

Abstract
The Geometroidea is a large superfamily of Lepidoptera in species composition and contains numerous economically important pest species that cause great loss in crop and forest production. However, understanding of mitogenomes remains limited due to relatively fewer mitogenomes previously reported for this megadiverse group. Here, we sequenced and annotated nine mitogenomes for Geometridae and further analyzed the mitogenomic evolution and phylogeny of the whole superfamily. All nine mitogenomes contained 37 mitochondrial genes typical in insects, and gene organization was conserved except for Somatina indicataria. In S. indicataria, the positions of two tRNAs were rearranged. The trnR was located before trnA instead of after trnA typical in Lepidoptera, whereas the trnE was detected rarely on the minority strand (N‐strand). This trnR‐trnA‐trnN‐trnS1‐ trnE ‐ trnF newly recognized in S. indicataria represents the first gene rearrangement reported for Geometroidea and is also unique in Lepidoptera. Besides, nucleotide composition analyses showed little heterogeneity among the four geometrid subfamilies involved herein, and overall, nad6 and atp8 have higher nucleotide diversity and Ka/Ks rate in Geometridae. In addition, the taxonomic assignments of the nine species, historically defined by morphological studies, were confirmed by various phylogenetic analyses based on the hitherto most extensive mitogenomic sampling in Geometroidea.
Keywords: gene rearrangement, Geometridae, mitochondrial genome, phylogeny
In this study, we determined nine mitochondrial genomes for the Geometridae for the first time. A tRNA gene rearrangement was recognized in S. indicataria, which represents the first gene rearrangement reported for Geometroidea and is also unique in Lepidoptera. The mitochondrial genomes of the Geometridae were comparatively analyzed, and various phylogenetic analyses were conducted based on the hitherto most extensive mitogenomic sampling in Geometroidea, which could facilitate the understanding of Geometroidea phylogeny.
1. INTRODUCTION
A typical arthropod mitochondrial genome (mitogenome) is circular and generally consists of 13 protein‐coding genes (PCGs), two ribosomal RNA genes (rRNAs), and 22 transfer RNA genes (tRNAs) (Cameron, 2014; Curole & Kocher, 1999). Besides, several noncoding elements, including the control region that regulates the replication and transcription of the mitogenome, are present (Boore, 1999). The mitogenome is characterized by a series of features such as cellular abundance, absence of introns, and a lack of extensive recombination, and thus, they represent one of the important molecular markers, such as the standard cox1 barcode sequence, used in studies on species identification and population genetics of insects, especially for the megadiverse Lepidoptera (Hajibabaei et al., 2006; Hebert et al., 2003, 2004). In recent years, with the decline of sequencing cost, increasing numbers of the whole mitogenomes have been sequenced and widely used in not only species identification and delimitation but also phylogeny and population genetics of the Lepidoptera and other insect groups (e.g., Du et al., 2019; Timmermans et al., 2014; Yang et al., 2015; Yang, Song, et al., 2019). In addition, mitochondrial gene arrangement also represents one kind of important information to infer evolutionary relationships of insects. For instance, the gene arrangement of trnM‐trnI‐ trnQ (the gene underlined is located on the minority strand) is regarded as a synapomorphy for Ditrysia in contrast to some other groups of Lepidoptera such as Adeloidea and Nepticuloidea and ancestral insect orders with the trnM‐ trnQ ‐trnI instead (Timmermans et al., 2014). However, although Lepidoptera is one of the species‐rich orders in insects, gene rearrangement events have been less reported for this group in comparison with some other orders especially the Hemiptera (e.g., Thao et al., 2004) and Hymenoptera (e.g., Tang et al., 2019) albeit with relatively lower species diversity.
The Geometroidea is one of the largest superfamilies in Lepidoptera and includes more than 24,000 described extant species (van Nieukerken et al., 2011). As leaf feeders, they feed on multiple kinds of typically woody plants, thus often causing a huge loss in agricultural and forest production (Li et al., 2018; Mitter et al., 2017). Three families, i.e., Geometridae, Uraniidae, and Sematuridae, had been defined for Geometroidea before (Mitter et al., 2017). Later, molecular evidence strongly suggested the inclusion of Epicopeiidae historically from Drepanoidea (Bazinet et al., 2013; Regier et al., 2013; Yang, Zhang, et al., 2019), and a new family Pseudobistonidae (Rajaei et al., 2015; Wang et al., 2019). To date, the relationship among the five families has been recovered as ((Uraniidae, Geometridae), (Sematuridae, (Epicopeiidae, Pseudobistonidae))) by most of the previous studies, but this topology needs to be confirmed because the clade consisting of Sematuridae, Epicopeiidae, and Pseudobistonidae has been either lowly supported (Rajaei et al., 2015; Wang et al., 2019) or sparsely sampled (Murillo‐Ramos et al., 2019) in previous studies.
In Geometroidea, mitogenomes of approximately 27 species from three families have been sequenced to date (GenBank, November 2021). This number is obviously disproportional relative to the huge species diversity of this superfamily. Moreover, among the three families, the reported mitogenomes of both the Epicopeiidae and Uraniidae were represented by one species. Given their wide application in the molecular systematics of insects, this situation will hinder the progress of investigating Geometroidea systematics using mitogenome data. Based on the existing mitogenomes, comparative analysis among geometroid members or/and deep phylogenetic analyses (e.g., Du et al., 2019; Yang et al., 2013; Yang, Song, et al., 2019) was conducted, which greatly further our understanding of phylogeny of this superfamily and related groups. In addition, the mitogenome sequences were also used to infer the phylogenetic relationships of three closely‐related Biston species in Geometridae, suggesting the existence of the budding speciation in these species (Cheng et al., 2017). In terms of gene arrangement, all the mitochondrial genomes published available show identical gene organization that is typical in Lepidoptera, and no gene rearrangement events have been reported for Geometroidea to date.
In this study, the mitogenomes of nine additional geometrid species were sequenced, annotated, and comparatively analyzed, aiming to increase the reported mitogenome diversity of Geometroidea and to improve our understanding of mitogenome evolution in this superfamily. Also, these data can provide mitogenome data for other studies on molecular systematics of Geometroidea. Among the nine species, Somatina indicataria showed a gene rearrangement of trnR‐trnA‐trnN‐trnS1‐ trnE ‐ trnF relative to the trnA‐trnR‐trnN‐trnS1‐trnE‐ trnF typical in Lepidoptera, which represents the first gene rearrangement reported for Geometroidea and is also unique in Lepidoptera.
2. MATERIALS AND METHODS
2.1. Samples, DNA extraction, and mitogenome sequencing
Adult moths were collected by light trap, at Mountain Jigongshan and Lushan country of Henan Province in China, from July to August 2020. Each specimen was identified through morphology and by blasting the standard mitochondrial cox1 barcode on the GenBank database. After identification, nine species from Geometridae of Geometroidea were selected, of which six from Ennominae, two from Geometrinae, and one from Sterrhinae, mainly because of the high species diversity of Ennominae and lack of existing mitogenomes of Geometrinae and Sterrhinae. Detailed specimen information is shown in Table S1, and voucher specimens are deposited in the Biology Laboratory of Zhoukou Normal University, China. In phylogenetic analyses, two other mitogenome sequences were retrieved from transcriptomes of Mania lunus (SRR1695439) and Calledapteryx dryopterata (SRR1021601) available on GenBank, which represented the Sematuridae and Uraniidae, respectively, together with other available mitogenomes to perform phylogenetic analyses of the Geometroidea.
Total genomic DNA was extracted from thoracic tissue isolated from a single specimen using DNeasy tissue kit (Qiagen, Germany), following the manufacturer's instructions. Nine libraries (each for one species) were constructed with TruSeq DNA PCR‐Free Sample Preparation Kit (Illumina, United States), and sequencing was conducted using an Illumina HiSeq 2500 platform with a strategy of 150 paired‐ends.
2.2. Mitogenome assembly and annotation
Raw sequences were checked for quality control using the FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Then, the Adapter Removal 2 (Schubert et al., 2016) and SOAPec 2.0.1 in SOAPdenovo 2.01 software package (Luo et al., 2012) were employed to filter these raw sequences to obtain clean paired reads. Next, we assembled mitogenome from clean paired reads using the Geneious R11 (Kearse et al., 2012). In this analysis, the “map to reference” strategy was selected to map all cleaned reads to an “anchor” of standard mitochondrial cox1 barcoding sequence that was amplified earlier using insect primer pair Lco1490 (F) and Hco2198 (R) (Folmer et al., 1994). After iteration up to 100 times with custom sensitivity, a target contig sequence with high coverage was generated. Lastly, the MEGA X (Kumar et al., 2018) was used to check the beginning and end of the contig sequence to circularize a complete mitochondrial genome after deleting the overlapping sequence.
The mitogenome sequence was annotated using MITOS2 webserver (Donath et al., 2019) with invertebrate genetic code. Gene boundaries were confirmed by aligning the gene sequence of the new mitogenome with that of previously reported geometrid mitogenomes available on GenBank with MEGA X (Kumar et al., 2018). The circular maps of the nine mitogenomes generated in this study were comparatively present using the CGView Comparison Tool (Grant et al., 2012). In addition, two species, Mania lunus and Calledapteryx dryopterata, belonging to Sematuridae and Uraniidae, respectively, were added to the phylogenetic analyses of this study. Mitogenomes of the two species were assembled using the same methods with that of the nine species, from their transcriptomes deposited on GenBank (accession numbers SRR1695439 and SRR1021601).
2.3. Sequence alignment and analyses
A total of 38 mitogenomes of Geometroidea were compiled and analyzed, including nine newly sequenced in the present study, two retrieved from transcriptomes publicly published, and 27 downloaded from GenBank. In addition, mitogenomes of 13 species from Noctuoidea, Bombycoidea, Lasiocampoide, Drepanoidea, and Mimallonoidea that represent the close relatives of the Geometroidea were selected as outgroup sequences in phylogenetic analyses (Table 1).
TABLE 1.
The species used in phylogenetic analyses.
| Superfamily | Family | Subfamily | Species | GenBank accession number | Mitogenome size (bp) |
|---|---|---|---|---|---|
| Geometroidea | Geometridae | Ennominae | Abraxas suspecta | NC_034804 | 15,537 |
| A. latifasciata | MK962622 | 15,794 | |||
| Apocheima cinerarium | NC_024824 | 15,722 | |||
| A. cinerarius | KR478686 | 15,661 | |||
| Biston thoracicaria | MN956510 | 15,538 | |||
| B. panterinaria | NC_020004 | 15,517 | |||
| B. perclara. | NC_030769 | 15,493 | |||
| B. suppressaria | NC_027111 | 15,628 | |||
| B. thibetaria | NC_030632 | 15,485 | |||
| Ectropis grisescens | MW337302 | 15,794 | |||
| E. obliqua | NC_036717 | 16,535 | |||
| Hypomecis punctinalis | MK903031 | 15,648 | |||
| Milionia basalis | MN495623 | 15,901 | |||
| Semiothisa cinerearia | MK880228 | 15,523 | |||
| Hydatocapnia marginata | MZ902340 | 15,615 | |||
| Luxiaria mitorrhaphes | MZ902343 | 15,340 | |||
| Menophra senilis | MZ902337 | 15,250 | |||
| Ophthalmitis albosignaria | MZ902339 | 15,559 | |||
| Amraica recursaria | MZ902338 | 15,582 | |||
| Cotta incongruaria | MZ902341 | 15,487 | |||
| Celenna sp. | KM244697 | 15,403 | |||
| Erannis ankeraria | NC_047212 | 15,250 | |||
| Jankowskia athleta | NC_027948 | 15,534 | |||
| Phthonandria atrilineata | NC_010522 | 15,499 | |||
| Larentiinae | Dysstroma truncata | KJ508061 | 15,828 | ||
| Hydrelia parvulata | MN962739 | 15,407 | |||
| Pasiphila chloerata | MN598218 | 15,602 | |||
| Operophtera brumata | NC_027723 | 15,748 | |||
| Geometrinae | Iotaphora admirabilis | NC_056092 | 16,140 | ||
| Pingasa rufofasciata | MZ902335 | 15,064 | |||
| Lophophelma iterans | MZ902342 | 15,545 | |||
| Sterrhinae | Idaea simplicior | MN715151 | 15,950 | ||
| I. effusaria | MN646772 | 16,161 | |||
| Somatina indicataria | MZ902336 | 15,723 | |||
| Epicopeiidae | Epicopeia hainesii | MK033610 | 15,395 | ||
| Sematuridae | Mania lunus | SRR1695439 | |||
| Uraniidae | Calledapteryx dryopterata | SRR1021601 | |||
| Uraniinae | Lyssa zampa | MW435592 | 15,314 | ||
| Noctuoidea | Erebidae | Erebinae | Eudocima phalonia | KY196412 | 15,575 |
| Arctiinae | Hyphantria cunea | GU592049 | 15,481 | ||
| Noctuidae | Hadeninae | Mythimna separata | KM099034 | 15,329 | |
| Nolidae | Risobinae | Risoba prominens | KJ396197 | 15,343 | |
| Notodontidae | Phalerinae | Phalera flavescens | JF440342 | 15,659 | |
| Bombycoidea | Sphingidae | Macroglossinae | Ampelophaga rubiginosa | KT153024 | 15,282 |
| Sphinginae | Sphinx morio | KC470083 | 15,299 | ||
| Saturniidae | Saturniinae | Eriogyna pyretorum | FJ685653 | 15,327 | |
| Bombycidae | Bombycinae | Bombyx mori | GU966614 | 15,656 | |
| Endromidae | Prismostictoides unihyala | MF100146 | 15,355 | ||
| Lasiocampoidea | Lasiocampidae | Dendrolimus kikuchii | MF100138 | 15,382 | |
| Drepanoidea | Drepanidae | Drepaninae | Drepana arcuata | KJ508053 | 15,302 |
| Mimallonoidea | Mimallonidae | Lacosoma valva | KJ508050 | 16,108 |
Note: The species with newly sequenced mitogenome was emphasized in bold.
Among 37 mitochondrial genes, 13 PCGs were individually aligned using the MUSCLE method in the TranslatorX online platform (Abascal et al., 2010) after the sequences were translated with an invertebrate genetic code. Two rRNAs and 22 tRNAs were independently aligned with Q‐INS‐i algorithm as implemented in the MAFFT online platform (Katoh et al., 2019). Further, the aligned tRNA and rRNA sequences were filtered using ClipKIT (Steenwyk et al., 2020) to delete ambiguously aligned sites with the kpic‐gappy algorithm.
Nucleotide composition was calculated using the MEGA X (Kumar et al., 2018). Strand asymmetry was calculated according to the formulas: AT skew = [A − T]/[A + T] and GC skew = [G − C]/[G + C] (Perna & Kocher, 1995). The DAMBE 5.3.74 (Xia, 2013; Xia et al., 2003) was used to conduct tests of substitutional saturation of different data partitions based on the Iss (i.e., index of substitutional saturation) statistics. For this method, if Iss is positively smaller than Iss.c (critical Iss), the indicated sequences may have experienced little substitutional saturation (Xia & Lemey, 2009). Nucleotide diversity and the ratio of nonsynonymous substitution (Ka) to synonymous substitution (Ks) for PCGs were calculated using DNASP 5.0 (Librado & Rozas, 2009). The effective number of codon (ENC) was calculated using CodonW 1.4.2 (Peden, 2000).
2.4. Phylogenetic analyses
To test the phylogenetic implication of the eleven newly generated mitogenomes, various phylogenetic analyses were performed based on the five following datasets: (1) PCG12: first and second codon positions of 13 PCGs; (2) PCG123: all codon positions of 13 PCGs; (3) PCG12R: first and second codon positions of 13 PCGs plus 24 RNAs; (4) PCG123R: all codon positions of 13 PCGs plus 24 RNAs; (5) PCGAA: amino acid sequences of 13 PCGs.
Maximum likelihood (ML) analyses were conducted using IQ‐TREE 2.0.4 (Nguyen et al., 2015) under the partitioning schemes and corresponding substitution models (Tables S2 and S3) determined by ModelFinder (Kalyaanamoorthy et al., 2017). Branch supports (BS) were calculated using 1000 ultrafast bootstrap replicates (Hoang et al., 2018). Bayesian inference (BI) analyses were performed with MrBayes 3.2.6 (Ronquist et al., 2012) with the partitioned models (Tables S4 and S5) determined by PartitionFinder 2.1.1 (Lanfear et al., 2017). Twelve processors were used to perform two independent runs each with six chains (five heated and one cold) simultaneously for at least 500,000 generations sampled every 100 generations. Convergences were considered to be reached when the estimated sample size (ESS) value was above 200 established by Tracer 1.7 (Rambaut et al., 2018) and the potential scale reduction factor (PSRF) approached 1.0 (Ronquist et al., 2012). The first 25% of samples were discarded as burn‐in and the remaining trees were used to calculate posterior probabilities (PP) in a 50% majority‐rule consensus tree.
3. RESULTS AND DISCUSSION
3.1. General mitogenome feature and gene rearrangement
Eight complete and one nearly complete mitogenomes were generated and annotated for nine geometrid species, which increased the reported mitogenome diversity, especially for the Geometrinae and Sterrhinae. In the nearly complete genome (P. rufofasciata), we failed to assemble the partial sequences of the control region that is characterized by highly biased base composition. The eight completely sequenced mitogenomes ranged from 15,250 bp (M. senilis) to 15,723 bp (S. indicataria) in size, which are comparable to other reported geometrid mitogenomes (Table 1). All newly generated mitogenomes have been submitted to GenBank with the accession numbers shown in Table 1. The annotation information of mitogenomes sequenced herein is summarized in Table S6. In the two mitogenomes retrieved from transcriptomes, some fragments or genes of RNAs were not assembled, but the 13 PCGs were completely annotated and used only in subsequent phylogenetic analyses.
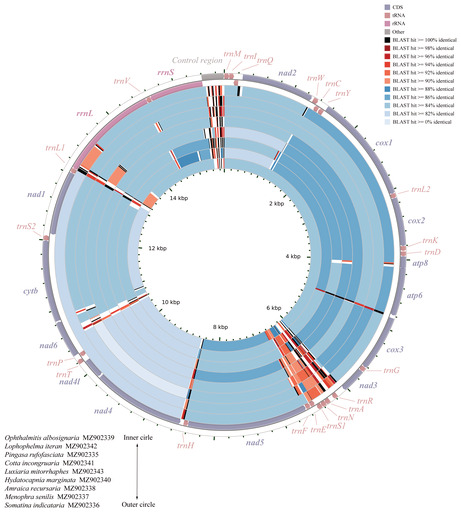
All mitogenomes contained 37 mitochondrial genes typical in insects (Figure 1), and these 37 genes, except for S. indicataria, showed identical gene organization to other reported geometrid mitogenomes, which are also typical of Lepidoptera (Cameron, 2014; Wu et al., 2016). In the mitogenome of S. indicataria, the positions of two tRNAs were arranged. The trnR was located before trnA instead of after trnA typical in Lepidoptera, whereas the trnE was translocated from the routinely recognized majority strand (J‐strand) to the minority strand (N‐strand). On the other hand, two long intergenic sequences (121 bp and 61 bp) were present before and after trnR, respectively, which were also important features distinct from other reported geometrid mitogenomes. To compare mitogenome evolution, mitochondrial gene rearrangement events previously reported for Lepidoptera were summarized and illustrated in Figure 2. Comparative analysis showed that two rearrangement clusters can be recognized in this order. One includes three tRNAs of trnM, trnI, and trnQ. The gene arrangement trnM‐trnI‐ trnQ is recognized in most lepidopteran members, in contrast to the trnI‐ trnQ ‐trnM in some nonditrysian lineages of Lepidoptera such as the Hepialoidea (Cao et al., 2012). Another is the gene cluster including six tRNAs between nad3 and nad5 genes. In this gene cluster, eleven kinds of gene rearrangements have been reported across seven superfamilies. In the S. indicataria, the trnR is located after trnA, similar to the only Parasa consocia of Limacodidae in Lepidoptera, whereas the trnE was detected rarely on the N‐strand. To confirm this result, we had methodologically reassembled the mitogenome from the high‐throughput sequencing data using Geneious R11 or other software. Moreover, before sequencing, the library was constructed using a single specimen of S. indicataria. Overall, the trnR‐trnA‐trnN‐trnS1‐ trnE ‐ trnF recognized in this study represents the first gene rearrangement event reported for Geometroidea and is also unique in Lepidoptera, which broadens our understanding of gene rearrangement in Geometroidea and Lepidoptera.
FIGURE 1.
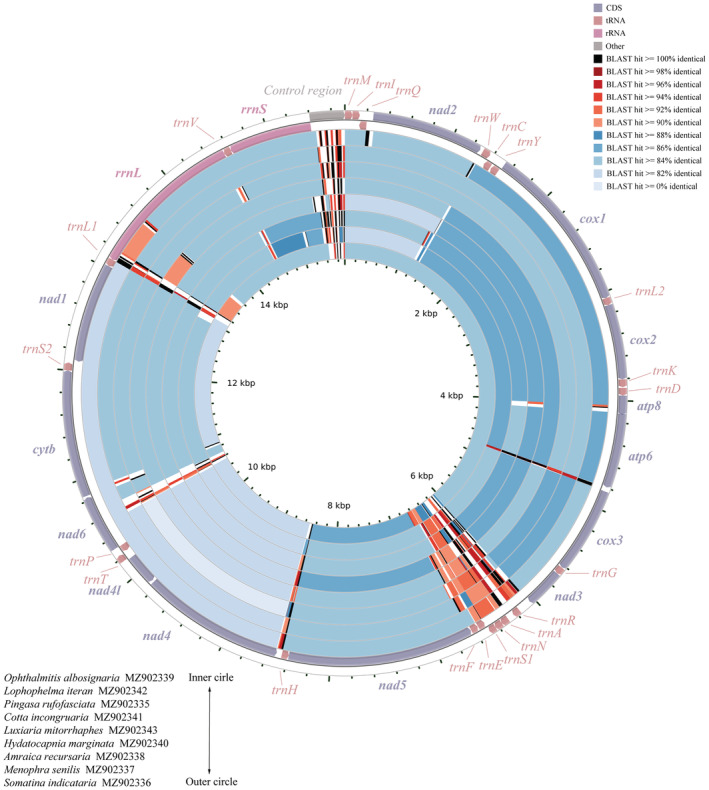
Circular diagram of the nine mitogenomes sequenced in this study. Different color is marked to show the nucleotide identity of BLAST hits relative to the reference mitogenome of Somatina indicataria at the outer circle.
FIGURE 2.
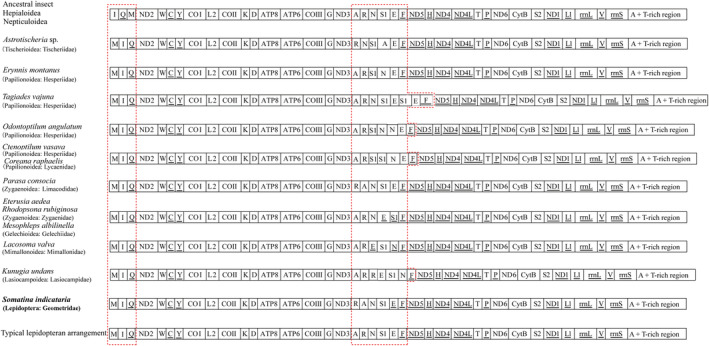
Gene arrangements of reported lepidopteran mitogenomes relative to ancestral insect mitogenome. Red rectangles indicate the two gene clusters with gene rearrangement. The gene with underline is located on the N‐strand. The taxon and its classification with gene rearrangement reported in this study are marked in bold.
3.2. Nucleotide composition
The A + T content of eight completely sequenced mitogenomes (Table 2) was highly biased, showing 80.9% (M. senilis) to 82% (S. indicataria), similar to other insect mitogenomes (Boore, 1999). The AT skews were negligible in size (<0.05), but the S. indicataria showed a significantly low AT skew (<0.01); by contrast, the GC skews were moderate and comparable below zero between −0.21031 (A. recursaria) and −0.17447 (C. incongruaria). Overall, the negligible AT skew and moderate GC skew detected in the eight mitogenomes are similar to other Lepidoptera and most insect species (Wei et al., 2010).
TABLE 2.
Nucleotide composition of nine newly determined mitogenomes for Geometridae.
| Subfamily | Species | Mitogenome size (bp) | A% | G% | C% | T% | AT% | AT skew | GC skew |
|---|---|---|---|---|---|---|---|---|---|
| Ennominae | Menophra senilis | 15,250 | 41.0 | 7.7 | 11.4 | 39.9 | 80.9 | 0.01329 | −0.19381 |
| Amraica recursaria | 15,582 | 41.8 | 7.5 | 11.4 | 39.3 | 81.1 | 0.03055 | −0.21031 | |
| Ophthalmitis albosignaria | 15,559 | 41.8 | 7.5 | 11.1 | 39.6 | 81.4 | 0.02684 | −0.19613 | |
| Hydatocapnia marginata | 15,615 | 41.8 | 7.5 | 11.4 | 39.3 | 81.1 | 0.03150 | −0.20421 | |
| Cotta incongruaria | 15,487 | 41.5 | 7.7 | 10.9 | 39.9 | 81.4 | 0.01999 | −0.17447 | |
| Luxiaria mitorrhaphes | 15,340 | 41.0 | 7.7 | 11.2 | 40.0 | 81.1 | 0.01262 | −0.18567 | |
| Geometrinae | Pingasa rufofasciata | 15,064 | 41.2 | 8.0 | 11.6 | 39.2 | 80.4 | 0.02469 | −0.18253 |
| Lophophelma iterans | 15,545 | 42.5 | 7.7 | 11.0 | 38.8 | 81.3 | 0.04534 | −0.17481 | |
| Sterrhinae | Somatina indicataria | 15,723 | 41.2 | 7.4 | 10.6 | 40.9 | 82.0 | 0.00395 | −0.17976 |
Among the four subfamilies of Geometridae (Figure 3a), the A + T contents ranged from 80.21% (Larentiinae) to 82.04% (Sterrhinae), showing little heterogeneity in nucleotide composition, which is in contrast to some insect groups generally at the same taxonomic levels (Liu et al., 2018; Nie et al., 2020; Song et al., 2016; Tang et al., 2019; Yang et al., 2018). Among the three codon positions within the 13 PCGs, the lowest A + T content was found for the second codon position, followed by the first and third codon positions, in accordance with most groups of insects, such as Zygaenoidea of Lepidoptera (Yang, Song, et al., 2019; Yang, Zhang, et al., 2019) and Cimicomorpha of Hemiptera (Yang et al., 2018). Overall, rRNAs showed a higher A + T content than PCGs and tRNAs. The AT skew and GC skew are commonly used for evaluating the nucleotide composition of insect mitogenomes (Perna & Kocher, 1995; Wei et al., 2010). In Geometridae, negligible AT skews and negative GC skews were recognized (Figure 3b, Table S7), and four geometrid subfamilies consistently showed that the second codon positions of 13 PCGs and rRNAs had the lowest values of AT skew and GC skew, respectively, a feature commonly present in other lepidopteran families (Cameron & Whiting, 2008) such as the Tortricidae recently reported by Yang et al. (2021). The ENC is routinely regarded between 20 and 61 and is negatively correlated with codon usage bias. The ENC = 20 indicates an absolute bias toward a synonymous codon, whereas ENC = 61 indicates the neutral codon usage (Wright, 1990). In the reported mitogenomes of Geometridae, the ENC values (Figure 3c) ranged from 30.4 to 35.53 and have almost no difference among the four subfamilies involved in this study but overall exhibiting codon usage bias to some extent. Moreover, the positive correlation between the ENC and GC3s (Figure 3d) indicates that the genomic G + C content is a significant factor in determining codon bias among geometrid species (Hershberg & Petrov, 2008; Plotkin & Kudla, 2011).
FIGURE 3.
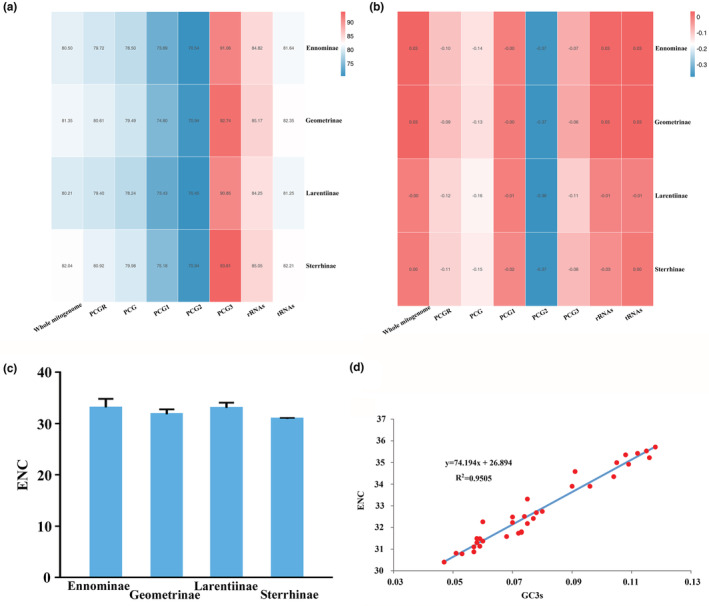
Nucleotide composition of the geometrid mitogenomes. (a) A + T content. (b) AT skew. (c) The averaged effective number of codons (ENC) of four geometrid subfamilies. (d) Scatter plot of the GC content of 3rd codon sites versus ENC.
3.3. Mitochondrial gene variation of Geometridae
Nucleotide diversity is commonly used for identifying regions with high nucleotide divergence and could provide guidelines for selecting species‐ or group‐specific markers used in molecular evolutionary studies, especially for taxa with high morphological similarity (Jia et al., 2010; Ma et al., 2020). To evaluate the variation patterns of 13 PCGs of Geometridae, nucleotide diversity was calculated through sliding window analysis for each PCG. The results (Figure 4a) showed a variable nucleotide diversity both within and among PCGs. At the gene level, the average values of nucleotide diversity varied from 0.114 (cox1) to 0.199 (nad6). The gene with nucleotide diversity next to nad6 was atp8, followed by cytb, cox3, nad4l, and nad4. The average values of nucleotide diversity for 13 PCGs can be also indicated by the sliding window analysis. The genes or gene regions with higher levels of nucleotide diversity identified herein could provide potential marker candidates for population genetics and species delimitation in Geometridae. In addition, the values of Ka, Ks, and Ka/Ks were calculated to compare the evolutionary patterns of 13 PCGs. As shown in Figure 4b, the atp8 and cox1 exhibit the highest and lowest Ka/Ks rates, respectively, indicating the 13 PCGs have different evolutionary rates. Notably, the Ka/Ks values for all PCGs were lower than one, indicating that they are evolving under purifying selection and are suitable for investigating phylogenetic relationships within the Geometridae.
FIGURE 4.
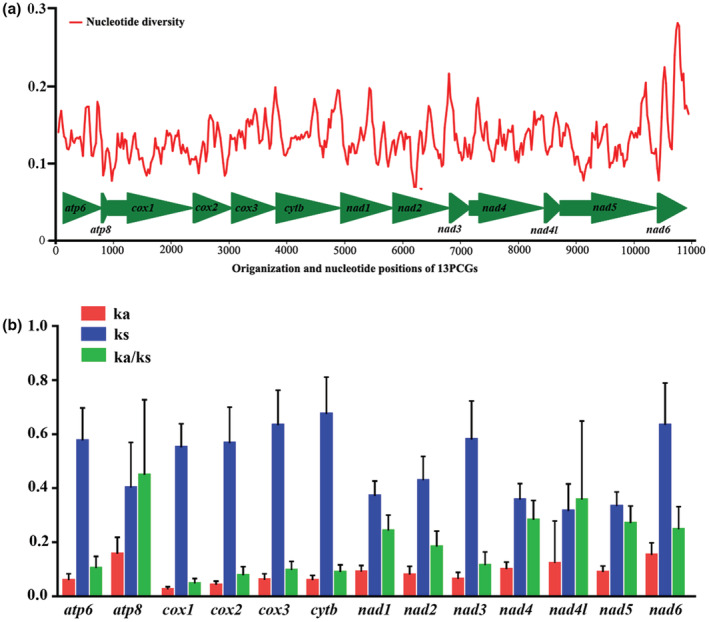
Gene variation of 13 PCGs in Geometridae. (a) The sliding window analysis shows the value of nucleotide diversity. (b) The Ka, Ks, and Ka/Ks of each PCG among geometrid representatives. Ka—nonsynonymous substitution; Ks—synonymous substitution.
3.4. Phylogenetic analyses of Geometroidea
Tests of substitution saturation (Table 3) showed all Iss values in the first and second coding positions of 13 PCGs and 22 tRNA were significantly lower than Iss.c values for both symmetrical and asymmetrical topologies. By contrast, the third coding positions of 13 PCGs and two RNAs showed evolutionary saturation to some extent, indicating that they had a faster evolutionary rate and might contain phylogenetic noise information (Owen et al., 2015). Thus, in subsequent phylogenetic analyses, five datasets associated with the inclusion and exclusion of the third coding positions and RNA sequences were considered to test the stability of topologies.
TABLE 3.
Saturation tests of different data partitions.
| NumOTU | Iss | Iss.cSym | PSym | Iss.cAsym | PAsym | |
|---|---|---|---|---|---|---|
| PCG1s | 32 | 0.286 | 0.809 | 0.0000 | 0.554 | 0.0000 |
| PCG2s | 32 | 0.165 | 0.809 | 0.0000 | 0.554 | 0.0000 |
| PCG3s | 32 | 0.595 | 0.809 | 0.0000 | 0.554 | 0.0000 |
| rRNAs | 32 | 0.768 | 0.797 | 0.4301 | 0.531 | 0.0000 |
| tRNAs | 32 | 0.424 | 0.777 | 0.0000 | 0.496 | 0.0455 |
Note: Two‐tailed tests were used.
According to recent molecular investigations (Bazinet et al., 2013; Rajaei et al., 2015; Regier et al., 2013; Wang et al., 2019; Yang, Zhang, et al., 2019), five families are included in Geometroidea. The present study sampled four families, including the Uraniidae represented in the mitogenome‐based phylogenetic investigation for the first time. Based on the hitherto most extensive mitogenomic sampling, our various resulting trees (Figures 5 and 6) showed generally the same topologies especially in terms of family‐ and subfamily‐level relationships. The relationships among four families were recovered as Geometridae + (Epicopeiidae + (Uraniidae + Sematuridae)) or Geometridae + (Sematuridae + (Epicopeiidae + Uraniidae)), with either Epicopeiidae or Sematuridae being sister to Uraniidae depending on different datasets. In detail, in ML analyses, the datasets PCG123R and PCG123 yielded the Uraniidae + Sematuridae, whereas other datasets showed Epicopeiidae + Uraniidae. In Bayesian analyses, the same situation was recovered. In terms of dataset selection, the results above indicate that the third coding positions of 13 PCGs have a deep effect on the tree topologies under both inference methods, whereas RNA genes did not. In addition, the tree based on PCGAA dataset showed a similar topology with that of PCG12R and PCG12 datasets. These results overall indicate that the third coding positions of 13 PCGs contain high phylogenetic informativeness although they may have experienced some substitution saturation (Yang et al., 2021). Regardless of the Pseudobistonidae with no mitogenome available, the close relationship between Epicopeiidae and Sematuridae is recovered by multilocus data in previous molecular studies (Rajaei et al., 2015; Wang et al., 2019). However, the placement of Uraniidae remains controversial, being closer to the Epicopeiidae and Sematuridae rather than to Geometridae as recovered by most of the previous multilocus molecular studies but with low to moderate supports (e.g., Rajaei et al., 2015; Wang et al., 2019). In this study, based on mitogenome evidence for the first time, the relationships among Sematuridae, Epicopeiidae, and Uraniidae remained unresolved, indicating that further investigation is necessary based on extensive sampling of these families (Mitter et al., 2017).
FIGURE 5.
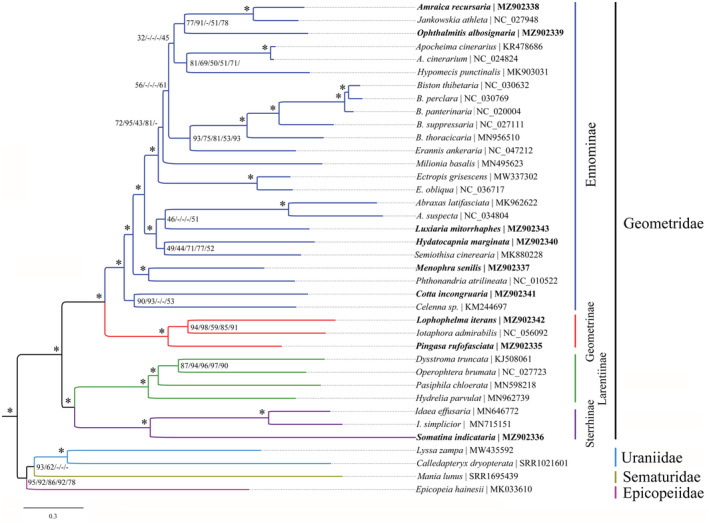
Maximum likelihood (ML) tree inferred from IQ‐TREE method based on PCG123R dataset. The species with newly sequenced mitogenome is emphasized in bold. Numbers separated by slash (/) on node represent the bootstrap values based on the PCG123R, PCG123, PCG12R, PCG12, and PCGAA datasets, respectively. The “*” on node represents bootstrap values ≥90 for all datasets. The “‐” represents an unrecovered node in ML tree of the corresponding dataset.
FIGURE 6.
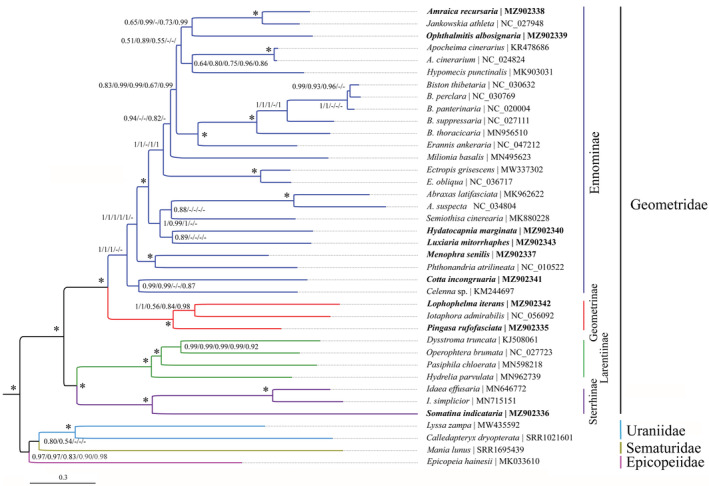
Bayesian tree inferred from MrBayes method based on PCG123R dataset. The species with newly sequenced mitogenome is emphasized in bold. Numbers separated by slash (/) on node represent the posterior probabilities based on the PCG123R, PCG123, PCG12R, PCG12, and PCGAA datasets, respectively. The “*” on node represents posterior probabilities ≥0.95 for all datasets. The “‐” represents unrecovered node in Bayesian tree of corresponding dataset.
In Geometridae, eight subfamilies are currently recognized (Murillo‐Ramos et al., 2019; Sihvonen et al., 2011). The present study sampled representatives of four subfamilies and their relationships were consistently recovered as (Sterrhinae + Larentiinae) + (Geometrinae + Ennominae) with strong supports. This topology is accordant with that of a multilocus study (Murillo‐Ramos et al., 2019) regardless of other four subfamilies with no mitogenome available.
The nine mitogenomes sequenced in the present study represented nine species of three subfamilies of the Geometridae, of which one belongs to Sterrhinae, two from Geometrinae, and the remaining six from Ennominae. Their taxonomic assignments were confirmed using mitogenome evidence for the first time, which provide support for previous morphological studies (Han & Xue, 2011; Jiang et al., 2011, 2012, 2014; Kuzmin & Beljaev, 2021; Sihvonen & Kaila, 2004; Walia, 2015).
AUTHOR CONTRIBUTIONS
Weili Ding: Conceptualization (equal); software (equal); validation (equal); writing – original draft (lead). Haizhen Xu: Methodology (equal); software (equal). Zhipeng Wu: Methodology (equal); software (equal). Lizong Hu: Conceptualization (equal); methodology (equal); writing – review and editing (equal). Li Huang: Conceptualization (equal); methodology (equal); writing – review and editing (equal). Mingsheng Yang: Conceptualization (equal); funding acquisition (equal); validation (equal); writing – review and editing (supporting). Lili Li: Conceptualization (equal); funding acquisition (equal); project administration (lead); supervision (lead); validation (equal); writing – review and editing (equal).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
Supporting information
Table S1.
Table S2.
Table S3.
Table S4.
Table S5.
Table S6.
Table S7.
ACKNOWLEDGMENTS
This work was supported by the Natural Science Foundation of China (31702046), the Postdoctoral Science Foundation of China (2021M692244), the Young Backbone Teacher Guiding Foundation in Colleges and Universities of Henan Province (2020GGJS211).
Ding, W. , Xu, H. , Wu, Z. , Hu, L. , Huang, L. , Yang, M. , & Li, L. (2023). The mitochondrial genomes of the Geometroidea (Lepidoptera) and their phylogenetic implications. Ecology and Evolution, 13, e9813. 10.1002/ece3.9813
Contributor Information
Mingsheng Yang, Email: yms-888@163.com.
Lili Li, Email: lililizksy@sohu.com.
DATA AVAILABILITY STATEMENT
All mitogenome sequences generated in this study were deposited in the GenBank under accession numbers MZ902335–MZ902343.
REFERENCES
- Abascal, F. , Zardoya, R. , & Telford, M. J. (2010). TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Research, 38, 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazinet, A. L. , Cummings, M. P. , Mitter, K. T. , & Mitter, C. (2013). Can RNA‐seq resolve the rapid radiation of advanced moths and butterflies (Hexapoda: Lepidoptera: Apoditrysia)? An exploratory study. PLoS One, 8, e82615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boore, J. L. (1999). Animal mitochondrial genomes. Nucleic Acids Research, 27, 1767–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron, S. L. (2014). Insect mitochondrial genomics: Implications for evolution and phylogeny. Annual Review of Entomology, 59, 95–117. [DOI] [PubMed] [Google Scholar]
- Cameron, S. L. , & Whiting, M. F. (2008). The complete mitochondrial genome of the tobacco hornworm, Manduca sexta (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene, 408, 112–123. [DOI] [PubMed] [Google Scholar]
- Cao, Y. Q. , Ma, C. , Chen, J. Y. , & Yang, D. R. (2012). The complete mitochondrial genomes of two ghost moths, Thitarodes renzhiensis and Thitarodes yunnanensis: The ancestral gene arrangement in lepidoptera. BMC Genomics, 13, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, R. , Xue, D. , Anthony, G. , & Han, H. (2017). Complete mitochondrial genomes throw light on budding speciation in three Biston species (lepidoptera, Geometridae). Zoologica Scripta, 46, 73–84. [Google Scholar]
- Curole, J. P. , & Kocher, T. D. (1999). Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends in Ecology & Evolution, 14, 394–398. [DOI] [PubMed] [Google Scholar]
- Donath, A. , Jühling, F. , AlArab, M. , Bernhart, S. H. , Reinhardt, F. , Stadler, P. F. , Middendorf, M. , & Bernt, M. (2019). Improved annotation of protein‐coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Research, 47, 10543–10552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, Z. Y. , Hasegawa, H. , Cooley, J. R. , Simon, C. , Yoshimura, J. , Cai, W. , Sota, T. , & Li, H. (2019). Mitochondrial genomics reveals shared phylogeographic patterns and demographic history among three periodical cicada species groups. Molecular Biology and Evolution, 36, 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmer, O. , Black, M. , Hoeh, W. , Lutz, R. , & Vrijenhoek, R. (1994). DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology, 3, 294–299. [PubMed] [Google Scholar]
- Grant, J. R. , Arantes, A. S. , & Stothard, P. (2012). Comparing thousands of circular genomes using the CGView comparison tool. BMC Genomics, 13, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajibabaei, M. , Janzen, D. H. , Burns, J. M. , Hallwachs, W. , & Hebert, P. D. N. (2006). DNA barcodes distinguish species of tropical lepidoptera. Proceedings of the National Academy of Sciences of the United States of America, 103, 968–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, H. , & Xue, D. (2011). Fauna Sinica, Insecta, Vol. 53, Lepidoptera, Geometridae, Ennominae. Science Press. [Google Scholar]
- Hebert, P. D. N. , Penton, E. H. , Burns, J. M. , Janzen, D. H. , & Hallwachs, W. (2004). Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator . Proceedings of the National Academy of Sciences of the United States of America, 101, 14812–14817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert, P. D. N. , Ratnasingham, S. , & De Waard, J. R. (2003). Barcoding animal life: Cytochrome c oxidase subunit 1 divergences among closely related species. Proceedings of the Royal Society, 270, 96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg, R. , & Petrov, D. A. S. (2008). Election on codon bias. Annual Review of Genetics, 42, 287–299. [DOI] [PubMed] [Google Scholar]
- Hoang, D. T. , Chernomor, O. , von Haeseler, A. , Minh, B. Q. , & Vinh, L. S. (2018). UFBoot2: Improving the ultrafast bootstrap approximation. Molecular Biology and Evolution, 35, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, W. Z. , Yan, H. B. , Guo, A. J. , Zhu, X. Q. , Wang, Y. C. , Shi, W. G. , Chen, H. T. , Zhan, F. , Zhang, S. H. , Fu, B. Q. , Littlewood, D. T. J. , & Cai, X. P. (2010). Complete mitochondrial genomes of taenia multiceps, T. hydatigena and T. pisiformis: Additional molecular markers for a tapeworm genus of human and animal health significance. BMC Genomics, 11, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, N. , Rikio, S. , & Han, H. (2012). One new and one newly recorded species of the genus Amraica Moore, 1888 (Lepidoptera: Geometridae: Ennominae) from China, with diagnoses of the Chinese species. Entomological Science, 15, 219–231. [Google Scholar]
- Jiang, N. , Xue, D. , & Han, H. (2011). A review of Ophthalmitis Fletcher, 1979 in China, with descriptions of four new species (Lepidoptera: Geometridae, Ennominae). Zootaxa, 2735, 1–22. [Google Scholar]
- Jiang, N. , Xue, D. , & Han, H. (2014). A review of Luxiaria Walker and its allied genus Calletaera Warren (Lepidoptera, Geometridae, Ennominae) from China. Zootaxa, 3856, 73–99. [DOI] [PubMed] [Google Scholar]
- Kalyaanamoorthy, S. , Minh, B. Q. , Wong, T. K. F. , Haeseler, A. , & Jermiin, L. S. (2017). ModelFinder: Fast model selection for accurate phylogenetic estimates. Nature Methods, 14, 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , Rozewicki, J. , & Yamada, K. D. (2019). MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Briefings in Bioinformatics, 20, 1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse, M. , Moir, R. , Wilson, A. , Stones‐Havas, S. , Cheung, M. , Sturrock, S. , Buxton, S. , Cooper, A. , Markowitz, S. , Duran, C. , Thierer, T. , Ashton, B. , Meintjes, P. , & Drummond, A. (2012). Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics, 28, 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , Li, M. , Knyaz, C. , & Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35, 1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin, A. A. , & Beljaev, E. A. (2021). New records of geometrid moths of the subfamily Ennominae (lepidoptera: Geometridae) from the Amurskaya oblast, Russian Far East. Acta Biologica Sibirica, 7, 219–226. [Google Scholar]
- Lanfear, R. , Frandsen, P. B. , Wright, A. M. , Senfeld, T. , & Calcott, B. (2017). PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Molecular Biology and Evolution, 34, 772–773. [DOI] [PubMed] [Google Scholar]
- Li, Q. , Wang, X. , Chen, X. , & Han, B. (2018). Complete mitochondrial genome of the tea looper caterpillar, Ectropis obliqua (lepidoptera: Geometridae) with a phylogenetic analysis of Geometridae. International Journal of Biological Macromolecules., 114, 491–496. [DOI] [PubMed] [Google Scholar]
- Librado, P. , & Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25, 1451–1452. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Song, F. , Jiang, P. , Wilson, J. J. , Cai, W. , & Li, H. (2018). Compositional heterogeneity in true bug mitochondrial phylogenomics. Molecular Phylogenetics and Evolution, 118, 135–144. [DOI] [PubMed] [Google Scholar]
- Luo, R. , Liu, B. , Xie, Y. , Li, Z. , Huang, W. , Yuan, J. , He, G. , Chen, Y. , & Pan, Q. (2012). SOAPdenovo2: An empirically improved memory‐efficient short‐read de novo assembler. Gigascience, 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, L. Y. , Liu, F. F. , Chiba, H. , & Yuan, X. Q. (2020). The mitochondrial genomes of three skippers: Insights into the evolution of the family Hesperiidae (lepidoptera). Genomics, 112, 432–441. [DOI] [PubMed] [Google Scholar]
- Mitter, C. , Davis, D. R. , & Cummings, M. P. (2017). Phylogeny and evolution of lepidoptera. Annual Review of Entomology, 62, 265–283. [DOI] [PubMed] [Google Scholar]
- Murillo‐Ramos, L. , Brehm, G. , Sihvonen, P. , Hausmann, A. , Holm, S. , Ghanavi, H. R. , Õunap, E. , Truuverk, A. , Staude, H. , Friedrich, E. , Tammaru, T. , & Wahlberg, N. (2019). A comprehensive molecular phylogeny of Geometridae (lepidoptera) with a focus on enigmaticsmall subfamilies. Peer J, 7, e7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, L. T. , Schmidt, H. A. , von Haeseler, A. , & Minh, B. Q. (2015). IQ‐TREE: A fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Molecular Biology and Evolution, 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie, R. , Andújar, C. , Gómez‐Rodríguez, C. , Bai, M. , Xue, H. J. , Tang, M. , Yang, C. T. , Tang, P. , Yang, X. , & Vogler, A. P. (2020). The phylogeny of leaf beetles (Chrysomelidae) inferred from mitochondrial genomes. Systematic Entomology, 45, 188–204. [Google Scholar]
- Owen, C. L. , Marshall, D. C. , Hill, K. B. R. , & Simon, C. (2015). The phylogenetic utility of acetyltransferase (ARD1) and glutaminyl tRNA synthetase (QtRNA) for reconstructing Cenozoic relationships as exemplified by the large Australian cicada Pauropsalta generic complex. Molecular Phylogenetics and Evolution, 83, 258–277. [DOI] [PubMed] [Google Scholar]
- Peden, J. F. (2000). Analysis of codon usage. University of Nottingham, 90, 73–74. [Google Scholar]
- Perna, N. T. , & Kocher, T. D. (1995). Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. Journal of Molecular Evolution, 41, 353–358. [DOI] [PubMed] [Google Scholar]
- Plotkin, J. B. , & Kudla, G. (2011). Synonymous but not the same: The causes and consequences of codon bias. Nature Reviews. Genetics, 12, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaei, H. , Greve, C. , Letsch, H. , Stüning, D. , Wahlberg, N. , Minet, J. , & Misof, B. (2015). Advances in Geometroidea phylogeny, with characterization of a new family based on Pseudobiston pinratanai (Lepidoptera, Glossata). Zoologica Scripta, 44, 418–436. [Google Scholar]
- Rambaut, A. , Drummond, A. J. , Xie, D. , Baele, G. , & Suchard, M. A. (2018). Posterior summarisation in Bayesian phylogenetics using tracer 1.7. Systematic Biology, 67, 901–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regier, J. C. , Mitter, C. , Zwick, A. , Bazinet, A. L. , Cummings, M. P. , Kawahara, A. Y. , Sohn, J. C. , Zwickl, D. J. , Cho, S. , Davis, D. R. , Baixeras, J. , Brown, J. , Parr, C. , Weller, S. , Lees, D. C. , & Mitter, K. T. (2013). A large‐scale, higher‐level, molecular phylogenetic study of the insect order lepidoptera (moths and butterflies). PLoS One, 8, e58568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist, F. , Teslenko, M. , Mark, P. , Ayres, D. L. , Darling, A. , Höhna, S. , Larget, B. , Liu, L. , Suchard, M. A. , & Huelsenbeck, J. P. (2012). MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology, 61, 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert, M. , Lindgreen, S. , & Orlando, L. (2016). AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Research, 9, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sihvonen, P. , & Kaila, L. (2004). Phylogeny and tribal classification of Sterrhinae with emphasis on delimiting Scopulini (lepidoptera: Geometridae). Systematic Entomology, 29, 324–358. [Google Scholar]
- Sihvonen, P. , Mutanen, M. , Kaila, L. , Brehm, G. , Hausmann, A. , & Staude, H. S. (2011). Comprehensive molecular sampling yields a robust phylogeny for geometrid moths (lepidoptera: Geometridae). PLoS One, 6, e20356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, F. , Li, H. , Jiang, P. , Zhou, X. , Liu, J. , Sun, C. , Vogler, A. P. , & Cai, W. (2016). Capturing the phylogeny of holometabola with mitochondrial genome data and Bayesian site‐heterogeneous mixture models. Genome Biology and Evolution, 8, 1411–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenwyk, J. L. , Buida, T. J. , Li, Y. N. , Shen, X. X. , & Rokas, A. (2020). ClipKIT: A multiple sequence alignment trimming software for accurate phylogenomic inference. PLoS Biology, 18, e3001007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, P. , Zhu, J. , Zheng, B. , Wei, S. , Sharkey, M. , Chen, X. , & Vogler, A. P. (2019). Mitochondrial phylogenomics of the Hymenoptera. Molecular Phylogenetics and Evolution, 131, 8–18. [DOI] [PubMed] [Google Scholar]
- Thao, M. L. , Baumann, L. , & Baumann, P. (2004). Organization of the mitochondrial genomes of whiteflies, aphids and pysillids (Hemiptera: Sternorrhyncha). BMC Evolutionary Biology, 4, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans, M. J. T. N. , Lees, D. C. , & Simonsen, T. J. (2014). Towards a mitogenomic phylogeny of lepidoptera. Molecular Phylogenetics and Evolution, 79, 169–178. [DOI] [PubMed] [Google Scholar]
- van Nieukerken, E. J. , Kaila, L. , Kitching, I. J. , Kristensen, N. P. , Lees, D. C. , Minet, J. , Mitter, C. , Mutanen, M. , Regier, J. C. , Simonsen, T. J. , Wahlberg, N. , Yen, S. , Zahiri, R. , Adamski, D. , Baixeras, J. , Bartsch, D. , Bengtsson, B. , Brown, J. W. , Bucheli, S. , … Andreas, Z. (2011). Order Lepidoptera Linnaeus, 1758. Zootaxa, 3148, 212–221. [Google Scholar]
- Walia, V. K. (2015). Studies on genitalia of four type species of subfamily ennominae (lepidoptera: Geometridae). Indian Journal of Entomology., 77, 266. [Google Scholar]
- Wang, H. , Holloway, J. D. , Wahlberg, N. , Wang, M. , & Nylin, S. (2019). Molecular phylogenetic and morphological studies on the systematic position of Heracula iscivitta reveal a new subfamily of Pseudobistonidae (lepidoptera: Geometroidea). Systematic Entomology, 44, 211–225. [Google Scholar]
- Wei, S. , Shi, M. , Chen, X. , Sharkey, M. J. , van Achterberg, C. , Ye, G. Y. , & He, J. H. (2010). New views on strand asymmetry in insect mitochondrial genomes. PLoS One, 5, e12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, F. (1990). The ‘effective number of codons’ used in a gene. Gene, 87, 23–29. [DOI] [PubMed] [Google Scholar]
- Wu, Y. P. , Zhao, J. L. , Su, T. J. , Luo, A. R. , & Zhu, C. D. (2016). The complete mitochondrial genome of Choristoneura longicellana (lepidoptera: Tortricidae) and phylogenetic analysis of lepidoptera. Gene, 591, 161–176. [DOI] [PubMed] [Google Scholar]
- Xia, X. (2013). DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Molecular Biology and Evolution, 30, 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, X. , & Lemey, P. (2009). The phylogenetic handbook: A practical approach to phylogenetic analysis and hypothesis testing, second edition (pp. 615–630). Cambridge University Press. [Google Scholar]
- Xia, X. , Xie, Z. , Salemi, M. , Chen, L. , & Wang, Y. (2003). An index of substitution saturation and its application. Molecular Phylogenetics and Evolution, 26, 1–7. [DOI] [PubMed] [Google Scholar]
- Yang, H. , Li, T. , Dang, K. , & Bu, W. (2018). Compositional and mutational rate heterogeneity in mitochondrial genomes and its effect on the phylogenetic inferences of Cimicomorpha (Hemiptera: Heteroptera). BMC Genomics, 19, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, M. , Li, J. , Su, S. , Zhang, H. , Wang, Z. , Ding, W. , & Li, L. (2021). The mitochondrial genomes of Tortricidae: Nucleotide composition, gene variation and phylogenetic performance. BMC Genomics, 22, 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, M. , Song, L. , Shi, Y. , Li, J. , Zhang, Y. , & Song, N. (2019). The first mitochondrial genome of the family Epicopeiidae and higher‐level phylogeny of Macroheterocera (lepidoptera: Ditrysia). International Journal of Biological Macromolecules, 136, 23–132. [DOI] [PubMed] [Google Scholar]
- Yang, M. , Zhang, H. , Song, L. , Shi, Y. , & Liu, X. (2019). The complete mitochondrial genome of Mahanta tanyae compared with other zygaenoid moths (lepidoptera: Zygaenoidea). Journal of Asia‐Pacific Entomology, 22, 513–521. [Google Scholar]
- Yang, X. , Cameron, S. L. , Lees, D. C. , Xue, D. , & Han, H. (2015). A mitochondrial genome phylogeny of owlet moths (lepidoptera: Noctuoidea), and examination of the utility of mitochondrial genomes for lepidopteran phylogenetics. Molecular Phylogenetics and Evolution, 85, 230–237. [DOI] [PubMed] [Google Scholar]
- Yang, X. , Xue, D. , & Han, H. (2013). The complete mitochondrial genome of Biston panterinaria (lepidoptera: Geometridae), with phylogenetic utility of mitochondrial genome in the lepidoptera. Gene, 515, 349–358. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Table S2.
Table S3.
Table S4.
Table S5.
Table S6.
Table S7.
Data Availability Statement
All mitogenome sequences generated in this study were deposited in the GenBank under accession numbers MZ902335–MZ902343.