

Abstract
Like temperature and pressure, solution pH is an important environmental variable in biomolecular simulations. Virtually all proteins depend on pH to maintain their structure and function. In conventional molecular dynamics (MD) simulations of proteins, pH is implicitly accounted for by assigning and fixing protonation states of titratable sidechains. This is a significant limitation, as the assigned protonation states may be wrong and they may change during dynamics. In this tutorial, we guide the reader in learning and using the various continuous constant pH MD methods in Amber and CHARMM packages, which have been applied to predict pKa values and elucidate proton-coupled conformational dynamics of a variety of proteins including enzymes and membrane transporters.
1. Introduction
1.1. Background
Solution pH is tightly regulated in biological environments [1]. In normal adult cells, intracellular pHi is about 7.2 and extracellular pHe is about 7.4 [2]; however, in cancer cells, the pH gradient is reversed, which promotes cancer progression, metabolic adaptation, and metastasis [2, 3]. Virtually all proteins depend on pH to maintain their structure and function [1, 4]. A small change in pH can perturb the functions of many enzymes. For example, SARS coronavirus main proteases have the maximal cleavage activity in a narrow pH range around 7.0 [5]. Below the optimum pH, the active site collapses due to the protonation state change of a histidine [5, 6]. The function of human β-secretase 1 peaks at pH 4.5, [7]; at low or high pH the protonation state of the catalytic dyad changes, which results in the closure of the “flap” that lies above the active site and blockage of substrate entrance [8]. Solution pH is also an important parameter in the creation of dynamic, stimuli-responsive materials [9]. For example, pH triggers the formation of hydrogel films from the biopolymer chitosan [10]; addition of surfactants allows the chitosan film to be reconfigured via a second pH signal to achieve different mechanical properties [11].
In a conventional molecular dynamics (MD) simulation, solution pH is implicitly taken into account by fixing the protonation states of titratable sites according to the experimental pKa values of model compounds (or peptides) or traditional pKa calculations based on a static structure. Traditional pKa methods include empirical approaches, e.g., PropKa [12], Rosetta [13], Poisson-Boltzmann solvers MCCE [14], APBS/PDB2PQR [15], DelPhiPKa [16], H++ [17], and other variants [18]. These tools are fast; however, the predicted protonation states are often incorrect for deeply buried sites [19], enzyme active sites [20], transmembrane proteins [21], and cysteines or lysines in general [22–24]. Also, even assuming an accurate assignment of protonation states at the beginning of MD, the conformational changes sampled by the simulation may modify the electrostatic environment around the titratable groups, changing their protonation states. To account for the direct coupling between conformational dynamics and protonation state changes and to offer more accurate pKa predictions, constant pH MD methods have been developed.
Over the past two decades, much progress has been made in the development of constant pH MD methods. In a discrete (also known as stochastic) constant pH MD (DpHMD), the MD trajectory is periodically interrupted by the Metropolis Monte-Carlo (MC) sampling of protonation states [25–29]. Here, protonation and deprotonation of titratable groups is sampled discretely, assuming one of the two states. Besides the advantage that only physical (protonated and deprotonated) states are sampled, the DpHMD method based on the hybrid MD/MC scheme is conceptually simple and practically straightforward to implement. In contrast, the continuous constant pH MD (CpHMD) [30–35] treats the protonation states of ionizable sites using an auxiliary set of continuous titration coordinates based on an extended Hamiltonian λ-dynamics approach [36]. This method allows the system to escape local energy minima by transiently accessing the partially protonated states. Although sampling the unphysial intermediate states is a major caveat, the CpHMD method offers significantly faster convergence for coupled residues, and importantly it can be readily extended to all-atom simulations. The faster convergence arises from the fact that at every MD step the λ values of all titratable sites are updated, whereas in a typical DpHMD implementation, the protonation state of a single residue is attempted at each MC step.
Development of an all-atom DpHMD algorithm based on the hybrid MD/MC approach is challenging, as in explicit solvent a switch in protonation state leads to a large change in electrostatic energy, which results in a nearly complete rejection of the MC move [29]. This problem, which is due to the lack of overlap between explicit solvent configurations for the protonated and deprotonated states [29], has been tackled by introducing a free energy calculation [37] or a short non-equilibrium MD (neMD) [28, 29, 38] (see later discussion).
In an implicit-solvent constant pH scheme, sampling of both conformational dynamics and protonation states is performed using a continuum electrostatic model, typically a generalized Born (GB) model. Implementations of this scheme in the CHARMM package [39] include the GBMV-[40] and GBSW- [41] based CpHMD [30–32] methods. Implementations in the Amber package [42] include the OBC-GB [43] based DpHMD [26] and the GBNeck2-based CpHMD [44, 45] methods.
In a hybrid-solvent constant pH scheme, conformational sampling is conducted with explicit solvent, while a continuum model e.g., GB or Poisson-Boltzmann (PB), is used to sample protonation states, i.e., calculating the solvation forces on the λ particles for the CpHMD methods or the energy changes due to protonation-state changes for the DpHMD methods. Implementations of this scheme include the first DpHMD method based on PB/TIP3P [25] for GROMACS [46], the GBSW/TIP3P based CpHMD method [33] in CHARMM [39], and the OBC-GB/TIP3P based DpHMD method [27] in Amber [42]. Note, the GBSW/TIP3P based CpHMD method [33] was extended to the treatment of transmembrane systems [21] by incorporating a membrane GBSW model [47].
An all-atom or fully explicit-solvent constant pH scheme removes the dependence on a continuum model in that it samples both conformational and protonation states in explicit solvent. An all-atom CpHMD method called CPHMDMSλD [48] was implemented by the Brooks group in the BLOCK module of the CHARMM package [39] as a part of the multi-site λ-dynamics (MSλD) functionality [49]. Most recently, the Brooks group developed a standalone GPU program called basic lambda dynamics engine (BLaDE) [50], which enables GPU-accelerated MSλD and constant pH simulations. The first all-atom particle mesh Ewald (PME) CpHMD method was implemented by the Shen group in CHARMM [39], as an extension to the GBSW and hybrid-solvent CpHMD methods in the PHMD module [34, 35, 51]. To compensate for the net charge fluctuation due to proton titration, the Shen group introduced co-titrating ions [34] or water [51]. Most recently, the all-atom PME CpHMD method was implemented in Amber (version Amber22 [52]) by the Shen group [53]. A λ-dynamics based all-atom CpHMD implementation was also developed by the Grubmüller group [54, 55] in the GROMACS package [46], although the method has not been validated for a large number of proteins yet. The proper treatment of tautomer states and the use of PME for λ dynamics remain to be addressed.
To circumvent the aforementioned configuration overlap problem in a DpHMD framework, Bürgi and van Gunsteren used thermodynamic integration (TI) to calculate the titration free energy change for the MC move [37]; however, TI calculations are computationally costly and do not readily converge. Recently, an all-atom DpHMD method based on a hybrid neMD/MC scheme has been implemented in NAMD [56] by the Roux group [29, 38]. This approach, which was originally proposed by Stern [28], has the flavor of both TI and λ dynamics methods, as it utilizes a (λ) coupling parameter in a neMD trajectory to gradually change the protonation state thus allowing solvent to adjust [29, 38]. Analogous to the titratable ion or water approach of the Shen group [34, 51], the hybrid neMD/MC implementation of the Roux group used the chemical transformation between a counterion and water to maintain charge neutrality of the system [29]. Interestingly, while the Roux group reported that charge neutrality acts as a constraint and decreases the acceptance ratio in the MC steps [29, 38], the Shen group observed a slow down in convergence of protonation state sampling [35, 51]. Having reviewed the various constant pH methods, the remainder of the tutorial will focus on the CpHMD implementations developed by the Shen group.
While accurate prediction of protonation states or pKa values is a goal, a major application of constant pH MD is to elucidate the mechanisms of pH-dependent or proton-coupled conformational dynamics. One can argue that the latter can also be studied by conventional fixed-protonation-state MD using simulations with different protonation states. While this may well be true when the switch of one protonation state is involved, the problem becomes intractable when the number N of titratable sites is large because the number of possible protonation states increases as 2N. Furthermore, the identity of the titratable site responsible for proton-coupled conformational changes is often unclear, which makes the fixed-protonation-state approach unfeasible. Importantly, by running fixed-protonation-state MD one cannot obtain the pKa value for the conformational transition [21, 57–59], which can be compared with experiment to validate the microscopic details revealed by simulation.
Table 1 summarizes the applications of the implicit-, hybrid-, and fully explicit-solvent CpHMD methods developed in the Shen group. The implicit-solvent GBSW- and GBNeck2-CpHMD methods have been mainly applied to pKa predictions of proteins [23, 32, 45, 60, 61], while the hybrid-solvent and all-atom CpHMD methods have been additionally applied to elucidate the mechanisms of pH-dependent or proton-coupled conformational processes, e.g., protein unfolding [62], structure-function relationships of proteases and kinases [6, 8, 63–65], protein/ligand binding [66–68], conformational activation of transmembrane channels and transporters [21, 58, 69], and materials [10, 11]. A recent application of GBNeck2-CpHMD method is the prediction of nucleophilic cysteine and lysine sites for targeted covalent drug design [6, 22, 24, 65, 70]. In this tutorial, we will discuss GBNeck2-CpHMD in Amber [20, 45], hybrid-solvent [33], and particle-mesh Ewald (PME) all-atom CpHMD [35] in CHARMM [39].
Table 1. Applications of the continuous constant pH MD (CpHMD) methods.
GBSW-CpHMD [31, 32], hybrid-solvent [33] and membrane-enabled hybrid-solvent [21] CpHMD methods are implemented in CHARMM [39]. The GRF [34, 51] and PME [35] based all-atom CpHMD methods are also implemented in CHARMM [39] and they can be used with co-titrating ions [34] or water [51]. GBNeck2-CpHMD [44, 45] method is implemented in Amber18 [42]. The pH replica-exchange protocol [33] was used for all applications except for those with GBSW-CpHMD where the temperature replica-exchange protocol [32] was used. All implementations except for GBNeck2-CpHMD are for CPU computing.
| System | Topic | Method | Reference |
|---|---|---|---|
| Soluble proteins | |||
| Benchmark proteins | pKa calculations | GBSW | [32, 60] |
| SNase | Blind pKa predictions for 87 engineered mutant proteins | GBSW | [61] |
| Peptides and mini-proteins | pH-dependent folding mechanisms | GBSW | [71–73] |
| Benchmark proteins | pKa calculations | Hybrid | [33] |
| NTL9, BBL | Unfolded states and unfolding mechanism | Hybrid | [62, 74] |
| SNase | pKa calculation for a deeply buried lysine; proton-coupled opening of the site | Hybrid | [75] |
| Spider silk protein | pH sensing residues for dimerization | Hybrid | [57] |
| BACE1, BACE2, cathepsin D, renin, plasmepsin D | Acid/base roles of the catalytic dyad; binding-induced protonation state change of the in-hibitor; pH-dependent conformational dynamics; proton-coupled dynamics of binding site water; pH-dependent inhibitor binding free energy calculations | Hybrid | [8, 64, 66–68, 76, 77] |
| c-Src Kinase | Proton-coupled conformational change of the DFG motif | Hybrid | [63] |
| Benchmark proteins | pKa calculations for Asp/Glu/His | All-atom | [35] |
| Various enzymes | Prediction of proton donor and nucleophile and physical determinants | Hybrid | [20] |
| Benchmark proteins | pKa calculations for Asp/Glu/His/Cys | GBNeck2 | [44, 45] |
| Various kinases | Prediction and rationalization of reactive Lys and Cys | GBNeck2 | [22, 24, 70] |
| Coronavirus papain-like proteases | Protonation states of His/Cys; proton-coupled conformational dynamics | GBNeck2 | [65] |
| Coronavirus main proteases | Protonation states of His and Cys; proton-coupled conformational change of the binding site | GBNeck2 | [6] |
| Transmembrane proteins | |||
| Proton channel M2 | Proton-coupled channel opening/closing | Membrane-hybrid | [69] |
| Sodium/proton antiporter NhaA | Identification of proton binding residues; proton-coupled conformational changes | Membrane-hybrid | [21, 59] |
| Efflux pump AcrB | Identification of proton binding residues; proton-coupled conformational transitions | Membrane-hybrid | [58] |
| μ-opioid receptor | pKa calculation | Membrane-hybrid | [78, 79] |
| Materials | |||
| Various surfactants | pKa calculation in micelle | GBSW and hybrid | [80] |
| Fatty acid | pKa calculation in micelle and bilayer | All-atom | [81] |
| Fatty acid | pH-dependent micelle/bilayer formation | Hybrid | [82, 83] |
| Peptide | pH-dependent unfolding of β-sheets; phase transition pKa | All-atom | [84] |
| Polysaccharide chitosan | Glucosamine titration; pH-dependent dissociation of a model crystallite; phase transition pKa; pH-dependent interaction with a surfactant | All-atom | [10, 11] |
1.2. The continuous constant pH molecular dynamics framework
In the CpHMD framework [30, 31], each titratable residue is assigned a titration coordinate λi, which is bound between 0 and 1, representing the (physical) protonated and deprotonated states, respectively. To satisfy the bounds, λi is expressed as a function of θ in the work of Shen group,
| (1) |
where θi can take on any value and is the underlying coordinate that is assigned fictitious mass (see below). The functional form of λ (Eq. 1) is somewhat arbitrary. In fact, in the MSλD based CpHMD method [49] as well as the λ-dynamics based CpHMD implementation in GROMACS [54, 55], λ is expressed using different functional forms of θ.
Since λ continuously evolves between 0 and 1, in simulation analysis we apply cutoffs to define protonated (λP < 0.2) and deprotonated states (λU > 0.8). Having the titration coordinate in place, we use an extended Hamiltonian to allow the simultaneous propagation of spatial (real) and titration (virtual) coordinates:
| (2) |
where ra refers to the (x,y,z) coordinates of atom a, and θi refers to the titration coordinate of titratable residue i. The two first terms of Eq. 2 give the kinetic energies of real atoms and virtual λ particles. Uint represent the titration-independent bonded and non-bond energies. We note that the force field parameters of some residue types (e.g., carboxylates) may depend on the protonation state; however, in the CpHMD [31, 33, 35, 44, 45, 48] as well as the hybrid MD/MC based DpHMD [26, 27] implementations the bonded parameters are fixed in one of the protonation states. This is a limitation that needs to be addressed in the future.
The fourth term, Uhybr, describes the non-bond energies, which are dependent on both spatial and titration coordinates. For the particle-mesh Ewald (PME) all-atom CpHMD [35] Uhybr includes only Coulomb and van der Waals (vdW) energies, while for the implicit- and hybrid-solvent CpHMD methods Uhybr also includes the generalized Born (GB) energy:
| (3) |
The dependence of the electrostatic energy on the titration coordinates arises from the requirement that the partial atomic charges on the titrating residue are linearly interpolated between the values in the deprotonated and protonated states with respect to λ:
| (4) |
where qj refers to the partial charge of an atom j in the titrating residue i. As to vdW energies, the interactions involving a titratable proton are linearly attenuated with respect to λ:
| (5) |
where i is the index for the titratable proton, j is the index for all other atoms, and refers to the protonation independent vdW interaction energy. If both i and j are titratable protons, the interaction is linearly attenuated by the λ values of both:
| (6) |
Note, the vdW energy correction is small and typically neglected in constant pH methods except for the CpHMD methods implemented in CHARMM and Amber [31, 33, 35, 44, 45].
Finally, the last term in Eq. 2 only affects titratable groups and is consisted of three biasing potentials:
| (7) |
The first biasing potential Umod describes a potential of mean force (PMF) of model titration along the λ-coordinate, where the model represents a fully solvent-exposed amino acid, i.e., a blocked single amino acid or a small peptide in solution. According to the linear response theory, model PMF is (in GB solvent) or can be approximated (in explicit solvent) as a quadratic function:
| (8) |
Here A and B are parameters that can be determined through free energy calculation methods, such as thermodynamic integration (TI, see section Parameterization). By analogy, in the case of coupled double-site titration [31], e.g., histidine or carboxylic acid, the model PMF is second order in both λ and x, where x is a coordinate that represents the tautomer interconversion. The two-dimensional PMF can be written as a bivariate polynomial,
| (9) |
Here is bound between 0 and 1, where is the underlying unbound variable.
Ubarr, the second biasing potential in Eq. 7, describes a barrier or penalty potential in the center of the titration coordinate,
| (10) |
where β is a parameter that determines the height of the barrier. Adding the barrier potential serves to increase the sampling time at the end-point states (e.g., λ < 0.2 or λ > 0.8) and therefore minimizing the unphysical vdW and electrostatic interactions associated with mixed state (e.g., 0.2 ≤ λ ≤ 0.8).
UpH, the last term of Eq. 7 describes the pH dependence of the deprotonation free energy and is given by
| (11) |
where kB is the Boltzmann constant, T is the system temperature, and pKamod is the model pKa for the titratable residue i.
1.3. Scope
This tutorial mainly covers two CpHMD methods that have been extensively validated and applied to various real-life problems: 1) the hybrid-solvent CpHMD [33] in CHARMM [39] and 2) the GBNeck2 implicit-solvent CpHMD [44, 45] in Amber [42]. Currently, fully patched Amber 18 and 20 released versions are supported. GBNeck2-CpHMD patches to them can be downloaded from our cphmd-patches repository. Details about how to apply them are described on that repository page. The CHARMM hybrid-solvent CpHMD currently supports CPUs only, but the Amber GBNeck2-CpHMD supports both CPU or GPU computing. We strongly recommend running the CpHMD simulations via the pH replica-exchange protocol to significantly accelerate convergence of pKa’s and conformational sampling; however, if desired, a single pH simulation or a set of independent pH simulations can also be conducted.
In addition to the above two methods, we will briefly discuss the PME all-atom CpHMD method implemented in CHARMM for CPU computing [35] and in Amber for GPU computing (Harris, Liu, and Shen, unpublished data). We note, this tutorial does not cover the GBSW implicit-solvent CpHMD method in CHARMM [30–32, 85], which is available for both CPU and GPU computing. The GBSW-CpHMD method with the temperature replica-exchange protocol has been extensively validated for protein pKa predictions and pH-dependent conformational dynamics.
2. Prerequisites
CpHMD is a specialized MD technique, so the user should have some familiarity with MD simulations and especially with the Amber or CHARMM packages. Users having experience with other MD engines, such as GROMACS [86], NAMD[87], or OpenMM [88] are encouraged to go through the introductory tutorials for Amber or CHARMM to get started. Before running a CpHMD simulation, it is important to know the research objective. If the objective is to predict the pKa values of soluble proteins or their protonation states at a certain pH condition, we recommend the GPU-accelerated GBNeck2-CpHMD in Amber [45], as the pKa’s converge rapidly and the accuracies for titrating Asp, Glu, His, Cys, and Lys (in solvent-exposed and buried sites) have been validated using a large number of proteins (see references in Table 1). If the objective is to investigate the detailed proton-coupled conformational dynamics, proton transfer, protein-ligand binding/unbinding, or transmembrane proteins, we currently recommend the hybrid-solvent CpHMD [33], as the accuracy has been extensively validated in terms of pKa values and description of proton-dependent conformational dynamics (see example applications in Table 1), although speed is limited due to the use of CPUs. We note, a GPU-accelerated all-atom PME CpHMD implementation in Amber22 [52] has been recently released by us [53] and holds a promise to offer more accurate description of proton-coupled conformational dynamics for heterogeneous systems such as protein-ligand complexes and transmembrane proteins. Like all computational chemistry calculations, it is important to know the system of interest before attempting any simulations. With such knowledge, one an properly choose the salt concentration, temperature, pH range, and titratable residue types to use in CpHMD. Also, the research objective dictates the simulation length. For example, to predict protonation states, simulation can be run until the unprotonated fractions at all pH are converged, typically 10–50 ns per replica. If however, a large conformational transition is of interest, the simulation should be much longer.
2.1. Background knowledge
Knowledge of classical MD is required to understand what we can and cannot achieve through a MD simulation. The user should also understand the parameters used in energy minimization, heating, equilibration, and production runs. Of course, chemistry knowledge of pH, pKa, and protonation states and how they may impact the physical properties of the specific biological or material system is essential for obtaining meaningful results.
CpHMD simulations are run on a workstation or high-performance computing (HPC) cluster under a Linux operating system. The user is expected to have a good knowledge of Linux commands for accessing and editing files, submitting jobs to servers, and retrieving simulation result files. Shell script and preferably also Python skills are required for pre- and post-processing files. For simulations on a HPC cluster, the user is required to be familiar with the specific batch scheduler and queuing system, e.g., SLURM (Simple Linux Universal Resource Manager), SGE (Sun Grid Engine), or PBS (Portable Batch System). The user is expected to know commands for submitting and checking the status of jobs. The user is also expected to set environmental variables required for the specific MD engine. Nowadays, a CpHMD job can also be performed on a workstation with a single or multiple GPUs. In this case, the user is required to use commands such as ‘nvidia-smi’ to check the GPU status and ‘nvcc -version’ to check the CUDA version as well. The user can use any data visualisation tool(s), but matplotlib in Python and Jupyter notebooks are recommended in our analysis tool set.
2.2. Software/system requirements
For GBNeck2-CpHMD, Amber16 is the minimum version. Requirements for installing Amber can be found in the Amber website. For the GPU version (pmemd.cuda), a Nvidia GPU card with at least compute compatibility sm3.0 should be installed with the latest driver. A version of CUDA required by Amber needs to be installed. After successfully installing CUDA, it is important to set three environmental variables in Linux. CUDA_HOME and LD_LIBRARY_PATH must point to the directories where the desired CUDA version is installed. We also need to specify CUDA_VISIBLE_DEVICES to GPU device indices. For example,
$ export CUDA_HOME=/usr/local/cuda−10.1 $ export LD_LIBRARY_PATH=/usr/local/cuda−10.1/lib64 $ export CUDA_VISIBLE_DEVICES=0,1
These variables will direct the Linux system to use the CUDA 10.1 version installed in the /usr/local directory. In the example above two GPU cards are available, and the indices 0 and 1 for CUDA_VISIBLE_DEVICES indicate the first and second GPU to use, respectively. Following installation of Amber, the user should set the environmental variable AMBER-HOME to the Amber installation folder.
$ export AMBERHOME=/path_to_Amber_version_xx/
Before preparing CpHMD inputs of a protein system, we need to clean up and fix problems in the PDB file, e.g., removing non-protein atoms, adding missing residues and atoms, and fixing non-canonical amino acid residues. To do that, we can use the Perl script convpdb.pl in mmtsb tool set [89] or pdbfixer in OpenMM [88]. For example, the following commands can be used to fix the file 1vii.pdb downloaded from RCSB (www.rcsb.org) [90].
$ convpdb.pl −nohetero −fixcoo −renumber 1 −out amber −chain A −segname 1 vii.pdb
or
$ pdbfixer −−add−atoms=heavy −−keep−heterogens=none −−add−residues −−replace −nonstandard 1 vii.pdb
Alternatively, we can use a homology modeling software such as SWISS-MODEL [91] to fix those problems especially when the number of continuously missing residues is large [91].
The next step is to add capping ligands ACE and NH2 to the N- and C-terminus and to modify ASP, GLU and HIS to AS2, GL2, and HIP residues if these residue types are set as titratable. For AS2 and GL2, a library file phmd.lib and a force field modification file frcmod.phmd are included in Ambertools after version 19 [42], or we can load them to tLeap/Leap from the directory containing the two files. The last step is to use tLeap/Leap in Amber/ Ambertools to generate topology and parameter files. Our cphmd-prep tool set uses the pdbfixer tool for the first step and can do all the aforementioned steps to prepare GBNeck2-CpHMD compatible input files.
We use a Python-based asynchronous replica exchange implementation for GPU pH replica-exchange simulations. Scripts in cphmd-analysis are used for analyzing CpHMD related files, including convergence checking, pKa calculation, and replica-exchange profiling. Some simple trajectory based analysis such as RMSD can also be done, but the users can use any tools, e.g., CPPTRAJ [92], PTRAJ [92], VMD [93], and MDAnalysis [94].
A suggestion to use Python dependent packages is to use Anaconda or Miniconda. With them, the user can create a virtual environment specific for CpHMD related calculations. For example, after installing Anaconda or Miniconda, we can type
$ conda create −−name cphmd python=3.7
Note that we choose Python version 3.7 because the OpenMM package currently only supports up to Python 3.7. Activate the environment named ‘cphmd’ by
$ conda activate cphmd
Many packages can be installed through the conda and pip tool.
$ conda install −c omnia pdbfixer parmed $ conda install −c conda−forge ambertools mdanalysis matplotlib $ conda install −c anaconda numpy scipy pandas $ pip install f90nml
Whenever the user wants to do some calculations or analysis related to CpHMD, they can activate this ‘cphmd’ virtual environment and use the installed tools without causing conflict with other software.
For running the hybrid-solvent CpHMD [33] in CHARMM [39] you will need to follow the installation instructions in the CHARMM documentation. We suggest including the MPI and the Hamiltonian replica exchange options.
$ ./install.com gnu xxlarge M mpif90 +CMPI +REPDSTR +ASYNC_PME +GENCOMM
If you want to install CHARMM on a local workstation for building and preparing systems or for running a single pH simulation, you can use the following installation command.
$ ./install.com gnu xxlarge
3. Workflow for the hybrid-solvent CpHMD in CHARMM
The hybrid-solvent CpHMD method makes uses of explicit solvent for conformational dynamics and a GB model for propagating protonation states [33]. As all MD simulations, CpHMD consists of three stages: system preparation, equilibration, and production. We will explain each stage using a specific protein as an example. To follow along, the tutorial files for soluble proteins can be downloaded from our GitLab website cphmd-tutorial/hphmd_charmm and for transmembrane proteins, the tutorial files can be downloaded from cphmd-tutorial/memb_hphmd_charmm. We will use the pH replica-exchange protocol [33] in all the examples.
3.1. System Preparation

To demonstrate CpHMD simulations of soluble proteins without retaining the crystal waters, we use the mini-protein BBL as an example. The tutorial files for system preparation can be found in the directory bbl_sys_prep.
Step 1. Prepare a protein structure with dummy hydrogens.
First, we retrieve the coordinates of a protein structure from Protein Data Bank[90], e.g., BBL (PDB: 1w4h [95]). A homology modeling tool such as SWISS-MODEL[91] can be used to add any missing atomic coordinates. Next, we convert the PDB file from the standard PDB format to the CHARMM PDB format using the MMTSB tool command [89] convpdb.pl or a user supplied script followed by a shell command “sed” to change all histidine residue names to HSP to facilitate titration.
$ perl convpdb.pl −out charmm22 −segnames −renumber 1 1w4h.pdb | sed ” s/HSD/HSP/g” >start. pdb
Here, the output PDB file is formatted for CHARMM (-out charmm22); the segment names are included in the output (-segnames); the residue identification numbers (resid) are renumbered starting at 1; and the input file is “1w4h.pdb.” The CHARMM default tautomer for His is HSD. Changing HSD to HSP indicates to the CpHMD program that histidine is titratable (see later discussion of the parameter file). Although all resid have been renumbered starting from 1, this does not have to be the case. Next we add hydrogen atoms to the protein using the HBUILD facility[96] in CHARMM [39].
To allow titration of Asp and Glu sidechains, we place dummy hydrogens on both carboxylate oxygens. Note, the dummy atoms do not interact with each other and only one is turned on when Asp/Glu is protonated [31]. The dummy hydrogens are placed in the syn position, because it is considered more favorable than the anti position (see discussion in Ref [31]). To prevent the dummy hydrogens from moving to the anti position, in which case nonnative hydrogen bonds may form and a neutral dummy may lose the ability to gain charge, in the CpHMD specific force field parameter file, the C–O bond rotation energy barrier is increased from the standard 0.5 to 6.0 kcal/mol (see discussion in Ref [31]).
Following the addition of hydrogens, a brief energy minimization is performed to correct unfavorable positions. Here 50 steps of steepest descent (SD) and 10 steps of adopted basis Newton-Raphson (ABNR) are used, whereby a harmonic restraint with the force constant of 50 kcal/mol/Å2 is placed on the heavy atoms. In many experiments, the N- and C-terminal ends of the protein are truncated and require capping. For N-terminus we use CH3CO (patch ACE), and for C-terminus we use NH2 (patch CT2) or NHCH3 (patch CT3). In special cases, the free ionized forms, and COO–, are used and can be set titratable when CpHMD is turned on. The input file step1_add_h_prot.inp is an example of how to perform these steps on BBL and can be run with the following command.
$ charmm − i step1_add_h_prot.inp −o step1.out
The example input also checks for disulfide bonds using a sulfur-to-sulfur distance cutoff of 2.5 Å. After running any input file, make sure to visualize the output structure to confirm all the dummy hydrogens and additional features are correctly constructed.
Step 2. Prepare a solvated system
Now we move to protein solvation by first building a large water box of the desired size. This is done by generating a plane of small cubic water boxes along the x- and y-axes, and then stacking the planar water boxes along the z-axis. CHARMM toppar directory contains the cubic water box made of preequilibrated TIP3P waters. The size of the large water box is calculated based on the longest dimension of the protein and a padding space between the protein and the edges of the water box. We recommend having at least ~10 Å padding in x, y, and z directions. The large cubic water box is then reshaped to a truncated octahedron by removing waters in all eight corners. The input file step2a_make_waterbox.inp is used to construct the octahedron water box.
$ charmm − i step2a_make_waterbox.inp −o step2_make_waterbox.out
Once the water box is built, the protein is placed at the center and water molecules within 2.8 Å of the protein heavy atoms are deleted.
The solvated system is subject to energy minimization to release unfavorable contacts. First, the protein heavy atoms are fixed and the system undergoes energy minimization using SD and ABNR for 50 steps each. Next, a five-stage minimization is conducted with the harmonic constants of 100, 50, 25, 5, and 0 kcal/mol on the protein heavy atoms using 50 steps of SD and 100 steps of ABNR in each stage. The input file step2b_solvate.inp can be used.
$ charmm − i step2b_solvate.inp −o step2b_solvate.out
Again, always remember to visualize the system and make sure the system has been constructed to your expectations.
3.2. System preparation that includes crystal waters

In some cases the X-ray structure contains water molecules and it is desirable to keep them, e.g., those in the enzyme active site. A tutorial showing how to set up a system with crystal waters can be found in the directory crys_wat_system_prep. Here we use a small protein, staphylococcus nuclease (SNase), as an example, as the X-ray structure (PDB ID: 3hzx) contains crystal waters. To keep the crystal waters, just a few additional steps are needed. First, move all the water molecules into a separate PDB file and make sure the file is properly formatted with the correct column names and column spacing. In the tutorial the crystal water coordinates have been exported into the the file xwat_original.pdb; however, this file needs to be reformatted for use in CHARMM. The Python script mod_xwat.py reformats this file to xwat_.pdb. We should note that most error messages involving the handling of crystal waters are due to the PDB file not being properly formatted.
Since the crystal waters resolved in a X-ray structure do not contain hydrogen positions, they can be added using the CHARMM input file step1b_add_h_crys_wat.inp. With hydrogens added to the protein and crystal waters, the two sets of coordinates need to be combined using the CHARMM input file step1c_merge_prot_crys_wat.inp. The rest of the system preparation is very similar to that without crystal waters. The only difference to note is when you solvate the system you can merge the segids of the crystal waters (segid: XWAT) with the bulk water (segid: SOLV) using the following command.
JOIN XWAT SOLV renumber RENAME SEGID SOLV sele segid XWAT end
Keep in mind this needs to be done after the deletion of water that overlaps with the protein; otherwise you run the risk of accidentally removing some crystal waters. An example input file is step2b_solvate_crys_wat.inp.
3.3. System preparation for transmembrane proteins

Due to the presence of a lipid bilayer, the preparation of transmembrane proteins for CpHMD simulations requires additional steps. We divide the preparation into the initial and final stages. The initial preparation is for fixed-charged equilibration, whereas the final preparation is for CpHMD simulations. The related files can be accessed from the directories initial_system_prep and final_system_prep.
To begin the initial system preparation, a solvated protein-membrane system is built. We start by retrieving the protein structure from the database OPM (Orientations of Proteins in Membranes), which properly orients the protein in the lipid bilayer using theoretical calculations and experimental data.[97] Just like for soluble proteins, any missing residues need to be added. Here we adopt the standard protonation states, i.e., Asp(−), Glu(−), Cys(0), Lys(+), and Arg(+). As the solution or model pKa of His is 6.54 [98], which is within one unit of the physiological pH, special attention needs to be given to its protonation (singly or doubly protonated) and tautomeric state (HSD or HSE) by considering the nearby hydrogen bonding and/or salt-bridge interactions. Like soluble proteins, the N- and C-terminal ends may be capped or if necessary the charged forms may be used.
With the complete protein structure (all heavy atom positions are in place), we use the script step1_genpsf.inp to build hydrogen positions followed by a short energy minimization in the GB implicit-solvent model to adjust any energetically unfavorable hydrogen positions. Next, we build the rest of the system and equilibrate the lipid bilayer following the CHARMM-GUI protocol [99] for placing the lipids, solvating the system, and adding ions. The input files for these steps can be found in the directory step1-5. Below we will briefly go through the steps.
After the protein structure is prepared, we place it at the origin and estimate its cross section using the script step2.1_orient.inp. The next step is to add water to the pore of the protein using the script step2.2_pwat.inp. To determine the system dimensions the script step3.1_size.inp is used. This script also estimates the lipid type area and the number of lipids required to be placed between the protein boundary and the periodic boundary. Approximately 4 to 5 lipids should be placed between the protein and the periodic boundary. Additionally, the script calculates the thickness of the water layer above and below the lipid bilayer. With the dimensions of the system determined, the lipid heads using big dummy spheres are placed to estimate the positions of the lipids in the bilayer. This is accomplished by step3.2_packing.inp. Next, the dummy spheres are replaced with lipid molecules from a library of randomized conformations using the script step4.1_lipid.inp. In this example, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) is used, but if other lipids are desired, the libraries of randomized conformations of lipid molecules can be found in the CHARMM-GUI Archive - Individual Lipid Molecule Library.
After the lipid bilayer is built, a rectangular water box is generated using the script step4.2_waterbox.inp, which solvates the protein/membrane complex. Next, the number of ions required to achieve the desired salt concentration is calculated, and the ions are randomly placed in the slabs of water above and below the membrane. In the final step, all of the system components are combined using the script step5_assembly.inp.
We now relax the assembled protein/membrane system in six stages with gradually reduced harmonic restraints using the fixed-charge MD CHARMM-GUI scripts in the directory step6. Following the system relaxation, a much longer (100 ns) simulation in the NPT ensemble is required to fully equilibrate the lipid bilayer. The protein heavy atoms are harmonically restrained during the simulation. The progress of the bilayer equilibration is monitored by the calculation of three properties: surface area per lipid, bilayer thickness, and lipid order parameters [100, 101]. The system is considered equilibrated when these properties plateau with the simulation time. Since this can be performed on any scalable or GPU-enabled MD engine using a standard protocol, we leave this step up to the user.
Following the fixed-charge equilibration of the lipid bilayer, we move to the final system preparation. Here, we prepare the protein for CpHMD titration by adding dummy hydrogens. Example input files can be found in the directory final_system_prep. The first step to break the final snap shot from the bilayer equilibration into separate components (protein, bilayer, bulk water, pore water, and ions). We add hydrogens using the script step1a_add_h.inp. PSF and CRD files are generated for the rest of the system components with the following scripts: step1b_genpsf_memb.inp, step1c_genpsf_water_bulk.inp, step1d_genpsf_water_pwat.inp, step1e_genpsf_ion_sod.inp, and step1f_genpsf_ion_cla.inp. Once all the PSF and CRD files have been made, the rest of the system can be rebuilt using the script step2_assemble.inp.
3.4. Equilibration of soluble proteins

Equilibration of soluble proteins for CpHMD is performed at a single pH in several stages. An example input for the BBL protein is equil_hphmd.inp. Since CpHMD is turned on during equilibration, titratable residues must be chosen. In the example here, Asp, Glu, and His are allowed to titrate because of the pH range used in the production simulation. The crystal structure pH condition or physiological pH 7.4 can be used. We specify the physiological ionic strength of 0.15 M and temperature of 298 K. Before invoking the PHMD module for CpHMD simulation, we need to call GBSW using the following command.
GBSW HYBRID SGAMMA 0.005 NANG 50 CONC 0.15 − SELE protein END
Here, SGAMMA (default 0.005) is the non-polar surface tension coefficient, NANG (default 50) is the number of angular integration points, and CONC (default 0.15) is the ionic strength in M. Except for CONC, which the user may change according to the specific problem, the default parameters should be used.
Now we invoke PHMD and set related options using the following command.
PHMD PAR 23 WRI 25 PH 7.4 NPRI 5000 PHFRQ 5 MASS 10.0 BARR 2.0 BETA 5.0 TEMP 298 LAM − SELE RESN ASP. or. RESN GLU. or. RESN HSP end
Here PH is the simulation pH (default 7); NPRI is the frequency of printing lambda values (5000 means 10 ps); PHFRQ is the update frequency for the lambda coordinates (default 5); TEMP is the temperature for λ dynamics (default 298); MASS is the fictitious mass of the λ particles (default 10); BARR is the quadratic barrier height (default 2.0 and is overwritten by the values specified in the parameter file phmd.inp); BETA is the friction coefficient for lambda dynamics (default 5.0); LAM tells the PHMD module to output lambda values; and SELE specifies the types of residues allowed to titrate (default all residues in phmd.inp file). Except for PH, NPRI, and SELE options, the default parameters should be used.
The length of each stage depends on the system and should be adjusted accordingly. In the BBL example equil_hphmd.inp, the heating/equilibration stages were run for 20 ps, because the system is very small and stable. First, the system is heated in the NVT ensemble, and next four stages of equilibration are performed in the NPT ensemble with decreasing harmonic restraints on the heavy atom positions, e.g., 5.0, 1.0, 0.1, and 0 kcal/mol/Å2. The idea here is to slowly relax the system by removing any high energy contacts and to allow for the initialization of protonation states at the specified pH (e.g., 7). Note, the equilibration time for the mini-protein BBL is short, and one can adjust it accordingly for larger proteins.
3.5. Equilibration of transmembrane proteins

Now three short equilibration steps can be performed using CpHMD at a single pH. Example inputs are in the directory cphmd_equil. As for a soluble protein, the specified pH should be the pH used in the experiment (e.g., crystal growth, NMR, etc) or physiological pH. In the first step, the CpHMD simulation is performed in the NVT ensemble with a harmonic force constant of 1.0 kcal/mol/Å2 for 0.1 ns using step0.1_equil.inp. In the second step, the CpHMD simulation is performed in the NPT ensemble using step0.2_equil.inp. In the third step, the simulation (step0.3_equil.inp) continues in the NPT ensemble for 0.8 ns with all restraints are removed. We note, the membrane-enabled CpHMD simulation makes use of the membrane GBSW model [47].
GBSW HYBRID SGAMMA 0.0 NANG 50 CONC 0.15 − TEMP 310 TMEMB 30 MSW 2.5 − RCYLN 15 SELE solu END
The options TMEMB, MSM, and RCYLN are used to specify a low dielectric slab in the GBSW model with a high dielectric cylinder centered in the protein. TMEMB specifies the slab thickness (Å) and should be set to the average difference in the Z positions of the lipid C2 atoms from the top and bottom leaflets. This will vary for different lipid types. MSM specifies the half membrane switching length for the dielectric transition between the low-dielectric slab and the bulk solution; the default is 2.5 Å. Finally, RCYLN is the radius of a high dielectric water cylinder placed in the center of the protein; it should be set to an appropriate value such that the water cylinder covers the entire protein but does not overlap with lipids as much as possible. Although no positional restraints are placed on the protein, we recommend using a cylindrical restraint with a harmonic force constant of 0.1 kcal/mol/Å2 to prevent the protein from a lateral drift. This can be done using the MMFP facility in CHARMM [39].
3.6. Production

The CpHMD production stage is similar for soluble and transmembrane proteins. The only difference is that the GBSW command includes additional options for the lipid bilayer (see discussion of the equilibration steps) for transmembrane protein simulations. An example input for the production run of a soluble protein is prod_hphrex.inp. A similar production input for a transmembrane protein is prod_hphrex.inp. An example job submission file for a HPC cluster is hphrex.qsub.
To begin the production run one must choose a pH range and number of pH replicas. The proper choice of the pH range depends on the research objective. For example, the pH range of 1.5–6.5 was used in the hybrid-solvent CpHMD study of the pH-dependent structure function relationship of the endosomal protein BACE1 in which the catalytic aspartyl residues play a central role [8]. On the other hand, the pH range 4–8 was used in the membrane-enabled hybrid-solvent CpHMD study of the pH-dependent conformational activation of the transmembrane M2 channel, whereby the titration of four histidines modulates the conformation of the channel [69]. Next, one needs to choose the total number of pH replicas. The spacing of the pH conditions needs to be close enough such that sufficient exchange probabilities (e.g., at least 20%) are obtained. To obtain good overlap between potential energy distributions of neighboring replicas a pH spacing of 0.5 and sometimes even 0.25 is required. Other aspects to consider are the number of available CPUs on the HPC and how many CPUs can be allocated to each replica. Simulations with CHARMM do not scale well past 8 CPUs, so for a job using 16 replicas we recommend requesting a total of 128 CPUs with 8 CPUs for each replica. We recommend making exchange attempts every 1000 MD steps. A typical pH replica-exchange CpHMD simulation has an average exchange ratio of ~40%, but if the exchange ratios are too low, more replicas can be added to fill in the spacing between replicas. To achieve optimum performance in enhanced sampling, several replicas should move across all pH conditions.
To use pH-based replica exchange in CHARMM, one can use the REPD module.
REPD NREP 8 PHMD EXLM FREQ 5000 UNIT 17
Here, the REPD command initiates replica exchange, and NREP states the number of replicas to use (in this case 8). PHMD EXLM states this is a special type of Hamiltonian exchange (i.e., pH-based) and allows for the exchange of pH conditions of two adjacent replicas. FREQ states the frequency of an exchange attempt.
Then, to run several replicas in parallel, CHARMM reads in a rep.cmd file for each pH replica with the syntax, rep.cmd_0, rep.cmd_1, to rep.cmd_(NREPS), using the following command in the input file.
$ stream rep.cmd
Notice this command lacks the ‘_0’ portion of the name. In each of the rep.cmd file you will have the command that states the pH environment of the replica.
PHMD RESPH OLDPH @crysph NEWPH @ph PKATEMP @temp
Here OLDPH is the pH used in the equilibration and NEWPH is the pH of the replica. Finally, PKATEMP should be set to the same temperature of the simulation. The rest of the replica exchange set up follows a standard MD run in CHARMM. Production run inputs for BBL (prod_hphrex.inp and rep.cmd) can be used to run one 2 ns stage of pH-Replica Exchange CpHMD.
3.7. General settings for MD and CpHMD in CHARMM
For all simulations the protein is represented by CHARMM22/CMAP force field.[102, 103] Note, the current CpHMD methods are not compatible with the CHARMM36 protein force field. For solvent the CHARMM modified TIP3P water model is used. For ions or lipid molecules the CHARMM36 force fields are used [100]. The van der Waals energies are calculated with a switching function from 10 to 12 Å and a cutoff at 14 Å. All simulations are conducted under periodic boundary conditions. The PME method [104] is used to calculate the long-range electrostatic energies and forces with a real space calculation cutoff of 12 Å and 1 Å grid spacing, and a 6th-order spline interpolation. The non-bond neighbor list is updated heuristically. Additionally, the SHAKE [105] algorithm is used to constrain all bonds involving hydrogens to allow for a 2-fs time step. For NPT simulations, the temperature is maintained with the Nóse-Hoover thermostat [106, 107]. The pressure is maintained with the Langevin piston pressure-coupling algorithm [108]. For the CpHMD simulations, the default GBSW radii [109, 110] are used for the protein. For small molecules, the generic atomic radii in the GBSW file may be used upon user verification. A CpHMD parameter file phmd.inp contains the model reference pKa values and the parameters in the titration potential of mean force for Asp, Glu, His, Cys, and Lys. Parameters for additional titratable groups can be obtained by following the steps discussed in section Parameterization.
4. Workflow for all-atom PME CpHMD in CHARMM
Minor modifications to the workflow for hybrid-solvent CpHMD are needed to set up and run all-atom PME CpHMD simulations in CHARMM [35]. The example inputs for simulations of BBL are downloadable from ephmd_charmm. To maintain charge neutrality during the all-atom PME CpHMD simulation, titratable water molecules [51] (or co-ions [34] in an earlier version) should be added. Specifically, each acidic (Asp or Glu) or basic residue (His or Lys) is coupled with one titratable water that can convert to a hydroxide (TIPU) or hydronium (TIPP). Details are explained in Ref [51]. Together with salt ions, titratable water molecules are placed in the water box using the input script step3_add_ions_titr.inp.
To run an all-atom PME CpHMD simulation, make sure the command GBSW is NOT invoked before calling PHMD. The additional options need to be added in the PHMD command to specify the residue names of titratable water (TIPU and TIPP) and the total number as well as residue IDs of the coupled pairs. For example, in the BBL simulation, 3 Asp, 3 Glu, and 2 His are titratable, and they are coupled to 8 titratable water molecules.
PHMD PAR 23 WRI 25 PH 7.0 NPRI 5000 PHFRQ 5 MASS 10.0 BETA 5.0 TEMP 298 LAM − sele resn ASP. Or. resn GLU. or. TIPU. or. resn HSP. or. TIPP end − qcouple 8 − resi 4 resc 4789 − resi 16 resc 4790 − resi 20 resc 4791 − resi 36 resc 4792 − resi 37 resc 4793 − resi 39 resc 4794 − resi 17 resc 4771 − resi 41 resc 4772
Here resn TIPU and TIPP declare the two types of titratable water. The option qcouple 8 specifies a total number of 8 coupled pairs. Following qcouple, residue IDs for the titratable group (e.g., resi 4) and co-titrating water (e.g., resc 4789) are listed. Note, all co-titrating pairs need to be specified manually.
5. Workflow for implicit-solvent CpHMD in Amber
Here we discuss how to run GBNeck2-CpHMD [44, 45] simulations in Amber. The related files can be found in the GitLab directory cphmd-tutorial/gphmd_Amber.
5.1. Preparation of structure and input files

To simplify system preparation for GBNeck2-CpHMD simulations, we recommend the user to download and install a tool set CpHMD-prep. Once installed, the shell script cphmd_prep.sh can be called to prepare the structure and input files for running CpHMD.
Before explaining cphmd_prep.sh, we briefly explain the important CpHMD specific files and options in Amber. Similar to CpHMD simulations in CHARMM (section 3.1), we first convert the PDB file to the Amber format, cap the terminal groups, and change the names of titratable Asp, Glu, and His residues to AS2, GL2, and HIP, respectively. We then add dummy hydrogens to AS2 and GL2. Besides the standard Amber force field files, tleap requires two CpHMD specific files for building topology and parameters: frcmod.phmd, which specifies the modifications of bonded parameters for AS2 and GL2, and phmd.lib, which contains the definitions of AS2 and GL2. For GBNeck2-CpHMD, set the PBradii to mbondi3 in tleap (see later discussion of modifications). For running CpHMD, an additional file gbneck2_input.parm is needed, which contains the partial charges and van der Waals energy flags of the protonated/deprotonated forms of the titratable residues as well as the parameters of the model titration PMFs (see section 7).
To run CpHMD, the pmemd.cuda command should contain several CpHMD options. The option -phmdparm reads in the aforementioned CpHMD parameter file gbneck2_input.parm. The option -phmdin reads in a CpHMD job control file, which specifies various options for CpHMD simulations. We note, except for MaskTitrRes(:) (titratable residue types, e.g., ‘AS2’, ‘GL2’, ‘HIP’, ‘CYS’, ‘LYS’) and Mask-TitrResTypes (number of titratable residues, e.g., 5), the default settings in the phmdin file should be kept. Finally, if specified the option -phmdstrt reads in a CpHMD restart file. We should also point out that for CpHMD runs, the Amber mdin files are similar to those for the fixed-charge MD using GBNeck2 implicit solvent and Langevin dynamics. The two CpHMD specific flags are iphmd and solvph. For GBNeck2-CpHMD, iphmd should be set to 1 and solvph should be set to the desired pH condition.
Now we explain the shell script cphmd_prep.sh.
cphmd_prep. sh [−pdb|−−pdbid] | [− fil |−−filename] [−cha|−−chainid] [−mod|−−modelid] [−con|−−conc] [− tim|−−time] [−tem|−−temp] [−dph] [− phl|−−phlow] [−phu|−−phup] [− res|−−restype] [−run|−−runmode] [−h|−−help]
If specified, the option -pdb|–pdbid reads in a PDB ID and downloads the corresponding structure from RCSB. If no PDB ID is given, the option -fil|–filename reads in a PDB file. The option -cha|–chainid specifies the chain ID to include in simulations (default ‘A’). If specified, the option -mod|– modelid reads in a rotamer model ID (e.g., A or B) in the PDB file. The following options are related to the simulation: -con|–conc specifies the ionic strength in M (default 0.15); -tim|–time specifies the simulation length in ns for each pH condition (default 10); -tem|–temperature specifies the simulation temperature in K (default 300); -phl|–phlow specifies the lowest pH (default 6.5); -phu|–phup specifies the highest pH (default 9.0); -phi|–pHintvl specifies the pH interval for calculating the individual simulation pH conditions between phlow and phup; -res|–restype specifies the amino acid residue types allowed to be titratable (default ‘Asp Glu His Cys Lys’); and -rm|–runmode specifies the type of simulation (‘async’ for asynchronous pH replica exchange, ‘rex’ for synchronous pH replica exchange, and ‘ind’ for independent pH simulations). Finally, -t|–test offers a test option (‘t’ means temporary files will be kept for checking) and -h|–help displays the usage and options.
cphmd_prep.sh accepts either a PDB ID or a user-prepared PDB file and calls PDBFixer in OpenMM[88] to extract the desired chain, add missing heavy atoms or residues and the terminal capping groups (CH3CO and NH2). Note, ions, ligands, waters, and unspecified chains from the PDB file are removed. Once the PDB file is cleaned up, tleap in Amber (or Ambertools) is used to build the positions of hydrogen as well as dummy hydrogen atoms according to the specified titratable residue types. As required by GBNeck2-CpHMD, [44, 45] the GB input radii of HD1/HE2 of His, SG of Cys, OD1/OD2 of Asp, and OE1/OE2 of Glu are changed to 1.17, 2.00, 1.40, and 1.40 Å respectively, and the interactions between the dummy carboxylate hydrogens in Asp/Glu are excluded. cphmd_prep.sh automatically generates input files as well. Below we give two examples.
Example 1. Predict the pKa’s of the EGFR kinase given a PDB ID.
Here we wish to predict the pKa’s and the protonation states of His, Cys, and Lys at the physiological pH 7.4.
$ cphmd_prep.sh −pdb 5U8L −mod A −phl 5.5 −phu 9.5 −res ‘His Cys Lys’
Here 5U8L is the PDB ID. We omit chain ID, because 5U8L contains only one chain (default is chain A), but we specify the model A (there are two rotamer models in the PDB file). For PDB file containing NMR models, a specific model can be selected using the -m flag. The pH range is 5.5 to 9.5, which extends 2 pH units above and below the physiological pH 7.4. Since the (default) pH interval is 0.5 units, the total number of simulation pH conditions is 9. In this example, simulation length (-l|–simlength), temperature (-tp|–temperature), and ionic strength (-i|–ionic) are set to the respective default values of 10 ns, 300 K, and 0.15 M.
Upon execution, cphmd_prep.sh generates a file ‘cphmd_prep.log’ and a folder 5U8L, which contains the files listed in the aforementioned checklist. ‘cphmd_prep.log’ contains the information of the job control parameters and also the names of the files generated for the current job. Inside the folder 5U8L, there are multiple files with names like 5U8L_chainA_A_TAG.EXT with different tags TAG and extensions EXT. ‘5U8L’ is the PDB ID provided, ‘chainA’ refers to chain A, and ‘A’ is the rotamer model ID provided. Tags include mini for minimization, equil* for different equilibration steps, and prod for production. Extensions include pdb for the PDB file, rst7 for the Amber rst7 coordinate file, parm for the Amber parm7 parameter file, mdin for the Amber input file, and phmdin for CpHMD control parameter file. We can also specify multiple chains. For example, if we want to include both chain A and chain B, the command option -cha ‘A B’ can be used, and the file names will start with 5U8L_chainAB. All files in the folder ‘5U8L’ are also zipped as cphmd_inputs.zip.
Example 2. Predict the pKa’s of the protein Snase given a PDB file.
$ cphmd_prep.sh − fil 3BDC. pdb −mod A −phl 1.0 −phu 12.0 −tim 4
Here the script takes a PDB filename and the simulations are conducted in a wider pH range from 1.0 to 12.0. With a default interval of 0.5 units, this amounts to 23 simulation pH conditions. Since the model pKa’s of Asp/Glu are 3.7/4.3 and the model pKa of Lys is 10.4, this pH range typically allows the titration of all default residue types ‘Asp Glu His Cys Lys’. For this reason, the residue type (-type) is omitted. Another difference from Example 1 is that a much shorter simulation length of 4 ns (-tim 4) is specified. This is because only a rough estimate of the pKa values is desired. Similar files are generated as in Example 1; however, there are 14 extra Amber input files due to the additional pH conditions. All related files can be found here 3BDC.
Several additional files are generated by cphmd_prep.sh. apHREX.py is a Python script for running asynchronous pH replica exchange simulations if -run is not specified or specified as async. The related file cphmd.groupfile is an Amber style for specifying individual replica files in pH replica-exchange simulations. Finally, the file README contains all the commands for successfully running GBNeck2-CpHMD simulations for the current job.
5.2. Running GBNeck2-CpHMD simulations
Here we discuss the protocol of running GBNeck2-CpHMD simulations using the two examples discussed above. All commands for minimization, equilibration, and production can also be found in the file README.
Energy minimization
The energy minimization is performed with the input file ending with ‘_mini.mdin’, e.g., 3BDC_chainA_A_mini.mdin for SNase. During minimization, a harmonic restraint with the force constant of 50 kcal mol−1 Å−2 is placed on all protein heavy atoms, except for the terminal and capping residues where all atoms are allowed to move. The minimization lasts 1000 steps, using the steepest descent and conjugate gradient algorithms for the first and second 500 steps, respectively. To run minimization, we call pmemd.MPI or pmemd.cuda if Amber 20 [111] or a later version is used.
$ export name= ‘3 BDC_chainA_A’
$ mpirun −n 4
$AMBERHOME/ bin/pmemd.MPI −O
− i ${name} _mini. mdin −c ${name}. rst7
−p ${name}. parm7 −ref ${name}. rst7
−r ${name} _mini. rst7 −o ${name} _mini. out
Here, ‘-n 4’ specifies 4 CPU cores. Note, a large number of processors may be specified if hardware allows. The ‘-ref’ option specifies the reference coordinate file (3BDC_ModelA_A_fixed.rst7). The ‘-l’ option specifies the initial coordinate file. It is omitted here, since it the same as the reference file. If minimization runs error free, a timing info is given out and two files are generated: the file 3BDC_ModelA_A_fixed_mini.out contains the printed energies, and *_mini.rst7 is the restart coordinate file for the next step equilibration.
Equilibration
Following minimization, we perform a four-stage equilibration to relax the protein structure at physiological pH 7.5 (or the crystallization pH). Note, for implicit-solvent simulations heating is not required. During the four-stage equilibration (2000 steps each), the harmonic force constant on the heavy atoms is gradually reduced from 5.0, 2.0, 1.0, to 0.0 kcal/mol/Å2. CpHMD is turned on by setting the iphmd keyword to 1 (GBNeck2-CpHMD) in the Amber input file.
Using SNase as an example, the first equilibration stage is run with the following command:
$ export name= ‘3 BDC_chainA_A’
$ $AMBERHOME/bin/pmemd.cuda −O
− i ${name} _equil1.mdin −c ${name} _mini.rst7
−p ${name}.parm7 −ref ${name} _mini.rst7
−r ${name} _equil1.rst7 −o ${name} _equil1.out
−x ${name} _equil1.nc −phmdin ${name}.phmdin
−phmdparm gbneck2_input.parm
−phmdout ${name} _equil1.lambda
−phmdrestrt ${name} _equil1.phmdrst
$ sed − i ‘ s/QPHMDStart = .true. ,/QPHMDStart
= .false. , /g’ ${name}. phmdin
$ sed − i ‘ s /PHMDRST/PHMDSTRT/g ’
${name}.phmdrst
Here, 3BDC_chainA_A_mini.rst7 generated by the minimization step is used as the initial (-c) and reference (-ref) coordinates. The Amber job input file 3BDC_chainA_A_equil1.mdin file specifies simulation length, restraints, and other MD control parameters. 3BDC_chainA_A.phmdin contains job control parameters for GBNeck2-CpHMD. gbneck2_input.parm, which is a common file for GBNeck2-CpHMD simulations, contains the model pKa values and titration parameters. 3BDC_chainA_A_equil1.rst7 is the restart file that contains coordinates and velocities. 3BDC_chainA_A_equil1.phmdrst is the corresponding CpHMD restart file that contains the λ values and velocities. 3BDC_chainA_A_equil1.out contains the energy information. 3BDC_chainA_A_equil1.lambda contains the λ coordinates, although it is of little use since they are from the equilibration run. The two ‘sed’ commands modify the names for the next step.
For the rest of the equilibration stages, the same commands are used but file names should be changed. For example, we should replace ‘equil1’ and ‘mini1’ with ‘equil2’ and ‘mini2’ for the second equilibration step, and use 3BDC_chainA_A_equil1.rst7 as the initial and reference coordinate file. In addition, we need to use a new -phmdstrt flag and use 3BDC_chainA_A_equil1.phmdrst as a CpHMD restart file. After all four equilibration stages are completed, files with the names *equil4* are generated.
The above example uses pmemd.cuda, but we can also use pmemd.MPI if no GPU is available. The only modification is to replace ‘$AMBERHOME/bin/pmemd.cuda’ with ‘mpirun -n 16 $AMBERHOME/bin/pmemd.MPI’ in the command, assuming 16 cores can be used for the simulation.
Production
For production runs, we can use either independent pH, synchronous pH replica exchange, or asynchronous pH replica exchange. For independent pH runs, the simulation at different pH conditions are run independently either on the same or different machines. The running order is arbitrary, and we collect all the output files together for data analysis. The input files can be generated using cphmd_prep.sh with –run ind. For SNase, the production run is invoked with the following command:
$ export name= ‘3 BDC_chainA_A ’
$ $AMBERHOME/ bin/pmemd. cuda −O
− i ${name}_pH7.5 _prod. mdin
−c ${name} _equil4. rst7 −p ${name}. parm7
−r ${name}_pH7.5 _prod1. rst7
−o ${name}_pH7.5 _prod1. out
−x ${name}_pH7.5 _prod1. nc
−phmdin ${name}. phmdin
−phmdparm gbneck2_input. parm
−phmdstrt ${name} _equil4. phmdrst
−phmdout ${name}_pH7.5 _prod1. lambda
−phmdrestrt ${name}_pH7.5 _prod1. phmdrst
− inf pH7. 5. mdinfo
The restart files generated by equilibration step 4 are used as starting files for the production run. No reference file is needed. Along with *.out, *.rst7, *.nc (trajectory), and *.phmdrst files, λ coordinates are saved in 3BDC_chainA_A_fixed_pH*_prod1.lambda. For the command above, we again use pmemd.cuda but we can also run on CPUs with pmemd.MPI. All input files for this example can be found in gphmd_snase_phind.
pH replica-exchange simulations usually converge much faster and thus require less simulation time than independent pH simulations [23, 33, 45]. The traditional replica-exchange algorithm allows each replica to run in parallel and data exchange is synchronized. The pH replica-exchange protocol is executed similarly as the temperature replica-exchange protocol in Amber, i.e., we need a group file that contains the commands and inputs for every pH condition and then mpirun is called. The commands in the group file are quite similar to those for the independent pH runs. The input files can be generated using cphmd_prep.sh with the option -run rex. For example,
mpirun −np 64 $AMBERHOME/bin/pmemd. MPI −ng 16 −groupfile cphmd.groupfile
Note that we use 4 processes for a single pH replica and a total of 64 processes are needed (-np 64) given the number of pH conditions (-ng 16) in the Snase example command. For using mpirun for CpHMD, we should make sure that each pH replica runs on a single node so that there is only minimal data exchange between nodes if the whole job spreads across multiple node. Supposing we have a CPU cluster consisting of 64-core nodes, we should make sure the number of pH replicas is a factor of 64. This example uses pmemd.MPI to run on CPU clusters. If we have a GPU cluster, we can replace the ‘-np 64 $AMBERHOME/bin/pmemd.MPI -ng 16’ with ‘-np 1 $AMBERHOME/bin/pmemd.cuda.MPI -ng 16’. However, if the number of GPUs is less than 16, it might not work properly. The input files can be found in the gphmd_snase_phrex folder.
The traditional replica-exchange algorithm was designed for CPU based HPC clusters, where a large number of processors are available such that all replicas can be run in parallel. However, the latter condition cannot be always met for today’s GPU workstation or even HPC cluster, i.e., the number of GPUs is smaller than the number of replicas. Thus, we implemented an asynchronous pH replica-exchange algorithm, [65] in which each replica is run consecutively and data exchange is performed after all replicas are run. The users can download the related Python program async_ph_replica_exchange that allows an arbitrary number of pH replicas to be run on an arbitrary number of GPUs.
A Python script apHREX.py generated by cphmd_prep.sh with ‘-run async’ is called to run the asynchronous pH replica-exchange simulations for 3BDC.
$ export name= ‘3 BDC_chainA_A ’
$ python apHREX. py 2000
template_$ {name} _prod. mdin ${name}. parm7
gbneck2_input. parm ${name}. phmdin
${name}_cphmd ${name} _equil4. rst7
1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5
5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5
9.0 9.5 10.0 10.5 11.0 11.5 12.0
Output files, including the standard Amber outputs, lambda files, and trajectory files, are concatenated on the fly according to the pH conditions instead of replicas in the pHREX method. These files are stored in the 3BDC_chainA_A_cphmd subdirectory. The input files for this example can be found in gphmd_snase_aphrex.
Other relevant settings
For optimum performance in pKa prediction, the default settings in GBNeck2-CpHMD should be followed. Currently, the Amber ff14SB force field is recommended for proteins. A Langevin thermostat with the collision frequency of 1 ps−1 is used to maintain the temperature for λ dynamics. A non-bond cutoff of 999 Å (i.e., no cutoff) is used. Files should be saved infrequently to avoid IO that substantially slows down simulations on the GPUs. The default frequency of saving λ files every 1000 MD steps is designed for simulations of 10 ns per replica. For longer simulations, less frequent saving should be used. If analysis of conformational dynamics is desired, a saving frequency of every 10,000 MD steps (20 ps with a 2-fs time step) is recommended for a simulation of 10 ns per replica. However, if only pKa’s are desired, trajectory saving should be set to the frequency of restart file writing, e.g., 500,000 MD steps (1 ns with a 2-fs time step) or even larger depending on the wall clock time of the simulation (aiming at writing restart file once or twice a day). Writing to the Amber log file should be avoided or as infrequent as possible.
6. Post-simulation analysis
CpHMD simulations allow the user to determine the protonation states of all titratable sites at the simulation pH conditions, the pKa values, or pH- or proton-coupled conformational dynamics. Below we will discuss these aspects.
6.1. Determination of protonation states and pKa values
The lambda file contains the time evolution of λ (titration state) and x (tautomeric state) coordinates of titratable groups. These data allow us to determine the protonation states at specific pH, calculate the pKa values and tautomer states, and monitor convergence of protonation-state sampling. As an example, Fig. 2 shows the time evolution of λ for Cys481 in the BTK kinase at different pH conditions.
Figure 2. Time series of the λ value for Cys481 in the BTK kinase.
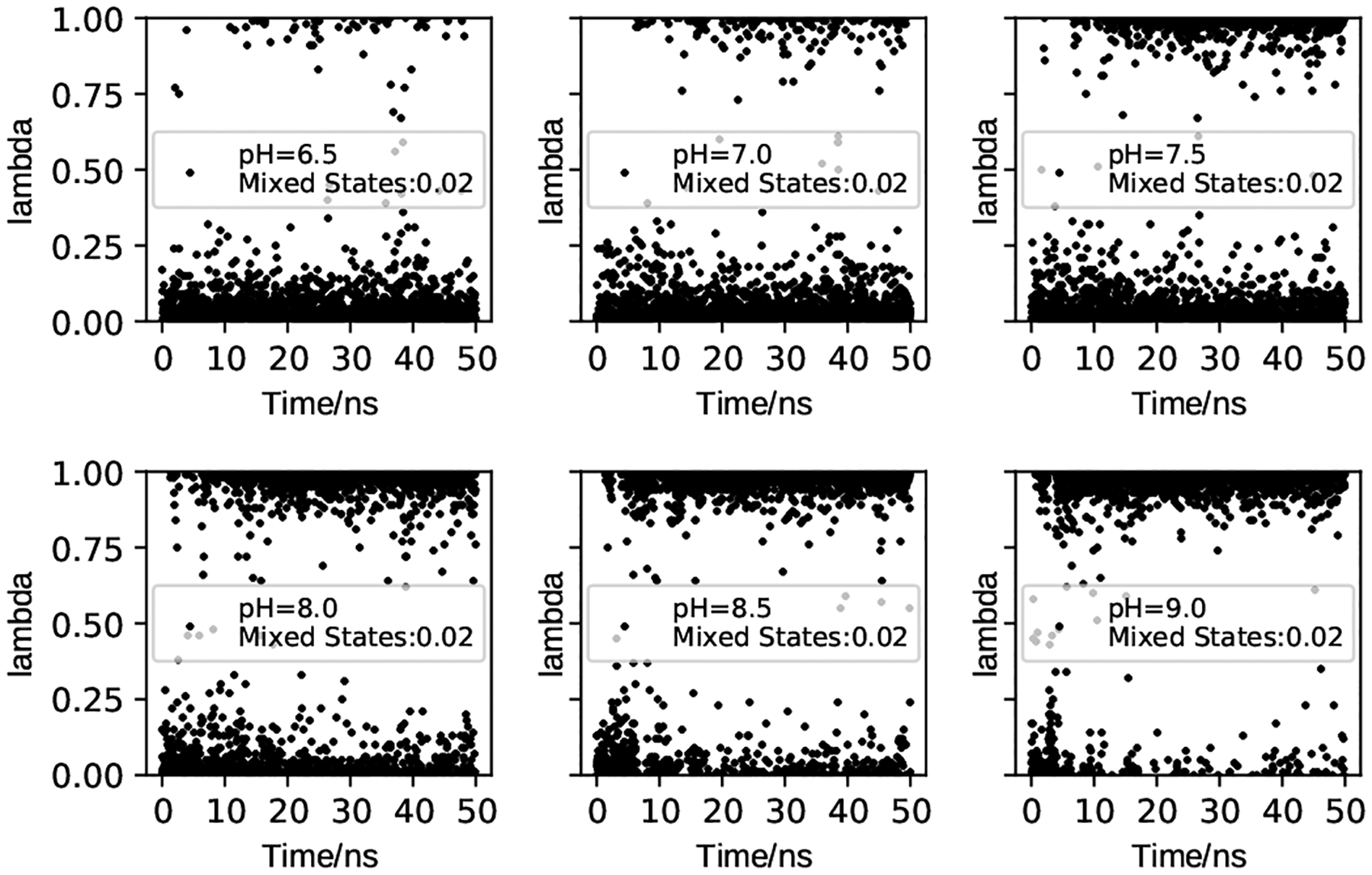
The data were saved every 20 ps (taken from Ref [112]). The pH conditions and fractions of mixed states (0.2 ≤ λ ≤ 0.8) are given. The plots show that the protonation-state sampling at all pH conditions converge after ~10 ns.
To facilitate analysis of the lambda files, we developed a Python tool cphmd_anal.py. This tool returns plots of the time series of unprotonated fractions and fitting of the unprotonated fractions to obtain pKa’s (see discussion below). For a trajectory at a specific pH, we can count the number of frames where a titratable residue i is in the protonated or unprotonated state, which is defined as λi < 0.2 or λi > 0.8, respectively. The unprotonated fraction of the residue (Si) is calculated as
| (12) |
where and are the number of protonated and unprotonated frames, respectively. Plotting the time series of the Si value informs convergence of the protonation state sampling (Fig. 1, step 5). Once convergence is reached, the pKa of residue i can be obtained by fitting the Si values at all simulation pH to the generalized Henderson-Hasselbalch equation,
| (13) |
where n is the Hill coefficient. The resulting best fit is referred to as the titration curve (Fig. 1, step 5). A significant deviation of n from 1 indicates cooperativity (n > 1) or anti-cooperativity (n < 1) with a neighboring residue. We note, in the past, λi < 0.1 and λi > 0.9 were used for defining the protonated and unprotonated states [31, 32]; however, our extensive studies (based on more than 100 proteins) showed that the calculated pKa value is not sensitive to the cutoff.
Figure 1.
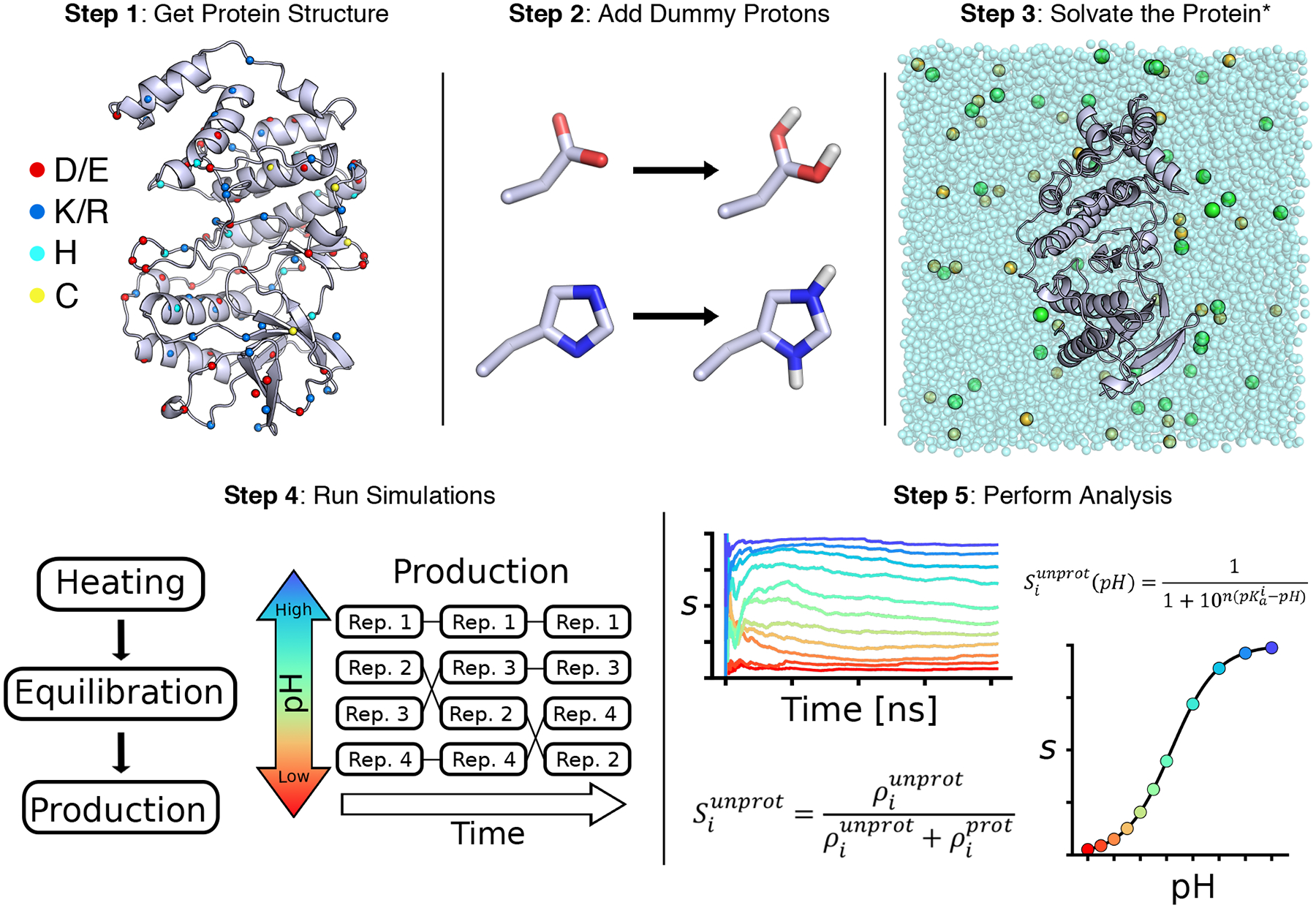
Overview of the CpHMD workflow
6.2. Analysis of proton-coupled conformational dynamics and rationalization of the calculated pKa values
A major application of CpHMD simulations is elucidation of pH-dependent or proton-coupled conformational dynamics, which in turn rationalizes the calculated pKa values. This practice can be best explained using an example. The pH replica-exchange GBNeck2-CpHMD simulations of the BTK kinase [112] gave a pKa of about 7.5 for Cys481, which is one unit lower than the model pKa of cysteine – the pKa value of an isolated cysteine fully exposed to solvent. Analysis of the pH replicas (i.e., trajectories at different pH conditions) revealed that Cys481 accepts hydrogen bonds (h-bond) from Asn481 and Thr410 when it is in the deprotonated thiolate state (Fig. 3a). Note, these h-bonds are absent in the crystal structure (PDB 3pj3). Following this observation, we plotted the deprotonated fractions of Cys481 at different pH conditions (Fig. 3b) and the occupancies of the h-bond formation between Cys481 and Asn484 or Thr410 (Fig. 3c). A comparison between the pH-dependent deprotonation and h-bond formation demonstrates that the two are correlated, i.e., deprotonation of Cys481 is coupled to the h-bond formation. It also suggests that the pKa downshift of Cys481 relative to the model value can be attributed to the stabilization of the deprotonated thiolate state by the h-bond formation with Asn484 and Thr410. We note, while implicit-solvent based CpHMD simulations have been successfully applied to pKa predictions and rationalization, hybrid-solvent and all-atom CpHMD simulations offer more accurate description of conformational dynamics. We refer the user to the studies listed in Table 1 as examples.
Figure 3. An example analysis of proton titration of a cysteine and coupling with conformational dynamics.
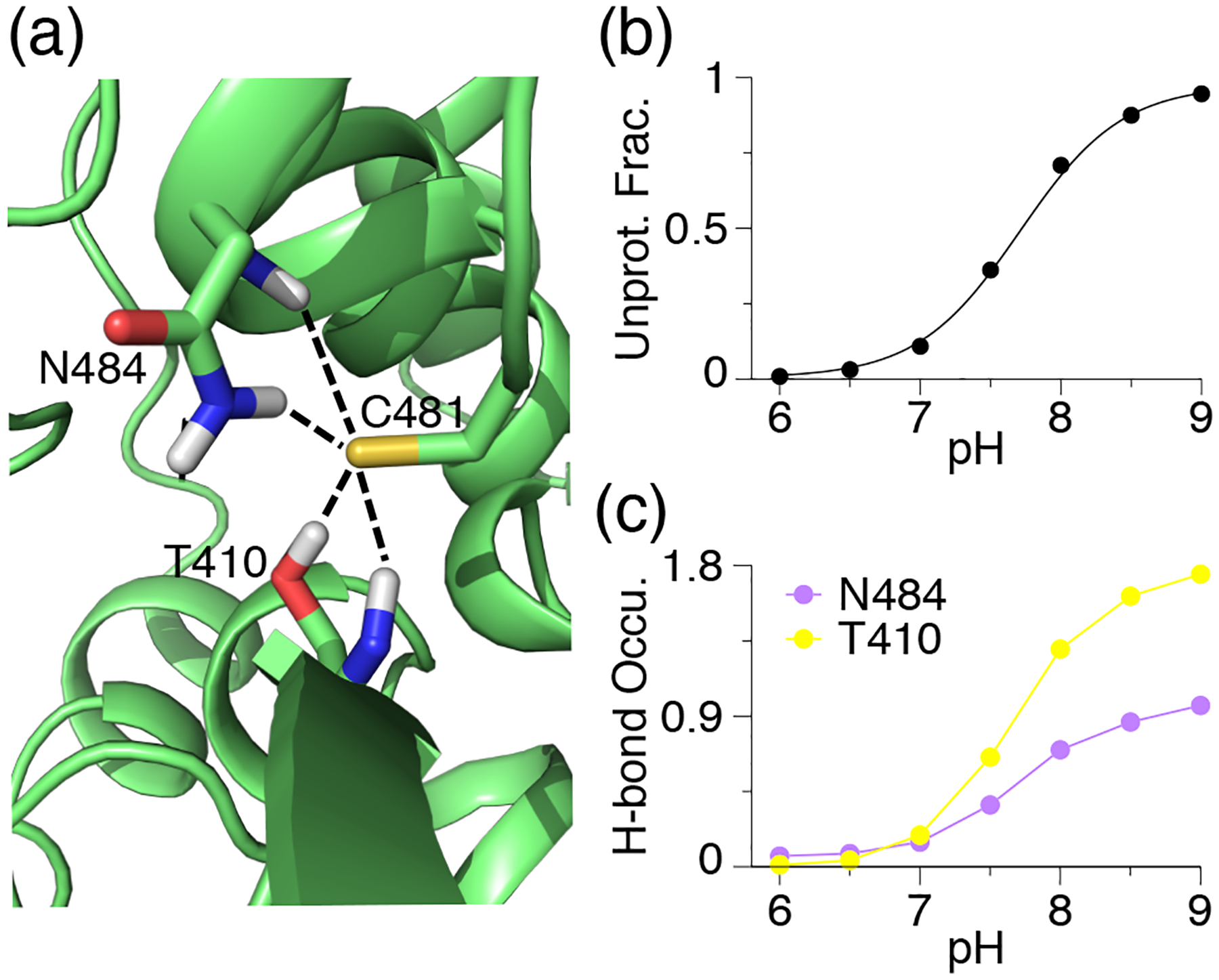
(a) A zoomed-in view of the structural environment of Cys481 in the BTK kinase taken from the trajectory at pH 9. The h-bonds with Asn484⋯ Cys481 and Thr410⋯ Cys481 are shown. The pH 9 replica is used. (b) The unprotonated fractions of Cys481 at different pH conditions. The titration curve represents the best fit to the Henderson-Hasselbalch equation. (c) Occupancy of the h-bond formation between Cys481 thiolate and Asn484 or Thr410 at different pH conditions. Reprinted with permission from Liu, Zhan et al.[112] Copyright 2021 American Chemical Society.
7. Parameterization of model titration
CpHMD is a relative free energy simulation approach, whereby the potential of mean force (PMF) of deprotonation of a titratable amino acid in the protein environment is calculated relative to that of a reference, i.e., a model compound or peptide in solution [30, 31]. Model compounds, which were used in the early development of CpHMD methods [30, 31, 35], are blocked single amino acid residues CH3CO-X-NH2 or CH3CO-X-CONH2, where X represents a titratable amino acid. In the development of the hybrid-solvent CpHMD [33] and GBNeck2-CpHMD [44, 45], model peptides CH3CO-AAXAA-NH2 were used to take advantage of recent experimental data [98, 113]. Since in the CpHMD simulation, a model PMF is subtracted, simulation of a model compound or peptide should return a zero PMF at the pH value equal to the model pKa. In other words, protonated and deprotonated states are sampled with equal probabilities (S ≈ 0.5). To ensure this is the case, the model PMF needs to be accurately determined.
To obtain the model PMF Umod, we use a free energy simulation method called thermodynamics integration, in which the mean forces at different values of θ (for single site titration) or θ and x for double site titration are calculated and then analytically integrated to obtain Umod. The single site titration model is applied to Cys and Lys. The latter has equivalent protons, so one is selected for titration. There are two types of double site titration model. His sidechains have two titratable nitrogens with different microscopic pKa’s, while carboxylate sidechains (Asp and Glu) have two titratable oxygens but identical microscopic pKa’s. These three titration models require different forms of Umod and methods of fitting.
7.1. Protocol of parameterization for single site model titration
For running TI simulations to obtain parameters, we set the options prlam to FALSE, prderiv to TRUE, and phtest to TRUE in the phmdin file, vph_theta to 0 in the phmdstrt file. For a single site titration model (e.g., Cys and Lys), we run CpHMD simulations at θi = (0.4, 0.6, 0.785, 1.0, 1.2, 1.4.). Each simulation is run for 10 ns, from which the mean forces 〈 ∂U/∂θ|θi〉 are calculated. Fitting of the mean forces to the following analytic form of ∂U/∂θ (Eq. 14) returns the two parameters A and B (Fig. 4).
Figure 4.
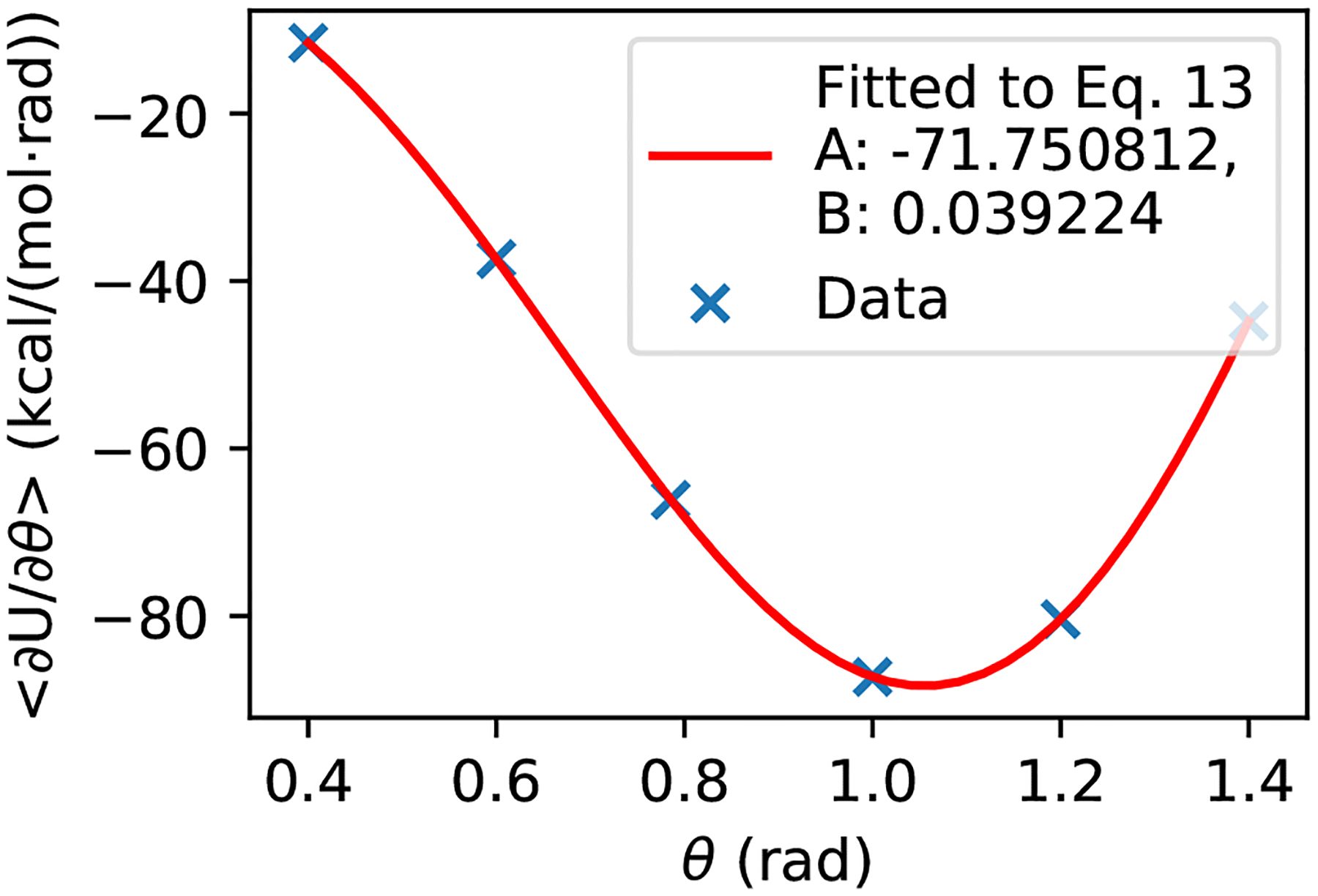
Example of fitting A and B parameters for a single site titration model.
| (14) |
As an example, Fig. 4 shows the fitting result for the GBNeck2-CpHMD titration of Cys model peptide. We note, related to error propagation. fitting should be performed in the θ space (and not in the transformed λ space) to minimize fitting errors.
Protocol of parameterization for histidine model titration
For a double site titration model with two different microscopic pKa’s (e.g. His), the two-dimensional PMF (Eq. 9) can be reduced to the following form [31],
| (15) |
where λ = sin2θ and x = sin2θx. The six parameters A0, B0, A1, B1, A10, and B10 are the parameters in the quadratic functions that describe the one-dimensional processes [31]. A0 and B0 correspond to HIP ⇌ HID, i.e., titration at Nϵ; A1 and B1 correspond to HIP ⇌ HIE, i.e., titration at Nδ; and A10 and B10 correspond to the tautomer interconversion HIE ⇌ HID.
Step 1. To obtain A0 and B0, we run TI simulations by fixing θx at 0 (λ = 0, HID, Nδ is protonated), varying θ at (0.0, 0.2, 0.4, 0.6, 0.785, 1.0, 1.2, 1.4, 1.5708), and fitting the resulting mean forces to the following one-dimensional function,
| (16) |
Step 2. To obtain A1 and B1, we run TI simulations by fixing θx at 1.5708 (λ = 1, HIE, Nδ is protonated), varying θ at (0.0, 0.2, 0.4, 0.6, 0.785, 1.0, 1.2, 1.4, 1.5708), and fitting to the following one-dimensional function,
| (17) |
Step 3. To obtain A10 and B10, we run TI simulations by fixing θ at 1.5708 (λ = 0, HIP, doubly protonated His), varying θx at (0.0, 0.2, 0.4, 0.6, 0.785, 1.0, 1.2, 1.4, 1.5708) and fitting the resulting mean forces to the following one-dimensional function,
| (18) |
Protocol of parameterization for carboxylic acid model titration
For double site titration models with two identical microscopic pKa’s (e.g., Asp and Glu), the two-dimensional PMF (Eq. 9) can be reduced to the following form [31],
| (19) |
where R1, …, R6 are parameters that can be determined via one-dimensional fitting.
Step 1. To generate the data for fitting, we run TI simulations at the combinations of θ value of 0.0, 0.4, 0.6, 0.785, 1.0, 1.2, or 1.4 and θx value of 0.0, 0.4, 0.6, 0.785, 1.0, 1.2, 1.4, or 1.5708.
Step 2. We obtain A and B parameters at each value of θ, A(θ) and B(θ), by fitting 〈∂U/∂θx〉 at different θx values to the derivative of the quadratic function,
| (20) |
where A(θ) and B(θ) are the θ-dependent parameters.
Step 3. We obtain A and B parameters at each value of θx, A(θx) and B(θx), by fitting 〈 ∂U/∂θ〉 at different θ values,
| (21) |
where A(θx) and B(θx) are the parameters.
Step 4. We obtain R1, R2, and R3 by fitting A(θi) to
| (22) |
Step 5. We set R4 = 0.5, because the two titrating sites are identical. We then obtain R5 by fitting A(θx) to
| (23) |
and obtain R6 by fitting B(θx) to
| (24) |
7.2. Scripts for TI simulations and parameterization
To facilitate TI simulations and parameter fitting, we implemented a Shell script heat_equil_prod_param.sh and a Python program cphmd_parm_fit.py. Here we use Cys to illustrate the usage.
$. /min.scr cys $. /heat_equil_prod_param.sh cys
After minimization, heating, equilibration, and production thermodynamic integration, we obtain ∂U/∂θ values saved in files with the extension .lambda as in the folder single (single site titration). We then call the Python script to fit the A and B parameters.
$ python cphmd_parm_fit.py single
Four files are generated: du.data, du.fit, du.png, and cphmd.parm. du.data contains the mean force data, du.fit contains the detailed fitting results, du.png is the plot of the fitting (Fig. 4), and cphmd.parm contains the A and B parameters that can be directly copied and pasted into the standard CpHMD parameter file. The instructions for parameterization of different models are described in the folder parameterization and the example results are included, as in the folders single, his, and carboxyl.
Funding Information
We acknowledge financial support from the National Institutes of Health (R01GM098818 and R01CA256557 to Shen and R44GM134756 to Harris) and National Science Foundation (CBET1932963 to Shen). We thank Zhi (Shane) Yue for contributing the input files for the membrane-enabled hybrid-solvent CpHMD simulations. We thank Paween Mahinthichaichan for providing comments on the input files for the membrane-enabled hybrid-solvent CpHMD simulations.
Footnotes
Potentially Conflicting Interests
The CpHMD-related software may be of interest to ComputChem LLC, for which J.S. is a founder and scientific advisor. J.S. also serves on the scientific advisory board of MatchPoint Therapeutics.
References
- [1].Casey JR, Grinstein S, Orlowski J. Sensors and Regulators of Intracellular pH. Nat Rev Mol Cell Biol. 2010; 11(1):50–61. 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- [2].Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: A Perfect Storm for Cancer Progression. Nat Rev Cancer. 2011; 11(9):671–677. 10.1038/nrc3110. [DOI] [PubMed] [Google Scholar]
- [3].White KA, Grillo-Hill BK, Barber DL. Cancer Cell Behaviors Mediated by Dysregulated pH Dynamics at a Glance. J Cell Sci. 2017; 130(4):663–669. 10.1242/jcs.195297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Roos A, Boron WF. Intracellular pH. Physiol Rev. 1981; 61:292–421. 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- [5].Tan J, Verschueren KHG, Anand K, Shen J, Yang M, Xu Y, Rao Z, Bigalke J, Heisen B, Mesters JR, Chen K, Shen X, Jiang H, Hilgenfeld R. pH-Dependent Conformational Flexibility of the SARS-CoV Main Proteinase (Mpro) Dimer: Molecular Dynamics Simulations and Multiple X-Ray Structure Analyses. J Mol Biol. 2005; 354(1):25–40. 10.1016/j.jmb.2005.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Verma N, Henderson JA, Shen J. Proton-Coupled Conformational Activation of SARS Coronavirus Main Proteases and Opportunity for Designing Small-Molecule Broad-Spectrum Targeted Covalent Inhibitors. J Am Chem Soc. 2020; p. 21883–21890. 10.1021/jacs.0c10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shimizu H, Tosaki A, Kaneko K, Hisano T, Sakurai T, Nukina N. Crystal Structure of an Active Form of BACE1, an Enzyme Responsible for Amyloid β Protein Production. Mol Cell Biol. 2008; 28(11):3663–3671. 10.1128/MCB.02185-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ellis CR, Shen J. pH-Dependent Population Shift Regulates BACE1 Activity and Inhibition. J Am Chem Soc. 2015; 137(30):9543–9546. 10.1021/jacs.5b05891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Li J, Wu S, Kim E, Yan K, Liu H, Liu C, Dong H, Qu X, Shi X, Shen J, Bentley WE, Payne GF. Electrobiofabrication: Electrically Based Fabrication with Biologically Derived Materials. Bio-fabrication. 2019; 11(3):032002. 10.1088/1758-5090/ab06ea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Morrow BH, Payne GF, Shen J. pH-Responsive Self-Assembly of Polysaccharide through a Rugged Energy Landscape. J Am Chem Soc. 2015; 137(40):13024–13030. 10.1021/jacs.5b07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tsai CC, Payne GF, Shen J. Exploring pH-Responsive, Switchable Crosslinking Mechanisms for Programming Reconfigurable Hydrogels Based on Aminopolysaccharides. Chem Mater. 2018; 30(23):8597–8605. 10.1021/acs.chemmater.8b03753. [DOI] [Google Scholar]
- [12].Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J Chem Theory Comput. 2011; 7(2):525–537. 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- [13].Kilambi KP, Gray JJ. Rapid Calculation of Protein pKa Values Using Rosetta. Biophys J. 2012; 103(3):587–595. 10.1016/j.bpj.2012.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Song Y, Mao J, Gunner MR. MCCE2: Improving Protein pKa Calculations with Extensive Side Chain Ro-tamer Sampling. J Comput Chem. 2009; 30:2231–2247. 10.1002/jcc.21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Unni S, Huang Y, Hanson RM, Tobias M, Krishnan S, Li WW, Nielsen JE, Baker NA. Web Servers and Services for Electrostatics Calculations with APBS and PDB2PQR. J Comput Chem. 2011; 32(7):1488–1491. 10.1002/jcc.21720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang L, Zhang M, Alexov E. DelPhiPKa Web Server: Predicting pKa of Proteins, RNAs and DNAs. Bioinformatics. 2016; 32(4):614–615. 10.1093/bioinformatics/btv607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Anandakrishnan R, Aguilar B, Onufriev AV. H++ 3.0: Automating pK Prediction and the Preparation of Biomolecular Structures for Atomistic Molecular Modeling and Simulations. Nucleic Acids Res. 2012; 40(W1):W537–W541. 10.1093/nar/gks375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Warwicker Jim. pKa Predictions with a Coupled Finite Difference Poisson-Boltzmann and Debye-Hückel Method: pKa Predictions with FD/DH. Proteins. 2011; 79(12):3374–3380. 10.1002/prot.23078. [DOI] [PubMed] [Google Scholar]
- [19].Alexov E, Mehler EL, Baker N, M Baptista A, Huang Y, Milletti F, Erik Nielsen J, Farrell D, Carstensen T, Olsson MHM, Shen JK, Warwicker J, Williams S, Word JM. Progress in the Prediction of pKa Values in Proteins. Proteins. 2011; 79(12):3260–3275. 10.1002/prot.23189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Huang Y, Yue Z, Tsai CC, Henderson JA, Shen J. Predicting Catalytic Proton Donors and Nucleophiles in Enzymes: How Adding Dynamics Helps Elucidate the Structure-Function Relationships. J Phys Chem Lett. 2018; 9:1179–1184. 10.1021/acs.jpclett.8b00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Huang Y, Chen W, Dotson DL, Beckstein O, Shen J. Mechanism of pH-Dependent Activation of the Sodium-Proton Antiporter NhaA. Nat Commun. 2016; 7(1):12940. 10.1038/ncomms12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu R, Yue Z, Tsai CC, Shen J. Assessing Lysine and Cysteine Reactivities for Designing Targeted Covalent Kinase Inhibitors. J Am Chem Soc. 2019; 141(16):6553–6560. 10.1021/jacs.8b13248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Harris RC, Liu R, Shen J. Predicting Reactive Cysteines with Implicit-Solvent-Based Continuous Constant pH Molecular Dynamics in Amber. J Chem Theory Comput. 2020; 16(6):3689–3698. 10.1021/acs.jctc.0c00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liu R, Zhan S, Che Y, Shen J. Reactivities of the Front Pocket N-Terminal Cap Cysteines in Human Kinases. J Med Chem. 2022; 65:1525–1535. 10.1021/acs.jmedchem.1c01186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Baptista AM, Teixeira VH, Soares CM. Constant- p H Molecular Dynamics Using Stochastic Titration. J Chem Phys. 2002; 117(9):4184–4200. 10.1063/1.1497164. [DOI] [Google Scholar]
- [26].Mongan J, Case DA, McCammon JA. Constant pH Molecular Dynamics in Generalized Born Implicit Solvent. J Comput Chem. 2004; 25(16):2038–2048. 10.1002/jcc.20139. [DOI] [PubMed] [Google Scholar]
- [27].Swails JM, York DM, Roitberg AE. Constant pH Replica Exchange Molecular Dynamics in Explicit Solvent Using Discrete Protonation States: Implementation, Testing, and Validation. J Chem Theory Comput. 2014; 10(3):1341–1352. 10.1021/ct401042b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Stern HA. Molecular Simulation with Variable Protonation States at Constant pH. J Chem Phys. 2007; 126(16):164112. 10.1063/1.2731781. [DOI] [PubMed] [Google Scholar]
- [29].Chen Y, Roux B. Constant-pH Hybrid Nonequilibrium Molecular Dynamics–Monte Carlo Simulation Method. J Chem Theory Comput. 2015; 11(8):3919–3931. 10.1021/acs.jctc.5b00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lee MS, Salsbury FR, Brooks CL III. Constant-pH Molecular Dynamics Using Continuous Titration Coordinates. Proteins. 2004; 56(4):738–752. 10.1002/prot.20128. [DOI] [PubMed] [Google Scholar]
- [31].Khandogin J, Brooks CL III. Constant pH Molecular Dynamics with Proton Tautomerism. Biophys J. 2005; 89(1):141–157. 10.1529/biophysj.105.061341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Khandogin J, Brooks CL III. Toward the Accurate First-Principles Prediction of Ionization Equilibria in Proteins†. Biochemistry. 2006; 45(31):9363–9373. 10.1021/bi060706r. [DOI] [PubMed] [Google Scholar]
- [33].Wallace JA, Shen JK. Continuous Constant pH Molecular Dynamics in Explicit Solvent with pH-Based Replica Exchange. J Chem Theory Comput. 2011; 7(8):2617–2629. 10.1021/ct200146j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wallace JA, Shen JK. Charge-Leveling and Proper Treatment of Long-Range Electrostatics in All-Atom Molecular Dynamics at Constant pH. J Chem Phys. 2012; 137(18):184105. 10.1063/1.4766352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Huang Y, Chen W, Wallace JA, Shen J. All-Atom Continuous Constant pH Molecular Dynamics With Particle Mesh Ewald and Titratable Water. J Chem Theory Comput. 2016; 12(11):5411–5421. 10.1021/acs.jctc.6b00552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kong X, Brooks CL III. Lambda-Dynamics: A New Approach to Free Energy Calculations. J Chem Phys. 1996; 105(6):10. 10.1063/1.472109. [DOI] [Google Scholar]
- [37].Bürgi R, Kollman PA, van Gunsteren WF. Simulating Proteins at Constant pH: An Approach Combining Molecular Dynamics and Monte Carlo Simulation. Proteins. 2002; 47(4):469–480. 10.1002/prot.10046. [DOI] [PubMed] [Google Scholar]
- [38].Radak BK, Chipot C, Suh D, Jo S, Jiang W, Phillips JC, Schulten K, Roux B. Constant-pH Molecular Dynamics Simulations for Large Biomolecular Systems. J Chem Theory Comput. 2017; 13(12):5933–5944. 10.1021/acs.jctc.7b00875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Brooks BR, Brooks CL III, MacKerell AD Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, et al. CHARMM: The Biomolecular Simulation Program. J Comput Chem. 2009; 30(10):1545–1614. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lee MS, Feig M, Salsbury FR, Brooks CL III. New Analytic Approximation to the Standard Molecular Volume Definition and Its Application to Generalized Born Calculations. J Comput Chem. 2003; 24(11):1348–1356. 10.1002/jcc.10272. [DOI] [PubMed] [Google Scholar]
- [41].Im W, Lee MS, Brooks CL III. Generalized Born Model with a Simple Smoothing Function. J Comput Chem. 2003; 24(14):1691–1702. 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- [42].Case DA, Ben-Shalom IY, Brozell SR, Cerutti DS, Cheatham TE III, Cruzeiro VWD, Darden TA, Duke RE, Ghoreishi D, Gilson MK, Gohlke H, Goetz AW, Greene D, Harris RC, Homeyer N, Huang Y, Izadi S, Kovalenko A, Kurtzman T, Lee TS, et al. AMBER 2018. San Francisco; 2018. [Google Scholar]
- [43].Onufriev A, Case DA, Bashford D. Effective Born Radii in the Generalized Born Approximation: The Importance of Being Perfect. J Comput Chem. 2002; 23(14):1297–1304. 10.1002/jcc.10126. [DOI] [PubMed] [Google Scholar]
- [44].Huang Y, Harris RC, Shen J. Generalized Born Based Continuous Constant pH Molecular Dynamics in Amber: Implementation, Benchmarking and Analysis. J Chem Inf Model. 2018; 58(7):1372–1383. 10.1021/acs.jcim.8b00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Harris RC, Shen J. GPU-Accelerated Implementation of Continuous Constant pH Molecular Dynamics in Amber: pKa Predictions with Single-pH Simulations. J Chem Inf Model. 2019; 59(11):4821–4832. 10.1021/acs.jcim.9b00754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Spoel DVD, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: Fast, Flexible, and Free. J Comput Chem. 2005; 26(16):1701–1718. 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- [47].Im W, Feig M, Brooks CL III. An Implicit Membrane Generalized Born Theory for the Study of Structure, Stability, and Interactions of Membrane Proteins. Biophys J. 2003; 85(5):2900–2918. 10.1016/S0006-3495(03)74712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Goh GB, Hulbert BS, Zhou H, Brooks CL III. Constant pH Molecular Dynamics of Proteins in Explicit Solvent with Proton Tautomerism: Explicit Solvent CPHMD of Proteins. Proteins. 2014; 82(7):1319–1331. 10.1002/prot.24499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Knight JL, Brooks CL. Multisite λ Dynamics for Simulated Structure–Activity Relationship Studies. J Chem Theory Comput. 2011; 7(9):2728–2739. 10.1021/ct200444f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hayes RL, Buckner J, Brooks CL III. BLaDE: A Basic Lambda Dynamics Engine for GPU-Accelerated Molecular Dynamics Free Energy Calculations. J Chem Theory Comput. 2021; 17(11):6799–6807. 10.1021/acs.jctc.1c00833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chen W, Wallace JA, Yue Z, Shen JK. Introducing Titratable Water to All-Atom Molecular Dynamics at Constant pH. Biophys J. 2013; 105(4):L15–L17. 10.1016/j.bpj.2013.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Case DA, Ben-Shalom IY, Brozell SR, Cerutti DS, Cheatham TE III, Cruzeiro VWD, Darden TA, Duke RE, Ghoreishi D, Gilson MK, Gohlke H, Goetz AW, Greene D, Harris RC, Homeyer N, Huang Y, Izadi S, Kovalenko A, Kurtzman T, Lee TS, et al. AMBER 2022. San Francisco; 2018. [Google Scholar]
- [53].Harris JA, Liu R, Vazquez Montelongo E, Martins de Oliveira V, Henderson JA, Shen J. GPU-Accelerated All-atom Particle-Mesh Ewald Continuous Constant pH Molecular Dynamics in Amber. bioRxiv. 2022; 10.1101/2022.06.04.494833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Donnini S, Tegeler F, Groenhof G, Grubmüller H. Constant pH Molecular Dynamics in Explicit Solvent with λ-Dynamics. J Chem Theory Comput. 2011; 7(6):1962–1978. 10.1021/ct200061r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Donnini S, Ullmann RT, Groenhof G, Grubmüller H. Charge-Neutral Constant pH Molecular Dynamics Simulations Using a Parsimonious Proton Buffer. J Chem Theory Comput. 2016; 12(3):1040–1051. 10.1021/acs.jctc.5b01160. [DOI] [PubMed] [Google Scholar]
- [56].Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable Molecular Dynamics with NAMD. J Comput Chem. 2005; 26(16):1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wallace JA, Shen JK. Unraveling a Trap-and-Trigger Mechanism in the pH-Sensitive Self-Assembly of Spider Silk Proteins. J Phys Chem Lett. 2012; 3(5):658–662. 10.1021/jz2016846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yue Z, Chen W, Zgurskaya HI, Shen J. Constant pH Molecular Dynamics Reveals How Proton Release Drives the Conformational Transition of a Transmembrane Efflux Pump. J Chem Theory Comput. 2017; 13(12):6405–6414. 10.1021/acs.jctc.7b00874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Henderson JA, Huang Y, Beckstein O, Shen J. Alternative Proton-Binding Site and Long-Distance Coupling in Escherichia Coli Sodium–Proton Antiporter NhaA. Proc Natl Acad Sci USA. 2020; 117(41):25517–25522. 10.1073/pnas.2005467117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wallace JA, Shen JK. Predicting pKa Values with Continuous Constant pH Molecular Dynamics. In: Methods Enzymol, vol. 466 Elsevier; 2009.p. 455–475. 10.1016/S0076-6879(09)66019-5. [DOI] [PubMed] [Google Scholar]
- [61].Wallace JA, Wang Y, Shi C, Pastoor KJ, Nguyen BL, Xia K, Shen JK. Toward Accurate Prediction of pKa Values for Internal Protein Residues: The Importance of Conformational Relaxation and Desolvation Energy. Proteins. 2011; 79(12):3364–3373. 10.1002/prot.23080. [DOI] [PubMed] [Google Scholar]
- [62].Yue Z, Shen J. pH-Dependent Cooperativity and Existence of a Dry Molten Globule in the Folding of a Miniprotein BBL. Phys Chem Chem Phys. 2018; 20(5):3523–3530. 10.1039/C7CP08296G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Tsai CC, Yue Z, Shen J. How Electrostatic Coupling Enables Conformational Plasticity in a Tyrosine Kinase. J Am Chem Soc. 2019; 141(38):15092–15101. 10.1021/jacs.9b06064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ma S, Henderson JA, Shen J. Exploring the pH-Dependent Structure–Dynamics–Function Relationship of Human Renin. J Chem Inf Model. 2021; 61(1):400–407. 10.1021/acs.jcim.0c01201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Henderson JA, Verma N, Harris RC, Liu R, Shen J. Assessment of Proton-Coupled Conformational Dynamics of SARS and MERS Coronavirus Papain-like Proteases: Implication for Designing Broad-Spectrum Antiviral Inhibitors. J Chem Phys. 2020; 153(11):115101. 10.1063/5.0020458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ellis CR, Tsai CC, Hou X, Shen J. Constant pH Molecular Dynamics Reveals pH-Modulated Binding of Two Small-Molecule BACE1 Inhibitors. J Phys Chem Lett. 2016; 7(6):944–949. 10.1021/acs.jpclett.6b00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Harris RC, Tsai CC, Ellis CR, Shen J. Proton-Coupled Conformational Allostery Modulates the Inhibitor Selectivity for β-Secretase. J Phys Chem Lett. 2017; 8(19):4832–4837. 10.1021/acs.jpclett.7b02309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Henderson JA, Harris RC, Tsai CC, Shen J. How Ligand Protonation State Controls Water in Protein–Ligand Binding. J Phys Chem Lett. 2018; 9(18):5440–5444. 10.1021/acs.jpclett.8b02440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Chen W, Huang Y, Shen J. Conformational Activation of a Transmembrane Proton Channel from Constant pH Molecular Dynamics. J Phys Chem Lett. 2016; 7(19):3961–3966. 10.1021/acs.jpclett.6b01853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Liu R, Verma N, Henderson JA, Zhan S, Shen J. Profiling MAP Kinase Cysteines for Targeted Covalent Inhibitor Design. RSC Med Chem. 2022; 13. 10.1039/D1MD00277E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Khandogin J, Chen J, Brooks CL III. Exploring Atomistic Details of pH-Dependent Peptide Folding. Proc Natl Acad Sci USA. 2006; 103(49):18546–18550. 10.1073/pnas.0605216103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Khandogin J, Brooks CL III. Linking Folding with Aggregation in Alzheimer’s. Proc Natl Acad Sci USA. 2007; 104:16880–16885. 10.1073/pnas.0703832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Khandogin J, Raleigh DP, Brooks CL III. Folding Intermediate in the Villin Headpiece Domain Arises from Disruption of a N-Terminal Hydrogen-Bonded Network. J Am Chem Soc. 2007; 129(11):3056–3057. 10.1021/ja0688880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Shen JK. A Method To Determine Residue-Specific Unfolded-State pKa Values from Analysis of Stability Changes in Single Mutant Cycles. J Am Chem Soc. 2010; 132(21):7258–7259. 10.1021/ja101761m. [DOI] [PubMed] [Google Scholar]
- [75].Shi C, Wallace JA, Shen JK. Thermodynamic Coupling of Protonation and Conformational Equilibria in Proteins: Theory and Simulation. Biophys J. 2012; 102(7):1590–1597. 10.1016/j.bpj.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ellis CR, Tsai CC, Lin FY, Shen J. Conformational Dynamics of Cathepsin D and Binding to a Small-Molecule BACE1 Inhibitor. J Comput Chem. 2017; 38(15):1260–1269. 10.1002/jcc.24719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Henderson JA, Shen J. Exploring the Catalytic Dyad Protonation States and Flap Dynamics of Malarial Plasmepsin II. J Chem Inf Model. 2021; 62:150–158. 10.1021/acs.jcim.1c01180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Vo QN, Mahinthichaichan P, Shen J, Ellis CR. How μ-Opioid Receptor Recognizes Fentanyl. Nat Commun. 2021; 12(1):984. 10.1038/s41467-021-21262-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Mahinthichaichan P, Vo QN, Ellis CR, Shen J. Kinetics and Mechanism of Fentanyl Dissociation from the μ-Opioid Receptor. JACS Au. 2021; 1(12):2208–2215. 10.1021/jacsau.1c00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Morrow BH, Wang Y, Wallace JA, Koenig PH, Shen JK. Simulating pH Titration of a Single Surfactant in Ionic and Nonionic Surfactant Micelles. J Phys Chem B. 2011; 115(50):14980–14990. 10.1021/jp2062404. [DOI] [PubMed] [Google Scholar]
- [81].Morrow BH, Eike DM, Murch BP, Koenig PH, Shen JK. Predicting Proton Titration in Cationic Micelle and Bilayer Environments. J Chem Phys. 2014; 141(8):084714. 10.1063/1.4893439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Morrow BH, Koenig PH, Shen JK. Atomistic Simulations of pH-Dependent Self-Assembly of Micelle and Bilayer from Fatty Acids. J Chem Phys. 2012; 137(19):194902. 10.1063/1.4766313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Morrow BH, Koenig PH, Shen JK. Self-Assembly and Bilayer–Micelle Transition of Fatty Acids Studied by Replica-Exchange Constant pH Molecular Dynamics. Langmuir. 2013; 29(48):14823–14830. 10.1021/la403398n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Cote Y, Fu IW, Dobson ET, Goldberger JE, Nguyen HD, Shen JK. Mechanism of the pH-Controlled Self-Assembly of Nanofibers from Peptide Amphiphiles. J Phys Chem C. 2014; 118(29):16272–16278. 10.1021/jp5048024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Arthur EJ, Brooks CL III. Parallelization and Improvements of the Generalized Born Model with a Simple sWitching Function for Modern Graphics Processors. J Comput Chem. 2016; 37(10):927–939. 10.1002/jcc.24280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from lap-tops to supercomputers. SoftwareX. 2015; 1–2:19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- [87].Phillips JC, Hardy DJ, Maia JDC, Stone JE, Ribeiro JV, Bernardi RC, Buch R, Fiorin G, Hénin J, Jiang W, McGreevy R, Melo MCR, Radak BK, Skeel RD, Singharoy A, Wang Y, Roux B, Aksimentiev A, Luthey-Schulten Z, Kalé LV, et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J Chem Phys. 2020; 153(4):044130. 10.1063/5.0014475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Eastman P, Swails J, Chodera JD, McGibbon RT, Zhao Y, Beauchamp KA, Wang LP, Simmonett AC, Harrigan MP, Stern CD, Wiewiora RP, Brooks BR, Pande VS. OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLoS Comput Biol. 2017; 13(7):e1005659. 10.1371/journal.pcbi.1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Feig M, Karanicolas J, Brooks CL III. MMTSB Tool Set: Enhanced Sampling and Multiscale Modeling Methods for Applications in Structural Biology. J Mol Graph Model. 2004; 22:2004. 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- [90].Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000; 28(1):235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018; 46(W1):W296–W303. 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Roe DR, Cheatham TE III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Comput. 2013; 9(7):3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- [93].Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996; 14(1):33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- [94].Michaud-Agrawal N, Denning EJ, Woolf TB, Beckstein O. MD-Analysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J Comput Chem. 2011; 32(10):2319–2327. 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Ferguson N, Sharpe TD, Schartau PJ, Sato S, Allen MD, Johnson CM, Rutherford TJ, Fersht AR. Ultra-Fast Barrier-Limited Folding in the Peripheral Subunit-Binding Domain Family. J Mol Biol. 2005; 353(2):427–446. 10.1016/j.jmb.2005.08.031. [DOI] [PubMed] [Google Scholar]
- [96].Brünger AT, Karplus M. Polar Hydrogen Positions in Proteins: Empirical Energy Placement and Neutron Diffraction Comparison. Proteins. 1988; 4(2):148–156. 10.1002/prot.340040208. [DOI] [PubMed] [Google Scholar]
- [97].Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: Orientations of Proteins in Membranes Database. Bioinformatics. 2006; 22(5):623–625. 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- [98].Thurlkill RL, Grimsley GR, Scholtz JM, Pace CN. pK Values of the Ionizable Groups of Proteins. Protein Sci. 2006; 15(5):1214–1218. 10.1110/ps.051840806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Jo S, Kim T, Im W. Automated Builder and Database of Protein/Membrane Complexes for Molecular Dynamics Simulations. PLoS ONE. 2007; 2(9):e880. 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr, Pastor RW. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J Phys Chem B. 2010; 114:7830–7843. 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Huang Y, Henderson JA, Shen J. Continuous Constant pH Molecular Dynamics Simulations of Transmembrane Proteins. In: Methods in Molecular Biology, vol. 2302 of Structure and Function of Membrane Proteins New York: Springer; 2021.p. 275–287. 10.1101/2020.08.06.239772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].MacKerell AD Jr, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Mich-nick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J Phys Chem B. 1998; 102(18):3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- [103].MacKerell AD, Feig M, Brooks CL III. Improved Treatment of the Protein Backbone in Empirical Force Fields. J Am Chem Soc. 2004; 126(3):698–699. 10.1021/ja036959e. [DOI] [PubMed] [Google Scholar]
- [104].Darden T, York D, Pedersen L. Particle Mesh Ewald: An W Log(N) Method for Ewald Sums in Large Systems. J Chem Phys. 1993; 98(12):4. 10.1063/1.464397. [DOI] [Google Scholar]
- [105].Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J Comput Phys. 1977; 23(3):327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- [106].Nosé S A Molecular Dynamics Method for Simulations in the Canonical Ensemblet. Mol Phys. 1984; 52:255–268. 10.1080/00268978400101201. [DOI] [Google Scholar]
- [107].Hoover WG. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys Rev A. 1985; 31(3):1695–1697. 10.1103/PhysRevA.31.1695. [DOI] [PubMed] [Google Scholar]
- [108].Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J Chem Phys. 1995; 103(11):4613–4621. 10.1063/1.470648. [DOI] [Google Scholar]
- [109].Nina M, Beglov D, Roux B. Atomic Radii for Continuum Electrostatics Calculations Based on Molecular Dynamics Free Energy Simulations. J Phys Chem B. 1997; 101(26):5239–5248. 10.1021/jp970736r. [DOI] [Google Scholar]
- [110].Chen J, Im W, Brooks CL III. Balancing Solvation and Intramolecular Interactions: Toward a Consistent Generalized Born Force Field. J Am Chem Soc. 2006; 128(11):3728–3736. 10.1021/ja057216r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Case DA, Metin Aktulga H, Belfon K, Ben-Shalom I, Brozell SR, Cerutti DS, Cheatham TE III, Cruzeiro VWD, Darden TA, Duke RE, Giambasu G, Gilson MK, Gohlke H, Goetz AW, Harris R, Izadi S, Izmailov SA, Jin C, Kasavajhala K, Kaymak MC, et al. Amber 2020. University of California, San Francisco; 2021. [Google Scholar]
- [112].Liu R, Zhan S, Che Y, Shen J. Reactivities of the Front Pocket N-Cap Cysteines in Human Kinases. J Med Chem. 2021; p. In press. 10.1101/2021.06.28.450170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Platzer G, Okon M, McIntosh LP. pH-dependent Random Coil 1H, 13C, and 15N Chemical Shifts of the Ionizable Amino Acids: A Guide for Protein pK a Measurements. J Biomol NMR. 2014; 60(2–3):109–129. 10.1007/s10858-014-9862-y. [DOI] [PubMed] [Google Scholar]