

Abstract
Neuroinflammation has been implicated in multiple brain disorders but the extent and the magnitude of change in immune-related genes (IRGs) across distinct brain disorders has not been directly compared. In this study, 1,275 IRGs were curated and their expression changes investigated in 2,467 postmortem brains of controls and patients with six major brain disorders, including schizophrenia (SCZ), bipolar disorder (BD), autism spectrum disorder (ASD), major depressive disorder (MDD), Alzheimer’s disease (AD), and Parkinson’s disease (PD). There were 865 IRGs present across all microarray and RNA-seq datasets. More than 60% of the IRGs had significantly altered expression in at least one of the six disorders. The differentially expressed immune-related genes (dIRGs) shared across disorders were mainly related to innate immunity. Moreover, sex, tissue, and putative cell type were systematically evaluated for immune alterations in different neuropsychiatric disorders. Co-expression networks revealed that transcripts of the neuroimmune systems interacted with neuronal-systems, both of which contribute to the pathology of brain disorders. However, only a few genes with expression changes were also identified as containing risk variants in genome-wide association studies. The transcriptome alterations at gene and network levels may clarify the immune-related pathophysiology and help to better define neuropsychiatric and neurological disorders.
Keywords: neuroimmune, neurological diseases, psychiatric disorders, cross-disorder analysis, transcriptome, gene expression
Introduction
Multiple lines of evidence support the notion that the immune system is involved in major “brain disorders,” including psychiatric disorders such as schizophrenia (SCZ) 1, bipolar disorder (BD) 2, and major depressive disorder (MDD)3, and brain development disorders such as autism spectrum disorder (ASD)4, and neurodegenerative diseases such as Alzheimer’s disease (AD)5, and Parkinson’s disease (PD)6. Patients with these brain diseases share deficits in cognition, blunted mood, restricted sociability, and abnormal behavior to various degrees. Transcriptome studies have identified expression alterations of immune-related genes (IRGs) in postmortem brains of AD7, PD8, ASD9, SCZ10–14 and BD10 separately. Cross-disorder transcriptomic studies further highlighted changes in IRGs15, 16. At the protein level, several peripheral cytokines showed reproducible disease-specific changes in a meta-analysis 17. Since brain dysfunction is considered the major cause of these disorders, studying immune gene expression changes in patient brains may reveal mechanistic connections between immune system genes and brain dysfunction. Most previous studies were limited to the analysis of individual disorders. There has not been a comprehensive comparison of the pattern and extent of inflammation-related changes in terms of immune constructs (subnetworks), neuro-immune interaction, genetic contribution, and relationship among diseases.
Neuroinflammation, an immune response taking place within the central nervous system (CNS), can be activated by psychological stress, aging, infection, trauma, ischemia, and toxins 18, 19. It is influenced by sex 20, age21 and genetics 22, which are known disease risk factors for both psychiatric and neurological diseases. The primary function of neuroinflammation is to maintain brain homeostasis through protection 23 and repair 24. Abnormal neuroinflammation activation could lead to dysregulation of mood 25, social behaviors 26, and cognitive abilities 27. Offspring who were fetuses when their mothers’ immune system was activated (MIA) showed dopaminergic hyperfunction 28, cognitive impairment 29, and behavioral abnormalities 30 as adults. Alternatively, acute and chronic neuroinflammation in adulthood can also alter cognition and behavior31–33.
Researchers have explored the underlying mechanism to explain how the neuroimmune system is activated. In animal models, both adult and developmental maternal immune activation in the periphery can increase pro-inflammatory cytokines in the brain, like what was found in humans with major mental illness14, 34. The interplay between CNS and peripheral immunity can be mediated in several ways. One way is inflammatory factors can increase permeabilization of the blood-brain barrier to enable the CNS to communicate with the peripheral immune system through two drainage systems, the glymphatic system35 and the meningeal lymphatic system36. A second way is environmental stimuli, such as stress, trauma, ischemia, and toxins, can promote the brain to release small molecules (e.g. cortisol) which trigger immune activation in both CNS and peripheral systems37. These small molecules also can increase the blood-brain barrier permeability, which enhances the shared immunity activities between peripheral blood and CNS38.
Previous studies identified immune gene dysregulations in brains of patients with several major brain disorders. For example, Gandal et al.16 found that up-regulated genes and splicing isoforms in SCZ, BD, and ASD were enriched in pathways such as inflammatory response and response to cytokines. One brain co-expression module up-regulated specifically in MDD was enriched for genes of cytokine-cytokine interactions, and hormone activity pathways15. The association of neurological diseases such as AD and PD with IRGs has also been reported7, 39. These studies examined the changes of immune system without going into details of specific subnetworks, the disease signature, or genetic versus environmental contribution.
The current boundary between neurological diseases and psychiatric disorders is primarily the presence of known pathology. The hypothesis of this study is that expression changes of specific subsets of IRGs constitute part of the transcriptome signatures that distinguishes diseases. Since tissue specificity, sex and genetics all could influence such transcriptome signatures, their effects were analyzed. Furthermore, the expectation is that neurological diseases and psychiatric disorders bear transcriptomic changes that may help to address how similar immunological mechanisms lead to distinct brain disorders.
To investigate immune-related signatures of transcriptome dysregulation in brains from patients with one of six neurological and psychiatric disorders, 1,275 genes known to be associated with neuroinflammation were selected and their expression across disorders was studied. Existing transcriptome data of 2,467 postmortem brain samples from donors with AD40–42, ASD43–46, BD43, 47–50, MDD49, 51, PD52–55, SCZ43, 47–50, 56, 57 and healthy controls (CTL) were collected and analyzed. Differentially expressed IRGs shared across disorders or specific to each disorder and their related co-expression modules were identified (Fig. S1). These genes and their networks and pathways provided important insight into how immunity may contribute to the risk of these neurological and psychiatric disorders, with a potential to refine disease classification.
Materials and Methods
Data description
Twenty-three studies of multiple brain disorders were obtained from the Gene Expression Omnibus (GEO), ArrayExpress, or directly from study authors (see Supplementary Table 1). Raw microarray gene expression data was obtained from 1,007 postmortem cortical brain samples(microarray) and 1,460 postmortem brain samples (RNA-seq). Each study was processed separately and analyzed according to the general workflow as described in Quality Control and Normalization.
Immune genes selection
Immune genes were collected from immune databases: Comparative Toxicogenomics Database58, ImmPort59, ImmunomeDB60, InnateDB61, ImmuneSigDB62, Gene Ontology database63, KEGG database64, and literature reviews65–67. The selected immune genes were included if they were found in more than two databases. Neuroimmunology and general immunology literature were consulted for the selection of immune genes. In total, 1,789 immune-related genes (IRGs) were included for analyses and 1,275 of them were detected across all microarray expression data.
Supplementary Table 2 lists the immune genes categorized by their reference databases and pathway annotation programs. Because there is a high degree of overlap among biological immune responses, genes with roles in multiple immune functions were assigned to more than one group.
Quality Control and Normalization
Known covariates included available biological and technical covariates for each study from GEO. Unknown covariates were estimated by R package sva for each study68. The lm function in R was used to adjust all the covariates (see Figure. S1). Case and control samples were from the same brain region in each cohort, but different cohorts utilized tissues from different brain regions. To minimize the influence of brain regions from different cohorts, a stringent correction procedure was applied to account for any possible influence of brain region on the results. By regressing out the variations attributable to brain regions, our analysis focused on the expression changes that occurred consistently across brain regions. The information on the various brain regions tested was shown in sTable 1. Outliers were defined as samples with standardized sample network connectivity Z scores < −2 and were removed. Both within and between studies batch effects were corrected with the ComBat function of the sva package in R. All available biological, technical, and unknown covariates except for the diagnostic group were regressed using lm function in R.
For all RNA-seq data, the R package sva was used to estimate unknown factors68. Picard Tools v1.13169 was used for quality check after read alignment. A matrix of “sequencing statistics” was generated and the first and second main components of this matrix were estimated. In subsequent studies, these sequencing statistics were employed as covariates. To address all the covariates, R’s lm function was used. Outliers were identified as previously described for microarray analyses.
Differential Gene Expression Analysis
Differential gene expression analysis of microarray data was calculated using a linear mixed-effects model in the nlme package in R, with diagnostic group and study treated as fixed effects and the unique subject as a random effect Spearman’s ρ was used to compare dIRGs meta-analysis fold change signatures across all disease pairs. There were 440 controls shared by the studies of SCZ and BD. The total number of controls (1,178) provided in the manuscript is after removing duplicates. To minimize the influence of overlapping controls, a statistical framework which collapsed results from multiple studies while accounting for any subjects overlapping between studies with a random effect term was used.
Differential gene expression of RNA-seq replication data was calculated using limma with empiric Bayes moderated t-statistics, including the all the known and unknown covariates based on log2(normalized FPKM) expression values.
Gene-level RNAseq Replication
The degree to which genes identified as differentially expressed in the discovery (microarray) datasets (FDR < 0.05) were replicated in the RNAseq data was evaluated as shown in Supplementary Table 4. For comparison, the background was restricted to the 865 genes present across all microarray and RNAseq datasets. Fisher’s exact test was used to calculate odds ratios and the statistical significance of overlap between microarray DGE genes and the RNAseq replication set (all genes with P<0.05 and concordant direction). Regression coefficients for each gene were calculated for each group and Spearman’s correlation was used to assess disturbance concordance between microarray data and RNA-seq data, as above.
Disorder Network Gene Co-Expression Network Mega-Analysis
Weighted Gene Co-Expression Network Analysis (WGCNA)70 was completed to place results from individual genes within their systems-level network architecture. Individual (covariate-regressed) expression datasets were combined using the 10,387 genes present across all studies. ComBat was used to mitigate batch effects, as shown in sFig. 1.
Disease-specific networks were generated by comparing networks constructed using patients from one of six brain disorders against networks constructed from healthy individuals. Then, a WGCNA integrated function was used to calculate module preservation statistics, and the Z summary score (Z score) was applied to evaluate whether a module was conserved or not. A disease-specific module was defined as z-score less than 10 in the all-expression matrix of controls and patients with other disorders.
Gene Set Enrichment
Gene set enrichment analysis was performed using clusterProfiler R packages71. Module functional enrichment of Gene Ontology pathways was assessed with GO-Elite v1.2.572 as well as with the gProfiler73 R package. Cell-type specific expression analysis of genes within each module was performed using the pSI package74 specificity index in R. Cell-type specific gene expression data was obtained from an RNAseq study of purified populations of neurons, astrocytes, oligodendrocytes, microglia, and endothelial cells derived from the adult human cerebral cortex75.
GWAS Enrichment
A set of GWAS summary statistics was compiled for several brain disorders, cognitive, and behavioral traits (Table S1). Summary statistics from GWAS meta-analyses of ASD, schizophrenia, bipolar disorder, and major depression were downloaded from the PGC website. GWAS of educational attainment, depressive symptoms, and neuroticism were obtained from the respective studies (Table S1). Gene-level analysis of GWAS results was performed by MAGMA v1.04, a gene-set annotation framework that accounts for linkage disequilibrium (LD) between SNPs76.
Code availability
The code of this work can be found at https://github.com/normacyyyyy/IRG-cross-disorder
Results
Expressions of immune-related genes were altered in brain disorders.
IRGs (1,789) were collected from curated databases including Comparative Toxicogenomics Database58, ImmPort59, ImmunomeDB60, InnateDB61, ImmuneSigDB77, Gene Ontology database with immune annotation63, KEGG database with immune annotation64, as well as additional literature reviews 65–67, followed by filtering based on expression profile in the human brains ( see Table S1, Fig. S1). Twenty-three transcriptomic datasets of multiple brain disorders from multiple brain regions from the Gene Expression Omnibus (GEO), ArrayExpress, or from the authors directly were compiled (see Table. S1). In total, transcriptome data of 2,467 postmortem brain samples from subjects with AD (n = 340 individuals), ASD (n = 103), BD (n = 188), MDD (n =87), PD (n = 97), SCZ (n = 474) and matched-CTL (n = 1,178) were collected. Individual datasets were subjected to rigorous quality control and normalization. Matched controls were employed to reduce confounding effects from diagnosis and covariates including age, sex, and batch (Fig. S3). Data sets derived from microarrays (n=1,007) were used as the discovery set and data sets derived from RNA-Seq were used as an independent replication set (Table. S1). Out of the 1,789 selected IRGs, 1,275 (71%) were detected across all the microarray data. The IRG curation procedure is summarized in a Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) workflow (Fig. S2). Table S2 lists the 1,275 detected IRGs categorized by their reference databases and pathway annotation programs.
After preprocessing the microarray data (methods and materials, Fig. S3), a whole transcriptome differential expression analysis was completed for each disorder using a linear mixed-effects model that accounted for sample overlap across studies. Filtering the differentially expressed genes (DEGs, FDR<0.05) for the 1,275 IRGs, dIRGs were identified in each of the six brain disorders (number of dIRGs in descending order of transcript identified as differentially expressed; AD: 638, ASD: 275, SCZ: 220, PD: 97, BD: 58, MDD: 27, Fig. 1A, Table S3). A conservative genome-wide threshold was used instead of study-wide threshold for significance to enable further evaluation of the relative enrichment of IRGs in all the DEGs under the same significance criteria. The dIRGs were significantly over-represented in the DEGs across disorders (adjust.p < 0.05, Table S3). The enrichment of IRGs in the DEGs supports the reported immune gene dysfunction in the transcriptomes of these six disorders, suggesting that inflammation-related changes may be a universal response to a variety of brain pathologies. Previous studies showed that drugs can also affect changes in gene expression78. To evaluate drug effect on gene expression of IRGs, Pearson correlation was used to assess the concordance of fold changes between case-control DEGs in each disorder from our study and DEGs induced by ten different drugs in the mouse and rat models downloaded from the Kaleidoscope78, 79. The case-control differences of the IRGs have only weak correlation (r < 0.3) with some of the changes induced by drugs, suggesting limited effect of drugs in our findings (Table S3).
Fig. 1. Differential expression of immune genes in six disorders.
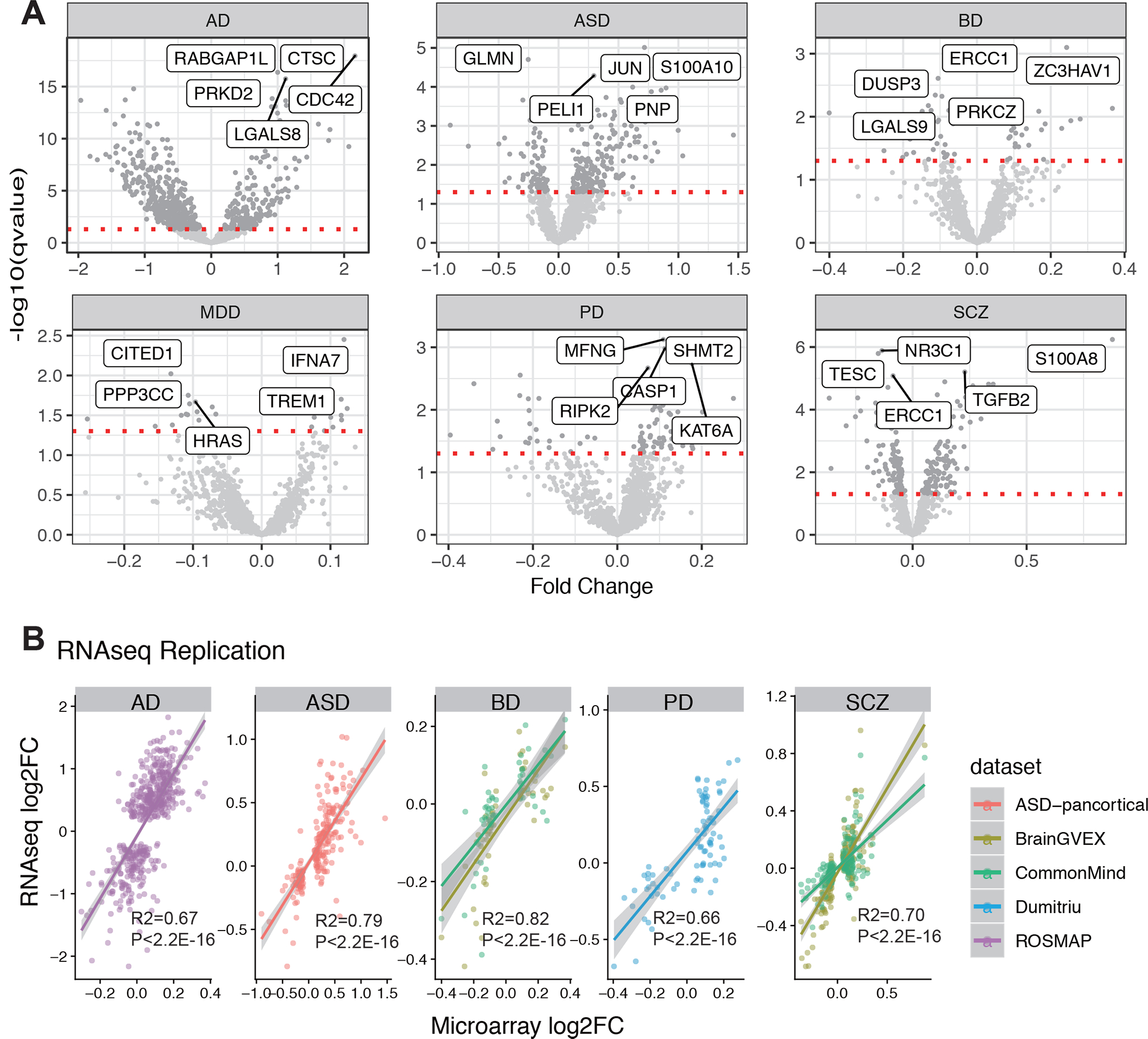
A. Volcano plot for each disorder. B. Effect size correlation between microarray data and RNA-seq data.
To replicate the findings, the independent RNA-seq datasets and processed data were used as detailed in the Methods and Materials. A significant overlap of dIRGs between discovery and replicate datasets was observed (Table S4). AD data achieved the highest replication rate (56%) while BD achieved the lowest (19%). Correlation effect sizes of case-control fold change between the microarray and RNA-seq results were compared without p-value cutoff to understand why some correlations between microarray and RNAseq data were low. High concordance of effect sizes of case-control fold change between the microarray and RNA-seq results for all IRGs was observed (R2 > 0.66, p.value < 2.2E-16, Fig. 1B, Table S4).
The changes of IRGs clustered by disorders and sex- and tissue-specific effects.
Hierarchical clustering was used based on the correlations of the fold changes of all the detected IRGs among different brain disorders, resulting in two distinct groups, one containing all psychiatric disorders (BD, SCZ, and MDD) and another containing neurological disorders AD and PD, plus ASD, which is considered a neurological disease (Fig. 2A, B, Fig. S4). The fold changes of IRGs were highly correlated between SCZ and BD (Spearman’s r = 0.75, p.value <0.001). When comparing the groups of psychiatric disorders and neurological disorders, a higher effect size (larger fold- change) in the expression of inflammatory related genes was detected in neurological disorders than in psychiatric disorders (t-test p.value < 2.2E-16, Fig. S4). RNA-seq data replicated the observation that larger immune-related dysregulation was present in neurological disorders than in psychiatric disorders (Fig. S4).
Fig. 2. Comparison of the effect size of differentially expressed IRGs among neuropsychiatric disease pairs.
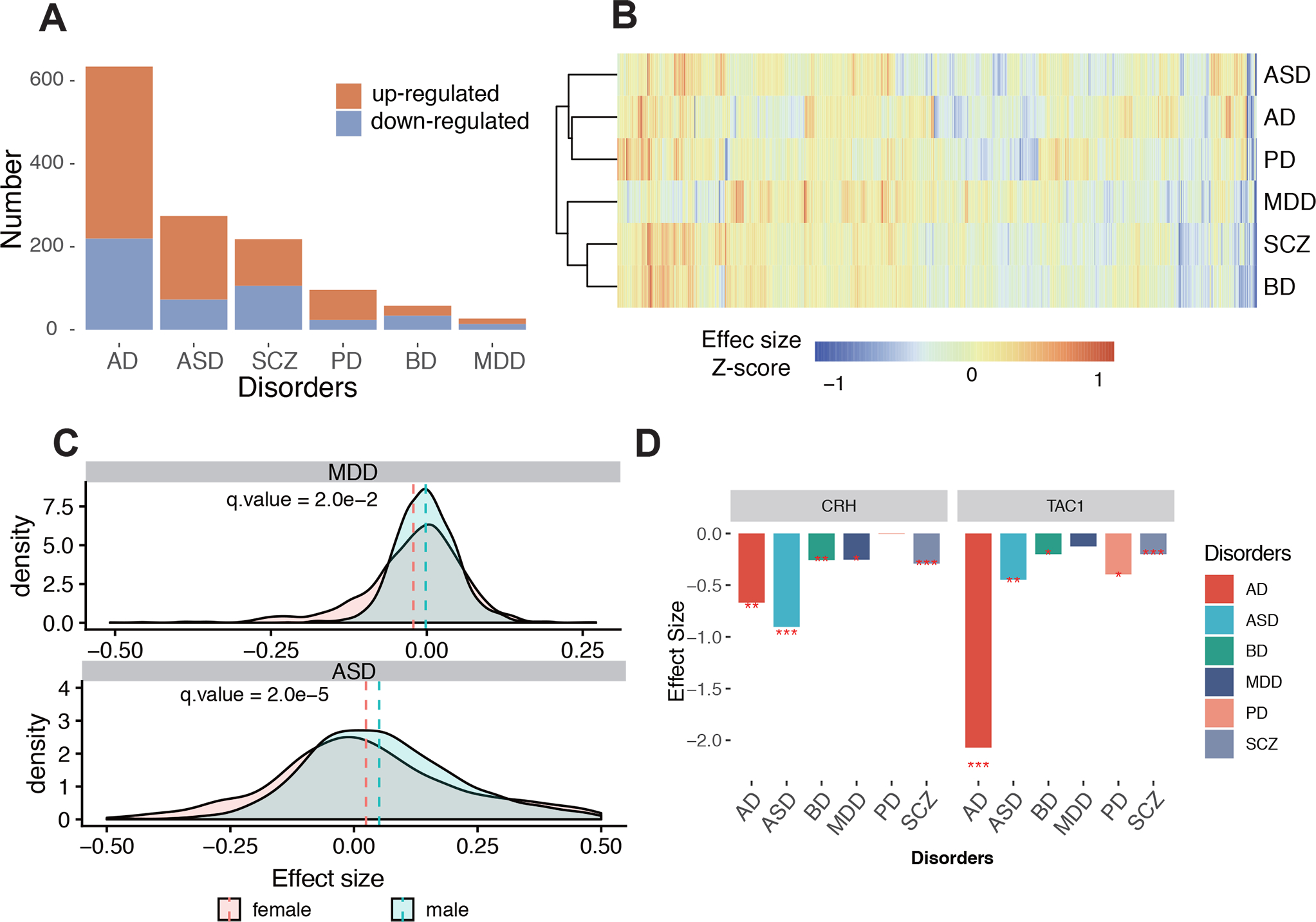
A. Numbers of up-regulated and downregulated dIRGs in the six disorders. Red represents up-regulated dIRGs, blue represents down-regulated dIRGs. B. Cluster tree of scaled effect size for all disorders based on 1,275 IRGs for their fold changes. C. Significant sex differences by effect size in ASD and MDD. The dash line indicated mean value of effect size. D. dIRGs shared across disorders: CRH and TAC1. *: fdr qvalue<0.05; **fdr qvalue<0.01; ***fdr qvalue<0.001
To test effects of sex on immune-related dysregulation, dIRGs were recomputed with the samples partitioned by sex. Comparing the effect sizes of the IRGs between male and female subgroups, significant sex differences were detected in ASD and MDD (Fig. 2C, Table S5; pASD = 0.003; pMDD = 4E-6), but not in other diseases. The IRGs showed larger magnitude of change in male ASD than in female ASD relative to corresponding controls, while the situation was the opposite for MDD with females having larger changes than males.
To investigate tissue specificity of IRG dysregulation, or more specifically, whether alterations of IRGs in the brain can be reflected in blood, the changes of IRGs in blood datasets of these six disorders were calculated (Table S1). The correlation of IRGs’ effect size showed negligible concordance (R2 from −0.24 to 0.11, P-value>0.05, Fig. S5), indicating that the majority of the changes of IRGs in the blood and brain do not overlap, implying distinct origins and/or cellular mechanisms. However, a few dIRGs showing consistent changes in brain and blood were identified (Table S6), such as S100A8 in SCZ. These genes may serve as candidates of disease peripheral biomarkers, which warrants a thorough investigation.
Innate immune genes are the most shared changes across all brain disorders.
Comparing the overlap of dIRGs across disorders, 26% of IRGs were dIRGs in two or more disorders (shared dIRGs, Fig. 3 A, B). Alterations of both adaptive and innate IRGs were found in each disease (Fig. S6, Table S3), but 68% of the shared dIRGs were classified as genes involving innate immune functions. To avoid bias caused by the number of IRGs in the two categories, the enrichment of dIRG was calculated and it was determined that they were significantly enriched in only innate IRGs (Fisher Exact Test OR > 2, qvalue <0.05). In the RNA-seq replicate data, there was a better replication in innate IRGs than in adaptive IRGs (Fig. S6, Table S4).
Fig. 3. Comparing dIRG-associated function across disorders.
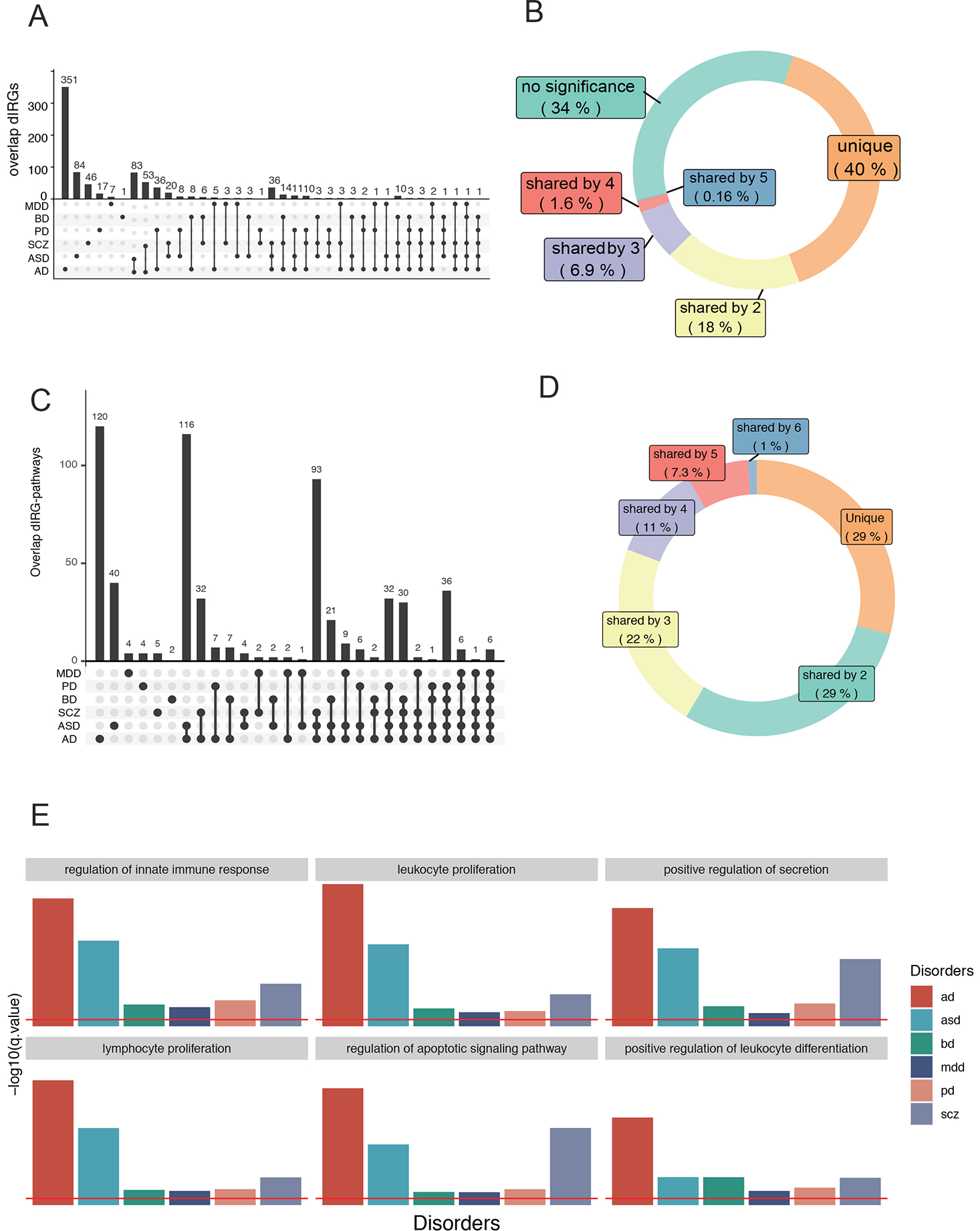
A. UpSet plot of dIRGs overlap between pairs of disorders. Dark cells and lines indicate that the set participates in the intersection. B. The doughnut chart shows the percentages of different IRGs types. C. UpSet plot of differential immune pathways overlap. The black dots and the black line show the overlapping dIRG-pathways between pairs of disorders. Cells that are dark indicate that it participates in the intersection. D. The doughnut chart shows the percentages of overlapping dIRG-pathways. E. Gene ontology enrichment analysis results of six pathways shared by dIRGs of all six disorders.
The two most shared dIRGs are Corticotropin-releasing hormone (CRH) and Tachykinin Precursor 1 (TAC1), which were differentially expressed in five of the six diseases (Fig. 2D). They are both involved in innate immunity according to the databases used and to the literature 58–61, 63, 64. CRH was downregulated in five of the six disorders; the exception was PD. CRH can regulate innate immune activation with neurotensin (NT), stimulating mast cells, endothelia, and microglia (57). TAC1 was down-regulated in five of the six disorders, the exception being MDD. TAC1 encodes four products of substance P, which can alter the immune functions of activated microglia and astrocytes 80. Independent RNA-seq data confirmed both findings of downregulated CRH and TAC1 in patient brains. These transcripts are also neuromodulators and have action on neurons, so they have roles in addition to immune functions.
To identify specific immune pathways that dIRGs are involved in, the enrichment of the dIRGs was tested in specific immune functions. Six dIRGs-enriched pathways were shared by all six disorders. “Regulation of innate immune response (GO:0045088)” was one of the six pathways (qvalue< 0.05, Table. S7, Fig. 3E). When a subset of the innate immune genes as defined by the GO database was used as input instead of all IRGs, hierarchical clustering resulted in the same clusters of psychiatric vs. neurological diseases (Fig. S7). This indicated that even though immune dysfunction was widespread in the six disorders, signature patterns in the subset innate immune genes were sufficient to differentiate neurological from psychiatric disorders.
Immune-related co-expression modules (IRMs) shared by diseases were related to brain development and aging.
To determine if neuroimmunity works in silos or co-operates with other functions, robust weighted gene co-expression network analysis (rWGCNA)70 was used to construct immune-related co-expression networks on the whole transcriptome. Sixteen brain co-expression networks were shared across disorders (Fig. 4A, Fig. S7) after adjusting the batch covariate. Three of the 16 networks were enriched for IRGs and were called immune-related co-expression modules (Fig. 4B, C; IRM4, IRG enrichment qvalue = 2.15E-2; IRM12, qvalue = 1.13E-04; IRM14, qvalue = 9.29E-12). These three IRMs were significantly associated with at least two disorders in the same direction (Fig. 4D). The Eigengene (hub gene) of IRM4 was negatively associated with all disorders (Fig. 4D). IRM4 was enriched for neuron markers (Fig. 4E). Two of the modules (IRM12 and IRM14) were enriched with microglia and endothelial marker genes (Fig. 4E), respectively, and were both positively associated with IRGs in AD and ASD (Fig. 4D). AD had the strongest association with all three IRMs (Fig. 4D). The endothelial-related IRM14 and neuron-related IRM4 were both enriched for tissue development (qvalue= 6.69E-5, Fig. 4F, Table S8) and neuron development (qvalue= 6.49E-11, Fig. 4F, Table S8). Additionally, the IRM4 was significantly enriched for late fetal cortical markers (qvalue= 1.921E-07, Table. S8).
Fig. 4. Shared immune-related coexpression modules.
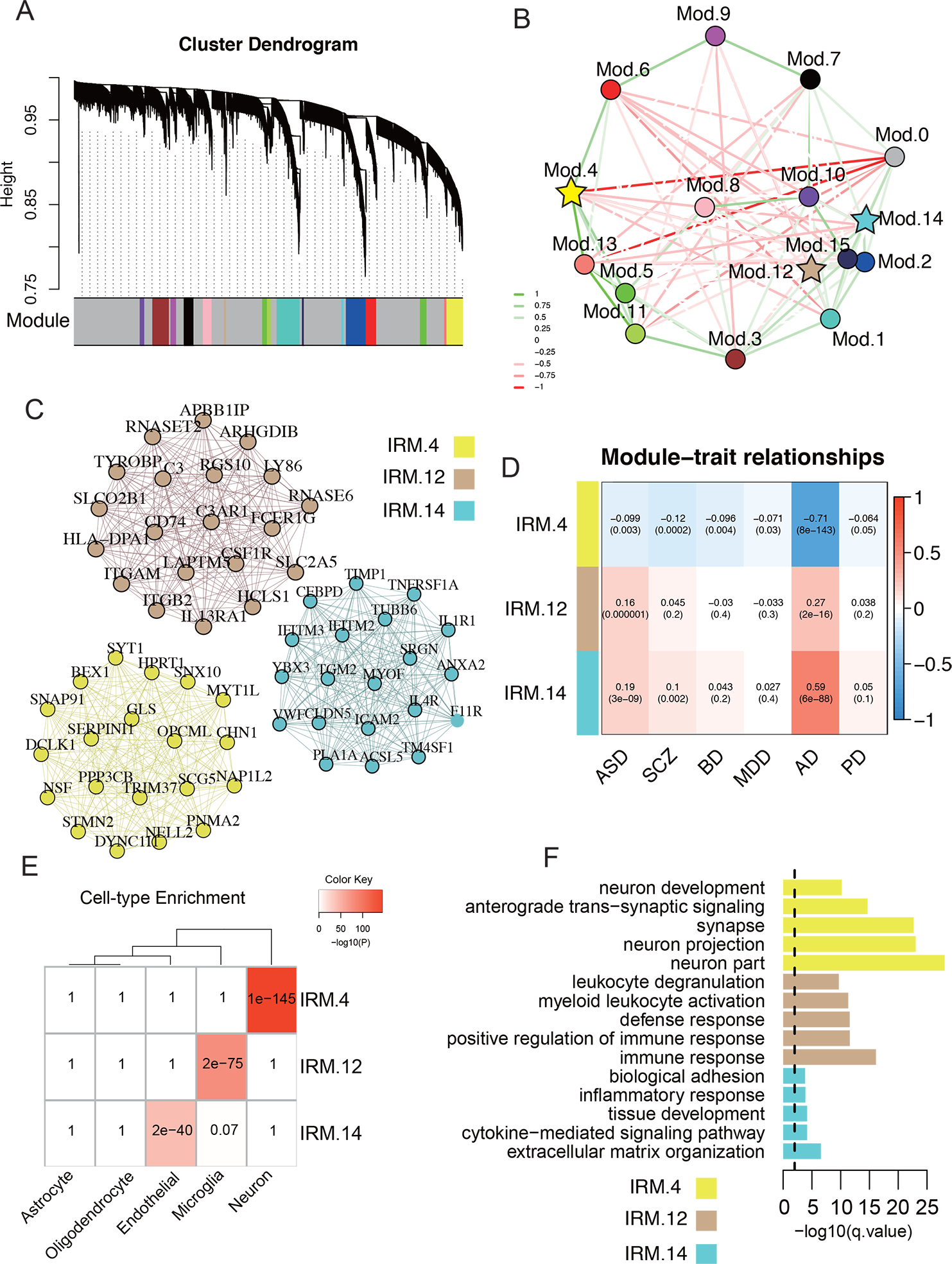
A. Robust gene dendrogram obtained by WGCNA. B. The multidimensional scaling plot demonstrates the relationship between modules. Modules highlighted by stars are enriched in immune genes (enrichment qvalue < 0.05). Edges are weighted by the strength of correlation between eigengenes of modules. C. The top 20 hub genes are plotted for the three IRM4, IRM12, and IRM14. A complete list of genes’ module membership (kME) is provided in data Table S8. Edges are weighted by the strength of the correlation between genes. D. Relationships of module eigengenes and diseases. Numbers in the table report the correlations of the corresponding module eigengenes and traits, with the p.values printed below the correlation coefficients r values. E. Cell marker enrichment of shared IRMs. F. Enrichment of the shared IRMs in pathways. Yellow: IRM4, Tan: IRM12, Cyan: IRM14
The influence of age on the IRMs was also assessed, showing that the age trajectories of these IRMs in cases had distinct patterns across disorders (Fig. S7D), which further illustrated the disease-specific temporal dynamics of these IRMs. For example, the neuron-related IRM-4 genes were continuously up-regulated in AD, with an inverted U-shaped curve peaking at age ~80, while PD showed a continuous downward trend in the same age range.
Disease-specific IRMs in AD, ASD, and PD imply distinct biological processes.
A search was completed for disease-specific IRMs for each disorder. rWGCNA was used to construct brain co-expression networks in the brains of each disorder and of controls, and they were compared against each other to identify disease-specific IRMs (Fig. 5A). Based on preservation results of one disease versus controls and against all other diseases (Fig. 5B, z-summary < 10), as well as immune gene enrichment results (Table S9; enrichment qvalue < 0.05), six disease-specific IRMs were identified, including one for AD, three for ASD, and two for PD. No disease-specific IRMs were detected for SCZ, BD, or MDD, which are considered psychiatric disorders.
Fig. 5. Disease-specific coexpression modules.
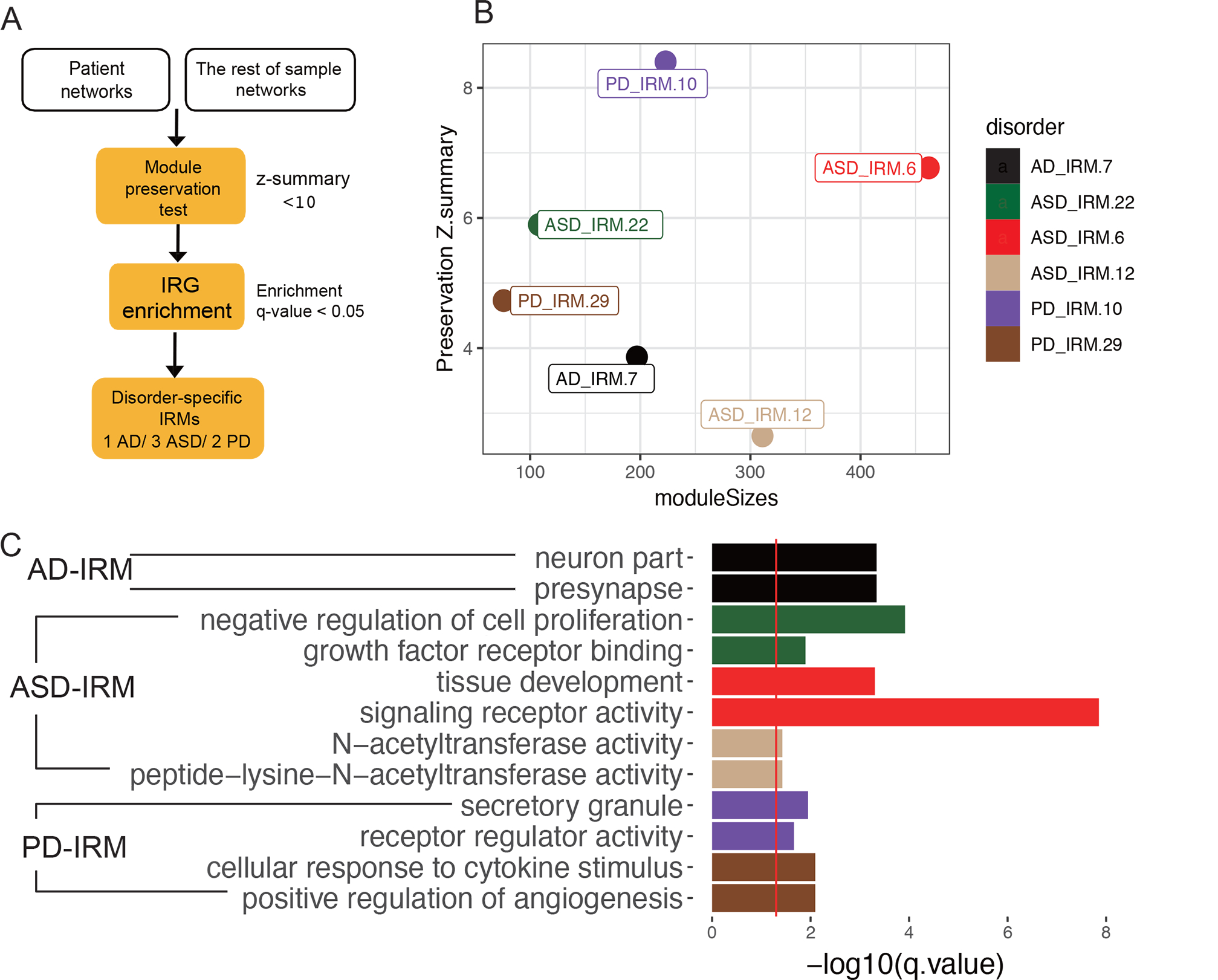
A. Workflow for identifying disease-specific IRMs. B. Module preservation plot of disease-specific IRMs. The median rank and Zsummary statistics of module preservation of disorder modules in background modules (y-axis) vs. module size (x-axis). C. Pathway enrichment of disease-specific immune modules.
The disease-specific IRMs were enriched for different functions (Fig. 5C, Table S9). The AD-specific IRM was enriched for neuron part (GO:0097458, qvalue= 4.57E-4) and presynapse (GO:0098793, qvalue = 4.57E-4). The PD-specific IRM was enriched for positive regulation of angiogenesis (GO:0045766, qvalue = 9.65E-06) and secretory granule (GO:0030141, qvalue= 6.31E-06). The ASD-specific IRMs were enriched for developmental biological processes such as negative regulation of cell proliferation (GO:0008285, qvalue= 1.21E-4) and growth factor receptor binding (GO:0070851, qvalue = 1.27E-02).
Common SNPs have a modest contribution to neuroimmune changes.
Tests were completed to determine whether IRGs, dIRGs, dIRG-enriched pathways, and IRMs were enriched for disease GWAS signals. A few dIRGs overlapping with genes that are in GWAS loci for all six diseases were detected, with AD having the largest number of genes (13 genes), and SCZ the second largest (5 genes), including C4A in SCZ (Table S10). However, none of these overlapping genes were significant after multiple testing. In contrast, a few dIRG-related pathways were significantly enriched in AD and SCZ GWAS signals and survived multiple testing corrections (Table S10). Among them, the amyloid precursor protein catabolic process was enriched in AD GWAS signals (qvalue= 3.9E-7). The leukocyte apoptotic process was enriched in SCZ GWAS signals (qvalue= 0.03). Only IRM-4 was enriched in SCZ GWAS signals (Table S10).
When two disorders have similar genetic risks, will they have similar changes of IRGs? In other words, is the genetic similarity between two disorders reflected by the similarity of expression changes of IRGs? The relationship between the effect-size correlation of dIRGs and the genetic correlation from the same pairs of disorders was assessed15. The genetic correlation was obtained from the Brainstorm Consortium81. Combining all the pairs of disorders, the correlation of these two kinds of correlations was modest but insignificant. (Fig. S8, Pearson’ r = 0.46, p.value = 0.08). Despite a small sample size of pairs of disorders (N=15), this analysis suggested that genetic factors had a minor contribution through affecting the IRGs to general brain disorders. This analysis captures collective contributions from all six disorders and cannot resolve individual contributions.
Discussion
This study focused on the neuroimmune changes represented by gene expression in multiple neuropsychiatric and neurological disorders. Transcriptome data of more than 2,000 brains from healthy controls and patients of six major brain disorders were used. This study showed that brain disorders have both shared and disease-specific immune-related changes by studying individual IRGs and related pathways and co-expression networks. In addition, the effects of biological factors such as tissue, sex, age, and cell type were evaluated. Four major findings of the neuroimmune system in brains of different neuropsychiatric disorders were highlighted: 1) the innate immune system carries more alterations than the adaptive immune systems in the six disorders; 2) the altered immune systems interact with brain-related biological pathways and networks contributing to the risk of disorders; 3) common SNPs have a limited contribution to immune-related disease risks, while the environmental contribution may be substantial; and 4) the expression profiles of dIRGs, particularly those of innate immune genes, clustered neurodevelopment disorder ASD with neurological diseases (AD and PD) instead of with psychiatric disorders (BD, MDD, and SCZ)
More than half of the shared dIRGs and dIRG-enriched pathways were related to the innate immune system. Dysregulation of the innate immune system is a common denominator for the six brain disorders in this study. Additionally, TLR1/2 mediates microglial activity, which could contribute to neuronal death through the release of inflammatory mediators82 Furthermore, innate immunity is critical in maintaining homeostasis in the brain. For example, the innate immune system has been reported to function in the CNS’s resilience83 and in synaptic pruning throughout brain growth84. When homeostasis is disrupted, abnormal innate immunity may impact a wide range of brain functions85.
The IRGs connected immunity with the neuronal processes and disease risk. The dIRGs were expected to be enriched for immune-related pathways since immune-related genes were selected as the focus in this study. However, these dIRGs cooperate with other brain-related biological processes. Such cooperative networks were revealed by the coexpression networks involving IRGs. The coexpression network analyses indicated that IRGs in brains were involved in brain developmental processes. Three brain universal IRMs were enriched with functions related to development in neurons, microglia, and endothelia. IRM4 is of particular interest for connecting neuron, immune system, and development to all six disorders tested in this study. The contribution of neuro-immune-development to SCZ and ASD is well accepted. Previous studies discovered the role of the immune system in the development of the CNS83, 86, 87. Abnormal immune activation during brain development can cause behavioral and neurochemical abnormalities relevant to disorders88–92. One additional disease-specific module, ASD-IRM6, is also associated with development, further implicating the importance of development in ASD risk. The connections between AD, PD, and neuro-immune-development through IRM4 remain unclear, as they are late-onset neurodegenerative diseases. The unique age trajectories of these IRM4 in AD patients suggested aging in the immune system was involved for the same set of genes. As another representative of the disease-shared module, IRM-14 was enriched with immune-related and tissue development-related pathways. This finding provides evidence supporting the hypothesis that immunity is involved in brain development93. The discovery of association of IRM-14 in these six disorders also suggests that a subset of immune-related genes of this specific network constitutes common ground for these different brain diseases. IRM-14 was enriched for cell markers of endothelial cell. The endothelial cell is involved when the vascular blood–brain barrier becomes more permeable to solutes with increasing degrees and duration of innate inflammation94. These findings indicated the importance of endothelial cells in the neuroimmune system.
This study showed that the disease-specific IRMs may influence expression of networked non-immune genes and contribute to the pathology of these diseases specifically. Six disease-specific IRMs were detected in AD, ASD, and PD, including a presynaptic-related AD-specific IRM, growth factor receptors-related ASD-specific IRMs, and a secretogranin-related PD-specific IRM. These disease-specific IRMs are supported by existing literature. Presynaptic proteins are known to be essential for synaptic function and to be related to cognitive impairments in AD95. Growth factor receptors96 and N-acetylcysteine97 are known to be involved in the etiology of ASD. Secretogranin may be a pivotal component of the neuroendocrine pathway and play an essential role in neuronal communication and neurotransmitter release in PD 98. Moreover, the immune system has been found to regulate presynaptic proteins99, EGFR100, and secretogranin98.
Microglia are highlighted in the immune changes in brains of AD and ASD in this study. Microglia are major participants in the brain’s immune system. This study showed that the IRM12 co-expression module was enriched for microglia genes and associated with inflammatory transcriptional change in AD and ASD but not the other four diseases. Does this suggest that microglial dysfunction contributes more to AD and ASD than to the other disorders? A PsychENCODE study showed the microglial module was upregulated in ASD and downregulated in SCZ and BD16, but the fold changes in SCZ and BD were much smaller than that in ASD. Larger sample size, or immune stratification may be needed to detect microglia contribution to other disorders such as SCZ and BD.
Debate exists over whether neuroinflammatory alterations in disorders are affected by genetics101, 102 or by the environment103, 104. This study offered support for both arguments. On the genetic risk side, the genetic contribution to neuroinflammation may be more relevant to AD and SCZ than the other four disorders. Several individual dIRGs are also GWAS signals such as IL6R in AD and C4A in SCZ. IL6R has been identified as a strong candidate gene of AD with both genetic and transcriptome supports105. C4A is a well-known SCZ GWAS locus and is up-regulated in SCZ (effect size=0.2, qvalue<0.05). Several dIRG-related pathways and co-expression modules were enriched in GWAS signals of neurological diseases AD and SCZ, respectively. The enrichment for AD was particularly strong, involving the amyloid precursor protein catabolic process in AD. A previous study identified this pathway under genetic control in AD106. However, this study did not detect statistically significant correlation between overall genetic risk and IRG changes between pairs of disorders in general. This IRG-subset result is in contrast with previous results on the whole transcriptome15 where a significant correlation (Spearman’s ρ =0.79) between expression changes and genetic correlation in pairs of disorders was reported. The genetic connection with immunity detected in the current study was likely indirect because no significant enrichment of dIRG in GWASs of ASD, PD, BD, and MDD was detected. On the environmental side, the environmental contribution to neuropsychiatric disease risk is strongly implicated through the current study with data suggesting that these brain diseases are related to stress, an environmental factor.
Unexpectedly, ASD, thought to be a neurodevelopmental disorder, was grouped with neurological diseases (AD and PD) instead of with psychiatric disorders (BD, MDD, and SCZ) based on changes of IRGs, particularly innate immune genes. Previous studies have reported that ASD patients exhibited more neurological and immunological problems107–110 compared to healthy people and to other brain disorders. As more etiologies are uncovered, the traditional classification of these diseases is increasingly challenged103. Furthermore, this study found that dIRGs change more in neurological diseases (AD, PD, and ASD) than in the psychiatric disorders (BD, SCZ, and MDD). This suggested that neuroimmune gene dysregulation was more severe in neurological diseases than in psychiatric disorders, led by AD. These findings suggest that different clusters of disorders may benefit from immune-related treatments strategies.
The two most shared dIRGs, TAC1 and CRH, have known effects on innate immune activation111, 112 and stress response113, 114. CRH is one of the most shared dIRGs across disorders. CRH has the core function of controlling the release of stress hormones. Studies have reported the relationship between immunity and stress115, 116 and showed patients with brain disorders had decreased cortisol responses to social stressors117. CRH and TAC1 are both consistently shown reduced in our microarray data (discovery) and RNA-seq data (validation). The differential expression of these two genes implied the dysregulation of the stress response in these major brain disorders. Chronic stress is known to lead to increased inflammation in the periphery118–121. Why TAC1 and CRH showed reduced expression? One possible explanation for the reduced expression is that prolonged stressful environment leads to abnormal cortisol regulation as shown by reduced GR and increased FKBP5, which could in turn lead to excessive accumulation of inflammatory factors in the brain. Another possible explanation is that changes in brain gene expression are dynamic and change over time. The current study is limited to the observation of expression level in postmortem brain tissue from patients likely at later stages of illness. The expression of CRH or TAC1 may have different expression patterns earlier on and/or vary with disease stages. Both possible mechanisms should be explored in future studies. One thing should be clear: increased inflammation does not mean increased activities of every gene in the systems.
The difference in numbers of dIRGs among disorders might be related to the sample size of each disease dataset. Transcriptome data from bulk tissues were used in this study, which did not reflect gene expression in specific brain cell types. The sva was used in this study to control hidden covariates, which minimized the cell type effects along with other covariates. The co-expression network analysis still suggested three cell types. Single-cell or deconvolutional data will be needed to uncover effects of subtypes of cells. Similarly, age and other factors were regressed out as well when detecting dIRGs. They could be studied specifically. Causal relationship between neuroinflammatory changes and disorders will need to be established.
In summary, this study provided a cross-disorder transcriptome study to explore neuroimmune system dysfunction in six neurological and psychiatric disorders. The fact that more than 60% of the IRGs had significantly altered expression in at least one of the six disorders indicated immune dysfunction widely exists in these brain disorders. The functional annotations of the dIRGs highlighted the shared dysfunction of innate immunity, and its ability to differentiate psychiatric disorders from neurological diseases. Disease-specific dIRGs and their associated pathways and coexpression modules may explain the distinct clinical features of each disorder. Therapeutics targeting different components of the systems may lead to distinct effects; some are general for all brain disorders while others could help specific disorders. A subset of patients may benefit from immune-related treatments.
Supplementary Material
Acknowledgments:
Published microarray datasets analyzed in this study are available on Gene Expression Omnibus (accession No. GSE28521, GSE28475, GSE35978, GSE53987, GSE17612, GSE12649, GSE21138, GSE54567, GSE54568, GSE54571, GSE54572, GSE29555, GSE5281, GSE28146, GSE20168, GSE20295, GSE54282, GSE68719 and GSE11223), ArrayExpress (accession no. E-MTAB-184), or directly from the study authors.
RNA-seq data (available on Synapse with accession numbers syn4590909 and syn4587609, with access governed by NIMH Repository and Genomics Resource) were generated as part of the PsychENCODE Consortium, supported by grants U01MH103339, U01MH103365, U01MH103392, U01MH103340, U01MH103346, R01MH105472, R01MH094714, R01MH105898, R21MH102791, R21MH105881, R21MH103877, and P50MH106934 awarded to Schahram Akbarian (Icahn School of Medicine at Mount Sinai), Gregory Crawford (Duke), Stella Dracheva (Icahn School of Medicine at Mount Sinai), Peggy Farnham (USC), Mark Gerstein (Yale), Daniel Geschwind (UCLA), Thomas M. Hyde (LIBD), Andrew Jaffe (LIBD), James A. Knowles (USC), Chunyu Liu (SUNY), Dalila Pinto (Icahn School of Medicine at Mount Sinai), Nenad Sestan (Yale), Pamela Sklar (Icahn School of Medicine at Mount Sinai), Matthew State (UCSF), Patrick Sullivan (UNC), Flora Vaccarino (Yale), Sherman Weissman (Yale), Kevin White (UChicago), and Peter Zandi (JHU).
RNA-seq data from the CommonMind Consortium used in this study (Synapse accession no. syn2759792) was supported by funding from Takeda Pharmaceuticals Company, F. Hoffman-La Roche and NIH grants R01MH085542, R01MH093725, P50MH066392, P50MH080405, R01MH097276, RO1-MH-075916, P50M096891, P50MH084053S1, R37MH057881, R37MH057881S1, HHSN271201300031C, AG02219, AG05138, and MH06692.
RNA-seq data from the ROSMAP were provided by the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago. Data collection was supported through funding by NIA grants P30AG10161 (ROS), R01AG15819 (ROSMAP; genomics and RNAseq), R01AG17917 (MAP), R01AG30146, R01AG36042 (5hC methylation, ATACseq), RC2AG036547 (H3K9Ac), R01AG36836 (RNAseq), R01AG48015 (monocyte RNAseq) RF1AG57473 (single nucleus RNAseq), U01AG32984 (genomic and whole exome sequencing), U01AG46152 (ROSMAP AMP-AD, targeted proteomics), U01AG46161(TMT proteomics), U01AG61356 (whole genome sequencing, targeted proteomics, ROSMAP AMP-AD), the Illinois Department of Public Health (ROSMAP), and the Translational Genomics Research Institute (genomic). Brain tissue for the study was obtained from the following brain bank collections: the Mount Sinai NIH Brain and Tissue Repository, the University of Pennsylvania Alzheimer’s Disease Core Center, the University of Pittsburgh NeuroBioBank and Brain and Tissue Repositories, the Harvard Brain Bank as part of the Autism Tissue Project (ATP), the Stanley Medical Research Institute, and the NIMH Human Brain Collection Core.
We thank Mingrui Yu, and Yan Xia for reviewing the manuscript.
Funding:
National Natural Science Foundation of China Nos. 82022024
National Natural Science Foundation of China Nos. 31970572
National Natural Science Foundation of China Nos. 31871276
the National Key R&D Project of China Grants No. 2016YFC1306000
the National Key R&D Project of China Grants No. 2017YFC0908701
Innovation-driven Project of Central South University Grant Nos. 2020CX003
NIH grants U01MH122591
NIH grants U01MH116489.
NIH grants R01MH110920.
Footnotes
Competing interests: Authors declare that they have no competing interests
Data and materials availability:
All data are available in the main text or the supplementary materials.
References and Notes
- 1.Parshukova D, Smirnova LP, Ermakov EA, Bokhan NA, Semke AV, Ivanova SA et al. Autoimmunity and immune system dysregulation in schizophrenia: IgGs from sera of patients hydrolyze myelin basic protein. J Mol Recognit 2019; 32(2): e2759. [DOI] [PubMed] [Google Scholar]
- 2.Benedetti F, Aggio V, Pratesi ML, Greco G, Furlan R. Neuroinflammation in Bipolar Depression. Front Psychiatry 2020; 11: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Resende R, Fernandes T, Pereira AC, De Pascale J, Marques AP, Oliveira P et al. Mitochondria, endoplasmic reticulum and innate immune dysfunction in mood disorders: Do Mitochondria-Associated Membranes (MAMs) play a role? Biochim Biophys Acta Mol Basis Dis 2020; 1866(6): 165752. [DOI] [PubMed] [Google Scholar]
- 4.Meltzer A, Van de Water J. The Role of the Immune System in Autism Spectrum Disorder. Neuropsychopharmacology 2017; 42(1): 284–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Webers A, Heneka MT, Gleeson PA. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol Cell Biol 2020; 98(1): 28–41. [DOI] [PubMed] [Google Scholar]
- 6.De Virgilio A, Greco A, Fabbrini G, Inghilleri M, Rizzo MI, Gallo A et al. Parkinson’s disease: Autoimmunity and neuroinflammation. Autoimmun Rev 2016; 15(10): 1005–1011. [DOI] [PubMed] [Google Scholar]
- 7.Lu Y, Li K, Hu Y, Wang X. Expression of Immune Related Genes and Possible Regulatory Mechanisms in Alzheimer’s Disease. Front Immunol 2021; 12: 768966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang X, Shao Z, Xu S, Liu Q, Liu C, Luo Y et al. Immune Profiling of Parkinson’s Disease Revealed Its Association With a Subset of Infiltrating Cells and Signature Genes. Front Aging Neurosci 2021; 13: 605970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horiuchi F, Yoshino Y, Kumon H, Hosokawa R, Nakachi K, Kawabe K et al. Identification of aberrant innate and adaptive immunity based on changes in global gene expression in the blood of adults with autism spectrum disorder. J Neuroinflammation 2021; 18(1): 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fillman SG, Sinclair D, Fung SJ, Webster MJ, Shannon Weickert C. Markers of inflammation and stress distinguish subsets of individuals with schizophrenia and bipolar disorder. Transl Psychiatry 2014; 4: e365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saetre P, Emilsson L, Axelsson E, Kreuger J, Lindholm E, Jazin E. Inflammation-related genes up-regulated in schizophrenia brains. BMC Psychiatry 2007; 7: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai HQ, Catts VS, Webster MJ, Galletly C, Liu D, O’Donnell M et al. Increased macrophages and changed brain endothelial cell gene expression in the frontal cortex of people with schizophrenia displaying inflammation. Mol Psychiatry 2020; 25(4): 761–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Catts VS, Sheedy D, McCrossin T, Kril JJ, Shannon Weickert C. Cortical grey matter volume reduction in people with schizophrenia is associated with neuroinflammation. Transl Psychiatry 2016; 6(12): e982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Volk DW. Role of microglia disturbances and immune-related marker abnormalities in cortical circuitry dysfunction in schizophrenia. Neurobiol Dis 2017; 99: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 2018; 359(6376): 693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018; 362(6420). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan N, Chen Y, Xia Y, Dai J, Liu C. Inflammation-related biomarkers in major psychiatric disorders: a cross-disorder assessment of reproducibility and specificity in 43 meta-analyses. Transl Psychiatry 2019; 9(1): 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.t Hart BA, den Dunnen WF. Commentary on special issue: CNS diseases and the immune system. J Neuroimmune Pharmacol 2013; 8(4): 757–759. [DOI] [PubMed] [Google Scholar]
- 19.Gendelman HE. Neural immunity: Friend or foe? J Neurovirol 2002; 8(6): 474–479. [DOI] [PubMed] [Google Scholar]
- 20.Mitra S, Chakrabarti N, Dutta SS, Ray S, Bhattacharya P, Sinha P et al. Gender-specific brain regional variation of neurons, endogenous estrogen, neuroinflammation and glial cells during rotenone-induced mouse model of Parkinson’s disease. Neuroscience 2015; 292: 46–70. [DOI] [PubMed] [Google Scholar]
- 21.Sparkman NL, Johnson RW. Neuroinflammation associated with aging sensitizes the brain to the effects of infection or stress. Neuroimmunomodulation 2008; 15(4–6): 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Owens T, Wekerle H, Antel J. Genetic models for CNS inflammation. Nat Med 2001; 7(2): 161–166. [DOI] [PubMed] [Google Scholar]
- 23.Dantzer R Neuroimmune Interactions: From the Brain to the Immune System and Vice Versa. Physiol Rev 2018; 98(1): 477–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nasr IW, Chun Y, Kannan S. Neuroimmune responses in the developing brain following traumatic brain injury. Exp Neurol 2019; 320: 112957. [DOI] [PubMed] [Google Scholar]
- 25.D’Mello C, Swain MG. Immune-to-Brain Communication Pathways in Inflammation-Associated Sickness and Depression. Curr Top Behav Neurosci 2017; 31: 73–94. [DOI] [PubMed] [Google Scholar]
- 26.Kelley KW, Bluthe RM, Dantzer R, Zhou JH, Shen WH, Johnson RW et al. Cytokine-induced sickness behavior. Brain Behav Immun 2003; 17 Suppl 1: S112–118. [DOI] [PubMed] [Google Scholar]
- 27.Lyman M, Lloyd DG, Ji X, Vizcaychipi MP, Ma D. Neuroinflammation: the role and consequences. Neurosci Res 2014; 79: 1–12. [DOI] [PubMed] [Google Scholar]
- 28.Ozawa K, Hashimoto K, Kishimoto T, Shimizu E, Ishikura H, Iyo M. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: a neurodevelopmental animal model of schizophrenia. Biol Psychiatry 2006; 59(6): 546–554. [DOI] [PubMed] [Google Scholar]
- 29.Vuillermot S, Weber L, Feldon J, Meyer U. A longitudinal examination of the neurodevelopmental impact of prenatal immune activation in mice reveals primary defects in dopaminergic development relevant to schizophrenia. J Neurosci 2010; 30(4): 1270–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lammert CR, Lukens JR. Modeling Autism-Related Disorders in Mice with Maternal Immune Activation (MIA). Methods Mol Biol 2019; 1960: 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Volk DW, Lewis DA. Early developmental disturbances of cortical inhibitory neurons: contribution to cognitive deficits in schizophrenia. Schizophr Bull 2014; 40(5): 952–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW et al. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation 2008; 5: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Volk DW, Chitrapu A, Edelson JR, Roman KM, Moroco AE, Lewis DA. Molecular mechanisms and timing of cortical immune activation in schizophrenia. Am J Psychiatry 2015; 172(11): 1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Purves-Tyson TD, Robinson K, Brown AM, Boerrigter D, Cai HQ, Weissleder C et al. Increased Macrophages and C1qA, C3, C4 Transcripts in the Midbrain of People With Schizophrenia. Front Immunol 2020; 11: 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 2012; 4(147): 147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015; 523(7560): 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morey JN, Boggero IA, Scott AB, Segerstrom SC. Current Directions in Stress and Human Immune Function. Curr Opin Psychol 2015; 5: 13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim SY, Buckwalter M, Soreq H, Vezzani A, Kaufer D. Blood-brain barrier dysfunction-induced inflammatory signaling in brain pathology and epileptogenesis. Epilepsia 2012; 53 Suppl 6: 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mukherjee S Immune gene network of neurological diseases: Multiple sclerosis (MS), Alzheimer’s disease (AD), Parkinson’s disease (PD) and Huntington’s disease (HD). Heliyon 2021; 7(12): e08518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Walker DG et al. Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiol Genomics 2007; 28(3): 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blalock EM, Buechel HM, Popovic J, Geddes JW, Landfield PW. Microarray analyses of laser-captured hippocampus reveal distinct gray and white matter signatures associated with incipient Alzheimer’s disease. J Chem Neuroanat 2011; 42(2): 118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013; 153(3): 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Psych EC, Akbarian S, Liu C, Knowles JA, Vaccarino FM, Farnham PJ et al. The PsychENCODE project. Nat Neurosci 2015; 18(12): 1707–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011; 474(7351): 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chow ML, Li HR, Winn ME, April C, Barnes CC, Wynshaw-Boris A et al. Genome-wide expression assay comparison across frozen and fixed postmortem brain tissue samples. BMC Genomics 2011; 12: 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garbett K, Ebert PJ, Mitchell A, Lintas C, Manzi B, Mirnics K et al. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol Dis 2008; 30(3): 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoffman GE, Bendl J, Voloudakis G, Montgomery KS, Sloofman L, Wang YC et al. CommonMind Consortium provides transcriptomic and epigenomic data for Schizophrenia and Bipolar Disorder. Sci Data 2019; 6(1): 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen C, Meng Q, Xia Y, Ding C, Wang L, Dai R et al. The transcription factor POU3F2 regulates a gene coexpression network in brain tissue from patients with psychiatric disorders. Sci Transl Med 2018; 10(472). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lanz TA, Reinhart V, Sheehan MJ, Rizzo SJS, Bove SE, James LC et al. Postmortem transcriptional profiling reveals widespread increase in inflammation in schizophrenia: a comparison of prefrontal cortex, striatum, and hippocampus among matched tetrads of controls with subjects diagnosed with schizophrenia, bipolar or major depressive disorder. Transl Psychiatry 2019; 9(1): 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iwamoto K, Bundo M, Kato T. Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum Mol Genet 2005; 14(2): 241–253. [DOI] [PubMed] [Google Scholar]
- 51.Chang LC, Jamain S, Lin CW, Rujescu D, Tseng GC, Sibille E. A conserved BDNF, glutamate- and GABA-enriched gene module related to human depression identified by coexpression meta-analysis and DNA variant genome-wide association studies. PLoS One 2014; 9(3): e90980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dumitriu A, Golji J, Labadorf AT, Gao B, Beach TG, Myers RH et al. Integrative analyses of proteomics and RNA transcriptomics implicate mitochondrial processes, protein folding pathways and GWAS loci in Parkinson disease. BMC Med Genomics 2016; 9: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y, James M, Middleton FA, Davis RL. Transcriptional analysis of multiple brain regions in Parkinson’s disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. Am J Med Genet B Neuropsychiatr Genet 2005; 137B(1): 5–16. [DOI] [PubMed] [Google Scholar]
- 54.Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med 2010; 2(52): 52ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Riley BE, Gardai SJ, Emig-Agius D, Bessarabova M, Ivliev AE, Schule B et al. Systems-based analyses of brain regions functionally impacted in Parkinson’s disease reveals underlying causal mechanisms. PLoS One 2014; 9(8): e102909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maycox PR, Kelly F, Taylor A, Bates S, Reid J, Logendra R et al. Analysis of gene expression in two large schizophrenia cohorts identifies multiple changes associated with nerve terminal function. Mol Psychiatry 2009; 14(12): 1083–1094. [DOI] [PubMed] [Google Scholar]
- 57.Narayan S, Tang B, Head SR, Gilmartin TJ, Sutcliffe JG, Dean B et al. Molecular profiles of schizophrenia in the CNS at different stages of illness. Brain Res 2008; 1239: 235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davis AP, Grondin CJ, Johnson RJ, Sciaky D, McMorran R, Wiegers J et al. The Comparative Toxicogenomics Database: update 2019. Nucleic Acids Res 2019; 47(D1): D948–D954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bhattacharya S, Dunn P, Thomas CG, Smith B, Schaefer H, Chen J et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci Data 2018; 5: 180015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Diaz-Ramos MC, Engel P, Bastos R. Towards a comprehensive human cell-surface immunome database. Immunol Lett 2011; 134(2): 183–187. [DOI] [PubMed] [Google Scholar]
- 61.Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R et al. InnateDB: systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res 2013; 41(Database issue): D1228–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Godec J, Tan Y, Liberzon A, Tamayo P, Bhattacharya S, Butte AJ et al. Compendium of Immune Signatures Identifies Conserved and Species-Specific Biology in Response to Inflammation. Immunity 2016; 44(1): 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.The Gene Ontology C The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 2019; 47(D1): D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000; 28(1): 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Birnbaum R, Jaffe AE, Chen Q, Shin JH, BrainSeq C, Kleinman JE et al. Investigating the neuroimmunogenic architecture of schizophrenia. Mol Psychiatry 2018; 23(5): 1251–1260. [DOI] [PubMed] [Google Scholar]
- 66.Yang H, Zhao K, Kang H, Wang M, Wu A. Exploring immune-related genes with prognostic value in microenvironment of breast cancer from TCGA database. Medicine (Baltimore) 2020; 99(14): e19561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang M, Wang X, Chen X, Zhang Q, Hong J. Novel Immune-Related Gene Signature for Risk Stratification and Prognosis of Survival in Lower-Grade Glioma. Front Genet 2020; 11: 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012; 28(6): 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Picard toolkit. 2019, Accessed Date Accessed 2019 Accessed.
- 70.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012; 16(5): 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zambon AC, Gaj S, Ho I, Hanspers K, Vranizan K, Evelo CT et al. GO-Elite: a flexible solution for pathway and ontology over-representation. Bioinformatics 2012; 28(16): 2209–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 2019; 47(W1): W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dougherty JD, Schmidt EF, Nakajima M, Heintz N. Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res 2010; 38(13): 4218–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016; 89(1): 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 2015; 11(4): e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011; 27(12): 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu X, Shukla R, Alganem K, Zhang X, Eby HM, Devine EA et al. Transcriptional profile of pyramidal neurons in chronic schizophrenia reveals lamina-specific dysfunction of neuronal immunity. Mol Psychiatry 2021; 26(12): 7699–7708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alganem K, Shukla R, Eby H, Abel M, Zhang X, McIntyre WB et al. Kaleidoscope: A New Bioinformatics Pipeline Web Application for In Silico Hypothesis Exploration of Omics Signatures. bioRxiv 2020: 2020.2005.2001.070805. [Google Scholar]
- 80.Spitsin S, Stevens KE, Douglas SD. Expression of substance P, neurokinin-1 receptor and immune markers in the brains of individuals with HIV-associated neuropathology. J Neurol Sci 2013; 334(1–2): 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brainstorm C, Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J et al. Analysis of shared heritability in common disorders of the brain. Science 2018; 360(6395). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lotz M, Ebert S, Esselmann H, Iliev AI, Prinz M, Wiazewicz N et al. Amyloid beta peptide 1–40 enhances the action of Toll-like receptor-2 and -4 agonists but antagonizes Toll-like receptor-9-induced inflammation in primary mouse microglial cell cultures. J Neurochem 2005; 94(2): 289–298. [DOI] [PubMed] [Google Scholar]
- 83.Morimoto K, Nakajima K. Role of the Immune System in the Development of the Central Nervous System. Front Neurosci 2019; 13: 916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nistico R, Salter E, Nicolas C, Feligioni M, Mango D, Bortolotto ZA et al. Synaptoimmunology - roles in health and disease. Mol Brain 2017; 10(1): 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Novellino F, Sacca V, Donato A, Zaffino P, Spadea MF, Vismara M et al. Innate Immunity: A Common Denominator between Neurodegenerative and Neuropsychiatric Diseases. Int J Mol Sci 2020; 21(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lenz KM, Nelson LH. Microglia and Beyond: Innate Immune Cells As Regulators of Brain Development and Behavioral Function. Front Immunol 2018; 9: 698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parab S, Quick RE, Matsuoka RL. Endothelial cell-type-specific molecular requirements for angiogenesis drive fenestrated vessel development in the brain. Elife 2021; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Goines P, Van de Water J. The immune system’s role in the biology of autism. Curr Opin Neurol 2010; 23(2): 111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bitanihirwe BK, Peleg-Raibstein D, Mouttet F, Feldon J, Meyer U. Late prenatal immune activation in mice leads to behavioral and neurochemical abnormalities relevant to the negative symptoms of schizophrenia. Neuropsychopharmacology 2010; 35(12): 2462–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Parboosing R, Bao Y, Shen L, Schaefer CA, Brown AS. Gestational influenza and bipolar disorder in adult offspring. JAMA Psychiatry 2013; 70(7): 677–685. [DOI] [PubMed] [Google Scholar]
- 91.Simanek AM, Meier HC . Association Between Prenatal Exposure to Maternal Infection and Offspring Mood Disorders: A Review of the Literature. Curr Probl Pediatr Adolesc Health Care 2015; 45(11): 325–364. [DOI] [PubMed] [Google Scholar]
- 92.Caccamo A, De Pinto V, Messina A, Branca C, Oddo S. Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J Neurosci 2014; 34(23): 7988–7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tanabe S, Yamashita T. The role of immune cells in brain development and neurodevelopmental diseases. Int Immunol 2018; 30(10): 437–444. [DOI] [PubMed] [Google Scholar]
- 94.Pais TF, Penha-Goncalves C. Brain Endothelium: The “Innate Immunity Response Hypothesis” in Cerebral Malaria Pathogenesis. Front Immunol 2018; 9: 3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reddy PH, Mani G, Park BS, Jacques J, Murdoch G, Whetsell W, Jr. et al. Differential loss of synaptic proteins in Alzheimer’s disease: implications for synaptic dysfunction. J Alzheimers Dis 2005; 7(2): 103–117; discussion 173–180. [DOI] [PubMed] [Google Scholar]
- 96.Russo AJ. Increased Epidermal Growth Factor Receptor (EGFR) Associated with Hepatocyte Growth Factor (HGF) and Symptom Severity in Children with Autism Spectrum Disorders (ASDs). J Cent Nerv Syst Dis 2014; 6: 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Durieux AM, Fernandes C, Murphy D, Labouesse MA, Giovanoli S, Meyer U et al. Targeting Glia with N-Acetylcysteine Modulates Brain Glutamate and Behaviors Relevant to Neurodevelopmental Disorders in C57BL/6J Mice. Front Behav Neurosci 2015; 9: 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li F, Tian X, Zhou Y, Zhu L, Wang B, Ding M et al. Dysregulated expression of secretogranin III is involved in neurotoxin-induced dopaminergic neuron apoptosis. J Neurosci Res 2012; 90(12): 2237–2246. [DOI] [PubMed] [Google Scholar]
- 99.Harris N, Fetter RD, Brasier DJ, Tong A, Davis GW. Molecular Interface of Neuronal Innate Immunity, Synaptic Vesicle Stabilization, and Presynaptic Homeostatic Plasticity. Neuron 2018; 100(5): 1163–1179 e1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sasada T, Azuma K, Ohtake J, Fujimoto Y . Immune Responses to Epidermal Growth Factor Receptor (EGFR) and Their Application for Cancer Treatment. Front Pharmacol 2016; 7: 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Network, Pathway Analysis Subgroup of Psychiatric Genomics C. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci 2015; 18(2): 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tylee DS, Sun J, Hess JL, Tahir MA, Sharma E, Malik R et al. Genetic correlations among psychiatric and immune-related phenotypes based on genome-wide association data. Am J Med Genet B Neuropsychiatr Genet 2018; 177(7): 641–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pape K, Tamouza R, Leboyer M, Zipp F. Immunoneuropsychiatry - novel perspectives on brain disorders. Nat Rev Neurol 2019; 15(6): 317–328. [DOI] [PubMed] [Google Scholar]
- 104.Costa-Pinto FA, Palermo-Neto J. Neuroimmune interactions in stress. Neuroimmunomodulation 2010; 17(3): 196–199. [DOI] [PubMed] [Google Scholar]
- 105.Haddick PC, Larson JL, Rathore N, Bhangale TR, Phung QT, Srinivasan K et al. A Common Variant of IL-6R is Associated with Elevated IL-6 Pathway Activity in Alzheimer’s Disease Brains. J Alzheimers Dis 2017; 56(3): 1037–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 2019; 51(3): 404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pan PY, Tammimies K, Bolte S. The Association Between Somatic Health, Autism Spectrum Disorder, and Autistic Traits. Behav Genet 2020; 50(4): 233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xiong J, Chen S, Pang N, Deng X, Yang L, He F et al. Neurological Diseases With Autism Spectrum Disorder: Role of ASD Risk Genes. Front Neurosci 2019; 13: 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Khan SA, Khan SA, Narendra AR, Mushtaq G, Zahran SA, Khan S et al. Alzheimer’s Disease and Autistic Spectrum Disorder: Is there any Association? CNS Neurol Disord Drug Targets 2016; 15(4): 390–402. [DOI] [PubMed] [Google Scholar]
- 110.Starkstein S, Gellar S, Parlier M, Payne L, Piven J. High rates of parkinsonism in adults with autism. J Neurodev Disord 2015; 7(1): 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Asadi S, Theoharides TC . Corticotropin-releasing hormone and extracellular mitochondria augment IgE-stimulated human mast-cell vascular endothelial growth factor release, which is inhibited by luteolin. J Neuroinflammation 2012; 9: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mashaghi A, Marmalidou A, Tehrani M, Grace PM, Pothoulakis C, Dana R. Neuropeptide substance P and the immune response. Cell Mol Life Sci 2016; 73(22): 4249–4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.He ZX, Yin YY, Xi K, Xing ZK, Cao JB, Liu TY et al. Nucleus Accumbens Tac1-Expressing Neurons Mediate Stress-Induced Anhedonia-like Behavior in Mice. Cell Rep 2020; 33(5): 108343. [DOI] [PubMed] [Google Scholar]
- 114.Claes SJ. Corticotropin-releasing hormone (CRH) in psychiatry: from stress to psychopathology. Ann Med 2004; 36(1): 50–61. [DOI] [PubMed] [Google Scholar]
- 115.Tsagarakis S, Grossman A. Corticotropin-releasing hormone: interactions with the immune system. Neuroimmunomodulation 1994; 1(6): 329–334. [DOI] [PubMed] [Google Scholar]
- 116.O’Kane M, Murphy EP, Kirby B. The role of corticotropin-releasing hormone in immune-mediated cutaneous inflammatory disease. Exp Dermatol 2006; 15(3): 143–153. [DOI] [PubMed] [Google Scholar]
- 117.Connors EJ, Shaik AN, Migliore MM, Kentner AC. Environmental enrichment mitigates the sex-specific effects of gestational inflammation on social engagement and the hypothalamic pituitary adrenal axis-feedback system. Brain Behav Immun 2014; 42: 178–190. [DOI] [PubMed] [Google Scholar]
- 118.Sinclair D, Fillman SG, Webster MJ, Weickert CS. Dysregulation of glucocorticoid receptor co-factors FKBP5, BAG1 and PTGES3 in prefrontal cortex in psychotic illness. Sci Rep 2013; 3: 3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sinclair D, Webster MJ, Fullerton JM, Weickert CS. Glucocorticoid receptor mRNA and protein isoform alterations in the orbitofrontal cortex in schizophrenia and bipolar disorder. BMC Psychiatry 2012; 12: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sinclair D, Fullerton JM, Webster MJ, Shannon Weickert C. Glucocorticoid receptor 1B and 1C mRNA transcript alterations in schizophrenia and bipolar disorder, and their possible regulation by GR gene variants. PLoS One 2012; 7(3): e31720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sinclair D, Tsai SY, Woon HG, Weickert CS. Abnormal glucocorticoid receptor mRNA and protein isoform expression in the prefrontal cortex in psychiatric illness. Neuropsychopharmacology 2011; 36(13): 2698–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials.