

Abstract

Indacenodithiophene (IDT) copolymers are a class of conjugated polymers that have limited long-range order and high hole mobilities, which makes them promising candidates for use in deformable electronic devices. Key to their high hole mobilities is the coplanar monomer repeat units within the backbone. Poly(indacenodithiophene-benzothiadiazole) (PIDTC16-BT) and poly(indacenodithiophene-thiapyrollodione) (PIDTC16-TPDC1) are two IDT copolymers with planar backbones, but they are brittle at low molecular weight and have unsuitably high elastic moduli. Substitution of the hexadecane (C16) side chains of the IDT monomer with isocane (C20) side chains was performed to generate a new BT-containing IDT copolymer: PIDTC20-BT. Substitution of the methyl (C1) side chain on the TPD monomer for an octyl (C8) and 6-ethylundecane (C13B) afford two new TPD-containing IDT copolymers named PIDTC16-TPDC8 and PIDTC16-TPDC13B, respectively. Both PIDTC16-TPDC8 and PIDTC16-TPDC13B are relatively well deformable, have a low yield strain, and display significantly reduced elastic moduli. These mechanical properties manifest themselves because the lengthened side chains extending from the TPD-monomer inhibit precise intermolecular ordering. In PIDTC16-BT, PIDTC20-BT and PIDTC16-TPDC1 side chain ordering can occur because the side chains are only present on the IDT subunit, but this results in brittle thin films. In contrast, PIDTC16-TPDC8 and PIDTC16-TPDC13B have disordered side chains, which seems to lead to low hole mobilities. These results suggest that disrupting the interdigitation in IDT copolymers through comonomer side chain extension leads to more ductile thin films with lower elastic moduli, but decreased hole mobility because of altered local order in the respective thin films. Our work, thus, highlights the trade-off between molecular packing structure for deformable electronic materials and provides guidance for designing new conjugated polymers for stretchable electronics.
Keywords: conjugated polymers, stretchable electronics, alkyl side chains, indacenodithiophene copolymers, organic field effect transistors
Introduction
Increased interest in deformable electronic devices such as e-skin, wearable sensors, stretchable organic photovoltaics, soft robotics, and stretchable organic field effect transistors warrants the development of intrinsically deformable electronic materials.1−4 π-Conjugated polymers (CPs) can be utilized as a semiconducting material within deformable devices because of their tunable electronic properties and wide variance of mechanical properties.5 As is the case for commodity polymers and their applications, a single CP often does not satisfy the various optoelectronic, semiconducting, or mechanical requirements imposed by a given deformable electronic application. Polymers with different elastic moduli, elongation at breaks, rheological properties, stress–strain profiles, solubilities, optoelectronic profiles, and hole mobilities are, thus, required to fulfill the various needs of certain applications. This warrants the further development of structure–property relationships for CP systems to guide the design of new intrinsically deformable CPs to address this need.
The structure of the backbone and solubilizing side chains of CPs dictates their materials properties.6 Alterations in the chemical structure of both the backbone and side chains impact the intrinsic electronic characteristics of CPs, their thermal properties, conformational order, and overall solid-state structures;7,8 changes to these properties directly influence materials properties of interest including hole mobility (μhole), elongation at break, and elastic modulus.9 These three metrics are among those considered when determining the application for which a particular CPs can be best utilized.10
Side chain substitution has been shown to impact the materials properties of semicrystalline CPs. For both poly(3-alkylthiophene) and diketopyrrolopyrrole (DPP)-based CPs, it has been shown that increasing the length or increasing branching of side chains results in increased extensibility and decreased elastic modulus.7,11,12 This is caused primarily because of a resultant decrease in both glass transition temperature (Tg) and the amount of load bearing interactions that occur between polymer backbones during deformation, with both effects affording these materials an enhanced ability to reorganize polymer chains during strain.5 It is important to consider, however, that the side chain modifications can also lead to changes in the overall polymer assembly and the resulting thin-film microstructures that manifest differences in μhole.8,13
CPs with limited long-range order are prime candidates for use in deformable electronics because their lower degree of crystallinity relative to semicrystalline CPs affords them the potential to dissipate strain energy through polymer chain rearrangement without the encumbrance of disrupting large crystalline domains and the concomitant decrease in μhole.14 Indacenodithiophene (IDT) copolymers are a class of donor–acceptor (D–A) CPs that are near amorphous due to the presence of four, often hexadecane (C16), side chains that flank the IDT monomer unit and inhibit long-range ordering in the crystalline structure for IDT-copolymer thin films.15 It has become well established that the backbone planarity of an IDT copolymer’s backbone gives rise to low energetic disorder and quasi-one-dimensional charge transport along the backbone.16,17 Poly(indacenodithiophene-benzothiazdiazole) (PIDTC16-BT) (Figure 1) is the most studied IDT copolymer and has displayed charge mobilities of up to 10 cm2 V–1 s–1.18 In terms of mechanical properties, PIDTC16-BT, like most polymers, is brittle at low molecular weight (Mn = 12 kg/mol) with a crack onset strain (CoS) of just 3%,19 extensible up to 22% of its original length at significantly higher molecular weight (Mw = 295 kg/mol),15 and it has elastic moduli ranging from 150 to 750 MPa depending on molecular weight.15,19 Similarly to PIDTC16-BT, poly(indacenodithiophene-thiopyrollodione) (PIDTC16-TPDC1) (Figure 1) has a highly coplanar backbone, limited long-range order, a CoS of 3%, an elastic modulus ranging between 110 and 410 MPa, and it displays μhole equal to that of PIDTC16-BT when processed under the same conditions.19 In contrast to both PIDTC16-BT and PIDTC16-TPDC1, the more torsioned IDT-copolymer poly(indacenodithiophene-benzopyrollodione) (PIDTC16-BPDC1) is much more extensible than PIDTC16-BT, displaying a CoS of 75% at low molecular weight (Mn = 14 kg/mol).19 The backbone torsion in PIDTC16-BPDC1 has the positive effect of generating a 360° side chain extension profile which allows the system to undergo plastic deformation, but the backbone disorder negatively impacts charge mobility.20 Comparing the mechanical and electronic properties of these IDT copolymers highlights the necessity of backbone planarity for low energetic disorder and thus high μhole as well as the impact that side chains have on the mechanical deformation of IDT copolymers. Therefore, altering the side chains of PIDTC16-BT and PIDTC16-TPDC1, which are both coplanar, is a potential strategy to endow each polymer system with increased extensibility and decreased elastic moduli without significantly altering their μhole.
Figure 1.

Structures of the BT-containing IDT copolymers which vary in the length of each side chain extending from the IDT monomer (left) and the TPD-containing IDT copolymers which have the same side chains at the IDT monomer and vary in side chain character at the TPD-position (right).
Five distinct IDT-copolymer systems are synthesized and investigated herein to determine the impact that side chain substitution has on the mechanical and electronic properties of planar IDT copolymers. The structures of these polymers are shown in Figure 1. There are two polymer samples with the PIDT-BT backbone structure. They differ in the length of the alkyl side chain on the IDT unit of backbone. These two polymers, which are referred to as the BT-containing IDT copolymers, are PIDTC16-BT and PIDTC20-BT due to the hexadecane (C16) and isodecane (C20) side chains covalently bound to the IDT monomer unit. Three polymer systems were synthesized with the PIDTC16-TPDC1 conjugated backbone structure. These systems, which are referred to as TPD-containing IDT copolymers, differ in the length and degree of branching of alkyl side chains stemming from the TPD monomer unit. These TPD-containing systems are referred to as PIDTC16-TPDC1, PIDTC16-TPDC8, and PIDTC16-TPDC13B due to the methyl (C1), octyl (C8), and (6-ethyl)-undecane (C13B) side chains that are covalently bound to the TPD monomer unit of each respective polymer. The “13B” notation of PIDTC16-TPDC13B signifies that the side chain contains 13-carbons in total and is branched. Each of the TPD-containing systems have IDT monomers with linear C16 side chains.
We observe that IDT copolymers with no or methyl side chains on their acceptor unit (PIDTC16-BT, PIDTC20-BT, and PIDTC16-TPDC1) display a (001) scattering peak in their GIWAXS profile that is representative of parallel alignment of polymer backbones with translational order within their thin films. For this arrangement of polymer backbones to occur, we hypothesize that it is necessary for the long and linear side chains stemming from the IDT unit to interdigitate with the side chains of neighboring chains. Fast scanning calorimetry (FSC) experiments also demonstrate that PIDTC16-BT, PIDTC20-BT, and PIDTC16-TPDC1 display liquid-crystalline-like ordering of the backbone which supports the GIWAXS evidence. The lengthened and branched side chains on the TPD-monomer unit in PIDTC16-TPDC8 and PIDTC16-TPDC13B prohibit interdigitation and side chain ordering, affecting in turn the backbone assembly. Due to the lack of side chain interdigitation, PIDTC16-TPDC8 and PIDTC16-TPDC13B seem to show improved ductility and decreased elastic moduli. However, while these polymers retain their backbone planarity, they display reduced μhole, an observation that provides further evidence for the importance of parallel alignment of IDT-copolymer backbones with translational order to achieve high μhole. In contrast, PIDTC16-BT, PIDTC20-BT, and PIDTC16-TPDC1 have higher μhole but are exceptionally brittle. These results demonstrate that side chain substitution of IDT copolymers affects their ability to interdigitate causing them to be more disordered. As a result, IDT copolymers with a too dense side chain attachment display reduced μhole, reduced elastic modulus, and increased elongation at break.
Experimental Methods
Full details are provided in the Supporting Information. A representative polymerization procedure is described below. All other polymerizations were carried out using a similar procedure.
Synthesis of PIDTC16-BT
Poly(4-methyl-7-(4,4,9,9-tetrahexadecyl-7-methyl-4,9-dihydro-s-indaceno[1,2-b:5,6-b′]dithiophen-2-yl)benzo[c][1,2,5]thiadiazole) was prepared as reported previously.19 A mixture of IDTC16 (58.2 mg, 0.05 mmol), BT-Br2 (14.7 mg, 0.05 mmol), tris(dibenzylideneacetone)dipalladium(0) (2.3 mg, 5 mol %), tris(o-anisyl)phosphine (1.8 mg, 10 mol %), cesium carbonate (81 mg, 0.25 mmol), and pivalic acid (2.6 mg, 0.025 mmol) in o-xylene (1 mL) was degassed and filled with nitrogen in a pressure reaction tube. The tube was sealed, and the mixture was heated at 100 °C for 16 h, then cooled to room temperature and precipitated into methanol (100 mL). The precipitate was filtered through a Soxhlet thimble and then purified by Soxhlet extraction with methanol, acetone, and hexanes. The hexanes fraction was collected, and the solvent was removed by rotary evaporation. The residue was dissolved in chloroform and precipitated into methanol. The precipitate was collected by filtration and dried under vacuum to afford a deep blue solid in 68% yield. (500 MHz, CDCl3, δ, ppm): 8.10 (s, 2H), 7.95 (s, 2H), 7.39 (s, 2H), 2.08 (s, 4H), 1.96 (s, 4H), 1.17 (m, 112H), 0.85 (t, 12H).
Results and Discussion
Five different IDT-copolymer samples were prepared using direct arylation polymerization (DArP) (Schemes S1 and S2). Unalkylated IDT- and TPD-monomers, and the alkylated monomers IDTC16 and TPDC1, were synthesized as previously reported.19 The monomer IDTC20 was synthesized by alkylating the IDT core with 1-bromoicosane instead of 1-bromohexadecane (Scheme S4). The TPDC8 and TPDC13B monomers were synthesized by alkylating the TPD core with 1-bromooctane and 6-(bromoethyl)-undecane, respectively, in place of methyl iodide (Scheme S3). Following our previously utilized procedures, direct arylation polymerization was used to synthesize PIDTC16-BT, PIDTC20-BT, PIDTC16-TPDC1, PIDTC16-TPDC8, and PIDTC16-TPDC13B.20 These polymers were subjected to 1H NMR spectroscopy (Figures S13–S20) and size exclusion chromatography at 30 °C to characterize their chemical structure and Mn. The degree of polymerization (DP) can be used to compare the size of each polymer system, given the fact that the mass of the D–A repeat unit varies greatly due to the variation in alkyl chain content in each system. Based on DP, all polymers can be designated as belonging to the low-molecular weight regime (Table 1).
Table 1. Size Exclusion Chromatography Results for Each Polymer System.
| Polymer Sample | Mn (g/mol) | Đ | Combined MW of Comonomer Units (u) | DP |
|---|---|---|---|---|
| PIDTC16-BT | 11000 | 1.8 | 1297 | 8 |
| PIDTC20-BT | 21000 | 2.8 | 1521 | 14 |
| PIDTC16-TPDC1 | 9000 | 1.7 | 1326 | 7 |
| PIDTC16-TPDC8 | 11000 | 1.8 | 1426 | 8 |
| PIDTC16-TPDC13B | 14000 | 2.1 | 1496 | 9 |
Grazing-incidence wide-angle X-ray scattering (GIWAXS) measurements were performed to assess the crystallinity of each polymer system’s thin films. Linecuts of the 2D GIWAXS scattering image (Figure S11) in the in-plane Qxy direction are shown in Figure 2. As is expected for IDT copolymers, each polymer studied displayed broad and diffuse crystallographic signals. The π-stacking peak (010) is expected in the 1.6–1.65 Å–1 region. Its absence and the broad peak at 1.2–1.5 Å–1 suggest disordered side chains, short coherence lengths, and reduced crystallinity21 for all the polymers and can therefore be considered to be lacking long-range crystalline order.17 The in-plane (100) signal can be seen for the TPD copolymers at 0.25 Å–1, indicating interlamellar spacing of ∼24–25 Å. Additionally, the out-of-plane (100) signal is observable in both the BT and TPD copolymers (Figure S12), suggesting that all polymers have some parallel alignment with the BT series being oriented more edge-on.
Figure 2.
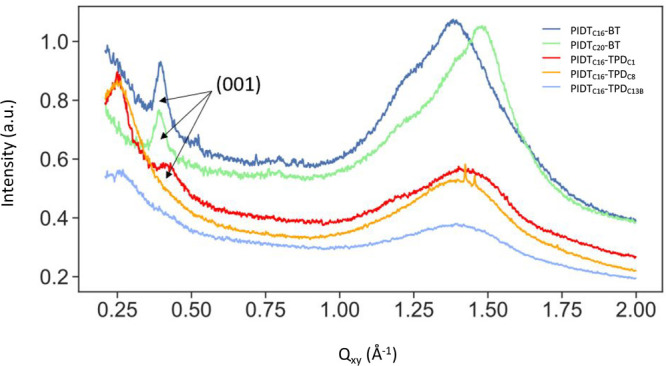
Linecuts of the 2D GIWAXS spectra in the Qxy direction of each IDT-copolymer is shown. The (001) signal is labeled for clarity.
PIDTC16-BT, PIDTC20-BT, and PIDTC16-TPDC1 displayed an in-plane (001) backbone signal, which indicates translational order of rigid IDT-copolymer segments (Figure 3a),21 while PIDTC16-TPDC8 and PIDTC16-TPDC13B do not show this peak. The (001) peak position of the BT-containing systems is at a slightly higher Q-value than observed for PIDTC16-TPDC1 due to slight differences in the backbone repeat-unit length. It has been observed in scanning tunneling microscopy measurements that side chains of PIDTC16-BT interdigitate tightly when aligned in parallel.22 The interdigitation of alkyl side chains is perhaps a necessary condition for the translational order, or precise intermolecular shifts, of IDT copolymer backbones, and thus for the appearance of the (001) reflection. Having long side chains on every monomer rather than every other monomer is effectively increasing the side chain attachment density, which has been shown to inhibit side chain interdigitation in CPs.23 Thus, it is reasonable to suggest that the absence of a (001) signal in PIDTC16-TPDC8 and PIDTC16-TPDC13B thin films is due to the long (and branched in the case of PIDTC16-TPDC13B) alkyl side chains extending from the TPD monomers, which disrupts the uniform pattern by which side chains extend away from the IDT copolymer backbone when only the IDT monomer is alkylated. The regular extension of alkyl chains from the IDT monomer unit alone provides sufficient space for interdigitation with side chains of neighboring, and parallelly aligned, macromolecules. Representations of interdigitating IDT copolymers with lower side chain attachment density and IDT copolymers with increased side chain attachment density are shown in Figure 3a,b, respectively. While the presence of the (001) signal in a thin film arises from just a fraction of polymer chains, the alignment of IDT copolymers is an arrangement that does impact the electronic properties of resultant thin films21 and may impact the mechanical properties of IDT-copolymer thin films studied herein.
Figure 3.

IDT copolymers with side chains on only the IDT subunit (blue block) are able to interdigitate side chains and show translational order between the chains (a), and IDT copolymers with side chains on the IDT-monomer and TPD-monomer cannot interdigitate their side chains and therefore do not align as precisely (b).
Ultraviolet–visible light absorption spectroscopy (UV–vis) and photoluminescence (PL) experiments were performed to elucidate how the optoelectronic properties of the IDT copolymers are impacted by side chain substitution and to probe the conformational order of each polymer system. UV–vis absorption and PL experiments were performed on each polymer. The UV–vis absorption spectra of all polymers in solution and thin films are shown in Figure 4. Relevant metrics are tabulated in Table 2. BT-containing IDT copolymers exhibit more pronounced redshifts in absorbance maximum than TPD-containing systems upon casting into thin films from solution, suggesting that they adopt a higher degree of planarization in the solid state. In solution, the BT-containing polymers have visible but less pronounced 0–0 transitions compared to the TPD-containing polymers, for which the transition is strongly pronounced. All polymers studied herein display a pronounced 0–0 transition in thin films. This 0–0 transition is indicative of J-aggregation behavior24 and has been observed in PIDTC16-BT and PIDTC16-TPDC1 previously.19,20 The Stokes shift of the TPD-containing IDT copolymers in solution is approximately half that of the BT-containing IDT copolymers. This suggests that the TPD-containing systems may in fact be more planar and rigid than the BT-containing systems,25 which is in agreement with previously performed density functional theory calculations comparing PIDTC16-BT to PIDTC16-TPDC1.19 The similarity in Stokes shift between PIDTC16-TPDC1, PIDTC16-TPDC8, and PIDTC16-TPDC13B suggests that the planarity and rigidity of the polymer backbone is not altered by side chain substitution. Within each IDT copolymer family, the optoelectronic properties are strikingly alike, suggesting that the side chain substitution at these positions minimally impacts the optoelectronic properties of IDT copolymers.
Figure 4.

Solution (a) and thin-film (b) UV–vis absorption spectra for each IDT-copolymer.
Table 2. Optoelectronic Properties of Each IDT-Copolymer System.
| Solution |
Thin film | ||||
|---|---|---|---|---|---|
| Sample | λmax.Absorbance (nm) | λmax.PL (nm) | Stokes shift (nm) | λmax.abs. (nm) | Δ Solution to thin film, absorbance (nm) |
| PIDTC16-BT | 662 | 708 | 46 | 674 | 12 |
| PIDTC20-BT | 656 | 705 | 49 | 676 | 20 |
| PIDTC16-TPDC1 | 598 | 624 | 26 | 606 | 8 |
| PIDTC16-TPDC8 | 604 | 626 | 22 | 608 | 4 |
| PIDTC16-TPDC13B | 604 | 626 | 22 | 612 | 8 |
To probe the thermal properties of these IDT copolymer systems, FSC was performed. The mechanical properties of CPs are known to be impacted by the melting temperatures (Tm) and glass transition temperatures (Tg) of the material,26,27 and possibly the side chain melting/softening.19 Intact crystallites and glassy morphologies both inhibit polymer chain reorganization during strain and lead to less extensible thin films.5 The Tg values for each polymer are extrapolated from the enthalpic recovery signatures during physical aging (Figures S1 and S2). A relatively small range of Tgs is found: 10 °C for PIDTC16-BT, 20 °C for PIDTC20-BT, 31 °C for PIDTC16-TPDC1, 4 °C for PIDTC16-TPDC8, and 0 °C for PIDTC16-TPDC13B. All the Tgs are below room temperature except for that of PIDTC16-TPDC1. For the four polymer systems which have Tgs lower than room temperature, this signifies that backbone reorganization is possible in these systems at ambient temperatures. Plastification of the backbone with increasing side chain length is consistent in the TPD series and is an expected relationship in CPs.28,29 Only a relatively small reduction in Tg between PIDTC16-TPDC8 and PIDTC16-TPDC13B is observed compared to the reduction found between PIDTC16-TPDC8 and PIDTC16-TPDC1. This can be attributed to the branching point slightly reducing the amount of side chain motion and therefore limiting backbone dynamics.30 The BT-containing polymers, however, display an increased Tg with increasing side chain length. This observation may be due to side chain interdigitation that affects the backbone and, hence, Tg; it is clear is that there is a critical side chain length above which the relation of Tg vs side chain length inverses.31 Moreover, the higher Tg found for PIDTC20-BT very likely is also a consequence of the larger molecular weight of this polymer.32
We note that the mechanical ductility of IDT copolymers is not strictly dependent on Tg but also on the side chain melting temperature.33 To induce side chain organization, and thus side chain interdigitation, the polymers were annealed above their glass transition temperatures. Figure 5 shows PIDTC16-BT and PIDTC20-BT exhibiting side chain melting endotherms at ∼20 and ∼50 °C, respectively, which will hinder their segmental backbone motion at temperatures between their Tg and this transition. There is no apparent side chain melting features in any of the TPD-containing polymers, which likely is due to the fact that the additional side chains on the TPD unit disrupt the packing of the R1 side chains (Figure 1). Without side chain ordering, there are fewer restrictions to mechanical deformations, which could lead to a decreased elastic modulus and larger strain at fracture for PIDTC16-TPDC8 and PIDTC16-TPDC13B.
Figure 5.

Side chain melting and liquid-crystalline-like ordering of IDT-based copolymers from fast scanning calorimetry. Heating traces of annealed (black) and reference (red) samples are shown, using the annealing temperatures listed on the right side of the panels (given in increments of 10 degrees). PIDT-BT copolymers show side chain melting and liquid-crystalline-like transitions upon annealing at low and high temperatures, respectively (see endotherms highlighted in red). PIDT-TPD copolymers show comparatively weak endotherms indicating limited side chain softening and liquid-crystalline-like ordering behavior.
In addition to side chain softening, high-temperature endotherms exist in PIDTC16-BT, PIDTC20-BT, and PIDTC16-TPDC1 which are assigned to liquid-crystalline-like order of the backbone.20 This ordering, which requires the parallel arrangement of neighboring polymer backbones is also apparent in the (001) signatures in GIWAXS measurements. Notably, these are not occurring in PIDTC16-TPDC8 and PIDTC16-TPDC13B. Figure S3 shows raw intensities of endotherms for each polymer to better identify the side chain transitions. The strong endotherms found for PIDTC16-BT, PIDTC20-BT, and PIDTC16-TPDC1 indicate more relative ordering, while PIDTC16-TPDC8 and PIDTC16-TPDC13B show very weak, ill-defined endotherms, indicative of limited ordering. Therefore, highest mobilities are expected in the polymers that exhibit strong liquid crystallinity.34 The increased side chain attachment density of PIDTC16-TPDC8 and PIDTC16-TPDC13B clearly results in decreased local ordering which will have a direct impact on the materials properties of their thin films and differentiate them from PIDTC16-BT, PIDTC20-BT, and PIDTC16-TPDC1.
Mid-IR charge modulation spectroscopy (CMS) was performed to probe the optical absorption of intrinsic, field-effect generated polarons, avoiding polaron interactions with dopant counterions.35 Mid-IR polaron absorption is dominated by two broad peaks: the lower energy peak “A” centered below 1000 cm–1 and the higher energy peak “B” centered at 2000–3000 cm–1. The intensity, spectral shape, and position of polaronic mid-IR transitions are sensitive to the nanoscale static energetic and structural disorder experienced by charge carriers.36,37 Generally, increases in the A/B peak ratio and associated red-shifting of the peaks correlate with polarons in environments with less static order. All materials tested in this study show a heavily red-shifted “A peak” (Figure 6). Notably, the full shape of peak A sits beyond the edge of detection (below ∼500 cm–1), as can be seen especially in the TPD-containing polymers where the polaron absorption continuously increases from ∼700 cm–1 to 500 cm–1. Nevertheless, the significantly red-shifted peak A starkly contrasting behavior is seen in classically semicrystalline polymers such as poly(3-hexylthiophene) (P3HT), where peak A is fully resolvable in the mid-IR spectral range at ∼700–1000 cm–1.38 The red-shifting of peak A observed in both the BT and TPD-containing materials suggests that polarons are significantly delocalized in these polymers, and, in conjunction with the lack of a strong π–π stacking signal observed in GIWAXS of these polymers (Figure 1), we interpret this delocalization as predominantly intrachain. While peak A is not fully resolvable in this experiment, and so quantitative comparisons of peak A position cannot be made, we do note subtle differences in peak B. While a more quantitative analysis is beyond the scope of this current work, polaron absorption spectra suggest strongly that polarons experience significant intrachain delocalization in both BT and TPD-containing materials, with possibly even greater intrachain delocalization in the TPD-containing materials. We note that our interpretation of CMS spectra in this work is based around qualitative assessment of the static energetic disorder of polymer chains and does explicitly include effects of changing dynamic energetic disorder (i.e., changing strengths of charge-phonon coupling). Dynamic energetic disorder can induce charge localization in organic materials, but we hypothesize that the dominant phonon mode responsible for charge-phonon coupling in organic semiconductors, the C=C intrachain stretching mode, is not expected to change significantly among the samples tested in this work.36,39 Therefore, we expect changes in dynamic energetic disorder to be minor among these materials. Based on our interpretation of significant intrachain polaron delocalization in both BT and TPD-containing materials, the differences in organic field effect transistor (OFET) performance among these materials can primarily be ascribed to differences in their respective thin film microstructure, which governs the ease of intermolecular transport.
Figure 6.

CMS spectra of the (a) BT-containing IDT copolymers and (b) TPD-containing IDT copolymers are shown.
Film-on-water thin film tensile measurements were performed to quantify and compare the mechanical properties of the IDT copolymers.40 The stress–strain curves of all polymer systems except PIDTC16-TPDC1 are shown in Figure 7 and their relevant mechanical metrics are tabulated in Table 3. Five samples were tested for each polymer and the thickness of the films ranged from 50 to 61 nm. PIDTC16-TPDC1 was too fragile to load onto the strain stage for reliable measurement (Figure S4). Comparing PIDTC16-BT to PIDTC20-BT, a slight increase in extensibility is observed when increasing the length of the alkyl side chains on the IDT subunit. The PIDTC16-BT sample is very brittle and fractured within 1% of strain at this low molecular weight. The PIDTC20-BT sample did elastically deform until fracture was observed at 2% strain. We suggest that the needed molecular reorganization is inhibited by the interdigitation of this polymer’s side chains, which leads to embrittlement of the material by preventing plastic flow.41,42 Since the Tg of PIDTC20-BT is higher than that of PIDTC16-BT, we ascribe the slightly larger elongation at break of PIDTC20-BT system compared to PIDTC16-BT to the former’s higher average DP. Moreover, we find that increasing the length of the IDT-unit side chain does not lead to a significant decrease in elastic modulus, supporting the view that the increase in side chain length does not have a significant plasticizing effect.
Figure 7.

Representative stress–strain curves obtained through film-on-water elongation measurements for all IDT copolymers except PIDTC16-TPDC1, which was too brittle to be tested. The colored ‘X’ on each line denotes the elongation at break of each material for clarity.
Table 3. Mechanical Properties and μhole of Each IDT Copolymer Are Tabulated.
| Polymer | Mn (kg/mol) | DP | Elastic modulus (MPa) | Avg. elongation at break (%) | μhole (cm2·V–1·s–1) | (001) reflection present |
|---|---|---|---|---|---|---|
| PIDTC16-BT | 11 | 8 | N.A.a | 0 ± 0 | 0.068 ± 0.020 | Yes |
| PIDTC20-BT | 21 | 14 | 520 ± 70 | 2 ± 1 | 0.023 ± 0.003 | Yes |
| PIDTC16-TPDC1 | 8 | 7 | N.A.b | N.A.b | 0.051 ± 0.030 | Yes |
| PIDTC16-TPDC8 | 11 | 8 | 14 ± 2 | 7 ± 2 | 0.007 ± 0.005 | No |
| PIDTC16-TPDC13B | 14 | 9 | 56 ± 5 | 16 ± 4 | 0.006 ± 0.002 | No |
The elastic moduli of PIDTC16-BT is effectively infinite because of the verticality of the stress–strain curve.
The PIDTC16-TPDC1 material was too fragile to be loaded onto the strain stage for measurement.
In contrast, both PIDTC16-TPDC8 and PIDTC16-TPDC13B were able to undergo plastic deformation during elongation, despite low molecular weight. As a result, these polymers displayed average elongation at break values of 7% and 16% for PIDTC16-TPDC8 and PIDTC16-TPDC13B, respectively. Side chain substitution at the TPD-position led to reductions in elastic moduli, with PIDTC16-TPDC8 and PIDTC16-TPDC13B displaying elastic moduli of 14 and 50 MPa, respectively. Because of the relatively small backbone Tg (4 and 0 °C), the elastic moduli, when compared to other reported polymers (e.g., DPP-based polymers have elastic moduli ∼150–480 MPa;7 P3HT has been reported to have elastic moduli ∼200–300 MPa43) are remarkably small despite these two polymers’ backbone rigidity. These attributes increase their suitability in wearable or e-skin devices due to a modulus which is more closely related to that of skin.44 We moreover suggest that the decreased elastic moduli of PIDTC16-TPDC8 and PIDTC16-TPDC13B and the ability to plastically deform are due to the lack of side chain order caused by the inclusion of longer side chains on the TPD unit.45 From a structural perspective, it is the increased density of alkyl side chains on PIDTC16-TPDC8 and PIDTC16B-TPDC13B that hinders side chain order (i.e., side chain interdigitation), which in turn provides these materials with a greater ability for backbone reorganization upon plastic deformation.23 Intriguingly, inclusion of the branched side chain leads to an increase in elastic modulus, suggesting that there may be increased interactions between the branched side chains on the TPD monomer and other side chains in the system. The higher elongation at break of PIDTC16-TPDC13B compared to PIDTC16-TPDC8 is potentially due to the larger and branched side chain on the TPDC13B monomer shielding the backbone more than the linear side chain of the TPDC8 monomer, enabling backbone motions. Moreover, by increasing the attachment density of alkyl side chains from every other to every monomer repeat unit, through inclusion of long alkyl side chains on the TPD unit, IDT copolymers can be made more extendable and make them feature a significantly decreased elastic moduli. This may occur because of limited or hindered side chain interdigitation and thus more facile reorganization of the polymer backbone during elongation.
OFETs were fabricated so that the electronic properties of each IDT copolymer could be assessed. These results are displayed in Table 3. Measurements were averaged from at least five transistors across three substrates and devices, and the film thickness ranged from 20 to 30 nm. We believe that the high standard deviation seen in μhole in some samples is a reflection inhomogeneities in film thickness. All polymer samples display threshold voltages similar to our previous reports on IDT copolymers.20 When assessing average μhole, lengthening of alkyl side chains decreases the μhole and on/off current ratio in every case. The μhole of PIDTC20-BT and PIDTC16-BT remain on the same order of magnitude. In contrast, the μhole of PIDTC16-TPDC8 and PIDTC16-TPDC13B are reduced by an order of magnitude compared to PIDTC16-TPDC1. It is observed that PIDTC16-TPDC1 and PIDTC16-BT have the same OFET characteristics, which corroborates our previous reports.19 The CMS results demonstrate that the BT-containing and TPD-containing IDT copolymers have similar static energetic disorder environments and, combined with limited crystalline order as found from GIWAXS, suggest similar intrachain polaron delocalization under static charge accumulation. If the electronic environment of polarons within the thin films are similar, then this indicates that the intramolecular charge transport may be unaffected by side chain substitution at the TPD-nitrogen position. Thus, one interpretation of decreases in μhole is that arrangements that enhance p-orbital overlap and facilitate intermolecular charge transport, including parallel chain alignment as found for PIDTC16-TPDC1, PIDTC20-BT and PIDTC16-BT, are hindered in PIDTC16-TPDC8 and PIDTC16-TPDC13B.21 It is also possible that side-chain interdigitation leads to subtle frequency shifts in low energy interchain vibrational modes that decrease dynamic disorder effects on intermolecular charge transport, though a detailed analysis of this effect remains outside the scope of this work.39 Accordingly, our data suggest that the inability of polymer side chains to interdigitate, and thus form two-dimensional aligned structures, may be responsible for the decrease in μhole of PIDTC16-TPDC8 and PIDTC16-TPDC13B.
Conclusion
Increasing the side chain attachment density of IDT copolymers from every other monomer unit to every monomer unit has widespread effects on electronic and mechanical properties of their thin films. With increased side chain attachment density comes an inability for side chains to interdigitate. This is supported by the absence of the (001) reflection in PIDTC16-TPDC8 and PIDTC16-TPDC13B and the lack of side chain ordering observed in FSC experiments, which support that the increased side chain attachment density is inhibiting side chain organization. Consequently, these systems display orders of magnitude less μhole than PIDTC16-BT, PIDTC20-BT, and PIDTC16-TPDC1, which do exhibit interdigitation of their side chains and thus display parallel and translational ordering of their backbones. In contrast, because of the lack of side chain interdigitation, PIDTC16-TPDC8 and PIDTC16-TPDC16 thin films can dissipate strain through plastic deformation, exhibiting greatly improved elongation at breaks and remarkably low elastic moduli. This suggests that side chain interdigitation is a major contributor to preventing polymer chain reorganization of IDT copolymers while under strain. Inhibition of interdigitation can endow planar IDT copolymers with reduced elastic moduli and the ability to deform plastically under strain.
Acknowledgments
Y.W. and X.G. thank the National Science Foundation under award number DMR-2047689 for supporting the thin-film mechanics characterization in this work. P.J.W.S, G.L., A.S., and C.K.L acknowledge the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES), under award DE-SC0020046 for the synthesis of the polymers as well as the GIWAXS and CMS characterization. J.O. and L.G. acknowledge National Science Foundation (NSF) under award number DMR-2104234 for the fabrication of OFETs. A.H.B. and N.S. are grateful for support by NSF under the award CHE-2108123. Part of this work was conducted at the UW Photonics Center and Research Training Testbeds supported by the Clean Energy Institute and the Molecular Analysis Facility, a National Nanotechnology Coordinated Infrastructure site at the University of Washington which is supported in part by the NSF (grant NNCI-1542101), the University of Washington, the Molecular Engineering and Sciences Institute, and the Clean Energy Institute.
Glossary
Abbreviations
- CPs
π-conjugated polymers
- μhole
hole mobility
- Tg
glass transition temperature
- IDT
indacenodithiophene
- C16
hexadecane alkyl chain
- PIDTC16-BT
poly(indacenodithiophene-benzothiazdiazole)
- Mn
number average molecular weight
- CoS
crack-onset-strain
- PIDTC16-TPDC1
poly(indacenodithiophene-thiopyrollodione)
- PIDTC16-BPDC1
poly(indacenodithiophene-benzopyrollodione)
- C20
isodecane alkyl chain
- C8
octyl alkyl chain
- C13B
(6-ethyl)-undecane
- DP
degree of polymerization
- D–A
donor–acceptor
- GIWAXS
grazing incidence wide-angle X-ray scattering
- UV–vis
ultraviolet–visible light absorbance
- PL
photoluminescence
- FSC
fast-scanning calorimetry
- Tm
melting temperature
- CMS
mid-IR charge modulation spectroscopy
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acspolymersau.2c00034.
Additional experimental details, materials, and methods. 1H NMR spectra for monomers and polymers, FSC data, tensile test results, OFET data, GIWAXS data, and UV–vis and fluorescence spectra (PDF)
Author Contributions
CRediT: Parker J.W. Sommerville conceptualization (lead), data curation (lead), formal analysis (lead), writing-original draft (lead); Alex H. Balzer conceptualization (supporting), data curation (supporting), formal analysis (supporting), writing-review & editing (supporting); Garrett Lecroy conceptualization (supporting), data curation (supporting), formal analysis (supporting), writing-review & editing (supporting); Lorenzo Guio data curation (supporting), formal analysis (supporting), writing-review & editing (supporting); Yunfei Wang data curation (supporting), formal analysis (supporting), investigation (supporting), writing-review & editing (supporting); Jonathan W Onorato data curation (supporting), formal analysis (supporting), investigation (supporting), writing-review & editing (supporting); Nadzeya A. Kukhta data curation (supporting), formal analysis (supporting), methodology (supporting), writing-review & editing (supporting); Xiaodan Gu data curation (supporting), investigation (supporting), supervision (supporting), writing-review & editing (supporting); Alberto Salleo data curation (supporting), funding acquisition (supporting), methodology (supporting), project administration (supporting), supervision (supporting), writing-review & editing (supporting); Natalie Stingelin data curation (supporting), formal analysis (supporting), methodology (supporting), supervision (supporting), writing-review & editing (supporting); Christine K. Luscombe conceptualization (lead), formal analysis (lead), funding acquisition (lead), project administration (lead), supervision (lead), writing-original draft (supporting), writing-review & editing (lead).
The authors declare no competing financial interest.
Supplementary Material
References
- Schara S.; Blau R.; Church D. C.; Pokorski J. K.; Lipomi D. J. Polymer Chemistry for Haptics, Soft Robotics, and Human-Machine Interfaces. Adv. Funct. Mater. 2021, 31 (39), 2008375. 10.1002/adfm.202008375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirringhaus H. 25th Anniversary Article: Organic Field-Effect Transistors: The Path Beyond Amorphous Silicon. Adv. Mater. 2014, 26 (9), 1319–1335. 10.1002/adma.201304346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benight S. J.; Wang C.; Tok J. B. H.; Bao Z. Stretchable and Self-Healing Polymers and Devices for Electronic Skin. Prog. Polym. Sci. 2013, 38 (12), 1961–1977. 10.1016/j.progpolymsci.2013.08.001. [DOI] [Google Scholar]
- St. Onge P. B. J.; Ocheje M. U.; Selivanova M.; Rondeau-Gagné S. Recent Advances in Mechanically Robust and Stretchable Bulk Heterojunction Polymer Solar Cells. Chem. Rec. 2019, 19 (6), 1008–1027. 10.1002/tcr.201800163. [DOI] [PubMed] [Google Scholar]
- Root S. E.; Savagatrup S.; Printz A. D.; Rodriquez D.; Lipomi D. J. Mechanical Properties of Organic Semiconductors for Stretchable, Highly Flexible, and Mechanically Robust Electronics. Chem. Rev. 2017, 117 (9), 6467–6499. 10.1021/acs.chemrev.7b00003. [DOI] [PubMed] [Google Scholar]
- Roth B.; Savagatrup S.; de los Santos N. V.; Hagemann O.; Carlé J. E.; Helgesen M.; Livi F.; Bundgaard E.; Søndergaard R. R.; Krebs F. C.; Lipomi D. J. Mechanical Properties of a Library of Low-Band-Gap Polymers. Chem. Mater. 2016, 28 (7), 2363–2373. 10.1021/acs.chemmater.6b00525. [DOI] [Google Scholar]
- Sugiyama F.; Kleinschmidt A. T.; Kayser L. V.; Rodriquez D.; Finn M.; Alkhadra M. A.; Wan J. M.-H.; Ramírez J.; Chiang A. S.-C.; Root S. E.; Savagatrup S.; Lipomi D. J. Effects of Flexibility and Branching of Side Chains on the Mechanical Properties of Low-Bandgap Conjugated Polymers. Polym. Chem. 2018, 9 (33), 4354–4363. 10.1039/C8PY00820E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges C. R.; Ford M. J.; Thomas E. M.; Gomez C.; Bazan G. C.; Segalman R. A. Effects of Side Chain Branch Point on Self Assembly, Structure, and Electronic Properties of High Mobility Semiconducting Polymers. Macromolecules 2018, 51 (21), 8597–8604. 10.1021/acs.macromol.8b01906. [DOI] [Google Scholar]
- Guo Z.; Shinohara A.; Pan C.; Stadler F. J.; Liu Z.; Yan Z.-C.; Zhao J.; Wang L.; Nakanishi T. Consistent Red Luminescence in π-Conjugated Polymers with Tuneable Elastic Moduli over Five Orders of Magnitude. Mater. Horiz. 2020, 7 (5), 1421–1426. 10.1039/D0MH00029A. [DOI] [Google Scholar]
- Higashihara T. Strategic Design and Synthesis of π-Conjugated Polymers Suitable as Intrinsically Stretchable Semiconducting Materials. Polym. J. 2021, 53 (10), 1061–1071. 10.1038/s41428-021-00510-1. [DOI] [Google Scholar]
- Moulton J.; Smith P. Electrical and Mechanical Properties of Oriented Poly(3-Alkylthiophenes): 2. Effect of Side-Chain Length. Polymer 1992, 33 (11), 2340–2347. 10.1016/0032-3861(92)90525-2. [DOI] [Google Scholar]
- Savagatrup S.; Printz A. D.; Rodriquez D.; Lipomi D. J. Best of Both Worlds: Conjugated Polymers Exhibiting Good Photovoltaic Behavior and High Tensile Elasticity. Macromolecules 2014, 47 (6), 1981–1992. 10.1021/ma500286d. [DOI] [Google Scholar]
- Ma Z.; Geng H.; Wang D.; Shuai Z. Influence of Alkyl Side-Chain Length on the Carrier Mobility in Organic Semiconductors: Herringbone vs. Pi-Pi Stacking. J. Mater. Chem. C 2016, 4 (20), 4546–4555. 10.1039/C6TC00755D. [DOI] [Google Scholar]
- Huang Y.-W.; Lin Y.-C.; Yen H.-C.; Chen C.-K.; Lee W.-Y.; Chen W.-C.; Chueh C.-C. High Mobility Preservation of Near Amorphous Conjugated Polymers in the Stretched States Enabled by Biaxially-Extended Conjugated Side-Chain Design. Chem. Mater. 2020, 32 (17), 7370–7382. 10.1021/acs.chemmater.0c02258. [DOI] [Google Scholar]
- Zheng Y.; Wang G. N.; Kang J.; Nikolka M.; Wu H.; Tran H.; Zhang S.; Yan H.; Chen H.; Yuen P. Y.; Mun J.; Dauskardt R. H.; McCulloch I.; Tok J. B. -H.; Gu X.; Bao Z. An Intrinsically Stretchable High-Performance Polymer Semiconductor with Low Crystallinity. Adv. Funct. Mater. 2019, 29, 1905340–1905340. 10.1002/adfm.201905340. [DOI] [Google Scholar]
- Wadsworth A.; Chen H.; Thorley K. J.; Cendra C.; Nikolka M.; Bristow H.; Moser M.; Salleo A.; Anthopoulos T. D.; Sirringhaus H.; McCulloch I. Modification of Indacenodithiophene-Based Polymers and Its Impact on Charge Carrier Mobility in Organic Thin-Film Transistors. J. Am. Chem. Soc. 2020, 142 (2), 652–664. 10.1021/jacs.9b09374. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Bronstein H.; Kronemeijer A. J.; Smith J.; Kim Y.; Kline R. J.; Richter L. J.; Anthopoulos T. D.; Sirringhaus H.; Song K.; Heeney M.; Zhang W.; McCulloch I.; DeLongchamp D. M. Molecular Origin of High Field-Effect Mobility in an Indacenodithiophene-Benzothiadiazole Copolymer. Nat. Commun. 2013, 4 (1), 2238–2238. 10.1038/ncomms3238. [DOI] [PubMed] [Google Scholar]
- Lamport Z. A.; Barth K. J.; Lee H.; Gann E.; Engmann S.; Chen H.; Guthold M.; McCulloch I.; Anthony J. E.; Richter L. J.; DeLongchamp D. M.; Jurchescu O. D. A Simple and Robust Approach to Reducing Contact Resistance in Organic Transistors. Nat. Commun. 2018, 9 (1), 5130. 10.1038/s41467-018-07388-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Tatum W. K.; Onorato J. W.; Zhang Y.; Luscombe C. K. Low Elastic Modulus and High Charge Mobility of Low-Crystallinity Indacenodithiophene-Based Semiconducting Polymers for Potential Applications in Stretchable Electronics. Macromolecules 2018, 51 (16), 6352–6358. 10.1021/acs.macromol.8b00898. [DOI] [Google Scholar]
- Sommerville P. J. W.; Li Y.; Dong B. X.; Zhang Y.; Onorato J. W.; Tatum W. K.; Balzer A. H.; Stingelin N.; Patel S. N.; Nealey P. F.; Luscombe C. K. Elucidating the Influence of Side-Chain Circular Distribution on the Crack Onset Strain and Hole Mobility of Near-Amorphous Indacenodithiophene Copolymers. Macromolecules 2020, 53 (17), 7511–7518. 10.1021/acs.macromol.0c00512. [DOI] [Google Scholar]
- Cendra C.; Balhorn L.; Zhang W.; O’Hara K.; Bruening K.; Tassone C. J.; Steinrück H.-G.; Liang M.; Toney M. F.; McCulloch I.; Chabinyc M. L.; Salleo A.; Takacs C. J. Unraveling the Unconventional Order of a High-Mobility Indacenodithiophene-Benzothiadiazole Copolymer. ACS Macro Lett. 2021, 10 (10), 1306–1314. 10.1021/acsmacrolett.1c00547. [DOI] [PubMed] [Google Scholar]
- Chen H.; Wadsworth A.; Ma C.; Nanni A.; Zhang W.; Nikolka M.; Luci A. M. T.; Perdigão L. M. A.; Thorley K. J.; Cendra C.; Larson B.; Rumbles G.; Anthopoulos T. D.; Salleo A.; Costantini G.; Sirringhaus H.; McCulloch I. The Effect of Ring Expansion in Thienobenzo[b]Indacenodithiophene Polymers for Organic Field-Effect Transistors. J. Am. Chem. Soc. 2019, 141 (47), 18806–18813. 10.1021/jacs.9b09367. [DOI] [PubMed] [Google Scholar]
- Kline R. J.; DeLongchamp D. M.; Fischer D. A.; Lin E. K.; Richter L. J.; Chabinyc M. L.; Toney M. F.; Heeney M.; McCulloch I. Critical Role of Side-Chain Attachment Density on the Order and Device Performance of Polythiophenes. Macromolecules 2007, 40 (22), 7960–7965. 10.1021/ma0709001. [DOI] [Google Scholar]
- Eder T.; Stangl T.; Gmelch M.; Remmerssen K.; Laux D.; Höger S.; Lupton J. M.; Vogelsang J. Switching between H- and J-Type Electronic Coupling in Single Conjugated Polymer Aggregates. Nat. Commun. 2017, 8 (1), 1641. 10.1038/s41467-017-01773-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkateshvaran D.; Nikolka M.; Sadhanala A.; Lemaur V.; Zelazny M.; Kepa M.; Hurhangee M.; Kronemeijer A. J.; Pecunia V.; Nasrallah I.; Romanov I.; Broch K.; McCulloch I.; Emin D.; Olivier Y.; Cornil J.; Beljonne D.; Sirringhaus H. Approaching Disorder-Free Transport in High-Mobility Conjugated Polymers. Nature 2014, 515 (7527), 384–388. 10.1038/nature13854. [DOI] [PubMed] [Google Scholar]
- Xie R.; Weisen A. R.; Lee Y.; Aplan M. A.; Fenton A. M.; Masucci A. E.; Kempe F.; Sommer M.; Pester C. W.; Colby R. H.; Gomez E. D. Glass Transition Temperature from the Chemical Structure of Conjugated Polymers. Nat. Commun. 2020, 11 (1), 893. 10.1038/s41467-020-14656-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z.; Cao Z.; Galuska L.; Zhang S.; Xu J.; Gu X. Glass Transition Phenomenon for Conjugated Polymers. Macromol. Chem. Phys. 2019, 220 (11), 1900062. 10.1002/macp.201900062. [DOI] [Google Scholar]
- Zhan P.; Zhang W.; Jacobs I. E.; Nisson D. M.; Xie R.; Weissen A. R.; Colby R. H.; Moulé A. J.; Milner S. T.; Maranas J. K.; Gomez E. D. Side Chain Length Affects Backbone Dynamics in Poly(3-Alkylthiophene)s. J. Polym. Sci., Part B: Polym. Phys. 2018, 56 (17), 1193–1202. 10.1002/polb.24637. [DOI] [Google Scholar]
- Zhang S.; Alesadi A.; Selivanova M.; Cao Z.; Qian Z.; Luo S.; Galuska L.; Teh C.; Ocheje M. U.; Mason G. T.; Onge P. B. J. S.; Zhou D.; Rondeau-Gagné S.; Xia W.; Gu X. Toward the Prediction and Control of Glass Transition Temperature for Donor-Acceptor Polymers. Adv. Funct. Mater. 2020, 30 (27), 2002221. 10.1002/adfm.202002221. [DOI] [Google Scholar]
- Cao Z.; Galuska L.; Qian Z.; Zhang S.; Huang L.; Prine N.; Li T.; He Y.; Hong K.; Gu X. The Effect of Side-Chain Branch Position on the Thermal Properties of Poly(3-Alkylthiophenes). Polym. Chem. 2020, 11 (2), 517–526. 10.1039/C9PY01026B. [DOI] [Google Scholar]
- Reimschuessel H. K. On the Glass Transition Temperature of Comblike Polymers: Effects of Side Chain Length and Backbone Chain Structure. J. Polym. Sci. Polym. Chem. Ed. 1979, 17 (8), 2447–2457. 10.1002/pol.1979.170170817. [DOI] [Google Scholar]
- Fox T. G.; Flory P. J. Second-Order Transition Temperatures and Related Properties of Polystyrene. I. Influence of Molecular Weight. J. Appl. Phys. 1950, 21 (6), 581–591. 10.1063/1.1699711. [DOI] [Google Scholar]
- Balar N.; Siddika S.; Kashani S.; Peng Z.; Rech J. J.; Ye L.; You W.; Ade H.; O’Connor B. T. Role of Secondary Thermal Relaxations in Conjugated Polymer Film Toughness. Chem. Mater. 2020, 32 (15), 6540–6549. 10.1021/acs.chemmater.0c01910. [DOI] [Google Scholar]
- McCulloch I.; Heeney M.; Bailey C.; Genevicius K.; Macdonald I.; Shkunov M.; Sparrowe D.; Tierney S.; Wagner R.; Zhang W.; Chabinyc M. L.; Kline R. J.; McGehee M. D.; Toney M. F. Liquid-Crystalline Semiconducting Polymers with High Charge-Carrier Mobility. Nat. Mater. 2006, 5 (4), 328–333. 10.1038/nmat1612. [DOI] [PubMed] [Google Scholar]
- Ghosh R.; Chew A. R.; Onorato J.; Pakhnyuk V.; Luscombe C. K.; Salleo A.; Spano F. C. Spectral Signatures and Spatial Coherence of Bound and Unbound Polarons in P3HT Films: Theory Versus Experiment. J. Phys. Chem. C 2018, 122 (31), 18048–18060. 10.1021/acs.jpcc.8b03873. [DOI] [Google Scholar]
- Ghosh R.; Pochas C. M.; Spano F. C. Polaron Delocalization in Conjugated Polymer Films. J. Phys. Chem. C 2016, 120 (21), 11394–11406. 10.1021/acs.jpcc.6b02917. [DOI] [Google Scholar]
- Ghosh R.; Luscombe C. K.; Hambsch M.; Mannsfeld S. C. B.; Salleo A.; Spano F. C. Anisotropic Polaron Delocalization in Conjugated Homopolymers and Donor-Acceptor Copolymers. Chem. Mater. 2019, 31 (17), 7033–7045. 10.1021/acs.chemmater.9b01704. [DOI] [Google Scholar]
- Chew A. R.; Ghosh R.; Pakhnyuk V.; Onorato J.; Davidson E. C.; Segalman R. A.; Luscombe C. K.; Spano F. C.; Salleo A. Unraveling the Effect of Conformational and Electronic Disorder in the Charge Transport Processes of Semiconducting Polymers. Adv. Funct. Mater. 2018, 28 (41), 1804142–1804142. 10.1002/adfm.201804142. [DOI] [Google Scholar]
- Troisi A. Charge Transport in High Mobility Molecular Semiconductors: Classical Models and New Theories. Chem. Soc. Rev. 2011, 40 (5), 2347–2358. 10.1039/c0cs00198h. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Galuska L. A.; Gu X. Water-Assisted Mechanical Testing of Polymeric Thin-Films. J. Polym. Sci. 2022, 60 (7), 1108–1129. 10.1002/pol.20210281. [DOI] [Google Scholar]
- Koch F. P. V.; Heeney M.; Smith P. Thermal and Structural Characteristics of Oligo(3-Hexylthiophene)s (3HT)n, n = 4–36. J. Am. Chem. Soc. 2013, 135 (37), 13699–13709. 10.1021/ja405792b. [DOI] [PubMed] [Google Scholar]
- O’Connor B.; Chan E. P.; Chan C.; Conrad B. R.; Richter L. J.; Kline R. J.; Heeney M.; McCulloch I.; Soles C. L.; DeLongchamp D. M. Correlations between Mechanical and Electrical Properties of Polythiophenes. ACS Nano 2010, 4 (12), 7538–7544. 10.1021/nn1018768. [DOI] [PubMed] [Google Scholar]
- Rodriquez D.; Kim J.-H.; Root S. E.; Fei Z.; Boufflet P.; Heeney M.; Kim T.-S.; Lipomi D. J. Comparison of Methods for Determining the Mechanical Properties of Semiconducting Polymer Films for Stretchable Electronics. ACS Appl. Mater. Interfaces 2017, 9 (10), 8855–8862. 10.1021/acsami.6b16115. [DOI] [PubMed] [Google Scholar]
- Griffin M. F.; Leung B. C.; Premakumar Y.; Szarko M.; Butler P. E. Comparison of the Mechanical Properties of Different Skin Sites for Auricular and Nasal Reconstruction. J. Otolaryngol. - Head Neck Surg. 2017, 46 (1), 33. 10.1186/s40463-017-0210-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savagatrup S.; Zhao X.; Chan E.; Mei J.; Lipomi D. J. Effect of Broken Conjugation on the Stretchability of Semiconducting Polymers. Macromol. Rapid Commun. 2016, 37 (19), 1623–1628. 10.1002/marc.201600377. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.