

Abstract
Cortical function critically depends on inhibitory/excitatory balance. Cortical inhibitory interneurons (cINs) are born in the ventral forebrain and migrate into cortex, where their numbers are adjusted by programmed cell death. Previously, we showed that loss of clustered gamma protocadherins (Pcdhγ), but not of genes in the alpha or beta clusters, increased dramatically cIN BAX-dependent cell death in mice. Here we show that the sole deletion of the Pcdhγc4 isoform, but not of the other 21 isoforms in the Pcdhγ gene cluster, increased cIN cell death in mice during the normal period of programmed cell death. Viral expression of the Pcdhγc4 isoform rescued transplanted cINs lacking Pcdhγ from cell death. We conclude that Pcdhγ, specifically Pcdhγc4, plays a critical role in regulating the survival of cINs during their normal period of cell death. This demonstrates a novel specificity in the role of Pcdhγ isoforms in cortical development.
Introduction:
In the cerebral cortex, inhibitory cortical neurons (cINs) are essential for sculpting, gating, and regulating neuronal excitation. Dysfunction or changes in the number of cINs is a hallmark of neurological disorders including epilepsy, schizophrenia, and autism (Chao et al., 2010; Lewis et al., 2005; Marín, 2012; Rossignol, 2011; Rubenstein & Merzenich, 2003; Verret et al., 2012). Ensuring the correct numbers of cINs are established during cerebral cortex development is an essential step in establishing proper brain function.
During embryonic development excess numbers of cINs are produced in the ventral forebrain within the medial and caudal ganglionic eminences (MGE and CGE). The young cINs then migrate dorsally into the cortex and form connections with locally produced excitatory neurons. Within the first two postnatal weeks of cortical development in mice, approximately 40% of cINs are eliminated by programmed cell death (Southwell et al., 2012; Wong et al., 2018). An intriguing feature of this developmental process of cell elimination is its timing and location. While most cINs are born in the ventral telencephalon between E11.5 and 16.5, their programmed cell death occurs postnatally (10–15 days later) and in the cortex, far from their birthplace. The timing of cIN programmed cell death correlates with several features of cortical development including the emergence of correlated activity and the development of cIN morphological complexity and synaptic connectivity (Ben-Ari et al., 2004; Connors et al., 1983; Priya et al., 2018; Seress et al., 1989; Tyzio et al., 1999; Yang et al., 2012). Indeed, changes in correlated neuronal activity have been associated with altered cIN survival. Increased correlated patterns of neuronal activity increased cIN survival while decreased cIN activity decreased cIN survival (Duan et al., 2020). Given the role of the clustered protocadherins in neuronal tiling, arborization, survival and axon targeting (Chen et al., 2012; Garrett et al., 2012, 2019; Katori et al., 2017; Lefebvre et al., 2008; Molumby et al., 2016; Mountoufaris et al., 2017; Prasad et al., 2008; Wang et al., 2002), we postulated that these diverse adhesion proteins could be required to establish initial cell-cell connectivity among neurons during early postnatal development and to regulate programmed cell death in the cortex.
The clustered protocadherins (Pcdhs) (Wu & Maniatis, 1999) are a set of fifty-eight cell surface homophilic-adhesion molecules that are tandemly arranged in three subclusters named alpha, beta, and gamma: Pcdha, Pcdhb, and Pcdhg (Wu et al., 2001). We have previously shown that the function of the isoforms in the Pcdhg cluster, but not those of the Pcdha or Pcdhb, are required for the survival of most cINs (Mancia Leon et al., 2020). Using transplantation to bypass neonatal lethality, we have also shown that the combined removal of three Pcdhγ isoforms (Pcdhγc3, Pcdhγc4, and Pcdhγc5) resulted in increased cell death. Within the Pcdhg cluster, the deletion of Pcdhγc4, but not that of other isoforms, is sufficient to cause neonatal lethality and increased cell death in the spinal cord (Garrett et al., 2019; Prasad et al., 2008). We wondered whether Pcdhγc4 also plays a unique and specific role in the regulation of cIN survival in the neocortex.
In the present study, we screened published single-cell RNA sequencing (scRNA-seq) datasets to determine whether Pcdhγc4 is expressed in cINs in the adult mouse cortex. The Pcdhγc4 was surprisingly enriched in the cIN population and largely absent from excitatory neurons in the adult cortex. In contrast, the Pcdhγc5 isoform was mainly expressed in excitatory neurons (Tasic et al., 2018; Yao et al., 2021). Next, we characterized the expression of Pcdhγc4 and Pcdhγc5 during early postnatal development in the cortex of mice. Using RNAScope, we observed that the expression of Pcdhγc5 was minimal at P5, increasing by P7. By P14, Pcdhγc5 showed preferential expression in excitatory neurons. Pcdhγc4 expression also increased from P5 to P7, and by P14 Pcdhγc4 was found enriched in the MGE-derived cINs. Using a series of knockout mice in which various of the Pcdhγ isoforms were deleted (Garrett et al., 2019), combined with heterochronic transplantation (Mancia Leon et al., 2020), we then showed that the 19 A- and B-type Pcdhγ isoforms, as well as two Pcdhγ C-type isoforms (Pcdhγc3 and Pcdhγc5), have at most a minimal effect on cIN survival. In contrast, the single deletion of Pcdhγc4 was sufficient to increase cell death dramatically among cINs derived from the MGE during the normal period of programmed cell death. Lastly, we showed that Pcdhγ-deficient cINs were rescued from excess cell death by the viral over-expression of the Pcdhγc4 isoform. We conclude that Pcdhγ diversity is not required for cIN survival; rather the expression of a single isoform, that of Pcdhγc4, is necessary and sufficient for the survival of most cINs.
Results:
Pcdhγ C-type expression in the mouse cortex during programmed cell death.
The Pcdhγ gene locus encodes 22 distinct Pcdhγ proteins, which are subclassified as A-, B-, or C-type isoforms. Previous work suggests that Pcdhγ and specifically the C-type isoforms play a key role in the regulation of cIN programmed cell death (Mancia Leon et al., 2020). This previous work suggested that either individual or multiple C-type γ-Pcdhs are required to maintain appropriate numbers of cINs in the cortex. In order to determine whether differential expression patterns of individual C-type γ-Pcdhs might be involved in cIN survival, we screened scRNA sequencing datasets to determine which C-type γ-Pcdhs are expressed in cIN. Interestingly, two scRNA sequencing datasets generated from adult animals (>P50) (Tasic et al., 2018; Yao et al., 2021) showed a distinct increase in the expression of Pcdhγc4 in GABAergic cells. In contrast, cells identified as glutamatergic excitatory neurons expressed higher levels of Pcdhγc5 (Fig. 1A–B). We next looked at the expression of Pcdhγc4 and Pcdhγc5 in the different sub-classes of both excitatory and inhibitory neurons (Fig.1C–D). Among inhibitory populations, interferon gamma-induced GTPase (Igtp), Neuron-derived neurotrophic factor (Ndnf), Parvalalbumin (Pvalb), Somatostatin (SST), and Vasoactive intestinal peptide-expressing (Vip) subtypes showed a higher expression of Pcdhγc4 and lower levels of Pcdhγc5. Among the excitatory neurons, higher levels of Pcdhγc5 expression was especially evident among deep and superficial glutamatergic neurons. Together, these scRNA-seq datasets indicate that the Pcdhγc4 and Pcdhγc5 genes are differentially expressed between cINs and excitatory neurons in the adult mouse cortex. Interestingly, expression of Pcdhγc3, which plays a unique role in promoting cortical dendrite arborization through its signaling partner Axin1 (Steffen et al., 2023), showed no distinct expression bias between GABAergic and Glutamatergic cells and was expressed at low levels similar to that of the other Pcdhγ genes (Fig. 1A–B).
Figure 1. Pcdhγ C-type expression in the mouse cortex during programmed cell death.

A-B. ScRNA-seq from previously generated dataset show expression bias of Pcdhγc4 in GABAergic cIN (A), and Pcdhγc5 in Glutamatergic neurons (B).Error bars represent the error of the mean.
C-D. Expression levels of Pcdhγc4 is higher in inhibitory neuron subtypes (C), while Pcdhγc5 expression is higher in Glutamatergic neuron subtypes (D). Error bars represent the error of the mean. interferon gamma-induced GTPase (Igtp), Neuron-derived neurotrophic factor (Ndnf), Parvalalbumin (Pvalb), Somatostatin (SST), and Vasoactive intestinal peptide-expressing (Vip).
E-F. RNA scope of Nkx2.1;Ai6 cortex during programmed cell death. Low magnification (E) and higher magnification (F) shows expression of Pcdhγc4 and Pcdhγc5 to be minimal at Pcdhγc5, Pcdhγc4 expression is increased by P7 and is expressed in MGE-derived ZsGreen+ cells (yellow arrows). At P14, expression of both Pcdhγc4 and Pcdhγc5 is high, but distinctly not in the same cells, with Pcdhγc4 being highly expressed in ZsGreen+ MGE-derived cINs. Scale bar low-magnification (G) = 100 um. Scale bar high-magnification (F) = 50 um.
G-H. IHC in P14 mouse cortex shows Pcdhγc5 association with excitatory neurons (Ctip2 and Cux1)(G), and not with GABAergic cIN (H). Scale bar top row = 50 um. Scale bar bottom row = 25 um.
In order to corroborate the above expression pattern histologically, we performed in-situ hybridizations using RNAscope, in combination with a reporter mouse in which MGE-derived cINs were labeled with a ZsGreen protein (Nkx2.1;Ai6 mouse line). Using validated probes for the Pcdhγc4 and Pcdhγc5 mRNA transcripts, we looked at multiple times during the period of programmed cell death (P5, P7, and P14). At P5, we found low expression of Pcdhγc4 and Pcdhγc5 in both ZsGreen+ or - cells. However, a subpopulation of ZsGreen+, MGE-derived cINs at P5 was associated with a higher Pcdhγc4 signal (Fig. 1E–F). By P7, both Pcdhγc4 and Pcdhγc5 were observed in the majority of cells, although the Pcdhγc4 signal associated with the ZsGreen+ neurons was more robust (Fig. 1E–F). At P14, when the period of programmed cIN cell death is largely over, we found a dramatic increase in Pcdhγc4 and Pcdhγc5 expression, consistent with previous reports of Pcdhγc5 expression beginning in the second postnatal week in rat cortex (Li et al., 2010). Cortical neurons at this time showed a clear preferential expression of either the Pcdhγc4 or the Pcdhγc5 isoform. ZsGreen+ neurons showed preferential expression of the Pcdhγc4 isoform, while many of the ZsGreen- cells showed lower or no expression of Pcdhγc4. Next, we aimed to validate these findings using IHC. While we were unable to procure a reliable antibody for Pcdhγc4, we did find an antibody that specifically labels the Pcdhγc5 isoform (validated using 3R2 mice lacking Pcdhγc5 gene; Garrett et al., 2019; data not shown). We co-stained for Pcdhγc5 and markers for upper-layer excitatory neurons (Cux1) and lower-layer excitatory neurons (Ctip2). At P14, Pcdhγc5 expression clearly surrounded neuronal nuclei expressing these excitatory transcription factor markers (Fig. 1G). To determine if Pcdhγc5 expression is lower in inhibitory neurons, we also stained P14 cortex for Pcdhγc5 and GABA (Fig. 1H). Consistent with the scRNA-seq data, GABA-expressing inhibitory neurons expressed low/background levels of Pcdhγc5. Together, these data indicate divergent expression of Pcdhγc4 isoforms in cINs and Pcdhγc5 isoforms in excitatory neurons, which is a pattern that emerges during the period of programmed cell death and is maintained in the adult cortex.
Pcdhγc4 deletion results in the elimination of most cINs
Our previous work and that of others has shown that the combined deletion of the three Pcdhγ C-type genes (Pcdhγc3, Pcdhγc4, and Pcdhγc5) results in increased cIN cell death during the period of naturally occurring cell death (Chen et al., 2012; Mancia Leon et al., 2020). Other studies have shown that constitutive genetic deletion of Pcdhγc4, but not that of Pcdhγc3 or Pcdhγc5, leads to neonatal lethality and increased interneuron cell death in the spinal cord (Garrett et al., 2019). To ascertain the function of Pcdhγc4 in the regulation of cIN programmed cell death, we used mice with constitutive deletions of the Pcdhγc4 isoform (PcdhγC4KO mice; Garrett et al., 2019). PcdhγC4KO mice were crossed to the Nkx2.1Cre; Ai14 MGE/preoptic area (POA) reporter mouse line to provide a genetically encoded fluorescent label for the cINs (Fig. 2A).
Figure 2 -. Genetic deletion of Pcdhγc4 increased cell death in MGE-derived cINs.
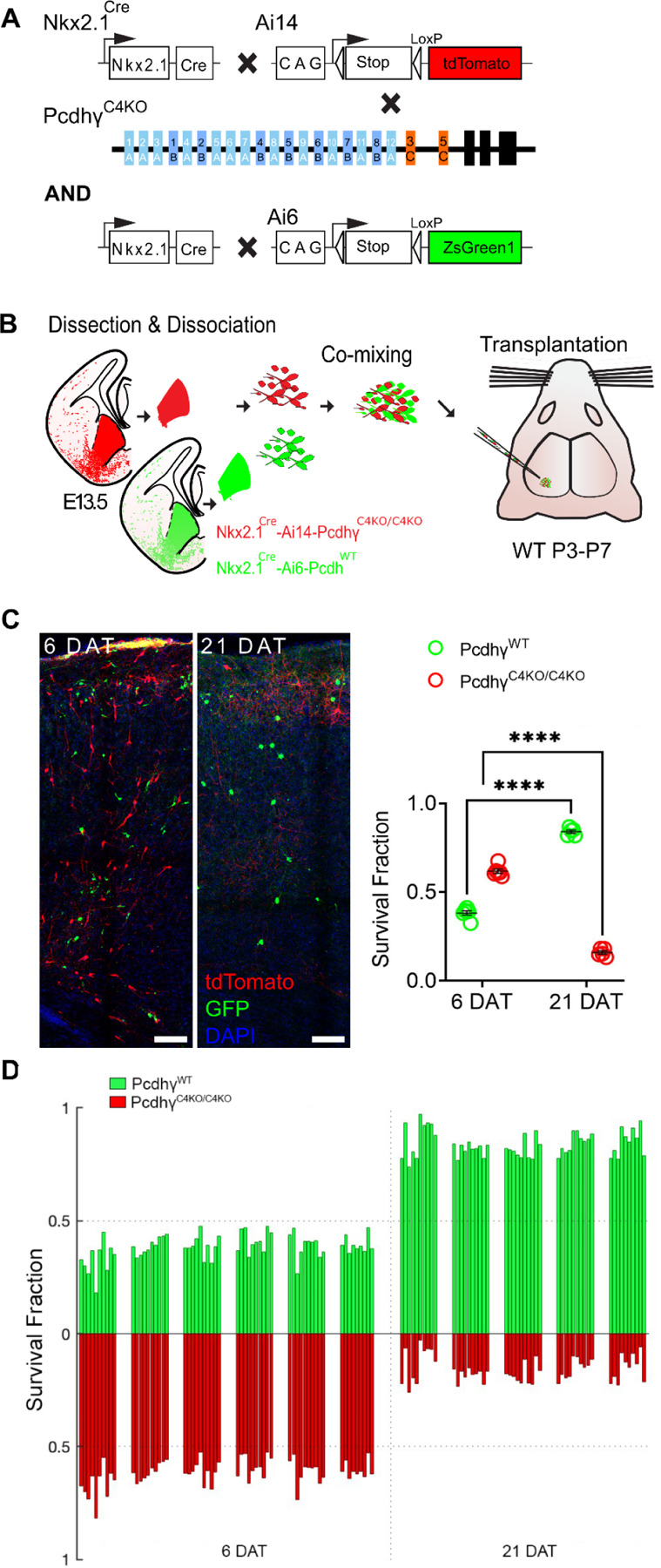
A. Diagram of genetic crosses between MGE/POA-specific reporter and PcdhγC4KO mice. PcdhγC4KO homozygous MGE cells were obtained from the Nkx2.1Cre; Ai14;PcdhγC4KO/C4KO embryos, whereas control cells were obtained from Nkx2.1Cre; Ai6 embryos.
B. Schematics of transplantation protocol. The MGEs from E13.5 PcdhγC4KO homozygous mutant or control embryos were dissected, dissociated, and mixed in similar proportions. The mixture of GFP+ (PcdhγWT) and tdTomato+ (PcdhγC4KO/C4KO) cells was grafted into the cortex of WT neonate mice.
C. Left - Confocal images from the cortex of 6 and 21 DAT mice. The transplanted cells are labeled with GFP (PcdhγWT) or tdTomato (PcdhγC4KO/C4KO). Right - Quantifications (shown as survival fraction) of surviving MGE-derived cINs at 6 and 21 DAT. Both the transplanted GFP and tdTomato-labeled cells undergo programmed cell death between 6 and 21 DAT, but the PcdhγC4KO/C4KO cells are eliminated at significantly higher rates.
D. Survival fraction quantification from (C) shown by the brain section (each bar) and separated by animals at 6 and 21 DAT.
Scale bar = 50 um, Nested-ANOVA, ****p =3.147e-10, n = 5 mice per time point and 10 brain sections quantified per mouse
Embryos homozygous for deletion of the Pcdhγc4 isoform develop normally with no apparent weight, size or brain abnormalities and are born in normal Mendelian ratios. However, these mice die perinatally (within 24 hours of birth) before the period of programmed cell death for cINs around P7. In order to bypass lethality and to study the role of Pcdhγc4 deletion in cIN survival postnatally, we utilized a co-transplantation method of cINs that are WT or mutant (Fig. 2B). This allowed us to compare the survival of mutant and WT cells within the same WT environment. The F2 generation of Nkx2.1Cre;Ai14;PcdhγC4KO/+ mice were bred to generate E13.5 embryos, homozygous for the PcdhγC4KO allele. In these embryos, MGE/POA-derived cells lacking Pcdhγc4 are fluorescently labeled with the tdTomato protein upon Cre-driven recombination of the Ai14 allele.
The MGE of embryos carrying the homozygous deletion of the Pcdhγc4 allele was microdissected. As a control, GFP-expressing cINs were derived from microdissected MGEs of Nkx2.1Cre;Ai6, in which MGE-POA-derived cells are fluorescently labeled with ZsGreen (Madisen et al., 2010) (Fig. 2A & B) or Gad67-GFP embryos (Supplementary Fig. 2A & B). The MGEs were dissociated and cells were mixed in similar proportions (GFP and tdTomato). The mixture of GFP cells with tdTomato cells was transplanted into the cortex of host neonatal mice (P3-P7). The homozygous PcdhγC4KO/C4KO cells were identified via tdTomato expression while cells carrying the PcdhγWT allele were identified via expression of GFP (Fig. 2C). Survival of the transplanted cells was analyzed at 6 and 21 days post-transplant (DAT), which corresponds to postnatal days (P) 0 and 15 for the transplanted cells, cellular ages that span the period of endogenous cIN programmed cell death. At 6 DAT, the proportion of the fluorescently labeled transplanted cells expressing GFP (PcdhγWT) was 38.2%, and the tdTomato-positive PcdhγC4KO/C4KO cells made up the balance, 61.8%, of all transplanted cells that survived. Importantly, there was no apparent change in the proportion of GFP to tdTomato cells between 4 and 6 DAT (data not shown), before the period of naturally occurring cell death for the transplanted cells. By 21 DAT, however, the proportion of GFP to tdTomato cells had shifted dramatically, indicating that one cell population was eliminated at higher rates. Indeed, the tdTomato-positive PcdhγC4KO/C4KO cell population dropped from 61.8 % (at 6 DAT) to 15.9% at 21 DAT. Conversely, the proportion of GFP-positive PcdhγWT cINs had increased from 38.2% (at 6 DAT) to 84.1% at 21 DAT. Note that the increase in the proportion of GFP-positive cells does not reflect an increase in survival, but rather that the tdTomato labeled cells were eliminated in much greater numbers during programmed cell death. Importantly, similar results were found in experiments where we co-transplanted PcdhγC4KO/C4KO with PcdhγWT Cells derived from Gad67-GFP embryos (Supplementary Fig. 2). These results suggest that cells that lack the Pcdhγc4 protein are more likely to be eliminated during the normal period of cell death than those cells that express the WT Pcdhγc4.
Deletion of all Pcdhγ, except Pcdhγc4, is sufficient for the survival of the majority of cINs.
We wondered whether Pcdhγc3 and Pcdhγc5 may also contribute to cIN survival. In our previous study, deletion of the C-type isoforms (Pcdhγc3, Pcdhγc4, and Pcdhγc5) resulted in the elimination of most cINs, to levels similar to those observed after the loss of function of the entire Pcdhγ cluster (Mancia Leon et al., 2020). These suggested that the 19 alternate A and B-type Pcdhγ isoforms do not significantly contribute to the survival of cINs. Here, we used the Pcdhγ1R1 mouse line (Garrett et al., 2019), which lacks all 19 A and B-type Pcdhγ isoforms, as well as Pcdhγc3 and Pcdhγc5, but retains Pcdhγc4 (Fig. 3A).
Figure 3 -. Most MGE-derived cINs survive the deletion of most Pcdhγ, except for Pcdhγc4.
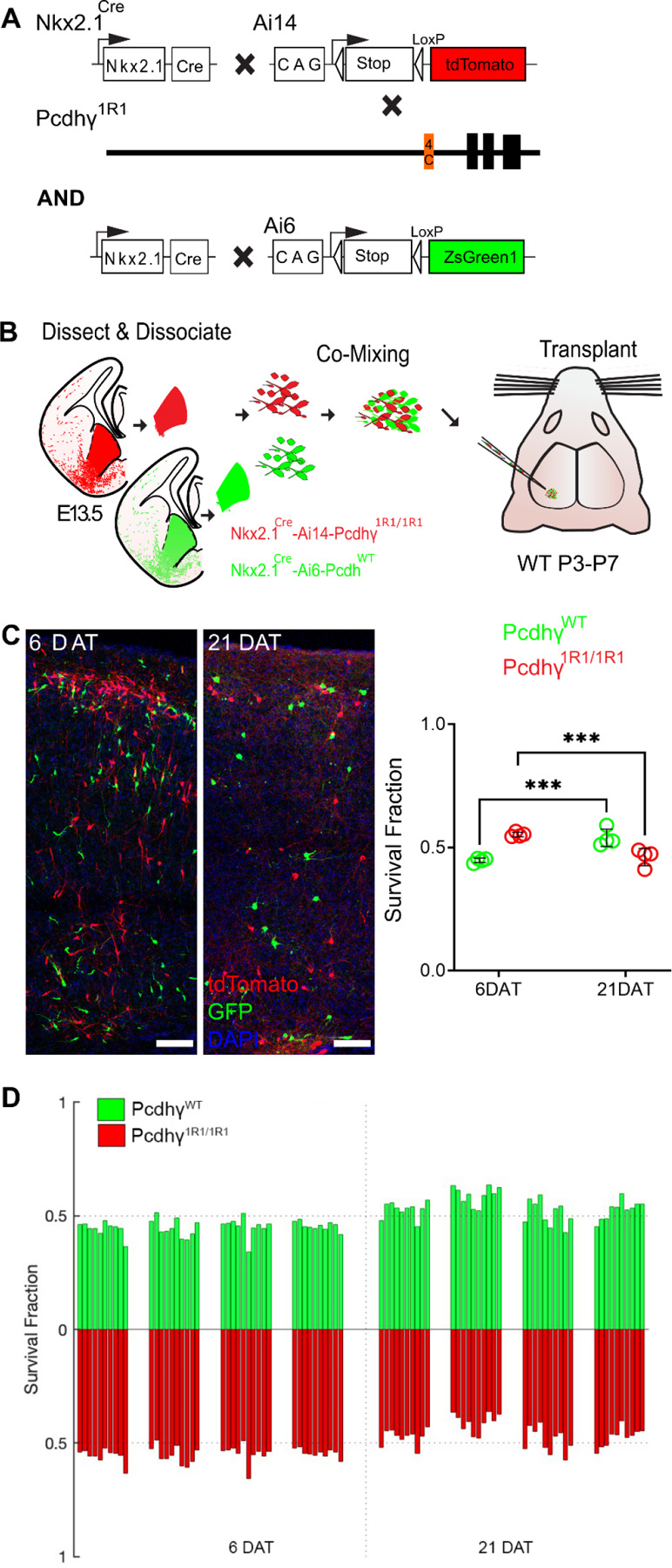
A. Diagram of genetic crosses between MGE/POA-specific reporter Nkx2.1Cre;Ai14 and Pcdhγ1R1 mice. Control cells were derived from Nkx2.1Cre;Ai6 mice while Pcdhγ1R1 mutant cells were derived from Nkx2.1Cre;Ai14; Pcdhγ1R1/1R1 mice.
B. Diagram of the transplantation protocol. The MGEs from E13.5 Pcdhγ1R1 homozygous or control embryos were dissected, dissociated, and mixed in similar proportions. The mixture of GFP+ (PcdhγWT) and tdTomato+ (Pcdhγ1R1/1R1) cells was grafted into the cortex of WT neonate mice.
C. Left - Confocal images from the cortex of 6 and 21 DAT mice. The transplanted cells are labeled with GFP (PcdhγWT) or tdTomato (Pcdhγ1R1/1R1). Right - Quantifications of the transplanted cells that survived at 6 and 21 DAT. Note that both the GFP or tdTomato labeled cells underwent programmed cell death between 6 and 21 DAT, but both cell types survived to similar levels.
D. Survival fraction quantification from (C) shown by the brain section (each bar) and separated by animals at 6 and 21 DAT.
Scale bar = 50 um, Nested-ANOVA, ***p = 0.0026, n = 4 mice per time point and 10 brain sections per mouse.
Mice homozygous for the Pcdhγ1R1 allele (Pcdhγ1R1/1R1) are born in normal mendelian ratios, develop normally and are fertile, as previously reported (Garrett et al., 2019). Pcdhγ1R1/R1 mice were crossed to the above Nkx2.1Cre; Ai14 mouse line to label MGE/POA-derived cINs with the tdTomato protein (Fig. 3A). As above, we used the co-transplantation to compare the survival of cINs solely expressing Pcdhγc4 to that of cINs expressing all 22 Pcdhγ isoforms. cIN precursor cells homozygous for the Pcdhγ1R1/R1 allele were obtained from E13.5 Nkx2.1Cre;Ai14;Pcdhγ1R1/R1 embryos. As a control, we used cIN precursor cells expressing WT Pcdhγ obtained from Nkx2.1Cre;Ai6 embryos (Fig. 3B). Survival of the transplanted cells was analyzed at 6 and 21 DAT and is represented as the proportion of GFP to tdTomato cells at these time points. At 6 DAT, roughly 44.7% of all transplanted cells were GFP-positive (WT Pcdhγ) while the remaining cells (55.2%) were tdTomato positive (Pcdhγ1R1/R1). As hypothiszed, the survival of the Pcdhγ1R1/1R1 tdTomato positive cells was remarkably similar to that of the GFP-labeled control cells (Fig. 3C–D). However, there was a small but significant drop in the Pcdhγ1R1/1R1 population (9.1%), as compared to a 45.9% decrease in the PcdhγC4KO/C4KO transplants (Fig. 2C–D). Similar results were observed when control cINs cells were obtained from the Gad67-GFP mouse line (Supplementary Fig. 3). Importantly, the proportion of GFP to tdTomato positive cells remained unchanged between 4 to 6 DAT (data not shown). Together, these data suggest that expression of Pcdhγc3 or Pcdhγc5 is not required for the survival of most cINs. Furthermore, these observations indicate that A- and B-type Pcdhγ isoforms are also not required for cIN survival. Given the small drop in cIN survival in Pcdhγ1R1/R1 cIN, we cannot completely rule out the possibility that Pcdhγc3 or Pcdhγc5 may make a small contribution to cIN survival. However, Pcdhγ1R1/R1 mice have been shown to express significantly decreased levels of Pcdhγc4 compared to WT mice (Garrett et al., 2019), which might explain the observed small, but statistically significant drop in the survival of the Pcdhγ1R1/1R1 cINs.
Exogenous expression of Pcdhγc4 in Pcdhγ knockout cINs rescues cINs from cell death.
The results above suggest that most cINs lacking expression of Pcdhγc4 are eliminated during the normal period of programmed cell death; in contrast, cINs expressing Pcdhγc4 as their only Pcdhγ isoform survive at nearly normal levels. We next asked whether cINs lacking expression of all twenty-two Pcdhγ isoforms could be rescued from cell death by re-introducing the Pcdhγc4 isoform. Since cINs lacking the function of all Pcdhγ isoforms are nearly eliminated when co-transplanted with WT cINs expressing Pcdhγ (see figure 10E in (Mancia Leon et al., 2020), we reasoned that a rescue effect would be most evident in these mutant cells.
Dissociated MGE cells from E13.5 of Nkx2.1Cre;Ai14;Pcdhγfcon3/fcon3 embryos were infected in suspension with lentivirus expressing the Pcdhγc4 isoform fused to GFP (Fig. 4A &B). The infected cells were transplanted into WT host neonate mice and survival was analyzed at 6 and 21 DAT. The transplanted cINs expressing lentivirus-driven Pcdhγc4 were identified via the co-expression of tdTomato and GFP (Fig. 4C). Pcdhγ mutant cells that expressed only tdTomato were identified as not infected. We compared the survival of Pcdhγc4-transduced (GFP+tdTomato+) to non-transduced (tdTomato+ GFP-) at 6 and 21 DAT. While both the Pcdhγc4-transduced and non-transduced cINs undergo a wave of cell death between the above time points, the fraction of tdTomato+GFP+ cells increased between 6 and 21 DAT (33% to 68%). In contrast, the fraction of tdTomato+GFP- cells decreased from 66% to 32% during the same period of time (Fig. 4 C). To control for possible non-specific effects of viral infection on cIN survival, we infected Nkx2.1Cre;Ai14 MGE cells that carry the WT Pcdhγ allele with lentivirus expressing GFP, and analyzed their survival at 6 and 21 DAT (Supplementary Fig. 4A &B). As above, we compared the survival of Pcdhγc4-transduced (tdTomato+GFP+) to non-transduced (tdTomato+GFP-) cells. At 6 DAT, the fraction of infected cells was approximately 50% and remained nearly constant between 6 and 21 DAT, indicating that infection with lentivirus and expression of GFP had no impact on cIN survival (Supplementary Fig. 4C & D). These results indicate that the introduction of Pcdhγc4 to cINs lacking endogenous Pcdhγ genes is sufficient to rescue the mutant cells from undergoing excessive apoptosis.
Figure 4 -. Lentiviral expression of Pcdhγc4 rescues cINs with Pcdhγ loss of function.
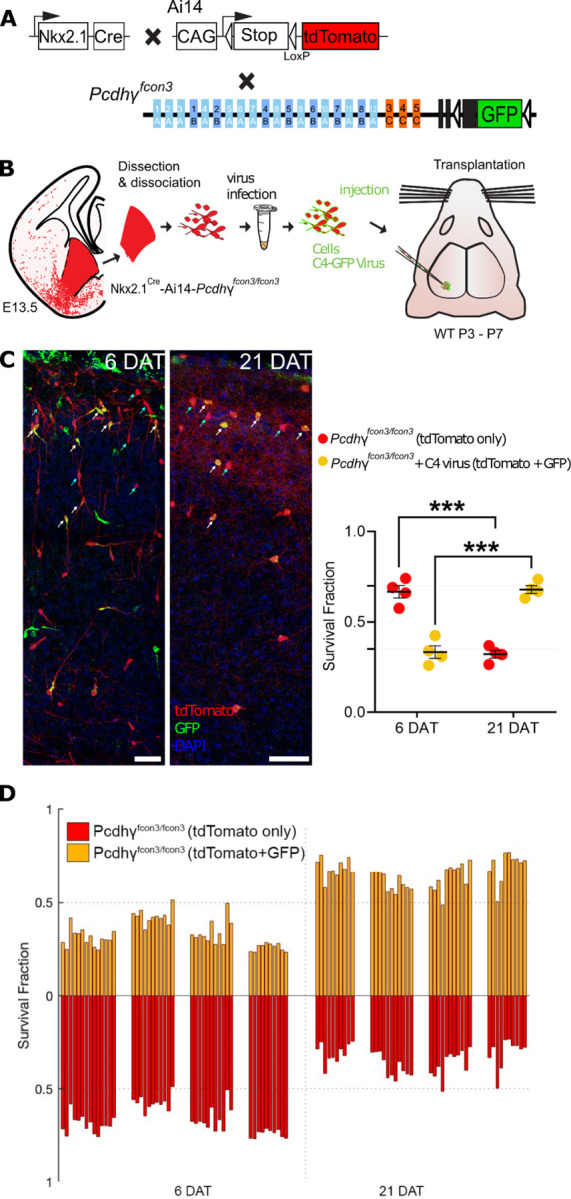
A. Diagram of mouse genetic crosses. Pcdhγfcon3 mice were crossed to the Nkx2.1Cre;Ai14 mouse line to generate embryos with loss of function of all Pcdhγ isoforms.
B. Schematic of the lentiviral infection and transplantation of MGE cIN precursors. The MGEs of Nkx2.1Cre;Ai14;Pcdhγfcon3/fcon3 embryos were dissected, dissociated, and infected in suspension with lentivirus carrying Pcdhγc4-GFP. The mixture of transduced and non-transduced cells was grafted into the cortex of WT neonate recipient mice.
C. Confocal images of the transplanted cINs in the cortex at 6 and 21 DAT. Notice the expression of GFP can be found near the cell surface including the cell processes, reflecting the putative location of the transduced Pcdhγc4-GFP protein. Quantifications of the tdTomato+GFP-(teal arrows) or tdTomato+GFP+ (yellow cells, white arrows) cells are shown as the fraction of cells from the total tdTomato+ cells at 6 and 21 DAT. The fraction of the Pcdhγc4-transduced yellow cells increases from 6 to 21DAT, while the fraction of non-transduced (tdTomato+ GFP-) decreases at equivalent time points.
D. Survival fraction quantification from (C) shown by the brain section (each bar) and separated by animals at 6 and 21 DAT.
Scale bar = 50 um, Nested-ANOVA, ****p = 0.0002, n = 4 mice per time point and 10 brain sections per mouse from one transplant cohort.
Discussion:
This study reveals the preferential expression of Pcdhγc4 in MGE-derived cINs, in contrast to excitatory neurons, which preferentially express the distinct Pcdhγc5 isoform. This segregated pattern of expression develops during the period of programmed cell death, which also coincides with other key maturational stages. Soon after migration, cINs must integrate into cortical circuits, forming nascent synapses with both inhibitory and excitatory neurons. During this period local inhibitory networks require increased interneuron-interneuron connectivity to facilitate synchronous activity which promotes interneuron survival (Duan et al., 2020). Opposing expression of Pcdhγc5 and Pcdhγc4 on excitatory and inhibitory neuron adhesion interfaces, respectively, could help segregate the two populations of neurons and be a key mechanism in the formation of initial cortical circuits.
Previous studies show that Pcdhγ C-type isoforms are required to regulate neuronal numbers during the critical window of programmed cell death (Chen et al., 2012; Mancia Leon et al., 2020). The timing of Pcdhγc4 enrichment during postnatal development suggests that this specific Pcdhγ isoform could be playing a key role in programmed cell death. Indeed a recent study of the spinal cord and retina reported that the deletion of Pcdhγc4, but not that of other Pcdhγ isoforms, leads to increased cell death and neonatal lethality (Garrett et al., 2019). However, cIN-programmed cell death occurs mostly postnatally, whereas cell death in the spinal cord takes place prenatally (Prasad et al., 2008; Wang et al., 2002). Since mice lacking Pcdhγc4 (PcdhγC4KO ) die soon after birth, we could not study the extent of cIN cell death directly in these mutant animals. We took advantage, therefore, of transplantation strategies to compare the survival of cells lacking Pcdhγc4 to cells that carry all Pcdhγ isoforms. PcdhγC4KO mice lack expression of Pcdhγc4 but express the 19 A and B type Pcdhγ isoforms as well as Pcdhγc3 and Pcdhγc5. Here, we show that most PcdhγC4KO cINs are eliminated during the period of programmed cell death, a result that is consistent with that found with the deletion of the entire Pcdhγ gene cluster or of the three Pcdhγ C-type isoforms (Carriere et al., 2020; Chen et al., 2012; Mancia Leon et al., 2020). The above finding also suggests that C-type isoforms Pcdhγc3 or Pcdhγc5 do not play a major role in the regulation of cIN survival; this is consistent with the relatively low expression of these isoforms seen in adult cINs. Indeed, when we studied the survival of cINs that solely express Pcdhγc4 using cells from Pcdhγ1R1 embryos, we found that the majority of cINs survived at levels similar to those of cINs that carry all Pcdhγ isoforms. However, transplanted Pcdhγ1R1 cINs did show a small but significant reduction in survival, compared to control cells. While we cannot completely exclude a role for other Pcdhγ isoforms, including Pcdhγc3 or Pcdhγc5 in the Pcdhγ1R1 cells, the observed reduction in survival is likely due to reduced levels of Pcdhγc4 expression (~10% of WT levels) in Pcdhγ1R1 mice (Garrett et al., 2019). Indeed, this low expression of Pcdhgc4 in Pcdhγ1R1 mice makes the survival of their cINs a more remarkable result. In order to test directly the function of Pcdhγc4, we used lentiviral driven expression of Pcdhγc4 in cINs that lack the function of all 22 Pcdhγ isoforms. Expression of Pcdhγc4 resulted in a dramatic increase in the survival of MGE-derived cINs when compared to cells lacking the function of the entire Pcdhγ cluster.
Our findings above suggest that Pcdhγ diversity is not required for the survival of cINs and show a unique and specific role for Pcdhγc4 in regulating the survival of cINs. Together, our results align with findings in the spinal cord and retina (Chen et al., 2012; Garrett et al., 2019), suggesting a potential general Pcdhγc4-driven mechanism that regulates the survival of local-circuit neurons across the CNS. While our data suggest that Pcdhγ diversity is dispensable for cIN survival, the diversity requirements for clustered Pcdhs in the Pcdha and Pcdhb clusters was not studied here: in all genotypes examined, these clusters remained intact. A recent study found that mutant mice lacking all call Pcdha, Pcdhb, and A- and B-type Pcdhg genes (thus retaining only Pcdhγc3, Pcdhγc4, and Pcdhγc5), also exhibited neonatal lethality and increased neuronal apoptosis (Kobayashi et al., 2023). These results are consistent with previous reports showing that Pcdhγc4 (uniquely among Pcdhγ isoforms) requires other “carrier” cPcdhs to be transported to the cell-surface (Thu et al. 2014). While the mutant mice used by Kobayahsi et al. (2022) retain the Pcdhγc3 and Pcdhγc5 isoforms, these are unlikely to act as sufficient carriers due to low embryonic expression of Pcdhγc3 and Pcdhγc5 (Kobayashi et al. 2022) and low intra-family affinity for cis-interactions required for carriers (Goodman et al., 2022). During normal development, it is also possible that Pcdhγc4 forms heterodimers, in cis, with members of Pcdha and/or Pcdhb clusters, providing cellular specificity that is important for cIN survival. In both scenarios, our findings suggest a unique role in which Pcdhγc4 regulates programmed cell death. However, the mechanisms by which the Pcdhγc4 isoform regulates apoptosis among young cINs, leading to the adjustment of local circuit neuron numbers, remains unknown. Presumably, this isoform has unique intracellular signaling partners; precedents for this possibility comes from a recent study showing that PcdhγC3 plays a unique role in promoting cortical pyramidal neuron dendritic arborization through intracellular interactions with Axin1 (Steffen et al., 2023).
Together this work, along with previous studies, suggests that the final number of cINs in the cerebral cortex is in part determined through a cell-or population-intrinsic mechanism involving cell-cell interactions among cINs of the same age (Southwell et al., 2012). Given the key role Pcdhγc4 plays in the survival of cINs and the fact that Pcdhγc4 is a cell-adhesion molecule that is involved in homophilic cell-cell recognition (Thu et al., 2014), initial cell-cell interactions within the cIN population could be mediated through Pcdhγc4. Other studies have shown that programmed cell death is also regulated at least in part through activity-dependent mechanisms (Duan et al., 2020; Wong et al., 2018, 2022). It remains undetermined what role, if any, Pcdhγc4 plays in the regulation of activity during the peak of programmed cell death. In this regard, it would be interesting to determine whether network activity is perturbed in cINs lacking Pcdhγc4. In addition, loss of vesicular GABA release from cINs and the resulting reduction in inhibition leads to their increased participation in network activity and concomitantly to increased cIN survival (Duan et al., 2020). Hence it would also be interesting to remove the function of vesicular GABA release and of Pcdhγ in cINs and determine their survival during programmed cell death.
Appropriate numbers of cINs are considered essential in the modulation of cortical function. This is ultimately adjusted by a period of programmed cell death once the young cINs have migrated into the cortical plate and have begun to make synaptic connections. Here, we identify the Pcdhγc4 isoform as an essential molecular component in the regulation of cIN programmed cell death. It is likely that some of the primary roles that Pcdhγc4 has in the adjustment of cIN cell number to match the number of excitatory neurons apply also to the human cerebral cortex. Recent work suggests that Pcdhγc4 plays a critical role in human brain development as mutations in this gene are associated with a neurodevelopmental syndrome with progressive microcephaly and seizures (Iqbal et al., 2021). An understanding of the cell-cell interactions that use Pcdhγc4 to regulate cIN cell death should give fundamental insights into how the cerebral cortex forms and evolves.
Methods
Animals.
All protocols and procedures followed the University of California, San Francisco (UCSF) guidelines and were approved by the UCSF Institutional Animal Care Committee. The following breeders were purchased from the Jackson Laboratory: Ai6, Ai14, Gad1-GFP, BAC-Nkx2.1-Cre (Nkx2.1Cre), and WT C57BL/6J. Pcdhγ-fcon3 (FCON3) mice were obtained from Joshua Sanes at Harvard University. Pcdhγ1R1/1R1(1R1), PcdhγC4KO/C4KO(C4KO) and Pcdhγ3R2/3R2(3R2) mice were transferred from the Weiner laboratory at the University of Iowa and re-derived, and bred at UCSF.
Pcdhγ loss of function mice were obtained by crossing Pcdhγfcon3/fcon3 mice with Nkx2.1Cre-Ai14-Pcdhγfcon3/+. Pcdhγ1R1/1R1(1R1) and PcdhγC4KO/C4KO(C4KO) mice were crossed to Nkx2.1Cre-Ai14 breeders to label the Nkx2.1 progenitor cells.
For cell transplantation experiments, GFP-expressing cIN in embryos was produced by crossing WT C57BL/6J to heterozygous mice expressing green fluorescent protein-expressing (GFP) driven by Gad1 or by crossing Nkx2.1Cre;Ai6 breeder to WT C57BL/6J mice. For all, tdTomato-expressing cells derived from MGE embryonic dissections we crossed the various mutant alleles to the Nkx2.1Cre-Ai14 line. Pcdh-γfcon3/fcon3 mutant embryonic tissue was obtained from embryos produced by crossing Nkx2.1Cre-Ai14-Pcdhγfcon3/+ mice with Pcdhγfcon3/fcon3 mice. Pcdhγ1R1/1R1 mutant tissue was obtained from embryos produced by crossing Nkx2.1Cre-Ai14-Pcdhγ1R1/1R1 breeders. PcdhγC4KO/C4KO mutant tissue was obtained from embryos produced by crossing Nkx2.1Cre-Ai14-PcdhγC4KO/+ breeders. Gad1-GFP, Nkx2.1Cre; Ai6 and Nkx2.1Cre-Ai14 offspring were genotyped under an epifluorescence microscope (Leica), and PCR genotyping was used to screen for Pcdhγfcon3/fcon3, Pcdhγ1R1/1R1 and PcdhγC4KO/C4KO alleles in tdTomato positive embryos or reporter negative embryos. All cell transplantation experiments were performed using wild type C57Bl/6 recipient mice P2 to P8. All mice were housed under identical conditions.
Plasmids.
The following plasmids were used in this work: pLenti-CAG-EGFP, pLenti-CAG-Pcdγc4-EGFP.
Timed pregnant mice.
In experiments requiring timed pregnant mice, the day when the sperm plug was observed was considered E0.5. Males were paired with females the night before and checked for plugs early the following day. MGE cells for transplantation were dissected from the fetal forebrain between E12.5 - E15.5 embryos as previously described (Southwell et al., 2012).
Transplantation.
For co-transplantation experiments, the concentration of cells of each genotype was determined using a hemocytometer; the GFP or tdTomato labeled cells were then mixed in similar proportions. To prepare the cells for transplantation, the cells suspension was concentrated by spinning in a table centrifuge for five minutes at 800g(rcf). The supernatant was removed and the resulting pellet was resuspended and mixed in a final volume of 1–6μL of Leibovitz L-15 medium (L15). This concentrated cell suspension was loaded into beveled glass micropipettes (~60–100 μm diameter, Wiretrol 5 μL, Drummond Scientific Company) prefilled with mineral oil and mounted on a microinjector as previously described (Wichterle et al., 1999). The viability and concentration of the cells in the glass micropipette was determined by using 100nl of cells diluted 200X in 10μL of L15 medium and 10μL trypan blue. The number of cells was then determined using a hemocytometer. Cells were injected into neonate mice P2 to P8. Prior to the injection of cells, the recipient mice (C57Bl/6) were anesthetized by hypothermia (~3–5 minutes) and positioned in a clay head mold that stabilizes the skull (Merkle et al., 2007). Micropipettes were positioned at an angle of 0 or 30 degrees from vertical in a stereotactic injection apparatus, and injected between 4–6mm A/P and 0.5 to1.5mm M/L and 0.8 to 0.5 mm in the Z direction. After the injections were completed, the transplanted mice were placed on a warm surface to recover from hypothermia. The mice were then returned to their mothers. Transplantation of cells involving the PcdhγC4KO/C4KO alleles was performed utilizing cells from MGEs that had been cryopreserved (Rodríguez-Martínez et al., 2017).
Tissue dissection and cell dissociation.
MGEs were dissected from E12.5 to E15.5 embryos as previously described (Southwell et al., 2012). Dissections were performed in ice-cold L15 medium and the dissected MGEs were kept in this medium at 4°C. After collecting and genotyping the embryos, MGE with the same genotype were pulled together and mechanically dissociated into a single cell suspension by repeated pipetting in L15 medium containing DNAse I (180μg/ml)using a P1000 pipette. For experiments involving cells from cryopreserved MGEs, cryovials were removed from −80°C, thawed at 37°C for 5 minutes and the content of each tube was resuspended in a 15mL falcon tube containing L15 medium at room temperature. The MGE were washed twice with L15 to remove residual DMSO and dissociated as above.
Cryopreservation of MGE in toto.
Dissected MGEs from each embryo were collected in 500 μL L15 medium and kept on ice until cryopreserved. MGEs were resuspended in 10% DMSO in L15 medium and cryopreserved as previously described (Rodríguez-Martínez et al., 2017). Vials were cooled to −80°C at a rate of −1°C/minute in a Nalgene™ Mr. Frosty Freezing Container and transferred the next day to liquid nitrogen for long term storage. Importantly, tissue for genotyping was collected from each embryo and labeled with a code name matching the codename of each cryotube used for the cryopreservation.
Cell counting.
GFP positive cells and tdTomato-positive cells were counted in all layers of the neocortex. Cells that did not display neuronal morphology were excluded from all quantifications. The vast majority of cells transplanted from the E13.5 MGE exhibited neuronal morphologies in the recipient brain. Cells at the injection site were not counted. Most cells migrated away from the injection site, but few remained trapped. Cells outside the cortex were not included in the quantification. In some injections, cells migrated to other parts of the brain, including the hippocampus and striatum. Images of co-transplanted cells were acquired on a SP8 (Leica) confocal microscope with a 10X magnification. Images of transplanted cells infected with lentivirus expressing the Pcdhγc4 were acquired on an SP8 confocal with a 10X magnification and digital zoom=2x. Only the region of the cortex containing GFP or tdTomato-positive cells was imaged. GFP and tdTomato-positive cells were counted using Neurolucida (MBF). For all experiments, transplanted cells were counted from coronal sections along the rostral-caudal axis and in at least 10 sections were counted per animal; only sections containing more than 10 cells per condition were used. Data is presented as the fraction of transplant-derived cells that express GFP and/or tdTomato. This fraction does not reflect the absolute number of cells, but their relative contribution to the overall population of transplant-derived cells at different DAT. For experiments involving the expression of lentiviral driven GFP or Pcdhγ in transplanted cINs, survival was determined by comparing the fraction of infected (tdTomato+GFP+) cells to the fraction of noninfected cells (tdTomato+GFP-) from the total number of transplanted cells (tdTomato+) in each brain section.
Viral vector subcloning.
All lentiviral plasmids were cloned from the backbone construct pLenti-CAG-ires-EGFP (addgene plasmid #122953). A Kozak sequence was added at the start of the coding region for all genes cloned. Plasmids were cloned using the Gibson Cloning Kit (NEB). The pLenti-CAG-EGFP construct was cloned by removing the ires sequence in between the BxtXI and BamHI restriction sites from the backbone construct. To clone the pLenti-CAG-Pcdhγc4-EGFP (fusion construct), the pLenti-CAG-ires-EGFP plasmid was digested with BamHI and BstXI to remove the IRES sequence. A PCR generated Pcdhγc4 coding sequence lacking the stop codon and containing a two amino acid (Ser, Arg) linker was cloned upstream of the GFP coding sequence. This construct was used to express Pcdhγc4 fused to GFP.
Viruses.
All lentivirus used in this study were made in the laboratory. Briefly viruses were produced in Lenti-X 293T cells (TakaraBio). Cells were grown to ≥90% confluency in maintenance media (DMEM/F-12, 15 mM HEPES, 2.5 mM GlutaMAX, 1% Pen/Strep, 10% FBS) in 15cm plates coated with Poly-D-lysine (Sigma-Aldrich P7405) at final concentration of 0.1mg per mL. Once cells reached the desired confluency, media was changed to DMEM/F-12 + 2% FBS. Cells were transfected using TransIT®-293 Transfection Reagent (Mirusbio) and Opti-MEM (Thermofisher). After 6 to 12 hours post-transfection, the media was changed to lentivirus production media (DMEM/F-12, 15 mM HEPES, 2.5 mM GlutaMAX, 1% Pen/Strep, 2% FBS) and 60μLof ViralBoost Reagent was added (ALSTEM) per 15cm plate. Virus supernatant was collected 48 hour post-transfection, filtered through a 45μm filter, and precipitated with Lentivirus Precipitation Solution (ALSTEM) overnight following manufacturer’s instructions. The viral pellet from two 15cm plates was concentrated in a final volume of 100μl for Pcdh constructs and 200μl for the control construct.
Viral infection of MGE precursor cells.
Following the dissociation of the MGEs, cells were concentrated by spinning in a table centrifuge for five minutes at 800g. The cell pellet was subsequently resuspended in an equal volume of Lentivirus and L15 medium. The cell-virus mix was incubated at 22–32°C at 1000rpm (190rcf) for 3 hours, mixing every 30 minutes.
Immunostaining.
P21 and older mice were fixed by transcardiac perfusion with 10mL of ice-cold PBS followed by 10mL of 4% formaldehyde/PBS solution; transcardiac perfusion of 5ml of either solution was used for P15 and younger mice. Brains were incubated overnight in the same fixative (12–24hrs) at 4°C, then rinsed with PBS and cryoprotected in 30% sucrose/PBS solution for 48 hours at 4°C. Unless otherwise stated, immunohistochemistry was performed on 30 μm floating sections in Tris Buffered Saline (TBS) solution containing 10% normal donkey serum, 0.5% Triton X-100 for all procedures on postnatal mice. All washing steps were done in 0.1% Triton X-100 TBS for all procedures. Sections were incubated overnight at 4°C with selected antibodies, followed by incubation at 4°C overnight in donkey secondary antibodies (Jackson ImmunoResearch Laboratories). Brain sections that had been transplanted with lentivirus infected MGE cells were incubated two nights in primary antibodies and overnight with secondary antibodies to enhance the viral expressed reporter GFP. For cell counting and post hoc examination of marker expression, sections were stained using chicken anti-GFP (1:2500, Aves Labs, GFP-1020, RRID:AB_10000240), rabbit anti-RFP (Rockland), rat anti-tdTomato (Kerafast). For staining with the Pcdhγc5 antibody, tissue fixation was performed as stated above. Sixteen um sections of cortical tissue were dried directly onto paraffin-subbed slides before being washed 3X for 5 min. each in TBS. Sections were incubated in a blocking solution with 2.5% BSA, 0.5% TritonX, in TBS for 2 hours at room temp. Slides were removed from the blocking buffer and washed again 3X for 5 min. Primary antibodies were diluted in 2.5% BSA 0.25% TritonX 0.02% in TBS overnight at 4°C. Antibodies used; Pcdhγc5 (Invitrogen, 1:150, RRID:AB_2724958), Ctip2 (Abcam, 1:200, PRID:AB_2064130), Cux1 (Santa Cruz Biotechnology, 1:100 AB_2261231), GABA ( Sigma-Aldrich,1:250, RRID:AB_477652). Sections were then washed 3X for 5 min. before being incubated with host-appropriate secondaries for 2 hours at room temp at a 1:500 dilution in.
RNAScope.
Brains of Nkx2.1Cre; Ai6 animals at multiple ages (P5, P7, and P14) were fixed, postfixed, and cryoprotected (as described in Immunostaining). Brains were cryosectioned and mounted directly onto microscope slides before being stored at −80 °C. The RNA scope experiments were completed as per the manufacturer’s instructions (RNAscope multiplex fluorescent reagent kit v2, Advanced Cell Diagnostics, Inc.). Sections were equilibrated to room temperature by one 10 min incubation at −20°C before a 10 min incubation at room temperature. Brain sections were dehydrated using increasing concentrations of Ethanol (50%, 70% and 100%) for 5 min incubations at room-temperature. Sections were then incubated with RNAscope Hydrogen Peroxide solution for 10 min. at room temperature, before 4 washes in distilled water. To perform the target retrieval step of the protocol, slides were incubated in distilled water for 10 seconds and then RNAscope 1X target retrieval buffer for 15 min. using an Oyster brand steamer at ~99°C. Sections were then washed for 15 seconds in distilled water before being transferred to 100% ethanol for 3 min. at room-temperature. Slides were then dried in an incubator at 60°C for 5 min. In order to reduce reagent usage, a hydrophobic barrier pen was used to surround the tissue. After the barrier was dry, tissue was incubated in ~5 drops of RNAscope Protease III, and incubated at 40°C for 30 min. using a HybEZ Humidity Control Tray and HybEZ oven.
Tissue was then incubated in the Pcdhγc4 (probe 835791-C3) and Pcdhγc5 (probe 1118501-C2) probes for 2 hours at 40°C. After incubation, Probes were hybridized with AMP1, AMP2, and AMP3 (in that order) through individual incubations at 40°C for 30 min (AMP3 incubation 15 min. incubation). Our probes were Channel-2 (Pcdhγc5) and Channel 3 (Pcdhγc4), so we skipped development HRP-C1 signal and continued directly to the development of the HRP-C2 stepRNAscope Multiplex FL v2 HRP-C2. Tissue was incubated with 3–4 drops of RNAscope multiplex v2 HRP-C2 solution for 15 min at 40°C, followed by a 2 min wash in 1X RNA scope wash buffer. Then, tissue was incubated with Cy3 (Thermo Fisher Scientific, 1:1000) diluted in TSA buffer (Advanced Cell Diagnostics, Inc.), followed by another 2 min wash in 1X Wash Buffer. This process was completed again using the RNAscope multiplex v2 HRP-C3 solution and Cy5 (Thermo Fisher Scientific, 1:1000). Slides were washed 3X in distilled water for 5 min., before being mounted using Fluoromount-G, DAPI mounting media.
Analysis of Previously Published Single Cell RNA Sequencing Datasets.
Data from Tasic et al., 2018 was obtained via the Broad Institute Single Cell Portal (https://singlecell.broadinstitute.org/single_cell/study/SCP6/a-transcriptomic-taxonomy-of-adult-mouse-visual-cortex-visp). Cells were grouped by GABAergic vs Glutamaterigc designation or cluster-type, as determined by the data generators. RPMK values for each cell in each cluster-type were averaged and plotted using PRISM. Data obtained from Yao et al., 2021 was obtained from the UCSC Cell Browser (https://cells.ucsc.edu/?ds=allen-celltypes+mouse-cortex+mouse-cortex-2019). Cells cells were grouped based on either GABAergic or Glutamatergic designations or by cluster-type determined by the data generators. Absolute transcript level Absolute transcript values for each cell in each cluster-type were averaged and plotted using PRISM.
Statistical Analysis.
Quantifications were performed by two different people, one of which was blinded to the genotype. For transplantation experiments, we estimated the average mutant and WT survival ratios in each mouse by counting the transplanted mutant and wild-type cells in each of ~10 sections. We used an ANOVA to determine whether the survival ratios in 6DAT mice were significantly different from those in 21DAT mice.
Supplementary Material
Acknowledgements
This work was supported by NIH Grant R01MH122478 to A.A.B., A.R.H., and M.P.S.; NIH Grants R01 NS028478 and R01 EY02517; and a generous gift from the John G Bowes Research Fund to A.A.B.; National Institutes of Health Grants R01 NS055272 and R21 NS090030 to J.A.W.; National Institutes of Health Grants R01NS116598; Hearing Research Inc.; the PBBR Breakthrough Fund; and the Coleman Memorial Fund. to A.R.H.; NIH Grants R01EY002874 and R01EY031059 to M.P.S. A.A.B. is the Heather and Melanie Muss Endowed Chair and Professor of Neurological Surgery at UCSF. M.P.S. is a recipient of the Research to Prevent Blindness Disney Award for Amblyopia Research.
References
- Ben-Ari Y., Khalilov I., Represa A., & Gozlan H. (2004). Interneurons set the tune of developing networks. Trends in Neurosciences, 27(7), 422–427. [DOI] [PubMed] [Google Scholar]
- Carriere C. H., Sing A. D., Wang W. X., Jones B. E., Yee Y., Ellegood J., Marocha J., Maganti H., Awofala L., Aziz A., Lerch J. P., & Lefebvre J. L. (2020). The gamma-Protocadherins regulate the survival of GABAergic interneurons during developmentally-regulated cell death. In bioRxiv (p. 2020.01.15.908087). 10.1101/2020.01.15.908087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao H.-T., Chen H., Samaco R. C., Xue M., Chahrour M., Yoo J., Neul J. L., Gong S., Lu H.-C., Heintz N., Ekker M., Rubenstein J. L. R., Noebels J. L., Rosenmund C., & Zoghbi H. Y. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature, 468(7321), 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W. V., Alvarez F. J., Lefebvre J. L., Friedman B., Nwakeze C., Geiman E., Smith C., Thu C. A., Tapia J. C., Tasic B., Sanes J. R., & Maniatis T. (2012). Functional Significance of Isoform Diversification in the Protocadherin Gamma Gene Cluster. Neuron, 75(3), 402–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors B. W., Benardo L. S., & Prince D. A. (1983). Coupling between neurons of the developing rat neocortex. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 3(4), 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Z. R. S., Che A., Chu P., Modol L., Bollmann Y., Babij R., Fetcho R. N., Otsuka T., Fuccillo M. V., Liston C., Pisapia D. J., Cossart R., & De Marco García N. V. (2020). GABAergic Restriction of Network Dynamics Regulates Interneuron Survival in the Developing Cortex. Neuron, 105(1), 75–92.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett A. M., Bosch P. J., Steffen D. M., Fuller L. C., Marcucci C. G., Koch A. A., Bais P., Weiner J. A., & Burgess R. W. (2019). CRISPR/Cas9 interrogation of the mouse Pcdhg gene cluster reveals a crucial isoform-specific role for Pcdhgc4. PLoS Genetics, 15(12). 10.1371/journal.pgen.1008554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett A. M., Schreiner D., Lobas M. A., & Weiner J. A. (2012). γ-protocadherins control cortical dendrite arborization by regulating the activity of a FAK/PKC/MARCKS signaling pathway. Neuron, 74(2), 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman K. M., Katsamba P. S., Rubinstein R., Ahlsén G., Bahna F., Mannepalli S., Dan H., Sampogna R. V., Shapiro L., & Honig B. (2022). How clustered protocadherin binding specificity is tuned for neuronal self-/nonself-recognition. eLife, 11. 10.7554/eLife.72416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal M., Maroofian R., Çavdarlı B., Riccardi F., Field M., Banka S., Bubshait D. K., Li Y., Hertecant J., Baig S. M., Dyment D., Efthymiou S., Abdullah U., Makhdoom E. U. H., Ali Z., Scherf de Almeida T., Molinari F., Mignon-Ravix C., Chabrol B., … Yigit G. (2021). Biallelic variants in PCDHGC4 cause a novel neurodevelopmental syndrome with progressive microcephaly, seizures, and joint anomalies. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 23(11), 2138–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katori S., Noguchi-Katori Y., Okayama A., Kawamura Y., Luo W., Sakimura K., Hirabayashi T., Iwasato T., & Yagi T. (2017). Protocadherin-αC2 is required for diffuse projections of serotonergic axons. Scientific Reports, 7(1), 15908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H., Takemoto K., Sanbo M., Hirabayashi M., Hirabayashi T., Hirayama T., Kiyonari H., Abe T., & Yagi T. (2023). Isoform requirement of clustered protocadherin for preventing neuronal apoptosis and neonatal lethality. iScience, 26(1), 105766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre J. L., Zhang Y., Meister M., Wang X., & Sanes J. R. (2008). gamma-Protocadherins regulate neuronal survival but are dispensable for circuit formation in retina. Development, 135(24), 4141–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis D. A., Hashimoto T., & Volk D. W. (2005). Cortical inhibitory neurons and schizophrenia. Nature Reviews. Neuroscience, 6(4), 312–324. [DOI] [PubMed] [Google Scholar]
- Li Y., Serwanski D. R., Miralles C. P., Fiondella C. G., Loturco J. J., Rubio M. E., & De Blas A. L. (2010). Synaptic and nonsynaptic localization of protocadherin-gammaC5 in the rat brain. The Journal of Comparative Neurology, 518(17), 3439–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L., Zwingman T. A., Sunkin S. M., Oh S. W., Zariwala H. A., Gu H., Ng L. L., Palmiter R. D., Hawrylycz M. J., Jones A. R., Lein E. S., & Zeng H. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature Neuroscience, 13(1), 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancia Leon W. R., Spatazza J., Rakela B., Chatterjee A., Pande V., Maniatis T., Hasenstaub A. R., Stryker M. P., & Alvarez-Buylla A. (2020). Clustered gamma-protocadherins regulate cortical interneuron programmed cell death. eLife, 9. 10.7554/eLife.55374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín O. (2012). Interneuron dysfunction in psychiatric disorders. Nature Reviews. Neuroscience, 13(2), 107–120. [DOI] [PubMed] [Google Scholar]
- Merkle F. T., Mirzadeh Z., & Alvarez-Buylla A. (2007). Mosaic organization of neural stem cells in the adult brain. Science, 317(5836), 381–384. [DOI] [PubMed] [Google Scholar]
- Molumby M. J., Keeler A. B., & Weiner J. A. (2016). Homophilic Protocadherin Cell-Cell Interactions Promote Dendrite Complexity. Cell Reports, 15(5), 1037–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountoufaris G., Chen W. V., Hirabayashi Y., O’Keeffe S., Chevee M., Nwakeze C. L., Polleux F., & Maniatis T. (2017). Multicluster Pcdh diversity is required for mouse olfactory neural circuit assembly. Science, 356(6336), 411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad T., Wang X., Gray P. A., & Weiner J. A. (2008). A differential developmental pattern of spinal interneuron apoptosis during synaptogenesis: insights from genetic analyses of the protocadherin-γ gene cluster. Development, 135(24), 4153–4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priya R., Paredes M. F., Karayannis T., Yusuf N., Liu X., Jaglin X., Graef I., Alvarez-Buylla A., & Fishell G. (2018). Activity Regulates Cell Death within Cortical Interneurons through a Calcineurin-Dependent Mechanism. Cell Reports, 22(7), 1695–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Martínez D., Martínez-Losa M. M., & Alvarez-Dolado M. (2017). Cryopreservation of GABAergic Neuronal Precursors for Cell-Based Therapy. PloS One, 12(1), e0170776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol E. (2011). Genetics and function of neocortical GABAergic interneurons in neurodevelopmental disorders. Neural Plasticity, 2011, 649325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein J. L. R., & Merzenich M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes, Brain, and Behavior, 2(5), 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seress L., Frotscher M., & Ribak C. E. (1989). Local circuit neurons in both the dentate gyrus and Ammon’s horn establish synaptic connections with principal neurons in five day old rats: a morphological basis for inhibition in early development. In Experimental Brain Research (Vol. 78, Issue 1). 10.1007/bf00230680 [DOI] [PubMed] [Google Scholar]
- Southwell D. G., Paredes M. F., Galvao R. P., Jones D. L., Froemke R. C., Sebe J. Y., Alfaro-Cervello C., Tang Y., Garcia-Verdugo J. M., Rubenstein J. L., Baraban S. C., & Alvarez-Buylla A. (2012). Intrinsically determined cell death of developing cortical interneurons. Nature, 491(7422), 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen D. M., Hanes C. M., Mah K. M., Ramos P. V., Bosch P. J., Hinz D. C., Radley J. J., Burgess R. W., Garrett A. M., & Weiner J. A. (2023). A unique role for Protocadherin γC3 in promoting dendrite arborization through an Axin1-dependent mechanism. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 10.1523/JNEUROSCI.0729-22.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B., Yao Z., Graybuck L. T., Smith K. A., Nguyen T. N., Bertagnolli D., Goldy J., Garren E., Economo M. N., Viswanathan S., Penn O., Bakken T., Menon V., Miller J., Fong O., Hirokawa K. E., Lathia K., Rimorin C., Tieu M., … Zeng H. (2018). Shared and distinct transcriptomic cell types across neocortical areas. Nature, 563(7729), 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thu C. A., Chen W. V., Rubinstein R., Chevee M., Wolcott H. N., Felsovalyi K. O., Tapia J. C., Shapiro L., Honig B., & Maniatis T. (2014). Generation of single cell identity by homophilic interactions between combinations of α, β and γ protocadherins. Cell, 158(5), 1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzio R., Represa A., Jorquera I., Ben-Ari Y., Gozlan H., & Aniksztejn L. (1999). The establishment of GABAergic and glutamatergic synapses on CA1 pyramidal neurons is sequential and correlates with the development of the apical dendrite. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 19(23), 10372–10382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L., Mann E. O., Hang G. B., Barth A. M. I., Cobos I., Ho K., Devidze N., Masliah E., Kreitzer A. C., Mody I., Mucke L., & Palop J. J. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell, 149(3), 708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Weiner J. A., Levi S., Craig A. M., Bradley A., & Sanes J. R. (2002). Gamma protocadherins are required for survival of spinal interneurons. Neuron, 36(5), 843–854. [DOI] [PubMed] [Google Scholar]
- Wichterle H., Garcia-Verdugo J. M., Herrera D. G., & Alvarez-Buylla A. (1999). Young neurons from medial ganglionic eminence disperse in adult and embryonic brain. Nature Neuroscience, 2(5), 461–466. [DOI] [PubMed] [Google Scholar]
- Wong F. K., Bercsenyi K., Sreenivasan V., Portalés A., Fernández-Otero M., & Marín O. (2018). Pyramidal cell regulation of interneuron survival sculpts cortical networks. Nature, 557(7707), 668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong F. K., Selten M., Rosés-Novella C., Sreenivasan V., Pallas-Bazarra N., Serafeimidou-Pouliou E., Hanusz-Godoy A., Oozeer F., Edwards R., & Marín O. (2022). Serotonergic regulation of bipolar cell survival in the developing cerebral cortex. Cell Reports, 40(1). 10.1016/j.celrep.2022.111037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q., & Maniatis T. (1999). A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell, 97(6), 779–790. [DOI] [PubMed] [Google Scholar]
- Yang J.-M., Zhang J., Yu Y.-Q., Duan S., & Li X.-M. (2012). Postnatal Development of 2 Microcircuits Involving Fast-Spiking Interneurons in the Mouse Prefrontal Cortex. Cerebral Cortex, 24(1), 98–109. [DOI] [PubMed] [Google Scholar]
- Yao Z., van Velthoven C. T. J., Nguyen T. N., Goldy J., Sedeno-Cortes A. E., Baftizadeh F., Bertagnolli D., Casper T., Chiang M., Crichton K., Ding S.-L., Fong O., Garren E., Glandon A., Gouwens N. W., Gray J., Graybuck L. T., Hawrylycz M. J., Hirschstein D., … Zeng H. (2021). A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell, 184(12), 3222–3241.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.