.1. Glycolysis Process
The glycolysis
tests for the chemical depolymerization of rigid PU foams were carried
out using two different configurations to provide the heat necessary
to sustain the reaction. The first configuration is based on a conventional
heat conduction apparatus comprising an MR Hei-Tec isomantle (Heidolph
Instruments, Schwabach, Germany) equipped with a 100 mL round-bottom
flask, a Pt 1000 temperature sensor, and mechanical stirring provided
by an overhead stirrer with a glass shaft equipped with a PTFE blade
end. The second configuration is based on a multi-modal lab-scale
MicroSYNTH microwave reactor (Milestone S.R.L., Sorisole, Italy) equipped
with hermetically sealed PTFE vessels of 100 mL, a magnetic stirring
mechanism, and a fiber optic temperature sensor. Comparisons will
be made by normalizing energy expenditures by the mass of the product.
A typical procedure to carry out a test involves the loading of the
cleavage agent (DEG, roughly 15 g) and the catalyst with preheating
of the system up to the working temperature. Subsequently, the PU
powder (roughly 23 g) is loaded step-wisely, according to its dissolution
in the reaction mixture. In the case of MW tests, PU is added together
with the cleavage agent in the reaction vessel in a single step. All
experiments were run at 200 °C, to balance catalyst activation
with minimal side product formation, and atmospheric pressure. According
to different preliminary tests, a mass feed ratio (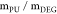 ) equal to 1.5 was employed in order to
obtain an adequate viscosity of the reaction mixture and a final product
with a suitable hydroxyl number for rigid PU foam production. This
factor has the strongest impact on the final percentage of substitution
of recycled polyol in the PU formulation. Catalysts were tested at
30 and 50 mmol/100gPU concentrations. According to several
preliminary experiments, reaction times were set to 4 h in the case
of conventional heating and 15 min with MW heating in order to obtain
glycolysis products with comparable properties.
) equal to 1.5 was employed in order to
obtain an adequate viscosity of the reaction mixture and a final product
with a suitable hydroxyl number for rigid PU foam production. This
factor has the strongest impact on the final percentage of substitution
of recycled polyol in the PU formulation. Catalysts were tested at
30 and 50 mmol/100gPU concentrations. According to several
preliminary experiments, reaction times were set to 4 h in the case
of conventional heating and 15 min with MW heating in order to obtain
glycolysis products with comparable properties.
2.2.2. Foam Preparation
The polyol mixtures obtained from the glycolysis of rigid PU wastes were tested to evaluate rigid PU foam production suitability and subsequently characterized for performance evaluation. 10 foams were produced, using three percentages of recycled polyol (namely, 15, 30, and 45%), and evaluated in comparison to equivalent products obtained from virgin polyols. In all the cases, the foams were prepared using a one-shot method involving the addition of the isocyanate to a premix comprising the different additives and reactants (polyols, catalysts, surfactants, and blowing agent). Upon vigorous mechanical agitation for about 10 s, the resulting mixture was poured in a pre-heated open mold to allow foam expansion. The obtained PU foams were subsequently heated at 70 °C for 24 h in order to complete the cross-linking reactions before the characterization phase.
2.3. Characterization
2.3.1. Glycolyzate Characterization
The aromatic amine content in the glycolysis product mixture was evaluated by reversed-phase HPLC. Specifically, a Waters e2695 separation model coupled with a Waters 2489 UV detector [Waters Corporation, Milford (MA), USA] with the double wavelength was employed. To perform the analysis, the following operating conditions were adopted:
column: hydrophobic C18 with a hydrophilic endcap (pore diameter: 300 Å, particle size: 5 μm, diameter x length: 4.6 × 250 mm)
eluent: Milli-Q ultrapure water (acidified with 0.05% of trifluoroacetic acid) and HPLC-grade methanol, starting condition 97% of water, with a gradient of 1%/min for 10 min
HPLC samples were prepared starting from glycolysis samples withdrawn from the reaction vessel and subsequently diluted with a 0.1 N potassium hydroxide solution in methanol in order to take into account the amphoteric character of amines. From this solution, after sonication and filtration using 0.2 μm PTFE filters, 10 μL was taken for injection in the HPLC column. The MDA calibration line and example of the chromatogram are shown in Figures S16 and S17.
Gel permeation chromatography (GPC) was employed to analyze the molecular weight distribution of the glycolysis products. For this analysis, a Jasco PU-980 pump model [JASCO, Easton (MD), USA] equipped with three Phenogel [Phenomenex, Torrance (CA), USA] size exclusion columns (particle diameter: 5 μm, pore diameter: 1000, 100, and 50 Å) and a JASCO RI-830 refractive index detector were used. Before injection, the glycolysis samples were dissolved in THF at the 0.04% w/w concentration and filtered using the 0.1 μm PTFE filter. Samples were eluted at 40 °C with a flowrate of 0.35 mL/min. Polystyrene standards [Fluka Analytical, Charlotte (NC), USA] were used to build a calibration line.
Qualitative investigation of the glycolysis products was carried out by Fourier-transform infrared spectroscopy (FT-IR) by looking at significant variations of urethane, alcoholic, and amine peaks. FT-IR spectra were collected using a Nicolet iS50 FT-IR spectrometer [Thermo Fisher Scientific, Waltham (MA), USA] using a Smart iTR attenuated total reflectance sampling accessory equipped with a diamond crystal. All the spectra were collected in the 4000–720 cm–1 wavenumber range.
The HV of the produced polyol mixture was evaluated according to the procedure reported in ASTM D4274-1147 using a Mettler Toledo G20S compact titrator [Mettler Toledo, Columbus (OH), USA], while the dynamic viscosity was measured at 25.0 ± 0.5 °C using a cone and plate viscometer [ATAC NμLine, Analytical Technology & Control Limited, Devizes (Wiltshire), UK] according to ASTM D4878-15.48
2.3.2. Foam Characterization
Scanning
electron microscopy (SEM) was employed for the analysis of the foam
morphology and to measure the average cell diameter according to ASTM
D3576-20.49 SEM micrographs were collected
using a Jeol 6490 SEM (Jeol Ltd., Tokyo, Japan) on gold-sputtered
samples with an accelerating voltage of 20 kV. SEM pictures are provided
in Supporting Information, Figures S8–S13.
Apparent foam density was measured according to ASTM D1622-2050 to comply with a fixed target reference density
of  for homogeneous comparison of the samples.
Mechanical tests were carried out using a Galdabini Sun2500 dynamometer
(Galdabini, Cardano al Campo, Italy) to evaluate compression resistance
according to ASTM D1621-16.51 Finally,
thermal conductivity was evaluated according to ASTM C177-1952 using a Holometrix Micromet Lambda 2300 V heat
flow meter apparatus (NETZSCH, Selb, Germany) at a mean temperature
of 10 °C with a temperature difference of 20 °C between
the plates. All the foams were tested 1 day after their preparation
to prevent any effect related to the ageing of the materials.
for homogeneous comparison of the samples.
Mechanical tests were carried out using a Galdabini Sun2500 dynamometer
(Galdabini, Cardano al Campo, Italy) to evaluate compression resistance
according to ASTM D1621-16.51 Finally,
thermal conductivity was evaluated according to ASTM C177-1952 using a Holometrix Micromet Lambda 2300 V heat
flow meter apparatus (NETZSCH, Selb, Germany) at a mean temperature
of 10 °C with a temperature difference of 20 °C between
the plates. All the foams were tested 1 day after their preparation
to prevent any effect related to the ageing of the materials.
3. Results and Discussion
3.1. Conventional and Microwave-Assisted Glycolysis
The characteristics of the products derived from the glycolysis processes were investigated by measuring specific properties in order to compare the combination of different catalysts and heating mechanisms. Viscosity (μ) was selected as the index of the extent of depolymerization of the PU waste, while the MDA content and HV were measured to assess the extent of secondary reactions and quantify the extent of reactive −OH groups of the products available for subsequent new PU foam preparation, respectively. Table 1 summarizes experimental data, viscosity, and MDA, categorized by the heating method and catalyst concentration.
Table 1. Properties of ConvH and MW Productsa.
| heating method | ConvH |
MW |
|||||
|---|---|---|---|---|---|---|---|
| heating time, [min] | 240 |
15 |
|||||
| catalyst | ccat, [mmol/100gPU] | μ, [mPa·s] | MDA, [% wt] | HV, [mgKOH/g] | μ, [mPa·s] | MDA, [% wt] | HV, [mgKOH/g] |
| KOAc | 30 | 2350 | 1.7 | 505 | 1840 | 2.3 | 500 |
| 50 | 2200 | 2.5 | 540 | 760 | 4.9 | 520 | |
| Sn(Oct)2 | 30 | 2540 | 1.3 | 540 | 2260 | 2.1 | 530 |
| 50 | 2300 | 1.9 | 550 | 1150 | 3.2 | 550 | |
| MEA | 30 | 8570 | 1.0 | 480 | N.A. | 0.20 | 510 |
| 50 | 7030 | 0.4 | 475 | 11840 | 0.30 | 530 | |
Maximum errors for viscosity measurements are ±80 cP, ±0.1%wt for MDA concentrations and ±15 mgKOH/g for HVs.
According to Table 1, the viscosity of the glycolyzate is highly influenced by both the type of catalyst and its concentration; the same can be said for the aromatic amine content. These two outputs are inversely related as the lower the viscosity, the higher the degree of depolymerization and, therefore, the higher the amount of byproducts formed at constant reaction time.
On the one hand, a glycolyzate with lower viscosity is desirable, as too viscous liquids are not easily handled and increase the energy expenditure of pumping equipment in industrial foam production facilities. On the other hand, to reduce the secondary undesired reaction, the MDA amount in the products must be kept as low as possible. Increasing the extent of the reaction as a consequence of the catalyst action has a parallel effect on both the MDA concentration and viscosity. Specifically, the increased depolymerization of PU decreases the product viscosity, but it is unavoidably accompanied by an increase in the MDA concentration. Such a conclusion is provided by the notable differences observed with the use of MEA; it is the least active catalyst tested, producing high viscosities although having very low MDA concentrations. This evidence is confirmed and cleared when examining MW glycolysis results. In the case of 30 mmol/100gPU, it was not possible to measure the viscosity as the PU was not completely dissolved and reacted. The low MEA activity can be explained by a transesterification mechanism proposed by Tang et al.53 according to which MEA may act through a non-ionic mechanism. This conclusion is supported by the fact that contrary to glycolysis with metal salts, glycolysis catalyzed by MEA through MW led to worse performances with respect to the same conventionally heated reaction.
In both the cases of KOAc and Sn(Oct)2, glycolysis proceeded quickly: MWs drastically improved the recycling process since 15 min of MW irradiation led to lower viscosities and higher MDA concentrations than 240 min through conventional heating. This result is very interesting since the reduction in reaction time is 94%. The higher MDA concentration found in MW glycolysis can be explained by considering two facts. First, MW glycolyzates have viscosities lower than those of their ConvH counterparts, which means that less than 15 min is required to reach the same viscosity as that obtained through ConvH. Second, metal salts such as KOAc and Sn(Oct)2 coupled with DEG provoke highly efficient dielectric heating and ionic polarization, providing the suitable conditions to carry out depolymerization reactions. Furthermore, Sn(Oct)2 and KOAc are equally capable of promoting transesterification as the obtained viscosities are comparable; however, the latter allows hydrolysis and pyrolysis to occur more than the former does. As a result, with Sn(Oct)2, a lower concentration of MDA is achieved together with a viscosity comparable to that of the KOAc-catalyzed product. Two proposed mechanisms by Murai et al.54 and Robins and Britain et al.55,56 can explain the higher selectivity of Sn(Oct)2 compared to that of KOAc. The former, possessing a divalent cation, produces a stable cyclic alkoxide that can coordinate the intermolecular exchange in the urethane group.57 The latter produces a less selective alkoxide but a more reactive acidic intermediate that promotes the hydrolysis reaction.
In addition to these considerations, FT-IR spectra can provide a clearer view. As seen from Figure 2, the characteristic peaks58−61 of the urethane structure can be monitored. Peaks at 1515 cm–1 (ν(C–C) aromatic ring) and 1130 cm–1 (ν(C–O) symmetric ether) can be used as a reference peak to study relative variations of other significant peaks. Lower transmittances of peaks at 1720 cm–1 (ν(C=O) urethane amide I), 1540 cm–1 (δ(N–H) urethane amide II), and 1230 cm–1 (ν(C–O) urethane) indicate that the polymer structure is less degraded from MEA than that in the other two cases. Shoulder-type peaks can be noticed at 1630 cm–1: those are more marked for KOAc rather than Sn(Oct)2 and MEA, testifying the presence of a higher concentration of free amino groups. The peak located between 3600 and 3100 cm–1 represents the presence of hydroxyl groups (ν(O–H)); they remain constant throughout the reaction because of the excess glycol, and transesterification of urethane through DEG does not consume any −OH group. The sharper this peak is, the higher the presence of aminic byproducts, as the peak underlying the hydroxyl bell at 3354 cm–1 is related to amino groups (ν(N–H) nonbonded). The peak at 2950 cm–1 (ν(C–H)) is constant as C–H and CH2 groups are not involved in reactions shown in the glycolysis process. Glycolysis reaction kinetics cannot be tracked through FT-IR spectra as it is a transesterification; therefore, no variations of functional groups would be noticed. Consequently, differences in transmittance have to be ascribed to secondary reactions (hydrolysis and pyrolysis) that consume the urethane bond which is represented by three vibrations, 1720, 1540, and 1230 cm–1. The higher the transmittance, the more the reactions proceeded. Peaks at 1515 and 1130 cm–1 are considered constant because the aromatic ring is not involved in any reaction shown in Figure 1, and the same happens for the ether bond. Polyols are freed from the reactions, and the only way of assessing the depolymerization degree, apart from the viscosity, is by measuring the molecular weight of the glycolysis product; this was done below. Figure 3 depicts the reactions involving the urethane bond of the polymer, as the said secondary reactions are noticeable through FT-IR whereas glycolysis with GPC.
Figure 2.

Infrared spectra of MW glycolysates. The catalyst concentration is 30 mmol/100gPU.
Figure 3.

Glycolysis and hydrolysis of the polyurethane (ϕ- is the substituent derived from the isocyanate polymer, R- is the substituent from the virgin polyol, and R′- and R″- are those derived from the glycolytic agent).
Figure S1 shows the same FT-IR spectra for the ConvH glycolysis products.
Finally, Figure 4 highlights the comparability of the ConvH and MW glycolysis products: despite the lower viscosity in the latter case, no additional peaks appear in the product obtained with MW-assisted glycolysis. This means that the reaction products have the same chemical groups but different concentrations. Noticeable differences in the spectra are due to a higher degree of depolymerization due to secondary reactions (hydrolysis and pyrolysis) developing carbon dioxide and therefore decreasing carbonyl groups and a higher byproduct concentration in the MW product leading to a lower transmittance of the carbonyl and amine peaks, 1720 and 1540 cm–1, respectively.
Figure 4.

Infrared spectra comparison between ConvH glycolysis and MW glycolysis. These curves refer to 30 mmol/100gPU KOAc as the catalyst.
Further validations to what is explained above can be found by considering GPC chromatograms of MW-assisted glycolysis products shown in Figure 6. GPC chromatograms of the conventionally heated-obtained glycolysis product are shown in Figure S2. A higher number of convolved peaks at lower retention times mean the presence of species with high molecular weight and, therefore, a lower degree of depolymerization. From the comparison between Figures 6 and S2, it is possible to assess both the lower extent of depolymerization with MEA through MW and vice versa, the greater extent of depolymerization obtained with KOAc and Sn(Oct)2. Table 2 summarizes the weight- and number-averaged molecular weights of the products. MW-assisted glycolysis with KOAc and Sn(Oct)2 shows a lower averaged molecular weight, Mw, with respect to the ConvH counterparts. This fact confirms viscosity data explained in Table 1; MW allows for a higher depolymerization degree. MEA glycolysis products instead have a higher Mw, when obtained with MWs rather than using the conventionally heated system. Higher molecular weights present in the product obtained with MEA shown in Figure 6 are due to the presence of partially dissolved PU (2370 Da) and due to parts of the polyester polyol used during pristine PU preparation (1570 and 1086 Da). Peaks indicating lower molecular weights instead are related to the polyether polyol (Figure S15) which is not affected by the glycolysis process. In products obtained with KOAc and Sn(Oct)2, only polyether polyol peaks can be identified, meaning that the polyester polyol is depolymerized as well during the glycolysis process. Also, the lower the molecular weight of the product, the higher the amount of freed polyol from the reaction. GPC allows us to confirm not only the proceeding of the main glycolysis reaction shown in Figure 3 but also the occurrence of another secondary reaction, the hydrolysis of the polyester polyol as schematized in Figure 5.
Figure 6.

GPC curves of the MW glycolysis. The catalyst concentration is 30 mmol/100gPU.
Table 2. Weight- and Number-Averaged Molecular Weights of Glycolysis Products. Đ Is the Polydispersity Indexa.
| glycolysis | heating | Mw, [Da] | Mn, [Da] | Đ = Mw/Mn, [-] |
|---|---|---|---|---|
| KOAc | ConvH | 1201 | 362 | 3.3 |
| KOAc | MW | 1014 | 273 | 3.7 |
| Sn(Oct)2 | ConvH | 1361 | 419 | 3.2 |
| Sn(Oct)2 | MW | 1100 | 374 | 2.9 |
| MEA | ConvH | 1537 | 468 | 3.3 |
| MEA | MW | 1613 | 398 | 4.1 |
Catalyst concentration is 30 mmol/100gPU.
Figure 5.

Hydrolysis of the polyester polyol yielding acidic compounds.
Due to the similarities in product properties according to the selected reaction times for the different heating mechanisms, further investigation was carried out with the aim to provide a simple estimation of the energy saving that can be obtained with the employment of MW heating in comparison to that obtained with ConvH, at least on a laboratory-scale equipment. The energy expenditure, EE, is calculated as the ratio between energy delivered to the system and total mass heated. In ConvH glycolysis, the energy given to the system is the summation of the product mass, heat capacity, and temperature increment (during the sampling time). The process is split into three phases: heating of the glycol, loading of PU, and reaction. Similarly, the energy given to the system in MW glycolysis is calculated by splitting the process in heating and reacting phases as PU is already loaded into the reaction vessel. In this case, also, a power profile (Figure S3) can be obtained from software to evaluate a heating efficiency. Table 3 gathers this information:
Table 3. Energy Expenditure Contributions.
| energy delivered | heating phase | loading phase | reacting phase | Expenditure |
|---|---|---|---|---|
| ConvH | 6.8 kJ | 8.0 kJ | 24.9 kJ | 0.31 kW h/kg |
| MW | 15.0 kJ | N.A. | 5.7 kJ | 0.17 kW h/kg |
The energy expenditure for the ConvH glycolysis is almost 2 times (1.8) of that necessary for the MW glycolysis, with an energy saving of 45%. Similar calculations can be done in relation to water consumption during the process. Considering an equal cooling water flowrate, the difference is only due to different process times: the water consumption is therefore decreased by 94%. The accuracy of these calculations will be validated during the scale-up of the MW process using a semi-pilot conventionally heated reactor.
In conclusion, products obtained with KOAc and Sn(Oct)2 showed the best performance from the glycolyzate properties’ point of view. A 30 mmol/100gPU catalyst concentration is sufficient to reach an acceptable viscosity (i.e., degree of depolymerization) keeping the byproduct concentration as low as possible. Sn(Oct)2 displayed a higher selectivity with respect to KOAc as it allows for comparable viscosity but a lower MDA content. In the case of MEA, conventional heating and 50 mmol/100gPU as the concentration are necessary to obtain a suitable product.
3.2. Preparation and Characterization of Foams
Once it was proved that the ConvH and MW products were comparable, new foams were prepared starting from MW products. Table 4 shows the results in terms of apparent densities and characteristic times recorded during the preparation. Foams with recycled polyol (RP) were named with this rationale: xRPy, where x is the substitution percentage (15, 30, and 45%) and y is a letter indicating the catalyst used during the glycolysis (K for potassium acetate, S for stannous octoate, and M for MEA). All products were tested; however, most promising results were obtained with products containing KOAc (series 15, 30, and 45RPK). Although Sn(Oct)2 allowed us to obtain a product with properties comparable with the ones obtained with KOAc, this catalyst is also a gel catalyst for the foam preparation. This fact led to a very reactive and difficult to process polyol, making it incompatible with the characteristic times of the preparation of new rigid PU foams (see foam 15RPS in Table 4). MEA (foam 15RPM) was worse in terms of glycolyzate properties; foams obtained with this product were brittle and chalky as further explained later. New rigid PU foams (with isocyanate index, INCO = 110) were produced with an increasing percentage of glycolyzate as a substitute for virgin polyol.
Table 4. Density and Characteristic Times of the Prepared Foams.
| foam | density, [kg/m3] | cream time, [s] | gel time, [s] | tack-free time, [s] |
|---|---|---|---|---|
| reference | 38 | 21 | 81 | 130 |
| 15RPK | 37 | 12 | 43 | 66 |
| 30RPK | 42 | 14 | 30 | 43 |
| 45RPK | 39 | 13 | 28 | 44 |
| 15RPS | 37 | 17 | 34 | 39 |
| 30RPS | 38 | 13 | 27 | 34 |
| 45RPS | 42 | 14 | 28 | 30 |
| 15RPM | 41 | 18 | 72 | 140 |
| 30RPM | 38 | 20 | 60 | 101 |
| 45RPM | 43 | 30 | 104 | 178 |
Measured HVs are reported in Table 1 and are found between 475 and 550 mgKOH/g. The cleavage agent, the feed ratio, and starting polyols affect the final HV of the product: DEG has an HV of 1057 mgKOH/g, higher than the starting polyols’ HV. However, the feed ratio of 1.5 kgPU/kgDEG had a diluting effect on the final product. Amine interferences (primary and secondary amines and higher fatty acids react with the reagent to form stable compounds47) may also occur. Overall, the products obtained were still suitable for the production of rigid PU foams. Therefore, foams were produced by substituting a part of the polyol mixture with the glycolyzate (at 0, 15, 30, and 45% with respect to the total amount of polyol, reference 15, 30, and 45RPK in Table 4). Figures 7–9 show the results related to the new foam preparation in terms of average cell diameter, compressive strength along the growth direction, and thermal conductivity for foams obtained with 30 mmol/100gPU KOAc-catalyzed glycolyzate. Substitution of part of virgin polyol with the recycled material allowed for better thermal and mechanical performances with respect to a reference foam. The percentage of substitution may reach 30% without having a property decay. As can be seen in Figures 7 and 8, higher compression strength resulted from a smaller cell diameter. The isocyanic backbone already contained in the glycolysis polyol due to rigid PU depolymerization may increase the miscibility with the isocyanate, leading to a finer cellular morphology.62,63
Figure 7.

Average cell diameter of the foam along with the substitution percentage.
Figure 9.

Thermal conductivity along with percentage substitution.
Figure 8.

Compression resistance along with percentage substitution.
The same considerations can be made in terms of thermal performances, as the lower the average cell diameter, the lower the thermal conductivity. However, this was not observed as thermal conductivities are comparable (Figure 9). This may be caused by a higher percentage of open cells.64 With 45% substitution (45RPK), a very slight and acceptable decrease (with respect to lower substitution percentages) in the properties was observed; this is due to a trade-off between two factors: glycolysis products containing aromatic rings that increase the miscibility with the isocyanate component and DEG which decreases the average functionality of the mixture. At 45% substitution (45RPK), DEG contribution became relevant, leading to a decrease in the mechanical performances due to lower cross-linking density in the foam. Best performing foams were obtained with the KOAc-catalyzed glycolysis product.
A higher compression strength can be explained considering that KOAc is commonly used as a trimerization catalyst. Although the produced foam is a rigid PU due to its isocyanate index, some isocyanurate structures can still be formed. Isocyanurate structure formation promoted by trimerization catalysts leads to higher mechanical performances.63
This was also confirmed by FT-IR spectra
shown in Figure S14 in which relative variations
of the peak at 1411
cm–1 (isocyanuric ring deformation) with respect
to the peak at 814 cm–1 ( of the isocyanate out of plane) are shown
as a function of the increasing substitution percentage of glycolyzate.
All data related to the foams obtained by substituting the virgin
polyol part with Sn(Oct)2- and MEA-catalyzed products are
presented in Figures S4–S7. Foams
obtained with an increasing amount of the Sn(Oct)2-catalyzed
glycolysis product (Figures S4 and S5)
showed no variations in thermal performances and an increased compression
strength, up to 25% higher than that of the reference with 45% substitution.
However, reactive mixtures were difficult to deal with. When stirring,
reaction times (foam 15RPS in Table 4) were drastically shortened due to the fact that Sn(Oct)2 is a strong gel catalyst used also during rigid PU preparation;
it can be seen how gel and tack-free times are very close to each
other, leading to an inadequate growth and maturation of the foam.
When increasing the glycolyzate content, the Sn(Oct)2 concentration
is increased as well: for this reason, maximum percentage substitution
is limited to 15%. Above 15%, foams obtained were dimensionally unstable.
of the isocyanate out of plane) are shown
as a function of the increasing substitution percentage of glycolyzate.
All data related to the foams obtained by substituting the virgin
polyol part with Sn(Oct)2- and MEA-catalyzed products are
presented in Figures S4–S7. Foams
obtained with an increasing amount of the Sn(Oct)2-catalyzed
glycolysis product (Figures S4 and S5)
showed no variations in thermal performances and an increased compression
strength, up to 25% higher than that of the reference with 45% substitution.
However, reactive mixtures were difficult to deal with. When stirring,
reaction times (foam 15RPS in Table 4) were drastically shortened due to the fact that Sn(Oct)2 is a strong gel catalyst used also during rigid PU preparation;
it can be seen how gel and tack-free times are very close to each
other, leading to an inadequate growth and maturation of the foam.
When increasing the glycolyzate content, the Sn(Oct)2 concentration
is increased as well: for this reason, maximum percentage substitution
is limited to 15%. Above 15%, foams obtained were dimensionally unstable.
Foams obtained with MEA (Figures S6 and S7) showed higher mechanical performances than the reference foam, up to 40% higher with 45% substitution, and no thermal property variations. Despite this, a brittle and chalky structure was observed. Even with MEA-catalyzed products, mixing problems were experienced as these have a higher viscosity; therefore, the maximum substitution percentage was 15%.
4. Conclusions
In this work, we assessed the feasibility of an MW-assisted glycolysis reaction to recover a polyol-like liquid product from rigid PU foam waste. Reaction conditions chosen allowed us to obtain a single-phase product, which eased its handling with respect to a split-phase process, suitable for rigid foam production. Among the three catalysts that were tested [potassium acetate, KOAc; stannous octoate, Sn(Oct)2; and monoethanolamine, MEA], products obtained with metal-based catalysts provided better properties than those obtained with MEA.
In all trials, except one, MWs drastically decreased the reaction time to reach a proper viscosity of the product, which decreases 94% going from 240 min, with a conventional-heated process, to 15 min. With MW heating, a simple energy balance showed that MW-assisted glycolysis is convenient with respect to the conventionally heated process as it allowed for an energy saving of 45% (0.17 kW h/kg with respect to 0.31 kW h/kg).
Foams obtained with metal-catalyzed products, in particular KOAc, are also the ones that provide best results. A substitution of virgin polyol up to 30% permits one to slightly reduce the thermal conductivity of the foam, coupled with an increase of roughly 20% in the compression strength. The maximum percentage of substitution with Sn(Oct)2 and the MEA-catalyzed product is 15% as the mixing process was complicated by high reactivity of the first product and high viscosity of the second one. However, the higher substitution percentage with respect to other studies was achieved also, thanks to the high scrap to solvent ratio that allows the HV of the product to be comparable to that of a conventional virgin polyol for rigid PU foam applications; this remarkably improves the circularity of the recycling process.
Acknowledgments
Riccardo Donadini gratefully acknowledges the financial support from the Foundation of Savings Bank of Padua and Rovigo.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c06297.
FT-IR spectra of ConvH glycolysis products, GPC chromatograms of ConvH glycolysis products, MW parameter profile, mechanical and thermal properties of foams obtained with Sn(Oct)2 and MEA, SEM images, FT-IR spectra of the foams, GPC chromatograms of virgin polyols, MDA calibration line, and MDA chromatograms (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bailey M. P.; Ondrey G.. Ultra freezers for safe storage of vaccines and valuable samples. https://www.chemengonline.com/ultra-freezers-for-safe-storage-of-vaccines-and-valuable-samples/?printmode=1 (accessed Nov 22, 2022).
- Polyurethane market size, share and trends analysis report. https://brandessenceresearch.com/chemical-and-materials/polyurethane-market-size (accessed Oct 17, 2022).
- Zia K. M.; Bhatti H. N.; Ahmad Bhatti I. Methods for Polyurethane and Polyurethane Composites, Recycling and Recovery: A Review. React. Funct. Polym. 2007, 67, 675–692. 10.1016/j.reactfunctpolym.2007.05.004. [DOI] [Google Scholar]
- Manzardo A.; Marson A.; Roso M.; Boaretti C.; Modesti M.; Scipioni A.; Lorenzetti A. Life Cycle Assessment Framework to Support the Design of Biobased Rigid Polyurethane Foams. ACS Omega 2019, 4, 14114–14123. 10.1021/acsomega.9b02025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paabo M.; Levin B. C. A Review of the Literature on the Gaseous Products and Toxicity Generated from the Pyrolysis and Combustion of Rigid Polyurethane Foams. Fire Mater. 1987, 11, 1–29. 10.1002/fam.810110102. [DOI] [Google Scholar]
- Reinerte S.; Avotina L.; Zarins A.; Cabulis U.; Viksna A. TG/DTA-FTIR as a Method for Analysis of Tall Oil Based Rigid Polyurethane Foam Decomposition Gaseous Products in a Low Oxygen Environment. Polym. Degrad. Stab. 2020, 180, 109313. 10.1016/j.polymdegradstab.2020.109313. [DOI] [Google Scholar]
- Yang W.; Dong Q.; Liu S.; Xie H.; Liu L.; Li J. Recycling and Disposal Methods for Polyurethane Foam Wastes. Procedia Environ. Sci. 2012, 16, 167–175. 10.1016/j.proenv.2012.10.023. [DOI] [Google Scholar]
- Martín A. J.; Mondelli C.; Jaydev S. D.; Pérez-Ramírez J. Catalytic Processing of Plastic Waste on the Rise. Chem 2021, 7, 1487–1533. 10.1016/j.chempr.2020.12.006. [DOI] [Google Scholar]
- Simón D.; Borreguero A. M.; de Lucas A.; Rodríguez J. F. Recycling of Polyurethanes from Laboratory to Industry, a Journey towards the Sustainability. Waste Manag. 2018, 76, 147–171. 10.1016/j.wasman.2018.03.041. [DOI] [PubMed] [Google Scholar]
- Kosloski-Oh S. C.; Wood Z. A.; Manjarrez Y.; de los Rios J. P.; Fieser M. E. Catalytic Methods for Chemical Recycling or Upcycling of Commercial Polymers. Mater. Horizons 2021, 8, 1084–1129. 10.1039/d0mh01286f. [DOI] [PubMed] [Google Scholar]
- Motokucho S.; Yamaguchi A.; Nakayama Y.; Morikawa H.; Nakatani H. Hydrolysis of aromatic polyurethane in water under high pressure of CO2. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 2004–2010. 10.1002/pola.28576. [DOI] [Google Scholar]
- Gerlock J.; Braslaw J.; Zinbo M. Polyurethane Waste Recycling. 1. Glycolysis and Hydroglycolysis of Water-Blown Foams. Ind. Eng. Chem. Process. Des. Dev. 1984, 23, 545–552. 10.1021/i200026a023. [DOI] [Google Scholar]
- Padhan R. K.Chemical Depolymerization of Polyurethane Foams via Combined Chemolysis Methods. In Recycling of Polyurethane Foams; Thomas S., Rane A. V., Kanny K., Thomas M. G., Eds.; Plastics Design Library; Elsevier, 2018, pp 89–96. [Google Scholar]
- Chuayjuljit S.; Norakankorn C.; Pimpan V. Chemical Recycling of Rigid Polyurethane Foam Scrap via Base Catalyzed Aminolysis. J. Met. Mater. Miner. 2002, 12, 19–22. [Google Scholar]
- Simioni F.; Modesti M. Controlled Degradation of Polyurethane for Recycling. Mat. Eng. 1991, 2, 127–144. [Google Scholar]
- Modesti M.; Simioni F. Chemical Recycling of Reinforced Polyurethane from the Automotive Industry. Polym. Eng. Sci. 1996, 36, 2173–2178. 10.1002/pen.10614. [DOI] [Google Scholar]
- Modesti M.; Simioni F.; Rienzi S. A. Recycling of Microcellular Polyurethane Elastomer Waste. J. Elastomers Plast. 1992, 24, 288–305. 10.1177/009524439202400403. [DOI] [Google Scholar]
- Troev K. A. A novel approach to recycling of polyurethanes: chemical degradation of flexible polyurethane foams by triethyl phosphate. Polymer 2000, 41, 7017–7022. 10.1016/s0032-3861(00)00054-9. [DOI] [Google Scholar]
- Molero C.; Mitova V.; Troev K.; Rodriguez J. F. Kinetics and Mechanism of the Chemical Degradation of Flexible Polyurethane Foam Wastes with Dimethyl H-Phosphonate with Different Catalysts. J. Macromol. Sci. Part A 2010, 47, 983–990. 10.1080/10601325.2010.506408. [DOI] [Google Scholar]
- Gama N.; Godinho B.; Marques G.; Silva R.; Barros-Timmons A.; Ferreira A. Recycling of Polyurethane by Acidolysis: The Effect of Reaction Conditions on the Properties of the Recovered Polyol. Polymer 2021, 219, 123561. 10.1016/j.polymer.2021.123561. [DOI] [Google Scholar]
- Gama N.; Godinho B.; Marques G.; Silva R.; Barros-Timmons A.; Ferreira A. Recycling of Polyurethane Scraps via Acidolysis. Chem. Eng. J. 2020, 395, 125102. 10.1016/j.cej.2020.125102. [DOI] [Google Scholar]
- Wang J.; Chen D.; Zhang Q. Analysis of Glycolysis Products of Polyurethane Fiber Waste with Diethylene Glycol. Fibers Polym. 2007, 8, 13–18. 10.1007/bf02908154. [DOI] [Google Scholar]
- Mellado M. M.; Rodriguez J. F.; Cerezo C.; Martinez A.. Process for Thew Recovery of Polyols from Polyurethane Foam Waste Using a Novel Catalyst, 2005.
- Sendijarevic V.Process for Chemical Recycling of Polyurethane-Containing Scrap, 2001.
- Ryntz R. A.; McRoberts T. M.. Closed-Loop Recycled Polyurethane Foam. Methods of Manufacture and Products Therefrom, 2013.
- Uekado K.; Yuasa A.. Method of Recycling Rigid Polyurethane Foam, 2001.
- Munzmay T.; Fuhrmann P.; Lamla F.; Meckel W.; Rasshofer W.. Process for the Glycolytic Decomposition of Polyurethane Plastics, US Patent 005,508,312 A, 1994.
- Simioni F.; Modesti M. Glycolysis of Flexible Polyurethane Foams. Cell. Polym. 1993, 12, 337–348. [Google Scholar]
- Modesti M.; Simioni F.; Munari R.; Baldoin N. Recycling of Flexible Polyurethane Foams with a Low Aromatic Amine Content. React. Funct. Polym. 1995, 26, 157–165. 10.1016/1381-5148(95)00031-a. [DOI] [Google Scholar]
- Lidström P.; Tierney J.; Wathey B.; Westman J. Microwave assisted organic synthesis-a review. Tetrahedron 2001, 57, 9225–9283. 10.1016/s0040-4020(01)00906-1. [DOI] [Google Scholar]
- Kappe C. O. Controlled Microwave Heating in Modern Organic Synthesis. Angew. Chem., Int. Ed. 2004, 43, 6250–6284. 10.1002/anie.200400655. [DOI] [PubMed] [Google Scholar]
- Rinaldi L.; Carnaroglio D.; Rotolo L.; Cravotto G. A Microwave-Based Chemical Factory in the Lab: From Milligram to Multigram Preparations. J. Chem. 2015, 2015, 1–8. 10.1155/2015/879531. [DOI] [Google Scholar]
- Hoang C. N.; Le T. T. N.; Hoang Q. D. Glycolysis of poly(ethylene terephthalate) waste with diethyleneglycol under microwave irradiation and ZnSO4·7H2O catalyst. Polym. Bull. 2019, 76, 23–34. 10.1007/s00289-018-2369-z. [DOI] [Google Scholar]
- Myren T. H. T.; Stinson T. A.; Mast Z. J.; Huntzinger C. G.; Luca O. R. Chemical and Electrochemical Recycling of End-Use Poly(Ethylene Terephthalate) (PET) Plastics in Batch, Microwave and Electrochemical Reactors. Molecules 2020, 25, 2742. 10.3390/molecules25122742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Fonseca R.; González-Marcos M. P.; González-Velasco J. R.; Gutiérrez-Ortiz J. I. Chemical Recycling of PET by Alkaline Hydrolysis in the Presence of Quaternary Phosphonium and Ammonium Salts as Phase Transfer Catalysts. WIT Trans. Ecol. Environ. 2008, 109, 511–520. 10.2495/wm080521. [DOI] [Google Scholar]
- Karayannidis G. P.; Achilias D. S. Chemical Recycling of Poly(Ethylene Terephthalate). Macromol. Mater. Eng. 2007, 292, 128–146. 10.1002/mame.200600341. [DOI] [Google Scholar]
- Alnaqbi M. A.; Mohsin M. A.; Busheer R. M.; Haik Y. Microwave Assisted Glycolysis of Poly(Ethylene Terephthalate) Catalyzed by 1-Butyl-3-Methylimidazolium Bromide Ionic Liquid. J. Appl. Polym. Sci. 2014, 132, 1–7. 10.1002/app.41666. [DOI] [Google Scholar]
- Chen F.; Wang G.; Shi C.; Zhang Y.; Zhang L.; Li W.; Yang F. Kinetics of Glycolysis of Poly(Ethylene Terephthalate) under Microwave Irradiation. J. Appl. Polym. Sci. 2013, 127, 2809–2815. 10.1002/app.37608. [DOI] [Google Scholar]
- Zeltins V.; Cabulis U.; Abolins A.; Gaidukovs S. Microwave Synthesis Of Polyols For Urethane Materials. IOP Conf. Ser. Mater. Sci. Eng. 2016, 111, 012015. 10.1088/1757-899x/111/1/012015. [DOI] [Google Scholar]
- Nikje M. M. A.; Mohammadi F. H. A. Polyurethane Foam Wastes Recycling under Microwave Irradiation. Polym. Plast. Technol. Eng. 2010, 49, 818–821. 10.1080/03602551003749635. [DOI] [Google Scholar]
- Alavi Nikje M. M.; Nikrah M.; Haghshenas M. Microwave Assisted ″Split-phase″ Glycolysis of Polyurethane Flexible Foam Wastes. Polym. Bull. 2007, 59, 91–104. 10.1007/s00289-007-0753-1. [DOI] [Google Scholar]
- Alavi Nikje M. M.; Nikrah M. Microwave-Assisted Glycolysis of Polyurethane Cold Cure Foam Wastes from Automotive Seats in ″Split-Phase″ Condition. Polym. Plast. Technol. Eng. 2007, 46, 409–415. 10.1080/03602550701242943. [DOI] [Google Scholar]
- Nikje M. M. A.; Nikrah M. Chemical Recycling and Liquefaction of Rigid Polyurethane Foam Wastes through Microwave Assisted Glycolysis Process. J. Macromol. Sci. Part A 2007, 44, 613–617. 10.1080/10601320701285003. [DOI] [Google Scholar]
- Mir Mohammad Alavi Nikje; Nikrah M.; Fateme Haji Aga Mohammadi Microwave-Assisted Polyurethane Bond Cleavage via Hydroglycolysis Process at Atmospheric Pressure. J. Cell. Plast. 2008, 44, 367–380. 10.1177/0021955x08090279. [DOI] [Google Scholar]
- Kiss G.; Rusu G.; Peter F.; Tănase I.; Bandur G. Recovery of Flexible Polyurethane Foam Waste for Efficient Reuse in Industrial Formulations. Polymers 2020, 12, 1533. 10.3390/polym12071533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grdadolnik M.; Drinčić A.; Oreški A.; Onder O. C.; Utroša P.; Pahovnik D.; Žagar E. Insight into Chemical Recycling of Flexible Polyurethane Foams by Acidolysis. ACS Sustain. Chem. Eng. 2022, 10, 1323–1332. 10.1021/acssuschemeng.1c07911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASTM D4274-16 . Standard Test Methods for Testing Polyurethane Raw Materials: Determination of Hydroxyl Numbers of Polyols, 2016; Vol. 1–10,.
- Standard ISO. 2884-2003 . Paints and Varnishes—Determination of Viscosity Using Rotary Viscometers, 2003;.
- ASTM D3576-04 . Standard Test Method Fo Cell Size of Rigid Cellular Plastics, 2010; Vol. 14, pp 1–5.
- ASTM D1622-20 . Standard Test Method for Apparent Density of Rigid Cellular Plastics, 2020; Vol. 1–5.
- ASTM D1621-16 . Standard Test Method for Compressive Properties of Rigid Cellular Plastics, 2016; Vol. 1–5,.
- ASTM C177-19 . Standard Test Method for Steady-State Heat Flux Measurements and Thermal Transmission Properties by Means of the Guarded-Hot-Plate Apparatus, 2019; Vol. 1–23,.
- Tang Z.; Wang L.; Yang J. Transesterification of rapeseed oil catalyzed by liquid organic amine in supercritical methanol in a continuous tubular-flow reactor. Eur. J. Lipid Sci. Technol. 2008, 110, 747–753. 10.1002/ejlt.200700256. [DOI] [Google Scholar]
- Murai M.; Sanou M.; Fujimoto T.; Baba F. Glycolysis of Rigid Polyurethane Foam under Various Reaction Conditions. J. Cell. Plast. 2003, 39, 15–27. 10.1177/002195503031021. [DOI] [Google Scholar]
- Robins J. Structural effects in metal ion catalysis of isocyanate-hydroxyl reactions. J. Appl. Polym. Sci. 1965, 9, 821–838. 10.1002/app.1965.070090303. [DOI] [Google Scholar]
- Britain J. W.; Gemeinhardt P. G. Catalysis of the Isocyanate-Hydroxyl Reaction. J. Appl. Polym. Sci. 1960, 4, 207–211. 10.1002/app.1960.070041112. [DOI] [Google Scholar]
- Molero C.; de Lucas A.; Rodríguez J. F. Activities of Octoate Salts as Novel Catalysts for the Transesterification of Flexible Polyurethane Foams with Diethylene Glycol. Polym. Degrad. Stab. 2009, 94, 533–539. 10.1016/j.polymdegradstab.2009.01.021. [DOI] [Google Scholar]
- Lobo H.; Bonilla J. V.. Handbook of Plastics Analysis (Plastics Engineering 68), 1st ed.; Lobo H., Bonilla J. V., Eds.; CRC Press, 2003; pp 656. [Google Scholar]
- Sharma B. K.Infrared Spectroscopy. Instrumental Methods of Chemical Analysis, 1st ed.; Goel Publishing House, 2005; pp 574. [Google Scholar]
- Dillon J. S.Infrared Spectroscopic Atlas of Polyurethanes, 1st ed.; Lancaster, 1991; pp 212. [Google Scholar]
- Socrates G.Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed.; John Wiley & Sons, Ltd., 2004; pp 364. [Google Scholar]
- Randall D.; Lee S.. The Polyurethanes Book, 1st ed.; John Wiley & Sons, Ed.; Chichester, 2002; pp 131. [Google Scholar]
- Modesti M.; Costantini F.; dal Lago E.; Piovesan F.; Roso M.; Boaretti C.; Lorenzetti A. Valuable Secondary Raw Material by Chemical Recycling of Polyisocyanurate Foams. Polym. Degrad. Stab. 2018, 156, 151–160. 10.1016/j.polymdegradstab.2018.08.011. [DOI] [Google Scholar]
- Kurańska M.; Pinto J. A.; Salach K.; Barreiro M. F.; Prociak A. Synthesis of Thermal Insulating Polyurethane Foams from Lignin and Rapeseed Based Polyols: A Comparative Study. Ind. Crops Prod. 2020, 143, 111882. 10.1016/j.indcrop.2019.111882. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.