

SUMMARY
The epidemic community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA) USA300 lineage has recently become a leading cause of hospital-associated bloodstream infections (BSI). Here, we leveraged this recent introduction into hospitals and the limited genetic variation across USA300 isolates to identify mutations that contribute to its success in a new environment. We found that USA300 BSI isolates exhibit altered virulence regulation. Using comparative genomics to delineate the genes involved in this phenotype, we discovered repeated and independent mutations in the transcriptional regulator sarZ. Mutations in sarZ resulted in increased virulence of USA300 BSI isolates in a murine model of BSI. The sarZ mutations derepressed the expression and production of the surface protein ClfB, which was critical for the pathogenesis of USA300 BSI isolates. Altogether, these findings highlight ongoing evolution of a major MRSA lineage and suggest USA300 strains can optimize their fitness through altered regulation of virulence.
Keywords: MRSA, GWAS, pathogenesis, bloodstream infections, gene regulation
Graphical Abstract
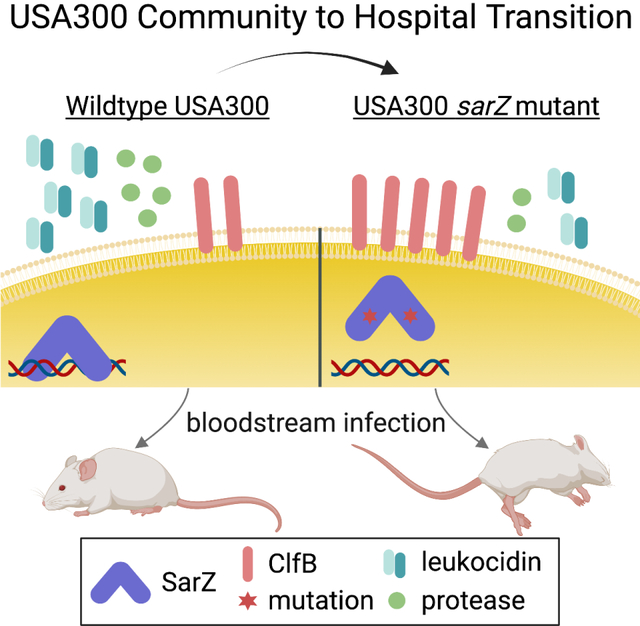
eTOC blurb
Staphylococcus aureus is a versatile pathogen. Dyzenhaus et al. show that USA300 clinical isolates from the bloodstream naturally acquire mutations in the transcription factor sarZ that increase their virulence in a murine model of bloodstream infection. This study highlights the power of genomics to discover new biology in infectious disease.
INTRODUCTION
Staphylococcus aureus is a diverse and versatile bacterium which asymptomatically colonizes about a third of the population, while also causing a wide variety of diseases1,2. These range from skin and soft tissue infections (SSTIs), which are most commonly associated with the community and otherwise healthy individuals, to more invasive and potentially deadly bloodstream infections (BSI)1,3. Increasingly, BSI and other invasive S. aureus infections (e.g., endocarditis, osteomyelitis, pneumonia) are hospital-associated4.
Historically, the lineages of methicillin-resistant S. aureus (MRSA) that cause community-associated (CA) SSTIs have been distinct from lineages responsible for hospital-associated (HA) invasive infections5. Traits distinguishing CA-MRSA isolates from HA-MRSA include the presence of the bi-component leukocidin known as LukSF-PV, or Panton-Valentine leukocidin toxin (PVL)6, and more generally increased production of toxins (e.g., PSMs, alpha-toxin and leukocidins) in CA-MRSA. Consequently, CA-MRSA are more cytotoxic than HA-MRSA in tissue culture models7–10. The increased toxin production in CA-MRSA largely results from a highly active accessory gene regulator (Agr), the quorum sensing system in S. aureus11,12. In contrast, HA-MRSA lineages often have mutations in the agr locus that attenuate its activity13,14. Counterintuitively, reduced cytotoxicity in HA-MRSA has been associated with increased lethality in a multi-center study of pneumonia8. This suggests that, while decreased in vitro cytotoxicity is a trait of HA-MRSA, other bacterial factors may be important in determining outcomes during infection in hospitalized patients. The finding that CA- and HA-MRSA have different interactions with host populations supports the idea that pinpointing differences in the two forms of MRSA could reveal pathways important in hospital-associated disease.
Since the divergence of HA-MRSA strains from CA-MRSA isolates happened decades ago, analysis of the genetic changes associated with the transition from community to hospital is complex. CA-MRSA, such as USA300, the predominant lineage in the US, are only distantly related to HA-MRSA, and were not linked to the healthcare environment15,16. However, USA300 has recently taken a foothold in hospitals and is now becoming one of the leading causes of nosocomial BSI4,16–18. Using cytotoxic attenuation as a proxy for hospital adaptation, our data and that of others indicate that many USA300 isolates from hospitalized patients are adapting to the hospital7,8. The reduction in cytotoxicity observed as “CA-USA300” isolates become “HA-USA300” could facilitate a better understanding of hospital adaptation in MRSA.
Our results show that USA300 BSI isolates exhibit a wide range of cytotoxic activity, with an overall decrease in in vitro cytotoxicity compared to contemporary CA-USA300 isolates from patients with SSTI. Comparative genomics identified uncharacterized mutations in the transcriptional regulator sarZ that were enriched among USA300 BSI isolates of low cytotoxicity. We show that these sarZ natural mutations rewired the expression of virulence factors in USA300, leading to increased pathogenesis in a murine model of BSI. Furthermore, we identified the surface protein ClfB as an important factor in the virulence of sarZ mutant USA300 BSI isolates in vivo. Collectively, these data demonstrate convergent evolution of sarZ mutations in USA300 BSI to alter the expression and production of virulence factors.
RESULTS
Altered Cytotoxicity of Bloodstream USA300 Isolates
To examine how USA300 BSI isolates differ in their virulence potential compared to CA-SSTI isolates, we screened a collection of MRSA clinical isolates collected from surveillance programs from two New York City health systems for their in vitro cytotoxicity to primary human neutrophils (hPMNs). Our collection of MRSA isolates included: 99 USA300 BSI isolates collected between 2014 and 2017; 35 USA300 SSTI isolates collected from 2014–2016; and 44 non-USA300 control HA-MRSA isolates (i.e., USA100 and USA500) collected contemporaneously. The USA300 BSI isolates were predominantly (75.5%) healthcare-associated owing to onset >48 hours after hospital admission and/or association with health care risk (e.g., recent hospitalization, transfer from a nursing home, healthcare interaction) (Table S1). The control strain AH-LAC19, a commonly used prototype CA-MRSA USA300 SSTI isolate was used to normalize the cytotoxic activity across experiments. FPR3757 and SF8300, two additional prototype SSTI isolates from early in the USA300 outbreak20,21, were also included in the analysis as they are commonly used as control strains.
Supernatants from cultures grown to early stationary phase were used to intoxicate hPMNs (Fig. 1A) and the percent dead hPMNs intoxicated with cultures grown in tryptic soy broth (TSB), normalized to AH-LAC, is displayed in Figure 1B. Control CA-MRSA USA300 strains and the majority of recent USA300 SSTI isolates were highly cytotoxic, while non-USA300 HA-MRSA isolates, USA100 and USA500, exhibited reduced cytotoxicity as previously observed7,8,22. However, USA300 BSI isolates were highly heterogeneous in their cytotoxicity, with some exhibiting cytotoxic levels like SSTI USA300 isolates, and others phenocopying the reduced cytotoxicity seen with the HA-MRSA isolates.
Figure 1. Phenotypic screen for cytotoxicity reveals altered virulence regulation in USA300 BSI isolates.
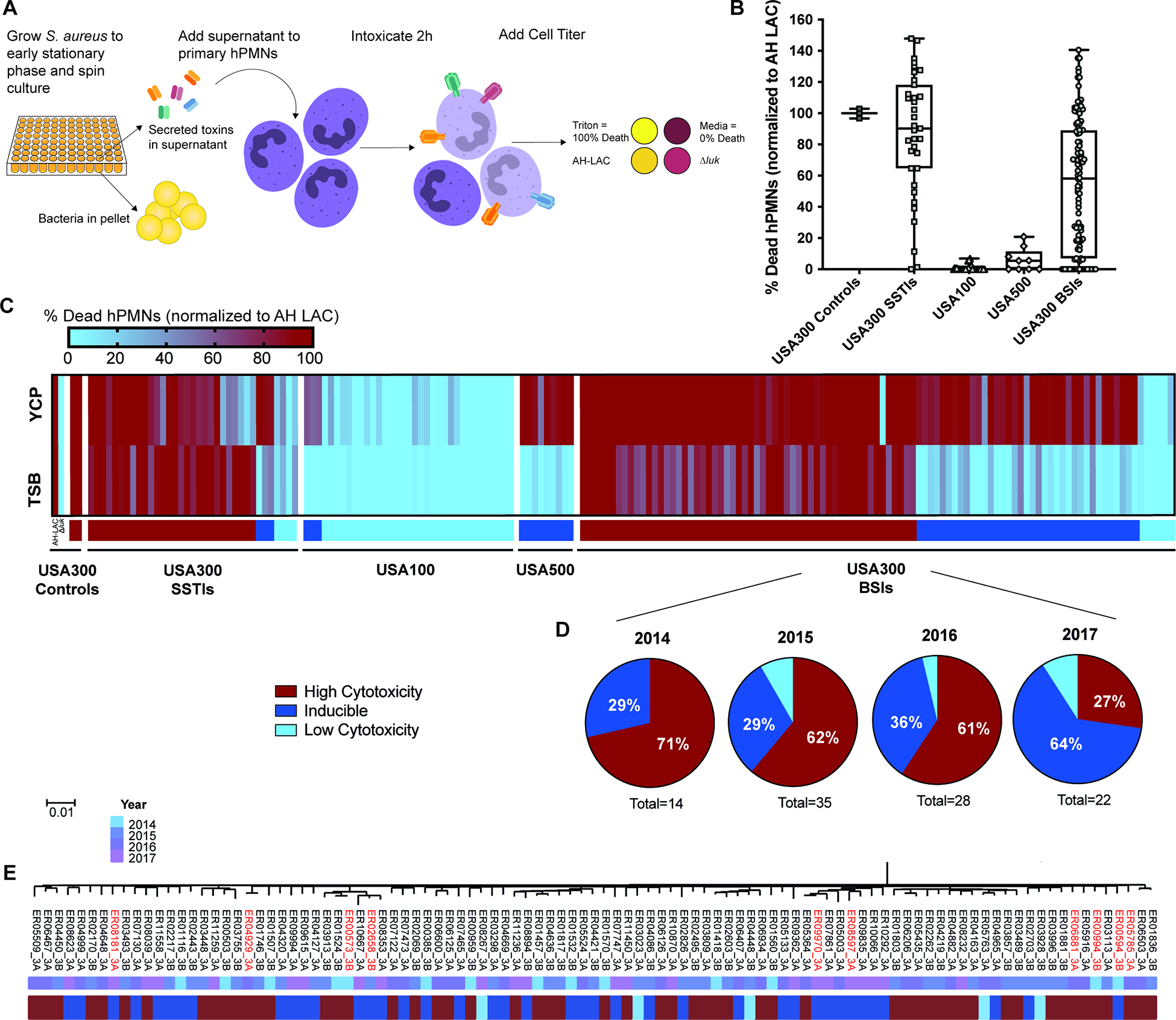
(A) Schematic of cytotoxicity screen. (B) Clinical S. aureus isolates grown in TSB were assessed for the ability of their supernatants to kill hPMNs. Percent death of 5% supernatants was normalized across experiments to control AH-LAC. Each point on the graph represents the mean cytotoxicity of a single isolate (n = 5–6 donors). Center line, median; box limits, upper and lower quartiles; whiskers; min and max. (C) Heat map depicting data from panel A, with the addition of 5% supernatant cytotoxicity data from bacteria grown in YCP media. Any normalized value above 100% is colored as 100%. Colored bars under heat map depict the cytotoxicity classification of each isolate: High Cytotoxicity = TSB >50%, Inducible = TSB <50% and YCP >40% increased from TSB value, Low Cytotoxicity = TSB <50% and not inducible. Strain Δluk is AH-LAC with deletion of all leukocidins59. Exact values depicted in heat map can be found in Table S2. (D) Proportions of USA300 BSI isolates classified by cytotoxicity, separated into groups by the year the isolate was collected. Total number of USA300 BSIs for each year is shown under each pie chart. (E) Maximum-likelihood phylogenetic tree produced from core genome SNVs identified from Parsnp whole-genome alignments of USA300 BSIs. Strains used for further phenotypic analysis are highlighted in red.
The idea that invasive USA300 BSI isolates would be evolving to be less virulent seemed counterintuitive, so we hypothesized that the observed decreased cytotoxicity was an indication of altered virulence regulation, rather than decreased virulence. We previously reported that the metabolite pyruvate can induce toxin production in vitro23. To test whether additional stimuli, in this case pyruvate, could induce toxin production in the low cytotoxic isolates, we screened our collection for cytotoxicity when grown in pyruvate containing media (YCP; Fig. 1C). Most non-cytotoxic USA300 BSI isolates were able to kill hPMNs when grown in YCP (38 of 44); we termed these strains ‘inducible’ USA300. Low cytotoxicity variants of the USA300-related MRSA lineage USA500 were also inducible (9 of 9). In contrast, low cytotoxic variants of the prototype HA-MRSA USA100 strains were largely uninducible (32 of 35), indicating that lineages more distantly related to USA300 are not inducible by pyruvate. Thus, we infer that USA300 BSI isolates have altered virulence regulation rather than an inability to be cytotoxic. Importantly, when considering the proportion of isolates with differing cytotoxicity phenotypes, we found that over time a large percentage (29–64%) of the USA300 BSI isolates consistently showed the low but inducible cytotoxic phenotype (Fig. 1D). Thus, regulation of virulence is altered among a substantial proportion of USA300 BSI isolates.
Genomic Comparison of Inducible and High Cytotoxicity Isolates
To understand the clonality and genetic basis for the inducible cytotoxicity phenotype, we sequenced the collection of USA300 BSI isolates and control MRSA strains. Each isolate was sequenced on Pacific Biosciences (PacBio) and Illumina platforms to generate finished-quality genomes. Of the 143 genomes, 128 (90%) were completely closed while the remainder yielded complete chromosomes in 1–3 contigs that could not be circularized due to repetitive regions that confounded alignment (Table S2). A phylogenetic tree of all BSI isolates depicts the close genetic relationship amongst the USA300 isolates despite their high degree of heterogeneity in cytotoxicity (Fig. S1). Notably, inducible USA300 BSI isolates did not cluster together in the phylogeny and were dispersed throughout the tree among highly cytotoxic USA300 strains (Fig. 1E). Thus, the inducible phenotype evolved repeatedly and independently, rather than by an outbreak or expansion of a single strain.
The close genetic relatedness of USA300 isolates enabled the implementation of comparative genomics to identify specific genes and mutations associated with the altered virulence of USA300 BSI isolates. We compared the genomes of 29 inducible isolates to 25 of their phylogenetically most closely related highly cytotoxic USA300 BSI isolates. The use of finished-quality sequences for all isolates enabled detection of the presence or absence of genes as well as high confidence single-nucleotide variants (SNVs) and insertions/deletions (indels). Given that the observed convergent evolution of the inducible phenotype could be due to distinct mutations in the same genes or pathways, we compared genetic differences between inducible and highly cytotoxic isolates in aggregate at the gene and operon level. Changes in three loci were statistically associated with the inducible phenotype: agr, lukSF/PV and sarZ (Fig. 2A), and together accounted for 27 of the 29 inducible strains (93%). The agr locus, an operon (agrBDCA) that encodes for a major regulator of virulence in S. aureus24,25, was mutated or disrupted in 31% of inducible USA300 BSI isolates compared to 8% in high cytotoxicity USA300. Inducible isolates had 10 distinct mutations in 9 different isolates, including promoter mutations, non-synonymous (NS) SNVs, and indels within genes (Fig. 2B–C). The agr locus is well known to contribute to the regulation of toxins through quorum sensing and has been shown to be mutated in most HA-MRSA strains8,12,13,24,26,27. This suggests that bloodstream USA300 isolates are evolving by mutating this important regulatory pathway, becoming like conventional HA-MRSA, both phenotypically and genetically.
Figure 2. Loci associated with inducible cytotoxicity USA300 BSI isolates.
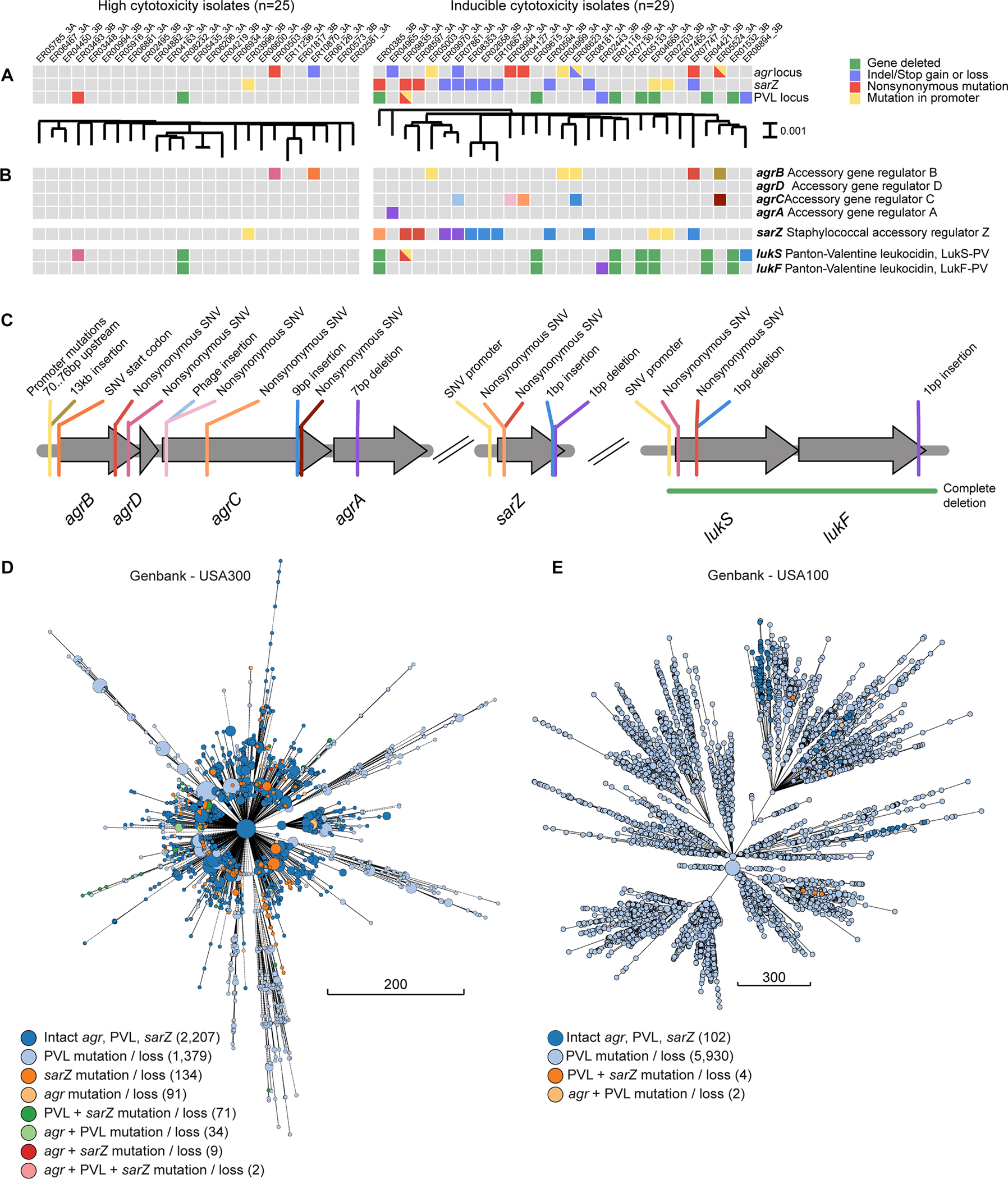
(A) Variant matrix for each of the operons that was significantly associated with the inducible phenotype (right) compared to the non-inducible isolates (left). The type of variant observed is indicated with a colored square. Between panel A and panel B, maximum-likelihood phylogenetic trees generated with parsnp are shown. (B) Same as panel A, but showing the genes in which the variants are observed. (C) Graphical view of the location and type of variants for each of the genes shown in panel B, with correspondingly colored lines. Core genome multilocus sequence typing (cgMLST) trees of (D) USA300 and (E) USA100 MRSA genomes deposited in Genbank between 2000 and 2020. Branch lengths correspond to the number of cgMLST locus differences. Genomes with ≤10 differences are collapsed into a single node. Nodes are sized proportionally to the number of genomes they represent and are colored according to which mutations or combinations of mutations were present.
Another locus associated with inducible cytotoxicity is lukSF/PV, which encodes for PVL28. This locus is present on the PVL phage, a genetic element that is found almost exclusively in CA-MRSA and has been previously reported to be lost in bloodstream isolates29–34. lukSF/PV was lost or mutated in 34% of inducible USA300 BSI isolates and 8% of the high cytotoxicity USA300. Most of these disruptions were loss of the toxin gene (Fig. 2B–C). Interestingly, the loss of the lukSF/PV loci was not due to excision of the phage, but rather due to recombination events at short direct repeats that delete large portions of the phage (Fig. S2). These observations provide further evidence that although the PVL toxin is important in the community setting it may be a liability in the nosocomial setting.
The third locus associated with inducible cytotoxicity in USA300 BSI isolates is sarZ, a transcriptional regulator thought to be involved in oxidative stress sensing35. sarZ has NS-SNVs or indels in 38% of inducible USA300 BSI isolates and zero in high cytotoxicity isolates (Fig. 2B–C). SarZ has been shown to be involved in virulence regulation in S. aureus35–39. However, the role of SarZ in USA300 virulence or the role of natural mutations in sarZ in bloodstream S. aureus isolates has not been described.
We examined the three loci associated with USA300 BSI inducible isolates broadly across other USA300 genomes in Genbank for the presence of the same genetic changes identified in our bloodstream isolates. Mutations or gene loss in all three loci were found: of 2,927 genomes, 1,486 carried at least one of our observed changes in lukSF/PV, 136 for agr, and 216 for sarZ (Fig. 2D). Like our inducible isolates, mutations were spread throughout the core genome multilocus sequence typing (cgMLST) tree, further supporting convergent evolution. Mutations in these loci are consistently detected over time in USA300, including in our most recent USA300 BSI isolates from 2021 (Fig. S3A–C). We additionally examined these loci in genomes from the common HA-MRSA lineage USA100 from Genbank (Fig. 2E, S3D) and our collection (Fig. S3B). As expected, the majority of USA100 strains in Genbank do not have PVL (5,936 of 6,038). Few have altered agr or sarZ loci as compared to the reference USA100 genome (2 and 4 of 6,038 respectively). This finding suggests that the agr and sarZ loci in the already hospital-adapted USA100 are not under the same evolutionary pressure as in USA300.
We were most interested in further examining the sarZ locus, as mutations in sarZ were most significantly associated with the inducible phenotype in our collection (38%) and their association with BSI is unknown. Importantly, our genomic analyses revealed that mutations in sarZ are not unique to BSI, but suggest they can be transmissible. Evidence of transmission of sarZ mutants is shown in Figures 2B and 2D, where there are clusters of closely related isolates with identical mutations in sarZ. The USA300 genomes in Genbank also revealed that isolates with mutations in sarZ are found in other invasive infections as well as carriage isolates (Fig. S3E). Notably, these data also show that the mutations in sarZ are not unique to New York City, but they can be found across the US and in other countries (Fig. S3F). Altogether, these data support the idea that sarZ is a target of evolution in USA300.
Contribution of SarZ to USA300 Cytotoxicity
SarZ is a MarR-family transcriptional regulator that responds to oxidative stress35,40. SarZ has a single critical cysteine, Cys13, that can be oxidized resulting in a conformational change that leads to loss of DNA binding, derepressing genes35,40. SarZ has been shown to regulate the oxidative stress response through changes in metabolism and virulence35. To confirm that disruption of sarZ alters in vitro cytotoxicity of USA300, we generated a sarZ isogenic mutant in AH-LAC (sarZ::bursa) and examined the mutant’s cytotoxic activity compared to wildtype (WT) AH-LAC. As a control, we used an isogenic AH-LAC mutant that lacks all leukocidins (Δluk), which is not cytotoxic when grown in TSB or YCP. We found that when grown in TSB, AH-LAC sarZ::bursa exhibited decreased cytotoxicity (Fig. 3A), but as with the USA300 BSI clinical isolates containing natural sarZ mutations, the phenotype disappeared when the strain was grown in YCP.
Figure 3. SarZ inducible-associated alleles lead to altered virulence regulation.
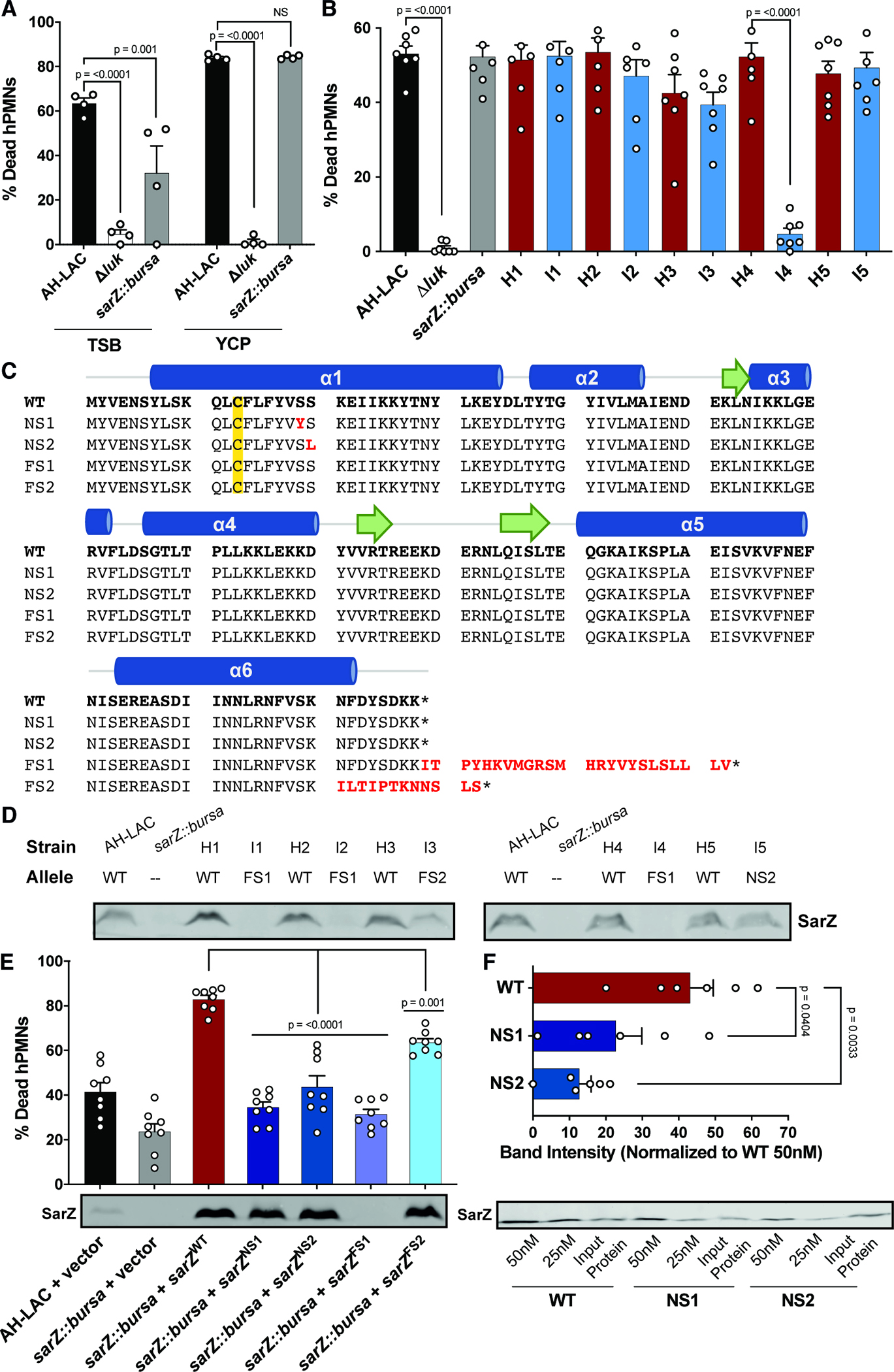
(A) Supernatants from AH-LAC and mutant strains grown to stationary phase in TSB or YCP media were assessed for their cytotoxicity to hPMNs (n = 4 donors). Data for 5% supernatant shown, full titration curves can be found in Fig. S4A/B. (B) Extracellular infection of hPMNs with AH-LAC strains or USA300 BSI isolates at MOI 50 (n = 7 donors). (C) Amino acid sequence of sarZ alleles. WT = wild type USA300 sarZ allele found in AH-LAC. The four mutant alleles found in USA300 BSI isolates are aligned below with differences highlighted in red. The major structural features of SarZ are shown above, where alpha helices are blue, beta sheets are represented by green arrows, and the critical cysteine (Cys13) is highlighted in yellow. (D) USA300 BSI isolates were grown to stationary phase and 15uL of whole cell lysates were run on a gel to detect SarZ by Western blot. (E) Supernatant cytotoxicity of AH-LAC sarZ::bursa strains complemented with the 5 different sarZ alleles, grown to stationary phase in TSB. Data from 1.25% supernatant shown, full titration curves can be found in Fig. S4C/D (n = 8 donors). SarZ Western blot shown below: AH-LAC sarZ::bursa complemented strains were grown to stationary phase and 5uL of whole cell lysates were run on a gel for Western blot. (F) Promoter pulldown (biotinylated Pssp) of WT compared to mutant SarZ purified protein (n=6). SarZ Western blot shows one representative experiment. Input Protein is 15ng of purified SarZ. Band intensity of pulldowns using 50nM and 25nM protein were normalized to Input Protein for each allele. Quantification of 25nM band intensity with WT 50nM = 100% is shown above. Statistical significance using a 2-way ANOVA with Sidak’s multiple comparisons test. Error bars indicate SEM. NS not significant.
We next investigated if sensing phagocytes could provide a more physiological signal to induce sarZ mutants to kill hPMNs. To this end, we infected hPMNs with live bacteria and monitored cell lysis. In addition to AH-LAC sarZ::bursa, we used five naturally occurring inducible USA300 sarZ variants. We paired these isolates with their most closely related high cytotoxicity USA300 BSI isolates with wildtype sarZ, agr and lukSF-PV loci (Table 1), as measured by core genome SNVs (Fig. S4E). Isolates were labeled I1-5 for inducible sarZ mutants, and H1-5 for their corresponding wildtype sarZ high cytotoxicity isolates. We found no difference in the proportion of dead hPMNs between AH-LAC and sarZ::bursa, indicating that the sarZ inactivated mutant can be induced to produce toxins and kill phagocytes during ex vivo infection (Fig. 3B). Moreover, we found no difference in hPMN death for clinical Pairs 1, 2, 3, and 5, showing that host stimuli can induce toxin expression and overcome the effects of mutations in naturally occurring sarZ mutants. Pair 4 was the exception, in that the inducible strain 4 (I4) showed significantly reduced ability to kill hPMNs. Further investigation into this isolate revealed a mutation unique to I4 in the lukAB locus; a single nucleotide deletion in lukA that leads to a frameshift and an early stop codon. This finding explains our discrepancy as LukAB is the main toxin responsible for killing hPMNs in this tissue culture model41,42.
Table 1. Clinical isolate pairs.
Inducible isolates with a mutation in sarZ are labeled as I1-I5. Closely related high cytotoxicity isolates that have a WT sarZ locus are labeled H1-H5. Isolates were paired based on relatedness.
| Strain | Pair | Cytotoxicity Classification | Label | sarZ Allele |
|---|---|---|---|---|
| ER00994.3B | 1 | High | H1 | WT |
| ER00594.3B | Inducible | I1 | FS1 | |
| ER00573.3B | 2 | High | H2 | WT |
| ER02658.3B | Inducible | I2 | FS1 | |
| ER06881.3A | 3 | High | H3 | WT |
| ER09970.3A | Inducible | I3 | FS2 | |
| ER05785.3A | 4 | High | H4 | WT |
| ER08181.3B | Inducible | I4 | FS1 | |
| ER04929.3A | 5 | High | H5 | WT |
| ER08597.3A | Inducible | I5 | NS2 |
As the sarZ::bursa strain mimicked the phenotype of the inducible USA300 BSI isolates, we hypothesized that the mutant sarZ alleles found in the inducible USA300 BSI isolates would disrupt SarZ production and/or function. There are four mutations found in the coding region of sarZ (Fig. 3C)40. Three isolates have NS-SNVs close to the critical Cys13 (termed NS1 and NS2), six isolates have a one-base pair frameshift deletion in the stop codon (termed FS1), and two isolates have a one-base pair frameshift insertion (termed FS2). Both the frameshift mutations are the result of indels within poly-A stretches (Fig. S4F). This phenomenon has been previously described as a mechanism by which S. aureus alters agr functionality43, suggesting a common strategy of altering virulence regulation across different genes.
To determine if isolates with mutations in sarZ can make protein, we evaluated production of SarZ via immunoblotting with the five USA300 BSI pairs and AH-LAC controls (Fig. 3D). As expected, SarZ was detected in WT AH-LAC and all the high cytotoxicity isolates. In contrast, no detectable SarZ was seen in AH-LAC sarZ::bursa or inducible isolates with the FS1 allele, suggesting that SarZ is not produced or is degraded with this mutation. Inducible isolates with the FS2 or NS2 allele do produce WT levels of SarZ, suggesting that the observed mutations disrupt SarZ function. Thus, the naturally occurring mutant alleles have distinct mechanisms that could lead to altered cytotoxicity phenotypes.
To further assess the function of the different sarZ alleles, we cloned the WT and the four mutant sarZ alleles where their expression was regulated by their native promoter into the multicopy plasmid pOS144 and performed complementation studies in AH-LAC sarZ::bursa. Expression of WT sarZ lead to increased cytotoxicity when compared to WT AH-LAC (Fig. 3E). In contrast, we found that each of the four mutant sarZ alleles exhibit reduced cytotoxicity compared to the WT complement strain. Interestingly, the cytotoxicity phenotype does vary; while the FS2 allele had an intermediate phenotype, the NS1, NS2 and FS1 had severely reduced activity. When evaluating production of SarZ by immunoblotting with these strains, we confirmed that the FS1 mutation ablates SarZ production. Since the NS1 and NS2 mutant proteins are produced but highly attenuated, we postulated that these mutations would lead to decreased binding of SarZ to DNA. To test this, we examined the ability of purified WT and the NS1 and NS2 mutated SarZ proteins to bind to promoters. Using biotinylated Pssp DNA, a promoter previously shown to be bound by SarZ36, we found that NS1 and NS2 SarZ indeed exhibit reduced binding as compared to WT SarZ (Fig. 3F).
USA300 sarZ Mutant Strains are More Virulent in Murine Models of BSI
Given that mutations in sarZ are found naturally in clinical isolates, we hypothesized that these mutations contribute to the pathogenesis of USA300 BSI. Thus, we investigated the virulence of USA300 BSI isolates in a murine BSI model. We used the five pairs of USA300 BSI isolates (Table 1) to compare inducible isolates with mutations in sarZ and high cytotoxicity isolates with a WT sarZ locus. To our surprise, the inducible isolates with mutations in sarZ were more virulent than the sarZ wildtype high cytotoxicity isolates (Fig. 4A). Infection with any sarZ mutants resulted in <15% survival two weeks post infection, while all isolates with a WT sarZ exhibited >50% survival. This striking lethality phenotype was apparent when comparing across all isolates and individual infection pairs (Fig. S5A). We used pairs 1 and 2 to infect mice at a lower dose and harvested organs 1-day post-infection to examine the bacterial burden. Mice infected with inducible sarZ mutant strains I1 and I2 harbored increased bacterial burden in multiple organs compared to their respective WT controls H1 and H2 (Fig. 4B). These data demonstrate that inducible USA300 sarZ mutants are more fit to survive and thrive in vivo.
Figure 4. Increased virulence in a murine BSI model due to mutations in sarZ.
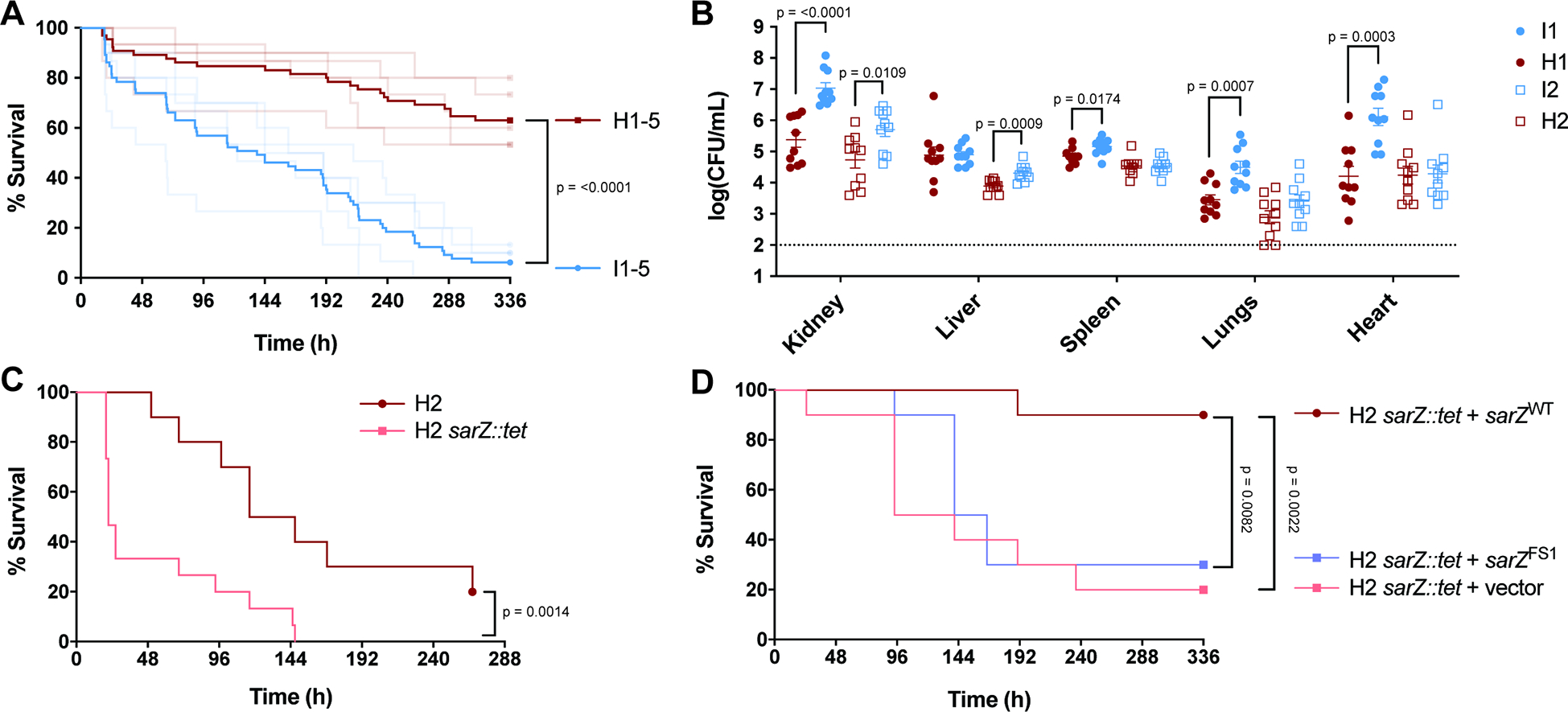
(A) Survival of mice infected i.v. (5×107 CFU) with USA300 BSI isolates. Inducible USA300 (I1-I5) that have mutations in sarZ are blue. Closely related high cytotoxicity USA300 (H1–5) that have a WT sarZ locus are red. Dark lines represent pooled data for the 5 inducible isolates and for the 5 high cytotoxicity isolates (n = 65). Faded lines represent survival data for each of the 10 isolates (n = 10–15). Data shown in faded lines is also depicted in Fig. S7, separated to show differences within pairs. (B) CFU burden of mice infected i.v. (1×107 CFU) with USA300 BSI isolates. Pairs 1 and 2 were used to infect mice and organs were harvested 1-day post-infection to determine CFU burden (n = 10). Statistical analysis using unpaired two-tailed t tests with Welch’s correction. (C) Survival of mice infected i.v. (7.5×107 CFU) with H2 and H2 sarZ::tet (n = 10–15). (D) Survival of mice infected i.v. (8×107 CFU) with H2 sarZ::tet complemented with WT and FS1 sarZ alleles (n = 10). Statistical analysis for survival curves done with the Log-rank (Mantel-Cox).
In addition to the sarZ mutations, the inducible clinical isolates contain other chromosomal SNVs and indels that could impact virulence (Fig. S4E). To directly evaluate the role of sarZ inactivation to the observed increased virulence in vivo, we infected mice with USA300 BSI high cytotoxic isolate H2 and an isogenic H2 sarZ::tet strain (Fig. 4C). Compared to the parental H2 strain, the H2 sarZ::tet strain was significantly more virulent in the BSI model. Importantly, the heightened virulence of the H2 sarZ::tet strain was attenuated when a WT copy of sarZ was expressed using a complementing plasmid, but not when the complementing plasmid contained the sarZ FS1 allele (Fig. 4D). While mutating sarZ in the contemporary USA300 BSI isolate background (H2) led to significantly increased virulence, this phenotype did not replicate when mutating sarZ in the reference SSTI isolate SF8300 (Fig. S5B). The increased virulence was also found to be specific to the bloodstream model of infection, as no advantage was seen for sarZ mutant isolates or isogenic mutants in in vitro growth, growth in whole human blood, or in a murine model of SSTI (Fig. S5C–E). Thus, mutation of sarZ increases the virulence potential of contemporary USA300 BSI isolates.
SarZ Regulates ClfB and Proteases in USA300
To understand how mutations in sarZ might be leading to increased virulence in murine BSI, we next determined the role of SarZ in the virulence regulatory network of USA300. Previous studies examining the SarZ regulon in other S. aureus strains suggest that SarZ alters expression of virulence factors, such as alpha-toxin and the protease SspA, and is intertwined with other regulators, including MgrA, Agr and SarA37,38. SarZ has also been found to be involved in oxidative stress responses by repressing genes such as ohr and msaB 35,45. To establish the SarZ regulon in USA300, the strains AH-LAC + vector, AH-LAC sarZ::bursa + vector, and AH-LAC sarZ::bursa + sarZWT were used. Since transcriptional differences observed utilizing laboratory-constructed mutants may not apply to clinical isolates with mutations elsewhere in the genome, we also constructed sarZ deletion (sarZ::tet) and complement strains in two clinical USA300 BSI isolates, I1 and H2. These two strains were chosen as one is an inducible isolate with the FS1 mutation (I1) and one is a high cytotoxicity isolate with a WT sarZ locus (H2) that are more distantly related than isolates within a pair – giving increased diversity of background mutations. sarZ::tet mutants were complemented using WT sarZ alleles at the native chromosomal site (sarZ::sarZWT). Deleting sarZ in the WT H2 isolate lead to decreased cytotoxicity, which was complemented by adding back the WT sarZ allele (Fig. S6B). There was no difference in cytotoxicity for I1 when sarZ was deleted (Fig. S6A), a result that was expected as this isolate has a nonfunctional sarZ allele (FS1; see Fig. 3D). In contrast, the I1 strain complemented with the WT sarZ allele (I1 sarZ::sarZWT) is significantly more cytotoxic than the sarZ::tet strain. After confirming that mutant strains exhibit altered virulence regulation, we used strains I1 and I1 sarZ::sarZWT for the RNA-seq as a comparison of the mutant FS1 allele versus a WT sarZ allele. We also used the H2 and H2 sarZ::tet strains as a comparison of a WT sarZ allele versus a mutated sarZ in a second BSI background, as the H2 sarZ::tet strains exhibit increased virulence in murine BSI (Fig. 4C).
Figure 5A shows the statistically differentially regulated genes in the AH-LAC background. We found many more differentially expressed genes in the SarZ overexpression strain. Accordingly, we focused our comparative analysis on 49 genes with significant and opposite expression changes in the sarZ::bursa and the overexpression sarZ strain background during either exponential or stationary growth, reasoning that these were most likely to reflect direct regulation by SarZ (Fig. 5B). To our surprise, we did not find differences in many of the previously described SarZ-regulated genes, except for sspA. This finding highlights the impact of genetic variation across S. aureus lineages on gene expression46. To focus our analysis on clinically relevant USA300 BSI variants, we further restricted the SarZ regulon to a subset of 16 genes that were also differentially expressed between H2 and H2 sarZ::tet and/or between I1 and I1 sarZ::sarZWT (Fig. 5C). This gave insight into the core genes that are regulated by SarZ in USA300 regardless of variations in strain backgrounds. In all three USA300 strain backgrounds (i.e., AH-LAC, I1 and H2), we found increased clfB expression and a decreased protease expression in the sarZ mutant strains. ClfB is a surface adhesin important in nasal colonization47,48 and is implicated in the pathogenesis of endocarditis and SSTI in rat and mouse models of infection, respectively49,50. clfB is expressed during exponential growth and the protein is rapidly degraded by the protease aureolysin during stationary phase51,52. In parallel, aureolysin (aur) and other proteases (sspABC) were found to be downregulated in the sarZ mutant stains. Proteases are involved in the degradation of surface proteins and secreted toxins, in turn impacting the virulence potential of S. aureus in murine models of infection53,54.
Figure 5. Characterization of the SarZ regulon in USA300.
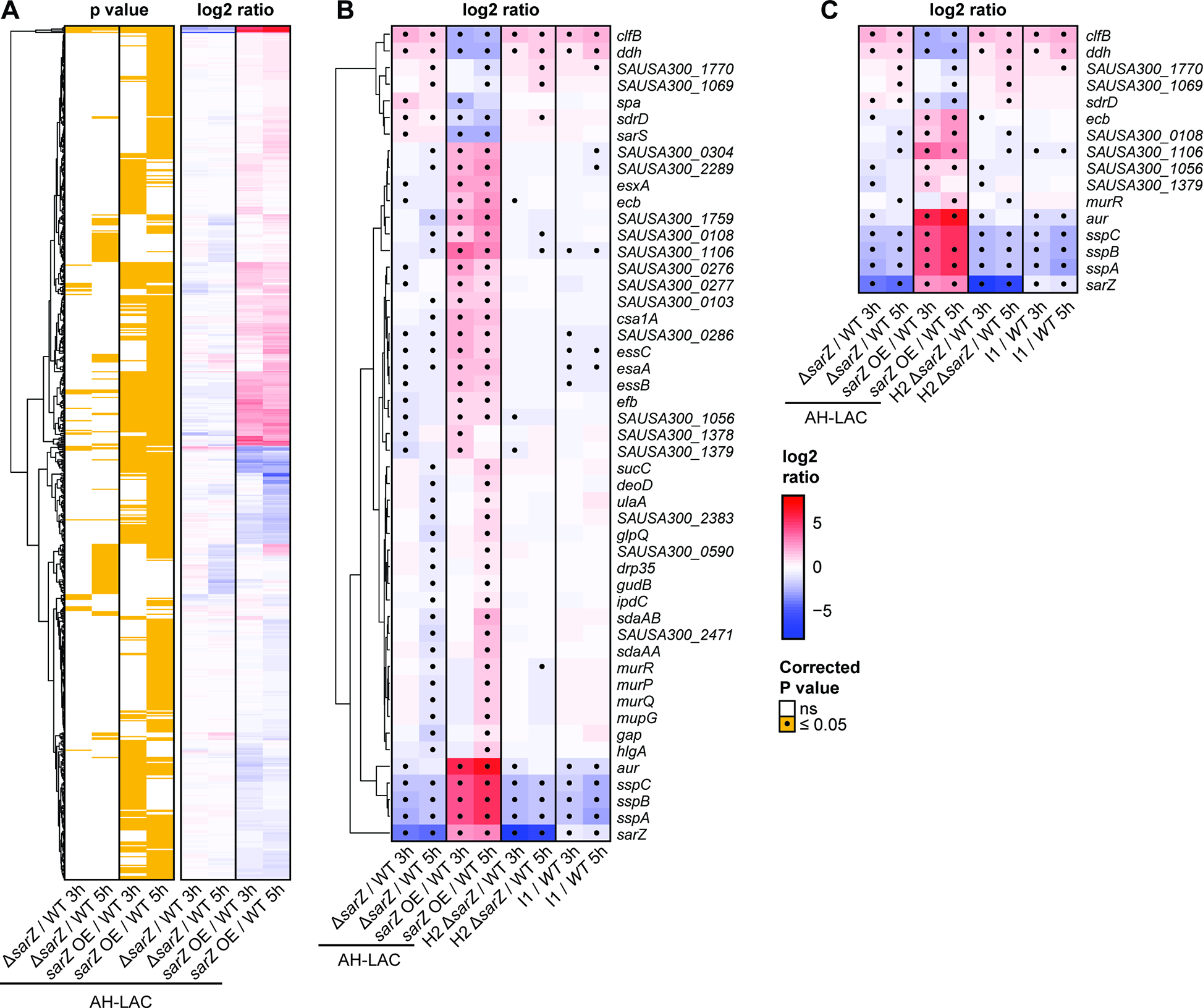
(A) Hierarchical clustering of 696 differentially expressed genes between the AH-LAC sarZ::bursa + vector transposon mutant (ΔsarZ), or the AH-LAC sarZ::bursa + sarZWT overexpression mutant (OE), compared to wild-type AH-LAC + vector (WT) during exponential (3h) and/or or stationary (5h) growth. Columns correspond to comparisons with labels indicated at the bottom. Log2 ratio color shades and intensity represent the difference in normalized log2 counts per million (CPM), with a color key shown on the far-right. Significant expression changes (FDR q<0.05) between three replicate experiments are highlighted in yellow in the matched panel on the left. (B) Same as panel A, but focusing on the 49 genes with opposite changes between the AH-LAC sarZ deletion and overexpression experiments, and including results for the sarZ loss-of-function mutants (H2 ΔsarZ and I1) compared to their (complemented) wild-type strains for two USA300 BSI isolates. Gene names and descriptions are shown on the right, and significant changes are marked by dots. (C) Same as B, after further restricting to the 16 genes with opposite regulation between sarZ deletion and overexpression experiments in AH-LAC and USA300 BSI isolate backgrounds.
To confirm that increases in clfB expression and decreases in protease expression are indeed phenotypes of the sarZ mutant strains, we isolated RNA from the five USA300 BSI pairs and performed qRT-PCR to quantify the levels of clfB, aur, and sspA. Overall, clfB expression was found to be increased in the inducible USA300 BSI isolates compared to closely related high cytotoxicity isolates (Fig. S6C). The protease expression data also supported what we learned from RNA-seq, where the inducible USA300 BSI isolates with mutations in sarZ exhibited decreased expression compared to high cytotoxicity isolates with a WT sarZ (Fig. S6D). We additionally examined expression of these genes in pair 2 when grown in 50% human serum (Fig. S6E). Under this more physiological condition, we found that the expression trends remained consistent. Altogether, these data show that inducible USA300 BSI isolates exhibit increased expression of clfB and decreased expression of proteases due to mutations of sarZ.
Increased ClfB in sarZ mutants contributes to virulence
Given that ClfB contributes to pathogenesis in preclinical models of infection49,50, and proteases influence the amount of ClfB protein on the surface of S. aureus52, we hypothesized that differential regulation of clfB in sarZ mutants would contribute to their increased lethality in the murine BSI model. First, to confirm direct regulation of clfB expression by SarZ, we used a promoter pull-down assay to assess if SarZ binds the clfB promoter (Fig. 6A). SarZ bound to the ssp promoter (positive control36) and the clfB promoter, but not to the purA promoter, a gene we found not to be regulated by SarZ. Moreover, clinical isolates with natural mutations in sarZ and isogenic sarZ mutant strains were found to produce increased levels of ClfB (Fig. 6B).
Figure 6. Increased ClfB in sarZ mutants contributes to lethality.
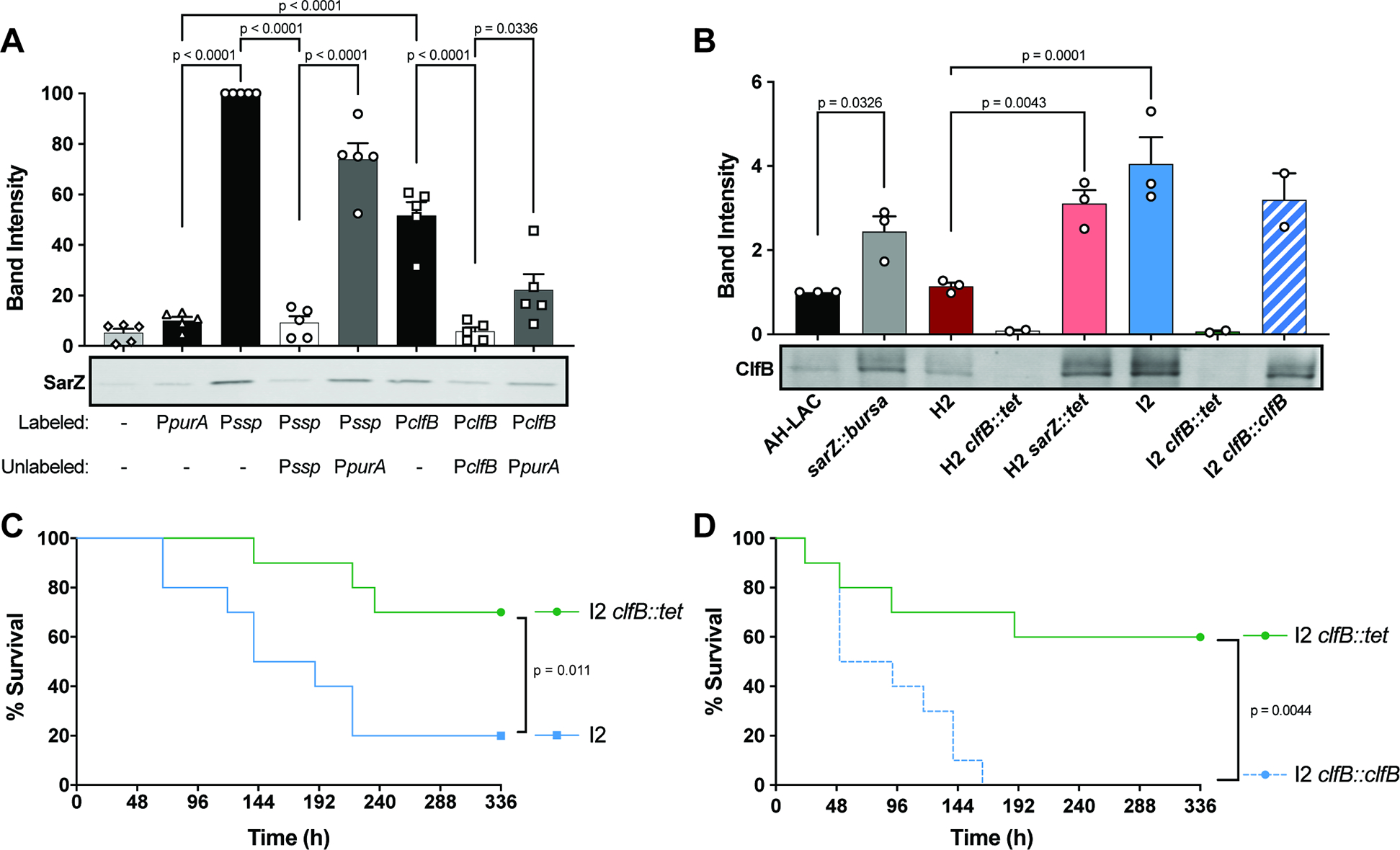
(A) Promoter pulldown of 50nM WT SarZ purified protein to biotinylated promoter DNA (n=5). SarZ Western blot shows one representative experiment. 15X unlabeled promoter DNA was used as a competitor. Quantification of band intensity normalized to Pssp = 100% is shown above. (B) ClfB Western blot and quantification of whole cell lysates grown 3h in TSB. Band intensity normalized to AH-LAC = 1 (n=2–3) (C) Survival of mice infected i.v. (1–2×107 CFU) with I2 and I2 clfB::tet (n = 10). (D) Survival of mice infected i.v. (3×107 CFU) with I2 clfB::tet and I2 clfB::clfB (n = 10). Statistical significance using a 2-way ANOVA with Sidak’s multiple comparisons test. Error bars indicate SEM. Statistical analysis for survival curves done with the Log-rank (Mantel-Cox).
To examine if the observed increased lethality of the USA300 BSI sarZ mutant strains is due to increase production of ClfB, we generated a clfB mutant strain in the hypervirulent USA300 BSI inducible strain I2 and infected mice with the WT and the clfB::tet isogenic pair. As expected (Fig. S5A), we observed high virulence by the parental I2 USA300 BSI sarZ strain where 80% of the mice succumbed to infection (Fig. 6C). In contrast, the I2 clfB::tet isogenic mutant exhibit decreased virulence, where only 30% mice succumbed to infection. Importantly, the attenuated virulence phenotype of the I2 clfB::tet isogenic strain was rescued by complementing clfB back into the native site in the chromosome (I2 clfB::clfB) (Fig. 6D). We next examined if the virulence attenuation upon deletion of clfB was specific to USA300 BSI strains defective in sarZ. We generated a clfB mutant in the USA300 BSI H2 background (H2 clfB::tet) and infected mice with a high dose of wildtype and the isogenic H2 clfB::tet strains. These experiments revealed that ClfB plays a minor role, if any, in the pathogenesis of USA300 BSI strains that harbor a wildtype sarZ in our murine model of BSI (Fig. S7). Collectively, these findings demonstrate that the increased lethality of USA300 BSI sarZ mutants is in part due to changes in regulation that lead to increased production of ClfB.
DISCUSSION
In this study, we leveraged the recent introduction of the USA300 lineage into the hospital, the genetic conservation across USA300 isolates, complete genome comparisons, and phenotypic analyses to discover mutations involved in the evolution of USA300. Three loci contributed to the altered cytotoxicity phenotype in USA300 BSI isolates: agr, lukSF/PV, and sarZ. Mutations in two of these loci, agr and lukSF/PV, have been previously implicated in hospital adaptation, indicating that USA300 BSI isolates use similar mechanisms as other HA-MRSA strains to thrive in this environment26,27,29,30. The advantage of reduced or altered cytotoxicity by HA-MRSA, including due to mutations in agr or the absence of lukSF/PV, is not fully understood. Hypotheses include that it leads to decreased inflammation and/or increased persistence and biofilm formation55,56. Our findings support that altered virulence regulation, including differential regulation of cytotoxicity, may be beneficial to HA-MRSA. We build on this by introducing a role for SarZ and specifically the SarZ-regulated adhesin ClfB in contributing to USA300 disease.
Our data demonstrate that naturally occurring mutations in sarZ among USA300 BSI isolates affect the function of the protein. Heightened lethality observed by USA300 BSI sarZ mutant strains in a murine BSI model suggest a virulence advantage to mutating sarZ. To understand how disrupting sarZ affects regulation of virulence genes that may contribute in murine BSI, we performed RNA-seq. This proved to be important, as despite the body of research describing the regulon of SarZ in other S. aureus backgrounds, there was little overlap of the transcriptional profile of USA300 with previously studied strains35,37,38,45. Whether this is due to differences in strain lineages, growth conditions, or the approaches used to define the SarZ regulon is unclear. Nevertheless, our RNA-Seq analysis uncovered that clfB is repressed by SarZ and genes coding for proteases are activated by SarZ in USA300. Given that proteases degrade ClfB on the surface of S. aureus52, the identification of clfB as a major target of SarZ suggested that ClfB might play an important role in the virulence of sarZ mutant isolates. Indeed, deletion of clfB in a representative inducible USA300 BSI sarZ mutant attenuated the hypervirulence of the parental strain. These data support a model whereby USA300 BSI sarZ mutant isolates exhibit enhanced virulence due to the increased production of an adhesin involved in pathogenesis, as well as low expression of proteases that are known to negatively regulate the levels of adhesins. Whether or not increased virulence due to ClfB in human BSI is a driver of sarZ mutation is unknown. It is possible that ClfB provides and advantage in hospitals by enhancing colonization48. As colonization is one of the major risk factors for invasive S. aureus infection57, enhanced colonization could help explain why we have found sarZ mutant isolates in BSIs. Additionally, the known role of ClfB in biofilm formation could also contribute to seeding of BSIs from indwelling devices58. Further research is needed to dissect the role of ClfB in the pathogenesis of USA300 BSI isolates.
Understanding the virulence mechanisms of USA300 in different environments may give us clues to the ways in which other lineages of MRSA adapt. It would be interesting to uncover the role of SarZ and ClfB more widely in HA-MRSA. As we found that the regulon of SarZ in USA300 differs from previously studied strains, it is unclear if mutations in sarZ would increase virulence in the same way as we have seen in USA300 BSI isolates. For example, the CC5 and CC30 HA-MRSA have different sarZ alleles than USA300 (Fig. S4G); how these alleles affect virulence in the context of a different genomic background is unknown. Additionally, other regulatory pathways in HA-MRSA could be changing the amount of ClfB and proteases. As we found that ClfB is important for the increased virulence of naturally occurring sarZ mutant strains, mutations in other regulators that increase ClfB could lead to a similar virulence advantage in other HA-MRSA.
At a more general level, these results highlight the power of microbial comparative genomics coupled with phenotypic analyses and in vivo experimentation as a foundation for examining the evolution of opportunistic pathogens. We expect that understanding the specific factors that allow S. aureus to thrive in clinically relevant environments will inform drug and vaccine design to combat this deadly pathogen.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Victor J. Torres (Victor.Torres@nyulangone.org).
Materials availability
Reagents and strains used and/or generated in this study will be made available on request upon completion of a Materials Transfer Agreement. Shipping fees might also be required.
Data and code availability
All genome data and assemblies are available in GenBank (see Table S2). Raw and processed mRNA sequence files are available at the NCBI Gene Expression Omnibus under accession no GSE185544. This information can also be found in the Key Resource Table
Custom-built pipelines for MRSA genome assembly and annotation are available in powerpak/pathogendb-pipeline github repository. Annotation of genetic changes between inducible and highly cytotoxic isolates and preparation of data matrices for GWAS was done with custom tools available in the mjsull/GWviz github repository. This information is included in the Key Resource Table. Additional methods and other relevant data can be found in Supplemental Information.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody Alexa Fluor 680 | Invitrogen | A-21057 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody Alexa Fluor 680 | Invitrogen | A-11011 |
| Anti-SarZ mouse polyclonal sera | This paper | N/A |
| Anti-ClfB rabbit polyclonal sera | Fleury et al. 201779 | N/A |
| Bacterial and virus strains | ||
| AH-LAC | Boles et al. 201019 | N/A |
| FPR3757 | Isolated from a patient (Diep et al. 2006)20 | N/A |
| SF8300 | Isolated from a patient (Diep et al. 2008)21 | N/A |
| AH-LAC Δluk | Blake et al. 201859 | N/A |
| AH-LAC sarZ::bursa | This paper | N/A |
| AH-LAC + vector | This paper | N/A |
| AH-LAC sarZ::bursa + vector | This paper | N/A |
| AH-LAC sarZ::bursa + sarZWT | This paper | N/A |
| AH-LAC sarZ::bursa + sarZNS1 | This paper | N/A |
| AH-LAC sarZ::bursa + sarZNS2 | This paper | N/A |
| AH-LAC sarZ::bursa + sarZFS1 | This paper | N/A |
| AH-LAC sarZ::bursa + sarZFS2 | This paper | N/A |
| I1 sarZ::tet | This paper | N/A |
| I1 sarZ::sarZWT | This paper | N/A |
| H2 sarZ::tet | This paper | N/A |
| H2 sarZ::sarZWT | This paper | N/A |
| H2 sarZ::tet + vector | This paper | N/A |
| H2 sarZ::tet + sarZWT | This paper | N/A |
| H2 sarZ::tet + sarZFS1 | This paper | N/A |
| SF8300 sarZ::tet | This paper | N/A |
| I2 clfB::tet | This paper | N/A |
| I2 clfB::clfB | This paper | N/A |
| H2 clfB::tet | This paper | N/A |
| BL21 pET15b-sarZ | This paper | N/A |
| BL21 pET15b-sarZNS1 | This paper | N/A |
| BL21 pET15b-sarZNS2 | This paper | N/A |
| Clinical isolates listed in Table S2 | This paper | N/A |
| Biological samples | ||
| LeukoPacks | New York Blood Center | N/A |
| SeraCare Normal Human Serum | Dba LGC Clinical Diagnostics | Ref# 27000-100 |
| Whole human blood | Torres Lab IRB | IRB # S12-01223 |
| Chemicals, peptides, and recombinant proteins | ||
| Lysostaphin | Smbi Products LLC | Cat# LSPN-50 |
| DNase (for bacterial whole cell lysates) | Promega | Cat# M6101 |
| RNase A | Promega | Cat# A7973 |
| HALT™ Protease Inhibitor | Thermo Scientific | Cat# 78438 |
| Sodium pyruvate | ACROS Organics | Cat# 113-24-6 |
| Chloramphenicol | Fisher BioReagents | Cat# BP904-100 |
| Erythromycin | Fisher BioReagents | Cat# BP920-25 |
| Tetracycline | Alfa Aesar | Cat# B21408 |
| Ampicillin | Fisher Scientific | Cat# BP1760 |
| Anhydrotetracycline | Thermo Scientific | Cat# AC233131000 |
| PCPA | Sigma-Aldrich | Cat# C6506 |
| IPTG | Research Products International | Cat# I5600025.0 |
| TiterMax® Gold | Titermax | Cat# G-20 |
| Avertin (2,2,2-tribromoethanol) | Sigma-Aldrich | Cat# T48402 |
| Streptokinase | Sigma-Aldrich | Cat# S313-250KU4 |
| Trypsin | Sigma-Aldrich | Cat# T4549 |
| DNase I (used in SK buffer) | Sigma-Aldrich | Cat# DN25-1G |
| RNase (used in SK buffer) | Alfa Aesar | Cat# J61996 |
| Dynabeads M-280 Streptavidin | Invitrogen | Cat# 11205D |
| Poly(dG:dC) naked | Invivogen | Cat# TLRLPGCN |
| His-SarZWT | This paper | N/A |
| His-SarZNS1 | This paper | N/A |
| His-SarZNS2 | This paper | N/A |
| Critical commercial assays | ||
| CellTiter 96® Aqueous One Solution | Promega | Cat# PR-G3581 |
| CytoTox-ONE™ Homogeneous Membrane Integrity Assay | Promega | Cat# PR-G7892 |
| RNA-STAT-60™ | Amsbio | Cat# CS-111 |
| TURBO DNA-free™ kit | Invitrogen | Cat# AM1907 |
| Ribo-Zero Plus rRNA Depletion Kit | Illumina | Cat# 20037135 |
| Stranded Total RNA Prep kit | Illumina | Cat# AM1907 |
| Gibson Assembly Master Mix | New England Biolabs | Cat# E2611L |
| DNeasy Blood and Tissue Kit | Qiagen | Cat# 69504 |
| Bugbuster | MilliporeSigma | Cat# 709215 |
| HisTrap HP column | Cytiva | Cat# 45-003-013 |
| SuperScript™ III First-Strand Synthesis System | Invitrogen | Cat# 1808004 |
| Deposited data | ||
| Genome assemblies | GenBank | See Table S2 |
| mRNA sequence files | NCBI Gene Expression Omnibus | Accession No. GSE185544 |
| Experimental models: Cell lines | ||
| NA | ||
| Experimental models: Organisms/strains | ||
| Swiss Webster Hsd (5 week old) | Envigo Inc | Hsd:ND4 |
| C57BL/6J (8 week old) | Jackson Laboratories | 000664 |
| Oligonucleotides | ||
| For primers used in cloning, see Table S4 | This paper | N/A |
| For primers used in qRT-PCR, see Table S4 | This paper | N/A |
| For primers used in promoter pulldowns, see Table S4 | This paper | N/A |
| Recombinant DNA | ||
| pOS1 | Schneedwind et al. 199244 | N/A |
| pIMAY | Monk et al. 201260 | Addgene plasmid #68939 |
| pIMAY* | Schuster et al. 201961 | Addgene plasmid #121441 |
| pET15b | Novagen | Cat# 69661-3 |
| Software and algorithms | ||
| HGAP3 | Chin et al. 201363 | https://www.pacb.com/products-and-services/analytical-software/smrt-analysis/ |
| Parsnp | Treangen et al. 201464 | http://github.com/marbl/harvest |
| etetoolkit | Huerta-Cepas et al. 201665 | http://etetoolkit.org/ |
| M.viridis | This paper | 10.5281/zenodo.7159034 |
| Nucdiff | Khelic et al. 201766 | https://github.com/uio-cels/NucDiff |
| GWviz | This paper | 10.5281/zenodo.7159034 |
| Roary | Page et al. 201567 | https://sanger-pathogens.github.io/Roary/ |
| operon-mapper | Taboada et al. 201868 | https://biocomputo.ibt.unam.mx/operon_mapper/ |
| Pyseer | Lees et al. 201870 | https://github.com/mgalardini/pyseer |
| NCBI datasets | Sayers et al. 202272 | https://www.ncbi.nlm.nih.gov/datasets/ |
| matplotlib | Hunter et al. 201775 | https://matplotlib.org/ |
| MLST | Seeman (Unpublished) | https://github.com/tseemann/mlst |
| cgMLST | Leopold et al. 201477 | https://www.cgmlst.org |
| Grapetree | Zhou et al. 201878 | https://github.com/achtman-lab/GrapeTree |
| PubMLST | Jolley et al. 201073 | https://pubmlst.org/ |
| cutadapt | Martin 201183 | https://github.com/marcelm/cutadapt/ |
| bowtie2 | Langmead et al. 201284 | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| htseq-count | Anders et al. 201585 | https://github.com/htseq/htseq |
| bioconductor | Huber et al. 201587 | https://bioconductor.org |
| limma | Ritchie et al. 201586 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Prism | GraphPad Inc | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal housing conditions.
For all models, animals received PicoLab® Rodent Diet 20 (LabDiet) and acidified water. Mice were housed 5 mice/cage under normal lighting cycle conditions (12hours ON/12 hours OFF) and temperature 70°F (±2°F).
Immunization and i.v. infections.
Female ND4 Swiss Webster mice (5-week-old, Envigo, Inc.).
Skin infections.
Female C57BL/6J mice (8-week-old, Jackson Laboratories).
Ethics Statement.
The study protocols were reviewed and approved by the Mount Sinai Hospital Institutional Review Board (IRB) for the collection and bacterial genome sequencing of discarded clinical bacteremia specimens by the Pathogen Surveillance Program (protocol HS# 13-00981) and chart reviews of BSI cases (protocol HS# 17-02246), as defined by DHHS regulations. A waiver of authorization for use and disclosure of protected health information (PHI) and a waiver of informed consent was approved for both protocols based on the criteria that the use or disclosure of PHI involved no more than minimal risk to the privacy of individuals, and because the research could not practically be conducted without the waiver and without access to and use of the PHI. The SSTI USA300 isolates were collected from a NYULH IRB approved protocol (S12-01223).
LeukoPaks were obtained from anonymous blood donors with informed consent from the New York Blood Center. Freshly isolated human whole blood was collected in accordance with a protocol approved by the NYU Grossman School of Medicine Institutional Review Board for Human Subjects (Torres Lab IRB number i14-02129). All donors provided written consent to participate in the study.
All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of NYU Grossman School of Medicine (Torres Lab IACUC number IA16-00050). All experiments were performed according to NIH guidelines, the Animal Welfare Act, and US federal law.
Bacterial strains and growth conditions.
S. aureus clinical isolates and laboratory strain AH-LAC were used for experiments. Bacteria were streaked on tryptic soy agar (TSA) and cultured in tryptic soy broth (TSB) or yeast-casamino acid with sodium pyruvate broth (YCP). Unless otherwise stated, cultures were grown at 37°C, shaking at 180 rpm, at a 45° angle, in 5mL media in 15mL conical tubes (Corning). Overnight cultures were started from single colonies. In the morning, cultures were diluted 1:100 and grown for a specified amount of time. Agar and broth were supplemented with antibiotics as needed to the following final concentrations: chloramphenicol to 10μg/mL, erythromycin to 5μg/mL and tetracycline to 4μg/mL, anhydrotetracycline 1μg/mL.
METHOD DETAILS
Construction of bacterial strains.
AH-LAC sarZ::bursa was generated by phage transduction using phage 80α lysate from the JE2 Nebraska Transposon Mutant Library NE567 strain. AH-LAC sarZ::bursa complement strains were made by cloning sarZ alleles into the pOS1 plasmid44 and electroporating into AH-LAC sarZ::bursa. sarZ, along with up- and down-stream intergenic regions, adding XmaI and NheI cut sites, were amplified from AH-LAC (WT allele), ER00385.B (NS1 allele), ER08597.3A (NS2 allele), ER00594.3B (FS1 allele) and ER09970.3A (FS2 allele) using primers sarZ-XmaI and sarZ-NheI.
Clinical isolate sarZ::tet strains were generated by cloning 800bp up- and down-stream of AH-LAC sarZ around the tetM tetracycline resistance gene and promoter from pJC1306 into the pIMAY plasmid (800bp up- and down-stream regions of sarZ have identical sequences in AH-LAC and clinical isolates)60. The following primers were used to amplify up- and down-stream regions of sarZ with 25bp homology to pIMAY or tetM for Gibson cloning: Up_sarZ_pIMAY_F, Up_sarZ_tet-R, Down_sarZ_tet_F, Down_sarZ_pIMAY_R. To amplify tetM: tetM_F, tetM_R. pIMAY was amplified using primers: pIMAY_F, pIMAY_R. The plasmid was electroporated into RN4220 and phage 80α lysate from the RN4220 strain was used for phage transduction of the plasmid into clinical isolates. In clinical isolates, the plasmid was integrated by growing in chloramphenicol at 37°C. Excision of plasmid by growth at 28°C with no antibiotic and plating on anhydrotetracycline. Mutants were selected for chloramphenicol sensitivity and tetracycline resistance. sarZWT complementation constructs were made as above, amplifying the WT locus from AH-LAC using primers Up_sarZ_pIMAY_F and Down_sarZ_pIMAY_R, phage transducing from RN4220 80α lysate into sarZ::tet strains and selecting for chloramphenicol sensitivity and tetracycline sensitivity. sarZWT and sarZFS1 complementation strains used for in vivo studies were made by electroporating the pOS1 plasmids with the corresponding alleles into ER00573.3B (H2) sarZ::tet. SF8300 sarZ::tet was generated by phage transduction using phage 80α lysate from ER00594.3B (I1) sarZ::tet.
To make ER02658.3B (I2) clfB::tet, a similar protocol to above was done using pIMAY*61. clfB up- and down-stream (800bp) were amplified with homology to tetM and pIMAY* digested with XhoI and XmaI: clfB_UP_pIMAY_F, clfB_UP_tet_R, clfB_DOWN_pIMAY_R and clfB_DOWN_tet_F. Construct was made using Gibson assembly and transformed into E. coli strain IM08B. Plasmid was electroporated into ER02658.3B. Integration and excision as above with selection on TSA tetracycline 20mM PCPA plates. clfB::clfB complementation constructs were made as above, amplifying the WT locus from AH-LAC using primers clfB_UP_pIMAY_F and clfB_DOWN_pIMAY_R, phage transducing from RN4220 80α lysate into the clfB::tet strain and selecting for chloramphenicol sensitivity and tetracycline sensitivity. ER00573.3B (H2) clfB::tet was generated by phage transduction using phage 80α lysate from a clfB::tet strain.
hPMN assays.
Primary human PMNs (hPMNs) were isolated from LeukoPaks of human blood samples as previously described62. For screen, S. aureus strains were grown in 150uL TSB or YCP in round bottom 96-well plates. Overnights were subcultured 1:100 and grown for 6h. Plates containing cultures were spun down to pellet bacteria and supernatants were pipetted off and frozen at −80°C. Supernatants were thawed at 4°C and added to 2×105 hPMNs diluted in RPMI 1640 supplemented with 10% FBS at 5% supernatant/well. hPMNs were intoxicated for 2h at 37°C and 5% CO2. hPMN viability was determined using CellTiter 96® Aqueous One Solution (Promega) after 1.5h incubation at 37°C and 5% CO2. Absorbance was measured at 492nm using a PerkinElmer Envision 2103 Multilable reader (PerkinElmer). For other cytotoxicity assays, bacterial overnights and subcultures (1:100 dilutions) were grown in 15mL tubes with 5mL specified media (either TSB or YCP). After a 5h subculture, bacteria were pelleted and supernatants were filtered (0.2-micron, PES membrane, Corning) and frozen at −80°C. hPMNs were intoxicated for 1h, using serial dilutions of filter supernatants ranging from 0.08%–20%.
For extracellular infections, bacteria were subcultured 5h in 5mL TSB, washed and diluted in RPMI 1640 with 0.1% HSA and 0.01M Hepes. Bacteria were added to 2×105 hPMNs at a MOI of 50. After a 2h infection at 37°C and 5% CO2, hPMN viability was determined by LDH release (CytoTox-ONE™ Homogeneous Membrane Integrity Assay, Promega), measured using the PerkinElmer Envision plate reader.
Genome sequencing, assembly, and annotation.
Clinical isolates selected for whole-genome sequencing were subcultured on TSA plates with 5% sheep blood under nonselective conditions. DNA was extracted from single colonies using the DNeasy Blood and Tissue Kit (Qiagen). Genomes were sequenced on the Pacific Biosciences (PacBio) RS-II platform and assembled with HGAP3 version 2.2.063. Additional Illumina sequencing of the same DNA samples was performed to address indel errors associated with homopolymer errors in PacBio Assemblies. PacBio assembly finishing and polishing with Illumina read data was then done as previously described, to produce finished-quality assemblies26. Finally, each genome was annotated using prokka using Staphylococcus aureus USA300_FPR3757 as a reference.
Phylogenetic analyses.
Maximum-likelihood phylogenetic trees were constructed using parsnp version 1.5.164 and visualized using etetoolkit65 and M.viridis (https://github.com/mjsull/m.viridis).
Genome-wide association (GWAS) analysis.
We selected 29 out of 40 inducible cytotoxicity isolates for comparative genomics, excluding 11 isolates that had growth defects and/or had borderline cytotoxicity when retested in a secondary screen. We additionally selected 29 out of the 56 high cytotoxicity isolates for comparison. After excluding isolates obtained from the same patients or suspected outbreaks, 25 high cytotoxicity and 29 inducible toxicity isolates were kept. Each genome was then compared to ER00503, the first high cytotoxicity (i.e. wild-type) isolate in our dataset, using nucdiff66, and SNP, small indels, and large insertions or deletions were annotated using GWviz (https://github.com/mjsull/GWviz).
Next, we performed a GWAS analysis between genetic changes at the gene or operon level, and the cytotoxicity phenotype. To this end, matched genes between isolates were identified using roary67 and used to create matrices with genes in rows and isolates in columns. Operons were annotated using operon-mapper68 and added as additional rows to these matrices. To identify gene or operon-level changes associated with the observed change in cytotoxicity phenotype we classified genes/operons in each isolate as functional or non-functional in a decreasing scale of severity based on (1) gene presence/absence alone, or by also considering the presence of (2) stop loss, stop gain and/or frameshift mutations, (3) nonsynonymous SNVs, and (4) changes in the promoter region defined as ≤100bp upstream of a coding sequence. An operon was considered non-functional if at least one of the genes in the operon met the loss-of-function definition. The code used to generate the four matrices is available at (https://github.com/mjsull/GWviz/utility).
Pyseer69,70 was then used to determine the loss-of-function events in each matrix that were significantly associated with the change in phenotype. Finally, we considered which combination of significantly associated loss events (pyseer p < 0.05) most likely explained the inducible phenotype. This was done using a simple scoring scheme that added 1 point for a loss event observed in an inducible isolate and subtracted 1 point for a loss event observed in a non-inducible isolate across all 4 matrices, and ranking genes and operons by their aggregate score.
Easyfig71 was used to compare and visualize loci of interest.
Analysis of S. aureus genomes in Genbank.
A total of 45,270 S. aureus genomes from human biosamples and their associated metadata were downloaded in April 2022 using NCBI datasets version 13.7.072. Genomes were deduplicated such that only the first genome per biosample was retained for further analysis. In addition, biosamples and genomes originating from the Mount Sinai Health System were removed, leaving 26,792 genomes after filtering. The sample collection date, collection location, and host disease were obtained from the biosample metadata where available. The multilocus sequence type (MLST) of each genome was determined using mlst (https://github.com/tseemann/mlst) and PubMLST73, and used to assign the USA group of each isolate as defined in Davie et al.74.
To obtain a comprehensive list of mutations seen in the inducible cytotoxicity isolates, the sarZ, agr, and PVL locus sequences from the 29 inducible cytotoxicity isolates were aligned against the locus sequences from the ER00573_3B reference isolate using BLASTn. Subsequently, each of the 26,792 Genbank genomes were compared at the same loci and all isolates with at least one change identical to those observed in the isolates with inducible cytotoxicity were considered as mutants. This process was repeated for 1,296 S. aureus genomes collected by the Mount Sinai Pathogen Surveillance Program (MS-PSP) between 2014 and 2021. Graphs of mutation frequencies over time were generated using matplotlib75. For all the deduplicated Genbank isolates, cgMLST schemes for each were determined using chewBBACA (version 2.8.5)76 and the cgMLST scheme available at cgmlst.org77. cgMLST trees were visualized using GrapeTree78.
SarZ purification.
We followed the methods from Ballal, A, et al. to clone and purify six-His tag-SarZ36. Full length sarZWT was amplified from AH-LAC, adding NdeI and BamHI cut sites and was cloned into pET15b, using primers sarZ_NdeI and sarZ_STOP_BamHI. The plasmid was transformed in E. coli strain BL21-DE3g. The expression strain was grown in 400mL LB broth with 100μg/mL ampicillin, at 37°C 250rpm until the OD600 reached 0.6. Culture was induced with 1mM IPTG and grown for 4h at 30°C 250rpm. Culture was spun down (10,000rpm, 4°C, 15 min) and resuspended in 20mM Tris pH 7.5, 300mM NaCl, 10% glycerol. Cells were lysed in 1x protease inhibitor by sonication and addition of 1x bugbuster, and centrifuged 10,000rpm, 30min, 4°C. Supernatant was filtered through a 0.2μm filter and 5mM Imidazole and 200mM NaCl were added, final pH 7.4. The protein was purified using a HisTrap HP column on an AKTA, eluting with a linear gradient, elution buffer 20mM Na2HPO4, 500mM NaCl, 400mM Imidazole pH 7.4. Purified protein was dialyzed in 10% glycerol in TBS and purity was assessed by Coomassie and concentration determined by nanodrop.
To purify SarZNS1 and SarZNS2, the above protocol was repeated, using DNA from ER00385.B (NS1 allele), ER08597.3A (NS2 allele) to amplify sarZ and clone into pET15b. Growth of expression strains was repeated as for SarZWT, but on a smaller scale, growing strains in 250mL LB broth with 100μg/mL amplicillin, at 37°C 250rpm until the OD600 reached 0.6. Culture was induced with 1mM IPTG and grown for overnight at 16°C 250rpm. Purification for mutant alleles was repeated as for SarZWT.
Production of anti-SarZ sera.
To make anti-SarZ sera, five female ND4 Swiss Webster mice (5-week-old, Envigo, Inc.) were immunized subcutaneously with 100μg purified His-SarZ in 1:1 10% glycerol, 1X TBS and TiterMax® Gold. Mice were boosted after 2 weeks and 4 weeks. At 7 weeks, serum was collected by cardiac puncture and Western blotting was performed to confirm specificity for SarZ.
Western blotting.
For SarZ Western blots, bacteria were subcultured in 5mL TSB for 5h and normalized to OD600=1.4. Cultures were washed in PBS and lysed to make whole cell lysates with 100 μg/ml lysostaphin, 40 u/ml DNase, 40 μg/ml RNase A, 1 × HALT™ Protease Inhibitor in lysis buffer (10 mM MgCl2, 1 mM CaCl2 in 50 mM Tris, pH 7.5). Whole cell lysates were boiled for 10 minutes in a SDS buffer. Samples were run in an SDS 12% polyacrylamide gel and transferred onto a nitrocellulose membrane. Membrane was blocked with 5% milk in PBS + 0.1% Tween (PBS-T) with 1:5000 human IgG. Anti-SarZ sera was added 1:10,000 and incubated overnight at 4°C. Membrane was washed with PBS-T and the secondary antibody goat anti-mouse Alexa 608 was added at 1:25,000 in PBS-T with 5% milk. Membrane was washed again in PBS-T and imaged on the Odyssey Infrared Imaging System (LI-COR Biosciences).
ClfB Western blots were generated by the same protocol with the following alterations: bacteria were subcultured for 3h, anti-ClfB sera79 was added 1:3,000, and the secondary used was goat anti-rabbit Alexa 608. Quantification of bands was done using Image Studio.
Promoter Pull Down.
Methods based on previously published work80. Biotin labeled and unlabeled promoters for genes of interest were amplified and purified: ssp (pSsp_F and pSsp_R or pSsp_biotin_R), clfB (pClfB_F and pClfB_R or pClfB_biotin_R). As was the control promoter for purA (pPurA_F and pPurA_R or pPurA_biotin_R). Primer sequences can be found in Table S4. 25μL M-280 Streptavidin Dynabeads (Invitrogen) were washed in wash buffer (2M NaCl, 1mM EDTA, 10mM Tris, pH 7.5) and incubated with 800fmol biotin labeled DNA for 30min at room temperature on a rotisserie. Beads were washed 3X and resuspended in SarZ binding buffer (25mM Tris-Cl, pH 7.5, 0.1mM EDTA, 75mM dithiothreitol, and 10% glycerol). For competition experiments 15X unlabeled DNA was added. Purified SarZ protein (50nM or 25nM as indicated) was mixed with Poly(dG:dC) (Invitrogen) and then added to beads and incubated 30min 30°C shaking at 550rpm. Beads were washed 2X with SarZ binding buffer and resuspended in 40uL SDS buffer. Beads were boiled for 10min and supernatant was collected for Western blotting. SarZ Western blots to detect protein pulled down on promoter DNA were generated as described in ‘Western blotting’, loading 15μL of sample. Blots were not blocked with human IgG, as this assay only used purified protein. Band intensity was quantified using Image Studio. For assays examining the ability of mutant SarZ to bind DNA, 1ng/μL SarZ was boiled in SDS and 15μL was loaded onto the same gel as pull-down samples as a control for detection using polyclonal anti-SarZ sera.
Murine intravenous infection.
Bacteria were subcultured in 5mL TSB for 3h, washed 2x in PBS and normalized to indicated CFU/mL. 5-week-old female ND4 Swiss Webster mice (Envigo, Inc.) were anesthetized intraperitoneally with 300μL avertin (2,2,2-tribromoethanol), dissolved in tert-Amyl-alcohol and diluted to final concentration 2.5% in saline. 100uL of bacteria was injected intravenously by retro-orbital injection. For survival experiments, mice were monitored daily for up to 2-weeks and euthanized upon severe signs of mortality or excessive weight loss. Specifically, we use the following criteria: 30% weight loss or animals experiencing a constellation of acute disease signs (i.e., ruffled fur, dyspnea, self-imposed isolation, rapid breathing, twitching, trembling, abnormal posture/positioning, inability to reach for food/water), as approved by our IACUC. To determine CFU-burden, mice were euthanized 1-day post-infection and organs were collected into 1mL PBS. Organs were bead beaten using a FastPrep, diluted and plated on TSA to enumerate CFU.
Murine skin infection.
Bacteria were subcultured in 5mL TSB for 3h, washed 2x in PBS and normalized to indicated CFU/mL. 8-week-old female C57BL/6J mice (Jackson Laboratories) were anesthetized intraperitoneally with 300μL avertin (2,2,2-tribromoethanol), dissolved in tert-Amyl-alcohol and diluted to final concentration 2.5% in saline. Flanks of mice were shaved, and one flank was injected subcutaneously with 50uL bacteria. Lesions were measured on day 2 and 3 post-infection. The width and length of lesions were measured and multiplied to get an approximate area of lesions. On day 3 post-infection, mice were euthanized, and skin punch biopsies (8mm in diameter) were homogenized in 1mL PBS. Samples were diluted and plated on TSA to enumerate CFU.
Growth curves.
S. aureus strains were grown overnight in 150uL TSB in round bottom 96-well plates. Cultures were spun down and supernatant was removed. Pellets were diluted 1:100 in PBS, followed by a 1:100 dilution in 150uL TSB into a honeycomb plate (final dilution 1:10,000). OD600 was measured every 30min using a BioScreenC: temperature 37°C, experiment length 24h, shaking continuous, amplitude maximum, speed normal, filter 600nm brown.
Survival in whole blood.
Protocol based on previously published work81,82. Bacteria were subcultured for 3h in 5mL TSB, spun down 400rpm 5min and OD normalized. Freshly isolated heparinized venous blood from 6 human donors was infected at an MOI of 1 (2×107 CFU/mL bacteria in 100uL blood) X5 tubes/donor. Samples were incubated in 2mL tubes at a 90° angle on a tube rotator (Thermo Fisher) at speed 10, 37°C. At 0h, 1h, 2h, 4h and 6h, a tube from each donor was combined 1:1 with SK buffer (2% saponin in PBS plus 200 U/mL streptokinase, 1mg/mL trypsin, 0.02 mg/mL DNase 1 and 0.1mg/mL RNase on ice). Note that the SK buffer was made fresh at each time point. Samples were incubated shaking at 37°C for 10min followed by rocking for 10min at 4°C. Samples were diluted and plated on TSA to enumerate CFU.
RNA extraction for RNA-seq and qRT-PCR.
Bacteria were subcultured for 3h and/or 5h in 5mL TSB, spun down, and resuspended in 1mL RNA STAT-60™ (Amsbio). Samples were bead beaten using FastPrep and spun down. Upper phase was collected and 200μL chloroform was added. 0.5mL isopropanol was added to aqueous phase to precipitate RNA. RNA was washed with 70% ethanol, air dried, and resuspended in RNase free water. 2500ng of RNA was DNase treated (TURBO DNA-free™ kit, Invitrogen Ambion).
For serum qRT-PCR, bacteria were subcultured for 3h in 5mL TSB, spun down 4000rpm 5min and resuspended in 650uL PBS. 100uL of bacteria were added to 1mL of 50% normal human serum (SeraCare) in RPMI and incubated 1h 37°C rolling on a rotator. Cultures were spun down, resuspended in 1mL RNA STAT-60™ and above protocol was followed.
RNA-seq profiling.
For each sample, approximately 250ng of RNA was first depleted of bacterial rRNAs using enzymatic ribodepletion with the Illumina Ribo-Zero Plus rRNA Depletion Kit (Illumina, Catalog no. 20037135) according to the manufacturer’s protocol. RNA-seq libraries were then prepared using the Illumina Stranded Total RNA Prep kit, with the depleted rRNA material as input and following the manufacturer’s protocol. Barcoded libraries were quantified by Qubit, and visualized on the Agilent Bioanalyzer, prior to equimolar pooling and sequencing on the NovaSeq SP platform in a 2×100nt paired-end format.
Differential gene expression analysis.
Raw reads were first trimmed by removing Illumina adapter sequences from 3’ ends using cutadapt83, and by removing 3′ read sequences if more than 20 bases with Q≥20 were present. Paired-end reads were then mapped to the FPR3757 (NC_007793) reference genome using Bowtie284, and htseq-count85 was used to produce strand-specific transcript count summaries. Raw fragment (i.e., paired-end read) counts were then combined into a numeric matrix, with genes in rows and samples in columns, and used as input for differential gene expression analysis with the limma R package86 in Bioconductor87. Normalization factors were computed on the data matrix using the weighted trimmed mean of M-values (TMM) method, followed by voom88 mean-variance transformation in preparation for Limma linear modeling. Only genes with expression levels ≥1 FPKM (fragments per kb per million reads) in at least 50% of samples, and a length ≥200 bp, were retained for further analysis. Filtered data were fitted to a design matrix containing all sample groups, and pairwise comparisons were performed between the groups of interest. Finally, eBayes adjusted P-values were corrected for multiple testing using the Holm method and used to select genes with significant expression differences (q ≤ 0.05).
qRT-PCR.
DNase treated samples were converted to cDNA using SuperScript™ III First-Strand Synthesis System (Thermo Scientific). 1uL of cDNA was used with TaqMan™ probes and Universal PCR Master Mix (Thermo Scientific) to detect rpoB, clfB, sspA and aur using QuantStudio™ 3 System. Genes normalized to rpoB housing keep gene and reported as 2−ΔCT. The probes used where: PrimerTime 5’ Hex/3’ BHQ-1 and Primer/probe sequences: rpoB_F, rpoB_R, rpoB_probe, clfB_F, clfB_R, cflB_probe, sspA_F, sspA_R, sspA_probe, aur_F, aur_R, aur_probe.
QUANTIFICATION AND STATISTICAL ANALYSIS
Prism software (GraphPad, Inc.) was used to perform statistical analysis. One-way or two-way ANOVA was used for multiple comparisons as indicated for cytotoxicity, hPMN infection, CFU-burden experiments, and relevant in vitro experiments. Unpaired t tests with Welch’s correction were used for qRT-PCR data. For survival curves, significance was determined using the log rank (Mantel-Cox) test. Results were corrected for multiple comparisons using the Bonferroni-corrected threshold. Precision measures, n values and statistical tests used are indicated in all figure legends. P =< 0.05 was used to determine significance. P values are reported in figures.
Supplementary Material
Supplementary Table 2. Isolate Information, related to Figure 1. See excel file.
Highlights.
Bloodstream infection USA300 isolates have evolved altered virulence regulation
Altered virulence is associated with mutations in the transcription factor SarZ
Mutating sarZ leads to hypervirulence in a murine bloodstream infection model
ClfB contributes to the hypervirulence of sarZ mutant USA300 isolates
ACKNOWLEDGEMENTS
We thank members of the Torres, Shopsin and van Bakel laboratories for insightful discussions and comments on this manuscript. We thank the NYU Langone Genome Technology Center (GTC) for expert support and BEI Resources for providing the transposon mutant NR-47110. pIMAY* was a gift from Dr. Angelika Grundling (Addgene plasmid # 121441; http://n2t.net/addgene:121441; RRID:Addgene_121441) and the anti-ClfB serum was a gift from Dr. Joan Geoghegan. Graphical abstract was created with BioRender.com
This work was supported in part by the NIH-National Institute of Allergy and Infectious Diseases award numbers R01s AI099394, AI105129 to V.J.T., AI137336, AI140754 to B.S. and V.J.T., and AI119145 to H.V.B. The GTC is partially supported by the Isaac Perlmutter Cancer Center Support Grant P30CA016087 from the NIH-National Cancer Institute. The work reported in this paper was also supported by the Office of Research Infrastructure of the National Institutes of Health (NIH) under award numbers S10OD018522 and S10OD026880. V.J.T. is a Burroughs Wellcome Fund Investigator in the pathogenesis of infectious diseases.
Footnotes
DECLARATION OF INTERESTS
V.J.T. is an inventor on patents and patent applications filed by New York University, which are currently under commercial license to Janssen Biotech Inc. Janssen Biotech Inc. provides research funding and other payments associated with a licensing agreement.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Tong SY, Davis JS, Eichenberger E, Holland TL, and Fowler VG Jr. (2015). Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 28, 603–661. 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wertheim HFL, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, and Nouwen JL (2005). The role of nasal carriage in Staphylococcus aureus infections. The Lancet Infectious Diseases 5, 751–762. 10.1016/s1473-3099(05)70295-4. [DOI] [PubMed] [Google Scholar]
- 3.Uhlemann AC, Otto M, Lowy FD, and DeLeo FR (2014). Evolution of community- and healthcare-associated methicillin-resistant Staphylococcus aureus. Infect Genet Evol 21, 563–574. 10.1016/j.meegid.2013.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Souli M, Ruffin F, Choi SH, Park LP, Gao S, Lent NC, Sharma-Kuinkel BK, Thaden JT, Maskarinec SA, Wanda L, et al. (2019). Changing Characteristics of Staphylococcus aureus Bacteremia: Results From a 21-Year, Prospective, Longitudinal Study. Clin Infect Dis 69, 1868–1877. 10.1093/cid/ciz112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thurlow LR, Joshi GS, and Richardson AR (2012). Virulence strategies of the dominant USA300 lineage of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA). FEMS Immunol Med Microbiol 65, 5–22. 10.1111/j.1574-695X.2012.00937.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tenover FC, and Goering RV (2009). Methicillin-resistant Staphylococcus aureus strain USA300: origin and epidemiology. J Antimicrob Chemother 64, 441–446. 10.1093/jac/dkp241. [DOI] [PubMed] [Google Scholar]
- 7.Laabei M, Uhlemann AC, Lowy FD, Austin ED, Yokoyama M, Ouadi K, Feil E, Thorpe HA, Williams B, Perkins M, et al. (2015). Evolutionary Trade-Offs Underlie the Multi-faceted Virulence of Staphylococcus aureus. PLoS Biol 13, e1002229. 10.1371/journal.pbio.1002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rose HR, Holzman RS, Altman DR, Smyth DS, Wasserman GA, Kafer JM, Wible M, Mendes RE, Torres VJ, and Shopsin B (2015). Cytotoxic Virulence Predicts Mortality in Nosocomial Pneumonia Due to Methicillin-Resistant Staphylococcus aureus. J Infect Dis 211, 1862–1874. 10.1093/infdis/jiu554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voyich JM, Braughton KR, Sturdevant DE, Whitney AR, Said-Salim B, Porcella SF, Long RD, Dorward DW, Gardner DJ, Kreiswirth BN, et al. (2005). Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J Immunol 175, 3907–3919. 10.4049/jimmunol.175.6.3907. [DOI] [PubMed] [Google Scholar]
- 10.Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, Kennedy AD, Dorward DW, Klebanoff SJ, Peschel A, et al. (2007). Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med 13, 1510–1514. 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
- 11.He L, Meng H, Liu Q, Hu M, Wang Y, Chen X, Liu X, and Li M (2018). Distinct virulent network between healthcare- and community-associated Staphylococcus aureus based on proteomic analysis. Clin Proteomics 15, 2. 10.1186/s12014-017-9178-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rudkin JK, Edwards AM, Bowden MG, Brown EL, Pozzi C, Waters EM, Chan WC, Williams P, O’Gara JP, and Massey RC (2012). Methicillin resistance reduces the virulence of healthcare-associated methicillin-resistant Staphylococcus aureus by interfering with the agr quorum sensing system. J Infect Dis 205, 798–806. 10.1093/infdis/jir845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeLeo FR, Kennedy AD, Chen L, Bubeck Wardenburg J, Kobayashi SD, Mathema B, Braughton KR, Whitney AR, Villaruz AE, Martens CA, et al. (2011). Molecular differentiation of historic phage-type 80/81 and contemporary epidemic Staphylococcus aureus. Proc Natl Acad Sci U S A 108, 18091–18096. 10.1073/pnas.1111084108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pruneau M, Mitchell G, Moisan H, Dumont-Blanchette E, Jacob CL, and Malouin F (2011). Transcriptional analysis of antibiotic resistance and virulence genes in multiresistant hospital-acquired MRSA. FEMS Immunol Med Microbiol 63, 54–64. 10.1111/j.1574-695X.2011.00830.x. [DOI] [PubMed] [Google Scholar]
- 15.Popovich KJ, Weinstein RA, and Hota B (2008). Are community-associated methicillin-resistant Staphylococcus aureus (MRSA) strains replacing traditional nosocomial MRSA strains? Clin Infect Dis 46, 787–794. 10.1086/528716. [DOI] [PubMed] [Google Scholar]
- 16.Seybold U, Kourbatova EV, Johnson JG, Halvosa SJ, Wang YF, King MD, Ray SM, and Blumberg HM (2006). Emergence of community-associated methicillin-resistant Staphylococcus aureus USA300 genotype as a major cause of health care-associated blood stream infections. Clin Infect Dis 42, 647–656. 10.1086/499815. [DOI] [PubMed] [Google Scholar]
- 17.Tenover FC, Tickler IA, Goering RV, Kreiswirth BN, Mediavilla JR, Persing DH, and Consortium M (2012). Characterization of nasal and blood culture isolates of methicillin-resistant Staphylococcus aureus from patients in United States Hospitals. Antimicrob Agents Chemother 56, 1324–1330. 10.1128/AAC.05804-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tickler IA, Goering RV, Mediavilla JR, Kreiswirth BN, Tenover FC, and Consortium HAI (2017). Continued expansion of USA300-like methicillin-resistant Staphylococcus aureus (MRSA) among hospitalized patients in the United States. Diagn Microbiol Infect Dis 88, 342–347. 10.1016/j.diagmicrobio.2017.04.016. [DOI] [PubMed] [Google Scholar]
- 19.Boles BR, Thoendel M, Roth AJ, and Horswill AR (2010). Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One 5, e10146. 10.1371/journal.pone.0010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF, et al. (2006). Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. The Lancet 367, 731–739. 10.1016/s0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 21.Diep BA, Palazzolo-Ballance AM, Tattevin P, Basuino L, Braughton KR, Whitney AR, Chen L, Kreiswirth BN, Otto M, DeLeo FR, and Chambers HF (2008). Contribution of Panton-Valentine leukocidin in community-associated methicillin-resistant Staphylococcus aureus pathogenesis. PLoS One 3, e3198. 10.1371/journal.pone.0003198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mekonnen SA, Palma Medina LM, Glasner C, Tsompanidou E, de Jong A, Grasso S, Schaffer M, Mader U, Larsen AR, Gumpert H, et al. (2017). Signatures of cytoplasmic proteins in the exoproteome distinguish community- and hospital-associated methicillin-resistant Staphylococcus aureus USA300 lineages. Virulence 8, 891–907. 10.1080/21505594.2017.1325064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harper L, Balasubramanian D, Ohneck EA, Sause WE, Chapman J, Mejia-Sosa B, Lhakhang T, Heguy A, Tsirigos A, Ueberheide B, et al. (2018). Staphylococcus aureus Responds to the Central Metabolite Pyruvate To Regulate Virulence. mBio 9. 10.1128/mBio.02272-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Novick RP (2003). Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol Microbiol 48, 1429–1449. 10.1046/j.1365-2958.2003.03526.x. [DOI] [PubMed] [Google Scholar]
- 25.Novick RP, and Geisinger E (2008). Quorum sensing in staphylococci. Annu Rev Genet 42, 541–564. 10.1146/annurev.genet.42.110807.091640. [DOI] [PubMed] [Google Scholar]
- 26.Altman DR, Sullivan MJ, Chacko KI, Balasubramanian D, Pak TR, Sause WE, Kumar K, Sebra R, Deikus G, Attie O, Rose H, Lewis M, Fulmer Y, Bashir A, Kasarskis A, Schadt EE, Richardson AR, Torres VJ, Shopsin B, & Bakel H. van. (2018). Genome Plasticity of agr-Defective Staphylococcus aureus during Clinical Infection. Infection and Immunity 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vance G, Fowler J, George Sakoulas, McIntyre Lauren M., Meka Venkata G., Arbeit Robert D., Cabell Christopher H., Stryjewski Martin E., Eliopoulos George M., Reller L. Barth, Corey G. Ralph, Tiffanny Jones, Natalie Lucindo, Yeaman Michael R., Bayer Arnold S. (2004). Persistent Bacteremia Due to Methicillin-Resistant Staphylococcus aureus Infection Is Associated with agr Dysfunction and Low-Level In Vitro Resistance to Thrombin-Induced Platelet Microbicidal Protein. The Journal of Infectious Disease 190, 1140–1149. [DOI] [PubMed] [Google Scholar]
- 28.Spaan AN, van Strijp JAG, and Torres VJ (2017). Leukocidins: staphylococcal bi-component pore-forming toxins find their receptors. Nat Rev Microbiol 15, 435–447. 10.1038/nrmicro.2017.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Binh An Diep HAC, Chang Richard F., Sensabaugh George F., and Perdreau-Remington Franc ¸oise (2006). Roles of 34 Virulence Genes in the Evolution of Hospital- and Community-Associated Strains of Methicillin-Resistant Staphylococcus aureus. The Journal of Infectious Disease 193, 1495–1503. [DOI] [PubMed] [Google Scholar]
- 30.Ellington MJ, Hope R, Ganner M, Ganner M, East C, Brick G, and Kearns AM (2007). Is Panton-Valentine leucocidin associated with the pathogenesis of Staphylococcus aureus bacteraemia in the UK? J Antimicrob Chemother 60, 402–405. 10.1093/jac/dkm206. [DOI] [PubMed] [Google Scholar]
- 31.François Vandenesch TN, Enright Mark C., Lina Gerard, Nimmo Graeme R., Heffernan Helen, Liassine Nadia, Bes Michèle, Greenland Timothy, Reverdy Marie-Elisabeth, and Etienne Jerome (2003). Community-Acquired Methicillin-Resistant Staphylococcus aureus Carrying Panton-Valentine Leukocidin Genes: Worldwide Emergence. Emerging Infectious Diseases journal 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guimaraes MA, Ramundo MS, Americo MA, de Mattos MC, Souza RR, Ramos-Junior ES, Coelho LR, Morrot A, Melo PA, Fracalanzza SE, et al. (2015). A comparison of virulence patterns and in vivo fitness between hospital- and community-acquired methicillin-resistant Staphylococcus aureus related to the USA400 clone. Eur J Clin Microbiol Infect Dis 34, 497–509. 10.1007/s10096-014-2253-1. [DOI] [PubMed] [Google Scholar]
- 33.Huang H, Flynn NM, King JH, Monchaud C, Morita M, and Cohen SH (2006). Comparisons of community-associated methicillin-resistant Staphylococcus aureus (MRSA) and hospital-associated MSRA infections in Sacramento, California. J Clin Microbiol 44, 2423–2427. 10.1128/JCM.00254-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jun Kaneko TK, Narita Sachiko, Tomita Toshio, Kamio Yoshiyuki (1998). Complete nucleotide sequence and molecular characterization of the temperate staphylococcal bacteriophage wPVL carrying Panton–Valentine leukocidin genes. Gene 215, 57–67. [DOI] [PubMed] [Google Scholar]
- 35.Chen PR, Nishida S, Poor CB, Cheng A, Bae T, Kuechenmeister L, Dunman PM, Missiakas D, and He C (2009). A new oxidative sensing and regulation pathway mediated by the MgrA homologue SarZ in Staphylococcus aureus. Mol Microbiol 71, 198–211. 10.1111/j.1365-2958.2008.06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ballal A, Ray B, and Manna AC (2009). sarZ, a sarA family gene, is transcriptionally activated by MgrA and is involved in the regulation of genes encoding exoproteins in Staphylococcus aureus. J Bacteriol 191, 1656–1665. 10.1128/JB.01555-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaito C, Morishita D, Matsumoto Y, Kurokawa K, and Sekimizu K (2006). Novel DNA binding protein SarZ contributes to virulence in Staphylococcus aureus. Mol Microbiol 62, 1601–1617. 10.1111/j.1365-2958.2006.05480.x. [DOI] [PubMed] [Google Scholar]
- 38.Tamber S, and Cheung AL (2009). SarZ promotes the expression of virulence factors and represses biofilm formation by modulating SarA and agr in Staphylococcus aureus. Infect Immun 77, 419–428. 10.1128/IAI.00859-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villet RA, Truong-Bolduc QC, Wang Y, Estabrooks Z, Medeiros H, and Hooper DC (2014). Regulation of expression of abcA and its response to environmental conditions. J Bacteriol 196, 1532–1539. 10.1128/JB.01406-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poor CB, Chen PR, Duguid E, Rice PA, and He C (2009). Crystal structures of the reduced, sulfenic acid, and mixed disulfide forms of SarZ, a redox active global regulator in Staphylococcus aureus. J Biol Chem 284, 23517–23524. 10.1074/jbc.M109.015826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dumont AL, Nygaard TK, Watkins RL, Smith A, Kozhaya L, Kreiswirth BN, Shopsin B, Unutmaz D, Voyich JM, and Torres VJ (2011). Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol Microbiol 79, 814–825. 10.1111/j.1365-2958.2010.07490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ventura CL, Malachowa N, Hammer CH, Nardone GA, Robinson MA, Kobayashi SD, and DeLeo FR (2010). Identification of a novel Staphylococcus aureus two-component leukotoxin using cell surface proteomics. PLoS One 5, e11634. 10.1371/journal.pone.0011634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Novick K.T.a.R. (2006). A slipped-mispairing mutation in AgrA of laboratory strains and clinical isolates results in delayed activation of agr and failure to translate δ- and α-haemolysins. Molecular Microbiology 59, 1519–1530. 10.1111/j.1365-2958.2005.04986.x. [DOI] [PubMed] [Google Scholar]
- 44.Olaf Schneewind PM, and Fischette Vincent A. (1992). Sorting of Protein A to the Staphylococcal Cell Wall. Cell 70, 267–281. [DOI] [PubMed] [Google Scholar]
- 45.Pandey S, Sahukhal GS, and Elasri MO (2019). The msaABCR Operon Regulates the Response to Oxidative Stress in Staphylococcus aureus. J Bacteriol 201. 10.1128/JB.00417-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Copin R, Shopsin B, and Torres VJ (2018). After the deluge: mining Staphylococcus aureus genomic data for clinical associations and host-pathogen interactions. Curr Opin Microbiol 41, 43–50. 10.1016/j.mib.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mulcahy ME, Geoghegan JA, Monk IR, O’Keeffe KM, Walsh EJ, Foster TJ, and McLoughlin RM (2012). Nasal colonisation by Staphylococcus aureus depends upon clumping factor B binding to the squamous epithelial cell envelope protein loricrin. PLoS Pathog 8, e1003092. 10.1371/journal.ppat.1003092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wertheim HF, Walsh E, Choudhurry R, Melles DC, Boelens HA, Miajlovic H, Verbrugh HA, Foster T, and van Belkum A (2008). Key role for clumping factor B in Staphylococcus aureus nasal colonization of humans. PLoS Med 5, e17. 10.1371/journal.pmed.0050017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Entenza JM, T.J.F., NI Eidhin D, Vaudaux P, Francioli P, and P. Moreillion (2000). Contribution of Clumping Factor B to Pathogenesis of Experimental Endocarditis due to Staphylococcus aureus. Infection and Immunity 69, 5443–5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lacey KA, Mulcahy ME, Towell AM, Geoghegan JA, and McLoughlin RM (2019). Clumping factor B is an important virulence factor during Staphylococcus aureus skin infection and a promising vaccine target. PLoS Pathog 15, e1007713. 10.1371/journal.ppat.1007713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Déirdre NíEidhin SP, PatriceFrancois, Pierre Vaudaux, Magnus Hoök and Foster Timothy J. (1998). Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin of Staphylococcus aureus. Molecular Microbiology 30, 245–257. [DOI] [PubMed] [Google Scholar]
- 52.McAleese FM, Walsh EJ, Sieprawska M, Potempa J, and Foster TJ (2001). Loss of clumping factor B fibrinogen binding activity by Staphylococcus aureus involves cessation of transcription, shedding and cleavage by metalloprotease. J Biol Chem 276, 29969–29978. 10.1074/jbc.M102389200. [DOI] [PubMed] [Google Scholar]
- 53.Kolar SL, Ibarra JA, Rivera FE, Mootz JM, Davenport JE, Stevens SM, Horswill AR, and Shaw LN (2013). Extracellular proteases are key mediators of Staphylococcus aureus virulence via the global modulation of virulence-determinant stability. Microbiologyopen 2, 18–34. 10.1002/mbo3.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zielinska AK, Beenken KE, Mrak LN, Spencer HJ, Post GR, Skinner RA, Tackett AJ, Horswill AR, and Smeltzer MS (2012). sarA-mediated repression of protease production plays a key role in the pathogenesis of Staphylococcus aureus USA300 isolates. Mol Microbiol 86, 1183–1196. 10.1111/mmi.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Painter KL, Krishna A, Wigneshweraraj S, and Edwards AM (2014). What role does the quorum-sensing accessory gene regulator system play during Staphylococcus aureus bacteremia? Trends Microbiol 22, 676–685. 10.1016/j.tim.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 56.Tuchscherr L, Pollath C, Siegmund A, Deinhardt-Emmer S, Hoerr V, Svensson CM, Thilo Figge M, Monecke S, and Loffler B (2019). Clinical S. aureus Isolates Vary in Their Virulence to Promote Adaptation to the Host. Toxins (Basel) 11. 10.3390/toxins11030135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Williams RE, Jevons MP, Shooter RA, Hunter CJ, Girling JA, Griffiths JD, and Taylor GW (1959). Nasal staphylococci and sepsis in hospital patients. Br Med J 2, 658–662. 10.1136/bmj.2.5153.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abraham NM, and Jefferson KK (2012). Staphylococcus aureus clumping factor B mediates biofilm formation in the absence of calcium. Microbiology (Reading) 158, 1504–1512. 10.1099/mic.0.057018-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blake KJ, Baral P, Voisin T, Lubkin A, Pinho-Ribeiro FA, Adams KL, Roberson DP, Ma YC, Otto M, Woolf CJ, et al. (2018). Staphylococcus aureus produces pain through pore-forming toxins and neuronal TRPV1 that is silenced by QX-314. Nat Commun 9, 37. 10.1038/s41467-017-02448-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Monk IR, Shah IM, Xu M, Tan MW, and Foster TJ (2012). Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio 3. 10.1128/mBio.00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schuster CF, Howard SA, and Grundling A (2019). Use of the counter selectable marker PheS* for genome engineering in Staphylococcus aureus. Microbiology (Reading) 165, 572–584. 10.1099/mic.0.000791. [DOI] [PubMed] [Google Scholar]
- 62.DuMont AL, Yoong P, Surewaard BG, Benson MA, Nijland R, van Strijp JA, and Torres VJ (2013). Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infect Immun 81, 1830–1841. 10.1128/IAI.00095-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10, 563–569. 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 64.Treangen TJ, Ondov BD, Koren S, and Phillippy AM (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol 15, 524. 10.1186/s13059-014-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huerta-Cepas J, Serra F, and Bork P (2016). ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol Biol Evol 33, 1635–1638. 10.1093/molbev/msw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khelik K, Lagesen K, Sandve GK, Rognes T, and Nederbragt AJ (2017). NucDiff: in-depth characterization and annotation of differences between two sets of DNA sequences. BMC Bioinformatics 18, 338. 10.1186/s12859-017-1748-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, Fookes M, Falush D, Keane JA, and Parkhill J (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Taboada B, Estrada K, Ciria R, and Merino E (2018). Operon-mapper: a web server for precise operon identification in bacterial and archaeal genomes. Bioinformatics 34, 4118–4120. 10.1093/bioinformatics/bty496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lees JA, Galardini M, Bentley SD, Weiser JN, and Corander J (2018). pyseer: a comprehensive tool for microbial pangenome-wide association studies. Bioinformatics 34, 4310–4312. 10.1093/bioinformatics/bty539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sullivan MJ, Petty NK, and Beatson SA (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sayers EW, Bolton EE, Brister JR, Canese K, Chan J, Comeau DC, Connor R, Funk K, Kelly C, Kim S, et al. (2022). Database resources of the national center for biotechnology information. Nucleic Acids Res 50, D20–D26. 10.1093/nar/gkab1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jolley KA, and Maiden MC (2010). BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11, 595. 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.David MZ, Taylor A, Lynfield R, Boxrud DJ, Short G, Zychowski D, Boyle-Vavra S, and Daum RS (2013). Comparing pulsed-field gel electrophoresis with multilocus sequence typing, spa typing, staphylococcal cassette chromosome mec (SCCmec) typing, and PCR for panton-valentine leukocidin, arcA, and opp3 in methicillin-resistant Staphylococcus aureus isolates at a U.S. Medical Center. J Clin Microbiol 51, 814–819. 10.1128/JCM.02429-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hunter JD (2007). Matplotlib: A 2D graphics environment. Comput Sci Eng 9, 90–95. Doi 10.1109/Mcse.2007.55. [DOI] [Google Scholar]
- 76.Silva M, Machado MP, Silva DN, Rossi M, Moran-Gilad J, Santos S, Ramirez M, and Carrico JA (2018). chewBBACA: A complete suite for gene-by-gene schema creation and strain identification. Microb Genomics 4. ARTN 000166 10.1099/mgen.0.000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Leopold SR, Goering RV, Witten A, Harmsen D, and Mellmann A (2014). Bacterial Whole-Genome Sequencing Revisited: Portable, Scalable, and Standardized Analysis for Typing and Detection of Virulence and Antibiotic Resistance Genes. Journal of Clinical Microbiology 52, 2365–2370. 10.1128/Jcm.00262-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou ZM, Alikhan NF, Sergeant MJ, Luhmann N, Vaz C, Francisco AP, Carrico JA, and Achtman M (2018). GrapeTree: visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res 28, 1395–1404. 10.1101/gr.232397.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fleury OM, McAleer MA, Feuillie C, Formosa-Dague C, Sansevere E, Bennett DE, Towell AM, McLean WHI, Kezic S, Robinson DA, et al. (2017). Clumping Factor B Promotes Adherence of Staphylococcus aureus to Corneocytes in Atopic Dermatitis. Infect Immun 85. 10.1128/IAI.00994-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sause WE, Balasubramanian D, Irnov I, Copin R, Sullivan MJ, Sommerfield A, Chan R, Dhabaria A, Askenazi M, Ueberheide B, et al. (2019). The purine biosynthesis regulator PurR moonlights as a virulence regulator in Staphylococcus aureus. Proc Natl Acad Sci U S A 116, 13563–13572. 10.1073/pnas.1904280116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Beavers WN, DuMont AL, Monteith AJ, Maloney KN, Tallman KA, Weiss A, Christian AH, Toste FD, Chang CJ, Porter NA, et al. (2021). Staphylococcus aureus Peptide Methionine Sulfoxide Reductases Protect from Human Whole-Blood Killing. Infect Immun 89, e0014621. 10.1128/IAI.00146-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thomer L, Emolo C, Thammavongsa V, Kim HK, McAdow ME, Yu W, Kieffer M, Schneewind O, and Missiakas D (2016). Antibodies against a secreted product of Staphylococcus aureus trigger phagocytic killing. J Exp Med 213, 293–301. 10.1084/jem.20150074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17, 3. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 84.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 12, 115–121. 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15, R29. 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 2. Isolate Information, related to Figure 1. See excel file.
Data Availability Statement
All genome data and assemblies are available in GenBank (see Table S2). Raw and processed mRNA sequence files are available at the NCBI Gene Expression Omnibus under accession no GSE185544. This information can also be found in the Key Resource Table
Custom-built pipelines for MRSA genome assembly and annotation are available in powerpak/pathogendb-pipeline github repository. Annotation of genetic changes between inducible and highly cytotoxic isolates and preparation of data matrices for GWAS was done with custom tools available in the mjsull/GWviz github repository. This information is included in the Key Resource Table. Additional methods and other relevant data can be found in Supplemental Information.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody Alexa Fluor 680 | Invitrogen | A-21057 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody Alexa Fluor 680 | Invitrogen | A-11011 |
| Anti-SarZ mouse polyclonal sera | This paper | N/A |
| Anti-ClfB rabbit polyclonal sera | Fleury et al. 201779 | N/A |
| Bacterial and virus strains | ||
| AH-LAC | Boles et al. 201019 | N/A |
| FPR3757 | Isolated from a patient (Diep et al. 2006)20 | N/A |
| SF8300 | Isolated from a patient (Diep et al. 2008)21 | N/A |
| AH-LAC Δluk | Blake et al. 201859 | N/A |
| AH-LAC sarZ::bursa | This paper | N/A |
| AH-LAC + vector | This paper | N/A |
| AH-LAC sarZ::bursa + vector | This paper | N/A |
| AH-LAC sarZ::bursa + sarZWT | This paper | N/A |
| AH-LAC sarZ::bursa + sarZNS1 | This paper | N/A |
| AH-LAC sarZ::bursa + sarZNS2 | This paper | N/A |
| AH-LAC sarZ::bursa + sarZFS1 | This paper | N/A |
| AH-LAC sarZ::bursa + sarZFS2 | This paper | N/A |
| I1 sarZ::tet | This paper | N/A |
| I1 sarZ::sarZWT | This paper | N/A |
| H2 sarZ::tet | This paper | N/A |
| H2 sarZ::sarZWT | This paper | N/A |
| H2 sarZ::tet + vector | This paper | N/A |
| H2 sarZ::tet + sarZWT | This paper | N/A |
| H2 sarZ::tet + sarZFS1 | This paper | N/A |
| SF8300 sarZ::tet | This paper | N/A |
| I2 clfB::tet | This paper | N/A |
| I2 clfB::clfB | This paper | N/A |
| H2 clfB::tet | This paper | N/A |
| BL21 pET15b-sarZ | This paper | N/A |
| BL21 pET15b-sarZNS1 | This paper | N/A |
| BL21 pET15b-sarZNS2 | This paper | N/A |
| Clinical isolates listed in Table S2 | This paper | N/A |
| Biological samples | ||
| LeukoPacks | New York Blood Center | N/A |
| SeraCare Normal Human Serum | Dba LGC Clinical Diagnostics | Ref# 27000-100 |
| Whole human blood | Torres Lab IRB | IRB # S12-01223 |
| Chemicals, peptides, and recombinant proteins | ||
| Lysostaphin | Smbi Products LLC | Cat# LSPN-50 |
| DNase (for bacterial whole cell lysates) | Promega | Cat# M6101 |
| RNase A | Promega | Cat# A7973 |
| HALT™ Protease Inhibitor | Thermo Scientific | Cat# 78438 |
| Sodium pyruvate | ACROS Organics | Cat# 113-24-6 |
| Chloramphenicol | Fisher BioReagents | Cat# BP904-100 |
| Erythromycin | Fisher BioReagents | Cat# BP920-25 |
| Tetracycline | Alfa Aesar | Cat# B21408 |
| Ampicillin | Fisher Scientific | Cat# BP1760 |
| Anhydrotetracycline | Thermo Scientific | Cat# AC233131000 |
| PCPA | Sigma-Aldrich | Cat# C6506 |
| IPTG | Research Products International | Cat# I5600025.0 |
| TiterMax® Gold | Titermax | Cat# G-20 |
| Avertin (2,2,2-tribromoethanol) | Sigma-Aldrich | Cat# T48402 |
| Streptokinase | Sigma-Aldrich | Cat# S313-250KU4 |
| Trypsin | Sigma-Aldrich | Cat# T4549 |
| DNase I (used in SK buffer) | Sigma-Aldrich | Cat# DN25-1G |
| RNase (used in SK buffer) | Alfa Aesar | Cat# J61996 |
| Dynabeads M-280 Streptavidin | Invitrogen | Cat# 11205D |
| Poly(dG:dC) naked | Invivogen | Cat# TLRLPGCN |
| His-SarZWT | This paper | N/A |
| His-SarZNS1 | This paper | N/A |
| His-SarZNS2 | This paper | N/A |
| Critical commercial assays | ||
| CellTiter 96® Aqueous One Solution | Promega | Cat# PR-G3581 |
| CytoTox-ONE™ Homogeneous Membrane Integrity Assay | Promega | Cat# PR-G7892 |
| RNA-STAT-60™ | Amsbio | Cat# CS-111 |
| TURBO DNA-free™ kit | Invitrogen | Cat# AM1907 |
| Ribo-Zero Plus rRNA Depletion Kit | Illumina | Cat# 20037135 |
| Stranded Total RNA Prep kit | Illumina | Cat# AM1907 |
| Gibson Assembly Master Mix | New England Biolabs | Cat# E2611L |
| DNeasy Blood and Tissue Kit | Qiagen | Cat# 69504 |
| Bugbuster | MilliporeSigma | Cat# 709215 |
| HisTrap HP column | Cytiva | Cat# 45-003-013 |
| SuperScript™ III First-Strand Synthesis System | Invitrogen | Cat# 1808004 |
| Deposited data | ||
| Genome assemblies | GenBank | See Table S2 |
| mRNA sequence files | NCBI Gene Expression Omnibus | Accession No. GSE185544 |
| Experimental models: Cell lines | ||
| NA | ||
| Experimental models: Organisms/strains | ||
| Swiss Webster Hsd (5 week old) | Envigo Inc | Hsd:ND4 |
| C57BL/6J (8 week old) | Jackson Laboratories | 000664 |
| Oligonucleotides | ||
| For primers used in cloning, see Table S4 | This paper | N/A |
| For primers used in qRT-PCR, see Table S4 | This paper | N/A |
| For primers used in promoter pulldowns, see Table S4 | This paper | N/A |
| Recombinant DNA | ||
| pOS1 | Schneedwind et al. 199244 | N/A |
| pIMAY | Monk et al. 201260 | Addgene plasmid #68939 |
| pIMAY* | Schuster et al. 201961 | Addgene plasmid #121441 |
| pET15b | Novagen | Cat# 69661-3 |
| Software and algorithms | ||
| HGAP3 | Chin et al. 201363 | https://www.pacb.com/products-and-services/analytical-software/smrt-analysis/ |
| Parsnp | Treangen et al. 201464 | http://github.com/marbl/harvest |
| etetoolkit | Huerta-Cepas et al. 201665 | http://etetoolkit.org/ |
| M.viridis | This paper | 10.5281/zenodo.7159034 |
| Nucdiff | Khelic et al. 201766 | https://github.com/uio-cels/NucDiff |
| GWviz | This paper | 10.5281/zenodo.7159034 |
| Roary | Page et al. 201567 | https://sanger-pathogens.github.io/Roary/ |
| operon-mapper | Taboada et al. 201868 | https://biocomputo.ibt.unam.mx/operon_mapper/ |
| Pyseer | Lees et al. 201870 | https://github.com/mgalardini/pyseer |
| NCBI datasets | Sayers et al. 202272 | https://www.ncbi.nlm.nih.gov/datasets/ |
| matplotlib | Hunter et al. 201775 | https://matplotlib.org/ |
| MLST | Seeman (Unpublished) | https://github.com/tseemann/mlst |
| cgMLST | Leopold et al. 201477 | https://www.cgmlst.org |
| Grapetree | Zhou et al. 201878 | https://github.com/achtman-lab/GrapeTree |
| PubMLST | Jolley et al. 201073 | https://pubmlst.org/ |
| cutadapt | Martin 201183 | https://github.com/marcelm/cutadapt/ |
| bowtie2 | Langmead et al. 201284 | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| htseq-count | Anders et al. 201585 | https://github.com/htseq/htseq |
| bioconductor | Huber et al. 201587 | https://bioconductor.org |
| limma | Ritchie et al. 201586 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Prism | GraphPad Inc | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||