

Abstract
The RNA binding protein ELAVL4/HuD regulates the translation and splicing of multiple Alzheimer’s disease (AD) candidate genes. We generated ELAVL4 knockout (KO) human induced pluripotent stem cell-derived neurons to study the effect that ELAVL4 has on AD-related cellular phenotypes. ELAVL4 KO significantly increased the levels of specific APP isoforms and intracellular phosphorylated tau, molecular changes that are related to the pathological hallmarks of AD. Overexpression of ELAVL4 in wild-type neurons and rescue experiments in ELAVL4 KO cells showed opposite effects and also led to a reduction of the extracellular amyloid-beta (Aβ)42/40 ratio. All these observations were made in familial AD (fAD) and fAD-corrected neurons. To gain insight into the molecular cascades involved in neuronal ELAVL4 signaling, we conducted pathway and upstream regulator analyses of transcriptomic and proteomic data from the generated neurons. These analyses revealed that ELAVL4 affects multiple biological pathways linked to AD, including those involved in synaptic function, as well as gene expression downstream of APP and tau signaling. The analyses also suggest that ELAVL4 expression is regulated by insulin receptor-FOXO1 signaling in neurons. Taken together, ELAVL4 expression ameliorates AD-related molecular changes in neurons and affects multiple synaptic pathways, making it a promising target for novel drug development.
Keywords: Alzheimer’s disease (AD), ELAVL4, HuD, RNA-binding protein
1. Introduction
ELAVL4 (other name: HuD) is a member of the Hu/ELAV-like family of RNA-binding proteins (RBPs) that is predominantly expressed in neurons and, at lower levels, in the pancreas and testis (Hinman and Lou, 2008). In addition to ELAVL4, the Hu family consists of two RBPs that are enriched for expression in neurons—i.e., HuB (ELAVL2) and HuC (ELAVL3)—and the ubiquitously expressed HuR (ELAVL1) (Bronicki and Jasmin, 2013). By post-transcriptionally processing (pre-) mRNAs, RBPs add an additional layer to gene regulation and produce a more diverse assortment of mRNAs and proteins (Nussbacher et al., 2019). Hu proteins contain three RNA recognition motifs (RRMs) that directly interact with AU-rich elements (RRM1 and RRM2) and poly-A tails (RRM3) of specific target mRNAs (Bolognani et al., 2010; Park et al., 2000). Through protein-protein interactions mediated by RRM3 and the hinge region that separates RRM2 and RRM3, Hu proteins can recruit additional proteins to mRNA-protein complexes (Bronicki and Jasmin, 2013; Fujiwara et al., 2012; Fukao et al., 2009). By directly binding mRNAs, Hu proteins are involved in multiple aspects of posttranscriptional gene regulation, including mRNA polyadenylation, alternative splicing, trafficking, turnover, and translation (Pascale et al., 2008).
As for human diseases associated with ELAVL4/HuD, links have been established with both dementia and type 2 diabetes mellitus (T2DM). As for dementia, it was already reported in 1986 that high titers of ELAVL4 autoantibodies had been found in the sera from a subset of patients with small-cell lung tumors who also developed a paraneoplastic syndrome characterized by dementia and other neurological disabilities (Graus et al., 1986; Szabo et al., 1991). In this respect, it was demonstrated that the autoantibodies produced against ELAVL4 ectopically expressed by lung tumor cells induced neuronal loss and inflammatory infiltrates in the brain, leading to the observed neurological symptoms (Graus et al., 1986; Nussbacher et al., 2019; Szabo et al., 1991). In addition, mice with a prenatal ELAVL4 knock-out showed deficits in hippocampus-dependent learning and memory (DeBoer et al., 2014), while it was also shown that upon learning and memory tasks, ELAVL4 expression is upregulated in the hippocampus of mice and rats (Pascale et al., 2004; Quattrone et al., 2001). However, mice that constitutively overexpress ELAVL4 in their forebrain also have impaired hippocampus-dependent learning and memory (Bolognani et al., 2007a). At the molecular level, ELAVL4 has been shown to strengthen synapses of individual dendritic branches by stabilizing the mRNA and increasing the expression of proteins with important synaptic functions, such as CAMK2A and BDNF (Sosanya et al., 2015; Vanevski and Xu, 2015). Additionally, ELAVL4 stabilizes and upregulates the expression of GAP43, another protein with a key role in hippocampal neuronal plasticity (Bolognani et al., 2007b). Based on all these findings, it was concluded that ELAVL4 functions as an essential regulator of synaptic plasticity during learning and memory tasks in mature neurons, likely having roles in both neurodevelopment and in the adult brain (Bolognani et al., 2007a; Deschenes-Furry et al., 2006; Lee et al., 2015).
The most common form of dementia is the progressive neurodegenerative disorder Alzheimer’s disease (AD) (Patterson, 2018). The two hallmark neuropathological lesions that are found in AD patients’ brains are extracellular neuritic plaques consisting of amyloid-β (Aβ)—which is generated through the processing of Amyloid Precursor Protein (APP)—and intracellular neurofibrillary tangles containing excessively phosphorylated tau protein (Jack et al., 2018). As for Aβ, the longer form consisting of 42 amino acids (Aβ42) is more prone to aggregate into plaques than the most common form with 40 amino acids (Aβ40), and familial AD mutations either raise the Aβ42 levels or lower the Aβ40 levels, each of which leads to a higher Aβ42/40 ratio. Therefore, an increased Aβ42/40 ratio is considered to be indicative of more neurotoxic Aβ (Bentahir et al., 2006; Hansson et al., 2019; Liu et al., 2021; Scheuner et al., 1996). In addition to being involved in regulating hippocampal memory-related processes in general, decreased expression of neuronal Hu proteins (including ELAVL4) has been reported in the hippocampus of AD patients (Amadio et al., 2009; Wang et al., 2018). Furthermore, ELAVL4 has been shown to bind and stabilize many mRNAs encoding proteins that have been specifically implicated in AD. As such, ELAVL4 binds and stabilizes the mRNAs—leading to increased protein expression—of both APP and BACE1, a β-secretase that is involved in amyloidogenic APP to Aβ processing (Kang et al., 2014). However, ELAVL4 can also promote the non-amyloidogenic processing of APP by stabilizing the mRNA and upregulating the expression of the α-secretase ADAM10 (Marchesi et al., 2016). Neuronally expressed Hu proteins—including ELAVL4—have also been reported to regulate the alternative splicing of APP mRNA (Fragkouli et al., 2017). Moreover, ELAVL4 has been shown to bind and stabilize the mRNAs of neprilysin, an Aβ degrading enzyme (Lim and Alkon, 2014), and neuroserpin, an extracellular enzyme that can both negatively and positively regulate Aβ clearance (Kinghorn et al., 2006; Subhadra et al., 2013). In addition to Aβ-related targets, ELAVL4 can directly bind, stabilize and upregulate the expression of tau (mRNA) in neuronal cells (Aranda-Abreu et al., 1999). Further, ELAVL4 stabilizes the mRNA and hence upregulates the expression of acetylcholinesterase (ACHE) (Deschenes-Furry et al., 2007), which provides another link with AD as ACHE inhibitors are currently the most often used drugs for symptomatic treatment of the disease (Hogan, 2014). Lastly, a recent co-expression network analysis of RNAseq data generated from human postmortem brain samples identified ELAVL4 as one of ten ‘hub genes’ in AD-related synaptic pathways (Hu et al., 2020).
Information on how ELAVL4 expression and function is regulated is scarce. Some evidence comes from work on its contribution to T2DM. Firstly, silencing of ELAVL4 expression increases apoptosis of pancreatic beta cells—that are functionally impaired in diabetes—implying that ELAVL4 is an important regulator of beta cell function and survival (Juan-Mateu et al., 2017). Together with the finding that ELAVL4 expression is markedly reduced in beta cells of diabetic patients (Hong et al., 2020), this indicates that ELAVL4 deficiency contributes to diabetes etiology. Furthermore, it was demonstrated in beta cells that the expression of ELAVL4 is regulated by insulin receptor-FOXO1 signaling (Lee et al., 2012), which is the only molecular mechanism for ELAVL4 expression regulation described thus far.
In this study, we determined the effects that ELAVL4 knock-out and overexpression have on AD-related phenotypes—i.e., APP (isoform) levels, Aβ42/40 ratio, and (phosphorylated) tau levels—in human induced pluripotent stem cell-derived glutamatergic cortical neurons. Subsequently, to gain insight into the molecular cascades underlying ELAVL4 signaling in human neurons, we analyzed RNA sequencing and proteomic data from neurons derived from the different cell lines generated in this study.
2. Materials and Methods
2.1. Cell culture
Induced pluripotent stem cells (iPSCs) harboring the APP London mutation (V717I) (fAD) and isogenic CRISPR corrected lines (fADcorr) were reported previously (Muratore et al., 2014; Muratore et al., 2017). iPSCs were maintained in Stemflex medium (ThermoFisher) on plates coated with growth factor reduced Matrigel (Corning). Differentiation to induced neurons (iNs) was performed as described previously (Lagomarsino et al., 2021; Wu et al., 2022; Zhang et al., 2013). Cells resulting from this differentiation protocol have been deeply characterized by scRNAseq, and virtually all cells (>98%) express markers of postmitotic neuronal fate (Lagomarsino et al., 2021). Briefly, iPSCs were plated at 95K/cm2 in mTeSR1 media one-day prior to viral transduction. Ultrahigh titer lentiviruses were obtained from Alstem for plasmids FUdeltaGW-rtTA (Addgene #19780), pTet-O-Ngn2-puro (Addgene #52047), and DRH-307 (Addgene #112670), and were transduced at MOIs 5, 2, and 2, respectively. On day in vitro (DIV) 1, differentiation was induced by Doxycycline (2μg/ml). On DIV 2, puromycin (5 μg/ml) was added to select for cells expressing Ngn2. On DIV 4, iNs were plated on Poly-L-Ornithine laminin-coated plates and cultured in Neurobasal medium containing Glutamax, dextrose, NEAA, B27, Doxycycline, puromycin, BDNF, CNTF, and GDNF. Culturing media was conditioned starting at DIV 18, and media, protein, and RNA samples were harvested at DIV 21.
2.2. CRISPR-Cas9 knock out
single guide RNAs targeting exons common to all human ELAVL4 splicing isoforms were designed using the Broad CRISPR design tool (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design) (figure 1A). The top three sgRNAs were cloned into the pXPR-003 (Addgene #52963) backbone and transfected with SpCas9 plasmid (Addgene #78166) into M17D cells using FuGENE HD (Promega) to assess sgRNA knockdown (KD) efficiency by qPCR. The most efficient M17D KD of ELAVL4 was observed using sgRNA B1, which was subsequently co-transfected with SpCas9 into fAD and fADcorr iPSCs using Lipofectamine2000. Two days post-transfection, iPSCs were selected with puromycin (5 μg/ml) and blasticidin (4 μg/ml) for enrichment. Editing efficiency in the resulting polyclonal lines was assessed with the GeneArt Genomic Cleavage Detection Kit (ThermoFisher). Monoclonal cell lines were obtained by limiting dilution and examined by sequencing over the sgRNA target region. After monoclonal selection, the cellular karyotype was assessed using the NanoString nCounter CNV codeset and visualized using copy number package in R (Nilsen et al., 2012).
Figure 1.

Validation of ELAVL4 knock out (KO), over expression (OE), and rescue in iPSCs and iNs.
(A) Interaction sites for the primary antibody, qPCR primers, and sgRNA for ELAVL4 used in this study projected on a schematic representation of ELAVL4. (B) Overview of the cell lines and lentiviral conditions generated/used in this study. (C) Full KO of ELAVL4 mRNA in fADcorr iNs (P < 0.0001, t-test). (D) 18.6- and 25.5-fold increase in ELAVL4 mRNA upon ELAVL4 sv2 and ELAVL4 sv1 OE expression in fADcorr iNs respectively (P < 0.001 and P < 0.0001, Dunnett’s following a significant ANOVA). (E) Full KO of ELAVL4 mRNA in fAD iNs (P<0.0001, t-test). (F) 13.5- and 12.4-fold increase in ELAVL4 mRNA upon ELAVL4 sv2 and ELAVL4 sv1 OE expression in fADcorr iNs respectively (P < 0.0001, Dunnett’s following a significant ANOVA). (G) Representative image of immunocytochemistry of ELAVL4 rescue with ELAVL4-sv1 in fADcorr ELAVL4 KO iNs, stained with the nuclear DNA stain Hoechst, the neuronal marker βIII-tubulin and ELAVL4, mCHerry signal from the overexpression plasmid, scale bars: 10 μm. (H) Example western blots are shown for fADcorr iNs and fAD iNs, the pre-synaptic marker SYN1 is not significantly affected by manipulating the expression levels of ELAVL4. (I) Full KO of ELAVL4 protein in fADcorr iNs (P < 0.0001, t-test). (J) 20.8- and 19.5-fold increase in ELAVL4 protein upon ELAVL4 sv2 and ELAVL4 sv1 OE expression in fADcorr iNs respectively (P < 0.0001, Dunnett’s following a significant ANOVA). (K) Full KO of ELAVL4 protein in fAD iNs (P<0.0001, t-test). (L) 22.4- and 22.3-fold increase in ELAVL4 mRNA upon ELAVL4 sv2 and ELAVL4 sv1 OE expression in fADcorr iNs respectively (P < 0.0001, Dunnett’s following a significant ANOVA).
2.3. Lentiviral overexpression of ELAVL4
The EIF1α promoter of pLVX-EF1α-IRES-mCherry (Takara #631987) was switched to a shorter CAG promoter (Genewiz) by ClaI and NotI digestion for higher viral packaging efficiency and elevated expression in neurons (pLVX-CAG-IRES-mCherry). The two most common isoforms of ELAVL4 (splicing variants 1 or 2 (sv1 or sv2) in the postnatal brain arise from alternative splicing of ELAVL4 pre-mRNA in the region that codes for the hinge between RRM2 and RRM3 (Bronicki and Jasmin, 2013). cDNA for ELAVL4 sv1 (Origene #RC218612) and sv2 (Genscript #OHu28682D) was amplified by PCR using primers 5’-CTCTAGAGCCACCATGGTTATGA-3’ (FW) and 5’-ATATGGATCCTCAGGACTTGTGGG-3’ (REV). Amplified cDNA and pLVX-CAG-IRES mCherry vector were digested by XbaI and BamHI-HF and ligated using T4-ligase. The ligated product was transformed in stable competent E. coli (NEB #C3040H) and plated on ampicillin plates. For lentivirus production, 90% confluent HEK293T cells in 15 cm culture plates were transfected with 24 μg pLVX-CAG-ELAVL4-IRES-mCherry, 12 μg envelope plasmid (Addgene #12259), and 18 μg packaging plasmid (Addgene #12260) using Lipofectamine2000. Media collected at 24H and 36H was pooled and concentrated with Lenti-X-Concentrator (Takara), virus particles were resuspended in PBS buffer.
2.4. Quantitative reverse transcription PCR
Samples for qPCR were prepared using a Fast SYBR Green Cells-to-CT Kit (ThermoFisher) according to manufacturer’s guidelines, measured using three technical replicates on a ViiA 7 System (Applied Biosystems), and normalized to GAPDH values during comparative Ct analysis (Livak and Schmittgen, 2001). Samples (n=3) from three replicate experiments were normalized to control conditions in the same experiment.
2.5. Western blotting
iN protein lysates were extracted using RIPA buffer (Abcam) containing complete protease inhibitor (Sigma) and phosphatase stop (Roche). Protein concentrations were determined by BCA (ThermoFisher), and samples were prepared with 4X sample buffer (LI-COR). Sample separation was performed using 4–12% Bis-Tris gels (ThermoFisher) in MOPS running buffer. Subsequently, samples were transferred onto nitrocellulose membranes. Membranes were blocked in blocking buffer (LI-COR) and probed overnight with primary antibodies at 4°C. Secondary anti-mouse and/or anti-rabbit antibodies (LI-COR), where appropriate, were incubated for 1 hour at room temperature (RT), and blots were scanned using the Odyssey CLx Imaging System (LI-COR). The primary antibodies used were: RαTau (DAKO, A0024), MαPhospho tau Thr181 (Thermo Fisher, MN1050), RαSYN1 (EMD-Millipore, 574777), MαAPP (Sigma-Aldrich, MAB348), RαELAVL4 (Proteintech, 24992-1-AP), and MαGAPDH (Proteintech, 60004-1-Ig). Samples (n=3) from three replicate experiments were normalized to control conditions in the same experiment.
2.6. Aβ-ELISA
A triplex electrochemiluminescence assay kit (K15200E-2, V-PLEX Aβ Peptide Panel 1 kit, (Meso Scale Diagnostics (MSD)) was used to simultaneously measure levels of Aβ38, Aβ40, and Aβ42 in conditioned media following manufacturer’s instructions. Samples (n=3) from three replicate experiments were normalized to control conditions in the same experiment.
2.7. RNA sequencing
Total RNA of DIV21 iNs was purified using PureLink RNA mini kits (ThermoFisher). RNA-Seq Library preparation and Illumina HiSeq 2×150bp sequencing was performed by Genewiz, Inc. Trimmomatic was used for trimming the beginning and ending bases from each read and identifying and trimming adapter sequences from the reads (Bolger et al., 2014). For quantifying gene level transcript abundances, we (pseudo)aligned the trimmed reads to a human reference genome (GRCh38) using Kallisto (Bray et al., 2016). Detection of differentially expressed genes (DEGs) was performed with Limma-voom using the count data (transcripts per million) obtained from Kallisto. P-values were corrected for multiple testing using the false discovery rate (FDR) method (Benjamini and Hochberg, 1995).
2.8. Immunostaining
DIV 21 iNs were fixed with 4% Paraformaldehyde solution at 4°C for 20 min at RT, blocked in blocking buffer (2% donkey serum, 0.1% Triton in PBS) for 1 hour at RT, and incubated with primary antibodies at 4°C overnight. The next day, secondary antibodies were incubated for 1 hour at RT followed by Hoechst DNA stain for 10 minutes at RT. Three 10 minutes PBS wash steps were performed between each step.
2.9. Phosphoproteomics and protein expression profiling
On DIV21, iN protein samples were collected in 4°C 1% SDS in H2O. Following cell lysis, samples were centrifuged at maximum speed (~21,000 × g) for 10 minutes at 4°C to pellet any cell debris. Protein concentrations were determined using a BCA assay (PierceTM). Proteins were reduced with 10 mM DTT for 1 hour at 56°C, then alkylated with 55 mM Iodoacetamide (IAA, Sigma) for 1 hour at RT on a rotator protected from light. SP3 beads were washed three times with milliQ water and 500ug beads were added per sample. Acetonitrile was added in 1:1 ratio, and the sample was incubated for 8 minutes. After incubating an additional 2 minutes on the magnetic rack, the supernatant was removed, and the beads were washed twice with 70% ethanol and once with 100% acetonitrile. After letting the beads air dry for 30 seconds to remove residual acetonitrile, 10ug of trypsin in 200ul of 50mM HEPES buffer was added for on bead digestion. Beads were sonicated for 1 minute and incubated o/n on the rotator. The next day, the beads were incubated on the magnetic rack for 2 minutes, and the supernatant containing digested peptides was transferred into a new tube. Peptides were dried down using a speedvac and then lyophilized.
To enable multiplexing and peptide quantification, samples were labeled using isobaric Tandem Mass Tags (TMT, ThermoFisher). Lyophilized samples were resuspended in 50 mM HEPES buffer (pH 7.4), and TMT was resuspended in anhydrous acetonitrile. 400 μg of TMT was added per sample to a final volume of 50 μl. After allowing the labeling reaction to proceed for 1 hour at RT on a platform shaker at 400 rpm, the reaction was quenched by adding 3.2 μl of 5% Hydroxylamine in HPLC-grade water. After an additional 15 minutes on the shaker, samples were pooled and dried by vacuum centrifugation.
For phosphoserine and phosphothreonine mass spectrometry (MS) analysis, samples were then resuspended in 400 μl of buffer (100 mM Tris-HCl, 0.3% NP-40, pH 7.4), and phosphopeptides were enriched using High-Select™ Fe-NTA phosphopeptide enrichment kits (Thermo). The phosphopeptide eluent was reduced in volume to 1–5 μl using vacuum centrifugation and resuspended by adding 10 μl of 5% acetonitrile in 0.1% formic acid. Subsequently, the sample was loaded onto a 50 μm i.d. capillary (Polymicro technologies) liquid chromatography (LC) column containing 5 μm C18 beads and a tip fritted with a mixture of Lithisil (PQ corp), Tetramethylammonium (TFA) Silicate (Sigma), and Formamide (Sigma). Phosphopeptide enrichment flow-through was preserved for use in a global proteomics run, where 2 μl of 1:500 diluted sample in 0.1% acetic acid was loaded onto a similarly constructed 50 μm i.d. capillary LC column. The column was placed in line with an HPLC, and the following gradient was run at a flow rate of 2 μl/min (A = 0.1% acetic acid, B = 80% MeCN in 0.1% acetic acid): 0–4 min, 0–14% B; 4–50 min, 14–42% B; 50–57 min, 42–60% B; 57–60 min, 60–100% B, 60–68 min, 100% B, 68–69min, 100–0%B; 69–75 min, 0% B. The samples were analyzed on a ThermoFisher Q-Exactive Plus mass spectrometer. The instrument was operated in data-dependent acquisition mode, with the top 15 most abundant precursors with charge +2 or greater selected for fragmentation and dynamic exclusion set to 15 seconds. Precursors were isolated with a 0.4 m/z window and fragmented at 33 NCE via HCD. Precursor scan settings were set to AGC = 3e6, maximum IT = 50 ms, and resolution of 70,000. MS2 scan settings were set to AGC = 1e5, maximum IT = 300 ms, and resolution of 35,000. The total acquisition time was 75 minutes per sample. MS datafiles were searched on MASCOT version 2.4 with fixed modifications for NEM alkylation on cysteines (+125.047 Da), addition of TMT 6-plex to N-termini and lysine residues (+229.163 Da), oxidation of methionine residues (+15.995 Da) and phosphorylation on tyrosine, threonine, and serine residues (+79.966 Da). Precursor tolerance was 10 ppm, fragment tolerance was 15 mmu, two missed cleavages were allowed. Peptides were considered to be positively identified if they had an ion score of at least 25. Peptides were discarded if they had TMT reporter ion intensities less than 1000 in any one channel, if they did not contain a phospho- or TMT-modification for the phosphoproteomics runs, or if they had isolation interference of more than 40%. To control for technical variation between channels, values were normalized to the median of each channel of the global proteomics runs. Differential expression analysis for peptides and phophopeptides were performed using limma-voom in R. P-values were corrected for multiple testing using the FDR method (Benjamini and Hochberg, 1995).
2.10. Enrichment analyses
By performing enrichment analyses, we aimed to gain insight into the molecular mechanisms that underlie the functional outcome of manipulating ELAVL4 expression in neurons. First, we used Ingenuity Pathway Analysis (IPA) (http://www.ingenuity.com; QIAGEN Bioinformatics, Redwood City, CA, USA) to assess the global enrichment of canonical pathways within the generated transcriptomic and proteomic data. IPA uses the Ingenuity Knowledge Base, a combined repository of data from publicly accessible databases and data that are manually curated through systematically reviewing published literature, and currently, 712 canonical pathways have been annotated by IPA. For pathway enrichment, we analyzed all differentially expressed genes (DEGs), differentially expressed proteins (DEPs), and differentially phosphorylated proteins (DPPs) in the different neuronal lines with a FDR-corrected P-value (FDR P) < 5.0 × 10−2 and a fold change (FC) ≥ |1.20|. Peptides and phosphorylated peptides were mapped to one master protein, and the lowest P-value and the corresponding FC per protein was used for analysis. P-values for enriched canonical pathways were corrected using the FDR method (Benjamini and Hochberg, 1995).
Subsequently, we used IPA to perform ‘upstream regulator’ analyses (Kramer et al., 2014), using the same lists of DEGs, DEPs and DPPs (i.e., with FDR P < 5.0 × 10−2 and FC ≥ |1.20|). Upstream regulators can be any type of molecule—e.g., proteins, hormones, drugs and chemical compounds, and transcription factors—that can affect the expression of ‘target’ genes/proteins. For these analyses, we specifically focused on four upstream regulators, i.e., APP and tau, the key components of the main pathological lesions in AD (Jack et al., 2018), and the insulin receptor (INSR) and FOXO1, the only known transcriptional regulators of ELAVL4 expression (in pancreatic beta cells) (Lee et al., 2012). IPA calculates P-values that reflect the enrichment of targets of the upstream regulator in an input list (in this study our RNAseq and phosphoproteomics data), i.e., the amount of overlap between all known targets of the upstream regulator and the number of targets that are found in the input list. Furthermore, based on the direction of the expression changes of the input genes/proteins (up- or downregulated), IPA calculates an activation score (Z-score) that reflects the activation state of the upstream regulator. Z-scores ≥ 2 or ≤ −2 – reflecting predicted activation or inhibition of the upstream regulator-dependent, downstream effects on target gene expression, respectively – are considered significant.
3. Results
3.1. Generation of ELAVL4/HuD knock out, overexpression and rescue induced neurons
We used CRISPR-Cas9 to generate two monoclonal ELAVL4 KO induced pluripotent stem cell (iPSC) lines. Both lines were derived from an early onset AD patient carrying the APP London mutation (Muratore et al., 2014). One line harbored the APPV717I mutation (to be referred to as ‘fAD’), while in the second, isogenic line, the mutation was corrected to wild type (WT) APP (referred to as fAD-corrected or ‘fADcorr’) (Muratore et al., 2017). We sequenced the genomic region around the binding site of the sgRNA and selected the fADcorr ELAVL4 KO clone B1c2, which has 27 nucleotide and 46 nucleotide deletions, and the fAD ELAVL4 KO B1c20 containing a 123 nucleotide deletion with a 23 nucleotide insertion and a one nucleotide deletion, for “induced neuron” (iN) differentiation (Figure 1B). Monoclonal unedited CRISPR control lines (‘CRC’) that had been subjected to the same process were also obtained. Common CNV analysis in the monoclonal lines using the Nanostring nCounter Human CNV codeset indicated that the editing process did not result in large chromosomal abnormalities (Supplementary Figure 1). In iPSCs, ELAVL4 expression is low and not readily detectable by either qPCR or Western blotting. Hence, to confirm loss of ELAVL4 protein levels in the KO, we transduced the iPSCs with lentivirus encoding NGN2 to produce iNs. The used iN protocol yields a homogeneous population of cells with neuronal morphologies and transcriptional profiles consistent with layer 2/3 excitatory projection neurons within two weeks (Srikanth et al., 2018; Zhang et al., 2013). On iN differentiation DIV21, RNA and protein were collected and subjected to qPCR and Western blotting using ELAVL4-specific primers and antibodies. Full KO of both mRNA and protein was observed in fADcorr ELAVL4 KO (clone B1c2) and fAD ELAVL4 KO (clone B1c20) (Figures 1C, 1H, and 1I and Figures 1E, 1H, and 1K, respectively). Overexpression (OE) and rescue of ELAVL4 expression in the ELAVL4 KO lines was performed by lentiviral transduction of ELAVL4 splice variant (sv) 1 (ENST00000371823.8) or sv2 (ENST00000371824.7) cDNA on DIV6 and resulted in a sustained 10- to 20-fold increased mRNA levels on DIV21 (Figures 1D,1F). Lentiviral rescue of ELAVL4 expression was confirmed in ELAVL4 KO iNs by immunofluorescent staining. Only lentivirus infected, i.e., mCherry positive, iNs express ELAVL4 (Figure 1G). Western blotting showed that sv2 was the most predominantly expressed splice variant in DIV21 iNs (Figure 1H). Protein expression of the presynaptic neuronal marker SYN1 was not affected by ELAVL4 KO, OE, or rescue in iNs (Figure 1H).
3.2. ELAVL4 expression affects APP phenotypes in induced neurons
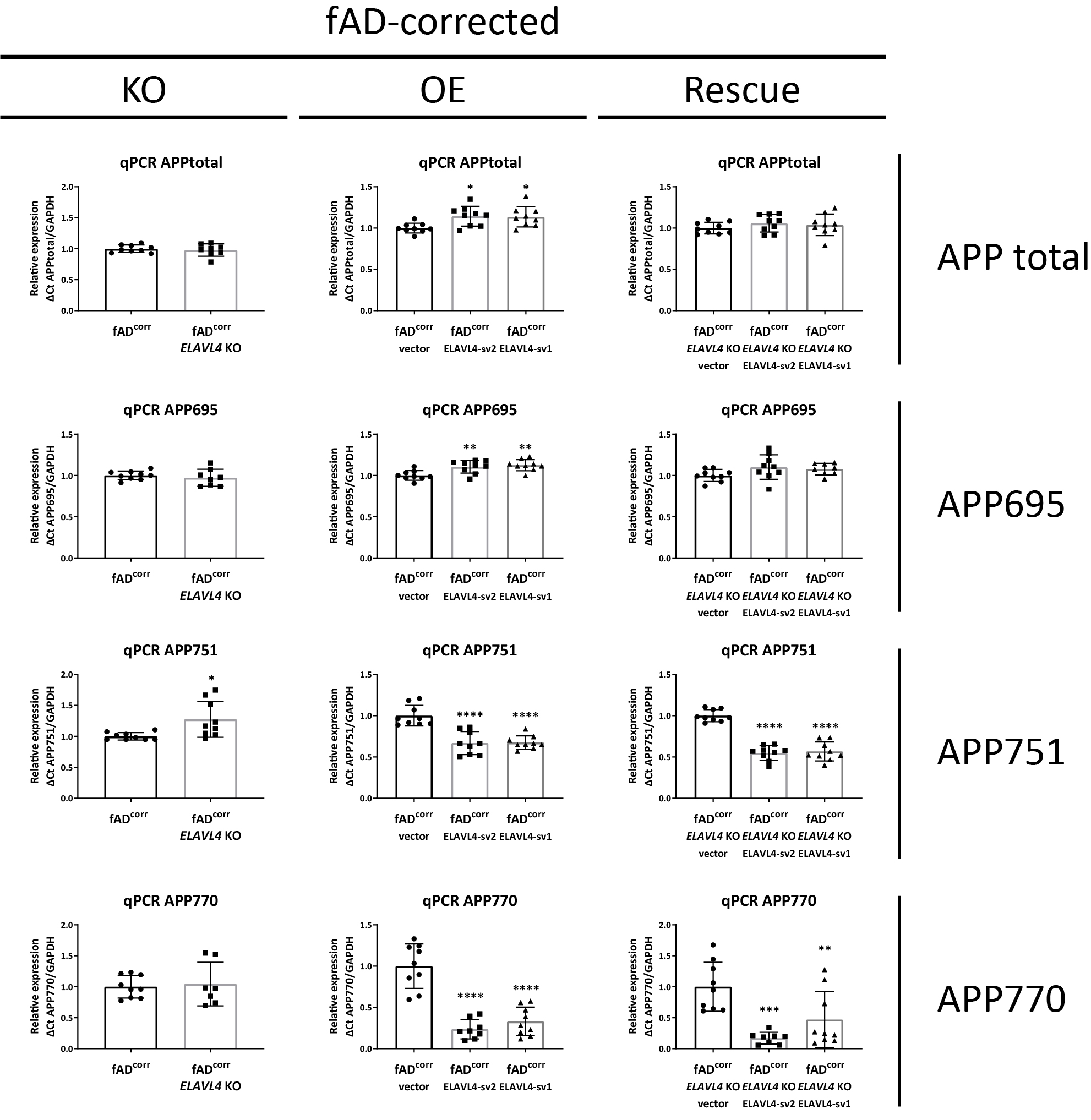
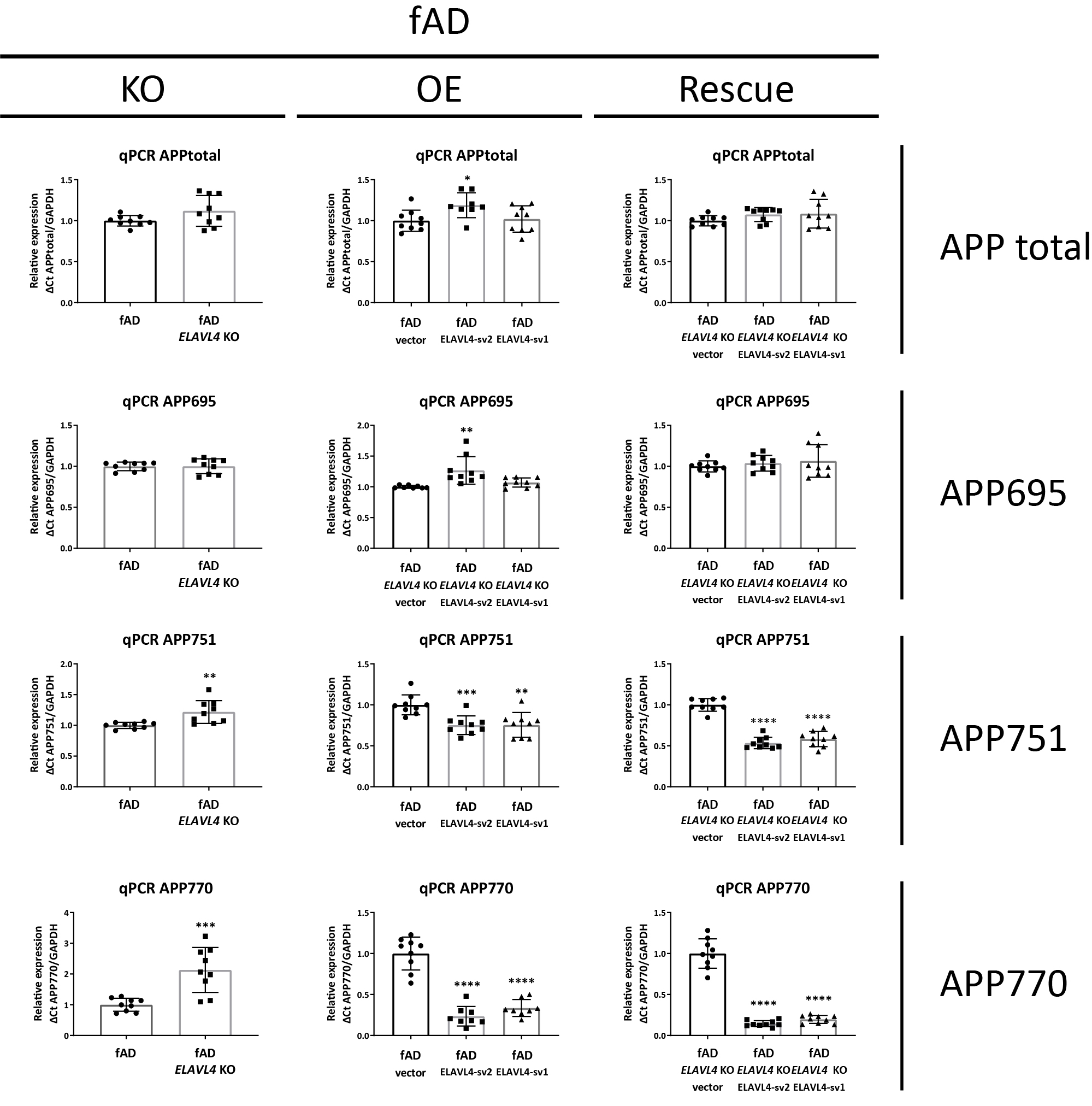
After we successfully manipulated ELAVL4 expression in iNs, we examined the effect of these manipulations on APP mRNA, protein, and processing. Alternative splicing of APP is known to result in three major isoforms: APP751 and APP770, which are ubiquitously expressed, and the predominantly neuronally expressed APP695 isoform (Wang et al., 2017). In both the fADcorr and fAD cell lines, loss-of-function of ELAVL4 resulted in the increased formation of longer APP splice variants (increased APP751 and APP770 isoforms) (Table 1, Supplementary Figures 2 and 3). OE and rescue of ELAVL4 expression in both ELAVL4 KO lines had the opposite effect: increased APP695 mRNA and reduced APP751 and APP770 mRNA (Table 1, Supplementary Figures 2 and 3). In the OE experiments, we observed an increase in total APP mRNA while in the KO and rescue experiment, total APP mRNA levels were not significantly affected (Table 1, Supplementary Figures 2 and 3). At the protein level, we observed lower APP expression after KO of EALVL4 in fAD neurons and increased APP protein expression in neurons with rescued or overexpressed ELAVL4 (Table 1, Figures 2A–D and 3A–D). We previously had found, and confirmed here, that fAD neurons express a higher Aβ42/40 ratio (Muratore et al., 2014). When we measured Aβ species that result from APP processing, we found that ELAVL4 KO lowered Aβ38 and Aβ40 levels in the culture media from fADcorr iNs and Aβ38, Aβ40, and Aβ42 levels in media of fAD iNs (Supplementary Figures 4 and 5). On the other hand, while OE of ELAVL4 sv2 increased the extracellular levels of Aβ38 and Aβ40, all other OE and rescue conditions did not affect the levels of the Aβ species measured (Supplementary Figures 4 and 5). However, the ratio of Aβ42/40 was consistently reduced in the ELAVL4 OE and rescue experiments (Table 1, Figures 2F,G and 3F,G).
Table 1.
Effects of ELAVL4 expression manipulation on APP and tau phenotypes
| APP Phenotypes | Tau Phenotypes | ||||
|---|---|---|---|---|---|
|
|
|||||
| RNA | Protein | Processing | RNA | Protein | |
|
| |||||
| Knock out | |||||
|
fADcorr
ELAVL4 KO |
Total ↓ 97.9 % n.s. | Total ↑100.1 % n.s. | Aβ42/40 ↑ 104.1 % n.s. | ↑ 111.9 % **** | Total tau ↑ 124.8 % * |
| 695 ↓ 97.2 % n.s. | Aβ42 ↓ 90.0 % n.s. | P-tau181 ↑ 105.2 % * | |||
| 751 ↑ 127.6 % * | Aβ40 ↓ 86.4 % * | ||||
| 770 ↑ 104.4 % n.s. | Aβ38 ↓ 90.8 % n.s. | ||||
|
fAD
ELAVL4 KO |
Total ↑112.0 % n.s. | Total ↓ 79.7 % ** | Aβ42/40 ↓ 97.7 % n.s. | ↓ 93.2 % * | Total tau ↑ 106.8 % n.s. |
| 695 ↑100.2 % n.s. | Aβ42 ↓ 84.3 % *** | P-tau181 ↑ 104.4 % * | |||
| 751 ↑ 121.9 % ** | Aβ40 ↓ 86.6 % ** | ||||
| 770 ↑ 213.5 % *** | Aβ38 ↓ 82.2 % ** | ||||
| Over Expression | |||||
|
fADcorr
ELAVL4-sv2 |
Total ↑ 114.3 % * | Total ↑ 114.7 % ** | Aβ42/40 ↓ 90.1 % ** | ↑ 103.9 % n.s. | Total tau ↓ 94.0 % n.s. |
| 695 ↑ 110.4 % ** | Aβ42 ↓ 94.2 % n.s. | P-tau181 ↓ 89.3 % * | |||
| 751 ↓ 66.8 % **** | Aβ40 ↑ 104.6 % n.s | ||||
| 770 ↓ 23.7 % **** | Aβ38 ↑ 108.9 % n.s | ||||
|
fAD
ELAVL4-sv2 |
Total ↑ 118.9 % * | Total ↑ 210.9 % **** | Aβ42/40 ↓ 87.4 % **** | ↑105.5 % n.s. | DAKO ↓ 99.7 % n.s. |
| 695 ↑ 126.8 % ** | Aβ42 ↓ 85.4 % n.s | P-tau181 ↓ 90.6 % * | |||
| 751 ↓ 75.2 % *** | Aβ40 ↓ 97.4 % n.s | ||||
| 770 ↓ 23.5 % **** | Aβ38 ↓ 96.2 % n.s. | ||||
|
fADcorr
ELAVL4-sv1 |
Total ↑ 113.6 % * | Total ↓ 85.1 % ** | Aβ42/40 ↓ 88.1 % *** | ↑ 110.0 % * | Total tau ↑ 100.4 % n.s. |
| 695 ↑ 112.5 % ** | Aβ42 ↑ 110.7 % n.s | P-Tau181 ↓ 83.7 % *** | |||
| 751 ↓ 67.5 % **** | Aβ40 ↑ 127.1 % ** | ||||
| 770 ↓ 33.1 % **** | Aβ38 ↑ 122.4 % ** | ||||
|
fAD
ELAVL4-sv1 |
Total ↑102.1 % n.s. | Total ↑ 145.9 % n.s. | Aβ42/40 ↓ 83.9 % **** | ↑ 114.0 % * | Total tau ↑ 119.4 % ** |
| 695 ↑ 107.0 % | Aβ42 ↓ 94.4 % n.s | P-tau181 ↓ 83.3 % **** | |||
| 751 ↓ 75.5 % ** | Aβ40 ↑ 111.5 % n.s. | ||||
| 770 ↓ 33.6 % **** | Aβ38 ↑ 103.6 % n.s. | ||||
| Rescue in ELAVL4 KO | |||||
|
fADcorr
ELAVL4 KO ELAVL4-sv2 |
Total ↑ 105.7 % n.s. | Total ↑ 140.8 % **** | Aβ42/40 ↑ 100.8 % n.s | ↓ 94.2 % n.s | DAKO ↓ 86.7 % n.s. |
| 695 ↑ 110.1 % n.s. | Aβ42 ↓ 85.0 % n.s. | P-tau181 ↓ 78.4 % **** | |||
| 751 ↓ 54.7 % **** | Aβ40 ↓ 84.6 % n.s. | ||||
| 770 ↓ 17.0 % *** | Aβ38 ↓ 96.0 % n.s. | ||||
|
fAD
ELAVL4 KO ELAVL4-sv2 |
Total ↑ 107.6 % n.s. | Total ↑ 277.6 % ** | Aβ42/40 ↓ 89.1 % *** | ↑106.3 % n.s. | DAKO ↓ 85.7 % n.s. |
| 695 ↑ 103.9 % n.s. | Aβ42 ↓ 92.4 % n.s | P-tau181 ↓ 85.2 % *** | |||
| 751 ↓ 53.5 % **** | Aβ40 ↑ 103.6 % n.s. | ||||
| 770 ↓ 14.5 % **** | Aβ38 ↑ 112.6 % n.s. | ||||
|
fADcorr
ELAVL4 KO ELAVL4-sv1 |
Total ↑ 103.9 % n.s. | Total ↑ 110.7 % n.s. | Aβ42/40 ↓ 93.0 % * | ↓ 95.4 % n.s | DAKO ↓ 91.6 % n.s. |
| 695 ↑ 107.7 % n.s. | Aβ42 ↓ 95.4 % n.s. | P-tau181 ↓ 70.4 % **** | |||
| 751 ↓56.7 % **** | Aβ40 ↑ 104.4 % n.s | ||||
| 770 ↓ 47.0 % ** | Aβ38 ↓ 99.6 % n.s. | ||||
|
fAD
ELAVL4 KO ELAVL4-sv1 |
Total ↑ 108.6 % n.s. | Total ↑ 241.5 % * | Aβ42/40 ↓ 88.4 % *** | ↑ 111.3 % ** | Total tau ↓ 89.1 % n.s. |
| 695 ↑ 106.6 % n.s. | Aβ42 ↓ 95.3 % n.s | P-tau181 ↓ 81.8 % **** | |||
| 751 ↓ 58.3 % **** | Aβ40 ↑ 108.9 % n.s. | ||||
| 770 ↓ 19.7 % **** | Aβ38 ↑ 113.3 % n.s. | ||||
NOTE. Summarized data. For all conditions: per condition, three replicates from three independent differentiations were used. Total protein determined by BCA was used to normalize measured values for individual Aβ species. For KO conditions: changes relative to CRISPR control line, P-value calculated with ↑ tests. For OE and rescue conditions: changes relative to empty vector control, P-values calculated with Dunnett’s multiple comparison that followed after ANOVA test.
P < 0.05
P < 0.01
P < 0.001
P < 0.0001
Figure 2.
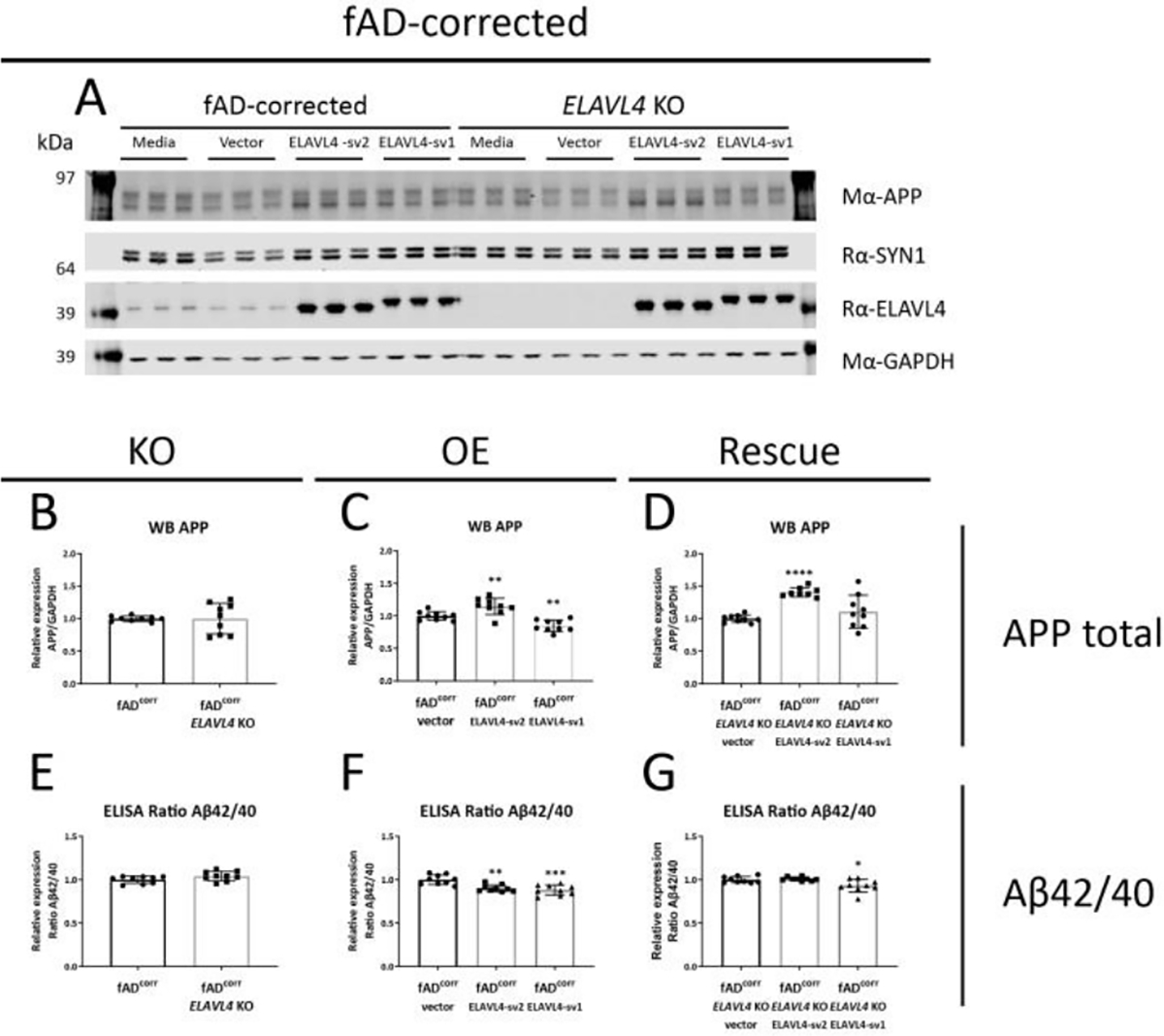
ELAVL4 affects APP protein levels and Aβ42/40 ratios in fADcorr iNs.
(A) Example western blot and (B-D) quantifications are shown for ELAVL4 knock out (KO) overexpression (OE) and rescue of ELAVL4 in fAD-corrected iNs. In the KO, changes relative to CRISPR control line that underwent mock targeting with an empty sgRNA vector transfected alongside with Cas9, and monoclonal subclones were isolated and analyzed in parallel to subclones where ELAVL4 was targeted. For OE and rescue conditions, changes relative to empty vector OE plasmid in the CRISPR control or ELAVL4 KO iNs respectively. At day 21 of differentiation, 48h condition media was collected and cells were lysed. (E-G) Aβ40 and Aβ42 levels were measured via multiplexed ELISA (MSD), and normalized to the fAD-corrected CRISPR control for each differentiation. Quantifications (B-G) are from 3 independent differentiations with n=3. In the KO experiments P-values were calculated with t tests, while in the OE and rescue experiments, P-values were calculated with Dunnett’s multiple comparison that followed after a significant ANOVA test, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Figure 3.
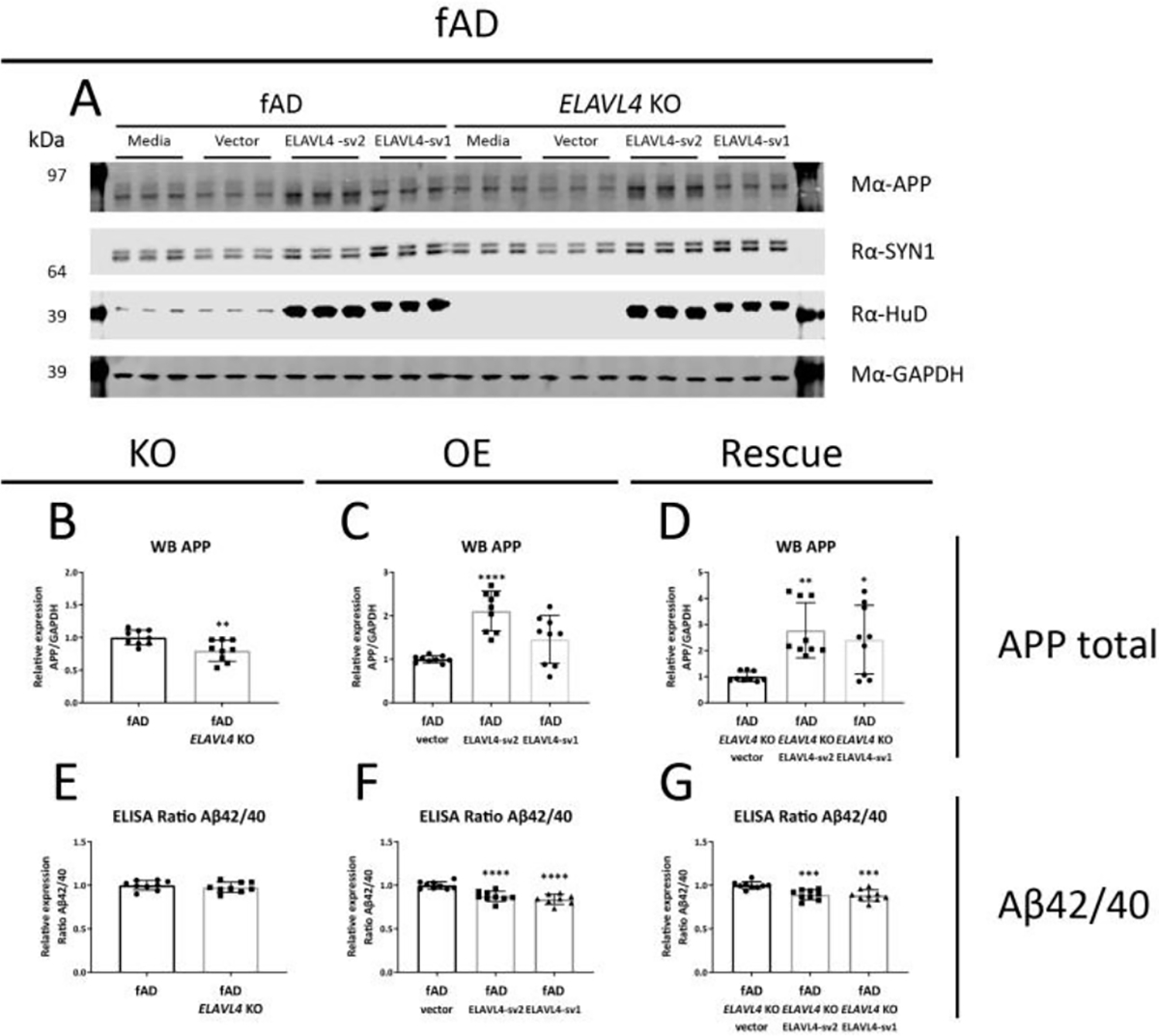
ELAVL4 affects APP protein levels and Aβ42/40 ratios in fAD iNs.
(A) Example western blot and (B-D) quantifications are shown for ELAVL4 knock out (KO) overexpression (OE) and rescue of ELAVL4 in fAD iNs. In the KO, changes relative to CRISPR control line that underwent mock targeting with an empty sgRNA vector transfected alongside with Cas9, and monoclonal subclones were isolated and analyzed in parallel to subclones where ELAVL4 was targeted. For OE and rescue conditions, changes relative to empty vector OE plasmid in the CRISPR control or ELAVL4 KO iNs respectively. At day 21 of differentiation, 48h condition media was collected and cells were lysed. (E-G) Aβ40 and Aβ42 levels were measured via multiplexed ELISA (MSD), and normalized to the fAD-corrected CRISPR control for each differentiation. Quantifications (B-G) are from 3 independent differentiations with n=3. In the KO experiments P-values were calculated with t tests, while in the OE and rescue experiments, P-values were calculated with Dunnett’s multiple comparison that followed after a significant ANOVA test, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
3.3. ELAVL4 expression affects tau phenotypes in induced neurons
We further evaluated the effect of ELAVL4 manipulation on AD-relevant phenotypes by examining tau, a protein that aggregates in the other pathological hallmark of AD: neurofibrillary tangles. The levels of tau mRNA increase and decrease in ELAVL4 KO iNs in fADcorr and fAD backgrounds, respectively (Table 1, Figures 4A and 5A). Overexpression of ELAVL4 sv1 increased tau mRNA in both backgrounds, and rescue with ELAVL4 sv1 increased tau mRNA in the fAD ELAVL4 KO iNs (Table 1, Figures 4B,C and 5B,C). The elevated phosphorylation of tau protein (pTau) facilitates intracellular tangle formation. We previously had found, and confirmed here, that fAD neurons express higher pTau protein levels (Muratore et al., 2014). ELAVL4 KO, OE, and rescue of ELAVL4 did not significantly alter total tau protein levels in most cells, with the exception of increased tau protein in the fADcorr ELAVL4 KO iNs and ELAVL4 sv2 OE in fAD iNs (Table 1, Figures 4G–I and 5G–I). However, ELAVL4 KO did consistently increase tau phosphorylation, specifically phosphorylation on residue threonine 181 (Thr181) in both the fADcorr and fAD cell lines (Table 1, Figures 4J and 5J). On the other hand, increasing ELAVL4 levels through OE and rescue experiments had the opposite effect in both fADcorr and fAD iNs, reducing phosphorylated Thr181 tau compared to total tau (Table 1, Figures 4K,L and 5K,L).
Figure 4.
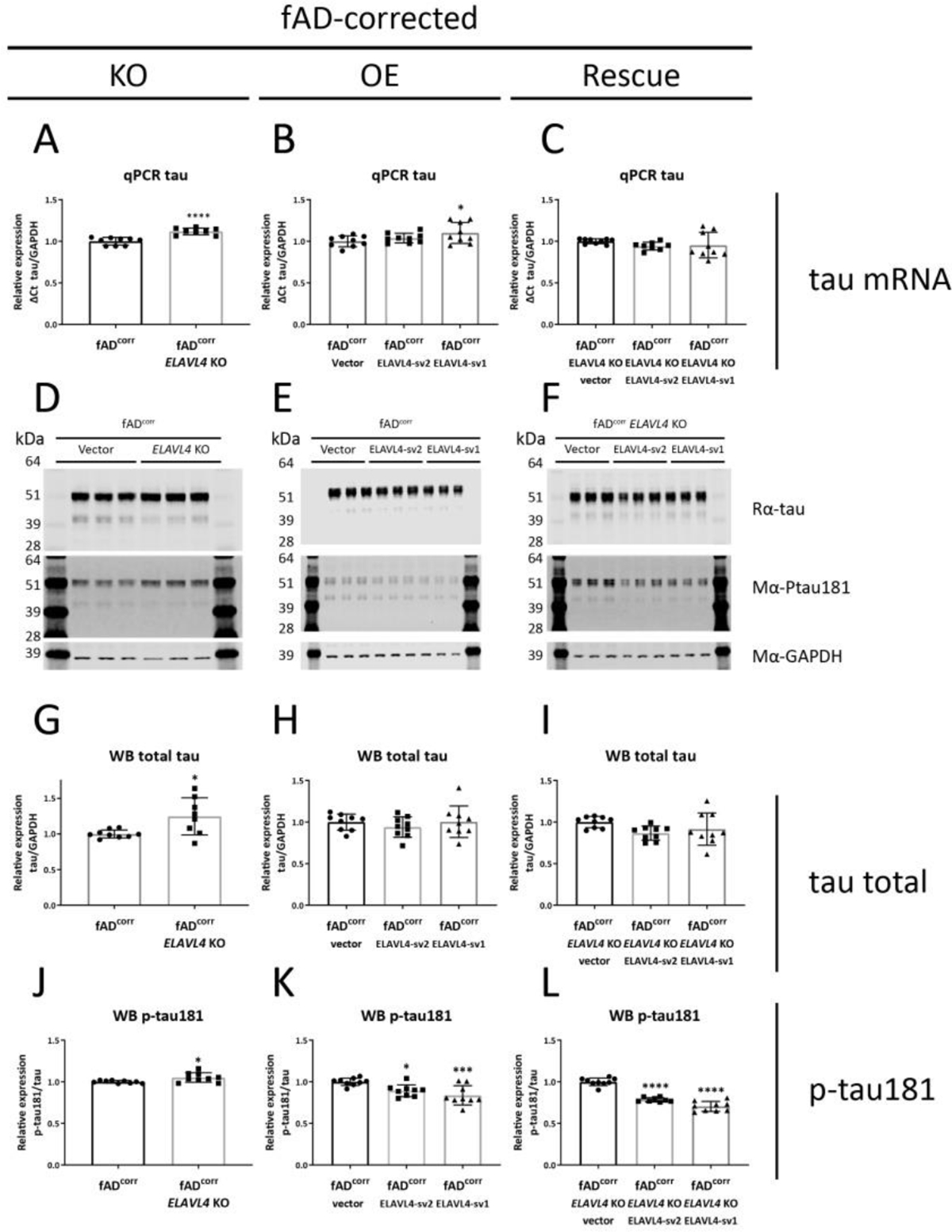
Increased ELAVL4 expression reduces tau phosphorylation in fAD-corrected iNs.
(A-C) Quantification of tau mRNA in iNs from ELAVL4 knock out (KO) overexpression (OE) and rescue in fAD-corrected induced neurons (iNs). In the KO, changes relative to CRISPR control line that underwent mock targeting with an empty sgRNA vector transfected alongside with Cas9, and monoclonal subclones were isolated and analyzed in parallel to subclones where ELAVL4 was targeted. For OE and rescue conditions, changes relative to empty vector OE plasmid in the CRISPR control or ELAVL4 KO iNs respectively. At day 21 of differentiation, cells were lysed and protein and RNA samples were harvested. (D-F) example blots and (G-L) quantifications tau and p-tau181 protein in ELAVL4 KO, OE, and rescue fAD-corrected iNs. Quantifications (A-C,G-L) are from 3 independent differentiations with n=3. In the KO experiments P-values were calculated with t tests, while in the OE and rescue experiments, P-values were calculated with Dunnett’s multiple comparison that followed after a significant ANOVA test, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Figure 5.
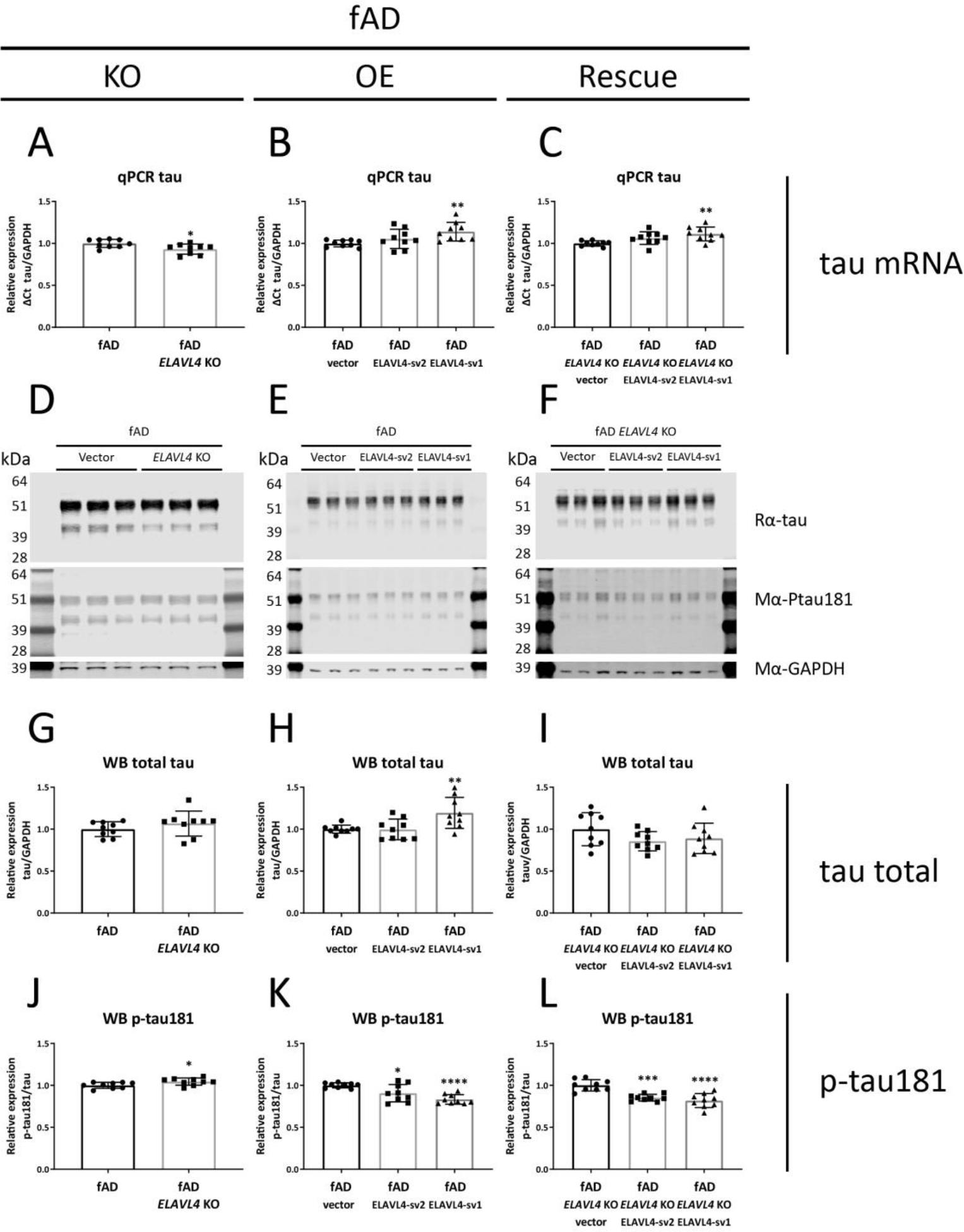
Increased ELAVL4 expression reduces tau phosphorylation in fAD iNs.
(A-C) Quantification of tau mRNA in iNs from ELAVL4 knock out (KO) overexpression (OE) and rescue in fAD induced neurons (iNs). In the KO, changes relative to CRISPR control line that underwent mock targeting with an empty sgRNA vector transfected alongside with Cas9, and monoclonal subclones were isolated and analyzed in parallel to subclones where ELAVL4 was targeted. For OE and rescue conditions, changes relative to empty vector OE plasmid in the CRISPR control or ELAVL4 KO iNs respectively. At day 21 of differentiation, cells were lysed and protein and RNA samples were harvested. (D-F) example blots and (G-L) quantifications tau and p-tau181 protein in ELAVL4 KO, OE, and rescue fAD iNs. Quantifications (A-C,G-L) are from 3 independent differentiations with n=3. In the KO experiments P-values were calculated with t tests, while in the OE and rescue experiments, P-values were calculated with Dunnett’s multiple comparison that followed after a significant ANOVA test, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
3.4. Canonical pathway and upstream regulator analysis results
After confirming that decreasing or increasing ELAVL4 expression affects AD-related molecular changes in iNs, we aimed to gain further insight into the molecular cascades involved in ELAVL4 signaling through conducting analyses of transcriptomic and proteomic data from iNs derived from the different cell lines that we had generated. For further analysis, we defined 10 sets of differentially expressed genes (DEGs) generated through comparing total RNA sequencing data from 48 samples of iNs that included 3 biological replicates for each of 16 conditions (Figure 6A, Table 3). First, we analyzed the DEGs in fADcorr ELAVL4 KO iNs compared to fADcorr iNs with WT ELAVL4 (Table 3.1, Figure 6B), the DEGs in fADcorr iNs in which ELAVL4 was overexpressed (combined analysis of ELAVL4 sv1 and sv2 OE) compared to fADcorr iNs with WT ELAVL4 (Table 3.2, Figure 6C), and the genes that were differentially expressed in the opposite direction (DEGs-OD) in the fADcorr ELAVL4 sv1+sv2 OE iNs and the fADcorr ELAVL4 KO (Table 3.3). Then, we analyzed the DEGs from the rescue experiments: the DEGs in fADcorr ELAVL4 KO iNs with ELAVL4 sv1+sv2 rescue compared to fADcorr ELAVL4 KO iNs (Table 3.4, Figure 6D), as well as the DEGs-OD in the fADcorr ELAVL4 sv1+sv2 rescue iNs and the fADcorr ELAVL4 KO iNs (Table 3.5). Subsequently, we performed the same five KO, OE, and rescue analyses based on RNAseq data from fAD iNs (Table 3.6–3.10, Supplementary Figure 6A–C). In addition to RNAseq data, we analyzed four sets of differentially expressed proteins (DEPs) and differentially expressed phosphoproteins (DPPs): DEPs and DPPs in fAD ELAVL4 KO iNs compared to fAD iNs (Table 3.11–12, Figure 7A,B), and DEPs and DPPs in fAD ELAVL4 KO iNs with ELAVL4 sv2 rescue compared to fAD ELAVL4 KO iNs (Table 3.13–14, Figure 7C,D).
Figure 6.

RNAseq characterization of iPSC lines generated in this study.
(A) Purified RNA of cell lysates collected from iNs DIV21 were analyzed by RNAseq. Genes were selected that mark subsets of neuronal and glial cells and a heat map was created using the pheatmap package in R. HKG, housekeeping gene iPSC/NPC, induced pluripotent stem cell/neural progenitor cell. (B) Differentially expressed genes (DEGs) in fADcorr ELAVL4 KO iNs compared to fADcorr iNs (C) DEGs in fADcorr iNs in which ELAVL4 was overexpressed (combined analysis of ELAVL4 sv1 and sv2 OE) compared to fADcorr iNs. (D) DEGs from the rescue experiments: the DEGs in fADcorr ELAVL4 KO iNs with ELAVL4 sv1+sv2 rescue compared to fADcorr ELAVL4 KO iNs. DEGs with FDR corrected P-values < 0.05 and fold changes ≥ |1.20| are indicated in red. Volcano DEGs plots were generated using the EnhancedVolcano package in R.
Table 3.
Canonical pathway analysis of RNAseq and (phospho)proteomics ELAVL4 KO, overexpression, and rescue neuronal cell lines, using IPA.
| RNAseq | phosphoprot eomics | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| fAD-corrected | fAD | fAD | |||||||||
| (1) 6260 DEGs ELAVL4 KO in fADcorr | (6) 3661 DEGs ELAVL4 KO in fAD | (11) 40 DEPs ELAVL4 KO in fAD | (12) 48 DPPs ELAVL4 KO in fAD | ||||||||
| Canonical Pathway | P-value | Canonical Pathway | P-value | Canonical Pathway | P-value | Canonical Pathway | P-value | ||||
| Axonal Guidance Signaling | 1,00E-19 | Synaptogenesis Signaling Pathway | 3,16E-11 | Sirtuin Signaling Pathway | 6,76E-05 | CDK5 Signaling | 2,29E-04 | ||||
| Synaptogenesis Signaling Pathway | 1,00E-13 | Axonal Guidance Signaling | 2,75E-08 | Mitochondrial Dysfunction | 5,25E-04 | UVB-Induced MAPK Signaling | 2,29E-04 | ||||
| Molecular Mechanisms of Cancer | 2,51E-12 | Sperm Motility | 8,13E-07 | NAD Signaling Pathway | 4,47E-03 | UVC-Induced MAPK Signaling | 2,29E-04 | ||||
| p53 Signaling | 5,75E-09 | Calcium Signaling | 9,77E-07 | Protein Ubiquitination Pathway | 2,24E-02 | Apelin Cardio myocyte Signaling Pathway | 2,29E-04 | ||||
| Kinetochore Metaphase Signaling Pathway | 5,75E-09 | Neuropathic Pain Signaling In Dorsal Horn Neurons | 1,07E-05 | DNA Methylation and Transcriptional Repression Signaling | 2,51E-02 | 14-33-mediated Signaling | 2,34E-04 | ||||
| (2) 609 DEGs ELAVL4 sv1+2 OE fADcorr | (3) 207 DEGs-OD ELAVL4 sv1+sv2 OE in fADcorr vs. ELAVL4 KO in fADcorr | (7) 360 DEGs ELAVL4 sv1+2 OE in fAD | (8) 74 DEGs-OD ELAV L4 sv1+sv 2 OE in fAD vs. ELAVL 4 KO in fAD | ||||||||
| Canonical Pathway | P-value | Canonical Pathway | P-value | Canonical Pathway | P -value | Canonical Pathway | P -value | ||||
| Kinetochore Metaphase Signaling Pathway | 6,61E-09 | Kinetochore Metaphase Signaling Pathway | 5,01E-14 | Human Embryonic Stem Cell Pluripotency | 2,57E-08 | cAMP-mediated signaling | 1,12E-02 | ||||
| Cyclins and Cell Cycle Regulation | 9,33E-08 | Cell Cycle: G2/M DNA Damage Checkpoint Regulation | 9,12E-10 | Cyclins and Cell Cycle Regulation | 2,57E-08 | ||||||
| Cell Cycle: G2/M DNA Damage Checkpoint Regulation | 1,20E-07 | Mitotic Roles of Polo-Like Kinase | 3,16E-07 | Cell Cycle: G2/M DNA Damage Checkpoint Regulation | 1,66E-07 | ||||||
| Cell Cycle: G1/S Checkpoint Regulation | 3,80E-07 | Cyclins and Cell Cycle Regulation | 1,48E-06 | Mouse Embryonic Stem Cell Pluripotency | 2,09E-07 | ||||||
| Mitotic Roles of Polo-Like Kinase | 2,57E-05 | ATM Signaling | 5,75E-05 | Regulation of the Epithelial-Mesenchymal Transition Pathway | 1,07E-06 | ||||||
| (4) 2032 DEGs ELAVL4 sv1+sv2 rescue in fADcorr ELAVL4 KO | (5) 920 DEGs-OD ELAVL4 sv1+sv2 rescue in fADcorr KO vs. ELAVL4 KO in fADcorr | (9) 1253 DEGs ELAVL4 sv1+sv2 rescue in fAD ELA VL4 KO | (10) 383 DEGs-OD ELAV L4 sv1+sv 2 rescue in fAD KO vs. ELAVL 4 KO in fAD | (13) 67 DEPs ELAVL4 sv2 Rescue in fAD ELAVL4 KO | (14) 103 DPPs ELAVL 4 sv2 Rescue in fAD ELAVL4 KO | ||||||
| Canonical Pathway | P-value | Canonical Pathway | P-value | Canonical Pathway | P-value | Canonical Pathway | P-value | Canonical Pathway | P-value | Canonical Pathway | P-value |
| Axonal Guidance Signaling | 1,58E-11 | Axonal Guidance Signaling | 3,89E-09 | Axonal Guidance Signaling | 4,79E-09 | Axonal Guidance Signaling | 1,00E-03 | Protein Kinase A | 3,24E-06 | Semaphorin Signaling in Neurons | 5,62E-08 |
| Molecular Mechanisms of Cancer | 1,23E-09 | Hepatic Fibrosis / Hepatic Stellate Cell Activation | 5,62E-09 | Factors Promoting Cardiogenesis in Vertebrates | 3,39E-06 | Calcium Signaling | 1,00E-03 | Role of PKR in Interferon Induction and Antiviral Response | 4,37E-06 | Renin-Angiotensin Signaling | 5,13E-07 |
| Hepatic Fibrosis / Hepatic Stellate Cell Activation | 1,12E-08 | Human Embryonic Stem Cell Pluripotency | 1,70E-08 | Molecular Mechanisms of Cancer | 3,39E-06 | Hepatic Fibrosis Signaling Pathway | 2,40E-03 | 14-33-mediated Signaling | 5,01E-06 | Semaphorin Neuronal Repulsive Signaling Pathway | 1,55E-06 |
| Hepatic Fibrosis Signaling Pathway | 1,55E-07 | Hepatic Fibrosis Signaling Pathway | 1,32E-07 | Human Embryonic Stem Cell Pluripotency | 3,39E-06 | Hepatic Fibrosis / Hepatic Stellate Cell Activation | 1,15E-02 | BAG2 Signaling Pathway | 9,12E-06 | ERK/MAPK Signaling | 1,91E-06 |
| Breast Cancer Regulation by Stathminl | 2,09E-07 | Kinetochore Metaphase Signaling Pathway | 1,78E-07 | Cyclins and Cell Cycle Regulation | 3,39E-06 | Rho GDI Signaling | 1,15E-02 | Sirtuin Signaling Pathway | 1,12E-05 | LPS-stimulated MAPK Signaling | 3,98E-06 |
NOTE Pathways that were among the top 5 most enriched pathways more than once are highlighted in bold. P-values are FDR-corrected for multiple testing
Figure 7.
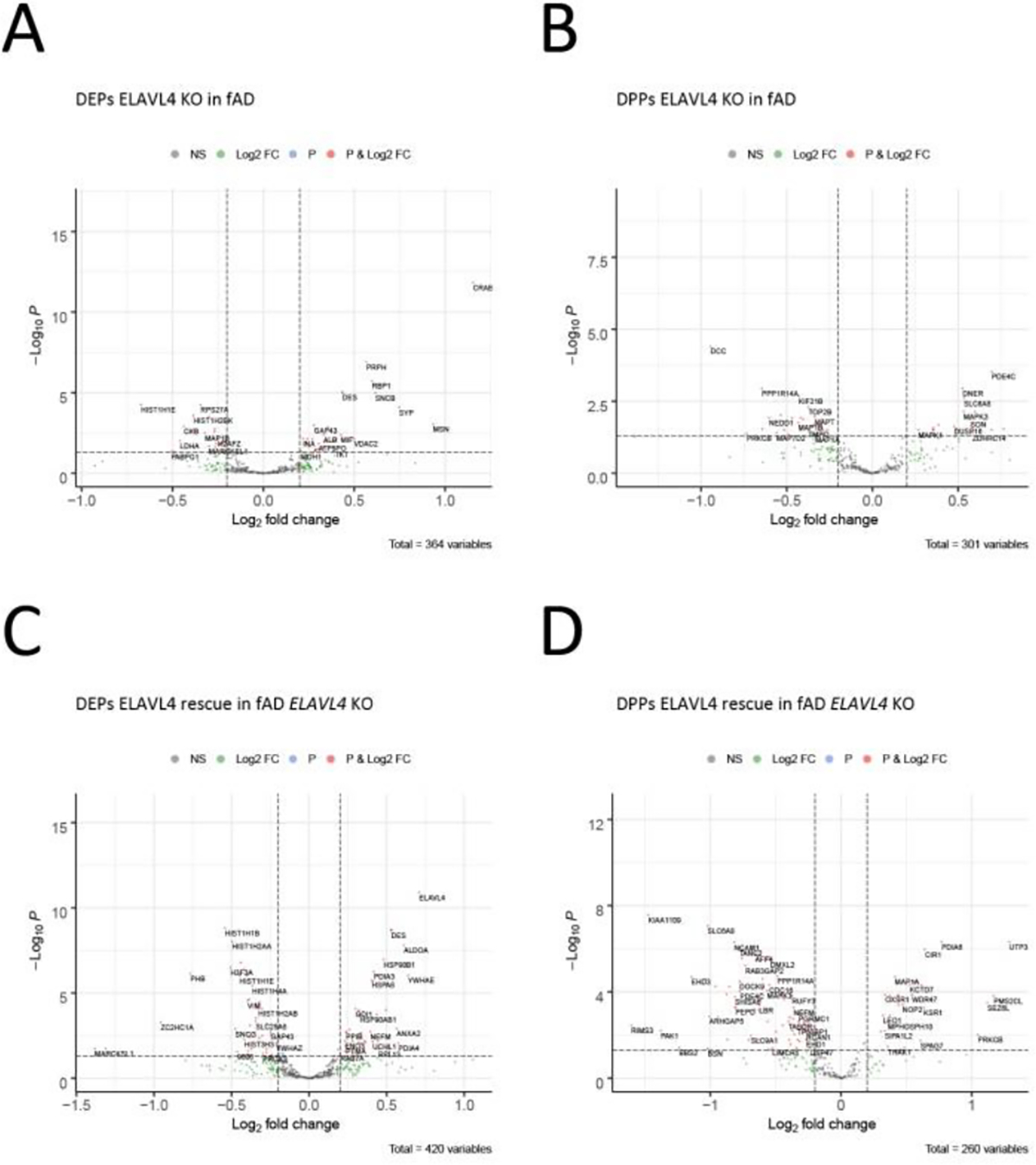
Proteomic characterization of iPSC lines generated in this study.
(A,C) Protein and (B,D) phosphoprotein quantification of DIV21 iNs from (A,B) ELAVL4 KO and (C,D) rescue in fAD induced neurons. Differentially expresses peptides (DEPs) and differentially phosporylated peptides (DPPs) with FDR corrected P-values < 0.05 and fold changes ≥ |1.20| are indicated in red. Volcano DEPs and DPPs plots were generated using the EnhancedVolcano package in R.
The data sets generated provide a rich resource for interrogating ELAVL4/HuD biology. Supplementary Table 1 includes the results of each of the above-described analyses, which can be readily probed for any gene or protein of interest. For example, as mentioned in the introduction, several genes have been described in the literature to be targets of ELAVL4, and in Table 2, we have listed these genes, with their expression changes in human neurons with ELAVL4 knock out or overexpression. Furthermore, the differential expression data can be searched for genes involved in AD-implicated molecular processes that may impact mitochondrial function, such as energy metabolism, mitochondrial dynamics and biogenesis, and autophagy (Ashleigh et al., 2022). In addition, the processed and raw (FASTQ) gene expression data is available through the gene expression omnibus (GSE202985).
Table 2.
Expression of ELAVL4 and ELAVL4 targets after ELAVL4 manipulation
| ELAVL4 knock out | ELAVL4 overexpression | ELAVL4 rescue in knock out | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||
| RNAseq | Protein | RNAseq | RNAseq | Protein | ||||||||||||
| fAD corrected | fAD | fAD corrected | fAD | fAD corrected | fAD | fAD corrected | fAD | |||||||||
| Gene | FC | FDR-P | FC | FDR-P | FC | FDR-P | FC | FDR-P | FC | FDR-P | FC | FDR-P | FC | FDR-P | FC | FDR-P |
| ELAVL4/HuD | −4,47 | 2,86E-37 | 3,24 | 8,13E-34 | N.A. | N.D. | 2,86 | 8,49E-33 | 2,62 | 2,25E-31 | 15,14 | 3,11E-48 | 9,40 | 1,13E-45 | 1,635 | 1,21E-11 |
| CAMK2A | 1,92 | 8,26E-12 | 1,10 | 2,42E-01 | N.A. | N.D. | −1,26 | 5,71E-02 | −1,29 | 3,13E-02 | −1,96 | 1,30E-10 | −1,53 | 1,43E-05 | N.A | N.D. |
| BDNF | 1,17 | 4,83E-03 | −1,19 | 1,55E-01 | N.A. | N.D. | −1,13 | 1,89E-01 | −1,04 | 7,80E-01 | −1,18 | 1,13E-02 | −1,15 | 3,58E-02 | N.A | N.D. |
| GAP43 | −1,07 | 4,00E-03 | 1,11 | 1,19E-04 | 1,21 | 1,05E-03 | 1,09 | 1,17E-02 | 1,12 | 1,07E-03 | 1,14 | 9,46E-06 | 1,07 | 3,02E-02 | −1,19 | 1,89E-03 |
| APP | 1,16 | 7,03E-13 | −1,06 | 4,32E-04 | N.A. | N.D. | 1,02 | 4,51E-01 | −1,10 | 8,38E-02 | −1,05 | 4,18E-03 | −1,03 | 1,16E-01 | N.A | N.D. |
| BACE1 | 1,13 | 6,71E-04 | 1,03 | 4,78E-01 | N.A. | N.D. | 1,08 | 1,26E-01 | −1,05 | 8,54E-01 | 1,01 | 8,06E-01 | 1,02 | 6,84E-01 | N.A | N.D. |
| ADAM10 | −1,10 | 1,69E-03 | −1,02 | 6,53E-01 | N.A. | N.D. | −1,10 | 2,73E-02 | −1,04 | 4,74E-01 | −1,22 | 5,78E-07 | −1,12 | 3,48E-03 | N.A | N.D. |
| NEP | −3,21 | 7,98E-08 | −1,23 | 3,39E-01 | N.A. | N.D. | −1,35 | 2,77E-01 | −1,14 | 7,80E-01 | −1,07 | 8,49E-01 | −1,26 | 4,01E-01 | N.A | N.D. |
| SERPINI1 | 1,29 | 3,55E-13 | 1,01 | 7,54E-01 | N.A. | N.D. | −1,04 | 4,02E-01 | −1,06 | 2,30E-01 | −1,01 | 7,64E-01 | −1,07 | 4,68E-02 | N.A | N.D. |
| TAU | −1,02 | 5,66E-01 | −1,02 | 4,84E-01 | 1,09 | 6,02E-01 | 1,15 | 1,48E-04 | 1,38 | 6,24E-01 | 1,16 | 2,85E-06 | 1,11 | 4,07E-04 | 1,08 | 5,07E-01 |
| ACHE | 1,02 | 6,09E-01 | −1,10 | 5,86E-03 | N.A. | N.D. | 1,18 | 2,15E-04 | 1,18 | 1,26E-04 | 1,30 | 1,63E-09 | 1,20 | 3,80E-06 | N.A | N.D. |
Abbreviations: FC, Fold change; FDR-P, FDR corrected P-value
In order to obtain a global view of the pathways that are altered following ELAVL4 modulation, we used IPA to conduct canonical pathway analyses of the above-described lists of DEGs, DEPs, and DPPs. When considering the top five most significantly enriched pathways for each comparison, 40 different pathways were enriched in the DEGs, DEPs, and DPPs (Table 3; full canonical pathway analysis results in Supplementary Table 2). Canonical pathways that were found to be among the top five enriched pathways in more than one comparison are (“X”=number of comparisons showing enrichment): “Axonal guidance signaling” (6X), “Cell Cycle: G2/M DNA Damage Checkpoint Regulation (4X)”, “Cyclins and Cell Cycle Regulation” (4X), “Kinetochore Metaphase Signaling Pathway” (4X), “Hepatic Fibrosis / Hepatic Stellate Cell Activation” (3X), “Hepatic Fibrosis Signaling Pathway” (3X), “Human Embryonic Stem Cell Pluripotency” (3X), “Molecular Mechanisms of Cancer” (3X), “14-3-3-mediated Signaling” (2X), “Calcium Signaling” (2X), “Mitotic Roles of Polo-Like Kinase” (2X), “Sirtuin Signaling Pathway” (2X), and “Synaptogenesis Signaling Pathway” (2X) (Table 3).
We then performed focused ‘upstream regulator’ analyses to identify regulators that influence the expression of multiple downstream targets, and we have used similar analyses previously to find upstream regulators for movement disorders (Klemann et al., 2018; Macri et al., 2018; Pagliaroli et al., 2019). We focused on assessing if altering ELAVL4 expression in neurons affects genes and proteins that are downstream transcriptional targets of four regulators. These regulators were APP and tau, the key components of the AD brain lesions, as well as the insulin receptor (INSR) and FOXO1, the only known transcriptional regulators of ELAVL4 expression (a mechanism that was demonstrated in pancreatic beta cells (Lee et al., 2012)). The analyses revealed that, with the exception of analysis 8 (DEGs-OD in the fAD ELAVl4 sv1+sv2 OE iNs versus the fAD ELAVL4 KO), all analyzed DEGs, DEPs and DPPs are significantly enriched for transcriptional targets downstream of APP signaling (Table 4). In analysis 1 (DEGs in the fADcorr ELAVL4 KO iNs and the fADcorr iNs), APP was also predicted to be activated (Z-score 2,36) (Table 4). In addition, half of the analyses (7/14) revealed a significant enrichment of transcriptional targets downstream of tau signaling (Table 4). Further, the upstream regulator analysis of the DEGs in both the fADcorr ELAVL4 KO and fAD ELAVL4 KO iNs (analyses 1 and 6) revealed that the INSR downstream targets are enriched and the INSR is predicted to be inhibited (Z-scores −3,14 and −3,15). In contrast, an enrichment of INSR targets and predicted activation of the INSR (Z-score 2,21) was identified in analysis 11 (DEPs in the ELAVL4 KO in fAD iNs) (Table 4). In 7 of the remaining 11 analyses, a significant enrichment INSR downstream targets was found. As for FOXO1, its downstream transcriptional targets were enriched and it was predicted to be inhibited in 3 OE analyses (analyses 2,3, and 7) and 3 rescue analyses (4,5 and 9), while FOXO1 target enrichment was identified in 3 of the remaining 8 analyses (Table 4).
Table 4.
Upstream regulator analyses of RNAseq and phosphoproteomics ELAVL4 KO, overexpression, and rescue neuronal cell lines, using IPA.
| RNAseq | phosphoproteomics | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||||
| fAD-corrected | fAD | fAD | ||||||||||||||||
| KO | (1) 6260 DEGs ELAVL4 KO in fADcorr | (6) 3661 DEGs ELAVL4 KO in fAD | (11) 40 DEPs ELAVL4 KO in fAD | (12) 48 DPPs ELAVL4 KO in fAD | ||||||||||||||
| Upstream regulator | Z-score | P-value | # Targets | X-score | P-value | # Targets | Z-score | P-value | # Targets | Z-score | P-value | # Targets | ||||||
| APP | 2,36 | 3,83E-10 | 315 | N.A. | 2,00E-06 | 180 | N.A | 3,90E-10 | 14 | N.A | 1,34E-02 | 6 | ||||||
| Tau | N.A. | 4,53E-06 | 172 | N.A. | 7,05E-03 | 91 | N.A | 2,80E-12 | 13 | N.A. | 6,92E-02 | 6 | ||||||
| INSR | −3,14 | 6,75E-06 | 155 | −3,15 | 9,29E-06 | 97 | 2,21 | 1,46E-05 | 7 | N.A. | N.S | N.A. | ||||||
| FOXO1 | N.A. | 1,15E-04 | 145 | N.A. | N.S | N.A. | N.A | 4,50E-02 | 3 | N.A. | N.S | N.A. | ||||||
| OE | (2) 609 DEGs ELAVL4 sv1+2 OE in fADcorr | (3) 207 DEGs-OD ELAVL4 sv1+sv2 OE in fADcorr vs. ELAVL4 KO in fADcorr | (7) 360 DEGs ELAVL4 sv1+2 OE in fAD | (8) 74DEGs-ODELAVL4 sv1+sv2 OE in fAD vs. ELAVL4 KO in fAD | ||||||||||||||
| Upstream regulator | Z-score | P-value | # Targets | Z-score | P-value | # Targets | Z-score | P-value | # Targets | Z-score | P-value | # Targets | ||||||
| APP | N.A. | 4,79E-03 | 37 | −2,40 | 1,05E-02 | 16 | N.A. | 1,05E-02 | 24 | N.A. | N.S | N.A. | ||||||
| Tau | N.A. | 3,65E-02 | 20 | N.A. | N.S | N.A. | N.A. | N.S | N.A. | N.A. | N.S | N.A. | ||||||
| INSR | N.A. | 4,26E-05 | 27 | N.A. | 8,52E-03 | 10 | N.A. | 5,10E-03 | 15 | N.A. | N.S | N.A. | ||||||
| FOXO1 | −3,31 | 1,14E-08 | 34 | −3,89 | 6,68E-10 | 21 | −3,63 | 2,06E-04 | 18 | N.A. | N.S | N.A. | ||||||
| Rescue | (4) 2032 DEGs ELAVL4 sv1+sv2 rescue in fADcorr ELAVL4 KO | (5) 920 DEGs-OD ELAVL4 sv1+sv2 Rescue in fADcorr KO vs. ELAVL4 KO in fADcorr | (9) 1253 DEGs ELAVL4 sv1+sv2 rescue in fAD ELAVL4 KO | (10) 383 DEGs-ODELAVL4 sv1+sv2 rescue in fAD KO vs. ELAVL4 KO in fAD | (13) 67 DEPs ELAVL4 sv2 rescue in fAD ELAVL4 KO | (14) 103 DPPs ELAVL4 sv2 rescue in fAD ELAVL4 KO | ||||||||||||
| Upstream regulator | Z-score | P-value | # Targets | Z-score | P-value | # Targets | Z-score | P-value | # Targets | Z-score | P-value | # Targets | Z-score | P-value | # Targets | Z-score | P-value | # Targets |
| APP | N.A. | 7,98E-04 | 106 | N.A. | 3,32E-03 | 53 | N.A. | 5,85E-05 | 76 | N.A. | 3,11E-02 | 23 | N.A. | 2,16E-12 | 20 | N.A. | 3,62E-03 | 11 |
| Tau | N.A. | 4,94E-04 | 65 | N.A. | N.S | N.A. | N.A. | N.S | N.A. | N.A. | N.S | N.A. | N.A. | 2,5E-17 | 20 | N.A. | 8,60E-08 | 14 |
| INSR | N.A. | 3,26E-04 | 60 | N.A. | 6,11E-03 | 29 | N.A. | 7,77E-03 | 36 | N.A. | N.S | N.A. | N.A. | 1,21E-02 | 5 | N.A. | N.S | N.A. |
| FOXO1 | −3,37 | 9,67E-08 | 71 | −2,96 | 1,26E-04 | 34 | −3,36 | 3,30E-08 | 52 | N.A. | 2,97E-02 | 13 | N.A. | N.S | N.A. | N.A. | N.S | N.A. |
NOTE IPA upstream regulator analyses results for APP, TAU, insulin receptor (INSR), and FOXO1 in the differentially expressed genes (DEGs) are shown. Significant P-values (P < 0.05) and Z-scores (Z ≤ −2 or Z ≥ 2) indicated in bold. In green: pathways are predicted to inhibited based on direction of differential gene expression. In magenta: pathways are predicted to be activated based on direction of differential gene expression.
4. Discussion
In this study, we showed that manipulating ELAVL4/HuD expression in iPSC-derived induced neurons (iNs) affects AD-relevant phenotypes in human neuronal cells. We found that increasing ELAVL4 expression in WT and ELAVL4 KO iNs ameliorated AD-associated molecular changes—i.e., reduced levels of specific APP isoforms, decreased Aβ42/40 ratio, and reduced levels of phosphorylated tau—from both familial AD (fAD) and fAD-corrected iPSCs. For APP splicing isoforms and phosphorylated tau levels, the opposite effect was observed in ELAVL4 KO cells.
Three main alternatively spliced isoforms of APP exist, i.e., APP751 and APP770 that are ubiquitously expressed and APP695, a predominantly neuronally expressed isoform (Wang et al., 2017). In both fAD-corrected and fAD iNs, knocking out ELAVL4 resulted in altered splicing of APP mRNA (increased APP751 and APP770), while overexpression and rescue of ELAVL4 expression in ELAVL4 KO cells had the opposite effect (decreased APP751 and APP770) and also led to an increase of APP695 levels. These findings are in line with the literature, where ELAVL4 was reported to regulate APP splicing, and a strong positive correlation between Hu protein expression and APP695 levels was found in the brain (Fragkouli et al., 2017). Specifically, ELAVL4 was found to modulate APP splicing by favoring exclusion of exons 7 and 8 that are coding for the longer APP isoforms (APP751 and APP770) (Fragkouli et al., 2017). In AD, a decrease of APP695 and an increase of the APP751/APP770 isoforms have been reported (Johnson et al., 1989; Moir et al., 1998), which suggests that ELAVL4 function is impaired in AD. Further, except for most of the OE experiments—in which we observed an increase in total APP mRNA—we found that total APP mRNA levels were not significantly affected by manipulating ELAVL4 expression. At the protein level, increased APP expression was seen in most experiments with ELAVL4 OE or rescued iNs, but lower APP expression was observed following ELAVL4 KO in fAD iNs. This being said, when we examined APP processing, we found that the Aβ42/40 ratio was reduced in almost all (7/8) of the ELAVL4 OE and rescue experiments, with higher Aβ42/40 ratios having been correlated to more severe AD pathology (Hansson et al., 2019; Kwak et al., 2020). Again, these findings are in line with ELAVL4 function being reduced in AD.
According to the Aβ cascade hypothesis, pathogenic Aβ oligomers trigger a signaling cascade that results in an increase in phosphorylated tau, synaptic dysfunction, and neurodegeneration in the brain of AD patients (Selkoe and Hardy, 2016). In this respect, increased CSF and plasma levels of tau phosphorylated at threonine 181 (Thr181) are used to predict elevated amounts of cerebral Aβ in the clinic (Fagan et al., 2009; Mielke et al., 2018). In keeping with this, our OE and rescue experiments indeed showed that upon upregulating ELAVL4 expression, not only the Aβ42/40 ratio but also the levels of intracellular tau phosphorylated at Thr181 were markedly decreased. Conversely, ELAVL4 KO also resulted in increased phosphorylated tau (at Thr181) levels. Interestingly, it was recently also found in iPSC-derived cerebral organoids carrying frontotemporal dementia-associated mutations in MAPT, ELAVL4 is increased in expression but mislocalized together with tau in stress granules, which results in impaired ELAVL4 function and altered synaptic signaling pathways (Bowles et al., 2021).
Taken together, our experimental findings provide new, additional evidence in human neurons for an important role of ELAVL4 in modulating AD pathogenesis. Specifically, increased expression and hence the activity of ELAVL4 in iNs seems to counteract the two main pathological hallmarks of AD, i.e., the formation of extracellular Aβ plaques (resulting from an increased Aβ42/40 ratio) and intracellular neurofibrillary tangles (resulting from an increase in Thr181-phosphorylated tau levels). Therefore, we submit that ELAVL4 KO iNs represent a novel cellular model of dysregulation of tau and Aβ that can be probed to further elucidate mechanisms of AD. In particular, our findings in rescued iNs—i.e., upon upregulation of ELAVL4 levels in ELAVL4 KO cells—indicate that novel approaches aimed at upregulating ELAVL4 expression levels may be beneficial therapeutically.
The database of RNA profiles (RNAseq) and protein levels ((phospho)proteomics) resulting from ELAVL4 modulation that we created provides a rich resource for us and others studying ELAVL4/HuD function to gain insights into the molecular events that result from manipulating ELAVL4 expression (KO, OE, or rescue) in human neurons. Analysing these data, our results for three already established ELAVL4 targets NEP, Tau, and ACHE follow a pattern that is consistent with previous findings, i.e., that ELAVL4 stabilizes the mRNA of these genes. For the other known targets listed in Table 2, this pattern is less consistent, which may be due to the fact that cell models different from human iNs or animal models and/or different experimental conditions were used to demonstrate that these genes were ELAVL4 targets.
Our unbiased canonical pathway analysis identified multiple pathways that are significantly enriched among the DEGs, DEPs, and DPPs linked to differential ELAVL4 expression. These included pathways that have been linked to AD previously. Specifically, we identified several enriched synaptic pathways, i.e., “axonal guidance signaling”, “semaphorin signaling in neurons”, and “synaptogenesis signaling”. Axonal guidance is generally associated with neurodevelopment. Indeed, Hu proteins—that include HuD/ELAVL4—are thought to be one of the earliest markers of neuronal differentiation, and in dentate granule cells from ELAVL4 OE mice, increased expression of axonogenesis-related genes has been observed (Perrone-Bizzozero et al., 2011). However, axonal guidance molecules can also stimulate or inhibit inflammatory responses and play a pivotal role in the inflammation of the nervous system associated with AD (Lee et al., 2019; Zhang et al., 2021). A second set of enriched pathways consisted of DNA damage pathways, including “UVB/C-induced MAPK signaling”, “ATM signaling”, and “p53 signaling”. Recently, we have shown that several synaptic pathways are impaired in the hippocampus of AD patients by analyzing transcriptomic data and have also linked these findings to somatic DNA damage (Soheili-Nezhad et al., 2021). In this respect, an impaired p53-mediated DNA damage response has been observed in AD patient brains (Farmer et al., 2020). Furthermore, aberrant re-entry into the cell cycle has been implicated in the pathogenesis of AD (Lee et al., 2009; Moh et al., 2011). This fits with our finding of an enrichment of pathways related to cell cycle regulation (“Cyclins and cell cycle regulation”, “Cell cycle: G2/M damage checkpoint regulation”, “Cell cycle: G1/S checkpoint regulation”, and “CDK5 signaling”). DNA damage and other stressors may also activate a senescence-like phenotype in neurons leading to inflammation and neuronal dysfunction (Fielder et al., 2017), and therefore, it is interesting that oxidative stress has been shown to induce posttranscriptional gene regulation by RBPs—including Hu protein family member HuR—of mRNAs involved in regulating cellular senescence (Abdelmohsen et al., 2008). The transcripts of several genes related to mitochondrial function are differentially expressed upon ELAVL4 manipulation, e.g., ATP5F1A and NDUFA1. In addition, Sirtuin and NAD signaling as well as mitochondrial dysfunction were in the top five enriched pathways in the differentially expressed proteins measured in ELAVL4 KO fAD neurons. Intriguingly, NAD and Sirtuin signaling have also been linked to both AD and aging/longevity (Bonda et al., 2011; Mouchiroud et al., 2013) through regulating the activity of FOXO1 (Giannakou and Partridge, 2004; Zhang et al., 2017), one of the upstream regulators for which we performed enrichment analyses. Lastly, some pathways that we identified are directly linked to phosphorylation of tau, i.e., BAG2 signaling (Qin et al., 2016), 14-3-3-mediated signaling (Foote and Zhou, 2012; Sluchanko and Gusev, 2011), and ERK/MAPK signaling (Cai et al., 2011).
Our upstream regulator analyses of the transcriptomic and proteomic data from most experiments that we performed—knocking out and/or overexpressing ELAVL4 in iNs—revealed that the differentially expressed genes/proteins are strongly enriched for transcriptional targets of both APP and tau. These findings suggest that ELAVL4 not only affects APP processing and tau phosphorylation but also signaling downstream from these two crucial AD-linked proteins. Furthermore, the results from our upstream regulator analyses of the transcriptomic data indicate that ELAVL4 expression in neurons may be regulated by insulin receptor (INSR)-FOXO1 signaling: in the ELAVL4 KO iNs, INSR is predicted to be inhibited, whereas FOXO1 is predicted to be inhibited in ELAVL4 OE/rescue cells. This is the same mechanism that regulates ELAVL4 expression in pancreatic beta cells (Lee et al., 2012). More specifically, upon binding insulin, the INSR is activated, and this leads to a downstream signaling cascade that results in phosphorylation of FOXO1, a transcription factor that represses ELAVL4 transcription. Phosphorylated FOXO1 translocates from the nucleus and loses its ability to regulate transcription, resulting in increased transcription and expression of ELAVL4 (Lee et al., 2012). Subsequently, as we demonstrated, increased ELAVL4 expression results in a decreased Aβ42/40 ratio and phosphorylated tau levels, thereby ameliorating the key AD-related molecular changes. In Figure 8, we have schematically represented the directionality of INSR-FOXO1 signaling putatively regulating ELAVL4 expression, with its subsequent effects on APP processing and tau phosphorylation.
Figure 8.
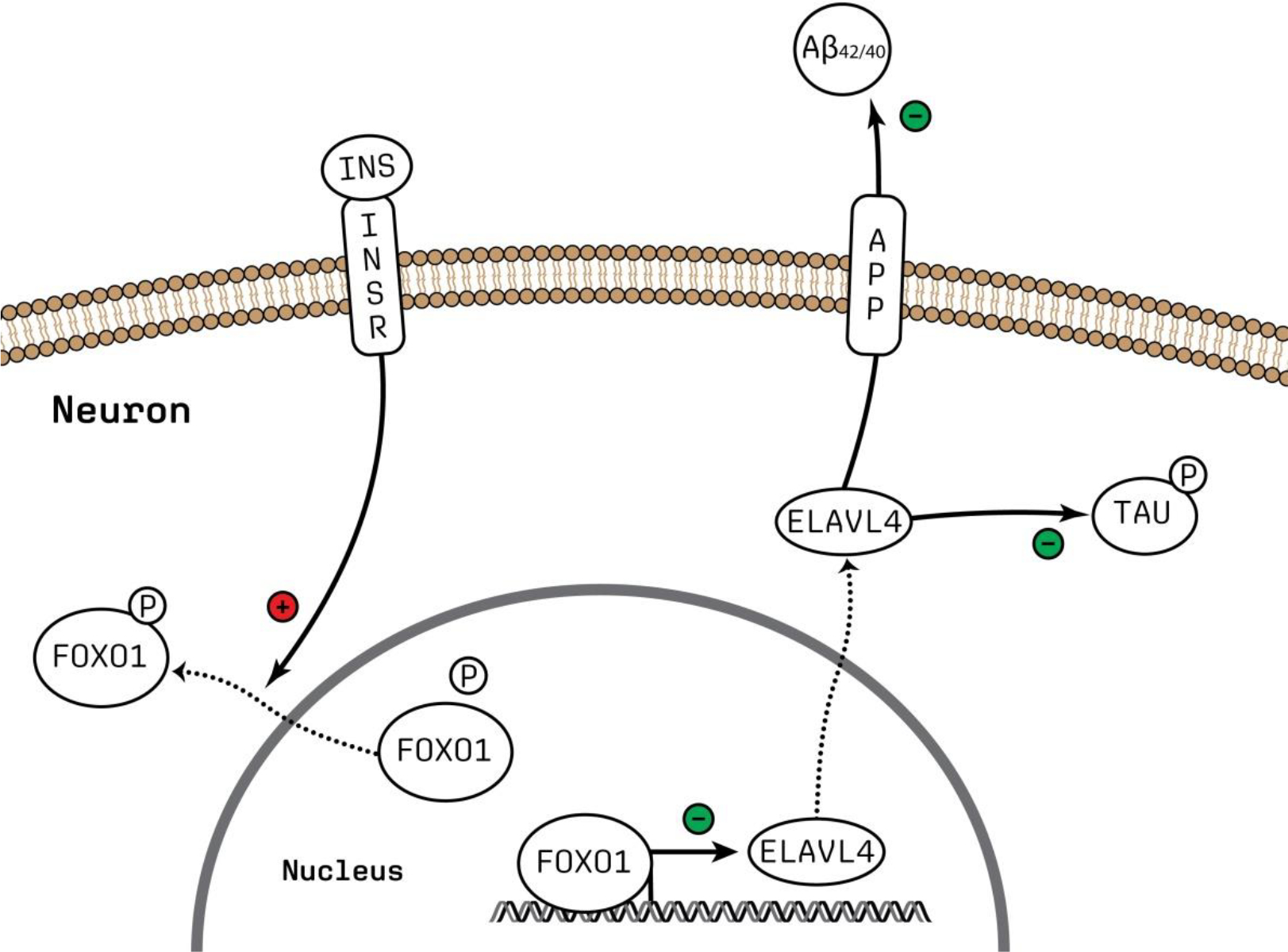
Schematic representation of INSR-FOXO1 signaling putatively regulating ELAVL4 expression and the downstream effects of ELAVL4 on APP processing and tau phosphorylation. The figure was made using Adobe Illustrator. Abbreviations: Aβ; amyloid beta, P; phosphate group.
As mentioned above, ELAVL4 is (also) an important regulator of pancreatic beta-cell function (Juan-Mateu et al., 2017), and ELAVL4 deficiency contributes to the etiology of diabetes type 2 (Hong et al., 2020). In this respect, it is interesting that insulin resistance (in the periphery and brain) has been implicated as a core mechanism underlying both diabetes and AD (Arnold et al., 2018). In the brain, insulin resistance was found to have a negative effect on synaptic plasticity, which in turn could contribute to AD progression (Ferrario and Reagan, 2018). Diabetes and AD are often comorbid, and Aβ peptides have been found to compete directly with insulin for INSR binding, leading to decreased INSR functioning and ultimately insulin resistance (Xie et al., 2002). Therefore, we could speculate that in AD, aggregated Aβ causes decreased INSR activity and subsequently decreased FOXO1 phosphorylation and ELAVL4 expression, which further exacerbates amyloidogenic APP processing, and hence AD pathology.
This study has some strengths and limitations. A particular strength is that we were able to generate and validate ELAVL4 KO cells as a human cell model of AD that could be used in further studies aimed at further elucidating the exact role of ELAVL4 in the brain in general, and in AD in particular. A limitation is that the iN system we used does not model the full complexity of the human brain, as neurons in the brain are interconnected with other brain regions and reside among other cell types. Therefore, more complex (3D) model systems including not only neurons but also e.g., astrocytes and microglia may be generated to study ELAVL4 function in an experimental setting that more closely resembles a functioning brain. Further, it is unlikely that ELAVL4 function in the AD brain is completely reduced to zero in the way that we have modeled with full ELAVL4 KO iNs. This being said, we also conducted OE and rescue experiments that showed effects opposite to those of the KO experiments, which strengthens the evidence that altered ELAVL4 expression leads to the observed phenotypic changes. Moreover, reproducing the observed effects of ELAVL4 manipulation in iPSC-derived neurons with different genetic backgrounds—e.g., neurons from patients with (other) fAD mutations in APP and/or PSEN1/2, and from sporadic AD patients—would strengthen our hypothesis that increasing ELAVL4 could be a novel therapeutical strategy for AD. Lastly, the putative ELAVL4 expression-regulating cascade in neurons that we proposed based on the analysis of the RNAseq data—i.e., the regulation of ELAVL4 expression by INSR-FOXO1 signaling—needs to be further studied and validated in follow-up experiments.
5. Conclusions
In conclusion, we show that increased ELAVL4/HuD expression ameliorates APP and tau pathology in neurons, irrespective of APP (V717I) mutation status. Furthermore, our transcriptomic data analyses suggest that ELAVL4 affects signaling downstream of APP and tau and that, in addition to pancreatic cells, INSR-FOXO1 signaling may regulate ELAVL4 expression in neurons, although the latter requires further experiments. Taken together with the results from published studies, increasing ELAVL4 expression and hence functional protein levels in the brain may be beneficial in AD—especially through the positive effects that ELAVL4 has on synaptic function—making it a promising target for novel drug development.
Supplementary Material
Supplementary figure 1. CRISPR/Cas9 editing did not result in large chromosomal abnormalities in iPSC lines generated in this study.
Common CNV analysis in the monoclonal lines using the Nanostring nCounter Human CNV codeset for (A) fAD corrected CRISPR control (B) fAD CRISPR control (C) fAD corrected ELAVL4 knock out B1c2 (D) fAD ELAVL4 knock out B1c20
{kind=link}
Supplementary figure 2. ELAVL4 affects APP mRNA splicing in fAD-corrected iNs.
Quantification of specific APP mRNA splice forms in iNs from ELAVL4 knock out (KO) overexpression (OE) and rescue in fAD-corrected induced neurons (iNs). In the KO, changes relative to CRISPR control line that underwent mock targeting with an empty sgRNA vector transfected alongside with Cas9, and monoclonal subclones were isolated and analyzed in parallel to subclones where ELAVL4 was targeted. For OE and rescue conditions, changes relative to empty vector OE plasmid in the CRISPR control or ELAVL4 KO iNs respectively. At day 21 of differentiation, cells were lysed and protein and RNA samples were harvested. Quantifications are from 3 independent differentiations with n=3. In the KO experiments P-values were calculated with t tests, while in the OE and rescue experiments, P-values were calculated with Dunnett’s multiple comparison that followed after a significant ANOVA test, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
{kind=link}
Supplementary figure 3. ELAVL4 affects APP mRNA splicing in fAD iNs.
Quantification of specific APP mRNA splice forms in iNs from ELAVL4 knock out (KO) overexpression (OE) and rescue in fAD induced neurons (iNs). In the KO, changes relative to CRISPR control line that underwent mock targeting with an empty sgRNA vector transfected alongside with Cas9, and monoclonal subclones were isolated and analyzed in parallel to subclones where ELAVL4 was targeted. For OE and rescue conditions, changes relative to empty vector OE plasmid in the CRISPR control or ELAVL4 KO iNs respectively. At day 21 of differentiation, cells were lysed and protein and RNA samples were harvested. Quantifications are from 3 independent differentiations with n=3. In the KO experiments P-values were calculated with t tests, while in the OE and rescue experiments, P-values were calculated with Dunnett’s multiple comparison that followed after a significant ANOVA test, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
{kind=link}
Supplementary figure 4. ELAVL4 affects extracellular Aβ species in fAD-corrected iNs
Quantification of extracellular Aβ species (Aβ38, Aβ40, and Aβ42) in culture media of ELAVL4 knock out (KO) overexpression (OE) and rescue in fAD-corrected induced neurons (iNs). Total protein determined by BCA was used to normalize measured values for individual Aβ species. In the KO, changes relative to CRISPR control line that underwent mock targeting with an empty sgRNA vector transfected alongside with Cas9, and monoclonal subclones were isolated and analyzed in parallel to subclones where ELAVL4 was targeted. For OE and rescue conditions, changes relative to empty vector OE plasmid in the CRISPR control or ELAVL4 KO iNs respectively. At day 21 of differentiation, 72 h conditioned media were collected and cells lysed. Quantifications are from 3 independent differentiations with n=3. In the KO experiments P-values were calculated with t tests, while in the OE and rescue experiments, P-values were calculated with Dunnett’s multiple comparison that followed after a significant ANOVA test, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
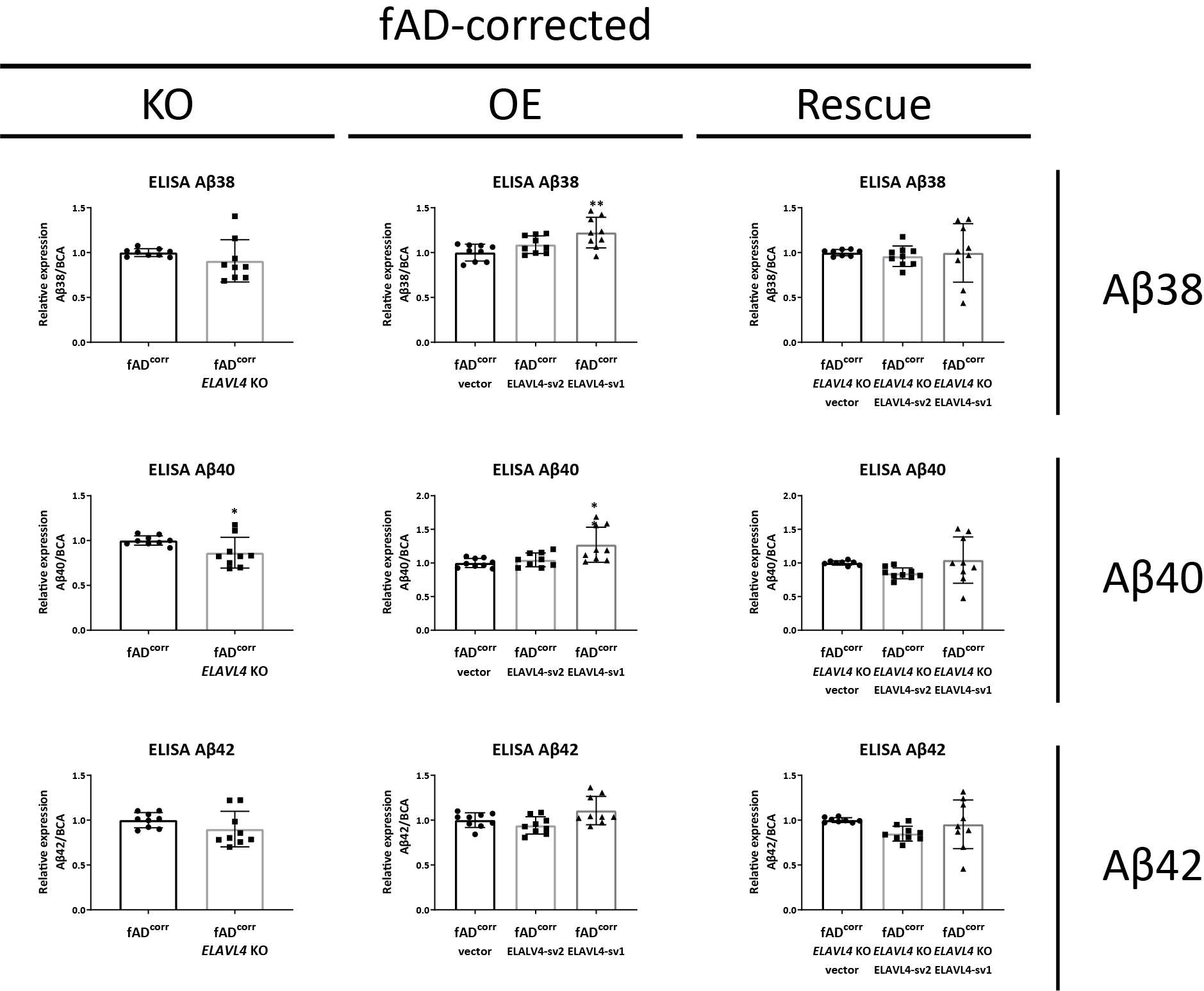{kind=link}
Supplementary figure 5. ELAVL4 affects extracellular Aβ species in fAD iNs.
Quantification of extracellular Aβ species (Aβ38, Aβ40, and Aβ42) in culture media of ELAVL4 knock out (KO) overexpression (OE) and rescue in fAD induced neurons (iNs). Total protein determined by BCA was used to normalize measured values for individual Aβ species. In the KO, changes relative to CRISPR control line that underwent mock targeting with an empty sgRNA vector transfected alongside with Cas9, and monoclonal subclones were isolated and analyzed in parallel to subclones where ELAVL4 was targeted. For OE and rescue conditions, changes relative to empty vector OE plasmid in the CRISPR control or ELAVL4 KO iNs respectively. At day 21 of differentiation, 72 h conditioned media were collected and cells lysed. Quantifications are from 3 independent differentiations with n=3. In the KO experiments P-values were calculated with t tests, while in the OE and rescue experiments, P-values were calculated with Dunnett’s multiple comparison that followed after a significant ANOVA test, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
{kind=link}
Supplementary figure 6. RNAseq characterization of fAD neurons derived from iPSC lines generated in this study.
(A) Differentially expressed genes (DEGs) in fAD ELAVL4 KO iNs compared to fAD iNs (B) DEGs in fAD iNs in which ELAVL4 was overexpressed (combined analysis of ELAVL4 sv1 and sv2 OE) compared to fAD iNs. (C) DEGs from the rescue experiments: the DEGs in fAD ELAVL4 KO iNs with ELAVL4 sv1+sv2 rescue compared to fAD ELAVL4 KO iNs. DEGs with FDR corrected P-values < 0.05 and fold changes ≥ |1.20| are indicated in red. Volcano DEGs plots were generated using the EnhancedVolcano package in R.
{kind=link}
Supplementary table 1. Full results of transcriptomic and proteomic differential expression analyses of iN neurons with ELAVL4 knock out, overexpression, or rescue
Supplementary table 2. Full results Ingenuity Pathway Analysis of transcriptomic and proteomic data
Highlights.
The RNA binding protein ELAVL4/HuD reduces the Aβ42/40 ratio in iPSC-derived neurons
ELAVL4 reduces tau phosphorylation in iPSC-derived neurons
ELAVL4 affects multiple biological pathways involved in synaptic function
Generated omics data provide a resource for interrogating ELAVL4 biology in neurons
Funding:
The research presented in this paper was supported by the Dutch charity foundation ‘Stichting Devon’. This work also was supported by NIH grants R01AG055909 and R01NS117446.
Footnotes
Declarations
Competing interests: G.P. is the director of Drug Target ID., Ltd. (Nijmegen, The Netherlands). The other authors have no competing interests to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Availability of data and material:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Abdelmohsen K, Kuwano Y, Kim HH, Gorospe M, 2008. Posttranscriptional gene regulation by RNA-binding proteins during oxidative stress: implications for cellular senescence. Biol Chem 389, 243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadio M, Pascale A, Wang J, Ho L, Quattrone A, Gandy S, Haroutunian V, Racchi M, Pasinetti GM, 2009. nELAV proteins alteration in Alzheimer’s disease brain: a novel putative target for amyloid-beta reverberating on AbetaPP processing. J Alzheimers Dis 16, 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda-Abreu GE, Behar L, Chung S, Furneaux H, Ginzburg I, 1999. Embryonic lethal abnormal vision-like RNA-binding proteins regulate neurite outgrowth and tau expression in PC12 cells. J Neurosci 19, 6907–6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SE, Arvanitakis Z, Macauley-Rambach SL, Koenig AM, Wang HY, Ahima RS, Craft S, Gandy S, Buettner C, Stoeckel LE, Holtzman DM, Nathan DM, 2018. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol 14, 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashleigh T, Swerdlow RH, Beal MF, 2022. The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimers Dement. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y, 1995. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 57, 289–300. [Google Scholar]
- Bentahir M, Nyabi O, Verhamme J, Tolia A, Horre K, Wiltfang J, Esselmann H, De Strooper B, 2006. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J Neurochem 96, 732–742. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B, 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognani F, Contente-Cuomo T, Perrone-Bizzozero NI, 2010. Novel recognition motifs and biological functions of the RNA-binding protein HuD revealed by genome-wide identification of its targets. Nucleic Acids Res 38, 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognani F, Qiu S, Tanner DC, Paik J, Perrone-Bizzozero NI, Weeber EJ, 2007a. Associative and spatial learning and memory deficits in transgenic mice overexpressing the RNA-binding protein HuD. Neurobiol Learn Mem 87, 635–643. [DOI] [PubMed] [Google Scholar]
- Bolognani F, Tanner DC, Nixon S, Okano HJ, Okano H, Perrone-Bizzozero NI, 2007b. Coordinated expression of HuD and GAP-43 in hippocampal dentate granule cells during developmental and adult plasticity. Neurochem Res 32, 2142–2151. [DOI] [PubMed] [Google Scholar]
- Bonda DJ, Lee HG, Camins A, Pallas M, Casadesus G, Smith MA, Zhu X, 2011. The sirtuin pathway in ageing and Alzheimer disease: mechanistic and therapeutic considerations. Lancet Neurol 10, 275–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowles KR, Silva MC, Whitney K, Bertucci T, Berlind JE, Lai JD, Garza JC, Boles NC, Mahali S, Strang KH, Marsh JA, Chen C, Pugh DA, Liu Y, Gordon RE, Goderie SK, Chowdhury R, Lotz S, Lane K, Crary JF, Haggarty SJ, Karch CM, Ichida JK, Goate AM, Temple S, 2021. ELAVL4, splicing, and glutamatergic dysfunction precede neuron loss in MAPT mutation cerebral organoids. Cell 184, 4547–4563 e4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L, 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Bronicki LM, Jasmin BJ, 2013. Emerging complexity of the HuD/ELAVl4 gene; implications for neuronal development, function, and dysfunction. RNA 19, 1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai T, Che H, Yao T, Chen Y, Huang C, Zhang W, Du K, Zhang J, Cao Y, Chen J, Luo W, 2011. Manganese induces tau hyperphosphorylation through the activation of ERK MAPK pathway in PC12 cells. Toxicol Sci 119, 169–177. [DOI] [PubMed] [Google Scholar]
- DeBoer EM, Azevedo R, Vega TA, Brodkin J, Akamatsu W, Okano H, Wagner GC, Rasin MR, 2014. Prenatal deletion of the RNA-binding protein HuD disrupts postnatal cortical circuit maturation and behavior. J Neurosci 34, 3674–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschenes-Furry J, Mousavi K, Bolognani F, Neve RL, Parks RJ, Perrone-Bizzozero NI, Jasmin BJ, 2007. The RNA-binding protein HuD binds acetylcholinesterase mRNA in neurons and regulates its expression after axotomy. J Neurosci 27, 665–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschenes-Furry J, Perrone-Bizzozero N, Jasmin BJ, 2006. The RNA-binding protein HuD: a regulator of neuronal differentiation, maintenance and plasticity. Bioessays 28, 822–833. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Shah AR, Aldea P, Roe CM, Mach RH, Marcus D, Morris JC, Holtzman DM, 2009. Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer’s disease. EMBO Mol Med 1, 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer KM, Ghag G, Puangmalai N, Montalbano M, Bhatt N, Kayed R, 2020. P53 aggregation, interactions with tau, and impaired DNA damage response in Alzheimer’s disease. Acta Neuropathol Commun 8, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrario CR, Reagan LP, 2018. Insulin-mediated synaptic plasticity in the CNS: Anatomical, functional and temporal contexts. Neuropharmacology 136, 182–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielder E, von Zglinicki T, Jurk D, 2017. The DNA Damage Response in Neurons: Die by Apoptosis or Survive in a Senescence-Like State? J Alzheimers Dis 60, S107–S131. [DOI] [PubMed] [Google Scholar]
- Foote M, Zhou Y, 2012. 14–3-3 proteins in neurological disorders. Int J Biochem Mol Biol 3, 152–164. [PMC free article] [PubMed] [Google Scholar]
- Fragkouli A, Koukouraki P, Vlachos IS, Paraskevopoulou MD, Hatzigeorgiou AG, Doxakis E, 2017. Neuronal ELAVL proteins utilize AUF-1 as a co-partner to induce neuron-specific alternative splicing of APP. Sci Rep 7, 44507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Fukao A, Sasano Y, Matsuzaki H, Kikkawa U, Imataka H, Inoue K, Endo S, Sonenberg N, Thoma C, Sakamoto H, 2012. Functional and direct interaction between the RNA binding protein HuD and active Akt1. Nucleic Acids Res 40, 1944–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukao A, Sasano Y, Imataka H, Inoue K, Sakamoto H, Sonenberg N, Thoma C, Fujiwara T, 2009. The ELAV protein HuD stimulates cap-dependent translation in a Poly(A)- and eIF4A-dependent manner. Mol Cell 36, 1007–1017. [DOI] [PubMed] [Google Scholar]
- Giannakou ME, Partridge L, 2004. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol 14, 408–412. [DOI] [PubMed] [Google Scholar]
- Graus F, Elkon KB, Cordon-Cardo C, Posner JB, 1986. Sensory neuronopathy and small cell lung cancer. Antineuronal antibody that also reacts with the tumor. Am J Med 80, 45–52. [DOI] [PubMed] [Google Scholar]
- Hansson O, Lehmann S, Otto M, Zetterberg H, Lewczuk P, 2019. Advantages and disadvantages of the use of the CSF Amyloid beta (Abeta) 42/40 ratio in the diagnosis of Alzheimer’s Disease. Alzheimers Res Ther 11, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinman MN, Lou H, 2008. Diverse molecular functions of Hu proteins. Cell Mol Life Sci 65, 3168–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan DB, 2014. Long-term efficacy and toxicity of cholinesterase inhibitors in the treatment of Alzheimer disease. Can J Psychiatry 59, 618–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y, Tak H, Kim C, Kang H, Ji E, Ahn S, Jung M, Kim HL, Lee JH, Kim W, Lee EK, 2020. RNA binding protein HuD contributes to beta-cell dysfunction by impairing mitochondria dynamics. Cell Death Differ 27, 1633–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu RT, Yu Q, Zhou SD, Yin YX, Hu RG, Lu HP, Hu BL, 2020. Co-expression Network Analysis Reveals Novel Genes Underlying Alzheimer’s Disease Pathogenesis. Front Aging Neurosci 12, 605961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R, Contributors, 2018. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14, 535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SA, Rogers J, Finch CE, 1989. APP-695 transcript prevalence is selectively reduced during Alzheimer’s disease in cortex and hippocampus but not in cerebellum. Neurobiol Aging 10, 755–760. [DOI] [PubMed] [Google Scholar]
- Juan-Mateu J, Rech TH, Villate O, Lizarraga-Mollinedo E, Wendt A, Turatsinze JV, Brondani LA, Nardelli TR, Nogueira TC, Esguerra JL, Alvelos MI, Marchetti P, Eliasson L, Eizirik DL, 2017. Neuron-enriched RNA-binding Proteins Regulate Pancreatic Beta Cell Function and Survival. J Biol Chem 292, 3466–3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MJ, Abdelmohsen K, Hutchison ER, Mitchell SJ, Grammatikakis I, Guo R, Noh JH, Martindale JL, Yang X, Lee EK, Faghihi MA, Wahlestedt C, Troncoso JC, Pletnikova O, Perrone-Bizzozero N, Resnick SM, de Cabo R, Mattson MP, Gorospe M, 2014. HuD regulates coding and noncoding RNA to induce APP-->Abeta processing. Cell Rep 7, 1401–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinghorn KJ, Crowther DC, Sharp LK, Nerelius C, Davis RL, Chang HT, Green C, Gubb DC, Johansson J, Lomas DA, 2006. Neuroserpin binds Abeta and is a neuroprotective component of amyloid plaques in Alzheimer disease. J Biol Chem 281, 29268–29277. [DOI] [PubMed] [Google Scholar]
- Klemann C, Xicoy H, Poelmans G, Bloem BR, Martens GJM, Visser JE, 2018. Physical Exercise Modulates L-DOPA-Regulated Molecular Pathways in the MPTP Mouse Model of Parkinson’s Disease. Mol Neurobiol 55, 5639–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A, Green J, Pollard J Jr., Tugendreich S, 2014. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak SS, Washicosky KJ, Brand E, von Maydell D, Aronson J, Kim S, Capen DE, Cetinbas M, Sadreyev R, Ning S, Bylykbashi E, Xia W, Wagner SL, Choi SH, Tanzi RE, Kim DY, 2020. Amyloid-beta42/40 ratio drives tau pathology in 3D human neural cell culture models of Alzheimer’s disease. Nat Commun 11, 1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagomarsino VN, Pearse RV 2nd, Liu L, Hsieh YC, Fernandez MA, Vinton EA, Paull D, Felsky D, Tasaki S, Gaiteri C, Vardarajan B, Lee H, Muratore CR, Benoit CR, Chou V, Fancher SB, He A, Merchant JP, Duong DM, Martinez H, Zhou M, Bah F, Vicent MA, Stricker JMS, Xu J, Dammer EB, Levey AI, Chibnik LB, Menon V, Seyfried NT, De Jager PL, Noggle S, Selkoe DJ, Bennett DA, Young-Pearse TL, 2021. Stem cell-derived neurons reflect features of protein networks, neuropathology, and cognitive outcome of their aged human donors. Neuron 109, 3402–3420 e3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EK, Kim W, Tominaga K, Martindale JL, Yang X, Subaran SS, Carlson OD, Mercken EM, Kulkarni RN, Akamatsu W, Okano H, Perrone-Bizzozero NI, de Cabo R, Egan JM, Gorospe M, 2012. RNA-binding protein HuD controls insulin translation. Mol Cell 45, 826–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HG, Casadesus G, Zhu X, Castellani RJ, McShea A, Perry G, Petersen RB, Bajic V, Smith MA, 2009. Cell cycle re-entry mediated neurodegeneration and its treatment role in the pathogenesis of Alzheimer’s disease. Neurochem Int 54, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WS, Lee WH, Bae YC, Suk K, 2019. Axon Guidance Molecules Guiding Neuroinflammation. Exp Neurobiol 28, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Lee JA, Kaang BK, 2015. Regulation of mRNA stability by ARE-binding proteins in synaptic plasticity and memory. Neurobiol Learn Mem 124, 28–33. [DOI] [PubMed] [Google Scholar]
- Lim CS, Alkon DL, 2014. PKCepsilon promotes HuD-mediated neprilysin mRNA stability and enhances neprilysin-induced Abeta degradation in brain neurons. PLoS One 9, e97756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Lauro BM, Wolfe MS, Selkoe DJ, 2021. Hydrophilic loop 1 of Presenilin-1 and the APP GxxxG transmembrane motif regulate gamma-secretase function in generating Alzheimer-causing Abeta peptides. J Biol Chem 296, 100393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD, 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Macri S, Spinello C, Widomska J, Magliozzi R, Poelmans G, Invernizzi RW, Creti R, Roessner V, Bartolini E, Margarit I, Glennon J, Laviola G, 2018. Neonatal corticosterone mitigates autoimmune neuropsychiatric disorders associated with streptococcus in mice. Sci Rep 8, 10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesi N, Amadio M, Colombrita C, Govoni S, Ratti A, Pascale A, 2016. PKC Activation Counteracts ADAM10 Deficit in HuD-Silenced Neuroblastoma Cells. J Alzheimers Dis 54, 535–547. [DOI] [PubMed] [Google Scholar]
- Mielke MM, Hagen CE, Xu J, Chai X, Vemuri P, Lowe VJ, Airey DC, Knopman DS, Roberts RO, Machulda MM, Jack CR Jr., Petersen RC, Dage JL, 2018. Plasma phospho-tau181 increases with Alzheimer’s disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimers Dement 14, 989–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moh C, Kubiak JZ, Bajic VP, Zhu X, Smith MA, Lee HG, 2011. Cell cycle deregulation in the neurons of Alzheimer’s disease. Results Probl Cell Differ 53, 565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir RD, Lynch T, Bush AI, Whyte S, Henry A, Portbury S, Multhaup G, Small DH, Tanzi RE, Beyreuther K, Masters CL, 1998. Relative increase in Alzheimer’s disease of soluble forms of cerebral Abeta amyloid protein precursor containing the Kunitz protease inhibitory domain. J Biol Chem 273, 5013–5019. [DOI] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, Guarente L, Auwerx J, 2013. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 154, 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratore CR, Rice HC, Srikanth P, Callahan DG, Shin T, Benjamin LN, Walsh DM, Selkoe DJ, Young-Pearse TL, 2014. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum Mol Genet 23, 3523–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratore CR, Zhou C, Liao M, Fernandez MA, Taylor WM, Lagomarsino VN, Pearse RV 2nd, Rice HC, Negri JM, He A, Srikanth P, Callahan DG, Shin T, Zhou M, Bennett DA, Noggle S, Love JC, Selkoe DJ, Young-Pearse TL, 2017. Cell-type Dependent Alzheimer’s Disease Phenotypes: Probing the Biology of Selective Neuronal Vulnerability. Stem Cell Reports 9, 1868–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen G, Liestol K, Van Loo P, Moen Vollan HK, Eide MB, Rueda OM, Chin SF, Russell R, Baumbusch LO, Caldas C, Borresen-Dale AL, Lingjaerde OC, 2012. Copynumber: Efficient algorithms for single- and multi-track copy number segmentation. BMC Genomics 13, 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbacher JK, Tabet R, Yeo GW, Lagier-Tourenne C, 2019. Disruption of RNA Metabolism in Neurological Diseases and Emerging Therapeutic Interventions. Neuron 102, 294–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliaroli L, Widomska J, Nespoli E, Hildebrandt T, Barta C, Glennon J, Hengerer B, Poelmans G, 2019. Riluzole Attenuates L-DOPA-Induced Abnormal Involuntary Movements Through Decreasing CREB1 Activity: Insights from a Rat Model. Mol Neurobiol 56, 5111–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Myszka DG, Yu M, Littler SJ, Laird-Offringa IA, 2000. HuD RNA recognition motifs play distinct roles in the formation of a stable complex with AU-rich RNA. Mol Cell Biol 20, 4765–4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascale A, Amadio M, Quattrone A, 2008. Defining a neuron: neuronal ELAV proteins. Cell Mol Life Sci 65, 128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascale A, Gusev PA, Amadio M, Dottorini T, Govoni S, Alkon DL, Quattrone A, 2004. Increase of the RNA-binding protein HuD and posttranscriptional up-regulation of the GAP-43 gene during spatial memory. Proc Natl Acad Sci U S A 101, 1217–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson C, 2018. World Alzheimer Report 2018: the state of the art of dementia research: new frontiers. Alzheimer’s Disease International (ADI): London, UK. [Google Scholar]
- Perrone-Bizzozero NI, Tanner DC, Mounce J, Bolognani F, 2011. Increased expression of axogenesis-related genes and mossy fibre length in dentate granule cells from adult HuD overexpressor mice. ASN Neuro 3, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Guo J, Zheng Q, Zhang H, 2016. BAG2 structure, function and involvement in disease. Cell Mol Biol Lett 21, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quattrone A, Pascale A, Nogues X, Zhao W, Gusev P, Pacini A, Alkon DL, 2001. Posttranscriptional regulation of gene expression in learning by the neuronal ELAV-like mRNA-stabilizing proteins. Proc Natl Acad Sci U S A 98, 11668–11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S, 1996. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med 2, 864–870. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J, 2016. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluchanko NN, Gusev NB, 2011. Probable participation of 14–3-3 in tau protein oligomerization and aggregation. J Alzheimers Dis 27, 467–476. [DOI] [PubMed] [Google Scholar]
- Soheili-Nezhad S, van der Linden RJ, Olde Rikkert M, Sprooten E, Poelmans G, 2021. Long genes are more frequently affected by somatic mutations and show reduced expression in Alzheimer’s disease: Implications for disease etiology. Alzheimers Dement 17, 489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosanya NM, Cacheaux LP, Workman ER, Niere F, Perrone-Bizzozero NI, Raab-Graham KF, 2015. Mammalian Target of Rapamycin (mTOR) Tagging Promotes Dendritic Branch Variability through the Capture of Ca2+/Calmodulin-dependent Protein Kinase II alpha (CaMKIIalpha) mRNAs by the RNA-binding Protein HuD. J Biol Chem 290, 16357–16371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth P, Lagomarsino VN, Pearse RV 2nd, Liao M, Ghosh S, Nehme R, Seyfried N, Eggan K, Young-Pearse TL, 2018. Convergence of independent DISC1 mutations on impaired neurite growth via decreased UNC5D expression. Transl Psychiatry 8, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subhadra B, Schaller K, Seeds NW, 2013. Neuroserpin up-regulation in the Alzheimer’s disease brain is associated with elevated thyroid hormone receptor-beta1 and HuD expression. Neurochem Int 63, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo A, Dalmau J, Manley G, Rosenfeld M, Wong E, Henson J, Posner JB, Furneaux HM, 1991. HuD, a paraneoplastic encephalomyelitis antigen, contains RNA-binding domains and is homologous to Elav and Sex-lethal. Cell 67, 325–333. [DOI] [PubMed] [Google Scholar]
- Vanevski F, Xu B, 2015. HuD interacts with Bdnf mRNA and is essential for activity-induced BDNF synthesis in dendrites. PLoS One 10, e0117264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Beckmann ND, Roussos P, Wang E, Zhou X, Wang Q, Ming C, Neff R, Ma W, Fullard JF, Hauberg ME, Bendl J, Peters MA, Logsdon B, Wang P, Mahajan M, Mangravite LM, Dammer EB, Duong DM, Lah JJ, Seyfried NT, Levey AI, Buxbaum JD, Ehrlich M, Gandy S, Katsel P, Haroutunian V, Schadt E, Zhang B, 2018. The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer’s disease. Sci Data 5, 180185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhou X, Li G, Zhang Y, Wu Y, Song W, 2017. Modifications and Trafficking of APP in the Pathogenesis of Alzheimer’s Disease. Front Mol Neurosci 10, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CI, Vinton EA, Pearse RV 2nd, Heo K, Aylward AJ, Hsieh YC, Bi Y, Adeleye S, Fancher S, Duong DM, Seyfried NT, Schwarz TL, Young-Pearse TL, 2022. APP and DYRK1A regulate axonal and synaptic vesicle protein networks and mediate Alzheimer’s pathology in trisomy 21 neurons. Mol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins R, 2002. Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci 22, RC221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Qi Z, Li J, Li M, Du X, Wang S, Zhou G, Xu B, Liu W, Xi S, Xu Z, Deng Y, 2021. Roles and Mechanisms of Axon-Guidance Molecules in Alzheimer’s Disease. Mol Neurobiol. [DOI] [PubMed] [Google Scholar]
- Zhang M, Zhang Q, Hu Y, Xu L, Jiang Y, Zhang C, Ding L, Jiang R, Sun J, Sun H, Yan G, 2017. miR-181a increases FoxO1 acetylation and promotes granulosa cell apoptosis via SIRT1 downregulation. Cell Death Dis 8, e3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, Xu W, Yang N, Danko T, Chen L, Wernig M, Sudhof TC, 2013. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1. CRISPR/Cas9 editing did not result in large chromosomal abnormalities in iPSC lines generated in this study.
Common CNV analysis in the monoclonal lines using the Nanostring nCounter Human CNV codeset for (A) fAD corrected CRISPR control (B) fAD CRISPR control (C) fAD corrected ELAVL4 knock out B1c2 (D) fAD ELAVL4 knock out B1c20
Supplementary figure 2. ELAVL4 affects APP mRNA splicing in fAD-corrected iNs.
Quantification of specific APP mRNA splice forms in iNs from ELAVL4 knock out (KO) overexpression (OE) and rescue in fAD-corrected induced neurons (iNs). In the KO, changes relative to CRISPR control line that underwent mock targeting with an empty sgRNA vector transfected alongside with Cas9, and monoclonal subclones were isolated and analyzed in parallel to subclones where ELAVL4 was targeted. For OE and rescue conditions, changes relative to empty vector OE plasmid in the CRISPR control or ELAVL4 KO iNs respectively. At day 21 of differentiation, cells were lysed and protein and RNA samples were harvested. Quantifications are from 3 independent differentiations with n=3. In the KO experiments P-values were calculated with t tests, while in the OE and rescue experiments, P-values were calculated with Dunnett’s multiple comparison that followed after a significant ANOVA test, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Supplementary figure 3. ELAVL4 affects APP mRNA splicing in fAD iNs.
Quantification of specific APP mRNA splice forms in iNs from ELAVL4 knock out (KO) overexpression (OE) and rescue in fAD induced neurons (iNs). In the KO, changes relative to CRISPR control line that underwent mock targeting with an empty sgRNA vector transfected alongside with Cas9, and monoclonal subclones were isolated and analyzed in parallel to subclones where ELAVL4 was targeted. For OE and rescue conditions, changes relative to empty vector OE plasmid in the CRISPR control or ELAVL4 KO iNs respectively. At day 21 of differentiation, cells were lysed and protein and RNA samples were harvested. Quantifications are from 3 independent differentiations with n=3. In the KO experiments P-values were calculated with t tests, while in the OE and rescue experiments, P-values were calculated with Dunnett’s multiple comparison that followed after a significant ANOVA test, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Supplementary figure 4. ELAVL4 affects extracellular Aβ species in fAD-corrected iNs
Quantification of extracellular Aβ species (Aβ38, Aβ40, and Aβ42) in culture media of ELAVL4 knock out (KO) overexpression (OE) and rescue in fAD-corrected induced neurons (iNs). Total protein determined by BCA was used to normalize measured values for individual Aβ species. In the KO, changes relative to CRISPR control line that underwent mock targeting with an empty sgRNA vector transfected alongside with Cas9, and monoclonal subclones were isolated and analyzed in parallel to subclones where ELAVL4 was targeted. For OE and rescue conditions, changes relative to empty vector OE plasmid in the CRISPR control or ELAVL4 KO iNs respectively. At day 21 of differentiation, 72 h conditioned media were collected and cells lysed. Quantifications are from 3 independent differentiations with n=3. In the KO experiments P-values were calculated with t tests, while in the OE and rescue experiments, P-values were calculated with Dunnett’s multiple comparison that followed after a significant ANOVA test, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Supplementary figure 5. ELAVL4 affects extracellular Aβ species in fAD iNs.
Quantification of extracellular Aβ species (Aβ38, Aβ40, and Aβ42) in culture media of ELAVL4 knock out (KO) overexpression (OE) and rescue in fAD induced neurons (iNs). Total protein determined by BCA was used to normalize measured values for individual Aβ species. In the KO, changes relative to CRISPR control line that underwent mock targeting with an empty sgRNA vector transfected alongside with Cas9, and monoclonal subclones were isolated and analyzed in parallel to subclones where ELAVL4 was targeted. For OE and rescue conditions, changes relative to empty vector OE plasmid in the CRISPR control or ELAVL4 KO iNs respectively. At day 21 of differentiation, 72 h conditioned media were collected and cells lysed. Quantifications are from 3 independent differentiations with n=3. In the KO experiments P-values were calculated with t tests, while in the OE and rescue experiments, P-values were calculated with Dunnett’s multiple comparison that followed after a significant ANOVA test, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Supplementary figure 6. RNAseq characterization of fAD neurons derived from iPSC lines generated in this study.
(A) Differentially expressed genes (DEGs) in fAD ELAVL4 KO iNs compared to fAD iNs (B) DEGs in fAD iNs in which ELAVL4 was overexpressed (combined analysis of ELAVL4 sv1 and sv2 OE) compared to fAD iNs. (C) DEGs from the rescue experiments: the DEGs in fAD ELAVL4 KO iNs with ELAVL4 sv1+sv2 rescue compared to fAD ELAVL4 KO iNs. DEGs with FDR corrected P-values < 0.05 and fold changes ≥ |1.20| are indicated in red. Volcano DEGs plots were generated using the EnhancedVolcano package in R.
Supplementary table 1. Full results of transcriptomic and proteomic differential expression analyses of iN neurons with ELAVL4 knock out, overexpression, or rescue
Supplementary table 2. Full results Ingenuity Pathway Analysis of transcriptomic and proteomic data
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.