

SUMMARY
As a non-invasive imaging technology, positron emission tomography (PET) plays a crucial role in personalized medicine, including early diagnosis, patient screening, and treatment monitoring. The advancement of PET research depends on the discovery of new PET agents, which requires the development of simple and efficient radiolabeling methods in many cases. As bioisosteres for halogen and carbonyl moieties, nitriles are important functional groups in pharmaceutical and agrochemical compounds. Here, we disclose a mild organophotoredox-catalyzed method for efficient cyanation of a broad spectrum of electron-rich arenes, including abundant and readily available veratroles and pyrogallol trimethyl ethers. Notably, the transformations not only are compatible with various affordable 12C and 13C-cyanide sources, but also could be applied to carbon-11 synthons to incorporate [11C]nitriles into arenes. The aryl [11C]nitriles can be further derivatized to [11C]carboxylic acids, [11C]amides, and [11C]alkyl amines. The newly developed reaction can serve as a powerful tool for generating new PET agents.
Keywords: Carbon-11, carbon-13, cyanation, dimethoxy arene, nitrile, photoredox catalysis, PET, radiolabeling, trimethoxy arene
Graphical Abstract

We developed a photoredox-catalyzed [11C]cyanide radiolabeling method to fulfill the need for a practical radiochemistry method to incorporate [11C]nitriles into (hetero)arenes. This highly efficient photoredox reaction produces aryl [11C], [12C] and [13C]nitriles using simple cyanide sources and organic photocatalysts. Under blue light (450–460nm) irradiation, naturally abundant and readily available veratroles as well as pyrogallol trimethyl ethers were site- and chemo-selectively converted into valuable methoxy aryl nitriles in up to 99% yield.
INTRODUCTION
Positron emission tomography (PET) is a commonly used functional imaging technique that employs radioactive probes to measure biological changes in a non-invasive manner. Advances in PET research often depends on the development of radiopharmaceuticals, the syntheses of which involve the introduction of radioisotopes into targeting ligands. As one of the most common elements in organic compounds, carbon has a PET isotope, 11C, that is widely used for radiolabeling bioactive molecules. With a short half-life of ~20 min, 11C-labeling reactions generally need to be highly efficient and have rapid kinetics.1 Indeed, various efficient 11C labeling methods have been developed over the past few decades, most of which rely on established synthons based on 11CH3I, 11CH3OTf, 11CO2, 11CO, and [11C]CN.
Among those labeled products, radiolabeled aryl [11C]nitriles have attracted great attention. This is because aryl nitriles are ubiquitous in natural products,2 pharmaceuticals,3,4 pigments, and agrochemical compounds.5 Aryl nitriles have been discovered as inhibitors that interact and bind to various protein residues such as cysteine,6 leucine,7 arginine,7–10 and glutamine residues.10 Moreover, aryl nitriles can serve as intermediates that can be efficiently transformed into other 11C-functional groups including amine, amide, ester, carboxylic acid, tetrazoles, aldehydes, and amidines11 within a half-life decay of 11C.12 Clearly, easy access to aryl [11C]nitriles and their derivatives could greatly benefit PET research by generating a spectrum of radiolabeled agents for evaluation.
Despite significant interest from a radiotracer development standpoint, there have been limited reports on efficient and convenient methods to generate aryl [11C]nitriles, especially general methods for site-selective (radio)cyanation of arenes. As shown in Scheme 1a, representative methods for 11C labeling of arenes were mainly accomplished by employing transition metals such as palladium catalysis13–16, Cu(I) catalysis17,18, or stoichiometric amounts of Cu(II)19,20, which typically required heating [11C]cyanide with the pre-activated aryl precursors to high temperature. The precursors are mostly electron-neutral and deficient aryl halide, aryl-palladium13,16, aryl boronic acids/esters19, and aryl stannanes20 that may involve multi-step synthesis and be complicated to prepare in certain situations. Using significant amounts of metal reagents during the labeling reaction could also complicate purification procedures due to residual metal elemental analysis being required as part of the quality control of radiopharmaceutical synthesis. A more direct radio-cyanation of arenes under metal-free conditions would fill a gap in current PET reaction labeling technology and would benefit the generation of new PET probe agents for biomedical study and drug development.
Scheme 1. Synthetic Approaches to 11C-Benzonitriles.

(a) Conventional transition metal-mediated 11C-cyanation.
(b) Photoredox-mediated 18F-fluorination by acridinium catalysts.
(c) This work: photoredox-catalyzed 11C-cyanation by RFTA catalyst or acridinium catalysts.
In light of the abundance of electron-rich arenes and aryl nitriles in therapeutics21 and given our recent progress in developing photoredox-mediated 18F-fluorination of electron-rich arenes22–24 (Scheme 1b), we explored whether organic photoredox catalysis could mediate 11C-cyanation on electron-rich arenes at the C–O bonds in a site-selective manner via arene cation radical intermediates (Scheme 1c).25 Recently, we found that organic photoredox-catalyzed cation radical-accelerated nucleophilic aromatic substitution (CRA-SNAr) reactions are possible with readily available aryl ether substrates, where the alkoxy group serves as the nucleofuge. Despite the success of these methods, the substrate scope and catalytic efficiency were somewhat limited due to catalyst decomposition by adventitious cyanide. In this study, we developed a photoredox system that not only greatly improves the catalytic efficiency (up to 99% yield), but also expands the reaction substrate scope to more electron-rich arenes (E1/2 < +1.50 V vs SCE)26 such as veratrole and pyrogallol trimethyl ether, derived from catechol, guaiacol, pyrogallol and syringol,27 which are important moieties and functional groups in natural products,28 synthetic drug candidates,28–34 and organic material structures.35,36 This photoredox-catalyzed transformation could be adapted to 11C- and 13C-cyanation for efficient late-stage (radio)labeling on a variety of readily available complex precursors with pyrogallol or veratrole cores.37
RESULTS AND DISCUSSION
Photoredox-catalyzed 12C-cyanation by catalyst S1
In a recent disclosure,25 substrate 1a (pyrogallol trimethyl ether) had low reactivity (18% isolated yield) in the CRA-SNAr cyanation reaction catalyzed by Mes-Acr-Ph+ catalyst (S2, E1/2 = +2.11 V vs SCE)38 due to the electron-rich structure of the trimethoxy functionality. Because one of our goals is to develop efficient new cyanation methods that are compatible with electron rich arenes, we used compound 1a as a model substrate to evaluate the catalytic efficiency. We first screened different photocatalysts using CRA-SNAr cyanation conditions. In brief, acetone cyanohydrin (4 equiv.), substrate 1a (0.1 mmol), and an excess amount of NaHCO3 (5 equiv.) were mixed with a catalytic amount (5 mol%) of photocatalysts and the mixtures were irradiated with 455 nm LEDs for 18 hours. The resulting C–O cyanation product 1b (2,6-dimethoxy benzonitrile) was then visualized by UV-HPLC equipped with a C18 column. As shown in Figure 1 (entries 1–7), riboflavin tetraacetate (RFTA, S1, E1/2 = +1.67 V vs SCE)39 resulted in the highest yield (59.7%, Figure 1, Entry 4) among the catalysts tested. We then screened other organic cyanide and inorganic cyanide sources and found the reaction yields of 2b were not greatly improved (See Table S3 and Table S5 in SI). However, compared with acetone cyanohydrin (ACH), trimethylsilyl cyanide (TMSCN) was found to give fewer byproducts based on HPLC analysis for both substrates 1a and 2a. Thus, TMSCN was then used for further optimization thereafter. It is noteworthy that acetone cyanohydrin remains a viable cyanide alternative as comparable reaction yields were obtained (Figure 1, Entries 13 & 14; Table S3, Entries 1 & 2). Using TMSCN as the cyanide source, the impact of solvents was then investigated for both 1a (Figure 1, Entries 8–12) and 2a (Table S4). We were pleased to find that the use of protic ethanol as the solvent afforded the highest reactivity, while acetonitrile could be employed as a non-protic co-solvent with little deleterious effect, which could aid in the solubility of certain substrates (Figure 1, Entry 12).
Figure 1. Cyanation Reaction Optimizationa.

TMSCN = trimethylsilylcyanide; ACN = acetone cyanohydrin. a The reactions were conducted in 0.1 mmol scale with photocatalyst (5.0 mol%), NaHCO3 (5.0 equiv), and the cyanide source (4.0 equiv) in solvent (1 mL), irradiated with 36 W blue LEDs (450 – 460 nm).
b Yields were determined by HPLC.
We then screened commonly used organic or inorganic basic or acidic additives as well as aqueous buffer solutions and identified that solid or aqueous sodium bicarbonate was optimal additive for the generation of free cyanide in situ, likely by improving the cyanide anion reactivity via its weak basicity and preventing the oxidation of the cyanide anion by the photoredox catalyst (Table S6). Similarly, buffer solutions with weak basicity were also tested using substrate 2a. High yields were observed for the reactions with slightly basic (pH = 8.0 ~ 9.0) buffers (20 μL) in the reaction solutions. No reaction was observed in the absence of the basic additives (Table S6). Heating a catalyst-free mixture of 2a, NaHCO3 and TMSCN in ethanol overnight generated almost none of the desired product (Figure 2, Entry 23). For the most reactive substrate 2a, the catalyst loading can be further reduced to 3.0 mol% without impacting the yield (Figure 2, Entry 17).
Figure 2. Cyanation Reaction Optimizationa.

a The reactions were conducted on a 0.1 mmol scale with photocatalyst (x mol%), NaHCO3 (b equiv.), and the cyanide source (c equiv.) in ethanol (1 mL), irradiated with two 36 W blue LED lights (450 – 460 nm).
b Yields were determined by HPLC on a Phenomenex, Kinetex® 5μm EVO C18 column based on the UV integrations with the standard curves’ correlation of compound 2a and 2b on the same HPLC.
c ln dark, reaction vial covered with aluminum foil.
Having identified the optimal reaction conditions, the substrate scope and functional group tolerance were further examined. As shown in Figure 3, various 5-position functionalized 1,2,3-trimethoxy benzenes were converted into the 2,6-dimethoxy aryl nitriles with minimal side reactions and remarkably high isolated yields. A series of functional groups such as tertiary amines (3a), bromides (4a), nitriles (5a), ketones (14a), esters (15a), benzoates (16a), and even alkenes (6a), were tolerated. Notably, a number of reactive benzylic functional groups such as benzyl chlorides (8a), nitriles (9a), alcohols (10a), ethers (11a), Boc protected amines (12a), and azides (13a), were also tolerated in the reaction regardless of potential competing SN2 and oxidization reactions, and provided the desired demethoxycyanation products with good to excellent isolated yields (61.0%–97.0%). The broad substrate scope made the 2,6-dimethoxy aryl nitrile moiety a good building block to analogs of a number of synthetic bioactive complex compounds.
Figure 3. Substrate Scope (simple molecules) for [12C]-Cyanation by Catalyst S1a.

a Isolated yields using standard reaction conditions (0.1 or 0.2 mmol) unless otherwise noted.
b 2.0 mmol scale reaction (followed general procedure 2 in the SI).
c MeCN/EtOH (v/v = 1/1, 0.1M) used as the solvent.
d Acetone cyanohydrin (ACH) was used as the CN source.
e Yields and product ratios were determined by the 1H NMR analysis.
f Yield was estimated by HPLC.
g 5 mmol scale reaction (followed general procedure 3 in the SI).
h 4 mmol scale reaction (followed general procedure 4 in the SI).
i From 1,3,5 trimethoxy benzene (26a).
j From 1,5-dimethoxy 2-isoproxy benzene (1a’).
k NaCN (3 equiv) and S1 (7.5 mol%) in H2O/EtOH (9/1) were used.
Single-site-activated substrates such as 18a, 19a, and 20a also produced the desired cyanation products with high yields site selectively. Notably, although unconverted 20a was observed when 2.5 equivalents of TMSCN was used in the standard conditions, a larger scale reaction with additional TMSCN (3.0–4.0 equiv.) or ACH (4.0 equiv.) and extended reaction times (96 h) increased yields to 95.0%. Penta-substituted substrate 21a favored the formation of 21b to 21c (21b:21c = 4:1).
The standard conditions were also successfully applied to 3,4-dimethoxy-N-boc-tyramine (22a), leading to 43% of the demethoxycyanation products and 37% of the C-H cyanation product at the C5 position. Additional examples (23a, 24a and 28a) involving the veratrole motif or 1,4-dimethoxy benzene (27a) also demonstrated the preferential formation of demethoxycyanation adducts over the C-H cyanation products. Ethoxy (29a) and isopropoxy (30a) leaving groups were unsurprisingly found to be less reactive than methoxy substrates. The electronic influence of the fluorine atom surprisingly resulted in a dramatic difference on the demethoxycyanation reactivity. The 2-fluorine-4-methoxy benzonitrile product (31b) was generated as the major cyanation product in over 80.0% yield. Methoxy naphthalenes (33a & 34a) afforded C-H cyanation products, which matched with our previous report on photoredox-catalyzed C-H cyanation.40 A small amount of demethoxycyanation products were observed for hetero arenes such as 2,3-dimethoxy pyridine (32a), methoxy naphthalene (35a), and 5-methoxy quinoline (36a), while only moderate C-H cyanation product formation was observed for 6-methoxy quinoline (37a).
With these promising results in hand, we subsequently performed site- and chemo-selective cyanation for late-stage functionalization (Figure 4) of existing natural product molecules bearing the electron-rich pyrogallol cores. Most of these natural products have relatively narrow therapeutic indices and increased antibiotic resistance. Efforts have been made to modify these molecules to reduce their toxicity and drug resistance. Selectively tuning one of the three methoxy groups may refine their structure-activity relationship (SAR), which has been relatively unexplored thus far. Among the tested molecules, N-Boc-glycine-podophyllotoxin (38a), was recently disclosed to possess potent anti-cancer activity.41 Our photoredox-catalyzed reaction introduced the nitrile group to 38a site-selectively, leading to 38b in 35.7% isolated yield. Colchicine (39a) is a natural product agent used to induce polyploidy in plant cells and an alternative agent for treating gout and inflammation in a variety of diseases.42–44 Alkoxyarene 39a was directly converted to the desired aryl nitrile analog (39b) in good yield (57.0%) by controlling the reaction at room temperature, which would greatly reduce side reactions such as photoisomerization and possible thermal decomposition.
Figure 4. Late-Stage Functionalization via [12C]-Cyanation by Catalyst S1a.

a Isolated yields using standard reaction conditions (0.1 or 0.2 mmol) unless otherwise noted.
b MeCN/EtOH (v/v = 1/1, 0.1M) used as the solvent.
c Reaction run at 22 °C for 60 h with 10 mol% S1 and 4 equiv of TMSCN.
d 0.05 mmol 38a with 5% mol S1, 4 equiv of TMSCN at 22 °C for 22 hours.
e S2 (10%) used as photocatalyst; HPLC estimated yield.25
Guanidine compounds have been found to be useful cardiac PET imaging agents.45 Under our standard reaction condition, we could introduce nitrile group onto the guanidine precursor (40b) in good yields (53.0%), suggesting the reaction may be used to produce 11C labeled benzyl guanidine analogs efficiently. Troxipide is a marketed drug for gastroesophageal reflux disease.46 Recent studies have found that troxipide’s analogs have anti-tumor properties.47 N-Boc-protected troxipide (41a) was functionalized with complete site selectivity to afford the corresponding aryl nitrile (41b) in 82.0% yield. Further Boc deprotection was also carried out without any impact on the nitrile group. Trimethoprim is a synthetic diaminopyrimidine antibiotic and a folate synthesis inhibitor used to treat infection48 and may also be used in combination with other drugs to treat pneumocystis pneumonia in HIV-infected patients.49 Site-selective cyanation of the trimethoprim diacetate 42b on the aromatic ring led to the nitrile product 42b with an excellent yield of 84.0%. The aryl nitrile analog of trimethoprim was successfully obtained by subsequent acetate deprotection in high yield.
To demonstrate the cyanation reaction can be used in preparative scale, we conducted larger scale reactions (200 mg-1.0 g) in round bottom flasks or flow devices (quartz chip). Scale-up reactions for 1a, 2a, 3a, 6a, 16a, 20a, 21a, 25a were performed using blue LED-irradiated round bottom flasks (Figure 3 and also see Figure S2 for the reaction setup). All substrates maintained similar yields after scale-up. The yields were further increased by adding an additional cyanide source and prolonging the irradiation time, except for 25a. Substrates 4a and 5a (2.0 mmol) were converted into the corresponding aryl nitriles in 88% (14 h) and 68% (24 h) isolation yield, respectively, in the flow devices (quartz chip, Scheme 2) with good catalytic efficiency. As a proof-of-principle example, acyl chloride 6d was generated in high yield from the crude photocyanation reaction mixture containing 6b, which was then converted to 6e, a nitrile analog of a cardio-cerebral vascular drug cinepazide in an overall 37.6% isolated yield. Similarly, acyl chloride 16d was be easily obtained by hydrolysis of ester 16b in basic methanol solution, which was then converted into 16e as a cyanide analog of trimebutine in 87.5% yield.
Scheme 2. Cyanation Scale Up and Synthetic Applications.

a. Followed general procedure 5 in the SI.
b The crude mixture was used without further purification.
Photoredox-catalyzed 13C-demethoxycyanation by catalyst S1
13C labeling is one of the stable isotope labeling techniques that can be utilized with mass spectrometry to help better understand cellular metabolism in living organisms.50 13C-enriched reagents have been used in hyperpolarized carbon-13 magnetic resonance imaging (MRI).51 These isotopologs provide chemical and spatial information, offering a probe for specific metabolic pathways. Despite progress, there remains a need to develop new and efficient l3C labeling methods for highly functionalized molecules.52,53 We also explored the use of our novel photoredox method to synthesize aryl [13C]nitriles. Compared with recently reported transition metal-catalyzed cyanide/[13C]cyanide exchange reactions,52,53 our method is metal-free, higher yielding, and affords 100% [13C]CN enriched products. For example, [13C]2b was isolated in a good yield (52%) when 10.0 mol% of S1 and 2.0 equiv of [13C]KCN were used. Encouragingly, the yield could be increased to up to 99% when 3.0 equivalents of [13C]KCN were added and irradiation time extended to 3 days. When [13C]TMSCN was used as the [13C]cyanide source, the corresponding carbon-13 enriched adducts [13C]1b, [13C]15b, [13C]39b, and [13C]42b were all forged in high isolated yields (Scheme 3).
Scheme 3. 13C-Cyanation of Methoxyarenes.

a Reactions were carried out with a cooling fan at 22 °C for 60 h.
b 72 h reaction with 3 equiv of K13CN.
Photoredox-catalyzed 11C-cyanation by catalyst S1
The ability to perform photoredox-catalyzed 11C-cyanation would be valuable since it could lead to new PET agents for various applications. Despite success in the aforementioned 12/13C-demethoxycyanation reactions, we perceived it could be challenging to directly perform 11C-radiocyanation by adopting these conditions because 1) only trace amounts of [11C]CN− can be used, compared with an excess of CN− in the 12/13C-demethoxycyanation; 2) the reaction requires fast kinetics (generally within 10 min) due to the short half-life of 11C, and 3) the best cyanide source, [11C]TMSCN, is difficult to generate rapidly in situ. Since TBA+[11C]CN− can be prepared by trapping [11C]HCN in weak basic solutions (See Section 5 in the SI), we initially explored 11C-demethoxycyanation conditions using TBA+[11C]CN.
Indeed, TBA+[11C]CN− reacted with 1a (0.03 mmol, 0.06 M) in the presence of S1 (2.0 mg, 12.2 mol%) to afford the expected product in 38% decay corrected radiochemical conversion (RCC) in 4 min (Figure 5, Entry 2). After an additional two min of LED irradiation, the resulting radioproduct increased to 50%. Previous studies from our groups demonstrated that laser irradiation (450 nm) improved RCCs in photoredox-catalyzed 18F-arene deoxyfluorination reactions22 and direct 18F-fluorination reactions of arenes.24 When we carried out the same reaction with 450 nm laser irradiation for 6 min, the RCC increased to 91.6±1.8% (Figure 5, Entry 1, N=3). For model substrate 1a, the laser irradiation time did not significantly affect the RCC (cf. 4 min, Entry 4; and 8 min, Entry 5). Replacing the N2 atmosphere with O2 did not change the RCC but led to more UV impurities (Figure 5, Entry 6). No reaction was observed in the absence of photocatalyst S1, confirming that the photocatalyst was necessary 11C-demethoxycyanation (Figure 5, Entry 10).
Figure 5. Optimization for Arene Demethoxy 11C-Radiocyanationa.

a The labeling reactions were conducted on a 0.03 mmol scale under 450 nm laser irradiation unless otherwise noted. The 11C-labeled product mixture aliquots were injected and analyzed by an HPLC equipped with both UV and radio detectors. All RCCs were decay corrected.
We assessed the substrate scope (Figure 6) using optimized labeling conditions in Figure 5, Entry 1. For most substrates, the RCCs of 11C-radiocyanation were high, correlating well with the photoredox-catalyzed 12C-cyanation. Although 5b was isolated in over 60% yield in the corresponding 12C-cyanation reaction, minimal formation of [11C]5b was observed in 11C labeling reaction. Similarly, compared with 94% yield of 4b, substrate 4a (0.03 mmol) was not reactive under standard 11C-labeling conditions. Interestingly, reducing the substrate loading from 0.03 mmol to 0.01 mmol led to greatly improved radiolabeling yields (26.1±5.2%, N=3). Aniline and mono-N-Boc protected aniline were unreactive substrates in this transformation, likely due to the high acidity of their respective cation radical species. In contrast, the doubly-protected 3,4,5-trimethoxy aniline (3a, 0.03 mmol) was converted into [11C]3b with a moderate RCC (36.5±2.4%, N=3). Decreasing 3a loading to 0.01 mmol resulted in an increase in RCC to 49.1% (N=1). Benzyl amines are compatible with the photoredox radiocyanation reactions, as N-Boc protected 3,4,5-trimethoxyl benzyl amine 12a (0.01 mmol) was converted into [11C]12b with an excellent RCC (89.0±4.5%, N=3).
Figure 6. Substrate Scope (simple molecules) for 11C-Cyanation by Catalyst S1a.

a Following general labeling procedure 1 or 2; the radiochemical conversion (RCC) was reported as average±SD% of reactions run in triplicate unless otherwise noted.
b From 1,5-dimethoxy 2-isoproxy benzene (1a’).
c Following the SN2 labeling reaction procedure in the SI.
d Blue LED irradiation.
e The labeling reaction was carried out with a flow device irradiated under a blue LED lamp.
f 0.05 mmol precursor was used.
g From 1,3,5 trimethoxy benzene (26a).
h Catalyzed by S3 (4-CzIPN, 2.4 mg).
i Catalyzed by S2 (Mes-Acr, 1.5 mg).
We investigated chemoselectivity between a typical SN2 radiolabeling reaction and the photoredox-catalyzed demethoxy radiocyanation using benzylic chloride substrate 8a. Thermal SN2 labeling conditions led to trimethoxybenzyl [11C]nitrile [11C]9a in a moderate RCC of 29.1% (N=1), and the photoredox-catalyzed radiocyanation conditions resulted in [11C]8b in a RCC of 61.9±3.9% (N=3), with no benzylnitrile product [11C]9a. Highly reactive 11C-labeled products such as benzyl chloride ([11C]8b), benzyl alcohol ([11C]10b), and benzyl azide ([11C]13b) could also serve as 11C-intermediates that can be conjugated with bioactive molecules with corresponding reactive counterparts, such as alcohols, carboxylic acids, dibenzylcyclooctyne, and strained alkenes, to synthesize additional radiolabeled bioactive agents.
Compared with the previous 12C-cyanation reactions, we observed greatly increased preference of demethoxy 11C-radiocyanation (leading to [11C]22b, [11C]23b, [11C]24b, [11C]27b, and [11C]28b) over the C-H 11C-radiocyanation reactions (leading to [11C]22c, [11C]23c, [11C]24c, [11C]27c, and [11C]28c). The use of methoxynaphthylene and methoxy quinoline substrates mainly led to C-H 11C-cyanation adducts ([11C]33c – [11C]37c) similar to that observed in the 12C-chemistry. Because LED light sources are more affordable than lasers, we also tested LED-promoted labeling under the standard conditions. For the most reactive substrate 2a, the RCC of [11C]2b was >90%, which was comparable to laser irradiation. However, laser irradiation was more efficient when less reactive substrate 20a was tested.
With these encouraging results on hand, we then explored late-stage 11C-radiocyanation of bioactive substrates. As shown in Figure 7, [11C]39b and [11C]42b were obtained in 62.2±6.1% (N=3) and 72.9±2.2% (N=7) RCCs, respectively. The protecting groups in [11C]42b were easily removed in 95.2% yield, leading to a close analog of a bioactive agent, trimethoprim. In addition to [11C]39b and [11C]42b, other 11C-labeled product, [11C]38b, [11C]40b, and [11C]41b were also obtained with moderate RCCs. The aryl [11C]-nitriles can also be further derivatized to other functional groups. For example, [11C]-amide [11C]1d could be obtained either from the isolated [11C]1b (76.3±14.0% RCC, N=3), or through a one-pot method (81.4±1.7% RCC, N=2) that will greatly shorten the synthesis from 1a. And the Am of [11C]1b was also determined to be 84.8± 7.0 GBq/μmol (N=4).
Figure 7. 11C-Cyanation of Bioactive Molecules by Catalyst S1a and [11C]Nitrile Hydrolysis.

a Following general labeling procedure 1 or 2; the radiochemical conversion (RCC) was reported as average±SD% of reactions run in triplicate unless otherwise noted.
b Blue LED irradiation.
c The labeling reaction was carried out with a flow device irradiated under a blue LED lamp.
d Catalyzed by S3 (4-CzIPN, 2.4 mg).
e Catalyzed by S2 (Mes-Acr, 1.5 mg).
f From Trimethoprim.
g Non-decay corrected isolated radiochemical yield (RCY) with 70 – 110 MBq nitrile product collected.
h 11.1 GBq TBA+[11C]CN− was used. (General labeling procedure 3 in the SI)
Overall, our results demonstrate the potential of using site-selective late-stage 11C-radiolabeling strategy for PET probe development based on existing natural products and drug candidates bearing electron-rich arene moieties. The low precursor loading in these reactions also simplified the purification process. Moreover, the corresponding carbon-12 authentic standards could be prepared using the same photoredox-catalyzed demethoxy 12C-cyanation reaction with moderate to very high yields. Automation of this newly developed method would greatly facilitate its adoption at different PET centers and is a current focus in our laboratories. Because TBA+[11C]CN− can be easily obtained from cyclotron/procab produced [11C]CN−, automation of the photoredox reaction would be key for this process.
Photoredox-catalyzed 11C-cyanation by catalysts S2 and S3
In addition to S1, we also explored the use of other catalysts for 11C-radiocyanation. We found S3 (4-CzIPN) could also carry out the demethoxy 11C-radiocyanation of 1a (0.006 mmol, 0.012 M), 2a (0.01 mmol, 0.02 M), and 41a (0.02 mmol, 0.04 M) in 73.1%, 95.1%, and 20.9% RCC, respectively (Figure 6). However, no desired 11C-radioproducts were detected when other bioactive molecules such as colchicine 39a and trimethoprim diacetate 42a were tested (Figure 7). We recently found that acridinium photocatalyst S2 could promote demethoxy 11C-cyanation of over 20 arenes with an array of functional groups. Even though the yields may be lower, photocatalyst S2 may have a broader substrate scope as it is more oxidizing than S1. To validate this hypothesis, we randomly selected 19 substrates in Figure 6 and Figure 7 along with new substrates 43a-65a for demethoxy 11C-radiocyanation reactions. As shown in Figure 8, although the RCCs were relatively lower for the highly electron rich arenes, S2 has a broader substrate scope than S1. For nitrile 5a, [11C]5b was the major radio product when using catalyst S2, while it was a minor product when catalyst S1 was used. Electron-rich substrates (43a, 44a, 46a, 47a, 50a, 57a, and 59a) were successfully labeled using S2, but failed to yield any significant product using S1 as the catalyst. For S2-catalyzed 11C-labeling reactions, the regioselectivity was identical to those obtained in 12C-cyanation conditions. Among other reactions tested, no C-H 11C-radiocyanation product was observed except for substrate 61a, a letrozole analog, which led to 18.1 % RCC of C-H cyanation product ([11C]61c) and 12.6% RCC of the demethoxy 11C-cyanation product ([11C]61b). Substrates 62a and 63a could also be labeled with 11C albeit in reduced RCCs, while substrate 49a, 60a, 64a, and 65a were all carefully examined and found to afford almost no desired 11C-radiolabeling products though they all demonstrated good reactivity when treated with TMSCN.25
Figure 8. Substrate Scope for [11C]-Cyanation by Catalyst S2a.

a The labeling reactions were conducted by using general labeling procedure 3 or 4; the RCC was reported as average±SD% of reactions run in triplicate unless otherwise noted.
b Blue LED irradiation.
c The labeling reaction was carried out with a flow device with LED irradiation.
d 1,3,5 trimethoxy benzene was used as the precursor.
e S1 used as catalyst (2.0 mg).
PET imaging study
In drug discovery, radiolabeled lead compounds or their analogs can be further evaluated in vivo to explore their bioactivity,54 and to measure the pharmacodynamics and pharmacokinetics of the drug candidates.55 Using our newly developed method, 11C-labeled aryl nitriles can be prepared for distribution studies.56 Previously, colchicine (39a) was reported to be excreted primarily through the biliary system, intestines, and kidneys with certain toxicity.57 In this report, we successfully synthesized a nitrile analog of colchicine ([11C]39b) using our 11C-cyanation catalyzed by S1. In a proof-of-concept study, we performed small animal PET study using [11C]39b to evaluate its distribution profile in vivo. As shown in Figure 9, [11C]39b showed apparent uptake in liver, intestines, and kidneys, which was similar to the reported patter of 39a (See detailed PET imaging ROIs analysis in Figure S8 in the SI).
Figure 9. Small animal PET Imaging Study of [11C]39b.
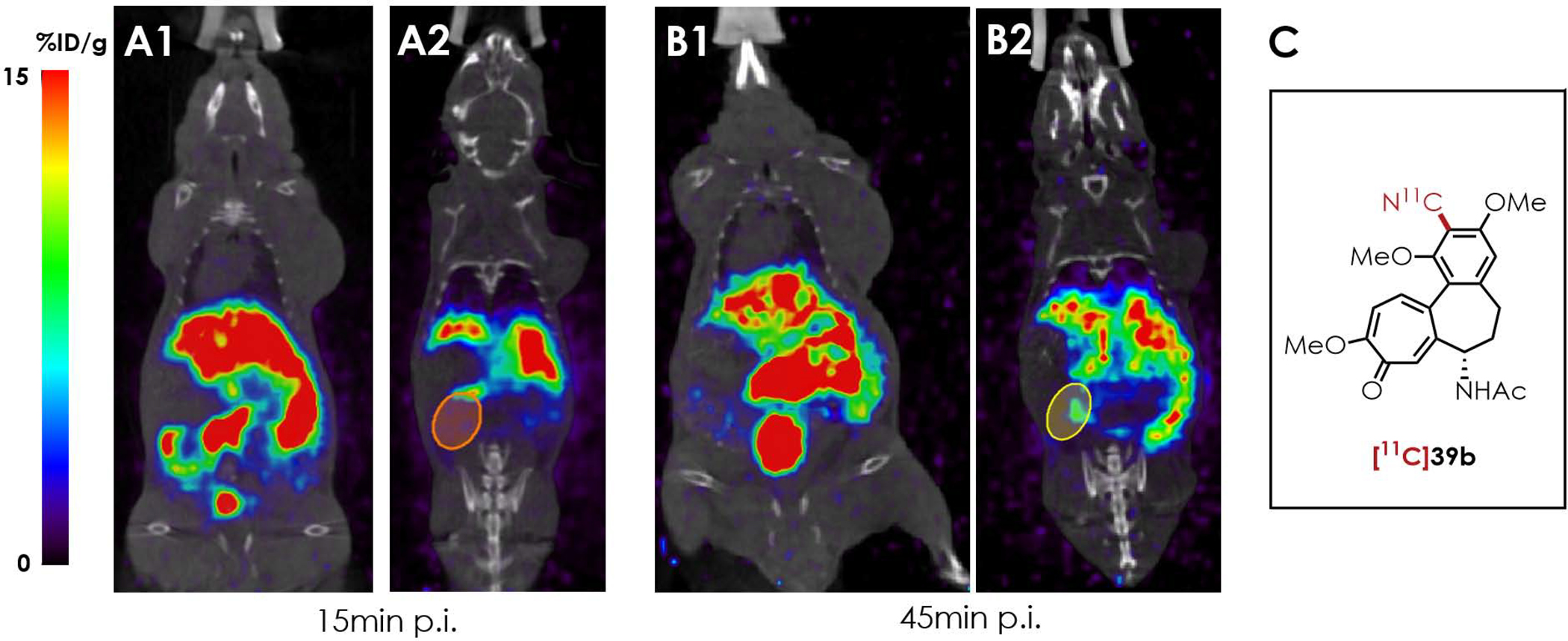
PET/CT image was acquired at 15 min (A1 and A2) and 45 min (B1 and B2) post intravenous tail vein injection of [11C]39b. A1 and B1 shows the liver coronal plane. A2 and B2 shows the kidney coronal plane with the kidney circled. (C) Chemical structure of [11C]39b.
CONCLUSIONS
We have developed a simple and efficient photoredox-catalyzed radiolabeling method for cyanation of a variety of electron-rich substrates with various functionalities. In particular, the reaction system based on RFTA (S1) could be used to synthesize electron rich aryl nitriles from [12C], [13C], and [11C]cyanide sources and easily available simple molecules, complex natural products and pharmaceuticals. The reaction system based on catalyst S2 affords lower yields but lends a broader substrate scope. Considering the importance of aryl nitriles and their ability to be further derivatized, our simple late-stage 11C-radiocyanation strategy enables the convenient transformation of off-the-shelf electron-rich aryl therapeutics into their aryl nitrile analogs as well as 11C-labeled radiopharmaceuticals for PET applications.
EXPERIMENTAL PROCEDURES
Full experimental procedures can be found in the supplemental information.
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, David A. Nicewicz (nicewicz@unc.edu) and Zibo Li (ziboli@med.unc.edu)
Materials availability
Reagents generated in this study will be made available on request, but we may require a payment and/or a completed materials transfer agreement if there is potential for commercial application.
Data and code availability
There is no dataset or code associated with this publication. Details of methods, experimental procedures, and characterization data are available in the supplemental information. Nuclear magnetic resonance (NMR) spectra are available.
Supplementary Material
THE BIGGER PICTURE.
Isotopic labeling is utilized to track bioavailability, metabolism and uptake studies of active pharmaceutical ingredients (API) in vivo. APIs with 11C isotopes are particularly useful for applications to PET imaging, though represents a significant synthetic challenge due to the short half-life (11C ~20 min). Though there are reports on developing PET agents based on alkyl [11C]nitriles, efficient and facile radio-cyanation of arenes, particularly electron-rich arenes, is an unmet need. The metal-free organic photoredox-catalyzed radiocyanation method reported herein introduces a [11C]nitrile with high radiochemical conversion (RCC), which can be further elaborated to functional groups such as [11C]carboxylic acids, [11C]amides, and [11C]alkyl amines. This strategy could result in easy access to new PET agents that are previously difficult to synthesize from readily available precursors.
Highlights.
Highly efficient photocatalyzed (radio)cyanation method
Site-selective and chemo-selective cyanation of various electron-rich arenes
Late-stage functionalization and aryl nitrile product derivatives
Mild conditions, affordable organocatalyst, versatile cyanide source and aryl substrates
ACKNOWLEDGMENTS
This work was supported in part by the National Institutes of Health (NIBIB) grants R01EB029451 (Z.L. and D.A.N.) and 5R01CA233904 (Z.L.), UNC LCCC pilot grant (Z.L. and D.A.N.), grant 1S10OD023611 (Z.L.), 1R01DK128447-01A1 (Z.W.) and the startup fund from UNC Department of Radiology, Biomedical Research Imaging Center (BRIC), and UNC Lineberger Comprehensive Cancer Center (Z.L.). We thank the University of North Carolina’s Department of Chemistry Mass Spectrometry Core Laboratory (CMSCL), especially Dr. Weatherspoon (CMSCL) and Dr. He Zhang (BRIC), for their assistance with mass spectrometry analysis. In addition, we thank Dr. Weiling Zhao (BRIC) for editing and proof reading the manuscript and thank Dr. Benjamin C. Giglio (BRIC) for the generous help on [11C]HCN production.
Footnotes
DECLARATION OF INTERESTS
The authors Z.L., D.A.N. and W.C. have filed a WO patent (patent applicant, the University of North Carolina at Chapel Hill, USA; inventors, Z.L, D.A.N and W.C.; patent no. WO 2020176804) related to part of the labelling methodology in this manuscript and is under review. Z.L. and D.A.N. are the cofounders of LED Radiofluidics. D.A.N. is a member of Chem’s advisory board. The remaining authors declare no competing interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCE
- 1.Ametamey SM, Honer M, and Schubiger PA (2008). Molecular imaging with PET. Chem Rev 108, 1501–1516. 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]
- 2.Xu Y, and Qu W (2021). [11C]HCN Radiochemistry: Recent Progress and Future Perspectives. European Journal of Organic Chemistry 2021, 4653–4682. 10.1002/ejoc.202100651. [DOI] [Google Scholar]
- 3.Fleming FF, Yao L, Ravikumar PC, Funk L, and Shook BC (2010). Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore. J Med Chem 53, 7902–7917. 10.1021/jm100762r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Wang Y, Li X, Yu Z, Song C, and Du Y (2021). Nitrile-containing pharmaceuticals: target, mechanism of action, and their SAR studies. RSC Med Chem 12, 1650–1671. 10.1039/d1md00131k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larock RC (1999). Comprehensive organic transformations : a guide to functional group preparations, 2nd Edition (Wiley-VCH; ). [Google Scholar]
- 6.Babine RE, and Bender SL (1997). Molecular Recognition of Proteinminus signLigand Complexes: Applications to Drug Design. Chem Rev 97, 1359–1472. 10.1021/cr960370z. [DOI] [PubMed] [Google Scholar]
- 7.Li JJ, Iula DM, Nguyen MN, Hu LY, Dettling D, Johnson TR, Du DY, Shanmugasundaram V, Van Camp JA, Wang Z, et al. (2008). Rational design and synthesis of 4-((1R,2R)-2-hydroxycyclohexyl)-2(trifluoromethyl)benzonitrile (PF-998425), a novel, nonsteroidal androgen receptor antagonist devoid of phototoxicity for dermatological indications. J Med Chem 51, 7010–7014. 10.1021/jm8009316. [DOI] [PubMed] [Google Scholar]
- 8.Okamoto K, Matsumoto K, Hille R, Eger BT, Pai EF, and Nishino T (2004). The crystal structure of xanthine oxidoreductase during catalysis: implications for reaction mechanism and enzyme inhibition. Proc Natl Acad Sci U S A 101, 7931–7936. 10.1073/pnas.0400973101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bohl CE, Gao W, Miller DD, Bell CE, and Dalton JT (2005). Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc Natl Acad Sci U S A 102, 6201–6206. 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou HB, Lee JH, Mayne CG, Carlson KE, and Katzenellenbogen JA (2010). Imaging progesterone receptor in breast tumors: synthesis and receptor binding affinity of fluoroalkyl-substituted analogues of tanaproget. J Med Chem 53, 3349–3360. 10.1021/jm100052k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan G, Zhang Y, and Wang J (2017). Recent Advances in the Synthesis of Aryl Nitrile Compounds. Advanced Synthesis & Catalysis 359, 4068–4105. 10.1002/adsc.201700875. [DOI] [Google Scholar]
- 12.Rappoport Z (1970). The chemistry of the cyano group (Interscience Pub.). [Google Scholar]
- 13.Zhao W, Lee HG, Buchwald SL, and Hooker JM (2017). Direct (11)CN-Labeling of Unprotected Peptides via Palladium-Mediated Sequential Cross-Coupling Reactions. J Am Chem Soc 139, 7152–7155. 10.1021/jacs.7b02761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nordeman P, Chow SY, Odell AF, Antoni G, and Odell LR (2017). Palladium-mediated (11)C-carbonylations using aryl halides and cyanamide. Org Biomol Chem 15, 4875–4881. 10.1039/c7ob01064h. [DOI] [PubMed] [Google Scholar]
- 15.Liu L, Xu Y, Shea C, Fowler JS, Hooker JM, and Tonge PJ (2010). Radiosynthesis and bioimaging of the tuberculosis chemotherapeutics isoniazid, rifampicin and pyrazinamide in baboons. J Med Chem 53, 2882–2891. 10.1021/jm901858n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee HG, Milner PJ, Placzek MS, Buchwald SL, and Hooker JM (2015). Virtually instantaneous, room-temperature [(11)C]-cyanation using biaryl phosphine Pd(0) complexes. J Am Chem Soc 137, 648–651. 10.1021/ja512115s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ponchant M, Hinnen F, Demphel S, and Crouzel C (1997). [11C]copper(I) cyanide: a new radioactive precursor for 11C-cyanation and functionalization of haloarenes. Applied Radiation and Isotopes 48, 755–762. 10.1016/S0969-8043(96)00299-0. [DOI] [Google Scholar]
- 18.Ma L, Placzek MS, Hooker JM, Vasdev N, and Liang SH (2017). [(11)C]Cyanation of arylboronic acids in aqueous solutions. Chem Commun (Camb) 53, 6597–6600. 10.1039/c7cc02886e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang L, Brooks AF, Makaravage KJ, Zhang H, Sanford MS, Scott PJH, and Shao X (2018). Radiosynthesis of [(11)C]LY2795050 for Preclinical and Clinical PET Imaging Using Cu(II)-Mediated Cyanation. ACS Med Chem Lett 9, 1274–1279. 10.1021/acsmedchemlett.8b00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makaravage KJ, Shao X, Brooks AF, Yang L, Sanford MS, and Scott PJH (2018). Copper(II)-Mediated [(11)C]Cyanation of Arylboronic Acids and Arylstannanes. Org Lett 20, 1530–1533. 10.1021/acs.orglett.8b00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haziq Qureshi M, R.W., and Marshall Christopher (2022). Top 200 Small Molecule Pharmaceuticals by Retail Sales in 2021. In https://njardarson.lab.arizona.edu/sites/njardarson.lab.arizona.edu/files/Top%20200%20Small%20Molecules%202021V3.pdf, ed.
- 22.Tay NES, Chen W, Levens A, Pistritto VA, Huang Z, Wu Z, Li Z, and Nicewicz DA (2020). (19)F- and (18)F-Arene Deoxyfluorination via Organic Photoredox-Catalysed Polarity-Reversed Nucleophilic Aromatic Substitution. Nat Catal 3, 734–742. 10.1038/s41929-020-0495-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen W, Wang H, Tay NES, Pistritto VA, Li KP, Zhang T, Wu Z, Nicewicz DA, and Li Z (2021). Arene radiofluorination enabled by photoredox-mediated halide interconversion. Nat Chem. 10.1038/s41557-021-00835-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen W, Huang Z, Tay NES, Giglio B, Wang M, Wang H, Wu Z, Nicewicz DA, and Li Z (2019). Direct arene C-H fluorination with (18)F(−) via organic photoredox catalysis. Science 364, 1170–1174. 10.1126/science.aav7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holmberg-Douglas N, and Nicewicz DA (2019). Arene Cyanation via Cation-Radical Accelerated-Nucleophilic Aromatic Substitution. Org Lett 21, 7114–7118. 10.1021/acs.orglett.9b02678. [DOI] [PubMed] [Google Scholar]
- 26.Popp JL, and Kirk TK (1991). Oxidation of methoxybenzenes by manganese peroxidase and by Mn3+. Arch Biochem Biophys 288, 145–148. 10.1016/0003-9861(91)90176-j. [DOI] [PubMed] [Google Scholar]
- 27.Luo H, Weeda EP, Alherech M, Anson CW, Karlen SD, Cui Y, Foster CE, and Stahl SS (2021). Oxidative Catalytic Fractionation of Lignocellulosic Biomass under Non-alkaline Conditions. J Am Chem Soc 143, 15462–15470. 10.1021/jacs.1c08635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li L, Jiang S, Li X, Liu Y, Su J, and Chen J (2018). Recent advances in trimethoxyphenyl (TMP) based tubulin inhibitors targeting the colchicine binding site. Eur J Med Chem 151, 482–494. 10.1016/j.ejmech.2018.04.011. [DOI] [PubMed] [Google Scholar]
- 29.Wen Z, Xu J, Wang Z, Qi H, Xu Q, Bai Z, Zhang Q, Bao K, Wu Y, and Zhang W (2015). 3-(3,4,5-Trimethoxyphenylselenyl)-1H-indoles and their selenoxides as combretastatin A-4 analogs: microwave-assisted synthesis and biological evaluation. Eur J Med Chem 90, 184–194. 10.1016/j.ejmech.2014.11.024. [DOI] [PubMed] [Google Scholar]
- 30.Liu R, Huang M, Zhang S, Li L, Li M, Sun J, Wu L, Guan Q, and Zhang W (2021). Design, synthesis and bioevaluation of 6-aryl-1-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazoles as tubulin polymerization inhibitors. Eur J Med Chem 226, 113826. 10.1016/j.ejmech.2021.113826. [DOI] [PubMed] [Google Scholar]
- 31.Yan J, Chen J, Zhang S, Hu J, Huang L, and Li X (2016). Synthesis, Evaluation, and Mechanism Study of Novel Indole-Chalcone Derivatives Exerting Effective Antitumor Activity Through Microtubule Destabilization in Vitro and in Vivo. J Med Chem 59, 5264–5283. 10.1021/acs.jmedchem.6b00021. [DOI] [PubMed] [Google Scholar]
- 32.Li W, Yin Y, Yao H, Shuai W, Sun H, Xu S, Liu J, Yao H, Zhu Z, and Xu J (2018). Discovery of novel vinyl sulfone derivatives as anti-tumor agents with microtubule polymerization inhibitory and vascular disrupting activities. Eur J Med Chem 157, 1068–1080. 10.1016/j.ejmech.2018.08.074. [DOI] [PubMed] [Google Scholar]
- 33.Wu CJ, Wu JQ, Hu Y, Pu S, Lin Y, Zeng Z, Hu J, and Chen WH (2021). Design, synthesis and biological evaluation of indole-based [1,2,4]triazolo[4,3-a] pyridine derivatives as novel microtubule polymerization inhibitors. Eur J Med Chem 223, 113629. 10.1016/j.ejmech.2021.113629. [DOI] [PubMed] [Google Scholar]
- 34.Friesen DE, Barakat KH, Semenchenko V, Perez-Pineiro R, Fenske BW, Mane J, Wishart DS, and Tuszynski JA (2012). Discovery of small molecule inhibitors that interact with gamma-tubulin. Chem Biol Drug Des 79, 639–652. 10.1111/j.1747-0285.2012.01340.x. [DOI] [PubMed] [Google Scholar]
- 35.Han J, Chiu A, Ganley C, McGuiggan P, Thon SM, Clancy P, and Katz HE (2021). 3,4,5-Trimethoxy Substitution on an N-DMBI Dopant with New N-Type Polymers: Polymer-Dopant Matching for Improved Conductivity-Seebeck Coefficient Relationship. Angewandte Chemie International Edition 60, 27212–27219. 10.1002/anie.202110505. [DOI] [PubMed] [Google Scholar]
- 36.Srivishnu KS, Banerjee D, Ramnagar RA, Rathod J, Giribabu L, and Soma VR (2021). Optical, Electrochemical, Third-Order Nonlinear Optical Investigations of 3,4,5-Trimethoxy Phenyl Substituted Non-Aqueous Phthalocyanines. Front Chem 9, 713–939. 10.3389/fchem.2021.713939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang Z, Ji X, and Lumb JP (2021). Total Synthesis of (S)-Cularine via Nucleophilic Substitution on a Catechol. Org Lett 23, 236–241. 10.1021/acs.orglett.0c04000. [DOI] [PubMed] [Google Scholar]
- 38.White AR, Wang L, and Nicewicz DA (2019). Synthesis and Characterization of Acridinium Dyes for Photoredox Catalysis. Synlett 30, 827–832. 10.1055/s-0037-1611744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muhldorf B, and Wolf R (2015). Photocatalytic benzylic C-H bond oxidation with a flavin scandium complex. Chem Commun (Camb) 51, 8425–8428. 10.1039/c5cc00178a. [DOI] [PubMed] [Google Scholar]
- 40.McManus JB, and Nicewicz DA (2017). Direct C-H Cyanation of Arenes via Organic Photoredox Catalysis. J Am Chem Soc 139, 2880–2883. 10.1021/jacs.6b12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao Y, Li D, Wei M, Du R, and Yan Z (2021). The ester derivatives obtained by C-ring modification of podophyllotoxin induce apoptosis and inhibited proliferation in PC-3M cells via down-regulation of PI3K/Akt signaling pathway. Bioorg Med Chem Lett 46, 128174. 10.1016/j.bmcl.2021.128174. [DOI] [PubMed] [Google Scholar]
- 42.Malik J, Javed N, Ishaq U, Khan U, and Laique T (2020). Is There a Role for Colchicine in Acute Coronary Syndromes? A Literature Review. Cureus 12, e8166. 10.7759/cureus.8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaul S, Gupta M, Bandyopadhyay D, Hajra A, Deedwania P, Roddy E, Mamas M, Klein A, Lavie CJ, Fonarow GC, and Ghosh RK (2021). Gout Pharmacotherapy in Cardiovascular Diseases: A Review of Utility and Outcomes. Am J Cardiovasc Drugs 21, 499–512. 10.1007/s40256-020-00459-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, The SHK, Xu XF, Ireland MA, Lenderink T, et al. (2020). Colchicine in Patients with Chronic Coronary Disease. N Engl J Med 383, 1838–1847. 10.1056/NEJMoa2021372. [DOI] [PubMed] [Google Scholar]
- 45.Zhao AY, Brooks AF, Raffel DM, Stauff J, Arteaga J, Scott PJH, and Shao X (2020). Fully Automated Radiosynthesis of [(11)C]Guanidines for Cardiac PET Imaging. ACS Med Chem Lett 11, 2325–2330. 10.1021/acsmedchemlett.0c00479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dewan B, and Balasubramanian A (2010). Troxipide in the management of gastritis: a randomized comparative trial in general practice. Gastroenterol Res Pract 2010, 758397. 10.1155/2010/758397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu N, Huo JL, Wang S, Yuan XH, and Liu HM (2020). Drug repurposing: Discovery of troxipide analogs as potent antitumor agents. Eur J Med Chem 202, 112471. 10.1016/j.ejmech.2020.112471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brogden RN, Carmine AA, Heel RC, Speight TM, and Avery GS (1982). Trimethoprim: a review of its antibacterial activity, pharmacokinetics and therapeutic use in urinary tract infections. Drugs 23, 405–430. 10.2165/00003495-198223060-00001. [DOI] [PubMed] [Google Scholar]
- 49.Masur H, Brooks JT, Benson CA, Holmes KK, Pau AK, Kaplan JE, National Institutes of, H., Centers for Disease, C., Prevention, and America, H.I.V.M.A.o.t.I.D.S.o. (2014). Prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: Updated Guidelines from the Centers for Disease Control and Prevention, National Institutes of Health, and HIV Medicine Association of the Infectious Diseases Society of America. Clin Infect Dis 58, 1308–1311. 10.1093/cid/ciu094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cocuron JC, Tsogtbaatar E, and Alonso AP (2017). High-throughput quantification of the levels and labeling abundance of free amino acids by liquid chromatography tandem mass spectrometry. J Chromatogr A 1490, 148–155. 10.1016/j.chroma.2017.02.028. [DOI] [PubMed] [Google Scholar]
- 51.Chokkathukalam A, Kim DH, Barrett MP, Breitling R, and Creek DJ (2014). Stable isotope-labeling studies in metabolomics: new insights into structure and dynamics of metabolic networks. Bioanalysis 6, 511–524. 10.4155/bio.13.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feng M, De Oliveira J, Sallustrau A, Destro G, Thuery P, Roy S, Cantat T, Elmore CS, Blankenstein J, Taran F, and Audisio D (2021). Direct Carbon Isotope Exchange of Pharmaceuticals via Reversible Decyanation. J Am Chem Soc 143, 5659–5665. 10.1021/jacs.1c01923. [DOI] [PubMed] [Google Scholar]
- 53.Reilly SW, Lam YH, Ren S, and Strotman NA (2021). Late-Stage Carbon Isotope Exchange of Aryl Nitriles through Ni-Catalyzed C-CN Bond Activation. J Am Chem Soc 143, 4817–4823. 10.1021/jacs.1c01454. [DOI] [PubMed] [Google Scholar]
- 54.Brown AK, Fujita M, Fujimura Y, Liow JS, Stabin M, Ryu YH, Imaizumi M, Hong J, Pike VW, and Innis RB (2007). Radiation dosimetry and biodistribution in monkey and man of 11C-PBR28: a PET radioligand to image inflammation. J Nucl Med 48, 2072–2079. 10.2967/jnumed.107.044842. [DOI] [PubMed] [Google Scholar]
- 55.Tago T, Furumoto S, Okamura N, Harada R, Adachi H, Ishikawa Y, Yanai K, Iwata R, and Kudo Y (2016). Structure-Activity Relationship of 2-Arylquinolines as PET Imaging Tracers for Tau Pathology in Alzheimer Disease. J Nucl Med 57, 608–614. 10.2967/jnumed.115.166652. [DOI] [PubMed] [Google Scholar]
- 56.Simeon FG, Lee JH, Morse CL, Stukes I, Zoghbi SS, Manly LS, Liow JS, Gladding RL, Dick RM, Yan X, et al. (2021). Synthesis and Screening in Mice of Fluorine-Containing PET Radioligands for TSPO: Discovery of a Promising (18)F-Labeled Ligand. J Med Chem 64, 16731–16745. 10.1021/acs.jmedchem.1c01562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ben-Chetrit E (2018). Colchicine. Textbook of Autoinflammation, 729–749. 10.1007/978-3-319-98605-0_40. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
There is no dataset or code associated with this publication. Details of methods, experimental procedures, and characterization data are available in the supplemental information. Nuclear magnetic resonance (NMR) spectra are available.