

Abstract
The mammalian liver harbors heterogeneous cell types that communicate via local paracrine signaling. Recent studies have delineated the transcriptomic landscape of the liver in nonalcoholic steatohepatitis (NASH) that provides insights into liver cell heterogeneity, intercellular crosstalk, and disease-associated reprogramming. However, the nature of intrahepatic signaling and its role in NASH progression remain obscure. Here we identified Cardiotrophin like cytokine factor 1 (CLCF1), a member of the IL-6 family cytokines, as a cholangiocyte-derived paracrine factor that was elevated in the liver from diet-induced NASH mice and NASH patients. AAV-mediated overexpression of CLCF1 in the liver ameliorated NASH pathologies in two diet-induced NASH models in mice, illustrating that CLCF1 induction may serve an adaptive and protective role during NASH pathogenesis. Unexpectedly, mRNA and protein levels of leukemia inhibitory factor receptor (LIFR), a subunit of the receptor complex for CLCF1, were markedly downregulated in NASH liver. Hepatocyte-specific inactivation of LIFR accelerated NASH progression in mice, supporting an important role of intrahepatic cytokine signaling in maintaining tissue homeostasis under metabolic stress conditions. Together, this study sheds light on the molecular nature of intrahepatic paracrine signaling during NASH pathogenesis and uncovers potential targets for therapeutic intervention.
Graphical Abstract
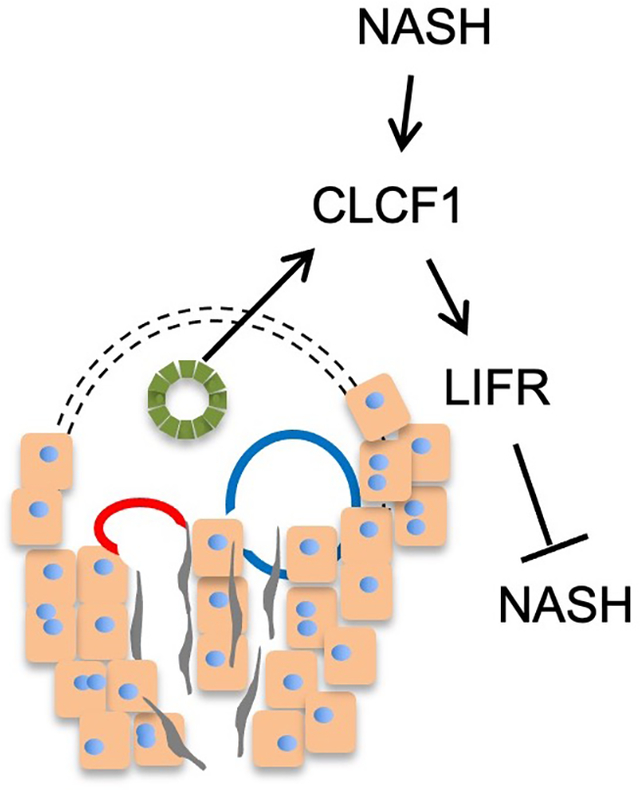
Introduction
Nonalcoholic fatty liver disease (NAFLD) represents a spectrum of metabolic liver disorders commonly associated with metabolic syndrome that affect over 25% of adults globally (1–4). Chronic fat accumulation in the liver, a condition known as hepatic steatosis, may exist in a clinically benign state, but frequently progresses to nonalcoholic steatohepatitis (NASH). The latter is a more severe metabolic liver disease that features persistent liver injury, lobular inflammation, and liver fibrosis. Patients with NASH are exposed to increased risk for end-stage liver disease, such as cirrhosis and hepatocellular carcinoma, and cardiovascular disease. While the regulatory framework that governs hepatic lipid metabolism has been elucidated in detail (5, 6), the pathophysiological mechanisms underlying the transition from steatosis to NASH and the progressive remodeling of the liver immune and stromal microenvironment during NASH remain to be established. The therapeutic options for NASH remain limited at present despite its prevalence and severe clinical sequelae.
A notable feature of NASH is the progressive remodeling of liver microenvironment involving stromal vascular and immune cell populations. Transcriptomic studies using liver biopsy samples have uncovered core gene signatures associated with human NASH, most prominently the pathways associated with hepatic metabolism, inflammatory response, and liver fibrosis (7–9). Remarkably, key molecular signatures of NASH were observed in diet-induced NASH in mice, suggesting that mouse and human NASH share common pathophysiological mechanisms (10, 11). The mammalian liver comprises heterogeneous cell types within its tissue microenvironment, including parenchymal hepatocytes and non-parenchymal cells (NPCs). Recent single-cell RNA sequencing (sc-RNAseq) studies have elucidated the transcriptomic landscape of major liver cell types, including hepatocyte, endothelial cell, hepatic stellate cell (HSC), cholangiocyte, and diverse immune cell types (12–14). A notable feature of liver cells is their spatial heterogeneity, which has been observed in hepatocytes (15, 16), endothelial cells (17), HSC (18, 19), and macrophages (20–23). Different cell types exhibit distinct expression patterns for genes encoding secreted ligands and membrane receptors that are highly conserved between mouse and human. These findings illustrate the complexity of pervasive cell-cell crosstalk and potential importance of intrahepatic paracrine signaling in maintaining tissue homeostasis and modulating disease progression (23).
The interleukin 6 (IL-6) family cytokines regulate diverse biological processes, including immune responses, development, metabolism, and tissue homeostasis (24–26). Hepatocyte-specific deletion of GP130 or STAT3, a transcription factor mediating downstream cytokine signaling, exacerbates liver injury in mice (27, 28). IL-6 exerts complex effects on hepatic and systemic metabolism and insulin sensitivity (29). Blockade of IL6 signaling using neutralizing antibodies accelerates liver fibrogenesis in MCD diet-fed db/db mice (30). Consistently, hepatocyte-specific inactivation of IL-6Ra accelerates HFD-induced liver injury and fibrosis (31). Recombinant ciliary neurotrophic factor (CNTF) improved diabetic parameters and reduced hepatic steatosis in db/db mice (32). Adenovirus-mediated cytokine receptor-like factor 1 overexpression suppresses type III collagen expression in mouse liver (33). A recent study demonstrated that a designer cytokine combining signaling features of IL-6 and CNTF elicits strong protective effects against diet-induced metabolic disorders in mice (34). Although most of IL-6 family cytokines have been shown to be beneficial to hepatic metabolism and liver fibrogenesis, oncostatin M (Osm) overexpression in mouse liver promotes development of severe fibrosis (35), illustrating complex action of the IL-6 family cytokines in hepatic metabolism and liver disease. In this study, we analyzed NASH-associated transcriptomic remodeling in diet-induced mouse liver and identified the IL-6 family cytokine Cardiotrophin like cytokine factor 1 (CLCF1) as a cholangiocyte-derived secreted factor that plays a protective role of NASH pathogenesis.
Methods
Animal studies
All animal studies were performed using procedures approved by Institutional Animal Care & Use Committee at the University of Michigan. Mice were maintained in a selective pathogen-free housing facility under 12-hr dark and 12-hr light cycle with free access to food and water. Mice were fed standard chow (Teklad 5001 Laboratory Diet), NASH diet that contains 40% fat (of which 18% was trans-fat), 22% fructose, and 2% cholesterol (D09100301, Research Diets Inc.), or a choline-deficient, amino acid-defined (0.1% methionine) high-fat diet (CDA-HFD, A06071309, Research Diets Inc.) to induce NASH, as previously described (23, 36). The mouse strain harboring Rosa26-floxed-STOP-flox-Cas9 knockin cassette (Rosa26-FSF-Cas9, The Jackson laboratory, strain #024857) was previously described (37). Hepatocyte-specific Cas9 transgenic mice were generated by crossing this strain with Albumin-Cre transgenic mice (38). Albumin Cre+/Rosa26-FSF-Cas9 (Alb-Cre+) and Cre−/Rosa26-FSF-Cas9 littermates were transduced with a recombinant adenovirus-associated virus (AAV) vector expressing two single guide RNAs (sgRNAs) targeting Lifr gene to generate hepatocyte-specific Lifr knockout (HepKO) and control mice, respectively. The sequence for sgRNAs used for Lifr targeting are: sgRNA1: GTTCAGCTATCGTCTCAAAT; sgRNA2: CATCTGACGATGCATGCAAA. For CLCF1 overexpression studies, adult male mice at 3–4 months of age were transduced with AAV-GFP or AAV-CLCF1 (1 × 10 11 genome copies/mouse) via tail vein injection.
NASH phenotype analysis
NASH phenotype in diet-induced NASH mice was analyzed as we previously described (39). Plasma AST and ALT levels were measured using assay kits from Teco Diagnostics (A524-150) and Wako (995-34791), respectively. For histological analysis, mouse liver was fixed in 3.7% formalin for overnight followed by paraffin-embedding. Liver sections were stained with hematoxylin and eosin (H&E) or Picrosirius (Sirius) red (Polysciences, #24901).
Primary hepatocyte culture
Primary hepatocytes were isolated from perfused wild-type mouse liver following collagenase type II digestion. Hepatocytes were cultured in DMEM containing 10% bovine serum and switched to serum-free medium prior to CLCF1 treatment (100 ng/ml, R&D 962-CL-050).
RNAscope in situ hybridization
Liver was fixed in 10% neutral-buffered formalin after dissection followed by formalin-fixed paraffin-embedded (FFPE) protocol. Liver sections were cut at 10μm for RNAScope and immunofluorescence staining. Briefly, liver sections were processed by dewaxing, rehydration, endogenous enzyme blockage (ACDbio, Hydrogen Peroxide) and target antigen retrieval. For RNAscope, protease Plus was applied to section for 10 minutes at 40 μC. Clcf1 RNA was hybridized with mm-Clcf1 probe (ACDbio, Cat#457971) for two hours at 40 μC. Cy3 (1:2000 diluted in TSA dilution buffer, RNAscope detection kit) was used for detection. Following RNAScope signal amplification, liver sections were blocked in 5% donkey serum prior to incubation with SPP1 antibody (R&D, AF808). A secondary antibody (R&D, anti-rabbit 488) and DAPI were then applied to section before mounting.
Immunofluorescence staining
For LIFR, GPNMB, and Desmin immunofluorescence staining, liver sections were permeabilized with 0.3% Triton X-100 in PBS and then blocked in 5% BSA. Primary antibody (LIFR: Abcam, Ab101228; GPNMB: R&D AF2330; Desmin: ThermoFisher, R8-9014-P1) was applied to liver sections overnight at 4 μC. Liver sections were then incubated in secondary antibody solution at room temperature for one hour followed by mounting (Vector Laboratories, H-1000). Images were taken with Leica SP5 Confocal Imaging System and SP8 LIGHTNING Confocal Microscope System.
Gene expression analysis
Total liver RNA was extracted using TRIzol method. Equal amounts of RNA were reverse transcribed to cDNA using MMLV-RT prior to quantitative PCR (qPCR) using the SYBER green method (Life Technologies). The qPCR primers are listed in Supplementary Table S2.
Immunoblotting analysis
To prepare protein lysates, liver samples were homogenized in a lysis buffer that contains 50 mM Tris (pH 7.5), 150 mM NaCl, 5 mM NaF, 25 mM β-glycerol phosphate, 1 mM sodium orthovanadate, 10% glycerol, 1% Triton X-100, 1 mM dithiothreitol (DTT), and freshly added protease inhibitors (Roche). Primary hepatocytes were harvested in lysis buffer containing 2% sodium dodecyl sulfate (SDS), 50 mM Tris-HCl (pH 6.8), 10 mM DTT, 10% glycerol, 0.002% bromophenol blue, and freshly added protease inhibitors. For immunoblots, antibodies against phosphorylation of STAT3 (9131), STAT3 (9139), cleaved caspase3 (9664), and phosphorylation of JNK (81E11) were purchased from Cell Signaling Technology. Antibodies for LIFR (sc-515337), HSP90 (sc-13119), and actin (sc-8432) were purchased from Santa Cruz Biotechnology. GPNMB antibody (AF2330) was purchased from R&D Systems. TREM2 antibody was a gift from Dr. Marco Colonna.
Data analysis
Published RNA sequencing (RNAseq) datasets were downloaded from the Gene Expression Omnibus database using the following accession numbers: bulk RNAseq analysis of livers from mice fed chow or NASH diet: GSE119340 (11); single-cell RNAseq analysis of liver cells from chow and NASH diet-fed mice: GSE129516 (23); bulk RNAseq analysis of livers from mice fed chow or CDA-HFD diet: GSE128940 (40); liver nuclei RNAseq analysis of chow, NASH, or CDA-HFD-fed mice: GSE162876 (41); human liver bulk RNAseq analysis of individuals with normal weight, NAFLD, or NASH: GSE126848 (42). For bulk RNAseq analysis, DESeq2 was used for raw counts normalization and differential expressed genes identification (43). Gene enrichment analysis was performed using web-based resource (www.metascape.org). Heatmap and bubble plots were generated by R. Bar plots and dot plots were produced by GraphPad.
Statistical analysis
The bar plots were presented as mean ± SEM. Student t test p-value was calculated to estimate difference between two groups. Correlation analysis was performed by GraphPad Simple Linear Regression.
Results
Dysregulation of IL-6 family cytokine signaling in NASH liver
Paracrine signaling between hepatocytes and non-parenchymal cells is important for tissue homeostasis and its disruptions contribute to progression of liver diseases. We previously performed RNA sequencing and quantitative proteomic analyses on healthy and diet-induced NASH mouse livers and delineated core molecular signatures characteristic of NASH, including lipid metabolism, inflammatory response, and liver fibrosis (11). Despite this, the nature of intrahepatic paracrine factors that influence NASH pathogenesis remains poorly understood. To address this, we performed differential gene expression analysis on the secretome genes encoding putative extracellular ligands and membrane receptors, as described in previous studies (23, 44). We identified a total of 210 ligands and 248 receptors that exhibited altered expression in the liver following diet-induced NASH (Figure 1A and Supplemental Table S1). Gene ontology analysis indicated that these differentially expressed genes are enriched for cytokine signaling and inflammatory response, notably those in the interleukin 6 (IL-6) family cytokines and receptors (Figure 1B).
Figure 1. NASH is linked to altered IL-6 family cytokine signaling in the liver.
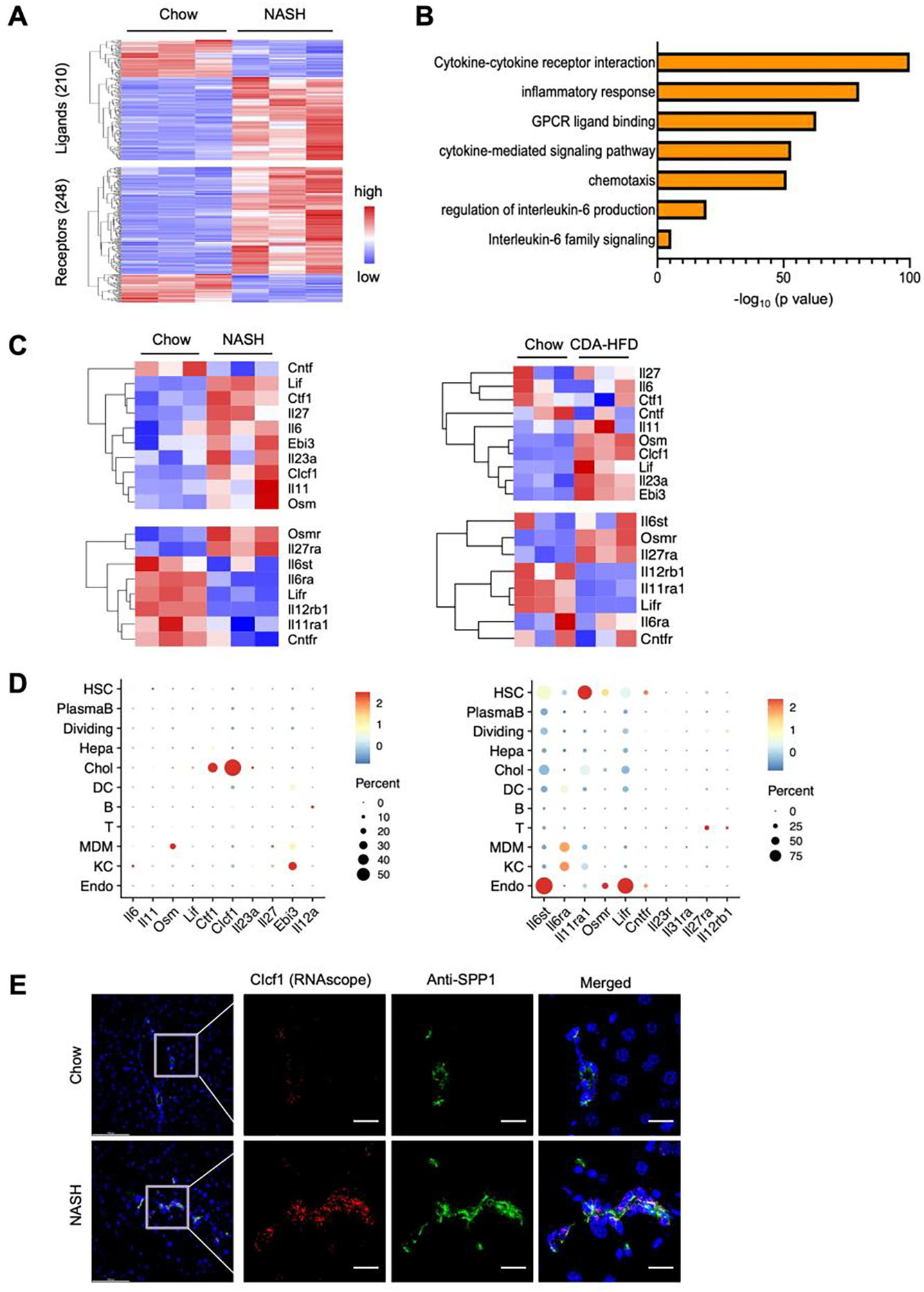
A) Heatmap of differentially expressed ligands (top) and receptors (bottom) based on bulk RNA sequencing analysis of the liver from mice fed chow or NASH diet for 20 weeks (GSE119340). Shown are genes that passed the filter of adjusted P-value < 0.05, absolute log2 (Fold change) > 0.5 and normalized count > 10 in at least one sample.
B) Pathway enrichment analysis of differential expressed ligands and receptors.
C) Heatmap of gene expression of IL-6 family members based on liver RNAseq data from mice fed chow or NASH diet for 20 weeks (left, GSE119340), and chow or CDA-HFD for 6 weeks (right, GSE128940).
D) Bubble plot of mRNA expression for IL-6 family ligands (left) and receptors (right) among different liver cell types based on single-cell RNAseq data (GSE129516).
E) RNAscope in situ hybridization (Clcf1) and immunofluorescence staining (anti-SPP1) of chow and NASH liver sections. Scale bar = 100μm.
The IL-6 family cytokines share a common signaling receptor subunit, glycoprotein 130 (gp130, encoded by Il6st), which activates downstream JAK/STAT signaling pathway to elicit biological effects in a cell type- and context-dependent manner (24–26). IL-6 is the prototypical cytokine in this family and is known to play an important role in hepatic metabolism, acute phase response, liver regeneration, and tumorigenesis (29). We therefore focused our analysis on members of the IL-6 family ligands and receptors that exhibit differential expression in healthy and NASH livers. Several IL-6 family ligands, including IL-6, Osm, Leukemia inhibitory factor (Lif), and Clcf1, exhibited increased mRNA expression in the liver upon NASH induction by NASH diet or choline-deficient L-methionine-defined high-fat diet (CDA-HFD) (Figure 1C). Interestingly, different subunits of the IL-6 family cytokine receptors displayed distinct patterns of NASH-associated alterations. Compared to control, mRNA expression of Osm receptor (Osmr) and Il27ra showed increased expression in NASH or CDA-HFD diet fed mouse livers. In contrast, hepatic expression of Lifr, Il12rb1, and Il11ra1 were downregulated in both models of diet-induced NASH. These results suggest that perturbations of intrahepatic signaling by the IL-6 family cytokines are a feature of NASH pathogenesis and may contribute to disease progression.
The mammalian liver contains heterogeneous cell types including hepatocytes and non-parenchymal cells. We recently performed single-cell RNAseq on liver cells isolated from chow or NASH diet-fed mice to delineate liver cell heterogeneity, cell-cell communication, and NASH-associated reprogramming at single-cell resolution (23). To elucidate the cellular landscape of IL-6 family cytokine signaling, we analyzed the distribution of ligand and receptor expression among individual liver cell types. While the transcript levels for most of the IL-6 family members were relatively low based on normalized single-cell RNAseq reads, abundant mRNA expression for Cardiotrophin like cytokine factor 1 (Clcf1) was observed in cholangiocytes, and to a lesser extent, in other liver cell types (Figure 1D). Expression of Cardiotrophin 1 (Ctf1), a cytokine closely related to Clcf1, was also observed in cholangiocytes. We next performed RNAscope in situ hybridization for Clcf1 coupled with immunofluorescence staining for Secreted Phosphoprotein 1 (SPP1), a cholangiocyte marker, on liver sections from mice fed chow or NASH diet. We observed a high degree of colocalization between Clcf1 mRNA and SPP1 protein (Figure 1E), indicating that Clcf1 is primarily expressed by cholangiocytes in the liver. More abundant Clcf1 hybridization signal was detected in NASH liver, likely due to expansion of biliary epithelial cells (ductular reaction) during NASH. CLCF1 transduces downstream signals through the gp130/LIFR receptor complex and STAT3 activation. The highest levels of Il6st and Lifr mRNA expression were observed in endothelial cells while their expression was also readily detected in other cell types, including hepatocytes. As such, CLCF1 is a cholangiocyte-derived cytokine that likely impacts on other cell types in the liver through paracrine signaling.
Diametrical regulation of hepatic CLCF1 and LIFR expression in mouse and human NASH
The highly restricted and abundant expression of Clcf1 in cholangiocytes raises the possibility that it may play a prominent role in intrahepatic IL-6 family cytokine signaling. Quantitative PCR (qPCR) analysis of hepatic gene expression confirmed that, compared to control, Clcf1 mRNA levels were elevated in the liver from NASH and CDA-HFD diet-fed mice by approximately 3-fold (Figure 2A). To our surprise, Lifr mRNA expression and protein levels were significantly lower in the NASH groups (Figure 2A–B). NASH-associated reprograming of hepatocyte transcriptome was recently analyzed using sorted fluorescence-labelled hepatocyte nuclei under normal and NASH diet conditions (41). Analysis of this RNAseq dataset revealed that Lifr mRNA expression in hepatocytes was markedly downregulated in both NASH and CDA-HFD models of diet-induced NASH (Figure 2C), suggesting that LIFR-mediated cytokine signaling in hepatocytes may be impaired during NASH. To clarify the cellular source of LIFR expression, we performed immunofluorescence staining on liver sections from chow and NASH diet-fed mice. We observed strong LIFR expression in a subpopulation of hepatocytes in chow-fed mouse liver, but to a much lesser extent, in NASH liver (Figure 2D). Interestingly, LIFR-positive hepatocytes were frequently found near the portal area, suggesting that physical proximity of ligand and receptor-expressing cells may facilitate CLCF1-LIFR paracrine signaling in the liver. We next examined whether NASH-associated dysregulations of Clcf1 and Lifr expression is linked to the severity of liver injury in a cohort of NASH diet-fed mice. Hepatic gene expression analysis indicated that Clcf1 mRNA levels are positively, while Lifr mRNA expression inversely, correlated with plasma ALT levels, a biomarker of liver injury (Figure 2E). In contrast, Il6st expression only showed modest association with plasma ALT levels.
Figure 2. Diametrical regulation of hepatic Clcf1 and Lifr expression during diet-induced NASH in mice.
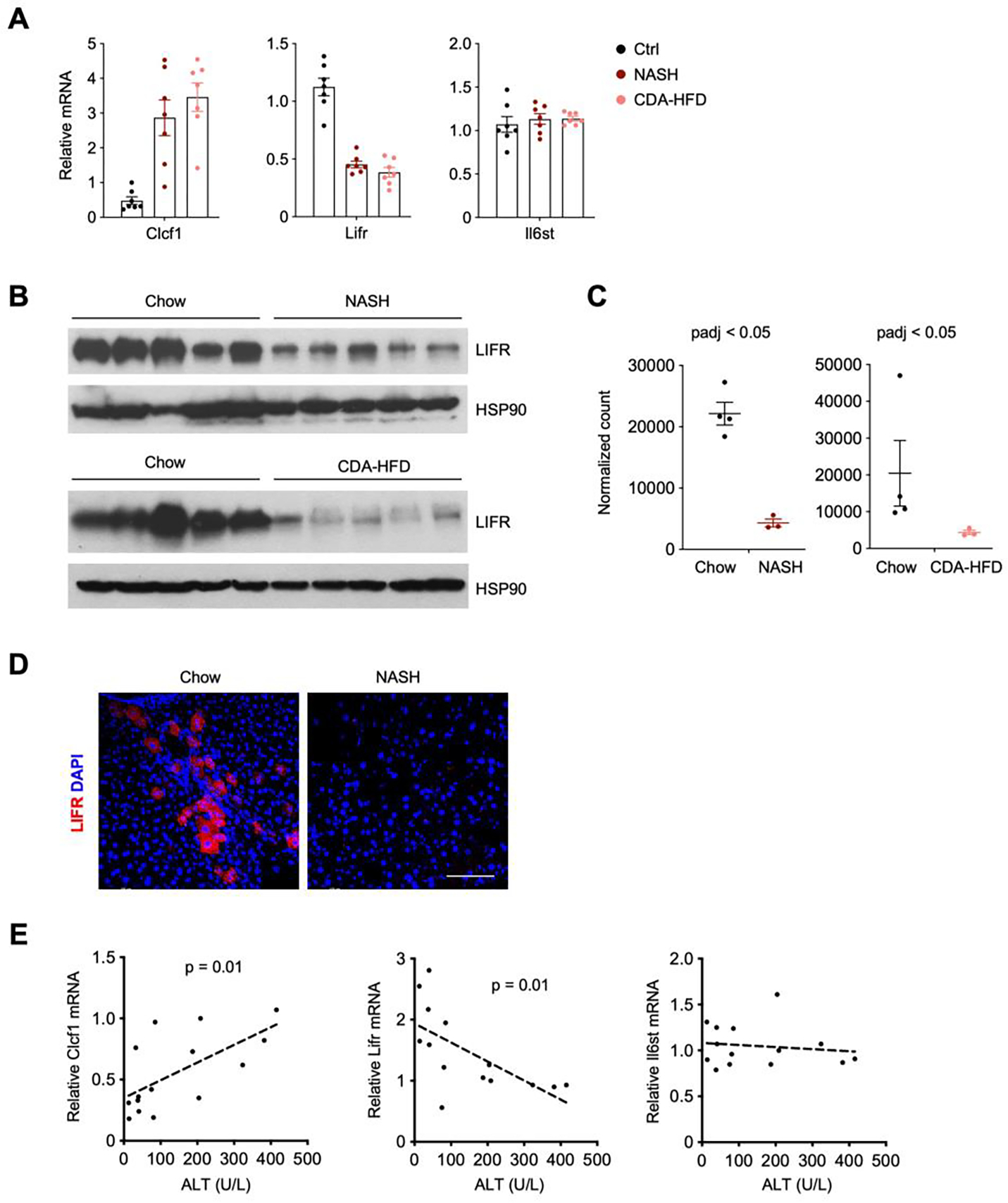
A) qPCR analysis of hepatic Clcf1, Lifr and Il6st expression in mice fed standard chow (n=7), NASH diet for 20 weeks (n=7), or CDA-HFD diet for 8 weeks (n=7).
B) Immunoblots of total liver lysates from mice fed chow, NASH or CDA-HFD diet.
C) Relative Lifr mRNA levels in hepatocyte nuclei isolated from mice fed chow, NASH or CDA-HFD diet (GSE162876).
D) Immunofluorescence staining of LIFR on liver sections from mice fed chow or NASH diet. Scale bar = 100μm.
E) Correlation of hepatic Clcf1, Lifr, and Il6st gene expression with plasma ALT levels in diet-induced NASH mice.
To determine whether Clcf1 and Lifr dysregulation occurs prior to the development of overt NASH pathologies, we examined their mRNA expression in a cohort of HFD-fed mice. HFD feeding typically induces obesity and hepatic steatosis without triggering core features of NASH such as liver injury and fibrosis. As shown in Figure 3A–B, Clcf1 mRNA levels are significantly correlated with obesity and hepatic fat content, while Lifr expression exhibited an opposite pattern of regulation. These results suggest that dysregulation of hepatic Clcf1/Lifr expression likely represents an early pathogenic event during the development of metabolic fatty liver disease. We next examined whether hepatic CLCF1/LIFR signaling may be disrupted in human NASH. We analyzed a liver RNAseq dataset from a cohort of normal weight individuals and patients with NAFLD or NASH (GSE126848) (42). As expected, mRNA levels of TREM2, a marker of NASH-associated macrophage (23), were markedly higher in the livers from NAFLD and NASH patients than that of normal weight control (Figure 3C). Hepatic expression of CLCF1 was significantly increased in NAFLD and NASH groups compared to healthy control. In contrast, LIFR and IL6ST mRNA levels were reduced in the disease groups. These findings illustrate that diametrical regulation of CLCF1 and its receptor subunits LIFR and IL6ST in the liver is a conserved molecular feature of mouse and human NASH.
Figure 3. Regulation of hepatic CLCF1 and LIFR expression in mouse and human NAFLD.
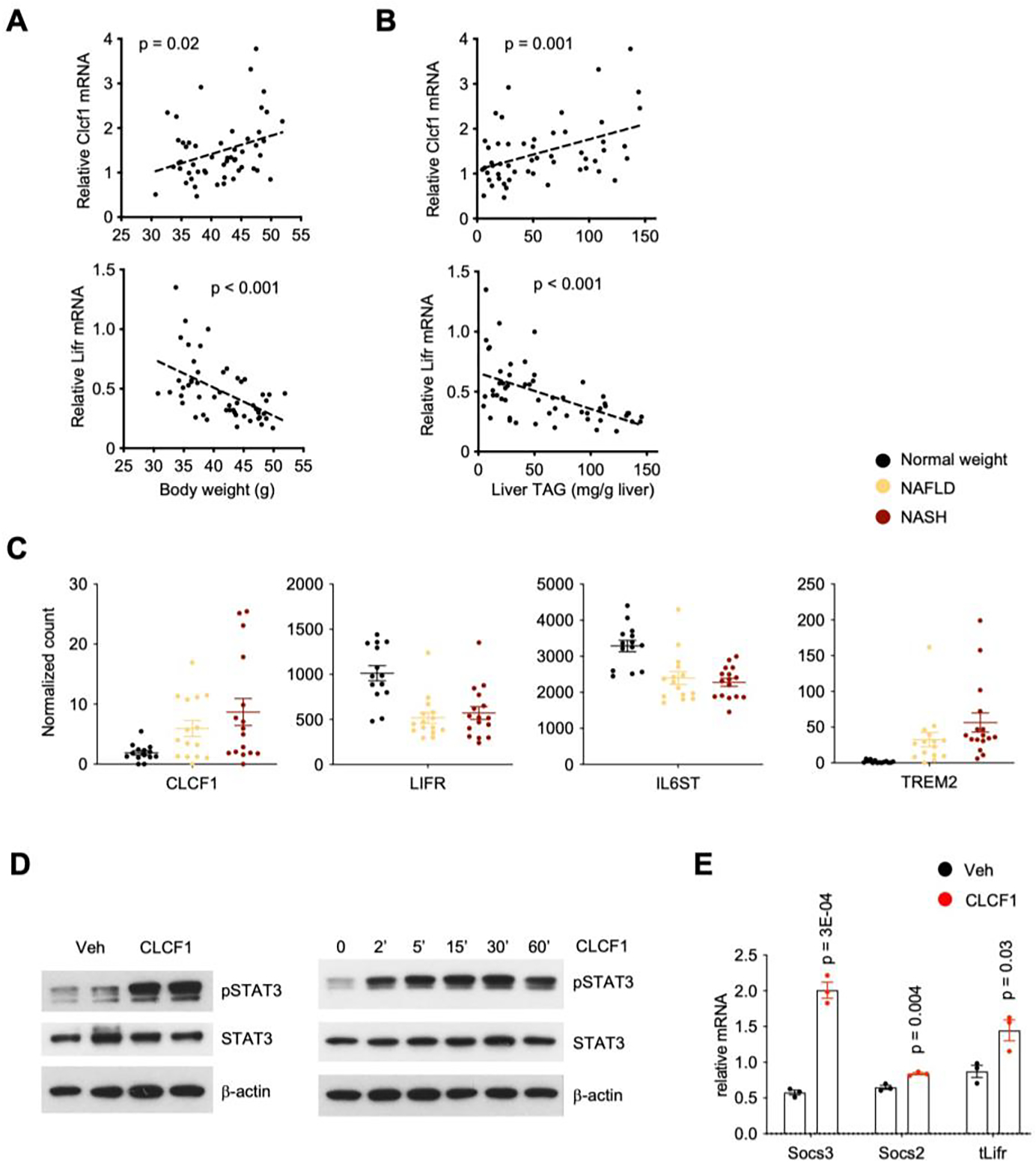
A-B) Correlation of hepatic Clcf1 and Lifr mRNA levels with body weight (A) and liver TAG content (B) in mice fed HFD for eight weeks.
C) Hepatic expression of CLCF1 and LIFR in individuals with normal weight (n=14), NAFLD (n=15), and NASH (n=16). Relative expression values were obtained from human liver RNAseq dataset GSE126848. P-value is derived from Wald test. Adjusted p-value in NASH vs normal weight is less than 0.05 for all panels.
D) Immunoblots of total protein lysates from primary hepatocytes treated with vehicle (Veh) or CLCF1 (100 ng/ml) for 15 mins.
E) qPCR analysis of gene expression in primary hepatocytes treated with vehicle or CLCF1 (100 ng/ml) for six hrs.
AAV-mediated CLCF1 overexpression protects mice from diet-induced NASH
The biological function of CLCF1 signaling in fatty liver disease has not been previously explored. To test whether CLCF1 is capable of triggering downstream signaling in hepatocytes, we treated cultured primary hepatocytes with recombinant CLCF1 and examined STAT3 phosphorylation and target gene expression. CLCF1 robustly stimulated tyrosine phosphorylation of STAT3, which reached peak levels approximately 30min following CLCF1 exposure (Figure 3D). mRNA expression of Suppressor of cytokine signaling 2 (Socs2) and Socs3 was increased in response to CLCF1 treatment (Figure 3E). These results support the presence of an intrahepatic signaling axis from cholangiocytes to the parenchymal hepatocytes in the liver.
It is possible that increased CLCF1 may act as a pathogenic factor that promotes the development of NASH pathologies. Alternatively, CLCF1 induction may be an adaptive response to reduced LIFR expression in NASH liver and therefore play a protective role in diet-induced NASH. To distinguish these possibilities, we examined the effects of hepatic CLCF1 overexpression on NASH progression in mice. We constructed a recombinant adenovirus-associated virus (AAV) expressing full-length mouse CLCF1 under the control of liver-specific thyroxine-binding globulin (TBG) promoter. We transduced wild type C57BL/6 mice with AAV-GFP or AAV-CLCF1 and subjected transduced mice to NASH diet feeding for a total of 20 weeks (Figure 4A). Measurements of plasma parameters revealed that, compared to control, mice transduced with AAV-CLCF1 exhibited significantly lower ALT and AST levels (Figure 4B), suggesting that CLCF1 overexpression ameliorates NASH-associated liver injury. Sirius red staining of liver sections showed that liver fibrosis was markedly attenuated in AAV-CLCF1 group (Figure 4C). Accordingly, we observed that CLCF1 decreased the abundance of Desmin-positive HSC and GPNMB-positive macrophages in the liver (Figure 4D). Hepatic gene expression analysis indicated that CLCF1 overexpression robustly stimulated STAT3 phosphorylation and mRNA expression of Socs2 and Socs3 (Figure 4E–F). Consistent with improved NASH, mRNA expression of genes involved in liver fibrosis (Col1a1, Col1a2, Col3a1, Col4a1, Col6a1, Mmp2, Acta2) and inflammatory response (Trem2, Gpnmb, Ccl2) was significantly reduced by CLCF1 (Figure 4G). Several genes involved in vascular endothelial function and hepatic lipid metabolism were also decreased in response to chronically elevated CLCF1. We did not observe significant differences between control and AAV-CLCF1 group under chow-fed condition (Figure S1), suggesting that CLCF1 signaling may be uniquely important during liver injury and disease progression.
Figure 4. AAV-mediated CLCF1 overexpression in the liver ameliorates NASH pathologies in mice fed NASH diet.
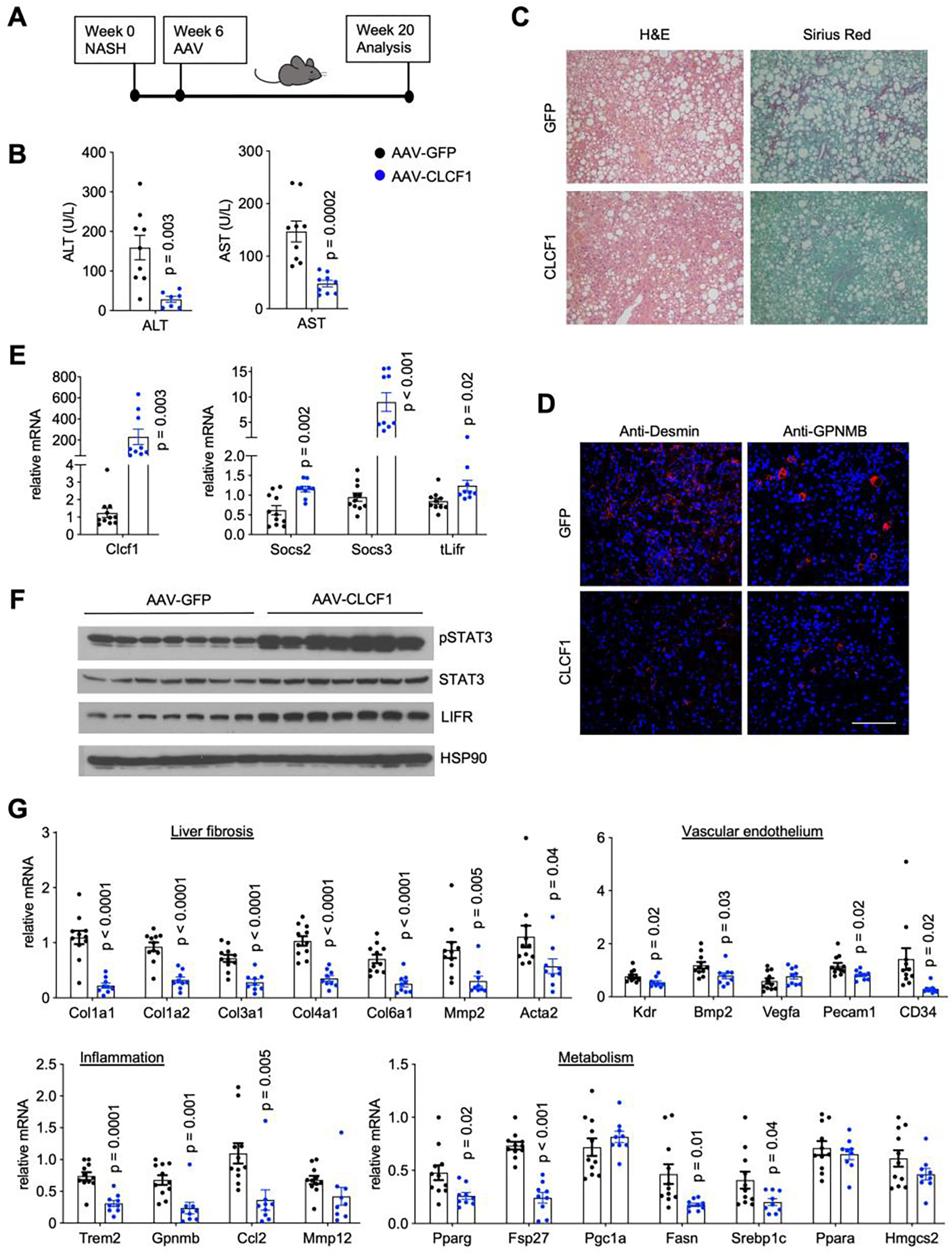
A) A schematic outline of study design.
B) Plasma ALT and AST levels in mice transduced with AAV-GFP (n=11) or AAV-CLCF1 (n=9) after 10 weeks of NASH-diet feeding.
C) Liver histology from transduced mice (H&E, left; Sirius Red staining, right).
D) Immunofluorescence staining of Desmin and GPNMB. Scale bar = 100μm.
E) qPCR analysis of hepatic genes involved in cytokine signaling.
F) Immunoblots of total liver lysates from transduced mice.
G) qPCR analysis of genes involved in liver fibrosis, vascular endothelium, inflammation, and hepatic metabolism in transduced mice.
Data in B), D) and F) represent mean ± SEM; two-tailed unpaired Student’s t-test.
To further establish the protective role of CLCF1, we performed overexpression studies using a second diet-induced NASH model. Mice were transduced with AAV-GFP or AAV-CLCF1 and subjected to CDA-HFD feeding for four weeks (Figure 5A). CDA-HFD feeding resulted in rapid development of major NASH pathologies in mice, including severe hepatic steatosis, inflammation, and liver fibrosis. Compared to AAV-GFP group, mice transduced with AAV-CLCF1 displayed markedly improved liver fibrosis as indicated by reduced collagen deposition (Figure 5B). CLCF1 overexpression appeared to have a modest effect on the severity of hepatic steatosis. While plasma ALT levels were comparable between two groups, AST levels trended to be lower in AAV-CLCF1 group (Figure 5C). The protective effects of CLCF1 on liver injury appeared less robust compared to the NASH group, likely due to the severe hepatotoxicity caused by CDA-HFD feeding. Analysis of hepatic gene expression revealed that CLCF1 overexpression stimulated STAT3 phosphorylation and expression of its downstream target gene Socs3 (Figure 5D–E). Consistent with histological findings, mRNA expression of genes involved in liver fibrosis and inflammation was significantly reduced in response to CLCF1 (Figure 5F). On the contrary, CLCF1 only moderately affected expression of several genes involved in hepatic glucose and lipid metabolism. Taken together, these overexpression studies illustrate a protective role of CLCF1 in diet-induced NASH progression.
Figure 5. AAV-mediated CLCF1 overexpression in the liver ameliorates NASH pathologies in mice fed CDA-HFD diet.
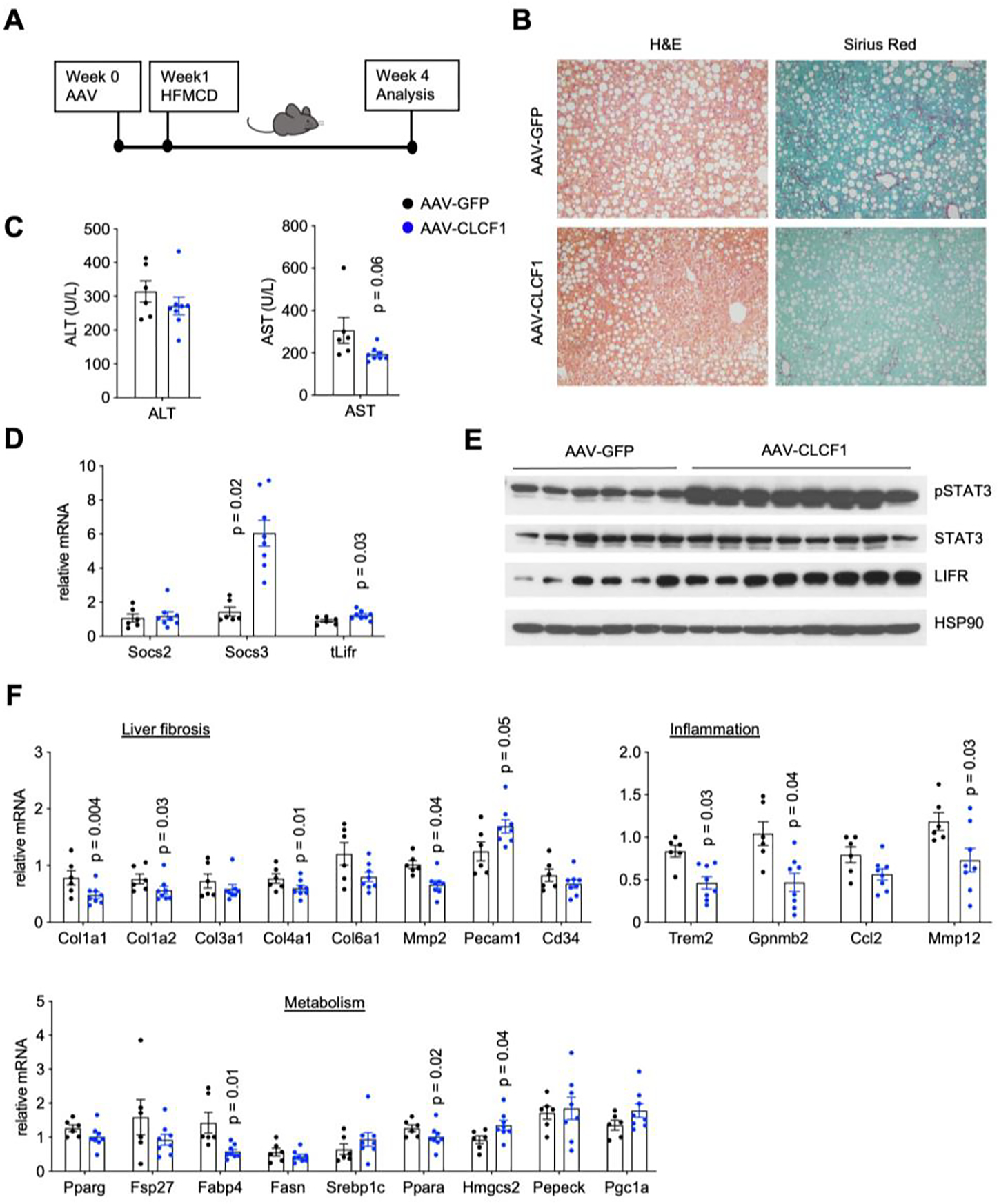
A) A schematic outline of study design.
B) Liver histology from transduced mice (H&E, left; Sirius Red staining, right).
C) Plasma ALT and AST levels in mice transduced with AAV-GFP (n=6) or AAV-CLCF1 (n=7) after six weeks of CDA-HFD diet feeding.
D) qPCR analysis of hepatic genes involved in IL-6 family cytokine signaling.
E) Immunoblots of liver lysates from transduced mice fed with CDA-HFD diet for six weeks.
F) qPCR analysis of genes involved in liver fibrosis, vascular endothelium, inflammation, and hepatic metabolism in transduced mice.
Data in C), D) and F) represent mean ± SEM; two-tailed unpaired Student’s t-test.
Hepatocyte-specific inactivation of LIFR exacerbates NASH pathogenesis
CLCF1 stimulates downstream signaling through the gp130/LIFR/CNTFR receptor complex (45, 46). As shown above, hepatic LIFR gene expression was downregulated in dietary models of NASH in mice and in human NASH. These observations led us to hypothesize that reduced hepatocyte LIFR expression may impair CLCF1 signaling in the liver and exacerbate NASH development. To test this, we used a CRISPR/Cas9-based approach to inactivate LIFR expression in hepatocytes in adult mice. We generated a mouse strain that expresses Cas9 specifically in hepatocytes by crossing Rosa26 floxed-STOP-floxed Cas9 (Rosa26-FSF-Cas9) knockin mice with Albumin-Cre (CRE) transgenic mice. To inactivate Lifr expression, we constructed a recombinant AAV vector expressing two single guide RNAs (AAV-sgRNAs) flanking exon 2 of Lifr gene, which contains the translation start codon (Figure 6A). We transduced Rosa26-FSF-Cas9/CRE+ (HepKO) and Rosa26-FSF-Cas9/CRE- (control) littermates with AAV-sgRNA to generate hepatocyte-specific Lifr knockout (HepKO) and control mice, respectively. As shown in Supplementary Figure S2A–C, AAV-sgRNA transduction resulted in Cas9-mediated deletion of exon 2 of Lifr gene and reduced LIFR protein levels in the liver. Given that endothelial cells and HSC also contribute to liver Lifr, we surmise that the efficiency of Cas9-mediated hepatocyte Lifr inactivation likely exceeds this estimate.
Figure 6. Hepatocyte-specific deletion of LIFR accelerates NASH progression.
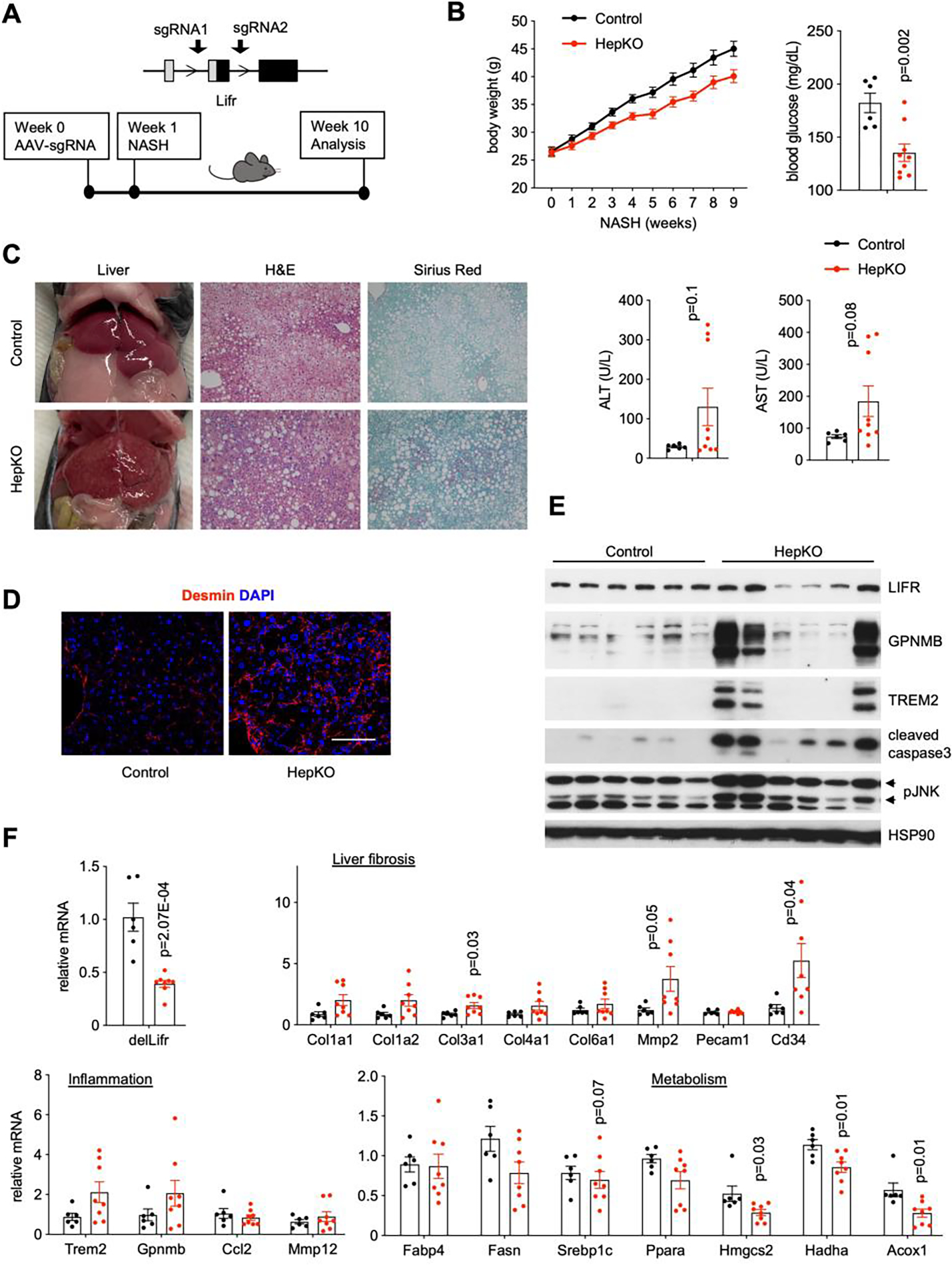
A) A diagram of Cas9-mediated Lifr deletion and study design. Albumin Cre negative/Rosa26-FSF-Cas9 (control) and Cre+/Rosa26-FSF-Cas9 (HepKO) littermates were transduced with AAV-sgRNAs targeting Lifr via tail vein injection. Transduced mice were subjected to NASH diet feeding for nine weeks.
B) Metabolic and NASH parameters of control (n=6) and HepKO (n=8) mice.
C) Liver appearance and histology in control and HepKO mice following ten weeks of NASH diet feeding.
D) Desmin immunofluorescence staining. Scale bar = 100μm.
E) Immunoblots of liver lysates from control and HepKO mice fed NASH diet.
F) qPCR analysis of hepatic gene expression.
Data in B) and F) represent mean ± SEM; two-tailed unpaired Student’s t-test.
Similar to CLCF1 overexpression, Lifr ablation elicited modest effects on liver injury and hepatic gene expression in chow-fed mice (Figure S2D–E). To determine the role of hepatocyte LIFR in diet-induced NASH, we subjected transduced Lifr HepKO and control mice to NASH diet feeding for ten weeks. Compared to control, mice transduced with AAV-sgRNAs gained significantly less weight and had lower blood glucose levels upon NASH diet feeding (Figure 6B). Plasma ALT and AST levels were elevated in the Lifr HepKO group. Lifr HepKO mouse livers appeared rough on surface, suggestive of more pronounced tissue fibrosis (Figure 6C). In support of this, Sirius red staining of liver sections revealed markedly increased collagen deposition in the HepKO group. Further, the abundance of Desmin-positive HSCs was markedly increased in the HepKO mouse livers (Figure 6D). Immunoblotting analyses indicated that hepatocyte-specific inactivation of LIFR results in elevated levels of phosphorylated JNK and cleaved caspase 3 (Figure 6E). Expression of genes involved in liver fibrosis and NASH-associated macrophages was also elevated in the HepKO group, albeit only reaching borderline statistical significance (Figure 6F). These results indicate that hepatocyte LIFR signaling is critical for preserving liver health under metabolic stress conditions.
Discussion
Intercellular crosstalk is a fundamental feature of mammalian tissue biology. In the liver, hepatocytes and non-parenchymal cells release secreted factors that act on their cognate receptors to elicit diverse biological effects. As such, intrahepatic paracrine signaling plays an important role in maintaining tissue homeostasis and health. A major unanswered question pertinent to NASH pathogenesis is how the landscape of cell-cell communication in the liver is altered in the disease state and whether its dysregulation of contributes to disease progression. By analyzing bulk and single-cell RNAseq datasets, we identified CLCF1 as a cholangiocyte-enriched secreted factor that is elevated in the liver during NASH pathogenesis in mice and humans. Among ten members of the IL-6 family cytokines, CLCF1 shows the most abundant expression based on normalized single-cell RNA sequencing reads and its expression is largely restricted to cholangiocytes. As such, it is likely that CLCF1 diffuses from the portal region to the rest of the liver parenchyma. Whether there exists a CLCF1 concentration gradient along the portal to central axis remains currently unknown. It is striking that only a subpopulation of hepatocytes exhibits high LIFR expression in the liver, raising the possibility that not all hepatocytes are equally responsive to CLCF1. Gain-of-function studies indicate that CLCF1 ameliorates the development of NASH pathologies in two models of diet-induced NASH in mice. AAV-mediated CLCF1 overexpression in the liver improved liver injury markers, attenuated expression of genes involved in inflammation, and reduced liver fibrosis. These findings suggest that induction of CLCF1 in NASH may represent an adaptive and protective response in the context of liver injury induced by NASH diet feeding.
Like other members of the IL-6 family cytokines, CLCF1 transduces downstream signal through the gp130/LIFR receptor complex. To our surprise, while gp130 expression remained comparable between control and NASH mouse livers, LIFR expression in hepatocytes is significantly downregulated upon diet-induced NASH. Importantly, reduced hepatic LIFR expression was also observed in NAFLD and NASH patients, suggesting that LIFR downregulation is a conserved feature of NASH in mice and humans. It is possible that elevated CLCF1 may serve as a compensatory mechanism to maintain signaling through gp130/LIFR receptor complex. Analysis of single-cell RNAseq data indicates that Lifr is expressed by multiple cell types in the liver, including most prominently in liver sinusoidal endothelial cells, HSCs, and hepatocytes. Previous studies have demonstrated that CLCF1 may regulate macrophage function and foam cell formation (47). At present, we cannot rule out CLCF1 signaling in non-parenchymal cells, including liver macrophages, may contribute to its protective effects. Nevertheless, our results using mice lacking LIFR signaling in hepatocytes support a critical role of LIFR signaling in hepatocytes during NASH pathogenesis. In fact, hepatocyte-specific inactivation of LIFR resulted in worsened liver injury, inflammation, and liver fibrosis. Interestingly, a recent study demonstrated that hepatocyte-specific deletion of LIFR promotes liver tumorigenesis (48), suggesting that hepatocyte LIFR signaling may protect against diet-induced NASH and NASH-associated liver cancer. Together, these results support the presence of a cholangiocyte to hepatocyte paracrine signaling pathway mediated by CLCF1 and its receptor gp130/LIFR in the liver.
Supplementary Material
Acknowledgements
We thank Michael Scales and Benjamin Allen for help with the RNAscope study and Xiaoling Peng for her technical support. This work was supported by NIH (DK102456 and DK118731 to J.D.L.). T.L. was supported by Patten Predoctoral Fellowship provided by the University of Michigan. H.K. was supported by an NRSA fellowship (FDK117615) and MSTP training grant (T32GM007863). L.Z. was supported by the American Heart Association Career Development Award.
Footnotes
References
- 1.Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021;184:2537–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rui L, Lin JD. Reprogramming of Hepatic Metabolism and Microenvironment in Nonalcoholic Steatohepatitis. Annu Rev Nutr 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, Ikramuddin S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020;323:1175–1183. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki A, Diehl AM. Nonalcoholic Steatohepatitis. Annu Rev Med 2017;68:85–98. [DOI] [PubMed] [Google Scholar]
- 5.Bence KK, Birnbaum MJ. Metabolic drivers of non-alcoholic fatty liver disease. Mol Metab 2021;50:101143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Santos-Baez LS, Ginsberg HN. Nonalcohol fatty liver disease: balancing supply and utilization of triglycerides. Curr Opin Lipidol 2021;32:200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arendt BM, Comelli EM, Ma DW, Lou W, Teterina A, Kim T, Fung SK, et al. Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n-3 and n-6 polyunsaturated fatty acids. Hepatology 2015;61:1565–1578. [DOI] [PubMed] [Google Scholar]
- 8.Govaere O, Cockell S, Tiniakos D, Queen R, Younes R, Vacca M, Alexander L, et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci Transl Med 2020;12. [DOI] [PubMed] [Google Scholar]
- 9.Lefebvre P, Lalloyer F, Bauge E, Pawlak M, Gheeraert C, Dehondt H, Vanhoutte J, et al. Interspecies NASH disease activity whole-genome profiling identifies a fibrogenic role of PPARalpha-regulated dermatopontin. JCI Insight 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clapper JR, Hendricks MD, Gu G, Wittmer C, Dolman CS, Herich J, Athanacio J, et al. Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am J Physiol Gastrointest Liver Physiol 2013;305:G483–495. [DOI] [PubMed] [Google Scholar]
- 11.Xiong X, Wang Q, Wang S, Zhang J, Liu T, Guo L, Yu Y, et al. Mapping the molecular signatures of diet-induced NASH and its regulation by the hepatokine Tsukushi. Mol Metab 2019;20:128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Moshe S, Itzkovitz S. Spatial heterogeneity in the mammalian liver. Nat Rev Gastroenterol Hepatol 2019;16:395–410. [DOI] [PubMed] [Google Scholar]
- 13.Ramachandran P, Matchett KP, Dobie R, Wilson-Kanamori JR, Henderson NC. Single-cell technologies in hepatology: new insights into liver biology and disease pathogenesis. Nat Rev Gastroenterol Hepatol 2020;17:457–472. [DOI] [PubMed] [Google Scholar]
- 14.Xiong X, Kuang H, Liu T, Lin JD. A single-cell perspective of the mammalian liver in health and disease. Hepatology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aizarani N, Saviano A, Sagar Mailly L, Durand S, Herman JS, Pessaux P, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019;572:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halpern KB, Shenhav R, Matcovitch-Natan O, Toth B, Lemze D, Golan M, Massasa EE, et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 2017;542:352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halpern KB, Shenhav R, Massalha H, Toth B, Egozi A, Massasa EE, Medgalia C, et al. Paired-cell sequencing enables spatial gene expression mapping of liver endothelial cells. Nat Biotechnol 2018;36:962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dobie R, Wilson-Kanamori JR, Henderson BEP, Smith JR, Matchett KP, Portman JR, Wallenborg K, et al. Single-Cell Transcriptomics Uncovers Zonation of Function in the Mesenchyme during Liver Fibrosis. Cell Rep 2019;29:1832–1847 e1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenthal SB, Liu X, Ganguly S, Dhar D, Pasillas MP, Ricciardelli E, Li RZ, et al. Heterogeneity of HSCs in a Mouse Model of NASH. Hepatology 2021;74:667–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krenkel O, Hundertmark J, Abdallah AT, Kohlhepp M, Puengel T, Roth T, Branco DPP, et al. Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity-related steatohepatitis. Gut 2020;69:551–563. [DOI] [PubMed] [Google Scholar]
- 21.Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019;575:512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, Bruni CM, et al. Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity 2020;52:1057–1074 e1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, Zhao XY, et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol Cell 2019;75:644–660 e645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giraldez MD, Carneros D, Garbers C, Rose-John S, Bustos M. New insights into IL-6 family cytokines in metabolism, hepatology and gastroenterology. Nat Rev Gastroenterol Hepatol 2021;18:787–803. [DOI] [PubMed] [Google Scholar]
- 25.Murakami M, Kamimura D, Hirano T. Pleiotropy and Specificity: Insights from the Interleukin 6 Family of Cytokines. Immunity 2019;50:812–831. [DOI] [PubMed] [Google Scholar]
- 26.Rose-John S Interleukin-6 Family Cytokines. Cold Spring Harb Perspect Biol 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng YR, Ma HD, Tsuneyama K, Yang W, Wang YH, Lu FT, Liu CH, et al. STAT3-mediated attenuation of CCl4-induced mouse liver fibrosis by the protein kinase inhibitor sorafenib. J Autoimmun 2013;46:25–34. [DOI] [PubMed] [Google Scholar]
- 28.Streetz KL, Tacke F, Leifeld L, Wustefeld T, Graw A, Klein C, Kamino K, et al. Interleukin 6/gp130-dependent pathways are protective during chronic liver diseases. Hepatology 2003;38:218–229. [DOI] [PubMed] [Google Scholar]
- 29.Schmidt-Arras D, Rose-John S. IL-6 pathway in the liver: From physiopathology to therapy. J Hepatol 2016;64:1403–1415. [DOI] [PubMed] [Google Scholar]
- 30.Yamaguchi K, Itoh Y, Yokomizo C, Nishimura T, Niimi T, Umemura A, Fujii H, et al. Blockade of IL-6 signaling exacerbates liver injury and suppresses antiapoptotic gene expression in methionine choline-deficient diet-fed db/db mice. Lab Invest 2011;91:609–618. [DOI] [PubMed] [Google Scholar]
- 31.Hou X, Yin S, Ren R, Liu S, Yong L, Liu Y, Li Y, et al. Myeloid-Cell-Specific IL-6 Signaling Promotes MicroRNA-223-Enriched Exosome Production to Attenuate NAFLD-Associated Fibrosis. Hepatology 2021;74:116–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sleeman MW, Garcia K, Liu R, Murray JD, Malinova L, Moncrieffe M, Yancopoulos GD, et al. Ciliary neurotrophic factor improves diabetic parameters and hepatic steatosis and increases basal metabolic rate in db/db mice. Proc Natl Acad Sci U S A 2003;100:14297–14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stefanovic L, Stefanovic B. Role of cytokine receptor-like factor 1 in hepatic stellate cells and fibrosis. World J Hepatol 2012;4:356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Findeisen M, Allen TL, Henstridge DC, Kammoun H, Brandon AE, Baggio LL, Watt KI, et al. Treatment of type 2 diabetes with the designer cytokine IC7Fc. Nature 2019;574:63–68. [DOI] [PubMed] [Google Scholar]
- 35.Matsuda M, Tsurusaki S, Miyata N, Saijou E, Okochi H, Miyajima A, Tanaka M. Oncostatin M causes liver fibrosis by regulating cooperation between hepatic stellate cells and macrophages in mice. Hepatology 2018;67:296–312. [DOI] [PubMed] [Google Scholar]
- 36.Xiong J, Liu T, Mi L, Kuang H, Xiong X, Chen Z, Li S, et al. hnRNPU/TrkB Defines a Chromatin Accessibility Checkpoint for Liver Injury and Nonalcoholic Steatohepatitis Pathogenesis. Hepatology 2020;71:1228–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 2014;159:440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Q, Sharma VP, Shen H, Xiao Y, Zhu Q, Xiong X, Guo L, et al. The hepatokine Tsukushi gates energy expenditure via brown fat sympathetic innervation. Nat Metab 2019;1:251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo L, Zhang P, Chen Z, Xia H, Li S, Zhang Y, Kobberup S, et al. Hepatic neuregulin 4 signaling defines an endocrine checkpoint for steatosis-to-NASH progression. J Clin Invest 2017;127:4449–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Widjaja AA, Singh BK, Adami E, Viswanathan S, Dong J, D’Agostino GA, Ng B, et al. Inhibiting Interleukin 11 Signaling Reduces Hepatocyte Death and Liver Fibrosis, Inflammation, and Steatosis in Mouse Models of Nonalcoholic Steatohepatitis. Gastroenterology 2019;157:777–792 e714. [DOI] [PubMed] [Google Scholar]
- 41.Loft A, Alfaro AJ, Schmidt SF, Pedersen FB, Terkelsen MK, Puglia M, Chow KK, et al. Liver-fibrosis-activated transcriptional networks govern hepatocyte reprogramming and intra-hepatic communication. Cell Metab 2021;33:1685–1700 e1689. [DOI] [PubMed] [Google Scholar]
- 42.Suppli MP, Rigbolt KTG, Veidal SS, Heeboll S, Eriksen PL, Demant M, Bagger JI, et al. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals. Am J Physiol Gastrointest Liver Physiol 2019;316:G462–G472. [DOI] [PubMed] [Google Scholar]
- 43.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang GX, Zhao XY, Meng ZX, Kern M, Dietrich A, Chen Z, Cozacov Z, et al. The brown fat-enriched secreted factor Nrg4 preserves metabolic homeostasis through attenuation of hepatic lipogenesis. Nat Med 2014;20:1436–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crisponi L, Buers I, Rutsch F. CRLF1 and CLCF1 in Development, Health and Disease. Int J Mol Sci 2022;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones SA, Jenkins BJ. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat Rev Immunol 2018;18:773–789. [DOI] [PubMed] [Google Scholar]
- 47.Pasquin S, Laplante V, Kouadri S, Milasan A, Mayer G, Tormo AJ, Savin V, et al. Cardiotrophin-like Cytokine Increases Macrophage-Foam Cell Transition. J Immunol 2018;201:2462–2471. [DOI] [PubMed] [Google Scholar]
- 48.Yao F, Deng Y, Zhao Y, Mei Y, Zhang Y, Liu X, Martinez C, et al. A targetable LIFR-NF-kappaB-LCN2 axis controls liver tumorigenesis and vulnerability to ferroptosis. Nat Commun 2021;12:7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.