


Abstract
Genome Information Broker (GIB) is a powerful tool for the study of comparative genomics. GIB allows users to retrieve and display partial and/or whole genome sequences together with the relevant biological annotation. GIB has accumulated all the completed microbial genome and has recently been expanded to include Arabidopsis thaliana genome data from DDBJ/EMBL/GenBank. In the near future, hundreds of genome sequences will be determined. In order to handle such huge data, we have enhanced the GIB architecture by using XML, CORBA and distributed RDBs. We introduce the new GIB here. GIB is freely accessible at http://gib.genes.nig.ac.jp/.
INTRODUCTION
Since the genome sequencing of Haemophilus influenzae was completed by The Institute for Genomic Research (TIGR) in 1995 (1), the complete genomes of more than 50 microbial species have been determined and published. Most of the data are disseminated in the universal flat file data format of the International Nucleotide Sequence Databank (INSD: DDBJ, EMBL and GenBank) as well as formats specific to the various genome sequencing groups. However, the data for an entire genome is not provided by the INSD as a single flat file but as multiple flat files of piece entries of the genome. A genome’s data are divided into multiple piece entries <350 kb each. Therefore, it is not easy to get a comprehensive view on a genome by use of INSD entries. In addition, genomic sequences and annotation are now too huge for users to retrieve even a subset of data and browse the sequence and annotation in a comprehensive and consistent way. It is critical, especially in the case of genome sequences that are as much as megabases in length, to have a tool that supports retrieval of information regarding a number of large genomes. Thus, we have developed a unified database of genomes with visualization tools that help browse the large amount of data.
The original version of GIB was developed as a browser for the Japan Escherichia coli genome project team in 1996 (2). At that time, GIB could handle only circular genomes. After that, linear genomes such as Borrelia burgdorferi were determined and published (3). We immediately improved GIB to handle linear genomes and added comparative capabilities to GIB. As a result of these improvements, GIB is now a powerful tool for not only searching single genome information but also comparing multiple genomes.
A number of genomes have been sequenced year by year and hundreds of genomes will be sequenced in the next couple of years. The previous version of GIB was based on HTML-like files with our original commands in a single platform. However, the amount of genomic data has become too huge for the HTML-like files to process. We have recently improved the system architecture of GIB based on distributed Relational Databases (RDBs) on multiple platforms and such technologies as Common Object Request Broker Architecture (CORBA) and extensible markup language (XML). We would like to introduce the new GIB system architecture here.
CONSTRUCTION OF THE DATABASE
The primary source of information for GIB is entries of the INSD. Whenever genome data are made pubic from the INSD, the data are immediately converted and implemented into GIB. We assemble flat files of the INSD piece entries into a single large flat file of the whole genome by using contiguous information in the CON entry of INSD corresponding to the genome. The assembled whole genome flat file is then implemented into the GIB database. The new version of GIB stores and processes information on any features described in the flat files in addition to CDS and RNA.
In addition, GIB has added functional category information other than in addition to the information in the flat files annotation. We use functional category information from TIGR (http://www.tigr.org/).
SYSTEM ARCHITECTURE
Figure 1 shows the system architecture of GIB. The hardware components of GIB are a World Wide Web server, three RDB servers and a homology search server.
Figure 1.

The new system architecture of GIB. The servers communicate each other by use of CORBA and XML.
The World Wide Web server of the new version of GIB is Apache (http://www.apache.org/) with PHP (http://www.php.net). PHP is the server-side HTML-embedded scripting language and is able to dynamically generate HTML page contents. It is a kind of generalization of the HTML-like file that we had developed specifically for the first version of GIB. Therefore, we moved smoothly from our proprietary HTML-like file to PHP technology. In addition, PHP has various libraries for connecting RDB servers, accessing CORBA objects and generating graphical images. By using PHP, the efficiency of development of new functionalities and database maintenance are drastically improved.
The new version of GIB uses PostgreSQL (http://www.postgresql.org/) for the Relational Database Management System (RDBMS) and is distributed over multiple PC Linux platforms. We are confident that GIB will continue to be a robust system for comparative genomics by distributing RDB servers on multiple platforms. We plan to increase the number of RDB servers according to the number of completed genomes.
The World Wide Web server communicates with the RDBs or the homology search servers by use of CORBA. Also, CORBA servers and client communicate with each other by use of XML. By using CORBA and XML, we expect that users will be able to access not only via a web browser but also directly to GIB databases through the Internet in the future.
SEARCHING AND VISUALIZATION OF GENOME DATABASE
In the GIB web page, keyword search, homology search and visualization of the data are available for genomic data. Both separate search of each genomes and comparison of multiple genomes are feasible. In the comparative genomes page, users can select multiple genomes that they are interested in. The selection made is recorded in the web browser at the users’ side by the HTML cookies and will be automatically called upon in the future session. This function simplifies repetitive queries.
In the case of a keyword search, users can search selected genome database(s) by ORF names, all the flat file features information such as feature key, feature qualifier and qualifier values, and region of location. Functional categories of ORFs are also used as a keyword for searching, although they are not always described in the flat file.
As a homology search engine, GIB implements BLAST2 (4) and FASTA3 (5). The target of the homology search is nucleotide sequences of any features including ORFs on condition that their location is given in the flat file and amino acid sequences translated from nucleotide sequences of ORF.
The visualization tools of the new GIB are based on PHP with GD library (http://www.boutell.com/gd/) that dynamically generates genome structure maps and chromosomes in PNG format. In the Genomic View page (Fig. 2), the genome structure map is displayed with ORFs colored by functional categories. The map is clickable to retrieve the Feature View page. Users can specify the region for Feature View page retrieval by moving the mouse cursor on the map. In the Feature View page (Fig. 3), the map of features in the specified region is displayed. The arrow images on the map are colored by features or functional categories and are also clickable. ORFs with same function are displayed in the same color on the map. Therefore, users can understand the distribution of the ORFs with the same function at a glance. If an arrow is selected, the user is able to immediately retrieve the feature information corresponding to the arrow in the Feature Information page. Furthermore, links to DBGET (http://www.genome.ad.jp/) and GTOP (http://spock.genes.nig.ac.jp/~genome/gtop.html) are available from the Feature Information page.
Figure 2.
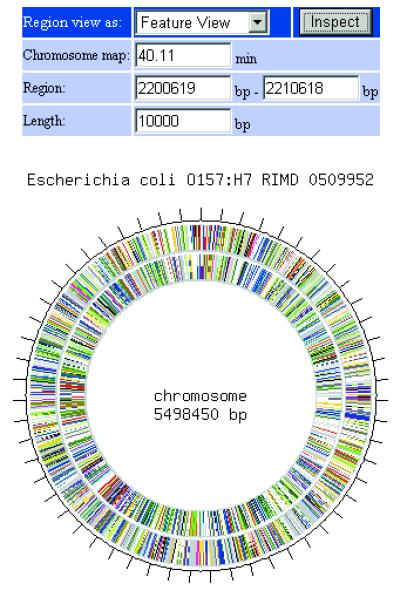
Image of Genomic View page on GIB. The map of genome structure is displayed with ORFs colored by functional categories.
Figure 3.
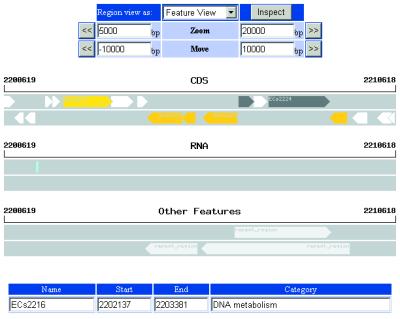
Image of Feature View page. The map of features, CDS, RNA and other features in specified region are displayed.
DATABASE ACCESS
GIB is freely accessible at http://gib.genes.nig.ac.jp/. Corrections, suggestions and feedback should be sent to gib-master@ddbj.nig.ac.jp.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Professors T. Gojobori and Y. Tateno of the Center for Information Biology and DNA Data Bank of Japan in the National Institute of Genetics for their useful suggestions.
REFERENCES
- 1.Fleischmann R.D., Adams,M.D, White,O., Clayton,R.A., Kirkness,E.F, Kerlavage,A.R., Bult,C.J., Tomb,J., Dougherty,B.A., Merrick,J.M. et al. (1995) Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science, 269, 496–512. [DOI] [PubMed] [Google Scholar]
- 2.Tamura T., Mori,H. and Sugawara,H. (1997) Genome Information Broker for large and small genomes. Trends Genet., 13, 498. [DOI] [PubMed] [Google Scholar]
- 3.Fraser C.M., Casjens,S., Huang,W.M., Sutton,G.G., Clayton,R., Lathigra,R., White,O., Ketchum,K.A., Dodson,R., Hickey,E.K. et al. (1997) Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature, 390, 580–586. [DOI] [PubMed] [Google Scholar]
- 4.Altschul S.F., Madden,T.L., Schaffer,A.A, Zhang,J., Zhang,Z., Miller,M. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearson W.R. and Lipman,D.J. (1988) Improved tools for biological sequence comparison. Proc. Natl Acad. Sci. USA, 85, 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]