

Abstract
Huntington’s disease (HD) is a neurodegenerative disorder caused by a CAG repeat expansion in the first exon of the gene huntingtin. There is no treatment to prevent or delay the disease course of HD currently. Oxidative stress and mitochondrial dysfunction have emerged as key determinants of the disease progression in HD. Therefore, counteracting mutant huntingtin (mHtt)-induced oxidative stress and mitochondrial dysfunction appears as a new approach to treat this devastating disease. Interestingly, mild mitochondrial uncoupling improves neuronal resistance to stress and facilitates neuronal survival. Mild mitochondrial uncoupling can be induced by the proper dose of 2,4-dinitrophenol (DNP), a proton ionophore that was previously used for weight loss. In this study, we evaluated the effects of chronic administration of DNP at three doses (0.5, 1, 5 mg/kg/day) on mHtt-induced behavioral deficits and cellular abnormalities in the N171–82Q HD mouse model. DNP at a low dose (1 mg/kg/day) significantly improved motor function and preserved medium spiny neuronal marker DARPP32 and postsynaptic protein PSD95 in the striatum of HD mice. Further mechanistic study suggests that DNP at this dose reduced oxidative stress in HD mice, which was indicated by reduced levels of F2-isoprostanes in the brain of HD mice treated with DNP. Our data indicated that DNP provided behavioral benefit and neuroprotective effect at a weight neutral dose in HD mice, suggesting that the potential value of repositioning DNP to HD treatment is warranted in well-controlled clinical trials in HD.
Keywords: Mitochondrial uncoupling; 2,4-dinitrophenol; Oxidative stress; DARPP32; Huntington’s disease
1. Introduction
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder, caused by an abnormal expansion of a CAG repeat in exon 1 of the gene encoding the huntingtin protein (Htt) (The Huntington’s disease Collaborative Research group, 1993). Individuals who have 36 CAG repeats or more in the huntingtin gene develop the clinical symptoms, including motor, cognition, and mental abnormalities that cause a progressive loss of functional capacity and shortened life span (Ross and Tabrizi, 2011). The mutation results in a polyglutamine tract at the N-terminal of the mutant Htt protein. The pathology of the disease has been attributed to toxic gain of functions for the mutant Htt as well as loss of beneficial functions of wild type Htt protein (Ross and Tabrizi, 2011). The major neuropathology in HD is the selective loss of medium spiny neurons in the caudate putamen of patients (Graveland et al., 1985; Reiner et al., 1988; Vonsattel et al., 1985). Despite remarkable progress in our understanding of this disease, the molecular logic connecting mutant Htt-mediated neuronal dysfunction and pathological symptoms remains unclear.
Accumulating data has indicated that oxidative stress plays a crucial role in HD pathogenesis (Kumar and Ratan, 2016). The two major factors that make the brain more prone to oxidative damage are high lipid concentrations and high energy requirements (Walker, 2007). Mutant Htt can also serve as the source of reactive oxygen species (ROS) (Rotblat et al., 2014). Moreover, the extensive oxidative DNA damage has also been reported in HD models (Browne, 2008; Gil-Mohapel et al., 2014; Goula et al., 2009). DNA oxidation was detected in serum, blood and leukocytes from HD patients (Chen et al., 2007; Hersch et al., 2006; Tunez et al., 2011). Impairment in the electron transport chain and mitochondrial dysfunction are the major mechanisms involved in the ROS mediated pathogenesis of HD (Sayre et al., 2008; Trushina and McMurray, 2007). HD patients showed an increased level of oxidative stress markers accompanied by a decrease in antioxidant status compared to healthy subjects (Chen et al., 2007; Montine et al., 1999). Both in vivo and in vitro studies have documented the protective role of various natural antioxidant products or synthetic entities in the prevention of HD (Johri and Beal, 2012; Stack et al., 2008). However, the neuroprotective treatment for HD patients has not yet been found.
Dietary restriction (DR) without malnutrition is a well-tested intervention in aging and models of neurodegenerative disorders (Arslan-Ergul et al., 2013; Hunt et al., 2006; Lopez-Lluch and Navas, 2016; Mattson, 2008; Sohal and Forster, 2014). We have demonstrated that HD mice maintained on a DR regimen exhibited significantly improved motor function and lessened brain pathology (Duan et al., 2003). Mild uncoupling increases metabolic inefficiency, effectively “restricting” caloric conversion into biological work; this is a mechanism for DR-mediated beneficial effects. Therefore, chemical mitochondrial uncoupler(s) may represent effective DR mimetic. The protonophore 2,4-dinitrophenol (DNP), which was extensively used as an obesity treatment in the 1930s (Colman, 2007), has been increasingly utilized as a putative DR mimetic. Later it was learned that DNP is a mitochondrial uncoupler (Geisler, 2011; Simkins, 1937). Promisingly, in flies and mice, DNP has been shown to increase lifespan, accompanied by decreases in oxidative damage (Caldeira da Silva et al., 2008; Padalko, 2005). In addition, chronic administration of DNP significantly improved learning and memory in mice (Geisler et al., 2016) and is beneficial in a stroke model by lowering ROS (Korde et al., 2005). In this study, we evaluated the effects of DNP on mutant Htt-associated abnormalities in a well-characterized N171–82Q HD mouse model. Our results indicate that chronic administration of appropriate low dose DNP improved motor function, preserved medium spiny neuronal identity, and reduced oxidative stress in HD mice, suggesting that mild mitochondrial uncoupling might be a new therapeutic strategy for HD.
2. Materials and methods
2.1. Mice
Male N171–82Q HD mice were mated to female B6C3F1/J mice (Jackson Laboratory, ME) to generate the experimental mice. Male mice were used in our current study, because we found gender-dependent phenotypic differences in N171–82Q HD mice, therefore we use male N171–82Q mice for this preclinical study as we did previously (Jiang et al., 2013; Jiang et al., 2012). Mice were randomly divided into six groups. DNP or placebo (1% sodium bicarbonate, pH 7.0) was orally given to the mice by gavage from 8 weeks old to the end of the study. Animals were housed under specific pathogen-free conditions with a reversed 12-h light/dark cycle maintained at 23 °C and provided with food and water ad libitum. All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by Johns Hopkins University Animal Care and Use Committee.
2.2. Drug preparation
2,4-DNP is slightly acidic and was dissolved by adding sodium bicarbonate. Briefly, DNP was added to water first and 90 mg/mL sodium bicarbonate solution was added at 1% to each dosing solution. Final drug concentrations were 0.5 mg/mL (for 5 mg/kg dose), 0.1 mg/mL (for 1 mg/kg dose) and 0.05 mg/mL (for 0.5 mg/kg dose). The DNP solution was stored at room temperature and kept from light. Fresh DNP solutions were made every three days.
2.3. Behavioral tests
Motor function was assessed on an 80-cm long and 5-mm wide square-shaped balance beam. The balance beam was mounted on 50-cm high supports. A bright light illuminated the start platform, and a darkened enclosed 1728 cm3 escape box (12 × 12 × 12 cm) was situated at the end of the beam. Disposable pads placed under the beam provided cushioning if a mouse fell off the beam. Mice were trained to walk across the beam twice at least 1 h prior to testing. If a mouse stopped during training, the tail was gently pressed to encourage movement. After the training trial, mice were left undisturbed for at least an hour before testing. The time for each mouse to traverse the balance beam was recorded with a 60-s maximum cut-off, and falls were scored as 60 s. In addition to the 5 mm balance beam, the tapered beam was also used to evaluate hindlimb function. To increase sensitivity of the task and to encourage the mice to run the beam reliably, the beam was angled at 17° from the horizontal such that the mouse was able to run uphill. Our apparatus also has a goal box, as the use of a goal box can be beneficial for expediting training and increasing the reliability of the test performance. As the beam is elevated above ground level, a soft landing area is essential beneath the beam, which in our laboratory consists of numerous bench pads. The traverse time when the mouse crossed the start line and ended was recorded.
2.4. Survival
Survival was monitored daily by experienced investigators (B.W and Q.P). The mice were considered at the end of life when they were unable to right themselves after being placed on their backs and initiate movement after being gently prodded for 30 s.
2.5. In vivo structural MRI acquisition and quantification of brain volume
In vivo structural MRI scans were performed on a horizontal 9.4T magnetic resonance imager (Bruker Biospin, Billerica) with a triple-axis gradient and an animal imaging probe. The detailed image capture and analysis were described in our previous study (Cheng et al., 2011). Briefly, mice were anesthetized with 1% isoflurane, respiration was monitored and the temperature was maintained during the entire scan. Images were acquired by a three-dimensional T2-weighted fast spin echo sequence with the following parameters: echo time (TE)/repetition time (TR) = 40/700 ms, resolution = 0.1 mm × 0.1 mm × 0.1 mm, echo train length = 4, number of average = 2 and flip angle = 40°. The imaging resolution and contrast were sufficient for automatic volumetric characterization of the mouse brains and substructures. The intensity-normalized images were submitted by the Diffeomap software to a linux cluster, which runs Large Deformation Diffeomorphic Metric Mapping (LDDMM). The transformations encode morphological differences between subject and template images and can be analyzed with deformation-based morphometry to detect regional changes in brain volume.
2.6. Protein extraction and Western blot analysis
Mouse brain tissues were collected at 4-h following the last DNP administration. Striatum was homogenized in RIPA buffer (Sigma) containing 50 mM Tris-HCl, pH 8.0, with 150 mM sodium chloride, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and freshly prepared protease inhibitor (1:100, Sigma) and/or phosphatase inhibitor (1:100, Sigma). Samples were then centrifuged at 14,000 ×g for 15 min at 4 °C and supernatant fractions were collected. Protein concentration was determined by the Micro BCA protein assay kit (Pierce Protein Research Products). Soluble proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked in the presence of 5% nonfat milk, and then incubated overnight with primary antibodies at 4 °C: rabbit anti-DARPP32 (1:2000, EMD Millipore, Cat# AB10518), mouse anti-beta actin (1:5000, Sigma, Cat# A2228), mouse anti-PSD95 (1:1000, Sigma, Cat#P246), mouse anti-HSP70 (1:5000, ThermoFisher Scientific, cat#MA3–028), and Rabbit anti-GRP78 (1:1000, EMD Millipore, cat#MABS474). The membrane was then exposed for 1 h to HRP-conjugated secondary antibody (1:3000; GE Healthcare) and immune-reactive proteins were visualized with a chemiluminescence-based detection kit according to the manufacturer’s protocol (ECL kit; Amersham Corp.).
2.7. Measurement of F2-isoprostanes using gas chromatograph-mass spectrometry
Brain tissue samples were snap frozen in liquid nitrogen and stored at −80 °C until analysis at the Vanderbilt University Eicosanoid Core Laboratory. Cerebral cortex samples were analyzed by gas chromatography and mass spectroscopy as we described previously (Milne et al., 2013). Esterified (bound) F2-Isoprostanes (F2-IsoP) quantity is reported in “ng F2-IsoP/gm tissue”.
2.8. Statistics
Data are expressed as the mean ± SD or SE. Statistical comparisons between groups were compared by One-way ANOVA with Holm-Sidak post-hoc test. Survival data were analyzed by Kaplan Meier analysis.
3. Results
3.1. DNP at low doses improves motor function in N171–82Q HD mice
N171–82Q HD mice exhibit adult-onset progressive motor deficits similar to those manifested in HD patients (Jiang et al., 2013; Jin et al., 2013; Masuda et al., 2008; Schilling et al., 1999). We examined whether peripheral (oral) administration of three ascending doses of DNP has any effect on motor function, which is impaired in the N171–82Q HD mice. We used a 5 mm square-shaped balance beam to assess the motor coordination of mice as we described previously (Jin et al., 2015; Peng et al., 2016). N171–82Q HD mice displayed significantly prolonged transverse time on the beam compared to the age-matched wild type control mice at 16 weeks of age, indicating impairment of motor coordination in the HD mice (Fig. 1A). DNP at all three tested doses (0.5, 1, 5 mg/kg) significantly improved the motor function of HD mice at both 16- and 22-weeks of age (~50-days and ~90-days on treatment), indicated by shorter transverse time on the beam in HD mice treated with DNP compared to that in HD mice on placebo (Fig. 1A–B). At 26 weeks of age (~120-days on treatment), low doses of DNP (0.5 and 1 mg/kg)-treated HD mice continuously exhibited improved motor function on the balance beam, whereas HD mice treated with a higher dose of DNP (5 mg/kg) had similar results to the placebo, suggesting lower doses are more advantageous (Fig. 1C). These results indicate that the protective effect of DNP is dose-dependent, and lower doses of DNP appear to have a long-term beneficial effect in HD mice. We also tested mice at 22-weeks and 26-weeks on the tapered beam tochallenge the hind limbs and the results were comparable to the 5-mm beam (data not shown).
Fig. 1.
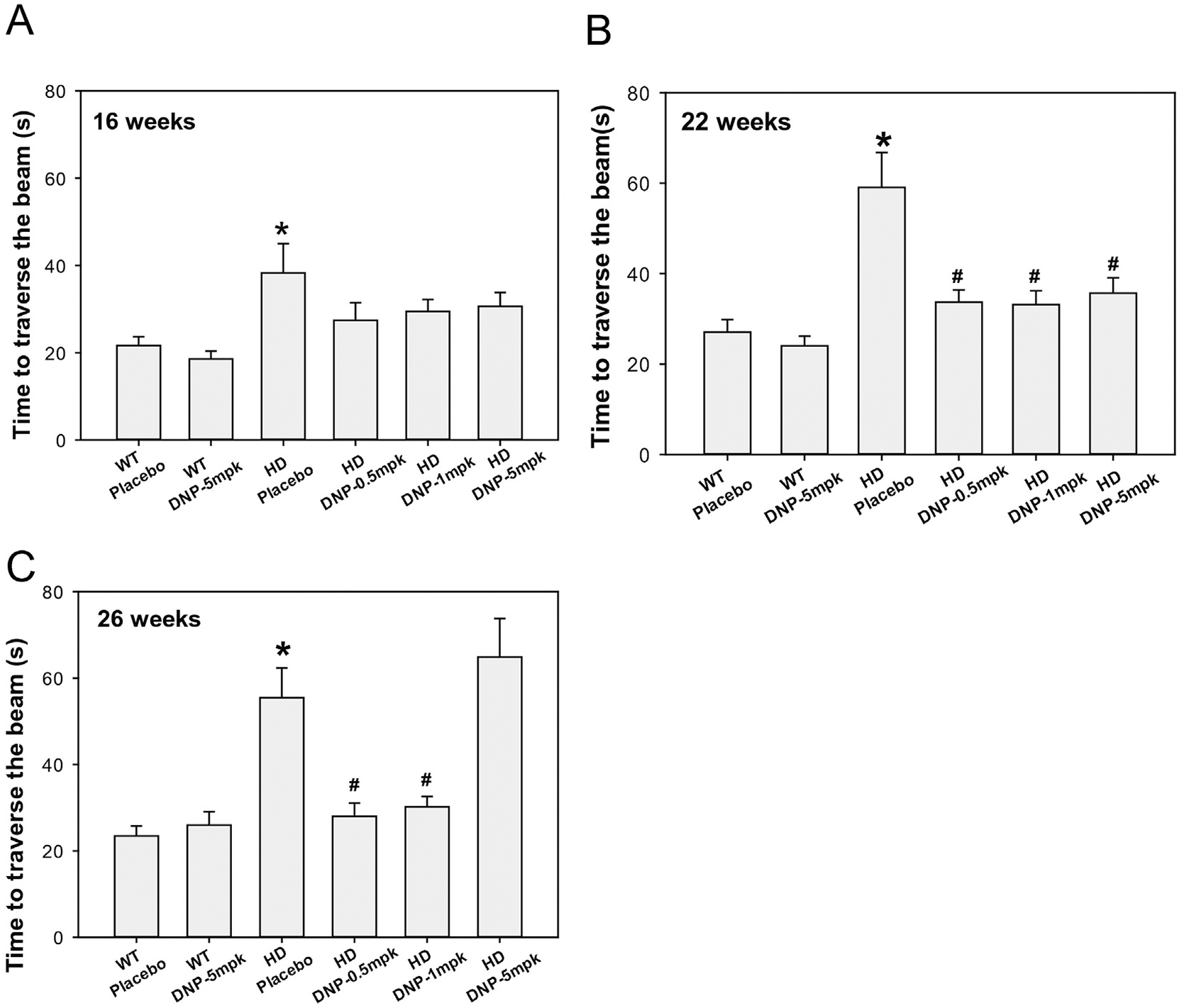
DNP improves motor function in N171–82Q HD mice. DNP was orally administered to mice daily at indicated doses from 8 weeks of age to the end of study. Mice were tested on a 5-mm square-shaped beam at 16-(A), 22-(B) and 26-(C) weeks of ages. n = 14–16 mice. The values are the mean and SEM. One-way ANOVA with Holm-Sidak post-hoc test was used. *p < 0.05 versus the values in wild type (WT)-placebo group; #p < 0.05 versus the HD-placebo group. Note that HD mice treated with DNP at 0.5 and 1 mg/kg (mpk) exhibited superior motor function, similar to the performance in WT mice at 26 weeks of age.
3.2. DNP preserves medium spiny neuronal marker protein DARPP32 and postsynaptic protein PSD95 in the striatum of N171–82Q HD mice
To investigate the underlying molecular mechanisms why motor function was improved by low dose of DNP in HD mice, we examined the medium spiny neuronal identity marker - dopamine- and cyclic AMP-regulated phosphoprotein of a molecular weight of 32 kDa (DARPP32). DARPP32 is downregulated in the HD striatal transcriptome, also in HD animal models, and these effects on transcription seem an essential feature of HD pathogenesis (Cha, 2007; Runne et al., 2008; Thomas et al., 2011; Young et al., 2013). We have demonstrated that DARPP32 protein levels declined in the striatum of N171–82Q HD mice (Jiang et al., 2013; Jiang et al., 2012; Jiang et al., 2014). We found that the DARPP32 protein levels were preserved by chronic administration of DNP at 1 mg/kg to equivalent levels in age-matched wild type mice (Fig. 2A), suggesting that DNP provided neuroprotective benefit in the HD mice.
Fig. 2.
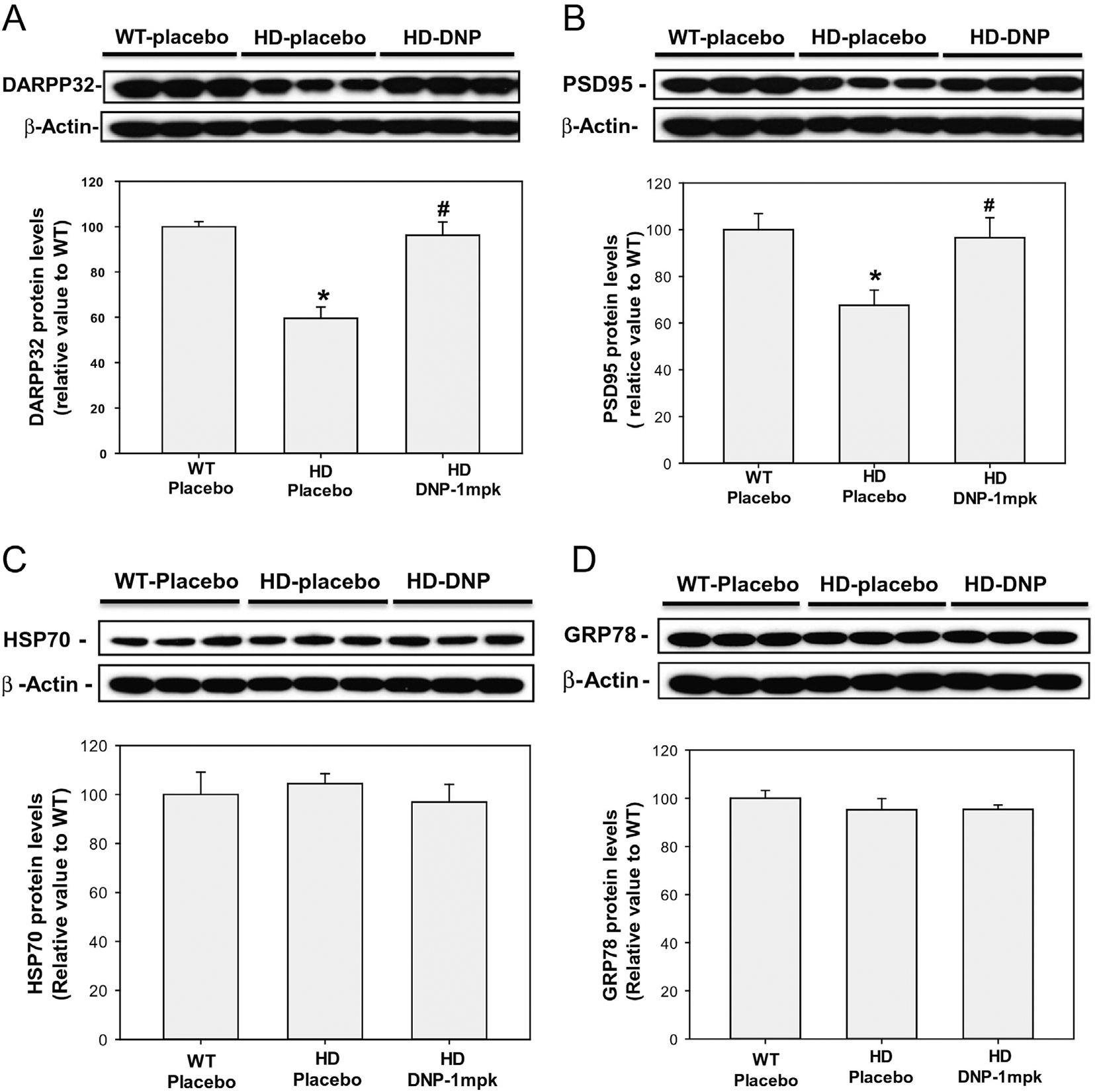
DNP preserves the protein levels of DARPP32 and PSD95 in the striatum of N171–82Q HD mice. DNP (1 mg/kg) was administered to mice daily from 8 weeks of age; at 26 weeks of age the mice were euthanized and the striatum was dissected for measurement of DARPP32 levels (A), PSD95 (B), HSP70(C), and GRP78 (D) by Western blotting. Top panels are representative blots and bottom panels are densitometry results from three mice in each group. The Y-axis values represent the ratio of targeted proteins (DARPP32 or PSD95) to beta-actin; the data were normalized to the ratio of WT-placebo group (100%). n = 3. *p < 0.05, compared to the value of WT- placebo group; #p < 0.05 compared to the value of HD- placebo group by one-way ANOVA with Holm-Sidak post-hoc tests.
PSD95 is a postsynaptic marker and this protein plays a role in anchoring NMDA receptors at the postsynaptic membrane and is required to sustain the molecular organization of the postsynaptic density and synaptic plasticity (Stein et al., 2003). PSD95 levels were reduced in HD mouse models, including full-length Htt knock-in mice (Valencia et al., 2013) and transgenic HD mice (Luthi-Carter et al., 2003), as well as the striatum of HD human brain (Fourie et al., 2014). It is postulated that ameliorating such synaptic changes delays clinical onset and/or prevents neurodegeneration. In fact, environmental enrichment significantly delayed the onset of deficit in PSD95 levels in HD mice (Nithianantharajah et al., 2008). We found that PSD95 protein levels were dramatically decreased in the striatum of N171–82Q HD mice, and DNP at 1 mg/kg significantly preserved the levels of PSD95 protein in the striatum of HD mice (Fig. 2B), indicating that DNP at this dose had beneficial effect in preserving postsynaptic density.
We next examined whether DNP at the protective dose induced cellular stress proteins in the mouse brain. We measured two inducible stress-responsive proteins - HSP70 and GRP78, which were up regulated in response to calorie restriction (Duan and Mattson, 1999; Manzanero et al., 2011; Mattson et al., 1999). We found that chronic administration of DNP at 1 mg/kg had no significant effect on the protein levels of HSP70 (Fig. 2C) or GRP78 (Fig. 2D) in the striatum of HD mice, suggesting that different molecular mechanisms were involved in the neuroprotective effect of DNP as it compares with those induced by calorie restriction.
3.3. DNP at the doses which improved motor function in HD mice has no significant effect on body weight, survival and brain atrophy in HD mice
In addition to neurological features, N171–82Q HD mice display body weight loss that is also observed in other HD mouse models as well as in HD patients (Carroll et al., 2015; Masuda et al., 2008; Schilling et al., 1999). We examined whether DNP at the doses used in this study affected body weight in mice. Wild type mice were given only the higher dose DNP (5 mg/kg), and HD mice were given three doses of DNP (0.5, 1, and 5 mg/kg). We found that DNP at all the doses used in our current study had no significant effect on body weight in either HD or in wild type mice (Fig. 3A). These results indicate that doses of DNP employed in the current study are weight neutral and considerably lower than what are historically used doses for obesity.
Fig. 3.
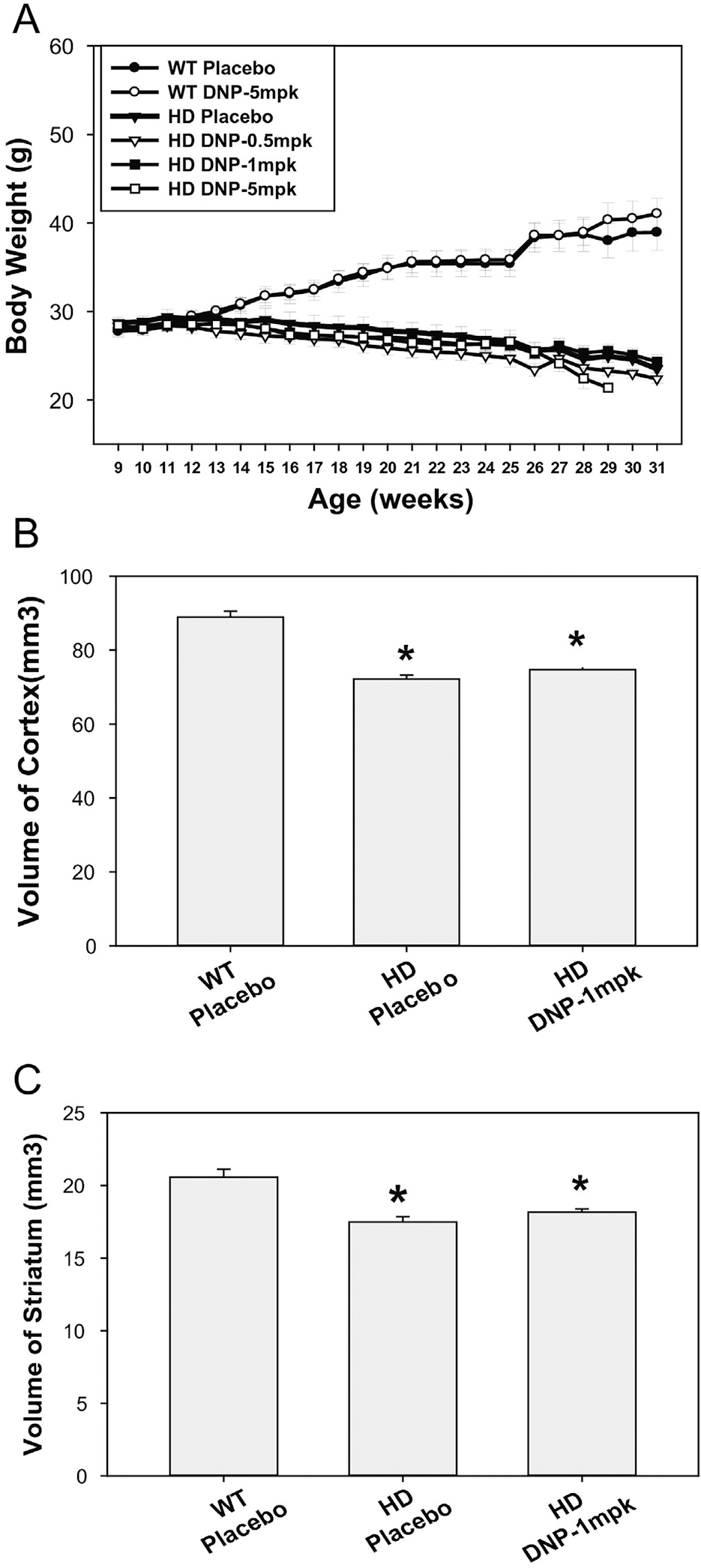
Effects of DNP on body weight and brain atrophy in HD mice. (A) Body weight was monitored from the beginning of study weekly. The values are the mean and SEM. n = 14–16. (B–C) Brain regional volumes were measured by structural MRI at 26 weeks of age, the values are mean and SE from 5 mice per group. *p < 0.001 versus WT-placebo group by one-way ANOVA with Holm-Sidak post-hoc tests.
N171–82Q mice have a shorter life span relative to wild type mice (Schilling et al., 1999); therefore we monitored the survival of HD mice treated with DNP. The life span in HD-placebo group was 220 ± 17 days (n = 15), HD mice treated with DNP at 0.5 mg/kg had an average lifespan as 206 ± 27 days (n = 15), and HD mice treated with DNP at 1 mg/kg had average lifespan at 234 ± 6 days (n = 15). HD mice treated with DNP at 5 mg/kg had a shorter lifespan compared to that in HD mice-placebo group (190 ± 13 days in HD-DNP at 5 mg/kg versus 220 ± 17 days in HD-placebo group, n = 15). DNP at all doses employed in this study did not significantly improve the survival of HD mice. To date, it is not precisely known why HD mice die; it can therefore be argued that motor scores and brain volume are better outcome measures versus survival.
The changes in brain volumes observed in N171–82Q HD-like mice strongly resemble those in human HD (Cheng et al., 2011). We next determined whether the improved motor function with the effective dose of DNP was associated with preventing brain atrophy. We conducted in vivo structural MRI scans and measured brain volumes at 26-weeks of age when significant brain atrophy was detected in N171–82Q HD mice (Jiang et al., 2013; Jiang et al., 2014). Based on the results on motor function assessment and survival, our neuroimaging study was focused on DNP at 1 mg/kg. N171–82Q mice showed significant volume loss in both the neocortex and striatum at this age. DNP at 1 mg/kg did not have significant effects on brain atrophy in HD mice (Fig. 3 B–C).
3.4. DNP reduces F2-isoprostanes levels in HD mouse brain
Oxidative stress has been implicated in HD (Browne, 2008; Browne and Beal, 2006; Chen et al., 2007). F2-isoprostanes are excellent biomarkers as well as potent mediators of oxidative stress in vivo in human (Milne et al., 2015). The literature data indicates that in vivo or postmortem cerebrospinal fluid and brain tissue levels of F2-isoprostanes are significantly increased in neurodegenerative diseases, including HD (Montine et al., 1999). We measured esterified F2-isoprostanes in the cerebral cortex by the most accurate method-gas chromatograph-mass spectrometry. We found that F2-isoprostanes levels were significantly elevated in HD mouse brains compared to those in age-matched littermate control mice. Chronic administration of DNP at 1 mg/kg dramatically reduced the F2-isoprostanes in the HD mouse brain (Fig. 4) and brought the levels of F2-isoprostanes in HD mouse brain to the wild type mouse levels.
Fig. 4.
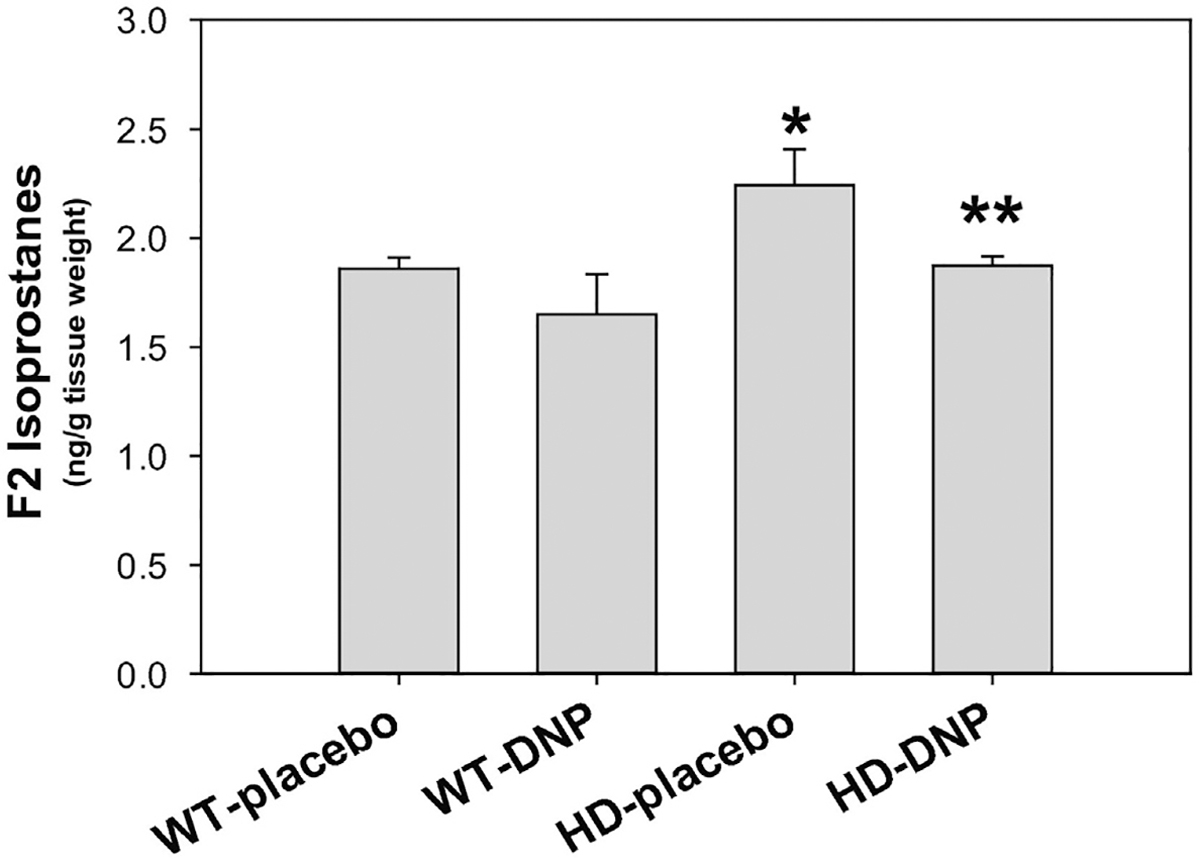
DNP attenuated oxidative stress in HD mouse brain. Cerebral cortex was dissected and F2-isoprostanes levels were measured by gas chromatography and mass spectrometry at 26 weeks old mice. Mice were chronically given placebo or DNP from 8 weeks of age. Data are Mean ± SE, n = 5 mice/group in WT-placebo, HD-placebo, HD-DNP and 3 mice in WT-DNP group. *p < 0.05 versus WT-placebo group, **p < 0.05 versus HD-placebo group by one-way ANOVA with Holm-Sidak post-hoc tests.
4. Discussion
In this study, we found that DNP at low doses had significantly improved motor function and preserved medium spiny neuronal identity in HD mice. The doses of DNP used in our study are equivalent to 50–60 times lower (2–5 mg/day) than the typical dose provided for obesity patients in the 1930′s (~300 mg/day). In order to demonstrate the potential for repurposing DNP for HD, both HD mice and wild type mice were treated with DNP higher dose (5 mg/kg), DNP at this dose was also weight neutral. All doses appeared to preserve motor function up to 22-weeks of age (~90-days of treatment), however the lower doses (0.5 and 1 mg/kg) rather than the high dose (5 mg/kg) consistently provided the protection in motor function at 26-weeks of age, implicating a possible hormetic effect (Geisler et al., 2016). Our results of F2-isprostane levels indicate that the effects of low dose of DNP reduced oxidative stress/ROS production by mild mitochondrial uncoupling while maintaining sufficient ATP production; while the long-term administration of higher dose of DNP (5 mg/kg), outside the hermetic zone, may result in insufficient energy production to support neuronal function, and abolishes the protective effect.
Mitochondria comprise the key components for energy demand/production coupling to coherently integrate cell respiration and energy metabolism to support cell survival, particularly neurons with high energy demand. Mitochondrial uncoupling proteins (UCPs) can be activated by free radicals, and their activity has a profound influence on neuronal function (Kim-Han and Dugan, 2005; Mailloux and Harper, 2011). By regulating mitochondrial biogenesis, calcium flux, free radical production and local temperature, neuronal UCPs can directly influence neurotransmission, synaptic plasticity and neurodegenerative processes (Mattson and Liu, 2003). Mitochondrial uncoupling reduces the production of ROS and consequent oxidative stress (Mailloux and Harper, 2011). Our results on F2-isoprostanes levels confirmed this is indeed the case. Prevention of overt production of ROS by DNP is significantly different than therapies attempting to clear ROS after they have been made using anti-oxidants (Enns et al., 2012; Smith and Murphy, 2010). Beyond the pharmacology of lowering ROS with DNP administration, there is a distinct difference between the acute and chronic actions of mitochondrial uncoupling. The acute effects of mitochondrial uncoupling prevent the reverse flow of electrons and lowering ROS (Korde et al., 2005), while chronic mitochondrial uncoupling triggers a cascade effect of cellular remodeling with mitochondrial biogenesis in neurons, such as induction of cAMP, lowering mTOR (De Felice and Ferreira, 2006; Liu et al., 2002; Sebollela et al., 2010). In either case, the initial mechanism of action is “non-genomic” with DNP being the transporter of a proton (H+) that is delivered to the mitochondrial matrix, causing a reduction in ATP production since the proton did not travel through ATP synthase (Geisler, 2011). As a result, there is a compensatory increase in beta-oxidation of lipids and glycolysis to reestablish the mitochondrial membrane potential through the electron transport system. In our current study, DNP was given chronically at weight-neutral doses over a long period of time, is sufficient to reduce ROS production and trigger cellular remodeling, thereby protecting neurons from mutant Htt toxicity.
DARPP32 is a cytosolic protein that is highly enriched in striatal medium spiny neurons. The protein is a pivotal integrator of dopamine signaling in MSNs neurons (Svenningsson et al., 2004). DARPP32 is also a fundamental component of the dopamine-signaling cascade (Fienberg et al., 1998; Greengard et al., 1999), and HD pathology is marked by extensive loss of medium spiny striatal neurons that express high levels of DARPP32; therefore, DARPP32 serves as a marker of neuronal loss or neuronal dysfunction in medium spiny neurons (Hickey et al., 2008; van Dellen et al., 2000). DNP at the low dose preserves DARPP32 protein levels in the striatum of HD mice, indicating that DNP protects medium spiny neurons from mutant Htt toxicity.
Aberrant synaptic plasticity (Cummings et al., 2007; Cummings et al., 2006; Hodgson et al., 1999; Murphy et al., 2000; Usdin et al., 1999) and altered expression of genes encoding synaptic proteins have been documented in various HD mouse models (Cha, 2007; Luthi-Carter et al., 2003; Sugars and Rubinsztein, 2003). The number of mitochondria in dendritic compartments correlates with the morphological plasticity of spines, which indicates that the postsynaptic distribution of mitochondria is essential and limiting for the support of synapses (Dorszewska, 2013). This indicates that mitochondria are more directly involved in synapse plasticity. PSD95 binds to neuronal cell adhesion molecules, which are assumed to form an intercellular bridge traversing the synaptic cleft (Ichtchenko et al., 1995; Irie et al., 1997; Song et al., 1999), interacts with microtubules or tubulin indirectly (Niethammer et al., 1998) and binds proteins that regulate the actin cytoskeleton (Furuyashiki et al., 1999), suggesting a major function of PSD-95 in normal synaptic plasticity. In the N171–82Q HD mouse striatum, we found a dramatic decrease of postsynaptic protein PSD95, and DNP preserved the PSD95 levels in HD mice, suggesting that the down-regulation of PSD-95 is a molecular mediator of HD pathogenesis and that amelioration of this PSD-95 deficit may also contribute to the beneficial effects of chronic administration of low dose DNP.
F2-Isoprostanes are unique prostaglandin-like products, which are formed via nonenzymatic, free radical-mediated peroxidation of polyunsaturated fatty acids, such as arachidonic acid (AA) (Hardy et al., 2011; Roberts and Morrow, 2002). The measurement of F2-IsoPs is currently the best available biomarker of lipid peroxidation and oxidative stress in vivo (Halliwell and Lee, 2010; Roberts and Milne, 2009). The level of F2-isoprostanes in HD patients was significantly higher than in the control group (Montine et al., 1999). Such stress contributes to neuronal dysfunction by damaging the main structures: DNA, proteins, and lipids. Previous studies suggest that there are increased immunostaining for malondialdehyde, 4-hydroxynonenal, and 15-F2t-isoprostanes in HD mice (Browne and Beal, 2006). Our current study using accurate gas chromatograph-mass spectrometry measured the F2-isoprostanes in the cerebral cortex of either placebo or DNP treated- HD mice. Our results suggest that increased oxidative stress in the N171–82Q mouse brain and DNP at low dose significantly reduced the oxidative stress.
It has been reported that DNP can cross the blood-brain barrier (Perry et al., 2013) and act directly on neurons to cause mitochondrial uncoupling and increase cellular resilience, similar to caloric restriction or moderate exercise improving brain bioenergetics when administered systemically (Caldeira da Silva et al., 2008), reversed diet-induced obesity, hypertriglyceridemia, and insulin resistance in diet-induced obesity (Goldgof et al., 2014; Perry et al., 2013). It is also possible that some of the peripheral responses to DNP are also beneficial for the brain protection.
Although the clinical use of DNP has been banned due to its side effects at high dose and narrow pharmacokinetic profile, DNP nevertheless continues to be used to improve appearance (Petroczi et al., 2015). DNP at low doses is protective against diet-induced obesity and hepatic steatosis, while improving glucose tolerance in mice and rats (Perry et al., 2015; Samuel et al., 2004). In addition, DNP has been reported to have neuroprotection and improved functional outcome in models of stroke (Korde et al., 2005; Liu et al., 2002), Parkinson’s disease (Wu et al., 2011), and an Alzheimer’s disease mouse model (Geisler et al., 2016). Combined with our current study, these findings suggest that DNP at appropriate weight-neutral doses may have clinical applications for neurodegenerative conditions. In conclusion, our data suggested that DNP preserved the molecular identity of medium spiny neurons, improved motor function, and reduced oxidative stress in HD mice. These findings provide validation for the merit of mild mitochondrial uncoupling as a potential therapeutic approach for HD.
Acknowledgements
We thank Jared LaBron for critical reading of the manuscript. This work was supported by Mitochon Pharmaceutical Inc and NIH R01 NS082338 to (W.D).
Abbreviations:
- DNP
2,4-dinitrophenol
- HD
Huntington’s disease
- mHtt
mutant huntingtin protein
- ROS
reactive oxygen species
- UCP
uncoupling protein
Footnotes
Conflict of interest
Dr. Duan reports receiving unrestricted research funds from Mitochon Pharmaceuticals, Inc. Drs. J. Geisler and R. Alonso are employees of Mitochon Pharmaceuticals, Inc. All other authors declare that they have no competing interests.
References
- Arslan-Ergul A, Ozdemir AT, Adams MM, 2013. Aging, neurogenesis, and caloric restriction in different model organisms. Aging Dis 4, 221–232. [PMC free article] [PubMed] [Google Scholar]
- Browne SE, 2008. Mitochondria and Huntington’s disease pathogenesis: insight from genetic and chemical models. Ann. N. Y. Acad. Sci. 1147, 358–382. [DOI] [PubMed] [Google Scholar]
- Browne SE, Beal MF, 2006. Oxidative damage in Huntington’s disease pathogenesis. Antioxid. Redox Signal. 8, 2061–2073. [DOI] [PubMed] [Google Scholar]
- Caldeira da Silva CC, Cerqueira FM, Barbosa LF, Medeiros MH, Kowaltowski AJ, 2008. Mild mitochondrial uncoupling in mice affects energy metabolism, redox balance and longevity. Aging Cell 7, 552–560. [DOI] [PubMed] [Google Scholar]
- Carroll JB, Bates GP, Steffan J, Saft C, Tabrizi SJ, 2015. Treating the whole body in Huntington’s disease. Lancet Neurol. 14, 1135–1142. [DOI] [PubMed] [Google Scholar]
- Cha JH, 2007. Transcriptional signatures in Huntington’s disease. Prog. Neurobiol. 83, 228–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CM, Wu YR, Cheng ML, Liu JL, Lee YM, Lee PW, Soong BW, Chiu DT, 2007. Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem. Biophys. Res. Commun. 359, 335–340. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Peng Q, Hou Z, Aggarwal M, Zhang J, Mori S, Ross CA, Duan W, 2011. Structural MRI detects progressive regional brain atrophy and neuroprotective effects in N171–82Q Huntington’s disease mouse model. NeuroImage 56, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman E, 2007. Dinitrophenol and obesity: an early twentieth-century regulatory dilemma. Regul. Toxicol. Pharmacol. 48, 115–117. [DOI] [PubMed] [Google Scholar]
- Cummings DM, Milnerwood AJ, Dallerac GM, Waights V, Brown JY, Vatsavayai SC, Hirst MC, Murphy KP, 2006. Aberrant cortical synaptic plasticity and dopaminergic dysfunction in a mouse model of Huntington’s disease. Hum. Mol. Genet. 15, 2856–2868. [DOI] [PubMed] [Google Scholar]
- Cummings DM, Milnerwood AJ, Dallerac GM, Vatsavayai SC, Hirst MC, Murphy KP, 2007. Abnormal cortical synaptic plasticity in a mouse model of Huntington’s disease. Brain Res. Bull. 72, 103–107. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Ferreira ST, 2006. Novel neuroprotective, neuritogenic and anti-amyloidogenic properties of 2,4-dinitrophenol: the gentle face of Janus. IUBMB Life 58, 185–191. [DOI] [PubMed] [Google Scholar]
- van Dellen A, Welch J, Dixon RM, Cordery P, York D, Styles P, Blakemore C, Hannan AJ, 2000. N-Acetylaspartate and DARPP-32 levels decrease in the corpus striatum of Huntington’s disease mice. Neuroreport 11, 3751–3757. [DOI] [PubMed] [Google Scholar]
- Dorszewska J, 2013. Cell biology of normal brain aging: synaptic plasticity-cell death. Aging Clin. Exp. Res. 25, 25–34. [DOI] [PubMed] [Google Scholar]
- Duan W, Mattson MP, 1999. Dietary restriction and 2-deoxyglucose administration improve behavioral outcome and reduce degeneration of dopaminergic neurons in models of Parkinson’s disease. J. Neurosci. Res. 57, 195–206. [DOI] [PubMed] [Google Scholar]
- Duan W, Guo Z, Jiang H, Ware M, Li XJ, Mattson MP, 2003. Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc. Natl. Acad. Sci. U. S. A. 100, 2911–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enns GM, Kinsman SL, Perlman SL, Spicer KM, Abdenur JE, Cohen BH, Amagata A, Barnes A, Kheifets V, Shrader WD, Thoolen M, Blankenberg F, Miller G, 2012. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol. Genet. Metab. 105, 91–102. [DOI] [PubMed] [Google Scholar]
- Fienberg AA, Hiroi N, Mermelstein PG, Song W, Snyder GL, Nishi A, Cheramy A, O’Callaghan JP, Miller DB, Cole DG, Corbett R, Haile CN, Cooper DC, Onn SP, Grace AA, Ouimet CC, White FJ, Hyman SE, Surmeier DJ, Girault J, Nestler EJ, Greengard P, 1998. DARPP-32: regulator of the efficacy of dopaminergic neurotransmission. Science 281, 838–842. [DOI] [PubMed] [Google Scholar]
- Fourie C, Kim E, Waldvogel H, Wong JM, McGregor A, Faull RL, Montgomery JM, 2014. Differential changes in postsynaptic density proteins in postmortem Huntington’s disease and Parkinson’s disease human brains. J. Neurodev. Disord. 2014, 938530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuyashiki T, Fujisawa K, Fujita A, Madaule P, Uchino S, Mishina M, Bito H, Narumiya S, 1999. Citron, a Rho-target, interacts with PSD-95/SAP-90 at glutamatergic synapses in the thalamus. J. Neurosci. 19, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler JG, 2011. Targeting energy expenditure via fuel switching and beyond. Diabetologia 54, 237–244. [DOI] [PubMed] [Google Scholar]
- Geisler JG, Marosi K, Halpern J, Mattson MP, 2016. DNP, mitochondrial uncoupling and neuroprotection: a little dab’ll do ya. Alzheimer’s & dementia: the journal of the Alzheimer’s Association Sep 4. pii: S1552–5260(16)32854-0. 10.1016/j.jalz.2016.08.001 (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Mohapel J, Brocardo PS, Christie BR, 2014. The role of oxidative stress in Huntington’s disease: are antioxidants good therapeutic candidates? Curr. Drug Targets 15, 454–468. [DOI] [PubMed] [Google Scholar]
- Goldgof M, Xiao C, Chanturiya T, Jou W, Gavrilova O, Reitman ML, 2014. The chemical uncoupler 2,4-dinitrophenol (DNP) protects against diet-induced obesity and improves energy homeostasis in mice at thermoneutrality. J. Biol. Chem. 289, 19341–19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goula AV, Berquist BR, Wilson DM 3rd, Wheeler VC, Trottier Y, Merienne K, 2009. Stoichiometry of base excision repair proteins correlates with increased somatic CAG instability in striatum over cerebellum in Huntington’s disease transgenic mice. PLoS Genet. 5, e1000749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveland GA, Williams RS, DiFiglia M, 1985. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 227, 770–773. [DOI] [PubMed] [Google Scholar]
- Greengard P, Allen PB, Nairn AC, 1999. Beyond the dopamine receptor: the DARPP-32/protein phosphatase-1 cascade. Neuron 23, 435–447. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Lee CY, 2010. Using isoprostanes as biomarkers of oxidative stress: some rarely considered issues. Antioxid. Redox Signal. 13, 145–156. [DOI] [PubMed] [Google Scholar]
- Hardy KD, Cox BE, Milne GL, Yin H, Roberts LJ 2nd., 2011. Nonenzymatic free radical-catalyzed generation of 15-deoxy-Delta(12,14)-prostaglandin J(2)-like compounds (deoxy-J(2)-isoprostanes) in vivo. J. Lipid Res. 52, 113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersch SM, Gevorkian S, Marder K, Moskowitz C, Feigin A, Cox M, Como P, Zimmerman C, Lin M, Zhang L, Ulug AM, Beal MF, Matson W, Bogdanov M, Ebbel E, Zaleta A, Kaneko Y, Jenkins B, Hevelone N, Zhang H, Yu H, Schoenfeld D, Ferrante R, Rosas HD, 2006. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2’dG. Neurology 66, 250–252. [DOI] [PubMed] [Google Scholar]
- Hickey MA, Kosmalska A, Enayati J, Cohen R, Zeitlin S, Levine MS, Chesselet MF, 2008. Extensive early motor and non-motor behavioral deficits are followed by striatal neuronal loss in knock-in Huntington’s disease mice. Neuroscience 157, 280–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR, 1999. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 23, 181–192. [DOI] [PubMed] [Google Scholar]
- Hunt ND, Hyun DH, Allard JS, Minor RK, Mattson MP, Ingram DK, de Cabo R, 2006. Bioenergetics of aging and calorie restriction. Ageing Res. Rev. 5, 125–143. [DOI] [PubMed] [Google Scholar]
- Ichtchenko K, Hata Y, Nguyen T, Ullrich B, Missler M, Moomaw C, Sudhof TC, 1995. Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell 81, 435–443. [DOI] [PubMed] [Google Scholar]
- Irie M, Hata Y, Takeuchi M, Ichtchenko K, Toyoda A, Hirao K, Takai Y, Rosahl TW, Sudhof TC, 1997. Binding of neuroligins to PSD-95. Science 277, 1511–1515. [DOI] [PubMed] [Google Scholar]
- Jiang M, Wang J, Fu J, Du L, Jeong H, West T, Xiang L, Peng Q, Hou Z, Cai H, Seredenina T, Arbez N, Zhu S, Sommers K, Qian J, Zhang J, Mori S, Yang XW, Tamashiro KL, Aja S, Moran TH, Luthi-Carter R, Martin B, Maudsley S, Mattson MP, Cichewicz RH, Ross CA, Holtzman DM, Krainc D, Duan W, 2012. Neuroprotective role of Sirt1 in mammalian models of Huntington’s disease through activation of multiple Sirt1 targets. Nat. Med. 18, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Peng Q, Liu X, Jin J, Hou Z, Zhang J, Mori S, Ross CA, Ye K, Duan W, 2013. Small-molecule TrkB receptor agonists improve motor function and extend survival in a mouse model of Huntington’s disease. Hum. Mol. Genet. 22, 2462–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Zheng J, Peng Q, Hou Z, Zhang J, Mori S, Ellis JL, Vlasuk GP, Fries H, Suri V, Duan W, 2014. Sirtuin 1 activator SRT2104 protects Huntington’s disease mice. Ann. Clin. Transl. Neurol. 1, 1047–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Albertz J, Guo Z, Peng Q, Rudow G, Troncoso JC, Ross CA, Duan W, 2013. Neuroprotective effects of PPAR-gamma agonist rosiglitazone in N171–82Q mouse model of Huntington’s disease. J. Neurochem. 125, 410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Peng Q, Hou Z, Jiang M, Wang X, Langseth AJ, Tao M, Barker PB, Mori S, Bergles DE, Ross CA, Detloff PJ, Zhang J, Duan W, 2015. Early white matter abnormalities, progressive brain pathology and motor deficits in a novel knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 24, 2508–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johri A, Beal MF, 2012. Antioxidants in Huntington’s disease. Biochim. Biophys. Acta 1822, 664–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim-Han JS, Dugan LL, 2005. Mitochondrial uncoupling proteins in the central nervous system. Antioxid. Redox Signal. 7, 1173–1181. [DOI] [PubMed] [Google Scholar]
- Korde AS, Pettigrew LC, Craddock SD, Maragos WF, 2005. The mitochondrial uncoupler 2,4-dinitrophenol attenuates tissue damage and improves mitochondrial homeostasis following transient focal cerebral ischemia. J. Neurochem. 94, 1676–1684. [DOI] [PubMed] [Google Scholar]
- Kumar A, Ratan RR, 2016. Oxidative stress and Huntington’s disease: the good, the bad, and the ugly. J Huntingtons Dis 5, 217–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Lu C, Wan R, Auyeung WW, Mattson MP, 2002. Activation of mitochondrial ATP-dependent potassium channels protects neurons against ischemia-induced death by a mechanism involving suppression of Bax translocation and cytochrome c release. J. Cereb. Blood Flow Metab. 22, 431–443. [DOI] [PubMed] [Google Scholar]
- Lopez-Lluch G, Navas P, 2016. Calorie restriction as an intervention in ageing. J. Physiol. 594, 2043–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthi-Carter R, Apostol BL, Dunah AW, DeJohn MM, Farrell LA, Bates GP, Young AB, Standaert DG, Thompson LM, Cha JH, 2003. Complex alteration of NMDA receptors in transgenic Huntington’s disease mouse brain: analysis of mRNA and protein expression, plasma membrane association, interacting proteins, and phosphorylation. Neurobiol. Dis. 14, 624–636. [DOI] [PubMed] [Google Scholar]
- Mailloux RJ, Harper ME, 2011. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 51, 1106–1115. [DOI] [PubMed] [Google Scholar]
- Manzanero S, Gelderblom M, Magnus T, Arumugam TV, 2011. Calorie restriction and stroke. Exp Transl Stroke Med 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda N, Peng Q, Li Q, Jiang M, Liang Y, Wang X, Zhao M, Wang W, Ross CA, Duan W, 2008. Tiagabine is neuroprotective in the N171–82Q and R6/2 mouse models of Huntington’s disease. Neurobiol. Dis. 30, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, 2008. Dietary factors, hormesis and health. Ageing Res. Rev. 7, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Liu D, 2003. Mitochondrial potassium channels and uncoupling proteins in synaptic plasticity and neuronal cell death. Biochem. Biophys. Res. Commun. 304, 539–549. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Pedersen WA, Duan W, Culmsee C, Camandola S, 1999. Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer’s and Parkinson’s diseases. Ann. N. Y. Acad. Sci. 893, 154–175. [DOI] [PubMed] [Google Scholar]
- Milne GL, Gao B, Terry ES, Zackert WE, Sanchez SC, 2013. Measurement of F2-isoprostanes and isofurans using gas chromatography-mass spectrometry. Free Radic. Biol. Med. 59, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne GL, Dai Q, Roberts LJ 2nd., 2015. The isoprostanes—25 years later. Biochim. Biophys. Acta 1851, 433–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine TJ, Beal MF, Robertson D, Cudkowicz ME, Biaggioni I, O’Donnell H, Zackert WE, Roberts LJ, Morrow JD, 1999. Cerebrospinal fluid F2-isoprostanes are elevated in Huntington’s disease. Neurology 52, 1104–1105. [DOI] [PubMed] [Google Scholar]
- Murphy KP, Carter RJ, Lione LA, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ, 2000. Abnormal synaptic plasticity and impaired spatial cognition in mice transgenic for exon 1 of the human Huntington’s disease mutation. J. Neurosci. 20, 5115–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer M, Valtschanoff JG, Kapoor TM, Allison DW, Weinberg RJ, Craig AM, Sheng M, 1998. CRIPT, a novel postsynaptic protein that binds to the third PDZ domain of PSD-95/SAP90. Neuron 20, 693–707. [DOI] [PubMed] [Google Scholar]
- Nithianantharajah J, Barkus C, Murphy M, Hannan AJ, 2008. Gene-environment interactions modulating cognitive function and molecular correlates of synaptic plasticity in Huntington’s disease transgenic mice. Neurobiol. Dis. 29, 490–504. [DOI] [PubMed] [Google Scholar]
- Padalko VI, 2005. Uncoupler of oxidative phosphorylation prolongs the lifespan of Drosophila. Biochemistry (Mosc) 70, 986–989. [DOI] [PubMed] [Google Scholar]
- Peng Q, Wu B, Jiang M, Jin J, Hou Z, Zheng J, Zhang J, Duan W, 2016. Characterization of behavioral, neuropathological, brain metabolic and key molecular changes in zQ175 knock-in mouse model of Huntington’s disease. PLoS One 11, e0148839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Kim T, Zhang XM, Lee HY, Pesta D, Popov VB, Zhang D, Rahimi Y, Jurczak MJ, Cline GW, Spiegel DA, Shulman GI, 2013. Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab. 18, 740–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Zhang D, Zhang XM, Boyer JL, Shulman GI, 2015. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347, 1253–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroczi A, Ocampo JA, Shah I, Jenkinson C, New R, James RA, Taylor G, Naughton DP, 2015. Russian roulette with unlicensed fat-burner drug 2,4-dinitrophenol (DNP): evidence from a multidisciplinary study of the internet, bodybuilding supplements and DNP users. Subst. Abuse Treat. Prev. Policy 10, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D’Amato CJ, Penney JB, Young AB, 1988. Differential loss of striatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. U. S. A. 85, 5733–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts LJ 2nd, Milne GL, 2009. Isoprostanes. J. Lipid Res. 50 (Suppl), S219–S223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts LJ 2nd, Morrow JD, 2002. Products of the isoprostane pathway: unique bioactive compounds and markers of lipid peroxidation. Cell. Mol. Life Sci. 59, 808–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Tabrizi SJ, 2011. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10, 83–98. [DOI] [PubMed] [Google Scholar]
- Rotblat B, Southwell AL, Ehrnhoefer DE, Skotte NH, Metzler M, Franciosi S, Leprivier G, Somasekharan SP, Barokas A, Deng Y, Tang T, Mathers J, Cetinbas N, Daugaard M, Kwok B, Li L, Carnie CJ, Fink D, Nitsch R, Galpin JD, Ahern CA, Melino G, Penninger JM, Hayden MR, Sorensen PH, 2014. HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc. Natl. Acad. Sci. U. S. A. 111, 3032–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runne H, Regulier E, Kuhn A, Zala D, Gokce O, Perrin V, Sick B, Aebischer P, Deglon N, Luthi-Carter R, 2008. Dysregulation of gene expression in primary neuron models of Huntington’s disease shows that polyglutamine-related effects on the striatal transcriptome may not be dependent on brain circuitry. J. Neurosci. 28, 9723–9731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI, 2004. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 279, 32345–32353. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Perry G, Smith MA, 2008. Oxidative stress and neurotoxicity. Chem. Res. Toxicol. 21, 172–188. [DOI] [PubMed] [Google Scholar]
- Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR, 1999. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 8, 397–407. [DOI] [PubMed] [Google Scholar]
- Sebollela A, Freitas-Correa L, Oliveira FF, Mendes CT, Wasilewska-Sampaio AP, Camacho-Pereira J, Galina A, Brentani H, Passetti F, De Felice FG, Dias-Neto E, Ferreira ST, 2010. Expression profile of rat hippocampal neurons treated with the neuroprotective compound 2,4-dinitrophenol: up-regulation of cAMP signaling genes. Neurotox. Res. 18, 112–123. [DOI] [PubMed] [Google Scholar]
- Simkins S, 1937. Dinitrophenol and desiccated thyroid in the treatment of obesity. JAMA 108, 2110–2117. [Google Scholar]
- Smith RA, Murphy MP, 2010. Animal and human studies with the mitochondriatargeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. 1201, 96–103. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Forster MJ, 2014. Caloric restriction and the aging process: a critique. Free Radic. Biol. Med. 73, 366–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JY, Ichtchenko K, Sudhof TC, Brose N, 1999. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc. Natl. Acad. Sci. U. S. A. 96, 1100–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack EC, Matson WR, Ferrante RJ, 2008. Evidence of oxidant damage in Huntington’s disease: Translational strategies using antioxidants. Ann. N. Y. Acad. Sci. 1147, 79–92. [DOI] [PubMed] [Google Scholar]
- Stein V, House DR, Bredt DS, Nicoll RA, 2003. Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J. Neurosci. 23, 5503–5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC, 2003. Transcriptional abnormalities in Huntington disease. Trends Genet. 19, 233–238. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P, 2004. DARPP-32: an integrator of neurotransmission. Annu. Rev. Pharmacol. Toxicol. 44, 269–296. [DOI] [PubMed] [Google Scholar]
- The Huntington’s Disease Collaborative Research Group, 1993. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983. [DOI] [PubMed] [Google Scholar]
- Thomas EA, Coppola G, Tang B, Kuhn A, Kim S, Geschwind DH, Brown TB, Luthi-Carter R, Ehrlich ME, 2011. In vivo cell-autonomous transcriptional abnormalities revealed in mice expressing mutant huntingtin in striatal but not cortical neurons. Hum. Mol. Genet. 20, 1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trushina E, McMurray CT, 2007. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 145, 1233–1248. [DOI] [PubMed] [Google Scholar]
- Tunez I, Sanchez-Lopez F, Aguera E, Fernandez-Bolanos R, Sanchez FM, Tasset-Cuevas I, 2011. Important role of oxidative stress biomarkers in Huntington’s disease. J. Med. Chem. 54, 5602–5606. [DOI] [PubMed] [Google Scholar]
- Usdin MT, Shelbourne PF, Myers RM, Madison DV, 1999. Impaired synaptic plasticity in mice carrying the Huntington’s disease mutation. Hum. Mol. Genet. 8, 839–846. [DOI] [PubMed] [Google Scholar]
- Valencia A, Sapp E, Kimm JS, McClory H, Ansong KA, Yohrling G, Kwak S, Kegel KB, Green KM, Shaffer SA, Aronin N, DiFiglia M, 2013. Striatal synaptosomes from Hdh140Q/140Q knock-in mice have altered protein levels, novel sites of methionine oxidation, and excess glutamate release after stimulation. J Huntingtons Dis 2, 459–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr., 1985. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 44, 559–577. [DOI] [PubMed] [Google Scholar]
- Walker FO, 2007. Huntington’s disease. Semin. Neurol. 27, 143–150. [DOI] [PubMed] [Google Scholar]
- Wu YN, Munhall AC, Johnson SW, 2011. Mitochondrial uncoupling agents antagonize rotenone actions in rat substantia nigra dopamine neurons. Brain Res. 1395, 86–93. [DOI] [PubMed] [Google Scholar]
- Young D, Mayer F, Vidotto N, Schweizer T, Berth R, Abramowski D, Shimshek DR, van der Putten PH, Schmid P, 2013. Mutant huntingtin gene-dose impacts on aggregate deposition, DARPP32 expression and neuroinflammation in HdhQ150 mice. PLoS One 8, e75108. [DOI] [PMC free article] [PubMed] [Google Scholar]