

Abstract
Chemogenetics is a technique for obtaining selective pharmacological control over a cell population by expressing an engineered receptor that is selectively activated by an exogenously administered ligand. A promising approach for neuronal modulation involves the use of “Pharmacologically Selective Actuator Modules” (PSAMs); these chemogenetic receptors are selectively activated by ultrapotent “Pharmacologically Selective Effector Molecules” (uPSEMs). To extend the use of PSAM/PSEMs to studies in nonhuman primates, it is necessary to thoroughly characterize the efficacy and safety of these tools. We describe the time course and brain penetrance in rhesus monkeys of two compounds with promising binding specificity and efficacy profiles in in vitro studies, uPSEM792 and uPSEM817, after systemic administration. Rhesus monkeys received subcutaneous (s.c.) or intravenous (i.v.) administration of uPSEM817 (0.064 mg/kg) or uPSEM792 (0.87 mg/kg), and plasma and cerebrospinal fluid samples were collected over 48 h. Both compounds exhibited good brain penetrance, relatively slow washout, and negligible conversion to potential metabolites—varenicline or hydroxyvarenicline. In addition, we found that neither of these uPSEMs significantly altered the heart rate or sleep. Our results indicate that both compounds are suitable candidates for neuroscience studies using PSAMs in nonhuman primates.
Keywords: chemogenetics, ligand-gated ion channels, rhesus macaque, pharmacokinetics, varenicline, uPSEMs
Graphical Abstract
INTRODUCTION
The most widely used chemogenetic system is currently the hM3Dq/hM4Di DREADDs, which are based on G-protein coupled receptors.1 However, DREADDs have several drawbacks. Because they work via second messengers, they have multiple mechanisms of action to facilitate or attenuate neuronal firing.2 Another drawback is that, despite there being several ligands to activate DREADDs, each has at least some degree of off-target binding.3–6 Another chemogenetic approach is based on engineered ligand-gated ion channels (LGICs). These ion channels directly influence the electrical properties of cell membranes using engineered “Pharmacologically Selective Actuator Modules” (PSAMs). These are channels that are chimeric LGICs comprising a mutated α7 nicotinic acetylcholine receptor (α7-nAChR) ligand binding domain, combined with the ion pore domain of either the excitatory cation-selective 5-HT3a receptor or the inhibitory chloride-selective glycine receptor (GlyR).7 PSAMs are selectively activated by ultrapotent “Pharmacologically Selective Effector Molecules” (uPSEMs).
We have previously shown that systemic administration of varenicline, a Food and Drug Administration (FDA)-approved agonist for one class of PSAMs, reduces neuronal activity in neurons expressing PSAM-GlyRs in rhesus monkeys.8 Given that uPSEMs have higher selectivity for PSAMs over varenicline, these compounds may be superior in research applications. The pharmacokinetics of systemically administered compounds can differ between rodents and primates.9 Thus, in order to expand the use of PSAMs in nonhuman primate (NHP) studies, information is needed regarding the pharmacokinetics of uPSEMs after systemic injection.
Here, in rhesus monkeys we measured the time course and brain penetrance of two new ultrapotent PSEM compounds, uPSEM792 and uPSEM817, that show promising binding specificity and efficacy profiles in in vitro studies.8 We also analyzed metabolite concentrations and monitored the occurrence of side effects associated with the use of these compounds by tracking physical activity, sleep, and heart rate. Overall, the uPSEMs were well tolerated.
RESULTS AND DISCUSSION
Pharmacokinetics of uPSEM817.
The maximal plasma concentration was quickly reached after s.c. administration (Tmax = 15 min). The maximum plasma concentration for a drug administered i.v. occurs at the time of administration; hence, a comparison of plasma Tmax for s.c. vs i.v. administration would not be meaningful. The Cmax was 71% higher after i.v. administration (11.78 vs 6.88 ng/mL for i.v. and s.c. administration, respectively; Table 1, Figure 1A). Similarly, the area under the curve (AUC) after i.v. administration (2.56 ng/(mL·h)) was 86% higher than that produced by s.c. administration (AUC, 1.38 ng/(mL·h); see Table 1). The uPSEM817 plasma concentration remained stable for approximately 3 h postinjection and dropped below 0.5 ng/mL by 24 h.
Table 1.
Pharmacokinetic Parameters of uPSEM817a
| uPSEM817 | hydroxyvarenicline | ||||
|---|---|---|---|---|---|
| route | plasma | CSF | plasma | CSF | |
| AUC (ng/mL·h) | s.c. | 1.382 | 0.799 | 0.556 | 0 |
| i.v. | 2.564 | 1.154 | 1.161 | 0.0015 | |
| Cmax (ng/mL) | s.c. | 6.883 | 2.67 | 0.553 | NA |
| i.v. | 11.78 | 2.06 | 0.874 | 0.05 | |
| Cmax (nM) | s.c. | 16.41 | 6.37 | 2.43 | NA |
| i.v. | 28.07 | 4.911 | 3.85 | 0.22 | |
| T max | s.c. | 15 min | 30 min | 60 min | NA |
| i.v. | 0 min | 90 min | 180 min | 90 min | |
Mean plasma and cerebrospinal fluid (CSF) values for uPSEM817 (0.064 mg/kg) with administration route via subcutaneous (s.c.) or intravenous (i.v.) injection. AUC: area under the curve; Cmax: maximal concentration; Tmax: time at maximal concentration. Not applicable (NA) indicates that hydroxyvarenicline was not detected in any of the samples. Parameters for varenicline are not listed, as this compound was not detected at any time point.
Figure 1.
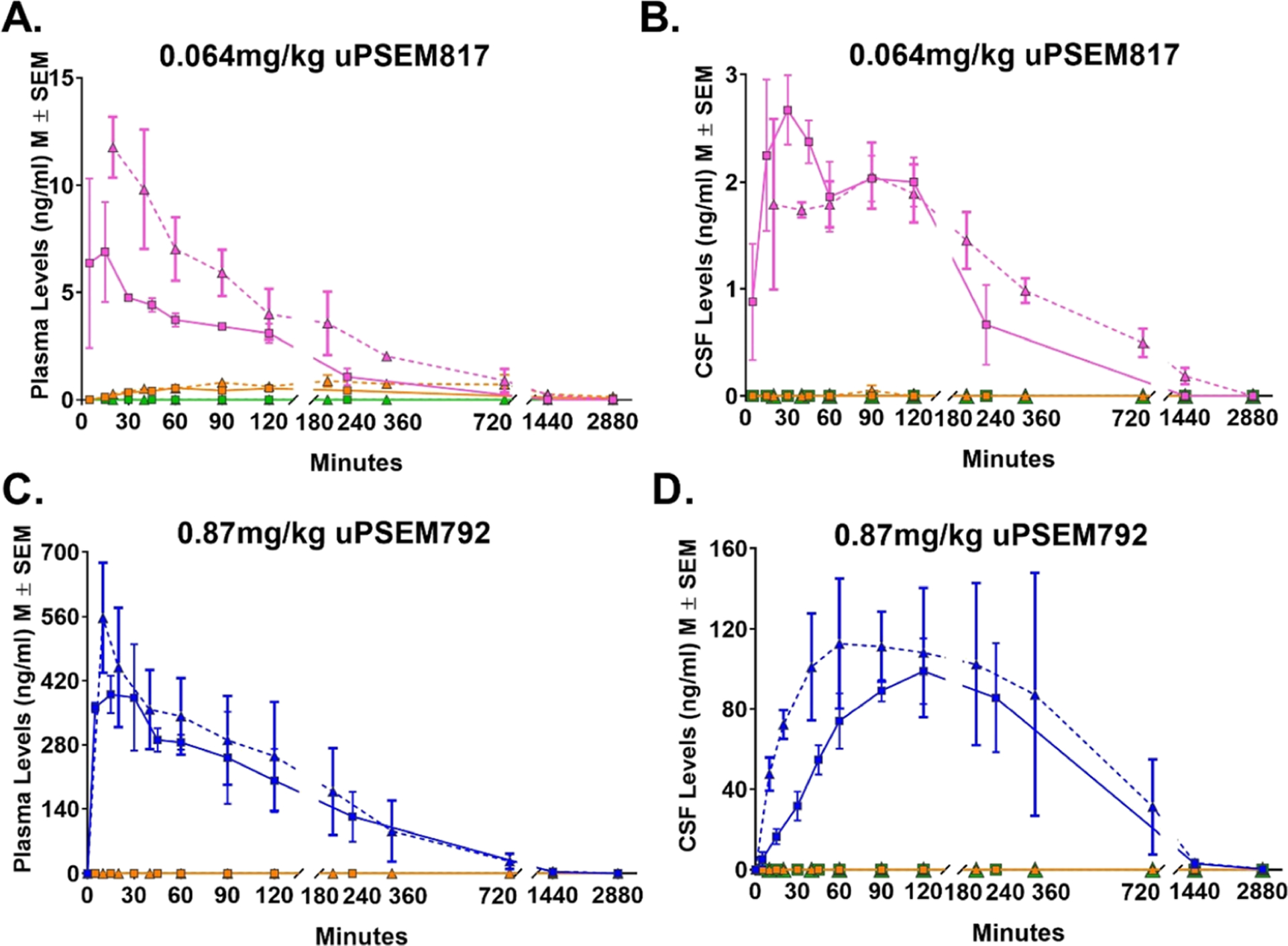
Time–concentration profiles of uPSEMs and metabolites. Levels of uPSEM817 (pink) and metabolites, hydroxyvarenicline (orange) or varenicline (green), in plasma (A) and CSF (B) after an intravenous (i.v.; triangle with dashed line; n = 2 rhesus monkeys) or subcutaneous (s.c.; square with solid line; n = 5 rhesus monkeys) administration of 0.064 mg/kg dose of uPSEM817. Panels C and D illustrate the levels of uPSEM792 (blue) and metabolites in plasma (C) and CSF (D) after i.v. or s.c. administration of 0.87 mg/kg dose of uPSEM792 in two rhesus monkeys.
Cmax in cerebrospinal fluid (CSF) (2.67 ng/mL, 6.37 nM) was reached within 30 min of the s.c. administration, whereas i.v. administration reached Cmax at 90 min (2.06 ng/mL, 4.91 nM; Table 1, Figure 1B). In the microdialysis experiment, we confirmed that uPSEM817 was not detectable in the brain before administration of uPSEM817. After s.c. injection, of uPSEM817, the concentrations of the compound in the brain microdialysates were 20.9, 20.1, and 8.2 ng/mL at 45, 85, and 125 min, respectively (corresponding to 49.8, 47.9, and 19.6 nM).
Following a single s.c. or i.v. administration of uPSEM817 (0.064 mg/kg), metabolic conversion of uPSEM817 to hydroxyvarenicline was detected in plasma with a very low Cmax of 0.55 ng/mL, 60 min after s.c. injections, and 0.87 ng/mL, 180 min after i.v. administration. Hydroxyvarenicline was only detected in a single CSF sample for one animal, 90 min postinjection for i.v. administration. Varenicline was not detected in either plasma or CSF at the limits of sensitivity for this assay. Thus, we tentatively conclude that hydroxyvarenicline was, for practical applications, not metabolized to varenicline.
Pharmacokinetics of uPSEM792.
The Tmax for plasma samples after s.c. administration of uPSEM792 (15 min) was similar to the Tmax for uPSEM817. The Cmax after s.c. administration (390.4 ng/mL) was 70% of the level measured after i.v. administration (557 ng/mL; Table 1, Figure 1C). However, the AUC after s.c. administration was modestly higher than that for i.v. administration (133.24 and 114.48 ng/(mL·h), respectively, Table 2). The uPSEM792 plasma concentration slowly declined over approximately 6 h post-injection and was still detectable at 48 h.
Table 2.
| uPSEM792 | |||
|---|---|---|---|
| route | plasma | CSF | |
| AUC (ng/mL·h) | s.c. | 133.24 | 74.124 |
| i.v. | 114.48 | 71.342 | |
| Cmax (ng/mL) | s.c. | 390.4 | 98.95 |
| i.v. | 557.0 | 112.7 | |
| Cmax (nM) | s.c. | 1405.6 | 356.26 |
| i.v. | 2005.4 | 405.76 | |
| T max | s.c. | 15 min | 120 min |
| i.v. | 0 min | 60 min | |
Mean plasma and CSF values for uPSEM792 (0.87 mg/kg) with administration route via subcutaneous (s.c.) or intravenous (i.v.) injection. AUC: area under the curve; Cmax: maximal concentration; Tmax: time at maximal concentration.
Hydroxvarenicline and varenicline were not detected. Note, however, that the lower limit of detection for hydroxyvarenicline for this assay was 0.5 ng/mL (higher than the detection limit of 0.1 ng/mL achieved for the samples collected after uPSEM817 administration).
The maximal concentration in the CSF (98.95 ng/mL, 356.26 nM) was reached within 120 min after s.c. administration, whereas i.v. administration resulted in a Tmax of 60 min (112.7 ng/mL, 405.76 nM; Table 2, Figure 1D). Collection of microdialysates in the putamen showed that uPSEM792 was not detectable in the brain prior to administration of the drug. After s.c. injection, the brain microdialysate concentrations of the compound were 41.9, 67.7, and 70.7 ng/mL at 45, 85, and 125 min, respectively, after injection (corresponding to 150, 243, and 254 nM).
Regardless of the administration route (s.c. or i.v.), we did not observe in vivo metabolism of uPSEM792 to hydroxyvarenicline or varenicline in plasma or CSF (Figure 1C,D).
Sleep disturbances and modulation of the heart rate are the side effects of varenicline.10–13 Therefore, we investigated whether uPSEMs have effects on the heart rate or sleep.
Heart Rate Measurements.
The analysis of the percent change from baseline heart rate revealed a significant main effect of time (F [7102] = 17.96, p < 0.001), such that the heart rate increased immediately after the injections and slowly came back down toward baseline over the 30 min of monitoring. The change in the heart rate was indistinguishable between uPSEM817 (0.064 mg/kg), uPSEM792 (0.87 mg/kg) and vehicle injections (Drug: F [2102] = 0.097, p = 0.91; Figure 2A).
Figure 2.
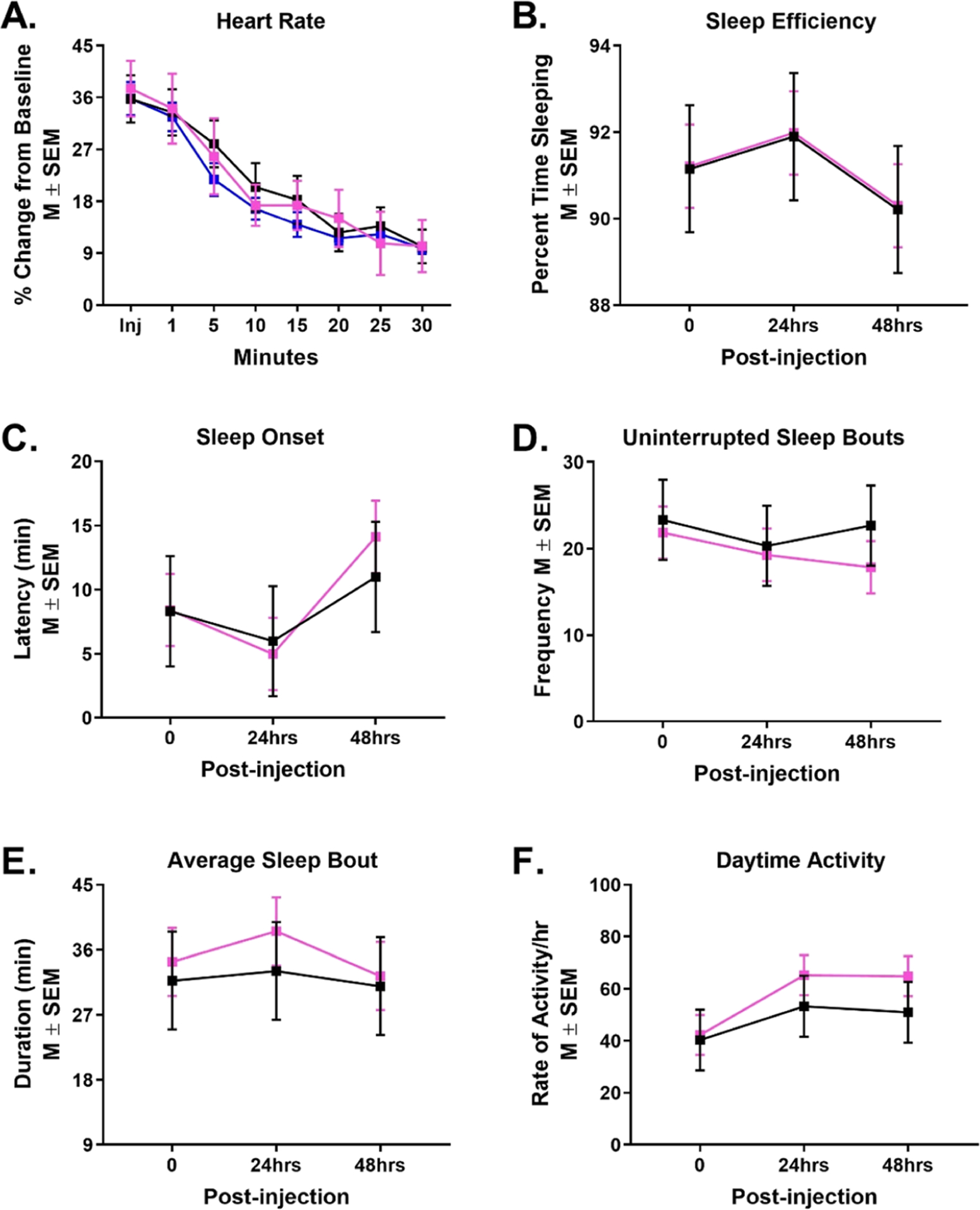
uPSEMs do not influence heart rate, sleep, or activity levels. Percent change from baseline heart rate over 30 min of monitoring in three rhesus monkeys (A) after a s.c. injection of uPSEM817 (0.064 mg/kg; pink), uPSEM792 (0.87 mg/kg; blue), or vehicle (black). Sleep efficiency (B), latency of sleep onset (C), number of uninterrupted sleep bouts (D), average sleep bout duration (E), and rate of activity/h. during the day (F) after s.c. administration of uPSEM817 (0.064 mg/kg) or vehicle in two rhesus monkeys. Note that activity monitoring for uPSEM792 was unable to be perform in the cohort of animals used in this study because of laboratory shutdowns during the COVID-19 pandemic.
Analysis of Daytime and Nighttime Activity.
The potential effects of uPSEM817 (0.064 mg/kg) on sleep were indirectly measured using actimetry, by the reduction of activity during night (lights-off) hours. The sleep efficiency, latency of sleep onset, number of uninterrupted sleep bouts, and average duration of sleep bouts did not differ across days of observation (Day: F [2, 23] = 0.84, p = 0.44; F [2, 24] = 1.92, p = 0.17; F [2, 24] = 0.19, p = 0.83; F [2, 23] = 0.31, p = 0.74, respectively). uPSEM817 did not impact any of these sleep measures either on the day of injection, nor 1 or 2-days postinjection (Drug: sleep efficiency F [2, 23.9] = 0.0005, p = 0.98; latency of sleep onset F [1, 24] = 0.06, p = 0.80; number of uninterrupted sleep bouts F [1, 24] = 0.44, p = 0.51; average duration of sleep bouts F [1, 23.8] = 0.50, p = 0.49; Figure 2B–E). The rate of activity per hour during the day also remained the same (F [1, 24] = 1.31, p = 0.26; Figure 2F).
Studies in mice have demonstrated that the uPSEMs activate their cognate PSAMs with low-nanomolar to sub-nanomolar potency and high selectivity.8 The intracerebral bioavailability of systemically administered compounds can differ significantly between rodent and primate species;9 thus our demonstration that the uPSEMs have good brain penetrance, relatively slow washout (half-lives of approximately 70 and 90 min for uPSEM817 and uPSEM792, respectively), and negligible conversion to varenicline or hydroxyvarenicline in rhesus monkeys suggests that they will be useful compounds for application to neuroscience studies in this species.
Two routes of administration were tested in our studies, i.v. and s.c. The i.v. route usually results in faster biodistribution of systemically administered compounds, but it is not always practical for NHP studies or may require sedation. Thus, s.c. administration may be preferable in some cases. We found similar results with i.v. and s.c. delivery, which would increase experimental flexibility when using these compounds for in vivo experiments in NHPs. The main difference was that i.v. delivery resulted in higher concentrations of the compounds. Both routes of administration resulted in stable concentrations (as measured in plasma and CSF) for 2–3 h after administration. For both compounds, the concentrations achieved in the brain seem likely to be sufficient to activate PSAMs.
Both uPSEMs tested in the present study (uPSEM792 and uPSEM817) have structural similarity to varenicline, an FDA-approved smoking cessation drug. Our analyses did not detect metabolism to varenicline when using doses of 0.064 mg/kg uPSEM 817 or 0.87 mg/kg uPSEM792. Hydroxyvarenicline, an intermediate compound, was detected at low levels in the plasma after uPSEM817 administration, but CSF concentration was negligible. The conversion of uPSEM817 to hydroxyvarenicline corresponds to de-alkylation of a propyl group. Hydroxyvarenicline was not detected in plasma or CSF after uPSEM792 administration, although because of technical difficulties detection of hydroxyvarenicline for the samples collected after PSEM792 assay (0.5 ng/mL) was less sensitive than the assay for the metabolite in uPSEM817 (0.1 ng/mL).
The doses used in the present study (0.064 mg/kg for uPSEM817, and 0.87 mg/kg for uPSEM792) were selected on the basis of the lowest effective doses measured in a mouse behavioral assay.8 Consequently, uPSEM792 was injected at a 14-fold higher dose than uPSEM817. However, the recorded peak plasma concentration of uPSEM792 was even larger than this difference, approximately 50-fold higher than uPSEM817. This may reflect potential saturation of clearance mechanisms with the higher uPSEM792 dose. In addition, uPSEM817 (cLog P: 2.9) is more hydrophobic than uPSEM792 (cLog P: 0.26), which may be associated with greater distribution to compartments outside of the plasma.
Effective brain access after systemic injection by crossing the blood–brain barrier (BBB) is a requirement for in vivo neuroscience applications of chemogenetics. It has been shown previously that neither of the uPSEMs are substrates for the P-glycoprotein efflux pump.8 Our results measuring CSF and parenchyma levels of uPSEM817 and uPSEM792 showed that these compounds have good brain bioavailability in monkeys. This is in agreement with previous rodent studies demonstrating brain penetrance of these compounds using in vivo positron emission tomography, in which binding of the radioligand 18F-ASEM to PSAM4-GlyR receptors was abolished by systemic injection of uPSEM817 or uPSEM792.8
Both uPSEMs had adequate BBB permeability as shown by their CSF:plasma ratios (uPSEM817 = 0.45 and uPSEM792 = 0.62). A previous study analyzing the pharmacokinetics of the structurally similar compound varenicline in rats reported a CSF:plasma ratio of 1.25,14 but this was based on the unbound fraction of the compound, which was not measured in our study. Therefore, the CSF:plasma ratios measured in our study may be an underestimate.
The concentrations of uPSEM817 (approx. 19 to 50 nM) and uPSEM792 (approx.150 to 250 nM) in the brain parenchyma recorded in our microdialysis experiments are more than sufficient to activate PSAMs, based on in vitro experiments in which the EC50s at PSAM4-GlyR channels were calculated as 0.3 and 2.3 nM for uPSEM817 and uPSEM792, respectively.8 Notably, the parenchyma:CSF ratios were markedly different between these two compounds. The peak uPSEM817 concentration in the microdialysate (50 nM) was approximately 8-fold higher than the peak concentration in the CSF (6.4 nM). Conversely, the peak uPSEM792 concentration in the microdialysate (255 nM) was approximately 65% of the peak concentration in the CSF (406 nM). This indicates that uPSEM817 may preferentially distribute to the parenchyma over the CSF. Several studies indicate that the parenchymal and CSF compartments can have distinct drug distribution properties.15,16 One caveat for the interpretation of the microdialysis results is the potential variability introduced by differences in the relative recovery correction factors for each microdialysis experiment; values reported for microdialysis are corrected values, based on a one-time recovery for the probe, done at the end of the experiment (when the probe may have reduced performance, possibly underestimating recovery). Also, microdialysis in the parenchyma measured the unbound fraction of drug, whereas CSF measurements were for total drug concentration. Although the concentrations measured in the brain are unlikely to activate nAChR receptors,8 our results suggest that 5-fold to 10-fold lower doses of uPSEMs could be used for in vivo chemogenetic experiments in NHPs to minimize the possibility of off-target effects.
Plasma levels of the uPSEMs reached maximal concentrations of 28 nM and 2 μM for intravenously administered uPSEM817 and uPSEM792, respectively. At similar concentrations, in vitro testing has shown that uPSEM817 does not activate either α4β2- or α7-nAChRs. In contrast, uPSEM792 could activate endogenous α4β2-nAChR (EC50 0.52 μM), but endogenous α7-nAChRs are not activated when exposed to 2 μM uPSEM792. However, neither uPSEM had significant effects on heart rate or sleep when compared to vehicle injections (Figure 2). Because our study was limited to the timing between uPSEM817 dosing and assessments of nighttime activities (6 h) future studies should be mindful of the potential sleep disturbances with uPSEM doses administered closer to light-off/night period. Off-target side effects could influence behavioral neuroscience study results, as has been shown recently with other chemogenetic ligands.3–6 While the search for off-target effects in our study was limited, the lack of identified adverse effects in this study and the evidence that considerably lower doses of uPSEMs are suitable for use in NHP further increase the attractiveness of uPSEMs for neuroscience chemogenetic studies.
The doses of uPSEM817 and uPSEM792 used in these studies were based on rodent studies8 and provide a starting point for PSAM-PSEM studies in NHPs. In vivo brain concentrations were not directly measured in past rodent dosing studies; thus it is not definitively known if the uPSEM792 concentrations in the brain parenchyma of rhesus monkeys were greater than with the corresponding weight-matched dose in mice. Nevertheless, there can be considerable pharmacokinetic differences between mice and primates. Thus it seems likely that uPSEM792 and uPSEM817 had much greater brain access in rhesus monkeys relative to mice. Although our previous work has demonstrated that the latest generation of PSAMs are effective for modulating neuronal activity in the rhesus monkey brain in vivo using varenicline,8 future studies should verify that these doses of uPSEM817 and uPSEM792 used in the present study activate PSAMs in NHPs in vivo. Moreover, it seems as even lower doses may be sufficient.
The combination of good brain penetrance, slow washout, negligible conversion to psychoactive compounds, and absence of peripheral side-effects at the doses tested in the present study leads us to conclude that uPSEM817 and uPSEM792 are well-suited for use in combination with PSAMs to modulate neuronal activity in normal and pathological brain circuits in NHP models. We are confident that the PSAM-PSEM technology will be a valuable tool for basic and translational neuroscience applications, and may be used, for example, to address questions related to Parkinson’s disease17 or epilepsy.18
METHODS
Six rhesus monkeys were used in this study; two from the National Institute of Mental Health (one 7-year-old female weighing 10.4 kg, and one 13-year-old male weighing 11.4 kg at the beginning of studies) and four from the Emory National Primate Research Center at Emory University (males, 4–5 years old and weighing 6.5–7.2 kg).
All monkeys were pair-housed indoors on a 12 h light/dark cycle, had free access to food and water, and received vegetables and fruit daily. All procedures conformed to the Institute for Laboratory Animal Research Guide and were performed under an Animal Study Proposal approved by the Animal Care and Use Committee of the National Institute of Mental Health or the Institutional Animal Care and Use Committee of Emory University and were in accordance with the United States Department of Agriculture (USDA) Animal Welfare Act.
Drugs and Formulation.
The uPSEM doses were selected based on the lowest effective doses in mice8 and excluded the mass of the counterion (see below). uPSEM817 tartrate was dissolved in sterile saline acidified to pH 4–5 with HCl, additional sterile saline was added to bring the concentration of the solution to 1 mg/mL, and the pH was then titrated back to neutral (pH 6–7) with NaOH. The acidification step was not followed when the uPSEM817 solution was prepared at Emory. The solution was passed through a 0.22 micron sterile filter prior to administration at a dose of 0.064 mg/kg (0.1 mg/kg before correction for the tartrate counterion). uPSEM792 hydrochloride was formulated and filtered per the protocol described above and was administered at a dose of 0.87 mg/kg (1.0 mg/kg before correction for the chloride counterion). The compounds used were obtained from Dr. Sternson’s lab (Janelia), Tocris (MN) or HelloBio (NJ).
uPSEM Intravenous Administration.
Intravenous administration challenges with uPSEM817 and uPSEM792 were done in two adult rhesus monkeys (NIMH cohort).
Surgeries.
The NIMH rhesus monkeys were implanted with indwelling subcutaneous access ports (Access Technologies, Illinois) for the collection of blood and CSF. The blood collection ports were connected to a catheter implanted into the femoral artery using a procedure similar to that previously described for femoral vein catheterization.19 The ports were flushed with saline and locked with taurolidine citrate solution (TCS, Access Technologies, Illinois). The CSF collection ports were implanted using a variation on a previously described technique,20 instead of a hemilaminectomy for access to the thecal sac, a laminotomy was performed. The CSF access port was flushed with saline and locked with dilute heparinized saline (10 USP/mL).
Intravenous uPSEM Injections and Sample Collection.
Baseline blood and CSF samples were obtained from the chronically implanted subcutaneous access ports. To clear the ‘dead space’ in the blood access port, 0.5 mL of blood was extracted and discarded. A sample of 2 mL of blood was then obtained and transferred to a 2 mL tube containing EDTA (3.5 mg) on ice. To clear the ‘dead space’ in the CSF access port, 0.25 mL of CSF was withdrawn and discarded. A 0.5 mL sample of CSF was collected and immediately transferred to dry ice. Either uPSEM817 (0.064 mg/kg) or uPSEM792 (0.87 mg/kg) was injected 30 min after baseline sample collection via a temporary catheter in the saphenous vein. Further blood and CSF samples were collected, per the procedure used for baseline collections, at the following time points post i.v. drug injection: 10, 20, 40, 60, 90, 120, 180, 360, 720 min, 24, and 48 h. All blood samples were centrifuged at 2500 rcf for 10 min within 60 min from time of collection, and the plasma was transferred to Eppendorf tubes for storage at −80 °C. CSF samples were transferred directly to the −80 °C freezer without thawing.
Subcutaneous uPSEM Administration.
Five adult rhesus monkeys participated in subcutaneous administration challenges (2 from the NIMH cohort, 3 from the Emory cohort). The three unoperated monkeys (Emory cohort) received a subcutaneous injection of uPSEM817 (0.064 mg/kg) and were then sedated with tiletamine HCL and zolazepam (Telazol, 3–5 mg/kg, im). CSF samples were obtained in a serial fashion (as explained below) from the cisterna magna, using a sterile 23-ga bevel-tipped needle by pressure difference and collected by gravity.21 CSF samples were collected in prechilled sterile Eppendorf tubes, immediately frozen on dry ice, and stored at −80 °C until the time of the assay.
Serial CSF taps were performed in sessions separated by 2 weeks. To reduce the risks associated with repeated spinal taps and given the time restrictions imposed by the anesthetized state, in each session, only 2–3 serial CSF taps were conducted per animal, so that not all time points were collected for all monkeys. Thus, after subcutaneous injection of uPSEM817 0.064 mg/kg, three individual CSF samples were available for analyses at 15, 30, 60, 90, 120, 1440, and 2880 min postinjection, and two individual CSF samples at 5, 45, and 240 min postinjection.
Blood samples were collected from the femoral vein immediately following each CSF tap. All blood samples were collected in prechilled 2 mL tubes containing EDTA (3.5 mg) and immediately placed on ice. Samples were centrifuged at 2500 rcf for 15 min in a refrigerated centrifuge (at 4 °C). Plasma was pipetted off and stored at −80 °C until assayed.
The same two adult rhesus monkeys used in the i.v. administration (NIMH cohort) were also used for the subcutaneous administration challenge of a 0.87 mg/kg dose of uPSEM792. All procedures were the same as described earlier. The post s.c. drug injection samples were collected at the same time points as uPSEM817 post s.c. drug collections.
Microdialysis.
In one monkey (Emory cohort), microdialysis experiments were conducted to measure the concentrations of uPSEM792 or uPSEM817 in the putamen before and after systemic injections of these compounds. Under isoflurane anesthesia and sterile conditions, the animal received a chronic recording chamber directed to the putamen, following previously described procedures.22,23 Prior to the microdialysis experiments, the putamen was identified with electrophysiological mapping, according to its stereotaxic location, depth in the dorso-ventral plane, and its relationship with other structures as well its characteristic neuronal firing activity.22–24
Thereafter, microdialysis probes (modified CMA-11, CMA Microdialysis, Kista, Sweden, 3 mm cuprophane membrane, molecular weight cut-off, 6 kD) were inserted into the putamen through a 22-gauge guide cannula. Two microdialysis experiments were performed, one each for uPSEM792 and uPSEM817. The probes were perfused with artificial cerebrospinal fluid (aCSF, CMA) at 2 μL/min. After probe insertion, two baseline (before injection) samples were collected 60 min apart (120 μL). uPSEM817 (0.064 mg/kg) or uPSEM792 (1 mg/kg) was then injected s.c. Five minutes after the injection, we started the collection of the post-injection samples, and three samples were collected (one sample every 40 min, 80 μL/sample). Following the experiment, the probe was removed from the brain and placed into a vial with a standard uPSEM solution, and an additional sample was collected for 40 min. This sample was used to calculate the recovery fraction of each microdialysis probe. We report the values corrected by this recovery fraction (11 and 7% for the uPSEM817 and uPSEM792 experiments respectively).
Analysis of Biological Samples.
All plasma, CSF, and microdialysis samples were assayed for the injected compound by Q2 solutions (Indianapolis, IN, see the Supporting Information for details). The plasma and CSF samples were also assayed for hydroxyvarenicline and varenicline. Both are potential metabolites of uPSEM817 and uPSEM792.25 Hydroxyvarenicline can convert to varenicline, and varenicline can directly interact with a variety of endogenous receptors. Given the negligible levels of these compounds detected in CSF samples after the uPSEM817 or uSPEM792 injections (see below), the microdialysis samples were not assayed for metabolites.
Heart Rate Monitoring.
Three adult rhesus monkeys (one female, two males) were used to determine whether uPSEM817 and uPSEM792 had any effect on heart rate, as previously shown with drugs that activate nAChRs (e.g., nicotine, varenicline.).12,13 Two monkeys were trained to voluntarily present a leg for heart rate monitoring via a wearable LED optical sensor (FitBit Inspire 2, San Francisco, CA or Polar OH1, Polar Electro Oy, Kempele, Finland) and one animal was monitored with an optical pulse-oximeter attached to its ear (SurgiVet Advisor Vital Signs Monitor by Smith Medical, Dublin, OH). For additional details see the Supporting Information. After recording the baseline heart rate, the monkeys received a subcutaneous injection of either 0.064 mg/kg uPSEM817, 0.87 mg/kg uPSEM792, or vehicle (phosphate-buffered saline, Corning, Corning, New York). The heart rate was recorded immediately after injection, at 1 min post-injection, and at 5 min intervals for 30 min post-injection. Each animal’s heart rate was monitored in this manner 2–3 times for each compound (uPSEM817, uPSEM792, vehicle) with at least 1 week between uPSEM compound administrations.
Day and Nighttime Activity Monitoring.
Because of limitations imposed by the laboratory shutdown at the start of the COVID-19 pandemic, these experiments were only carried out with uPSEM817. To determine whether the agent affected sleep or activity levels, the agent was injected in two adult male monkeys wearing accelerometers (Actical, Respironics Inc., Murrysville, PA) that were attached to the collars. Accelerometers do not provide a direct measure of sleep, instead “sleep” states are inferred by the absence of activity and “awake” states are inferred by threshold activity levels as defined below. The method has been previously been shown to effectively track putative sleep states.26 For five separate weeks, the monkeys’ activity levels were continuously measured from Friday to Monday morning. Accelerometers were placed on the monkeys’ collars on a Friday, and the monkeys were given an s.c. injection at 1 pm of either 0.064 mg/kg uPSEM817 (3 separate sessions per monkey) or vehicle (PBS; two separate sessions per monkey). Accelerometry data were then analyzed with the standard Actiware algorithm (Respironics),27 which defines “sleep” on a weighted, sliding average with a threshold optimized for night-time activity of nonhuman primates (the threshold for determining the onset of sleep was set to 14, instead of the standard setting of 2028) and sensitivity to detect night-time sleep disruption. Sleep efficiency was quantified as the total number of minutes slept each night as the percentage of the duration of the lights-off period (7 pm–7 am), as described previously.28,29 The number of bouts of uninterrupted sleep and their average duration was also calculated. The latency to fall asleep (sleep onset) was defined as time between lights off and the beginning of the first ≥3-min period of uninterrupted sleep. Daytime activity was calculated as the average activity/min starting when the animal was returned to their home cage on the day of injection until lights off (1 pm–7 pm). The same daytime activity period was compared for the day of injection, day 1 (24 h postinjection) and day 2 (48 h postinjection).
Statistical Analyses.
The area under the curve (AUC), maximal plasma concentration (Cmax), and time of maximal plasma concentration (Tmax), derived from plots of concentration over time, were calculated for uPSEM817, uPSEM792, varenicline, and hydroxyvarenicline, using GraphPad Prism 7.01 (GraphPad Software Inc., La Jolla, USA). The data are reported as means ± SEM. The molar mass used for calculations was 419.43 g/mol uPSEM817 tartrate salt, 277.75 g/mol for uPSEM792 hydrochloride, 211.26 g/mol for varenicline (Pubchem ID 5310966), and 227.26 g/mol for hydroxyvarenicline (M3b; Pubchem ID 29982201). The oil:water partition coefficient (cLog P) of uPSEM817 and uPSEM792 was calculated using Chem Draw Professional (PerkinElmer).
The change from baseline heart rate across the monitored post-injection time was calculated separately for the uPSEM817, uPSEM792, and vehicle experiments. Data were analyzed with a linear mixed model (LMM), with percent change from baseline heart rate as the outcome variable, drug injection (uPSEM817, uPSEM792, Vehicle) and time postinjection (0, 1, 5, 10, 15, 20, 25, 30 min post) as fixed effects, and individual monkey as a random effect to account for repeated sessions for each monkey.
Sleep efficiency, latency to fall asleep, number of uninterrupted sleep bouts, average duration of sleep bouts, and daytime activity levels were analyzed using a LMM with drug injection (uPSEM817, Vehicle) and day postinjection (0 = same day, 1, 2 days post) as fixed effects, and individual monkey as a random effect.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank David Ji for his contribution to the Actical data analysis, Damien Pitard for his assistance with the awake heart rate monitoring experiments, Charalotta Campbell of Q2 solutions for LC-MS analysis, and Xing Hu for help in the microdialysis experiments.
Funding
This work was funded by NIH-NINDS 1R21NS106346 (AG) and the NIH, Office of Research Infrastructure Program, P51 OD011132. M.A.G.E., B.J.R., G.P.F., and J.Y.S. were supported by the Intramural Research Program, NIMH, NIH, DHHS annual report ZIAMH 002619. Any opinions expressed are the authors’ own and do not necessarily represent views of the NIMH, NIH, and DHHS.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.2c00525.
Liquid chromatograph mass spectrometry (LC–MS); heart rate monitoring; LC gradients used for analysis of uPSEM and metabolites; mass spectrometer parameters used for analysis; and correspondence between optical pulse-oximeter devices (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschemneuro.2c00525
The authors declare the following competing financial interest(s): Author S.M.S. has issued patents on this technology and owns stock in Redpin Therapeutics, LLC, which is a biotech company focusing on therapeutic applications of chemogenetics. S.M.S. is a cofounder and consultant for Redpin Therapeutics. All the other authors (A.G., J.R., M.A.G.E., J.Y.S., G.P.F., B.J.R., and T.W.) declare no competing financial interests.
Contributor Information
Jessica Raper, Emory National Primate Research Center, Emory University, Atlanta, Georgia 30329, United States.
Mark A. G. Eldridge, Laboratory of Neuropsychology, National Institute of Mental Health, Bethesda, Maryland 20892, United States
Scott M. Sternson, Janelia Research Campus, Howard Hughes Medical Institute, Ashburn, Virginia 20147, United States; Department of Neurosciences, Howard Hughes Medical Institute, University of California, San Diego, La Jolla, California 92093, United States
Jalene Y. Shim, Laboratory of Neuropsychology, National Institute of Mental Health, Bethesda, Maryland 20892, United States
Grace P. Fomani, Laboratory of Neuropsychology, National Institute of Mental Health, Bethesda, Maryland 20892, United States
Barry J. Richmond, Laboratory of Neuropsychology, National Institute of Mental Health, Bethesda, Maryland 20892, United States
Thomas Wichmann, Emory National Primate Research Center, Emory University, Atlanta, Georgia 30329, United States; Department of Neurology, Emory University School of Medicine, Atlanta, Georgia 30322, United States; Morris K. Udall Center of Excellence for Parkinson’s Disease, Emory University, Atlanta, Georgia 30322, United States.
Adriana Galvan, Emory National Primate Research Center, Emory University, Atlanta, Georgia 30329, United States; Department of Neurology, Emory University School of Medicine, Atlanta, Georgia 30322, United States; Morris K. Udall Center of Excellence for Parkinson’s Disease, Emory University, Atlanta, Georgia 30322, United States.
REFERENCES
- (1).Armbruster BN; Li X; Pausch MH; Herlitze S; Roth BL Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Atasoy D; Sternson SM Chemogenetic Tools for Causal Cellular and Neuronal Biology. Physiol. Rev 2018, 98, 391–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Upright NA; Baxter MG Effect of chemogenetic actuator drugs on prefrontal cortex-dependent working memory in nonhuman primates. Neuropsychopharmacology 2020, 45, 1793–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ilg A-K; Enkel T; Bartsch D; Bähner F Behavioral effects of acute systemic low-dose clozapine in wild-type rats: Implications for the use of DREADDs in behavioral neuroscience. Front. Behav. Neurosci 2018, 12, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).MacLaren DA; Browne RW; Shaw JK; Krishnan Radhakrishnan S; Khare P; España RA; Clark SD Clozapine N-Oxide Administration Produces Behavioral Effects in Long-Evans Rats: Implications for Designing DREADD Experiments. eNeuro 2016, 3, ENEURO.0219–16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rodd ZA; Engleman EA; Truitt WA; Burke AR; Molosh AI; Bell RL; Hauser SR CNO administration increases dopamine and glutamate in the medial prefrontal cortex of wistar rats: Further concerns for the validity of the CNO-activated DREADD procedure. Neuroscience 2022, 491, 176. [DOI] [PubMed] [Google Scholar]
- (7).Sternson SM; Roth BL Chemogenetic tools to interrogate brain functions. Annu. Rev. Neurosci 2014, 37, 387–407. [DOI] [PubMed] [Google Scholar]
- (8).Magnus CJ; Lee PH; Bonaventura J; Zemla R; Gomez JL; Ramirez MH; Hu X; Galvan A; Basu J; Michaelides M; Sternson SM Ultrapotent chemogenetics for research and potential clinical applications. Science 2019, 364, No. eaav5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Syvänen S; Lindhe O; Palner M; Kornum BR; Rahman O; Långström B; Knudsen GM; Hammarlund-Udenaes M Species differences in blood-brain barrier transport of three positron emission tomography radioligands with emphasis on P-glycoprotein transport. Drug Metab. Dispos 2009, 37, 635–643. [DOI] [PubMed] [Google Scholar]
- (10).Savage RL; Zekarias A; Caduff-Janosa P Varenicline and abnormal sleep related events. Sleep 2015, 38, 833–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ashare RL; Lerman C; Tyndale RF; Hawk LW; George TP; Cinciripini P; Schnoll RA Sleep Disturbance During Smoking Cessation: Withdrawal or Side Effect of Treatment? J. Smok. Cessat 2017, 12, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Donny EC; Caggiula AR; Sweitzer M; Chaudhri N; Gharib M; Sved AF Self-administered and yoked nicotine produce robust increases in blood pressure and changes in heart rate with modest effects of behavioral contingency in rats. Pharmacol., Biochem. Behav 2011, 99, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jutkiewicz EM; Rice KC; Carroll FI; Woods JH Patterns of nicotinic receptor antagonism II: cardiovascular effects in rats. Drug Alcohol Depend. 2013, 131, 284–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rollema H; Shrikhande A; Ward KM; Tingley FD 3rd; Coe JW; O’Neill BT; Tseng E; Wang EQ; Mather RJ; Hurst RS; Williams KE; de Vries M; Cremers T; Bertrand S; Bertrand D Pre-clinical properties of the alpha4beta2 nicotinic acetylcholine receptor partial agonists varenicline, cytisine and dianicline translate to clinical efficacy for nicotine dependence. Br. J. Pharmacol 2010, 160, 334–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Aird RB A study of intrathecal, cerebrospinal fluid-to-brain exchange. Exp. Neurol 1984, 86, 342–358. [DOI] [PubMed] [Google Scholar]
- (16).Liu X; Van Natta K; Yeo H; Vilenski O; Weller PE; Worboys PD; Monshouwer M Unbound drug concentration in brain homogenate and cerebral spinal fluid at steady state as a surrogate for unbound concentration in brain interstitial fluid. Drug Metab. Dispos 2009, 37, 787–793. [DOI] [PubMed] [Google Scholar]
- (17).Vazey EM; Aston-Jones G New tricks for old dogmas: optogenetic and designer receptor insights for Parkinson’s disease. Brain Res. 2013, 1511, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lieb A; Weston M; Kullmann DM Designer receptor technology for the treatment of epilepsy. EBioMedicine 2019, 43, 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Graham ML; Rieke EF; Wijkstrom M; Dunning M; Aasheim TC; Graczyk MJ; Pilon KJ; Hering BJ Risk factors associated with surgical site infection and the development of short-term complications in macaques undergoing indwelling vascular access port placement. J. Med. Primatol 2008, 37, 202–209. [DOI] [PubMed] [Google Scholar]
- (20).Felice BR; Wright TL; Boyd RB; Butt MT; Pfeifer RW; Pan J; Ruiz JA; Heartlein MW; Calias P Safety evaluation of chronic intrathecal administration of idursulfase-IT in cynomolgus monkeys. Toxicol. Pathol 2011, 39, 879–892. [DOI] [PubMed] [Google Scholar]
- (21).Raper J; Morrison RD; Daniels JS; Howell L; Bachevalier J; Wichmann T; Galvan A Metabolism and Distribution of Clozapine-N-oxide: Implications for Nonhuman Primate Chemogenetics. ACS Chem. Neurosci 2017, 8, 1570–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bogenpohl J; Galvan A; Hu X; Wichmann T; Smith Y Metabotropic glutamate receptor 4 in the basal ganglia of parkinsonian monkeys: ultrastructural localization and electrophysiological effects of activation in the striatopallidal complex. Neuro-pharmacology 2013, 66, 242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Nanda B; Galvan A; Smith Y; Wichmann T Effects of stimulation of the centromedian nucleus of the thalamus on the activity of striatal cells in awake rhesus monkeys. Eur. J. Neurosci 2009, 29, 588–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).DeLong MR Putamen: activity of single units during slow and rapid arm movements. Science 1973, 179, 1240–1242. [DOI] [PubMed] [Google Scholar]
- (25).Obach RS; Reed-Hagen AE; Krueger SS; Obach BJ; O’Connell TN; Zandi KS; Miller S; Coe JW Metabolism and disposition of varenicline, a selective alpha4beta2 acetylcholine receptor partial agonist, in vivo and in vitro. Drug Metab. Dispos 2006, 34, 121–130. [DOI] [PubMed] [Google Scholar]
- (26).Qin DD; Feng SF; Zhang FY; Wang N; Sun WJ; Zhou Y; Xiong TF; Xu XL; Yang XT; Zhang X; Zhu X; Hu XT; Xiong L; Liu Y; Chen YC Potential use of actigraphy to measure sleep in monkeys: comparison with behavioral analysis from videography. Zool. Res 2020, 41, 437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Paquet J; Kawinska A; Carrier J Wake detection capacity of actigraphyduring sleep. Sleep 2007, 30, 1362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Cortes JA; Gomez G; Ehnerd C; Gurnsey K; Nicolazzo J; Bradberry CW; Jedema HP Altered activity-based sleep measures in rhesus monkeys following cocaine self-administration and abstinence. Drug Alcohol Depend. 2016, 163, 202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Barrett CE; Noble P; Hanson E; Pine DS; Winslow JT; Nelson EE Early adverse rearing experiences alter sleep-wake patterns and plasma cortisol levels in juvenile rhesus monkeys. Psychoneuroendocrinology 2009, 34, 1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.