

Summary
Despite much concerted effort to better understand severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral infection, relatively little is known about the dynamics of early viral entry and infection in the airway. Here we analyzed a single-cell RNA sequencing dataset of early SARS-CoV-2 infection in a humanized in vitro model, to elucidate key mechanisms by which the virus triggers a cell-systems-level response in the bronchial epithelium. We find that SARS-CoV-2 virus preferentially enters the tissue via ciliated cell precursors, giving rise to a population of infected mature ciliated cells, which signal to basal cells, inducing further rapid differentiation. This feedforward loop of infection is mitigated by further cell-cell communication, before interferon signaling begins at three days post-infection. These findings suggest hijacking by the virus of potentially beneficial tissue repair mechanisms, possibly exacerbating the outcome. This work both elucidates the interplay between barrier tissues and viral infections and may suggest alternative therapeutic approaches targeting non-immune response mechanisms.
Subject areas: Biological sciences, Immunology, Virology, Transcriptomics
Graphical abstract
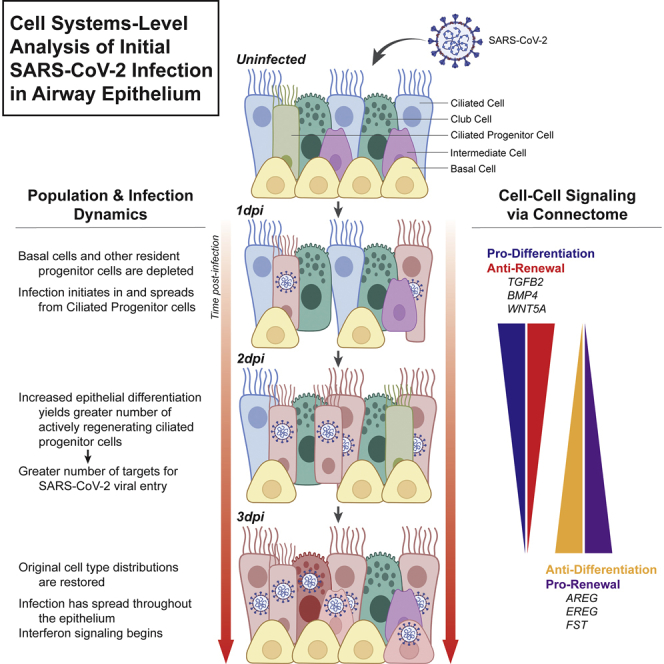
Highlights
-
•
scRNA-seq of initial SARS-CoV-2 infection reveals viral entry via ciliated progenitors
-
•
Basal cells are depleted as they are induced to differentiate on day 1 of infection
-
•
Infected ciliated cells express pro-differentiation signals mainly via TGFβ pathway
-
•
The virus can “hijack” tissue repair mechanisms, increasing viral load
Biological sciences; Immunology; Virology; Transcriptomics
Introduction
Since its first outbreak was reported on December 31, 2019 in Wuhan, China,1 COVID-19 has claimed over 1,077,303 lives in the United States, and over 6.6 million globally, as of December 2022.2,3 Despite recent rollout of vaccines against the virus and a declining case fatality rate in the United States, the pandemic has not abated globally and new, more infectious strains of the virus are still being identified.4,5,6 A major challenge to fight the virus remains, which is the lack of precise characterization of the onset of infection and early disease progression. This limited understanding of the early phases of disease has contributed to the massive loss of life.
The virus, SARS-CoV-2, enters the human host via the ACE2 receptor on ciliated cells of large airways, before spreading locally and systemically.7,8,9,10,11,12 Beyond this, little is known about downstream mechanisms leading to physiologic symptoms, including cough, pneumonia, progression onto acute respiratory distress syndrome (ARDS), and death.13,14
The initial interactions between airway epithelium and the virus are difficult to evaluate in vivo, as patient testing usually occurs in the days following infection. In addition, the SARS-CoV2 virus has a strong tropism for human cells and is not easily studied in typical animal models. To address these problems, in vitro models have been leveraged to observe early stages of infection dynamics in cultured human cells. One common model has utilized cultured human bronchial epithelial cells (HBECs) at an air-liquid interface (ALI). Bronchial cells differentiate into a native-like pseudostratified mucociliary epithelium, which can then be inoculated with virus. These models have proven to be remarkably accurate representations of proximal airway epithelial reaction to SARS-CoV-2 infection.12,15 They also have the added benefit of avoiding challenges associated with analyzing clinical samples, such as sample degradation and patient-to-patient variability.12,16
While the dramatic and unique immunological response to SARS-CoV-2 infection has been well documented,17,18,19,20,21,22 the timing of its initiation is still poorly characterized.23 Indeed, the inflammatory and immune response to the virus may not fully develop in the first few days following infection.24 These findings raise the question of how the first line of defense, the airway epithelium, responds to the infection over the first few days following exposure, in the absence of a full-fledged immune reaction. Combining biomimetic models of the bronchial epithelium with single-cell RNA sequencing (scRNA-seq) analysis methods can provide unprecedented information regarding the dynamic processes of viral infection and host cellular response. Identification and characterization of the cell types that comprise airway epithelium by scRNA-seq can lead to better molecular-level understanding of viral tropism, tissue damage, and the dynamics of compensatory tissue reorganization. Furthermore, scRNA-seq can provide a detailed view of changing cell-cell communication, reflected in expression patterns of cognate ligand-receptor pairs that are indicative of rapidly evolving cell-cell communication patterns.
In this work, we describe the first three days of SARS-CoV-2 infection in an ALI model of proximal airway epithelium, by performing a de novo analysis of a recently published single-cell gene expression dataset.12 By progressively refining cellular identities, we find that the virus preferentially enters via cells that are in the process of differentiating from basal-secretory intermediate cells or secretory cells into ciliated cells. As these infected ciliated progenitor cells further differentiate following viral infection, the viral infection spreads to more mature ciliated cells over time. Concurrently, viral infection and injury of ciliated epithelium also trigger a rapid enhancement of differentiation of basal cells, perhaps in order to restore the injured ciliated epithelial layer, leading to a substantial transient depletion of the resident progenitor cell populations. By the third day of infection, at the viral load examined, the renewal rate of basal cells is partially restored, thereby replenishing their numbers. These cell population dynamics are correlated with a transient increase of pro-differentiation cues followed by expression of pro-renewal cues for basal cells, which are all secreted by the population of infected ciliated cells. We propose a system-level mechanism that may underlie these processes and show that this mechanism creates a key vulnerability in airway epithelial tissue innate immunity during the first critical days of viral infection and replication. This work provides a high-resolution and nuanced perspective on dynamics of the proximal epithelial cell system from the onset of SARS-CoV-2 infection, thereby revealing fundamental properties of barrier-tissue response to viral entry and identifying routes for potential targeted interventions.
Results
Single-cell analysis of cell population dynamics during SARS-CoV-2 infection
scRNA-seq data were obtained from a previous manuscript, wherein ALI cultures of differentiated HBECs (Lonza) were inoculated with SARS-CoV-2 virus and harvested at 1 day post-infection (dpi), 2 dpi, and 3 dpi.12 The three infected time points and an uninfected mock condition were sequenced at the single-cell level (Figure 1A). In a de novo analysis, performed independently of work presented in the original manuscript, the scRNA-seq data were integrated across the time points (Seurat25), and all typical proximal airway epithelial cell types were identified, including basal cells, cycling basal cells, a small population of basal cells undergoing epithelial-mesenchymal transition (EMT, a known artifact of the ALI culture method26), intermediate cells (between basal and club phenotypes), club cells, goblet cells, ciliated cells, ionocytes, tuft cells, and pulmonary neuroendocrine cells (PNECs) (Figures 1B and 1C). These cell types were identified by top differentially expressed genes (Figure 1D).
Figure 1.

Single-cell analysis of cell population dynamics during SARS-CoV-2 infection
(A) Schematic of the experimental protocol; cells under four ALI conditions were collected and transcriptomically sequenced, including the first three days post-viral infection and an uninfected control condition, denoted as “Mock”. All wet-lab experimentation is documented in Ravindra et al.12
(B and C) UMAP analysis of scRNA-seq data aligned in Seurat, colored and labeled by (B) cell type and (C) infection condition, to demonstrate batch correction across samples.
(D) Heatmap of marker genes used for each cell type; columns represent average expression per cluster, and rows are unity normalized and colored by relative expression (see STAR Methods).
(E) Bar graph of cell type proportions per time point for the four largest clusters by cell number: basal, intermediate, club, and ciliated cells.
(F) UMAP results colored by pseudotime, as calculated using Monocle 3, split by time point, to demonstrate transient loss of progenitor cell populations in early infection time points.
(G and H) UMAP analysis of SARS-CoV-2 infected cells (blue) (G) aligned and (H) split by time point, to track the dynamics of the localized pockets of infected ciliated cells.
(I) Bar graphs of the proportion of infected cells per cell type at each time point for ciliated, club, intermediate, and basal cells. Cells were identified as infected if they expressed ≥10 exonic SARS-CoV-2 viral transcripts (see STAR Methods).
This initial cell type specification was used to study the epithelial cell population dynamics following SARS-CoV-2 infection. Strikingly, we observed a distinct, acute depletion of basal cells at 1 dpi, which recovered to near-control (mock) levels by 3 dpi (Figure 1E). More broadly, at 1 dpi, there was a profound depletion of all less-mature cells in the model, as identified by pseudotime,27 across all cell types at 1dpi (Figure 1F). This indicated a transient depletion of all population-specific resident progenitors, suggesting a phenotypic switch from renewal to differentiation, and an increased phenotypic flux toward mature cell types in the infected conditions. Like the basal cell cluster, population-specific resident progenitor cells also recovered their numbers by 3 dpi. Using viral transcripts to indicate SARS-CoV-2 infection (consistent with prior methods12), we ascertained that viral load initiates and concentrates in the ciliated cell cluster but, interestingly, primarily in a subset of cells in one region of the ciliated cluster that express markers of immaturity (Figures 1G and 1H). Longitudinal analysis of data on subsequent post-infection days suggests that the viral infection then spreads from this somewhat immature cell type to more mature cells, either by maturation of these infected cells or by viral replication and re-infection of more mature cells. By 3 dpi, nearly 25% of ciliated cells in the culture system are infected (Figure 1I). Overall, our initial observations using dynamic scRNA-seq demonstrate a transient depletion of basal and other progenitor cells upon infection, as well as greater SARS-CoV-2 tropism toward an immature subset of ciliated cells.
SARS-CoV-2 enters via ciliated progenitor cells
To gain a better perspective on SARS-CoV-2 infection dynamics in ciliated cells, we re-analyzed the ciliated cluster to gain a more discrete appreciation of the relevant subtypes (Figures 2A and 2B). While all cells in this cluster expressed canonical ciliated cell markers, four ciliated subtypes were identified and named, based on differential gene expression patterns. These subtypes include ciliated progenitors, defined by increased basal and secretory markers such as KRT5 and SCGB1A1, respectively; two mature ciliated clusters, with mature ciliated 1 defined by genes associated with immune response, such as HLA-DRB5; and mature ciliated 2 with uniquely high expression of genes associated with cilia motility, such as DLEC1. Finally, we defined a separate subtype of novel infected ciliated cells, which appear only in infected conditions, are not associated with any other clusters, and express genes unique to viral responsiveness, such as IFIT2 (Figure 2C).
Figure 2.

SARS-CoV-2 enters via ciliated progenitor cells
(A and B) UMAP of subset ciliated cells colored and labeled by (A) subtype (inset shows subtypes on Figure 1 embedding) and (B) time point, to demonstrate batch correction across samples.
(C) Heatmap of marker genes for each ciliated subtype; columns represent average expression per cluster, and rows are unity normalized and colored by relative expression (see STAR Methods).
(D) Bar graph of ciliated subtype proportions per time point.
(E) UMAP analysis results colored by pseudotime, as calculated using Monocle 3, split by time point, to demonstrate transient loss of ciliated progenitor cells in early infection time points.
(F and G) UMAP results for SARS-CoV-2 infected cells (blue) were (F) split by time point and (G) aligned.
(H) Bar graphs of the proportion of infected cells per ciliated subtype at each time point.
(I) Pie charts of proportion of total infected ciliated cells represented by each subtype per time point.
(J) Selected top gene ontology (GO) terms for differentially expressed genes (DEGs) between ciliated subtypes, plotted by significance (-log(p_val)), indicating the preferential association of viral-associated gene expression with ciliated progenitor cells.
(K) Heatmap of genes associated with the ciliated progenitor-associated GO term “Viral entry into host cell” with added known and putative receptors for SARS-CoV-2, demonstrating relatively high expression of these genes in ciliated progenitor cells; columns represent average expression per cluster, and rows are unity normalized and colored by relative expression (see STAR Methods).
(L) Heatmap of expression of viral entry genes in individual ciliated cells ordered by pseudotime, and labeled by ciliated subtype and infection status, demonstrating peak viral entry gene expression and infection in ciliated progenitor cells; columns represent expression levels of individual cells, and rows are unity normalized and colored by relative expression (see STAR Methods).
With this sub-clustering, we revisited the cell population dynamics analysis. Similar to basal cells in the bulk sample, the relative number of ciliated progenitor cells acutely decreased at 1 dpi compared to baseline (mock) and then recovered to near-control proportions by 3 dpi (Figure 2D). This loss of ciliated progenitor cells was also reflected in the pseudotime analysis, revealing that the least mature cells were depleted on infection and then recovered over time (Figure 2E). Viral load is concentrated first in novel infected cells at 1 dpi, followed by ciliated progenitor cells as their numbers are replenished (Figures 2F and 2G). By 3 dpi, nearly 25% of ciliated progenitor cells are infected, as well as nearly 100% of novel infected ciliated cells (Figure 2H). By 2 dpi and 3 dpi, when the cell populations begin to re-stabilize, ciliated progenitor cells account for 60% of all infected ciliated cells, with novel infected ciliated cells accounting for 28% at 3 dpi (Figure 2I).
Analysis of differentially expressed genes between ciliated cell subtypes using gene ontology (GO) revealed functional groupings of genes unique to each subtype (Figure 2J). Notably, this method further suggested that ciliated progenitors are the main hub of viral entry and transcription among ciliated cells. GO analysis also confirmed functional roles of the other ciliated subtypes, including antigen presentation in mature ciliated 1 cells, cilia function in mature ciliated 2 cells, and interferon production in novel infected cliated cells. By pulling the genes categorized in the “viral entry into host cell” GO category, as well as known and putative genes associated with specific entry of SARS-CoV-2, such as ACE2, TMPRSS2,10 Cathepsin L (CTSL), HMGB1,28 neuropilin-1 (NRP1),29 BSG,30 and FURIN,31 we observed that most of these genes are preferentially expressed in the ciliated progenitor population (Figure 2K). In addition, TMPRSS2, BSG, HMGB1, and CTSL also showed high expression in mature ciliated 1 cells.
Interestingly, the novel infected ciliated cells, which are nearly ubiquitously infected with SARS-CoV-2, did not significantly express any genes associated with viral entry, suggesting these cells arise from already infected ciliated cells. Indeed, these cells fall just after ciliated progenitor cells in the pseudotime developmental trajectory. Ciliated cells ordered by pseudotime confirm that expression of viral entry genes concentrates early in the ciliated cell differentiation trajectory (Figure 2L). These observations strongly argue that ciliated progenitor cells are the main point of entry for SARS-CoV-2.
Interferon response analysis
At 3 dpi, genes associated with interferon production and response are uniquely upregulated over all other time points (Figure 3A). Comparison of genes associated with interferon-related GO categories demonstrates that expression of most of these genes increases over time following infection, peaking around 3 dpi (Figure 3B). Specifically, IFIT1, IFIT2, IFIT3, and IFIT5 appear most strongly at 3 dpi (Figure 3C).
Figure 3.

Interferon response analysis
(A) Selected top GO terms for genes differentially expressed between bulk samples across time, plotted by significance.
(B) Heatmap of interferon gene list determined by all interferon-related GO terms in panel A, grouped by dendrogram to demonstrate expression trends over time; columns represent average expression per cluster, and rows are unity normalized and colored by relative expression (see STAR Methods).
(C) Dot plot of IFIT gene expression over time.
(D and E) Feature plots of interferon signaling score (as calculated using Seurat’s AddModuleScore) with the gene list in panel (B) in (D) bulk and (E) ciliated cell populations; a high score demonstrates greater relative expression of this group of genes, suggesting interferon-related gene expression maximized at 3 days post-infection (dpi), and primarily localized to a portion of mature ciliated 1 cells and club cells.
It is worth noting that these viral response signals appear as part of the innate immune response and are mounted by epithelium in the absence of immune cells.32 These genes mainly appear in the subsets of ciliated and club cells, which is consistent with previous findings,12,33 as well as some cells in PNEC, basal, and basal EMT populations (Figure 3D). Interferon-related gene expression is most concentrated in an infection-specific archetype of the mature ciliated 1 cell cluster and in the novel infected ciliated cells (Figure 3E). Taken together, this suggests that interferon secretion is not the immediate response of epithelial cells to infection with SARS-CoV-2 virus, leaving the possibility that the epithelium may undergo another defensive response first.
Analysis of signaling networks during infection
We then explored putative mechanisms underlying the transient enhancement of basal cell differentiation, followed by restoration of pro-renewal cues by 3 dpi. Mapping the single-cell data against the FANTOM5 database of ligand-receptor interactions allowed us to explore cell-cell communication networks as a function of time following infection (Figure 4A). We performed Wilcoxon rank-sum tests on all genes potentially involved in intercellular communication for each major cell type shown in Figure 4B, using the mock condition as a control for each post-infection time point.34 The resultant list of all ligands and receptors displayed study-relevant and statistically significant differential expression following viral infection. Each signaling mechanism having a significant perturbation of both ligand and receptor, vs. the mock control, was then plotted over time (Figure 4C).
Figure 4.

Analysis of signaling networks during infection
(A) To determine the set of ligands and receptors that were perturbed by infection, paired Wilcoxon rank-sum tests were performed, by bulk cell type, using mock cells as the reference, for each post-infection time point on all genes in the FANTOM5 database.
(B) This routine was performed with the ciliated subtypes grouped as a single cell type.
(C) All ligand-receptor interactions found to be significantly perturbed were then plotted over time, with the ciliated population broken into subtypes; particular focus was given to the novel infected ciliated cell population (red trapezoid), which does not exist in the mock condition and only appears post-infection.
(D–I) Longitudinal line plots of key ligands of interest, split and colored by cell type, over time. Ligand expression was calculated using a standard log-normalization technique (see STAR Methods).
(J) Heatmap of all genes of interest over time; columns represent average expression per cluster, and rows are unity normalized and colored by relative expression (see STAR Methods). See also Figure S1.
Interestingly, we found that novel infected ciliated cells, which harbor the greatest viral load, rapidly increased expression of TGFβ2 and BMP4, which can exert pro-differentiation and anti-proliferation effects on pulmonary basal cells (Figures 4D and 4E).35,36,37,38 In addition, basal cells (and virtually all observed cell types, except novel infected ciliated cells and mature ciliated 2 cells) expressed high and gradually increasing levels of transforming growth factor β (TGFβ)-receptor, TGFBR2, while maintaining high levels of BMP receptors, including BMPR1A (Figures S1A and S1B). The increase in BMP4 signaling was transient, peaking at 1 dpi, along with expression of its canonical target, ID3, in basal and other progenitor cells (Figure S1C). In contrast, TGFB2 expression increased more gradually and was more persistent, along with upregulation of TGFβ target genes CTGF, ITGAV, and THBS1 (thrombospondin) (Figures S1D–S1F). Interestingly, the proteins encoded by these genes have been shown to further promote TGFβ signaling, constituting putative positive feedback loops, which may explain their prolonged expression.39,40,41
The overall pro-differentiation signal was also likely enhanced by a transient increase in expression of a non-canonical WNT ligand, WNT5A, known to be antagonistic to canonical WNT signaling implicated in self-renewal of basal cells (Figure 4F).42,43,44,45 However, by 3 dpi, there was an increase of expression of a potent inhibitor of BMP and TGFβ signaling, follistatatin (FST), as well as a strong increase of expression of EGFR ligands AREG and EREG (Figures 4G–4I). AREG has been shown to promote basal cell self-renewal at the expense of differentiation,46 suggesting that these signals can curtail pro-differentiation signaling. By shutting off pro-differentiation cues and encouraging self-renewal, the system can prevent total exhaustion of the basal cell pool and support replenishment of basal and progenitor cells. Taken together, these data suggest that basal cells are initially being induced to differentiate by TGFβ and BMP signals arising from novel infected ciliated cells, potentially aided by antagonism of pro-renewal WNT signaling. By 3 dpi, these pro-differentiation signals are halted by inhibition of TGFβ and BMP signaling by FST, and pro-renewal of basal cells is restored by expression of AREG and EREG.
The interplay between BMP and TGFβ signaling can be complex, potentially leading to synergistic or antagonistic effects.47 However, our analysis indicates that both TGFβ2 and BMP4 signal through SMAD1/5 rather than SMAD2/3 signaling pathways (Figures S1G–S1J). Indeed, SMAD2/3 responsive genes SERPINE1 and PLAU were downregulated in putative TGFβ target cells, despite the evidence of TGFβ2 activation, with SMAD3 expression also decreasing over time (Figures S1K and S1L).48 Furthermore, we found a gradual upregulation of genes previously implicated as direct SMAD1/5 targets, BMPR2 and JAG1, consistent with more persistent TGFβ2, rather than more transient BMP-mediated signaling kinetics (Figures S1M and S1N).49 In contrast, in novel infected ciliated cells, PLAU expression transiently increased, suggestive of a divergent signaling program (Figure S1L).
These observations raised the question of how the pro-differentiation, and later pro-proliferation, signals were induced. Both TGFβ2 and BMP4 can be induced in response to tissue damage from acute or chronic injury.35,36,37,38 However, importantly, TGFβ signaling can also be induced by various viral infections.50 This could occur, for example, due to an increase of intracellular reactive oxygen species (ROS) triggered following viral entry, which has been observed in SARS-CoV-2 infection.51 We found evidence of the onset of oxidative stress in intermediate and basal cells, as indicated by an increase in expression of isoforms of glutathione S-transferase (GSTA1 and GSTP1) and thioreductin (TXN) (Figures S1O–S1Q). Interestingly, TXN underwent rapid downregulation, specifically in novel infected ciliated cells, suggesting a virus-mediated repression of the anti-oxidative stress mechanisms in these cells, consistent with prior results showing suppressive effects of various viral infections, including SARS-CoV-2, on Nrf2-mediated anti-oxidative stress responses.52 This view was corroborated by the surprising decrease of ACE2 in the novel infected ciliated cells (Figure S1R). Indeed, ACE2 has recently been implicated as a direct Nrf2 target, and thus its expression is expected to drop along with the expression of other Nrf2 targets, such as TXN.53,54,55
The delayed upregulation of the basal cell self-renewal-promoting factors, AREG and EREG, is of less clear origin. However, it is of interest that these genes are targets of Nrf2 and YAP/TAZ signaling.56,57 Even though we found that the components of the YAP/TAZ signaling network, YAP1 and WWTR1, were transiently downregulated (which could assist in basal cell differentiation58), they recovered by 3 dpi, which in the presence of enhanced Nrf2 signaling across different cell types could lead to elevated AREG and EREG expression (Figures S1S and S1T).46,59
A number of other signaling networks displayed dynamical alterations during the first few days following SARS-CoV-2 infection. In particular, we found that NOTCH1 increased gradually in basal and intermediate cells, which could further promote differentiation toward ciliated and possibly club cell lineages (Figure S1U).59 Trends in these and other signaling network perturbations are summarized in Figure 4P, as well as Figure S1.
Overall, these data suggest that a particularly high level of oxidative stress in novel infected ciliated cells, not mitigated by the Nrf2-mediated response, may have led to enhanced TFGβ signaling from novel infected ciliated cells to other cell targets, including basal cells. By deriving the source of this pro-differentiation signaling, we can complete the picture of the communication network that drives a brief period of rapid basal cell differentiation following viral infection.
Discussion
The results presented here suggest that SARS-CoV-2 infection can trigger a rapid and transient increase in basal and progenitor cell differentiation, enabled by expression of pro-differentiation signaling cues by virally infected cells (Figure 5). The cues are secreted by a newly described subset of infected ciliated cells (novel infected ciliated cells, or NICs), that cluster separately from other mature and progenitor ciliated cells and display a virtually 100% infection rate.
Figure 5.

Summary schematics of cell dynamics regulated by inferred cell-cell interactions
For a Figure360 author presentation of this figure, see https://doi.org/10.1016/j.isci.2023.106175.
(A) Signaling mechanisms influencing population dynamics during early viral infection. At 1dpi, TGFB2 and BMP4, primarily from novel infected ciliated (NIC) cells, promote differentiation and block self-renewal of basal cells (BC); WNT5A further blocks self-renewal in BC; this contributes to the observed depletion of progenitor cell populations, including BC and ciliated progenitor (CP) cells, while mature ciliated (MC) cells are largely maintained; increased differentiation also provides a wider window for viral entry via CP cells. By 3dpi, FST expressed by NIC and BC blocks aforementioned TGFB2 and BMP4 signaling, effectively blocking differentiation, while newly increased AREG and EREG signaling restores BC self-renewal; progenitor cell populations (indicated by cell size in the diagram), and patterns of homeostatic turnover are restored, simultaneously with upregulation of interferon-related signaling (see Figure 3).
(B and C) Model of CP vulnerability to infection. Comparison of models of viral infection via ciliated progenitor cells (B) vs viral infection via mature ciliated cells (C). The virus achieves greater overall cellular infection when leveraging the rapid epithelial turnover during early infection and viral entry into CP cells than if the viruses were to enter via MC cells alone, as is frequently assumed. This summarizes our findings and demonstrates the efficiency of the viral entry strategy leveraged by the SARS-CoV-2 virus in infecting human host airway epithelium. Images adapted from BioRender.
The process of differentiation transiently depletes basal cells, a key airway epithelial stem cell reservoir, while supporting the proportion of ciliated cells. As cellular proportions stabilize by 3 dpi, up-regulation of new anti-differentiation, pro-renewal cues, particularly those of the EGF receptor family, restores the renewal of basal and progenitor cells. Although this process can serve as a typical strategy to repair the bronchial tissue damage inflicted by the initial viral infection, it may also be compromised by the apparent tropism of the virus toward active progenitor cells rather than to stable mature cells. As a result, the tissue-repair process itself may open the door wider to infection by exposing increased progenitors for viral entry and subsequent progressive infection of mature epithelial cells. A similar viral entry and infection strategy targeting progenitor cells has been previously observed in placental infection by Zika virus and human cytomegalovirus.60,61
Our findings provide additional resolution to the identification of ciliated cells as the main entry point for SARS-CoV-2 and known patterns of viral tropism to other cell types of airway epithelium.12 In addition, our findings of preferential infectivity in proximal epithelial progenitors are paralleled by findings that SARS-CoV-2 preferentially infects ACE2-expressing alveolar type II (ATII) cells, which are progenitor cells of distal lung epithelium.7 We further confirm gene expression patterns consistent with interferon stimulation (i.e., IFIT and ISG), as described previously.12
Another interesting finding is the rapid downregulation of ACE2 receptor in novel infected ciliated cells. This is consistent with findings by Ravindra et al. (using the same dataset), where ACE2 expression did not correlate or increase with infection in any cell type.12 This is also consistent with previous findings that ACE2 expression decreases with SARS-CoV-1 infection62,63,64 and in an animal model of ARDS.65 ACE2 enables cellular entry of SARS-CoV-1 and HCoV-NL63,66,67,68 and though SARS-CoV-1 and SARS-CoV-2 both utilize cathepsins L (CTSL) and B (CTSB) in viral entry, HCoV-NL63 does not10,69,70 In our experiments, CTSL and CTSB were both differentially expressed by subsets of ciliated cells (Figure 2K). It has been shown that SARS-CoV-1 viral infectivity correlates with epithelial differentiation state in vitro, as well as ACE2 expression level and localization.71
Observed reduction in ACE2 gene expression in our dataset may be coupled with the demonstrated reduction in ACE2 protein content in SARS-CoV-2-infected cells.72,73,74 Observed instances of ACE2 reduction on viral infection are likely due to internalization following binding of the virus.75,76 ACE2 has also been found to be protective from experimental acute lung injury and pulmonary inflammation,62,77,78,79 which may further explain how loss of ACE2 during SARS-CoV-2 infection contributes to severe disease. From the viral perspective, a decrease of ACE2 expression may help limit the multiplicity of single-cell infection by the virus, thereby increasing viral yield by the cell due to an increased cell survival by avoidance of high MOI. This effect is observed and exploited in viral vaccine production80 and is supported by mathematical modeling.81 An increased viral yield, on a per-cell basis, may help spread the infection within the epithelial layers, increasing the overall pathogenicity of the virus.
These results add a new dimension to the discussion of age and gender dependence of COVID-19 progression. Children tend to be less susceptible to SARS-CoV-2 infection and develop less severe symptoms.82,83 Many hypotheses have been developed and investigated to understand this phenomenon. It is unclear whether expression of viral entry genes varies with age or what role that may play in age-related infectivity.84,85,86 However, known differences between the developing and mature innate immune systems of children and adults seem to contribute to observed differences in infectivity and disease severity. Children have shown greater numbers of innate immune cells and those pre-activated for innate immune response in the upper airway than adults.85,87,88 Critically, airway epithelial cells appear to be primed to respond to viral insult through enhanced viral sensing in the upper airways of children.85,88
This may support a hypothesis based on our findings that children and young people may experience reduced COVID-19 severity due to naturally higher epithelial regenerative capacities, allowing them to shed infected cells more quickly than adults. Homeostatic airway epithelial turnover and progenitor cell function decrease with age.89 In young people, rapid epithelial turnover in response to more rapidly sensing inhaled SARS-CoV-2 particles, as observed in our study, may be adequate to successfully block viral entry or shed and replace infected cells before systemic transmission, except in cases of exposure to sufficiently high viral loads. This protective mechanism would be greatly reduced in older populations.
The COVID-19 pandemic is not yet beaten, and the infection dynamics of the virus must be elucidated in order to treat remaining cases and fully vanquish this disease. By identifying systemic vulnerabilities and modeling offensive viral strategies, this work may open the door to potential points of intervention for the prevention and/or treatment of SARS-CoV-2 infection.
Limitations of the study
It must be noted that transcriptional analyses do not account for downstream translational or protein behaviors, which may vary from mRNA patterns, as has been demonstrated in ACE2.90,91 Experiments involving protein analyses must be employed to complete the stories outlined by transcriptional analyses. The findings of this study also would be strengthened by additional biological replicates, as the dataset analyzed only contains a single scRNA-seq sample per time point. Further experimental validation will strengthen this picture of SARS-CoV-2 infection dynamics in vitro and in vivo.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| scRNAseq data | (Ravindra et al., 2021)12 | GEO: GSE166766 |
| Original code | This manuscript | https://doi.org/10.6084/m9.figshare.19096955 |
| Software and algorithms | ||
| Seurat (v3.0) | (Butler et al., 2018;92 Satija et al., 2015;25 Stuart et al., 2019)93 |
https://satijalab.org/seurat/ |
| ComplexHeatmap (vX.X.X) | (Gu et al., 2016)94 | http://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| Connectome (v1.0.0) | (Raredon et al., 2022)95 | https://msraredon.github.io/Connectome/ |
| The Gene Ontology Resource + GO Enrichment Analysis | (Ashburner et al., 200096; Carbon et al., 202197; Mi et al., 2019)98 | http://geneontology.org/ |
| Monocle (v3) | (Cao et al., 2019)27 | https://cole-trapnell-lab.github.io/monocle3/ |
| ggplot2 (v3.3.5) | (Wickham, 2016)99 | https://ggplot2.tidyverse.org/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Andre Levchenko (andre.levchenko@yale.edu).
Materials availability
This study did not generate new unique reagents.
Method details
Generation and preprocessing of scRNAseq data
scRNAseq data of ALI cultures were obtained from Ravindra et al.12 Briefly, human bronchial epithelial cells (HBECs, Lonza) were cultured at an air-liquid interface (ALI) for 28 days to achieve full mucociliary differentiation. Cultures were then challenged at the apical surface with 104 plaque forming units (PFU) of SARS-CoV-2. An uninfected mock control and samples at 1dpi, 2dpi, and 3dpi were harvested with TrypLE Express Enzyme (ThermoFisher) and prepared with the Chromium Next GEM Single Cell 3’ Gel beads v3.1 kit (10X Genomics), at a target loading of 10,000 cells per sample. Libraries were generated using the Chromium Single Cell 3’ Library Kit v3.1 (10X Genomics) and sequenced on the NovaSeq 6000 using HiSeq 100 base pair reads and dual indexing, to an average depth of 31,383 reads per cell. Data were aligned and pre-processed using the 10x Genomics Cell Ranger pipeline and a combined human and SARS-CoV-2 genome. On average across samples, there were 10,000 to 15,000 counts per cell and 2,400 to 3,600 unique genes per sample. For more information, see original publication.12
Alignment and clustering of scRNAseq data
Count matrices and metadata were input to Seurat v3.0.25 Integration was performed across infection time points using the standard Seurat v3 integration pipeline to enable consistent cell type identification.92,93 The aligned object was further normalized, scaled, and clustered in UMAP space by PCA, regressing on percent mitochondrial reads and number of counts on the scale step. Resultant clusters were named as cell types based on unique canonical gene expression. Small clusters of doublets or partial cells were removed based on multiple cell marker expression and uniquely high read counts, or uniquely low read counts, respectively. Following cleaning and initial processing, all subsequent analyses were performed on a total of 75,245 cells across four conditions.
To further parcellate ciliated subtypes, all ciliated cells (21,510 cells) were subset and re-clustered following the same standard Seurat pipeline as above. Putative subtypes were identified and named based on functionality associated with differentially expressed genes (FindAllMarkers).
Downstream scRNAseq analyses
Most plotting was performed using Seurat and ggplot2.25 All heatmaps were generated using ComplexHeatmap94 and unity normalized by row to represent greatest and least expressive column condition, while preserving intermediate variation. Cell type and ciliated subtype population proportions were estimated by calculating the percent of each time point sample comprised by each cell population. While this method suffers potential quantitative error due to dissociation bias and capture efficiency, this error was reduced by simultaneous dissociation and GEM generation of samples. Cells were determined to be infected with SARS-CoV-2 by a previously described method.12 Briefly, single cells were considered infected if they contained ≥ 10 exonic SARS-CoV-2 viral transcripts, to control for viral cell surface attachment, ambient virus in the suspension, or read misalignment. Infected cell bar plots were generated by calculating the percent of each cell population at each time point that was determined to be virally infected, using the above metric for infection. Alternatively, infected cell circle plots were generated by calculating the cell population percent breakdown of all infected cells per infected time point.
Pseudotime analysis was performed using Monocle 3,27 where cells are aligned on a dimensionless time-scale based on diffusion of gene expression. The standard Monocle pipeline was performed on the data slot of the bulk sample and ciliated subset, calling alignment arguments where applicable to control for batch effects between time point samples.100 Calculated pseudotime scores were then transferred to metadata of the respective Seurat objects in order to plot on existing UMAP embeddings.
Gene ontological analysis was performed using the GO Enrichment Analysis function on the Gene Ontology web platform (geneontology.org).96,98,97 First differential gene analysis was performed (FindAllMarkers), with thresholds on min.pct = 0.5 and logfc.threshold = 0.5, among conditions of interest. These conditions included putative ciliated subtypes to differentiate functionality between clusters, and the bulk population across time points to identify global trends over the course of infection. Resultant list of DEGs was uploaded to generate lists of GO terms associated with biological processes of each condition. Top GO terms were curated to remove similar terms with comparable gene lists and irrelevant terms, and plotted by . Genes associated with particular GO terms were used for various plotting and scoring. Cell scoring of expression of gene lists was performed for genes associated with interferon-related GO terms (AddModuleScore).
In all instances where gene expression is quantified from scRNAseq data (i.e. line plots in Figures 4 and S1), we utilized a log-normalization technique that is standard in the Seurat analysis pipeline.25 In this case, expression level is determined from the number of transcripts of a given gene measured within a single cell, relative to the total number of transcripts sequenced for that cell. These transcripts are referred to as “counts” and this relation is as follows:
Cell-cell signaling vectors were mapped using Connectome v1.0.095 with a minimum ligand and receptor expression of 2.5% on sending and receiving cell respectively. Differential cell-cell signaling mechanisms were identified by performing a Wilcoxon Rank Sum test comparing populations in Mock and infected samples, for each time point, across all ligands and all receptors present in the FANTOM5 database with an adjusted p-value cutoff of 0.05. This routine was performed with the ciliated subtypes grouped as a single cell type in each sample, so as to preserve node architecture and make 1:1 connectomic comparisons possible. This provided a list of ligand-receptor mechanisms showing significant differential expression in at least one cell type-cell type vector in at least one time point comparison. This mechanism list was then used to filter the original complete connectomic edgelist to an edgelist of interest, which were considered significant. The R package ggplot299 was then used to create plots of longitudinal cell-cell signaling trends over time for both sending and receiving populations.
Quantification and statistical analysis
Gene ontology
To generate gene lists, differential gene analysis was performed (FindAllMarkers), with thresholds of min.pct = 0.5 and logfc.threshold = 0.5, among conditions of interest, to determine significantly differential genes. Top GO terms were plotted by , as calculated in the GO Enrichment Analysis software, to compare significance values among terms.
Connectome
Significantly perturbed cell-cell signaling mechanisms were identified by performing a Wilcoxon Rank Sum test comparing populations in Mock and infected samples, for each time point, across all ligands and all receptors present in the FANTOM5 database with a minimum ligand and receptor expression cutoff of 2.5% per cluster and an adjusted p-value cutoff of 0.05.
Acknowledgments
This work was supported by grants from the National Institutes of Health (U54 CA209992 (NCI) to A.L.; F32HL162428 to A.M.G.; F30HL143906, T32GM136651, and T32GM086287 to M.S.B.R.), as well as private research gifts and an unrestricted research gift from Humacyte Inc. We would like to acknowledge Drs. Craig Wilen, David van Dijk, and Akiko Iwasaki for helpful discussions and input on the interpretation of our results.
Author contributions
Conceptualization, A.M.G., M.S.B.R., M.P.K., and A.L.; Methodology, A.M.G., M.S.B.R., and M.P.K.; Software, A.M.G., M.S.B.R., and M.P.K.; Validation, M.S.B.R; Formal Analysis, A.M.G., M.S.B.R., M.P.K., and A.L.; Investigation, A.M.G., M.S.B.R., and M.P.K.; Resources, L.E.N. and A.L.; Data Curation, A.M.G.; Writing – Original Draft, A.M.G. and A.L.; Writing – Review & Editing, A.M.G., M.S.B.R., L.E.N., and A.L.; Visualization, A.M.G., M.S.B.R., and M.P.K.; Supervision, A.L.; Project Administration, A.L.; Funding Acquisition, L.E.N. and A.L.
Declaration of interests
L.E.N. is the CEO, founder, and shareholder in Humacyte, Inc, which is a regenerative medicine company. Humacyte produces engineered blood vessels from allogeneic smooth muscle cells for vascular surgery. L.E.N.’s spouse has equity in Humacyte, and L.E.N. serves on Humacyte’s Board of Directors. L.E.N. is an inventor on patents that are licensed to Humacyte and that produce royalties for L.E.N. L.E.N. has received an unrestricted research gift to support research in her laboratory at Yale. Humacyte did not influence the conduct, description, or interpretation of the findings in this report.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: February 10, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106175.
Supplemental information
Data and code availability
-
•
This paper analyzes existing, publicly available scRNAseq data. The Gene Expression Omnibus (GEO) accession number is listed in the key resources table.
-
•
All original code has been deposited at Figshare and is publicly available as of the date of publication. The DOI is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Wang C., Horby P.W., Hayden F.G., Gao G.F. A novel coronavirus outbreak of global health concern. Lancet. 2020;395:470–473. doi: 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World health organisation . World health organisation; 2022. COVID-19 Weekly Epidemiological Update. 30 November 2022. [Google Scholar]
- 3.Centers for Disease control and prevention (2022). COVID data tracker weekly Review, December 02, 2022.
- 4.Meng B., Kemp S.A., Papa G., Datir R., Ferreira I.A.T.M., Marelli S., Harvey W.T., Lytras S., Mohamed A., Gallo G., et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021;35:109292. doi: 10.1016/j.celrep.2021.109292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chia P.Y., Xiang Ong S.W., Chiew C.J., Ang L.W., Chavatte J.M., Mak T.M., Cui L., Kalimuddin S., Chia W.N., Tan C.W., et al. Virological and serological kinetics of SARS-CoV-2 Delta variant vaccine-breakthrough infections: a multi-center cohort study. Clin. Microbiol. Infect. 2021;28:612.e1–612.e7. doi: 10.1016/j.cmi.2021.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pulliam J.R.C., van Schalkwyk C., Govender N., von Gottberg A., Cohen C., Groome M.J., Dushoff J., Mlisana K., Moultrie H. Increased risk of SARS-CoV-2 reinfection associated with emergence of Omicron in South Africa. Science. 2022;376:eabn4947. doi: 10.1126/science.abn4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hou Y.J., Okuda K., Edwards C.E., Martinez D.R., Asakura T., Dinnon K.H., 3rd, Kato T., Lee R.E., Yount B.L., Mascenik T.M., et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell. 2020;182:429–446.e14. doi: 10.1016/j.cell.2020.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leung J.M., Yang C.X., Tam A., Shaipanich T., Hackett T.L., Singhera G.K., Dorscheid D.R., Sin D.D. ACE-2 expression in the small airway epithelia of smokers and COPD patients: implications for COVID-19. Eur. Respir. J. 2020;55 doi: 10.1183/13993003.00688-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaefer I.M., Padera R.F., Solomon I.H., Kanjilal S., Hammer M.M., Hornick J.L., Sholl L.M. In situ detection of SARS-CoV-2 in lungs and airways of patients with COVID-19. Mod. Pathol. 2020;33:2104–2114. doi: 10.1038/s41379-020-0595-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sungnak W., Huang N., Bécavin C., Berg M., Queen R., Litvinukova M., Talavera-López C., Maatz H., Reichart D., Sampaziotis F., et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020;26:681–687. doi: 10.1038/s41591-020-0868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ravindra N.G., Alfajaro M.M., Gasque V., Huston N.C., Wan H., Szigeti-Buck K., Yasumoto Y., Greaney A.M., Habet V., Chow R.D., et al. Single-cell longitudinal analysis of SARS-CoV-2 infection in human airway epithelium identifies target cells, alterations in gene expression, and cell state changes. PLoS Biol. 2021;19:30011433. doi: 10.1371/journal.pbio.3001143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arentz M., Yim E., Klaff L., Lokhandwala S., Riedo F.X., Chong M., Lee M. Characteristics and outcomes of 21 critically ill patients with COVID-19 in Washington state. JAMA. 2020;323:1612–1614. doi: 10.1001/jama.2020.4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X., et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu N., Wang W., Liu Z., Liang C., Wang W., Ye F., Huang B., Zhao L., Wang H., Zhou W., et al. Morphogenesis and cytopathic effect of SARS-CoV-2 infection in human airway epithelial cells. Nat. Commun. 2020;11:3910–3918. doi: 10.1038/s41467-020-17796-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang J., Hume A.J., Abo K.M., Werder R.B., Villacorta-Martin C., Alysandratos K.D., Beermann M.L., Simone-Roach C., Lindstrom-Vautrin J., Olejnik J., et al. SARS-CoV-2 infection of pluripotent stem cell-derived human lung alveolar type 2 cells elicits a rapid epithelial-intrinsic inflammatory response. Cell Stem Cell. 2020;27:962–973.e7. doi: 10.1016/j.stem.2020.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lucas C., Wong P., Klein J., Castro T.B.R., Silva J., Sundaram M., Ellingson M.K., Mao T., Oh J.E., Israelow B., et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584:463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi T., Ellingson M.K., Wong P., Israelow B., Lucas C., Klein J., Silva J., Mao T., Oh J.E., Tokuyama M., et al. Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature. 2020;588:315–320. doi: 10.1038/s41586-020-2700-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giamarellos-Bourboulis E.J., Netea M.G., Rovina N., Akinosoglou K., Antoniadou A., Antonakos N., Damoraki G., Gkavogianni T., Adami M.E., Katsaounou P., et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. 2020;27:992–1000.e3. doi: 10.1016/j.chom.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Z., Ren L., Zhang L., Zhong J., Xiao Y., Jia Z., Guo L., Yang J., Wang C., Jiang S., et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe. 2020;27:883–890.e2. doi: 10.1016/j.chom.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mathew D., Giles J.R., Baxter A.E., Oldridge D.A., Greenplate A.R., Wu J.E., Alanio C., Kuri-Cervantes L., Pampena M.B., D'Andrea K., et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369 doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maggi E., Canonica G.W., Moretta L. COVID-19: unanswered questions on immune response and pathogenesis. J. Allergy Clin. Immunol. 2020;146:18–22. doi: 10.1016/j.jaci.2020.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lieberman N.A.P., Peddu V., Xie H., Shrestha L., Huang M.L., Mears M.C., Cajimat M.N., Bente D.A., Shi P.Y., Bovier F., et al. In vivo antiviral host transcriptional response to SARS-CoV-2 by viral load, sex, and age. PLoS Biol. 2020;18 doi: 10.1371/journal.pbio.3000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Satija R., Farrell J.A., Gennert D., Schier A.F., Regev A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015;33:495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greaney A.M., Adams T.S., Brickman Raredon M.S., Gubbins E., Schupp J.C., Engler A.J., Ghaedi M., Yuan Y., Kaminski N., Niklason L.E. Platform effects on regeneration by pulmonary basal cells as evaluated by single-cell RNA sequencing. Cell Rep. 2020;30:4250–4265.e6. doi: 10.1016/j.celrep.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao J., Spielmann M., Qiu X., Huang X., Ibrahim D.M., Hill A.J., Zhang F., Mundlos S., Christiansen L., Steemers F.J., et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496–502. doi: 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei J., Alfajaro M.M., DeWeirdt P.C., Hanna R.E., Lu-Culligan W.J., Cai W.L., Strine M.S., Zhang S.M., Graziano V.R., Schmitz C.O., et al. Genome-wide CRISPR screens reveal host factors critical for SARS-CoV-2 infection. Cell. 2021;184:76–91.e13. doi: 10.1016/j.cell.2020.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cantuti-Castelvetri L., Ojha R., Pedro L.D., Djannatian M., Franz J., Kuivanen S., van der Meer F., Kallio K., Kaya T., Anastasina M., et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370:856–860. doi: 10.1126/science.abd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matusiak M., Schürch C.M. Expression of SARS-CoV-2 entry receptors in the respiratory tract of healthy individuals, smokers and asthmatics. Respir. Res. 2020;21:252. doi: 10.1186/s12931-020-01521-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shenoy A.T., Lyon De Ana C., Arafa E.I., Salwig I., Barker K.A., Korkmaz F.T., Ramanujan A., Etesami N.S., Soucy A.M., Martin I.M.C., et al. Antigen presentation by lung epithelial cells directs CD4(+) TRM cell function and regulates barrier immunity. Nat. Commun. 2021;12:5834. doi: 10.1038/s41467-021-26045-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chua R.L., Lukassen S., Trump S., Hennig B.P., Wendisch D., Pott F., Debnath O., Thürmann L., Kurth F., Völker M.T., et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020;38:970–979. doi: 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- 34.Ramilowski J.A., Goldberg T., Harshbarger J., Kloppmann E., Lizio M., Satagopam V.P., Itoh M., Kawaji H., Carninci P., Rost B., Forrest A.R.R. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat. Commun. 2015;6:7866. doi: 10.1038/ncomms8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saito A., Horie M., Nagase T. TGF-Beta signaling in lung health and disease. Int. J. Mol. Sci. 2018;19:2460. doi: 10.3390/ijms19082460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tadokoro T., Gao X., Hong C.C., Hotten D., Hogan B.L.M. BMP signaling and cellular dynamics during regeneration of airway epithelium from basal progenitors. Development. 2016;143:764–773. doi: 10.1242/dev.126656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aschner Y., Downey G.P. Transforming growth factor-beta: master regulator of the respiratory system in health and disease. Am. J. Respir. Cell Mol. Biol. 2016;54:647–655. doi: 10.1165/rcmb.2015-0391TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bartram U., Speer C.P. The role of transforming growth factor beta in lung development and disease. Chest. 2004;125:754–765. doi: 10.1378/chest.125.2.754. [DOI] [PubMed] [Google Scholar]
- 39.Mamuya F.A., Duncan M.K. aV integrins and TGF-ss-induced EMT: a circle of regulation. J. Cell Mol. Med. 2012;16:445–455. doi: 10.1111/j.1582-4934.2011.01419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murphy-Ullrich J.E., Suto M.J. Thrombospondin-1 regulation of latent TGF-beta activation: a therapeutic target for fibrotic disease. Matrix Biol. 2018;68–69:28–43. doi: 10.1016/j.matbio.2017.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lipson K.E., Wong C., Teng Y., Spong S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair. 2012;5:S24. doi: 10.1186/1755-1536-5-S1-S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caprioli A., Villasenor A., Wylie L.A., Braitsch C., Marty-Santos L., Barry D., Karner C.M., Fu S., Meadows S.M., Carroll T.J., Cleaver O. Wnt4 is essential to normal mammalian lung development. Dev. Biol. 2015;406:222–234. doi: 10.1016/j.ydbio.2015.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hussain M., Xu C., Lu M., Wu X., Tang L., Wu X. Wnt/beta-catenin signaling links embryonic lung development and asthmatic airway remodeling. Biochim. Biophys. Acta, Mol. Basis Dis. 2017;1863:3226–3242. doi: 10.1016/j.bbadis.2017.08.031. [DOI] [PubMed] [Google Scholar]
- 44.Serra R., Easter S.L., Jiang W., Baxley S.E. Wnt5a as an effector of TGF beta in mammary development and cancer. J. Mammary Gland Biol. Neoplasia. 2011;16:157–167. doi: 10.1007/s10911-011-9205-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim H.T., Yin W., Nakamichi Y., Panza P., Grohmann B., Buettner C., Guenther S., Ruppert C., Kobayashi Y., Guenther A., Stainier D.Y.R. WNT/RYK signaling restricts goblet cell differentiation during lung development and repair. Proc. Natl. Acad. Sci. USA. 2019;116:25697–25706. doi: 10.1073/pnas.1911071116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zuo W.L., Yang J., Gomi K., Chao I., Crystal R.G., Shaykhiev R. EGF-amphiregulin interplay in airway stem/progenitor cells links the pathogenesis of smoking-induced lesions in the human airway epithelium. Stem Cell. 2017;35:824–837. doi: 10.1002/stem.2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo X., Wang X.F. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009;19:71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y., Handley D., Kaplan T., Yu H., Bais A.S., Richards T., Pandit K.V., Zeng Q., Benos P.V., Friedman N., et al. High throughput determination of TGF beta 1/SMAD3 targets in A549 lung epithelial cells. PLoS One. 2011;6:e20319. doi: 10.1371/journal.pone.0020319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morikawa M., Koinuma D., Tsutsumi S., Vasilaki E., Kanki Y., Heldin C.H., Aburatani H., Miyazono K. ChIP-seq reveals cell type-specific binding patterns of BMP-specific Smads and a novel binding motif. Nucleic Acids Res. 2011;39:8712–8727. doi: 10.1093/nar/gkr572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mirzaei H., Faghihloo E. Viruses as key modulators of the TGF-beta pathway; a double-edged sword involved in cancer. Rev. Med. Virol. 2018;28 doi: 10.1002/rmv.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delgado-Roche L., Mesta F. Oxidative stress as key player in severe acute respiratory syndrome coronavirus (SARS-CoV) infection. Arch. Med. Res. 2020;51:384–387. doi: 10.1016/j.arcmed.2020.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olagnier D., Farahani E., Thyrsted J., Blay-Cadanet J., Herengt A., Idorn M., Hait A., Hernaez B., Knudsen A., Iversen M.B., et al. SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 2020;11:4938. doi: 10.1038/s41467-020-18764-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hawkes H.J.K., Karlenius T.C., Tonissen K.F. Regulation of the human thioredoxin gene promoter and its key substrates: a study of functional and putative regulatory elements. Biochim. Biophys. Acta. 2014;1840:303–314. doi: 10.1016/j.bbagen.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 54.Tonelli C., Chio I.I.C., Tuveson D.A. Transcriptional regulation by Nrf2. Antioxidants Redox Signal. 2018;29:1727–1745. doi: 10.1089/ars.2017.7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao S., Ghosh A., Lo C.S., Chenier I., Scholey J.W., Filep J.G., Ingelfinger J.R., Zhang S.L., Chan J.S.D. Nrf2 deficiency upregulates intrarenal angiotensin-converting enzyme-2 and angiotensin 1-7 receptor expression and attenuates hypertension and nephropathy in diabetic mice. Endocrinology. 2018;159:836–852. doi: 10.1210/en.2017-00752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reiss L.K., Fragoulis A., Siegl S., Platen C., Kan Y.W., Nautiyal J., Parker M., Pufe T., Uhlig U., Martin C., et al. Interplay between nuclear factor erythroid 2-related factor 2 and amphiregulin during mechanical ventilation. Am. J. Respir. Cell Mol. Biol. 2014;51:668–677. doi: 10.1165/rcmb.2013-0279OC. [DOI] [PubMed] [Google Scholar]
- 57.Zhang J., Ji J.Y., Yu M., Overholtzer M., Smolen G.A., Wang R., Brugge J.S., Dyson N.J., Haber D.A. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat. Cell Biol. 2009;11:1444–1450. doi: 10.1038/ncb1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao R., Fallon T.R., Saladi S.V., Pardo-Saganta A., Villoria J., Mou H., Vinarsky V., Gonzalez-Celeiro M., Nunna N., Hariri L.P., et al. Yap tunes airway epithelial size and architecture by regulating the identity, maintenance, and self-renewal of stem cells. Dev. Cell. 2014;30:151–165. doi: 10.1016/j.devcel.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Z., Lu W., Zhang Y., Zou F., Jin Z., Zhao T. The hippo pathway and viral infections. Front. Microbiol. 2019;10:3033. doi: 10.3389/fmicb.2019.03033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tabata T., Petitt M., Puerta-Guardo H., Michlmayr D., Wang C., Fang-Hoover J., Harris E., Pereira L. Zika virus targets different primary human placental cells, suggesting two routes for vertical transmission. Cell Host Microbe. 2016;20:155–166. doi: 10.1016/j.chom.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tabata T., Petitt M., Zydek M., Fang-Hoover J., Larocque N., Tsuge M., Gormley M., Kauvar L.M., Pereira L. Human cytomegalovirus infection interferes with the maintenance and differentiation of trophoblast progenitor cells of the human placenta. J. Virol. 2015;89:5134–5147. doi: 10.1128/Jvi.03674-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuba K., Imai Y., Rao S., Gao H., Guo F., Guan B., Huan Y., Yang P., Zhang Y., Deng W., et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Glowacka I., Bertram S., Herzog P., Pfefferle S., Steffen I., Muench M.O., Simmons G., Hofmann H., Kuri T., Weber F., et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010;84:1198–1205. doi: 10.1128/Jvi.01248-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Haga S., Yamamoto N., Nakai-Murakami C., Osawa Y., Tokunaga K., Sata T., Yamamoto N., Sasazuki T., Ishizaka Y. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc. Natl. Acad. Sci. USA. 2008;105:7809–7814. doi: 10.1073/pnas.0711241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wösten-van Asperen R.M., Lutter R., Specht P.A., Moll G.N., van Woensel J.B., van der Loos C.M., van Goor H., Kamilic J., Florquin S., Bos A.P. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J. Pathol. 2011;225:618–627. doi: 10.1002/path.2987. [DOI] [PubMed] [Google Scholar]
- 66.Hofmann H., Pyrc K., van der Hoek L., Geier M., Berkhout B., Pöhlmann S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. USA. 2005;102:7988–7993. doi: 10.1073/pnas.0409465102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.He L., Ding Y., Zhang Q., Che X., He Y., Shen H., Wang H., Li Z., Zhao L., Geng J., et al. Expression of elevated levels of pro-inflammatory cytokines in SARS-CoV-infected ACE2+ cells in SARS patients: relation to the acute lung injury and pathogenesis of SARS. J. Pathol. 2006;210:288–297. doi: 10.1002/path.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Padmanabhan P., Desikan R., Dixit N.M. Targeting TMPRSS2 and Cathepsin B/L together may be synergistic against SARS-CoV-2 infection. PLoS Comput. Biol. 2020;16 doi: 10.1371/journal.pcbi.1008461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang I.C., Bosch B.J., Li F., Li W., Lee K.H., Ghiran S., Vasilieva N., Dermody T.S., Harrison S.C., Dormitzer P.R., et al. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J. Biol. Chem. 2006;281:3198–3203. doi: 10.1074/jbc.M508381200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jia H.P., Look D.C., Shi L., Hickey M., Pewe L., Netland J., Farzan M., Wohlford-Lenane C., Perlman S., McCray P.B., Jr. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 2005;79:14614–14621. doi: 10.1128/JVI.79.23.14614-14621.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Blume C., Jackson C.L., Spalluto C.M., Legebeke J., Nazlamova L., Conforti F., Perotin J.M., Frank M., Butler J., Crispin M., et al. A novel ACE2 isoform is expressed in human respiratory epithelia and is upregulated in response to interferons and RNA respiratory virus infection. Nat. Genet. 2021;53:205–214. doi: 10.1038/s41588-020-00759-x. [DOI] [PubMed] [Google Scholar]
- 73.Onabajo O.O., Banday A.R., Stanifer M.L., Yan W., Obajemu A., Santer D.M., Florez-Vargas O., Piontkivska H., Vargas J.M., Ring T.J., et al. Interferons and viruses induce a novel truncated ACE2 isoform and not the full-length SARS-CoV-2 receptor. Nat. Genet. 2020;52:1283–1293. doi: 10.1038/s41588-020-00731-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ng K.W., Attig J., Bolland W., Young G.R., Major J., Wrobel A.G., Gamblin S., Wack A., Kassiotis G. Tissue-specific and interferon-inducible expression of nonfunctional ACE2 through endogenous retroelement co-option. Nat. Genet. 2020;52:1294–1302. doi: 10.1038/s41588-020-00732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.South A.M., Diz D.I., Chappell M.C. COVID-19, ACE2, and the cardiovascular consequences. Am. J. Physiol. Heart Circ. Physiol. 2020;318:H1084–H1090. doi: 10.1152/ajpheart.00217.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Verdecchia P., Cavallini C., Spanevello A., Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020;76:14–20. doi: 10.1016/j.ejim.2020.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Imai Y., Kuba K., Rao S., Huan Y., Guo F., Guan B., Yang P., Sarao R., Wada T., Leong-Poi H., et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sodhi C.P., Wohlford-Lenane C., Yamaguchi Y., Prindle T., Fulton W.B., Wang S., McCray P.B., Jr., Chappell M., Hackam D.J., Jia H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg(9) bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell Mol. Physiol. 2018;314:L17–L31. doi: 10.1152/ajplung.00498.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rey-Parra G.J., Vadivel A., Coltan L., Hall A., Eaton F., Schuster M., Loibner H., Penninger J.M., Kassiri Z., Oudit G.Y., Thébaud B. Angiotensin converting enzyme 2 abrogates bleomycin-induced lung injury. J. Mol. Med. (Limerick) 2012;90:637–647. doi: 10.1007/s00109-012-0859-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aggarwal K., Jing F., Maranga L., Liu J. Bioprocess optimization for cell culture based influenza vaccine production. Vaccine. 2011;29:3320–3328. doi: 10.1016/j.vaccine.2011.01.081. [DOI] [PubMed] [Google Scholar]
- 81.Rüdiger D., Kupke S.Y., Laske T., Zmora P., Reichl U. Multiscale modeling of influenza A virus replication in cell cultures predicts infection dynamics for highly different infection conditions. PLoS Comput. Biol. 2019;15:e1006819. doi: 10.1371/journal.pcbi.1006819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Swann O.V., Holden K.A., Turtle L., Pollock L., Fairfield C.J., Drake T.M., Seth S., Egan C., Hardwick H.E., Halpin S., et al. Clinical characteristics of children and young people admitted to hospital with covid-19 in United Kingdom: prospective multicentre observational cohort study. BMJ. 2020;370:m3249. doi: 10.1136/bmj.m3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Viner R.M., Mytton O.T., Bonell C., Melendez-Torres G.J., Ward J., Hudson L., Waddington C., Thomas J., Russell S., van der Klis F., et al. Susceptibility to SARS-CoV-2 infection among children and adolescents compared with adults: a systematic Review and meta-analysis. JAMA Pediatr. 2021;175:143–156. doi: 10.1001/jamapediatrics.2020.4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tao Y., Yang R., Wen C., Fan J., Ma J., He Q., Zhao Z., Song X., Chen H., Shi G., et al. Preliminary analyses of scRNA sequencing and immunohistochemistry of children's lung tissues indicate the expression of SARS-CoV-2 entry-related genes may not be the key reason for the milder syndromes of COVID-19 in children. Clin. Transl. Med. 2021;11:e300–e306. doi: 10.1002/ctm2.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Loske J., Röhmel J., Lukassen S., Stricker S., Magalhães V.G., Liebig J., Chua R.L., Thürmann L., Messingschlager M., Seegebarth A., et al. Pre-activated antiviral innate immunity in the upper airways controls early SARS-CoV-2 infection in children. Nat. Biotechnol. 2022;40:319–324. doi: 10.1038/s41587-021-01037-9. [DOI] [PubMed] [Google Scholar]
- 86.Muus C., Luecken M.D., Eraslan G., Sikkema L., Waghray A., Heimberg G., Kobayashi Y., Vaishnav E.D., Subramanian A., Smillie C., et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat. Med. 2021;27:546–559. doi: 10.1038/s41591-020-01227-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sacco K., Castagnoli R., Vakkilainen S., Liu C., Delmonte O.M., Oguz C., Kaplan I.M., Alehashemi S., Burbelo P.D., Bhuyan F., et al. Immunopathological signatures in multisystem inflammatory syndrome in children and pediatric COVID-19. Nat. Med. 2022;28:1050–1062. doi: 10.1038/s41591-022-01724-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yoshida M., Worlock K.B., Huang N., Lindeboom R.G.H., Butler C.R., Kumasaka N., Dominguez Conde C., Mamanova L., Bolt L., Richardson L., et al. Local and systemic responses to SARS-CoV-2 infection in children and adults. Nature. 2022;602:321–327. doi: 10.1038/s41586-021-04345-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Watson J.K., Sanders P., Dunmore R., Rosignoli G., Julé Y., Rawlins E.L., Mustelin T., May R., Clarke D., Finch D.K. Distal lung epithelial progenitor cell function declines with age. Sci. Rep. 2020;10:10490. doi: 10.1038/s41598-020-66966-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ferrario C.M., Jessup J., Chappell M.C., Averill D.B., Brosnihan K.B., Tallant E.A., Diz D.I., Gallagher P.E. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111:2605–2610. doi: 10.1161/CIRCULATIONAHA.104.510461. [DOI] [PubMed] [Google Scholar]
- 91.Wang G., Lai F.M.M., Lai K.B., Chow K.M., Kwan C.H.B., Li K.T.P., Szeto C.C. Discrepancy between intrarenal messenger RNA and protein expression of ACE and ACE2 in human diabetic nephropathy. Am. J. Nephrol. 2009;29:524–531. doi: 10.1159/000185629. [DOI] [PubMed] [Google Scholar]
- 92.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 95.Raredon M.S.B., Yang J., Garritano J., Wang M., Kushnir D., Schupp J.C., Adams T.S., Greaney A.M., Leiby K.L., Kaminski N., et al. Computation and visualization of cell-cell signaling topologies in single-cell systems data using Connectome. Sci. Rep. 2022;12:4187. doi: 10.1038/s41598-022-07959-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Carbon S., Douglass E., Good B.M., Unni D.R., Harris N.L., Mungall C.J., Basu S., Chisholm R.L., Dodson R.J., Hartline E., et al. Gene Ontology Consortium. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021;49:D325–D334. doi: 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mi H., Muruganujan A., Ebert D., Huang X., Thomas P.D. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019;47:D419–D426. doi: 10.1093/nar/gky1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wickham H. Springer; 2016. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- 100.Haghverdi L., Lun A.T.L., Morgan M.D., Marioni J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018;36:421–427. doi: 10.1038/nbt.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available scRNAseq data. The Gene Expression Omnibus (GEO) accession number is listed in the key resources table.
-
•
All original code has been deposited at Figshare and is publicly available as of the date of publication. The DOI is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.