

Abstract
Background:
Pancreatic cancer (PC) has an extremely high mortality rate, where obstructive jaundice due to cholestasis is a classic symptom. Conjugated bile acids (CBAs) such as taurocholic acid (TCA) have been reported to activate both the ERK1/2 and AKT signaling pathways via S1P receptor 2 (S1PR2) and promote growth of cholangiocarcinoma. Thus, we hypothesize that CBAs, which accumulate in cholestasis, accelerate PC progression via S1PR2.
Methods:
Murine Panc02-luc, and human AsPC-1, MIA PaCa2, and BxPC-3 cells were treated with TCA, S1PR2 agonist CYM5520, S1PR2 antagonist JTE-013, Sphingosine-1-phosphate (S1P), and functional S1P receptor antagonist (except S1PR2) FTY720. Bile duct ligation (BDL) was performed on liver implantation or intraperitoneal injection of Panc02-luc cells.
Results:
Panc02-luc and AsPC-1 cells predominantly expressed S1PR2, and their growth and migration were stimulated by TCA or CYM5520 in dose-dependent manner, which was blocked by JTE-013. This finding was not seen in PC cell lines expressing other S1P receptors than S1PR2. Panc02-luc growth stimulation by S1P was not blocked by FTY720. BDL significantly increased PC liver metastasis compared to sham. PC peritoneal carcinomatosis was significantly worsened by BDL confirmed by number of nodules, tumor weight, bioluminescence, Ki-67 stain, ascites, and worse survival compared to sham. CYM5520 significantly worsened PC carcinomatosis, whereas treatment with anti-S1P antibody or FTY720 also worsened the progression.
Conclusions:
CBAs accelerated growth of S1PR2 predominant PC both in vitro and in vivo. This finding implicates S1PR2 as a potential therapeutic target in metastatic S1PR2 predominant pancreatic cancer.
Introduction
Pancreatic cancer (PC) is the fourth leading cause of cancer death in the United States, with more than 57,600 estimated new cases and 24,640 estimated deaths in 20201. Prognosis is dismal, with a 9% five-year survival1. This is largely attributable to the fact that 80-85% of patients have advanced, unresectable disease at the time of diagnosis2. Thus, there is an ongoing need to identify effective systemic therapies for advanced PC, such as peritoneal carcinomatosis.
In patients with pancreatic head cancer, obstructive jaundice is the most common presentation, and the resulting cholestasis is associated with multiple metabolic abnormalities3. Cholestasis, the blockage of conjugated bile acid (CBA) flow, is not only a potential consequence of advanced pancreaticobiliary cancer but also a factor in cancer progression. As a matter of fact, bile acids have been recognized not only as detergents, but also as important signaling molecules involved in the regulation of metabolism4–6. For example, cholestasis is a well-known risk factor for the development of cholangiocarcinoma, and biliary obstruction has been shown to promote its progression in murine models7,8. Additionally, multiple studies have established a link between bile acids and the development of gastrointestinal cancer9. Bile acids activate the epidermal growth factor receptor (EGFR), resulting in increased expression of cyclooxygenase-2 (COX-2); they also decrease the expression of farnesoid X receptor (FXR), leading to increased development of hepatobiliary malignancies10,11. CBAs also promote cholangiocarcinoma growth through activation of the sphingosine-1-phosphate receptor 2 (S1PR2) via the extracellular regulated kinase (ERK)1/2 and protein kinase B (AKT) signaling pathways12.
Sphingosine-1-phosphate (S1P) is a bioactive lipid mediator which regulates cell proliferation, invasion, and angiogenesis in cancer cells13–15. For example, S1P is a key link in the relationship between chronic mucosal inflammation and colitis-associated cancer, which we previously reported16. Additionally, we have found that secreted S1P is associated with lymphangiogenesis and lymph node metastases, suggesting that it worsens cancer progression17–20. Finally, pancreatic cancer-derived S1P has been shown to activate pancreatic stellate cells which promote cancer cell growth and invasion21. S1P is generated by sphingosine kinases, SphK1 and SphK2, and functions either intracellularly, or via five different G-protein coupled receptors, S1PR-1-513,15. Our group reported that CBAs activate the cell proliferation and survival signaling pathways by acting as ligands for S1PR2 in hepatocytes6. We also discovered that CBAs activate S1PR2 in the nucleus of hepatocytes that epigenetically regulate lipid and sterol metabolism in the liver, indicating that bile acid signaling plays pivotal roles in the liver4. Notably, S1PR2 is activated specifically by taurocholic acid (TCA) and other CBAs, but not unconjugated bile acids22.
Unlike in cholangiocarcinoma, the link between obstructive jaundice in PC and activation of S1P receptors is still unclear. Based on our findings of S1PR2 stimulation by CBAs, we hypothesize that CBAs, which accumulate in obstructive jaundice, accelerate the progression of metastatic PC via S1PR2 signaling. In this study, we seek to first perform an in vitro characterization of S1P receptor expression in various human and murine pancreatic cells and determine the influence of CBAs on PC cell growth and migration. Secondly, we seek to investigate the effect of CBAs on PC progression using a murine model of obstructive jaundice in metastatic pancreatic cancer.
Materials and Methods
Cell culture
Panc02-luc cells, a murine pancreatic adenocarcinoma line which expresses the firefly luciferase 3 gene, were used in order to measure tumor growth using bioluminescence imaging (BLI), which is well-described23. Panc02-luc cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% FBS and 500 μg/mL of G418. Human pancreatic adenocarcinoma cell lines AsPC-1 and BxPC-3 (ATCC, Manassas, VA) were cultured in RPMI 1640 without phenol red with 10% FBS and 1% v/v Pen Strep (Thermo Fisher Scientific), and cell lines PANC-1 and MIA PaCa-2 (ATCC, Manassas, VA) were cultured in DMEM with 10% FBS and 1% v/v Pen Strep. Each cell line was cultured under sterile conditions, maintained at 37 °C in a mixture of 5% CO2 and 95% air.
RNA isolation and quantitative RT-PCR
Total cellular RNA was isolated using Trizol reagent (QIAGEN, Inc, Valencia, CA) and reverse-transcribed into first-strand complementary DNA (cDNA) using the High-Capacity cDNA Reverse Transcription Kit from Life Technologies. Messenger RNA (mRNA) levels of S1PRs were determined by real-time reverse transcriptase polymerase chain reaction (RT-PCR) using iQTM SYBR Green Supermix reagents and normalized using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an internal control.
Cell proliferation assays
WST assay was used to assess in vitro cell growth in response to bile acid taurocholate (TCA) from Sigma-Aldrich (St. Louis, MO), S1PR2 agonist CYM552024 (Sigma-Aldrich, St. Louis, MO), S1PR2 inhibitor JTE-013, or combinations of the above. Additionally, in vitro cell growth was assessed in response to S1P, with or without FTY720, a functional antagonist of S1P receptors 1, 3, 4, and 5. 2.5 x 103 Panc02-luc cells and 5 x 103 AsPC-1 cells were cultured in 96-well plates in serum-free medium and cultured overnight in serum-free medium. To determine the optimal concentration range of chemicals, Panc02-luc cells and AsPC-1 were serum-starved for 24 hours and then treated with different concentrations of TCA (0-100 μM) or CYM5520 for 48 hours. Viable cells were then quantified using the Cell Counting Kit-8 (CCK-8) from Dojindo Molecular Technologies, Inc. (Rockville, MD).
After the optimal reagent concentration was determined, cells were treated with either 100 μM of TCA, or 0.5 μM of CYM5520, each with and without 10 μM of JTE-013 (Cayman Chemical, Boston, MA). Other cells were treated with 100 nM S1P, with or without 5 μM of FTY720. Viable cell growth was assessed after 48 hours using CCK-8. Absorbance readings were done at A450nm with the Victor3 Multilabel Plate Counter (PerkinElmer, Waltham, MA).
Scratch migration assay
Panc02-luc (murine) or AsPC-1 (human) cells were plated at a density of 5 x 105 cells per well in six-well culture dishes and allowed to form a confluent monolayer. After a 24-hour period without serum, the cell monolayer was scratched with a sterile 200 μL pipette tip, rinsed with serum-free medium to remove detached cells, and photographed under the 10x objective of an Olympus 1X71 microscope (Olympus Corp., Center Valley, PA). Each dish, excluding controls, was then treated with either 100 μM TCA, 10 μM JTE-013, or a combination of both. Photographs were taken at 0 hours, 24 hours, and 48 hours to document cell migration. Images acquired for each treatment group were further analyzed using IPLab 4.0 imaging software (Scanalytics, Inc., Rockville, MD) to quantify cell migration.
Murine metastatic pancreatic cancer model
All animal studies were conducted in the Animal Research Core Facility at VCU School of Medicine in accordance with the institutional guidelines approved by the VCU Institutional Animal Care and Use Committee (IACUC), which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). Immunocompetent male C57Bl/6 mice between 8-12 weeks of age, weighing approximately 20-30 grams, were obtained from Jackson Laboratory (Bar Harbor, ME).
In the orthotopic liver metastasis model, 5 x 104 Panc02-luc cells in 20 μL Matrigel were inoculated into the left lobe of the liver (Fig. 1a). In the orthotopic peritoneal carcinomatosis model, 1 x 106 Panc02-luc cells in 1 mL of PBS were intraperitoneally injected. Three days after implantation, mice were weighed and tumor burden was determined by bioluminescence imaging (BLI) by the IVIS imaging system25 after d-luciferin intraperitoneal injection. The mice in each model were then randomized to two groups: sham laparotomy, or bile duct ligation (BDL) performed using our previously established technique26(Fig.1b).
Figure 1.
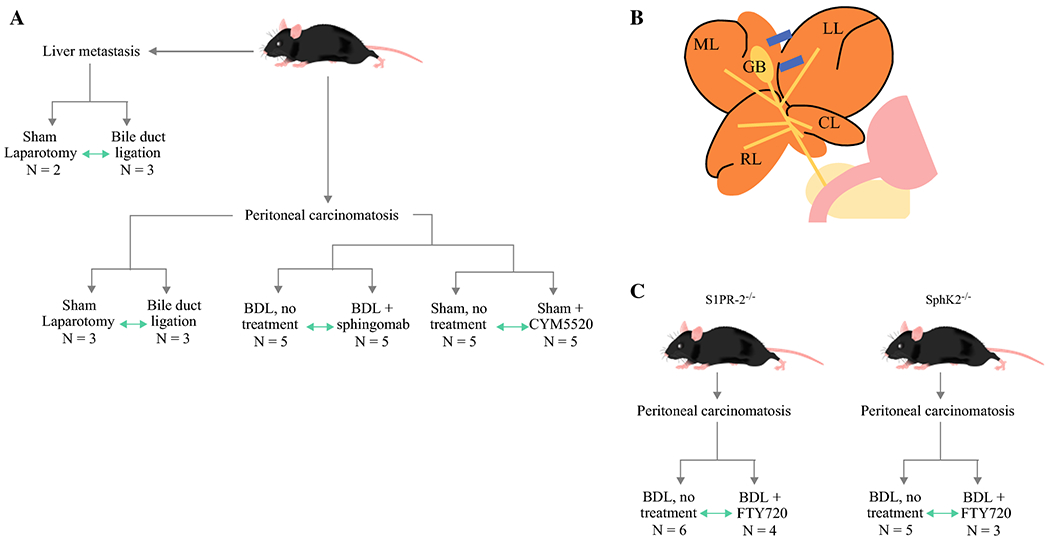
Study design and methods. a. Schematic outlining design of liver metastasis and peritoneal carcinomatosis models. Green arrows indicate direct comparison between treatment groups. b. Diagram indicating ligation position of LMHL: LL – left lobe, GB – gallbladder, ML – median lobe, RL – right lobe, CL – caudate lobe. c. Schematic of S1PR2 knockout mice and SphK2 knockout mice.
In the peritoneal carcinomatosis model, mice in the sham laparotomy group were further randomized to no treatment, or treatment with S1PR2 agonist CYM5520: 0.3 mg/kg CYM5520 in PBS was instilled intraperitoneally every 24 hours. A subset of mice in the BDL group was used in a separate study arm to assess the treatment effect of sphingomab (S1P neutralizing antibody). Mice were randomized to either 30 mg/kg of sphingomab (Lpath, San Diego, CA) in PBS injected intraperitoneally once per week, or to injections of PBS once per week as a control.
S1PR2 knockout mice (S1PR2−/−) and SphK 2 knockout mice (SphK2−/−) were a gift from Dr. R. Proia (NIDDK) and were used in a separate study arm to assess the treatment effect of FTY720, a functional antagonist of all S1P receptors except S1PR2 (Fig. 1C). As described above, Panc02-luc cells were injected into the peritoneum, and mice underwent BDL. Each group of mice (S1PR2−/− and SphK2−/−) was randomized to receive either 0.3 mg/kg of FTY720 dissolved in water with ethanol, administered orally once per day, or water/ethanol alone as a control.
Histopathologic analysis
Immediately following sacrifice, peritoneal carcinomatosis nodules were removed and fixed in 10% neutral buffered formalin for immunohistochemical analysis. Cell proliferation was determined by staining with rabbit monoclonal antibodies against Ki-67 (Dako), a nuclear protein expressed in proliferating cells.
Statistics
All data were expressed as the mean ± Standard Error. Data were analyzed for statistical significance with unpaired two-tailed Student’s t test. Survival analysis was performed using the Kaplan-Meier method and differences were assessed using the log-rank test with SPSS software (IBM SPSS statistics 22). P values < 0.05 were considered statistically significant in all analyses.
Results
S1PR2 is the predominant S1P receptor in Panc02-luc (murine) and AsPC-1 (human) pancreatic cancer (PC) cell lines, whereas it was not in MiaPaca2 and BxPC-3 cells.
First, we investigated the expression profiles of five S1P receptors in pancreatic cancer cell lines, Panc02-luc, AsPC-1, MIA PaCa-2 and BxPC-3, to identify the PC cell model that predominantly expresses S1PR2. Real-time RT-PCR was used to determine the mRNA expression levels of all five S1P receptors in both the murine and pancreatic adenocarcinoma cell lines. Figure 2 demonstrates S1PR2 to be the dominant S1P receptor expressed in murine Panc02-luc cells and human AsPC-1 cells (Fig. 2a). In the human BxPC-3 and MIA PaCa-2 cell lines, S1PR-5 was the dominantly expressed S1P receptor (Fig.2b). These results suggest that Panc02-luc and AsPC-1 cells are the appropriate models of “S1PR2 predominantly expressing” PC cells.
Figure 2.
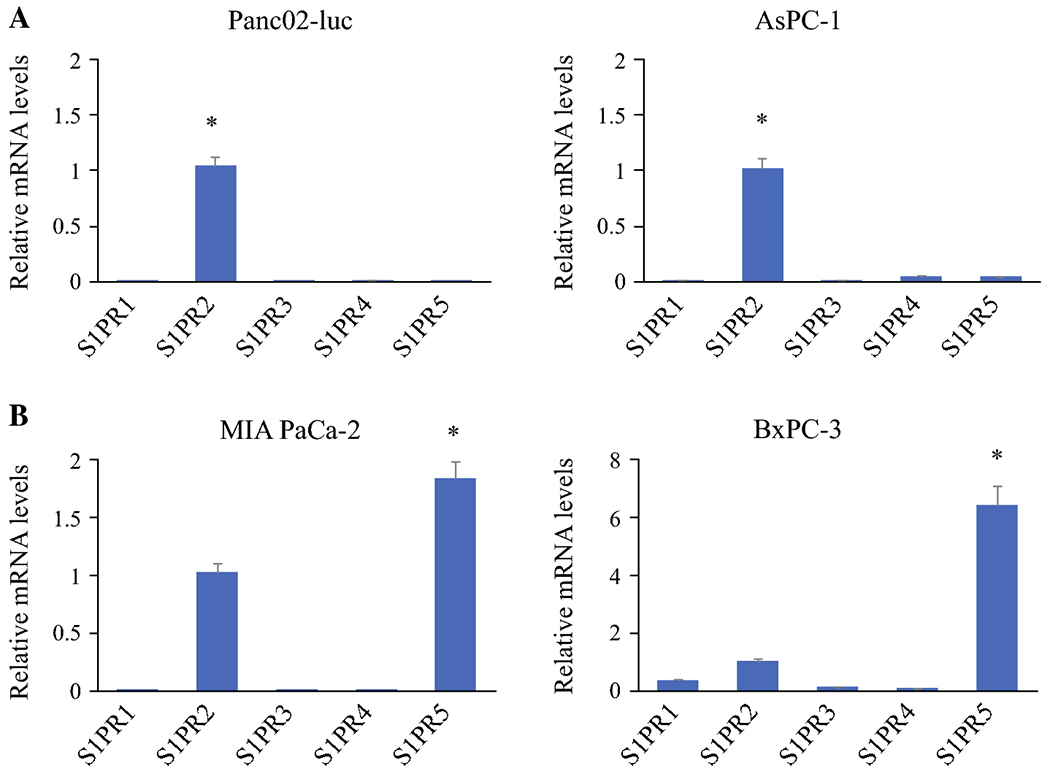
Differential expression of S1PRs in pancreatic cancer cells. mRNA levels of S1PR1, S1PR3, S1PR4 and S1PR5 relative to S1PR2 (designated =1) are shown. a. Murine Panc02-luc and AsPC-1 cells predominantly express S1PR2. b. Human MIA PaCa2 and BxPC-3 express higher levels of S1PR-5. mRNA levels of individual S1PRs were detected by real-time RTPCR and normalized using GAPDH as an internal control. *P<0.001, compared to S1PR2; n=6.
Taurocholic acid (TCA) stimulates pancreatic cancer cell growth via S1PR2 in S1PR2 predominant, but not the other PC cells
Based on our previous work that cholangiocarcinoma cells were growth stimulated with TCA via S1PR2, it was of interest whether that is the case in PC cells. As expected, TCA significantly stimulated the growth of S1PR2-expressing cell lines Panc02-luc and AsPC-1 in dose dependent manner (Fig. 3a). S1PR2 agonist, CYM5520, similarly stimulated the growth of those cells in dose-dependent manner, suggesting that growth stimulation of TCA is via S1PR2 signaling (Fig. 3b). In contrast, growth of MIA PaCa-2 and BxPC-3, which predominantly express S1PR-5, was not affected by TCA nor CYM5520 (Supplementary Fig. S1).
Figure 3.
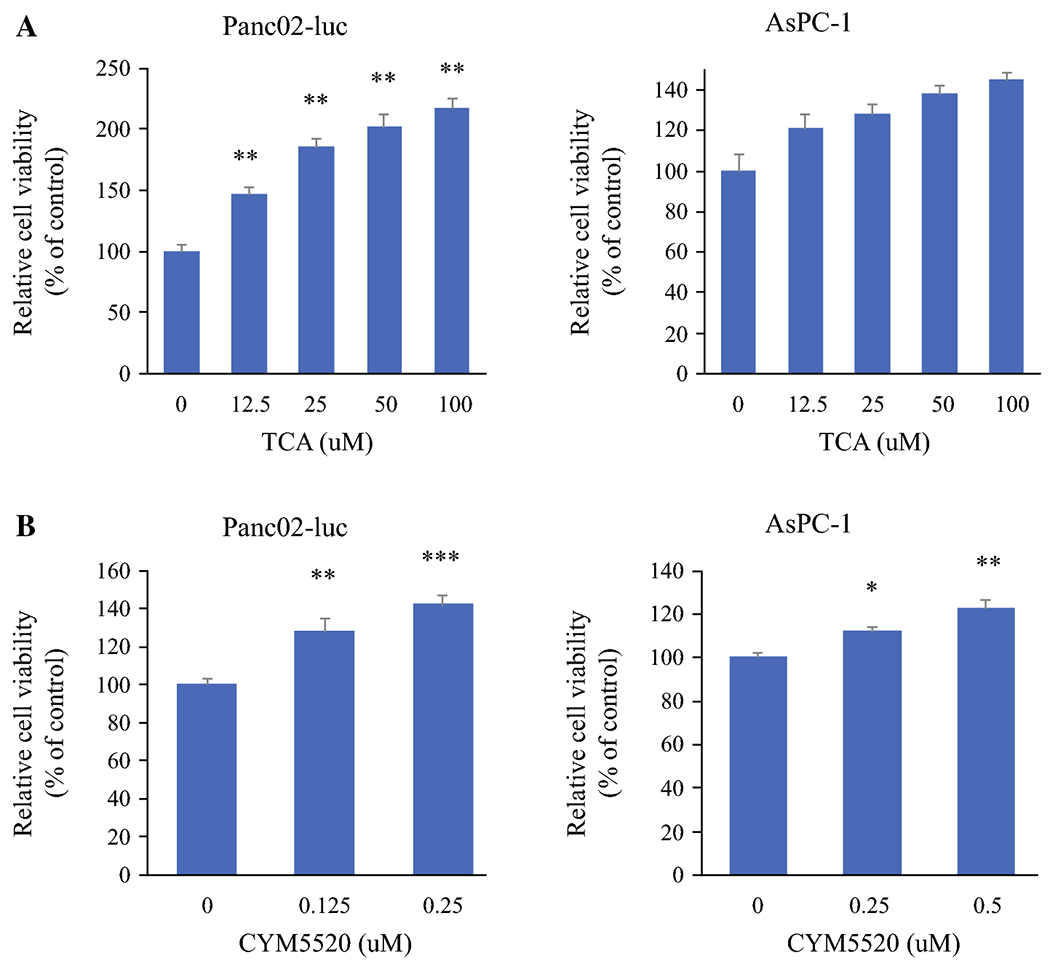
Effect of TCA and CYM5520 on cell proliferation in pancreatic cancer cells. Panc02-luc cells and AsPC-1 were serum starved for 24 hours and then treated with different concentrations of a. TCA (0-100uM), or b. CYM5520 (0-0.25 uM) for 48 hours. At the end of treatment, viable cells were quantified using CCK-8. Relative cell number, compared to control group, is shown. *P<0.05, **P<0.01; ***P<0.001, compared to vehicle control; n=8.
Further, cell growth of Panc02-luc and AsPC-1 was inhibited by S1PR2 antagonist, JTE-013, regardless of the presence or absence of TCA (Fig. 4a). Cell growth of Panc02-luc was stimulated by S1P, regardless of the presence or absence of FTY720, S1P receptor functional antagonist that spares S1PR2 (Fig. 4b). These results suggest that the growth stimulatory effect of TCA and S1P is indeed via S1PR2 in Panc02-luc and AsPC-1 cells.
Figure 4.
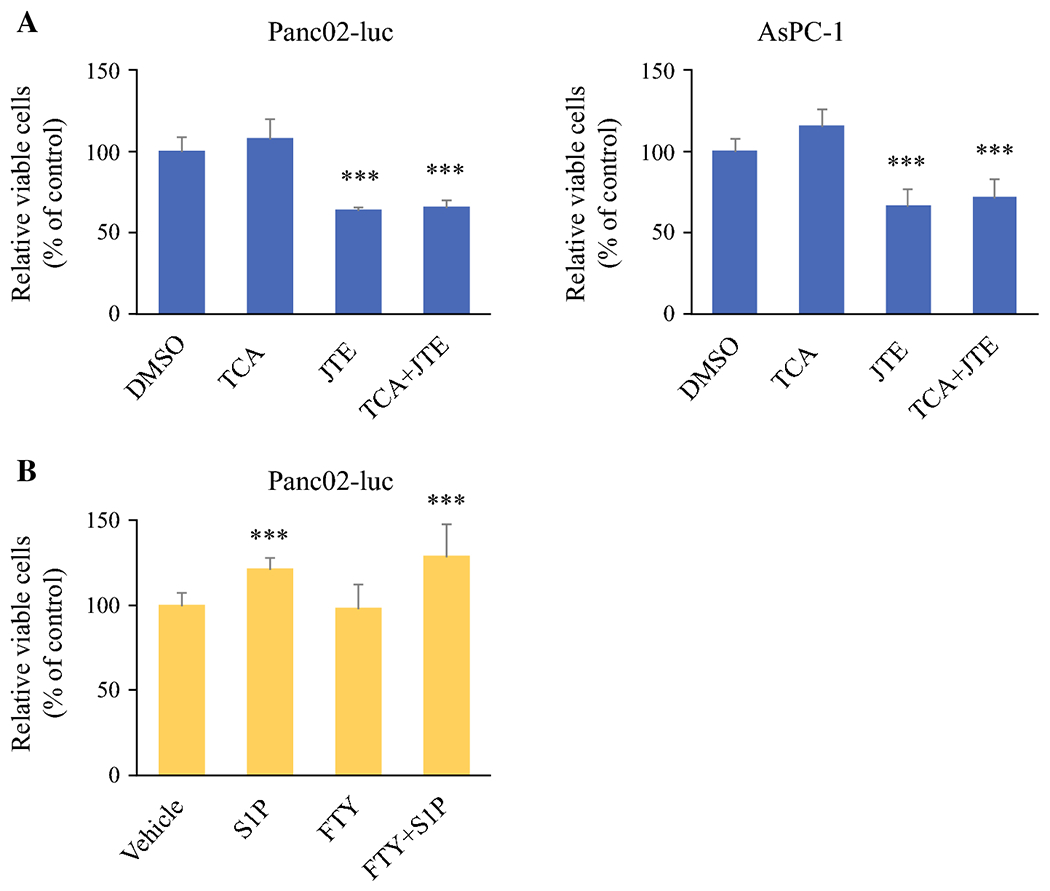
Effect of various treatments on murine and human pancreatic cancer cell viability. a. Panc02-luc and AsPC-1 cells demonstrated decreased cell viability in the presence of S1PR2 antagonist JTE-013, regardless of presence or absence of TCA. b. Panc02-luc cells demonstrated increased cell viability in the presence of S1P, regardless of presence or absence of FTY720. ***P<0.001, compared to vehicle control.
TCA induces PC cell migration via S1PR2
Given the growth stimulatory effect of TCA on S1PR2 predominant cells, it was of interest whether TCA have any effect on migration of these cells. In vitro scratch cell migration assays were performed with both Panc02-luc and AsPC-1 cells. Compared to controls, treatment with TCA produced significant cell migration or “wound closure” in both Panc02-luc and AsPC-1 cells (Fig. 5a) cell lines. Treatment with JTE-013 significantly inhibited cell migration, regardless of the presence or absence of TCA (Fig. 5b). These results suggest that TCA stimulate the migration of S1PR2 predominant cells via S1PR2 signaling, consistently in two cell lines.
Figure 5.
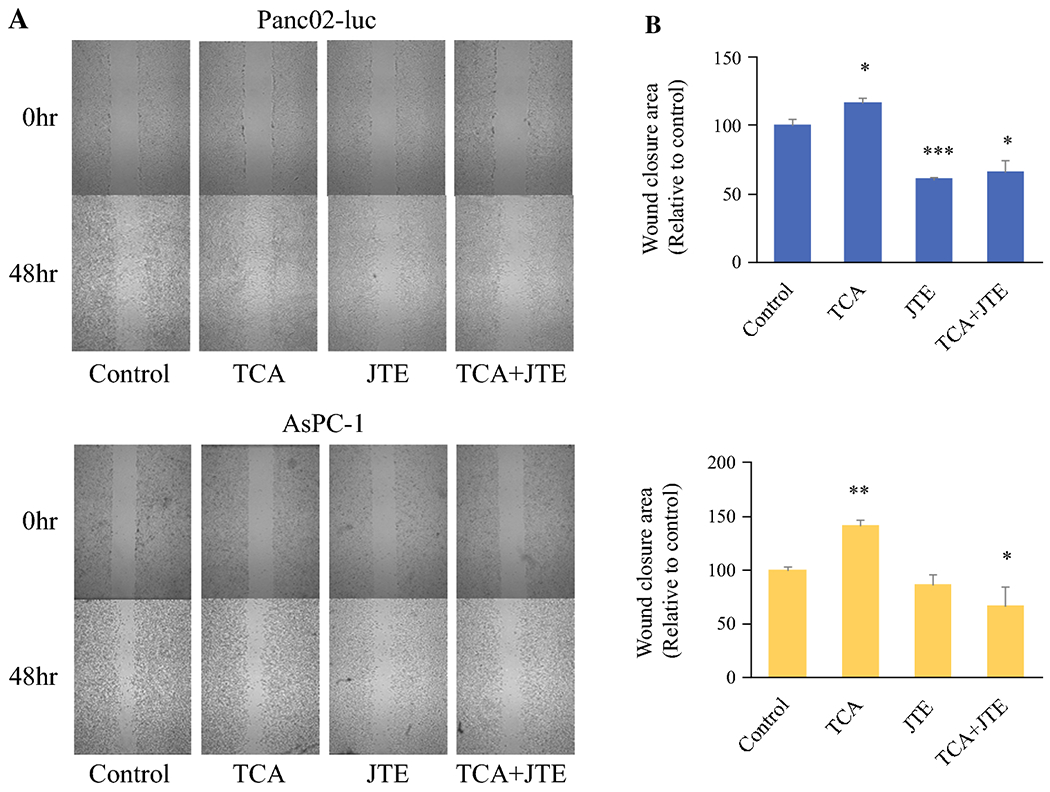
Effect of TCA and JTE-013 on murine and human pancreatic cancer cell migration. a. Murine Panc02-luc (top) and human AsPC-1 (bottom) photographs of cell migration at 0 and 48 hours in the presence of TCA, JTE-013, or combination. b. Graph of relative wound closure area in Panc02-luc (top) and AsPC-1 (bottom) cell plates. *P<0.05, **P<0.01, ***P<0.001, TCA and JTE groups compared to control; JTE+TCA group compared to TCA alone, n=6.
Cholestasis increased tumor burden in PC liver metastasis model
Given the in vitro results that TCA, one of the CBAs, stimulated growth and migration of S1PR2 predominant PC cells, it was of interest whether this translates to in vivo system. We inoculated murine Panc02-luc cells to the left lobe of immunocompetent male C57Bl/6 mice and bile duct ligation (BDL) was performed as we described previously26. Since inoculated Panc02-luc cells are fluorescent with intravenous administration of luciferin, tumor burden was assessed throughout the study using BLI. In mice who underwent pancreatic cancer cell implantation into the liver, solid tumor was revealed in the left hepatic lobe after sacrifice on day 14 (Fig. 6a). In the majority of subjects, inoculation of Panc02 cells in Matrigel generated a single bulk tumor without satellite lesions. Compared to mice who underwent sham laparotomy, BDL was associated with significantly higher tumor burden and rate of tumor growth as determined by BLI (Fig. 6b). This result suggest that cholestasis stimulate PC cell growth in vivo.
Figure 6.
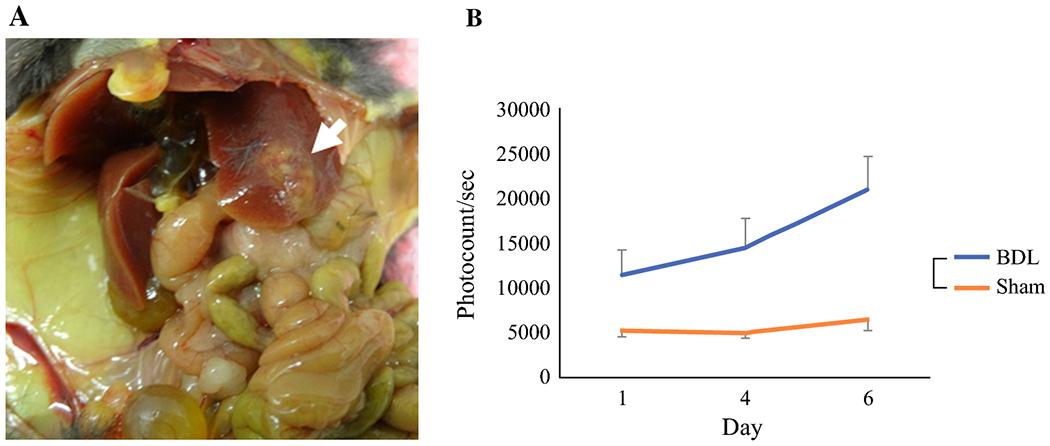
Liver metastasis model with implantation of 5 x 104 Panc02-luc cells into left lobe of liver. a. Solid tumor (white arrow) is seen in left hepatic lobe after sacrifice on day 14. b. Tumor burden determined by in vivo BLI. Mice that underwent BDL had increased tumor burden compared to sham laparotomy, n=5.
Cholestasis increased PC peritoneal tumor burden via S1PR2 signaling
The PC liver metastasis model did aggravate with cholestasis; however, given that BDL model physically obstructs the bile flow, there is a possibility that the effect of cholestasis on PC cell growth may be due to physical pressure by the obstruction rather than the chemical effect of CBA. To exclude this possibility, the effect of cholestasis on PC peritoneal carcinomatosis was investigated. Panc02-luc cells were injected into the peritoneum of immunocompetent male C57Bl/6 mice, in the same manner, we previously reported27. The presence of ascites as well as number and weight of peritoneal nodules was assessed after sacrifice on day 14 (Fig. 7a). Peritoneal metastatic nodules were noted to be scattered throughout the peritoneal cavity, mainly in the omentum, bowel mesentery and serosa, and peritoneum. Their location varied remarkably between animals such that detailed analyses, such as number of nodules on the liver surface, could not be assessed. Compared to mice who underwent sham laparotomy, BDL was associated with a statistically significant increase in number of nodules (mean 28.3 ± 2.17, compared to sham, mean 17 ± 0.82), increased in total weight of peritoneal nodules (mean 1.09 ± 0.20 g, compared with sham, mean 0.65 ± 0.15 g) as well as the percentage of mice with ascites (100% versus 33.3%) (Fig. 7b). Compared to sham, BDL was associated with a statistically significant increase in tumor burden and rate of tumor growth (Fig. 7c), increased Ki-67 expression (72% versus 53%) (Fig. 7d), and shorter survival (Fig. S2).
Figure 7.
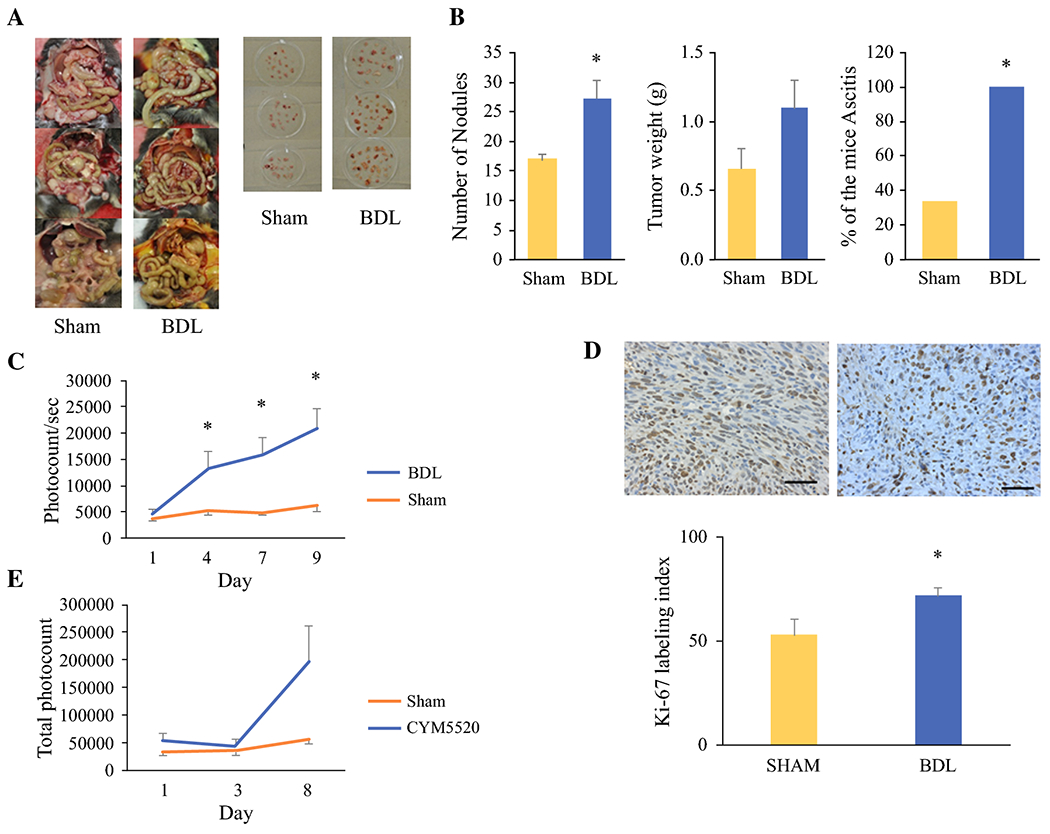
Peritoneal carcinomatosis model results. a. Post-sacrifice comparison of carcinomatosis and peritoneal nodules on day 14 after intraperitoneal injection. Jaundice is clearly visualized in the BDL group compared to sham laparotomy (left). BDL was associated with increased number and size of peritoneal nodules (right). b. Numbers of nodules, total weights of all nodules and percentage of the mice with ascites after BDL compared to sham laparotomy. c. In vivo BLI demonstrates increased tumor burden in mice following BDL. d. Paraffin-embedded tumor sections were immunostained with Ki-67 (top). Ki-67 labeling index is increased in mice following BDL (bottom). e. Effect of TCAs in BDL is mimicked by S1PR2 agonist CYM5520. Data are expressed as mean±SEM, n=5, *, P<0.05, compared with the control group.
In order to test whether worsening of PC carcinomatosis by cholestasis is due to S1PR2 signaling, the tumor burden of PC peritoneal carcinomatosis was compared between mice that underwent sham laparotomy with or without administration of S1PR2 agonist, CYM5520. Mice treated with CYM5520 demonstrated a marked increase in tumor burden and rate of tumor growth on BLI, compared to sham laparotomy alone (Fig. 7e). These results suggest that cholestasis generated by BDL aggravated PC peritoneal carcinomatosis by increasing the tumor burden via S1PR2 signaling.
Given that S1PR2 is a S1P receptor, it was of interest to investigate whether treatment with anti-S1P antibody that block S1P signaling will suppress growth stimulatory effect of cholestasis. Surprisingly, mice with peritoneal carcinomatosis treated with anti-S1P-antibody, sphingomab, showed decreased survival compared to no treatment (Fig. S3a). Further, sphingomab treatment demonstrated a significantly increased tumor burden by BLI on day 10 (Fig. S3b). This result indicates that the growth effect of cholestasis is not via S1P signaling. SphK2−/− mice are known to have higher level of S1P in the circulation due to compensation by SphK1. We found that treatment with FTY720 on SphK2−/− mice did not change the growth of PC cells (Fig. S4a). In S1PR2−/− mice treated with FTY720 compared with controls, there was a trend towards increased tumor burden by BLI, and mice who received FTY720 were found to have a higher average tumor weight after sacrifice on day 14 (Fig. S4b). These results suggest that S1PR2 in cancer cells and not the other cells are responsible for the PC cell growth.
Discussion
In this study, we demonstrated that the accumulation of CBAs, as occurs in obstructive jaundice, correlates with in vitro PC cell growth, and progression of metastatic pancreatic cancer in a murine model. Our in vitro results show that S1P receptor expression varies among human PC cell lines. Comparing the effects of TCAs, an S1PR2 agonist, and S1PR2 antagonist on these various cells suggests that activation of S1PR2 is the underlying mechanism at play. Both TCA and CYM5520 increased cell growth and migration in PC cells which predominantly expressed S1PR2, but neither had any effect on the other cells. Additionally, treatment with JTE-013 negated the effects of TCA on cell growth and migration. JTE-013 has been shown to inhibit activation of ERK1/2 and AKT by S1P and TCA6. In fact, structural modeling of the S1P receptors demonstrated that only S1PR2, and not the other S1P receptors, can accommodate TCA binding6.
Similar effects were noted in the in vivo portion of the study in our murine model of cholestasis in metastatic PC. In both our liver metastasis model and peritoneal carcinomatosis model, BDL was associated with increased tumor growth compared to sham. In the peritoneal carcinomatosis model, tumors in mice who had undergone BDL were found to have a higher Ki-67 index. This stimulating effect of cholestasis on tumor growth was mimicked in mice who had undergone sham but were then treated with CYM5520, again supporting the hypothesis that S1PR2 activation is the key step. This finding is in line with our previous work, demonstrating that CBAs activate the cell proliferation and survival signaling pathways primarily via S1PR2 in hepatocytes6. CBAs activate S1PR2 and upregulate expression of SphK2 in hepatocytes that regulate lipid and sterol metabolism4, indicating that bile acid signaling via S1PR2 and SphK2 play pivotal roles in the liver. CBAs also promote the growth and invasion of cholangiocarcinoma cells via activation of S1PR2 in vitro12, possibly via COX-2 expression28.
The development of an animal model which accurately mimics chronic cholestasis is associated with some challenges. We have utilized the left/median hepatic bile duct ligation (LMHL) model that the survival in our hands was 81% at day 1426 whereas the others reported 12% mortality at 28 weeks7. Thus, the mortality associated with BDL itself was a potential confounding factor in our survival analysis of the various treatments. While our study demonstrated decreased survival in mice who underwent BDL compared to sham, this may have been due in part to the portal hypertension and liver fibrosis that develops relatively rapidly in this model of obstructive jaundice26. Other animal models of cholestasis which have been described in the literature include bile injection, “knockout” genetic modification, chemical-induced, and viral-induced6,7,29,30, which are each associated with advantages and disadvantages. Insufficient bile production in our model prohibited bile quantification. Despite our previous experience with orthotopic implantation models26,27,31–41 and lack of access to transgenic models, we could not successfully generate a more physiologic pancreatic gland model. Differences between murine and human pancreas prohibited generation of consistent results. Further, it is of interest to investigate the duration of S1PR2 signaling and whether it is reversible, because many patients with obstructive jaundice are stented prior to therapy, and our data suggests that this practice would limit cancer progression. Unfortunately, we are unable to assess that due to technical feasibility in our current study. However, given that S1PR2 antagonist JTE-013 can reverse cholestatic liver injury, this approach can be a future consideration in our models to simulate reversal of cholestasis and S1PR2 signaling.
S1P links inflammation and cancer progression16,18,42–48. Since bile duct ligation causes inflammation, one may wonder whether tumor growth by bile duct ligation may be due to S1P production rather than cholestasis and signaling via S1PR2. To this end, we evaluated the effect of anti-S1P antibody sphingomab on tumor growth and survival, which demonstrated that this was not the case. Sphingomab has been shown to significantly reduce tumor progression with no significant toxicity15 in murine models49,50. The various S1P receptors not only have different functions compared to each other, but conflicting functions48, for example, S1PR2 has been described to play both pro- and anti-cancer roles. Tong et al. demonstrated that suppression of S1PR2 expression promotes esophageal squamous cell proliferation51. Regardless of the functions of the receptors, sphingomab blocks all S1P signaling. The humanized form of sphingomab, sonepcizumab/ASONEP, underwent phase II study in 2016 for patients with metastatic renal cell carcinoma52. Unexpectedly we found that sphingomab increased Panc02-luc tumor burden and worsened survival after BDL. Further, it is known that the cancer promoting effect of S1P is signaled mainly via S1PR1 and S1PR3, and that S1PR2 inhibits S1PR1 and S1PR313. Therefore, our results that an S1PR2 agonist aggravated cancer spread also suggest that worsening of cancer is due to cholestasis and not to S1P signaling.
There are theoretically three routes to target the SphK/S1P/S1PR signaling axis to block cancer progression: inhibitors of SphKs, antagonists of S1P receptors, and S1P-blocking antibodies49. To further explore the association between S1PR2 and PC progression, we evaluated the effects of BDL in S1PR2 knockout mice and SphK2 knockout mice with peritoneal carcinomatosis, with and without treatment with FTY720. In our previous studies using SphK deficient mice, we demonstrated that PC progression is decreased in SphK1−/− mice, due to decreased generation of S1P27, and that SphK2−/− mice caused compensation overexpression of SphK1 that result in high concentration of extracellular S1P4. Regarding S1P receptor deficiency, we found that S1PR2−/− mice had lower levels of jaundice after BDL compared to wild type mice53. This model eliminates any S1PR2 signaling in all the host cells. FTY720 is a structural analog of sphingosine, and is an immunomodulatory drug used in multiple sclerosis49. In vivo, it is phosphorylated by SphK2, and then acts as a functional antagonist at S1P receptors 1, 3, 4, and 5 (but not S1PR2) to inhibit lymphocyte egress from lymph nodes15,54. The dephosphorylated form has been reported to induce apoptosis in various types of cancer cells55–57, via S1P receptor-independent mechanisms49. We found that treatment with FTY720 negated the protective effect of S1PR2 deficiency, and S1PR2−/− mice treated with FTY720 had a significantly higher tumor weight compared with controls. This result suggests that indeed, S1PR2 expression in PC cells and not the other host cells are necessary for cholestasis-mediated PC carcinomatosis growth. In contrast, SphK2−/− mice in the same model showed no difference in tumor growth after FTY720 treatment compared to controls, which suggests that abundant circulating S1P does not increase PC cell growth via S1PR2.
In summary, we found that conjugated bile acids stimulate pancreatic cancer progression via S1PR2. Our findings suggest S1PR2 inhibition as a potential therapeutic target in S1PR2 predominant pancreatic cancer, but further studies are needed to translate this result to the clinical setting.
Supplementary Material
Synopsis:
We found that conjugated bile acids accelerated S1PR2-expressing pancreatic cancer cell growth both in vitro as well as in vivo in a murine model of obstructive jaundice. This suggests the S1PR2 as a potential therapeutic target in pancreatic cancer treatment.
Grant support
This research was supported by National Institutes of Health, USA grant number R01CA160688, R37CA248018, R01CA250412, R01CA251545, R01EB029596, as well as US Department of Defense BCRP grant number W81XWH-19-1-0674 and W81XWH-19-1-0111 to K. Takabe; VA Merit Award I01BX004033, Research Career Scientist Award (IK6BX004477), VA ShEEP Grant (1 IS1 BX004777), National Institutes of Health Grant R01 DK104893 and R01DK-057543 to H. Zhou; National Cancer Institute, cancer center support grant P30CA016056 to Roswell Park Comprehensive Cancer Center.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors have no potential conflicts of interest to disclose.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. Jan 2020;70(1):7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. Aug 13 2011;378(9791):607–620. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Gaag NA, Kloek JJ, de Castro SM, Busch OR, van Gulik TM, Gouma DJ. Preoperative biliary drainage in patients with obstructive jaundice: history and current status. J Gastrointest Surg. Apr 2009;13(4):814–820. doi: 10.1007/s11605-008-0618-4. [DOI] [PubMed] [Google Scholar]
- 4.Nagahashi M, Takabe K, Liu R, et al. Conjugated bile acid-activated S1P receptor 2 is a key regulator of sphingosine kinase 2 and hepatic gene expression. Hepatology. Apr 2015;61(4):1216–1226. doi: 10.1002/hep.27592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park MA, Zhang G, Norris J, et al. Regulation of autophagy by ceramide-CD95-PERK signaling. Autophagy. Oct 2008;4(7):929–931. doi: 10.4161/auto.6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Studer E, Zhou X, Zhao R, et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology. Jan 2012;55(1):267–276. doi: 10.1002/hep.24681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang H, Li TW, Peng J, et al. A mouse model of cholestasis-associated cholangiocarcinoma and transcription factors involved in progression. Gastroenterology. Jul 2011;141(1):378–388, 388 e371-374. doi: 10.1053/j.gastro.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Labib PL, Goodchild G, Pereira SP. Molecular Pathogenesis of Cholangiocarcinoma. BMC Cancer. Feb 28 2019;19(1):185. doi: 10.1186/s12885-019-5391-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernstein H, Bernstein C, Payne CM, Dvorak K. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J Gastroenterol. Jul 21 2009;15(27):3329–3340. doi: 10.3748/wjg.15.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dai J, Wang H, Shi Y, Dong Y, Zhang Y, Wang J. Impact of bile acids on the growth of human cholangiocarcinoma via FXR. J Hematol Oncol. Oct 12 2011;4:41. doi: 10.1186/1756-8722-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maroni L, Alpini G, Marzioni M. Cholangiocarcinoma development: the resurgence of bile acids. Hepatology. Sep 2014;60(3):795–797. doi: 10.1002/hep.27223. [DOI] [PubMed] [Google Scholar]
- 12.Liu R, Zhao R, Zhou X, et al. Conjugated bile acids promote cholangiocarcinoma cell invasive growth through activation of sphingosine 1-phosphate receptor 2. Hepatology. Sep 2014;60(3):908–918. doi: 10.1002/hep.27085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takabe K, Paugh SW, Milstien S, Spiegel S. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. Jun 2008;60(2):181–195. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takabe K, Spiegel S. Export of sphingosine-1-phosphate and cancer progression. J Lipid Res. Sep 2014;55(9):1839–1846. doi: 10.1194/jlr.R046656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sabbadini RA. Sphingosine-1-phosphate antibodies as potential agents in the treatment of cancer and age-related macular degeneration. Br J Pharmacol. Mar 2011;162(6):1225–1238. doi: 10.1111/j.1476-5381.2010.01118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang J, Nagahashi M, Kim EY, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. Jan 14 2013;23(1):107–120. doi: 10.1016/j.ccr.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aoyagi T, Nagahashi M, Yamada A, Takabe K. The role of sphingosine-1-phosphate in breast cancer tumor-induced lymphangiogenesis. Lymphat Res Biol. Sep 2012;10(3):97–106. doi: 10.1089/lrb.2012.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang WC, Nagahashi M, Terracina KP, Takabe K. Emerging Role of Sphingosine-1-phosphate in Inflammation, Cancer, and Lymphangiogenesis. Biomolecules. 2013;3(3). doi: 10.3390/biom3030408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagahashi M, Ramachandran S, Kim EY, et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res. Feb 1 2012;72(3):726–735. doi: 10.1158/0008-5472.CAN-11-2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takabe K, Yamada A, Rashid OM, et al. Twofer anti-vascular therapy targeting sphingosine-1-phosphate for breast cancer. Gland Surg. Aug 1 2012;1(2):80–83. doi: 10.3978/j.issn.2227-684X.2012.07.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bi Y, Li J, Ji B, et al. Sphingosine-1-phosphate mediates a reciprocal signaling pathway between stellate cells and cancer cells that promotes pancreatic cancer growth. Am J Pathol. Oct 2014;184(10):2791–2802. doi: 10.1016/j.ajpath.2014.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagahashi M, Yuza K, Hirose Y, et al. The roles of bile acids and sphingosine-1-phosphate signaling in the hepatobiliary diseases. J Lipid Res. Sep 2016;57(9):1636–1643. doi: 10.1194/jlr.R069286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Little EC, Wang C, Watson PM, Watson DK, Cole DJ, Camp ER. Novel immunocompetent murine models representing advanced local and metastatic pancreatic cancer. J Surg Res. Aug 2012;176(2):359–366. doi: 10.1016/j.jss.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Satsu H, Schaeffer MT, Guerrero M, et al. A sphingosine 1-phosphate receptor 2 selective allosteric agonist. Bioorg Med Chem. Sep 1 2013;21(17):5373–5382. doi: 10.1016/j.bmc.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun A, Hou L, Prugpichailers T, et al. Firefly luciferase-based dynamic bioluminescence imaging: a noninvasive technique to assess tumor angiogenesis. Neurosurgery. Apr 2010;66(4):751–757; discussion 757. doi: 10.1227/01.NEU.0000367452.37534.B1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aoki H, Aoki M, Yang J, et al. Murine model of long-term obstructive jaundice. J Surg Res. Nov 2016;206(1):118–125. doi: 10.1016/j.jss.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aoki H, Aoki M, Katsuta E, et al. Host sphingosine kinase 1 worsens pancreatic cancer peritoneal carcinomatosis. J Surg Res. Oct 2016;205(2):510–517. doi: 10.1016/j.jss.2016.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu R, Li X, Qiang X, et al. Taurocholate Induces Cyclooxygenase-2 Expression via the Sphingosine 1-phosphate Receptor 2 in a Human Cholangiocarcinoma Cell Line. J Biol Chem. Dec 25 2015;290(52):30988–31002. doi: 10.1074/jbc.M115.668277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Z, Wu Y, Wang B, et al. Intrahepatic cholestasis induced by α-naphthylisothiocyanate can cause gut-liver axis disorders. Environ Toxicol Pharmacol. Aug 2021;86:103672. doi: 10.1016/j.etap.2021.103672. [DOI] [PubMed] [Google Scholar]
- 30.Berntsen NL, Fosby B, Valestrand L, et al. Establishment of a surgical bile duct injection technique giving direct access to the bile ducts for studies of the murine biliary tree. Am J Physiol Gastrointest Liver Physiol. Mar 1 2018;314(3):G349–g359. doi: 10.1152/ajpgi.00124.2017. [DOI] [PubMed] [Google Scholar]
- 31.Katsuta E, DeMasi SC, Terracina KP, et al. Modified breast cancer model for preclinical immunotherapy studies. J Surg Res. Aug 2016;204(2):467–474. doi: 10.1016/j.jss.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katsuta E, Oshi M, Rashid OM, Takabe K. Generating a Murine Orthotopic Metastatic Breast Cancer Model and Performing Murine Radical Mastectomy. J Vis Exp. Nov 29 2018(141). doi: 10.3791/57849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katsuta E, Rashid OM, Takabe K. Murine breast cancer mastectomy model that predicts patient outcomes for drug development. J Surg Res. Nov 2017;219:310–318. doi: 10.1016/j.jss.2017.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katsuta E, Rashid OM, Takabe K. Clinical relevance of tumor microenvironment: immune cells, vessels, and mouse models. Hum Cell. Oct 2020;33(4):930–937. doi: 10.1007/s13577-020-00380-4. [DOI] [PubMed] [Google Scholar]
- 35.Kawaguchi T, Foster BA, Young J, Takabe K. Current Update of Patient-Derived Xenograft Model for Translational Breast Cancer Research. J Mammary Gland Biol Neoplasia. Jun 2017;22(2):131–139. doi: 10.1007/s10911-017-9378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okano M, Oshi M, Butash A, et al. Orthotopic Implantation Achieves Better Engraftment and Faster Growth Than Subcutaneous Implantation in Breast Cancer Patient-Derived Xenografts. J Mammary Gland Biol Neoplasia. Mar 2020;25(1):27–36. doi: 10.1007/s10911-020-09442-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oshi M, Okano M, Maiti A, et al. Novel Breast Cancer Brain Metastasis Patient-Derived Orthotopic Xenograft Model for Preclinical Studies. Cancers (Basel). Feb 14 2020;12(2). doi: 10.3390/cancers12020444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rashid OM, Nagahashi M, Ramachandran S, et al. An improved syngeneic orthotopic murine model of human breast cancer progression. Breast Cancer Res Treat. Oct 2014;147(3):501–512. doi: 10.1007/s10549-014-3118-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rashid OM, Nagahashi M, Ramachandran S, et al. Is tail vein injection a relevant breast cancer lung metastasis model? J Thorac Dis. Aug 2013;5(4):385–392. doi: 10.3978/j.issn.2072-1439.2013.06.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rashid OM, Takabe K. Animal models for exploring the pharmacokinetics of breast cancer therapies. Expert Opin Drug Metab Toxicol. Feb 2015;11(2):221–230. doi: 10.1517/17425255.2015.983073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Terracina KP, Aoyagi T, Huang WC, et al. Development of a metastatic murine colon cancer model. J Surg Res. Nov 2015;199(1):106–114. doi: 10.1016/j.jss.2015.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ikarashi M, Tsuchida J, Nagahashi M, et al. Plasma Sphingosine-1-Phosphate Levels Are Associated with Progression of Estrogen Receptor-Positive Breast Cancer. Int J Mol Sci. Dec 13 2021;22(24). doi: 10.3390/ijms222413367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miura K, Nagahashi M, Prasoon P, et al. Dysregulation of sphingolipid metabolic enzymes leads to high levels of sphingosine-1-phosphate and ceramide in human hepatocellular carcinoma. Hepatol Res. May 2021;51(5):614–626. doi: 10.1111/hepr.13625. [DOI] [PubMed] [Google Scholar]
- 44.Nagahashi M, Abe M, Sakimura K, Takabe K, Wakai T. The role of sphingosine-1-phosphate in inflammation and cancer progression. Cancer Sci. Dec 2018;109(12):3671–3678. doi: 10.1111/cas.13802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oshi M, Newman S, Tokumaru Y, et al. Intra-Tumoral Angiogenesis Is Associated with Inflammation, Immune Reaction and Metastatic Recurrence in Breast Cancer. Int J Mol Sci. Sep 13 2020;21(18). doi: 10.3390/ijms21186708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Satyananda V, Oshi M, Tokumaru Y, et al. Sphingosine 1-phosphate (S1P) produced by sphingosine kinase 1 (SphK1) and exported via ABCC1 is related to hepatocellular carcinoma (HCC) progression. Am J Cancer Res. 2021;11(9):4394–4407. https://www.ncbi.nlm.nih.gov/pubmed/34659894. [PMC free article] [PubMed] [Google Scholar]
- 47.Tsuchida J, Nagahashi M, Nakajima M, et al. Sphingosine Kinase 1 is Associated With Immune Cell-Related Gene Expressions in Human Breast Cancer. J Surg Res. Dec 2020;256:645–656. doi: 10.1016/j.jss.2020.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang P, Yuan Y, Lin W, Zhong H, Xu K, Qi X. Roles of sphingosine-1-phosphate signaling in cancer. Cancer Cell Int. 2019;19:295. doi: 10.1186/s12935-019-1014-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gupta P, Taiyab A, Hussain A, Alajmi MF, Islam A, Hassan MI. Targeting the Sphingosine Kinase/Sphingosine-1-Phosphate Signaling Axis in Drug Discovery for Cancer Therapy. Cancers (Basel). Apr 15 2021;13(8). doi: 10.3390/cancers13081898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Visentin B, Vekich JA, Sibbald BJ, et al. Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell. Mar 2006;9(3):225–238. doi: 10.1016/j.ccr.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 51.Tong S, Chen SC, Xu KY, Fang B, Wang SH, Wang JJ. 14-3-3zeta promotes esophageal squamous cell carcinoma invasion by repressing S1PR2 protein expression through NF-kappaB signaling. Arch Biochem Biophys. Apr 2 2018;643:7–13. doi: 10.1016/j.abb.2018.02.009. [DOI] [PubMed] [Google Scholar]
- 52.Pal SK, Drabkin HA, Reeves JA, et al. A phase 2 study of the sphingosine-1-phosphate antibody sonepcizumab in patients with metastatic renal cell carcinoma. Cancer. Feb 15 2017;123(4):576–582. doi: 10.1002/cncr.30393. [DOI] [PubMed] [Google Scholar]
- 53.Wang Y, Aoki H, Yang J, et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology. Jun 2017;65(6):2005–2018. doi: 10.1002/hep.29076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brinkmann V, Billich A, Baumruker T, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. Nov 2010;9(11):883–897. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- 55.Azuma H, Takahara S, Horie S, Muto S, Otsuki Y, Katsuoka Y. Induction of apoptosis in human bladder cancer cells in vitro and in vivo caused by FTY720 treatment. J Urol. Jun 2003;169(6):2372–2377. doi: 10.1097/01.ju.0000064938.32318.91. [DOI] [PubMed] [Google Scholar]
- 56.Bai LY, Chiu CF, Chiu SJ, Chu PC, Weng JR. FTY720 Induces Autophagy-Associated Apoptosis in Human Oral Squamous Carcinoma Cells, in Part, through a Reactive Oxygen Species/Mcl-1-Dependent Mechanism. Sci Rep Jul 17 2017;7(1):5600. doi: 10.1038/s41598-017-06047-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lucas da Silva LB, Ribeiro DA, Cury PM, Cordeiro JA, Bueno V. FTY720 treatment in experimentally urethane-induced lung tumors. J Exp Ther Oncol. 2008;7(1):9–15. https://www.ncbi.nlm.nih.gov/pubmed/18472638. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.